

Abstract
The standard practice in neuropathology is to diagnose Alzheimer’s Disease (AD) based on the distribution and abundance of neurofibrillary tangles and Aβ deposits. However, other significant abnormalities including neuroinflammation, gliosis, white matter degeneration, non-Aβ micro-vascular disease, and insulin-related metabolic dysfunction require further study to understand how they could be targeted to more effectively remediate AD. This review addresses non-Aβ and non-pTau AD-associated pathologies, highlighting their major features, roles in neurodegeneration, and etiopathic links to deficits in brain insulin and insulin-like growth factor signaling and cognitive impairment. The discussion delineates why AD with its most characteristic clinical and pathological phenotypic profiles should be regarded as a brain form of diabetes, i.e. Type 3 Diabetes, and entertains the hypothesis that Type 3 Diabetes is just one of the categories of insulin resistance diseases that can occur independently or overlap with one or more of the others, including Type 2 Diabetes, metabolic syndrome, and non-alcoholic fatty liver disease.
Key Phrases: Alzheimer’s disease, white matter degeneration, microvascular disease, insulin resistance, dementia, amyloid, neuroinflammation
Alzheimer’s Disease Fundamentals
Alzheimer’s Disease—Definition and Diagnostics Overview
Alzheimer’s disease (AD) is characteristically associated with progressive alterations in behavior, impairment of recent or short-term memory, and declines in executive functions and cognitive functions (McKhann et al., 2011). Structured longitudinal neuropsychological tests of memory, intellectual function and language are used to render a diagnosis of possible or probable AD. However, to increase diagnostic accuracy, clinical and neuropsychological testing are supplemented with laboratory and neuroimaging assessments (McKhann et al., 2011), including assays of amyloid precursor protein-amyloid beta 1–42 peptide (Aβ1–42) and phospho-Tau (pTau231) in cerebrospinal fluid (CSF) and serum (Blennow et al., 2015a, Olsson et al., 2016), magnetic resonance imaging (MRI) (Duncan et al., 2013, Pantano et al., 1999), functional MRI (fMRI), diffusion tensor imaging (DTI) (Amlien and Fjell, 2014), single-photon emission computed tomography (SPECT), positron emission tomography (PET), and magnetic resonance spectroscopy (MRS) (Jones and Waldman, 2004, Ewers et al., 2011).
Aging versus Alzheimer’s Disease
Aging leads to atrophy and reduced function of most organs throughout the body, including the brain (Double et al., 1996). Lifestyle choices have measurable positive and negative effects on aging such that, healthful eating habits and regular physical and mental exercise help preserve cognitive function (Daly et al., 2015, Rolandi et al., 2016, Madsen et al., 2015), whereas poor lifestyle choices accelerate physical and functional aging (Beydoun et al., 2014, Moreira, 2013). Furthermore, aging is the most consistent and dominant risk factor for neurodegeneration. One of the main differences between aging and neurodegeneration is that with aging, the brain exhibits modest degrees of atrophy and functional loss over a period of years, whereas in AD, the declines are swifter and relentless, driving formerly fully functioning people to eventual end-stage vegetative states. These concepts suggest that lifestyle measures that either curtail or exacerbate the aging process may also modulate risk and rates of developing AD as well as other neurodegenerative diseases.
Characteristic Neuropathology
Typically, neurodegeneration begins before it becomes clinically manifested, i.e. there is a pre-symptomatic period during which deficits are subtle or silent. Although the asymptomatic phase enables victims to continue functioning, the negative aspect is that the disease remains hidden during period when it may be reversible. The potential for early intervention is further challenged by social and family pressures that cause afflicted individuals to remain guarded and secretive about their cognitive deficits. Postmortem studies have shown that brain structures hit earliest by AD, including medial temporal and orbitofrontal regions, are functionally linked to process memory acquisition. With advancement of AD, neurodegeneration “spreads” to involve other brain regions, causing progressive destruction of corticolimbic circuitry.
At the tissue or histopathological level, progressive loss of neurons and synaptic terminals, mediated by apoptosis, necrosis, oxidative stress, and neuroinflammation, represent fundamental cellular pathologies that correspond with atrophy of corticolimbic structures. What distinguishes neuronal loss in AD from non-degenerative disease processes is the co-accumulation of three major proteins including abnormally phosphorylated-Tau, Aβ1–42, and ubiquitin (Nelson et al., 2012, Hyman et al., 2012, Montine et al., 2012). Tau is a major neuronal cytoskeletal protein that provides neurons with structure and enables them to form stable interconnections. Aberrant hyper-phosphorylation of tau through inappropriate activation of kinases such as glycogen synthase kinase-3β (GSK-3β), causes Tau fibrillization, aggregation, and ubiquitination, followed by stress activation of the unfolded protein response, loss of neuronal function, and ultimately cell death. Insoluble, fibrillary aggregates of hyper-phosphorylated and ubiquitinated Tau are major components of neurofibrillary tangles, dystrophic neurites, and neuropil threads which characteristically are present in AD, and progressively accumulate with increasing severity of neurodegeneration (Serrano-Pozo et al., 2011). Their presence in the brain is detectable by immunohistochemical staining with antibodies to phospho-Tau or ubiquitin, or by silver-based histochemical staining.
Aβ1–42 is a ~4 kD peptide generated by secretase cleavage of amyloid beta precursor protein (AβPP). Under normal circumstances, AβPP cleavage products, including Aβ1–42 are continuously cleared from the brain by transport into the general circulation (Ueno et al., 2014). In both aging and AD, Aβ1–42 accumulates in cortical and leptomeningeal vessel walls, cortical and sub-cortical perivascular spaces, plaques, and soluble oligomeric Aβ derived diffusible ligands (ADDLs) which are neurotoxic fibrils (Kalaria and Ballard, 1999, Viola and Klein, 2015). Like hyper-phosphorylated tau, insoluble Aβ1–42 deposits in vessels and plaques are ubiquitinated. Aβ1–42 accumulations in brain are readily detected by immunohistochemical staining for Aβ1–42 or ubiquitin, light microscopic imaging of Congo Red stained sections under polarized light, or fluorescence microscopy of thioflavin-S stained histological sections of brain.
Clinical Neuroimaging and Laboratory Studies Detect Aβ1–42 and pTau Accumulations in the Brain
Extensive postmortem investigations demonstrating progressive accumulations of pTau and Aβ1–42 associated lesions with advancing stages of AD-associated cognitive decline inspired the development of non-invasive tests to detect and monitor related abnormalities in living patients. Neuroimaging by positron emission tomography (PET) was developed to detect Aβ1–42 and pTau using F18 isotopically-labeled tracers (Fleisher et al., 2012, Cselenyi et al., 2012). PET imaging for Aβ emerged in about 2002. In 2012, after several iterations, its use as a diagnostic aid for AD won FDA approval (Yang et al., 2012). PET imaging with F18 isotopically labeled tracers for pTau (707 and 708) is used to detect pTau accumulations in brains of people suspected of having cognitive impairment or dementia due to AD (Zhang et al., 2012, Declercq et al., 2016). Non-invasive PET imaging of Aβ and pTau accumulations in the brain provide objective data about the nature and distribution of neurodegeneration, and opportunity to increase accuracy of the clinical diagnosis. Initially, these approached drew tremendous excitement because abundant signals reflecting Aβ and pTau accumulations were detected in selected AD cases, whereas those neuroimaging pathologies were virtually absent in healthy controls. Furthermore, the PET imaging for Aβ and pTau showed that the abnormal signals extended well beyond cortical-limbic structures (Shin et al., 2011, Braskie et al., 2010, Barrio et al., 2008, Small et al., 2006, Rowe et al., 2013a, Rowe et al., 2013b), a phenomenon that could potentially explain occurrences of more global deficits as AD progresses.
Cerebrospinal fluid (CSF) assays for pTau and Aβ were also developed based on the characteristic presence, distribution, and abundance of related lesions in AD brains (Blennow et al., 2015a, Olsson et al., 2016). CSF levels of pTau increase with AD progression, whereas Aβ levels increase in the early stages of AD but subsequently decline as brain clearance declines and brain levels increase. Reduced brain clearance of Aβ is also marked by lower serum levels (Blennow et al., 2015b). Together with monitoring the clinical course and assessing longitudinal changes in neuropsychological performance (McKhann et al., 2011), a reasonably accurate diagnosis can be made in most cases that are at intermediate to somewhat late stages of AD.
Limitations and Concerns Regarding Aβ and pTau as Dominant Diagnostic and Therapeutic Targets
The predominant distributions of atrophy in corticolimbic structures and in the parietal and temporal lobes are not specific to AD since other forms of dementia including Dementia with Lewy bodies and frontotemporal lobar degeneration often exhibit overlapping patterns of brain atrophy. Furthermore, AD variants and overlapping or complex forms of neurodegeneration can have non-standard and asymmetric distributions of atrophy. These factors limit the accuracy of AD diagnoses that are solely based on anatomical neuroimaging or macroscopic examination of the brain, justifying the need for histopathological studies, including the use of molecular marker-based analyses to distinguish among the various subtypes of neurodegeneration.
PET imaging to detect pTau and Aβ accumulations have become widely used as diagnostic aids for AD. However, postmortem studies have established that neither “biomarker” is specific for AD, as they both accumulate in several diseases (Hulette et al., 2009, Naasan et al., 2016, Jellinger, 2003, Washington et al., 2016, Chandra, 2015, Barrio et al., 2015). Correspondingly, with broadened use of F18-PET imaging of Aβ and pTau, significant positive signals have been detected in many conditions including other dementias (Engler et al., 2008, Berti et al., 2011), traumatic brain injury (Hong et al., 2014), and normal aging (Chetelat et al., 2013), indicating that Aβ nor pTau accumulations are not specific for AD. Therefore, non-invasive assays restricted to these bio-indices may not confer the diagnostic accuracy needed subject assignment in clinical therapeutic trials, and instead, the best diagnostic strategy is to utilize neuroimaging data, including PET studies in conjunction with the clinical profile and formal neuropsychological testing. In many respects, this scenario tells us that major abnormalities unrelated to pTau and Aβ also have important roles in the pathogenesis and progression of AD.
Major Brain Abnormalities Unrelated to Aβ and pTau in AD
Overview of the Problem
Although diagnostic criteria for rendering a neuropathologic diagnosis of AD have been streamlined to semi-quantitative assessments of neurofibrillary tangles and senile plaques in specific brain regions (Hyman et al., 2012), realistically, the nature and distribution of neurodegeneration are far broader. Major abnormalities not routinely considered despite their overwhelming presence in AD include: neuronal loss; neuroinflammation; gliosis; oxidative and nitrosative stress; white matter degeneration; vascular degeneration, particularly in white matter (Brun et al., 1995, Vinters, 2015), and blood-brain barrier disruption (Bridges et al., 2014, Grammas et al., 2011, Johanson et al., 2018). In addition, impairments in brain metabolism (glucose and oxygen utilization) (Hoyer, 1982, de Leon et al., 1983, Faulstich, 1991, Daulatzai, 2017), although recognized for decades and frequently assessed, have not been incorporated into the cluster AD biomarkers. Failure to consider these important aspects of AD, limits opportunity to fully understand the nature of disease and therefore optimally strategize in the development of forward-looking therapeutic interventions. For example, ignoring the degenerative changes that occur white matter and microvessels is problematic because these abnormalities occur early and their progression can have of greater overall negative impact on brain function than the burdens of neurofibrillary tangles and plaques. Limiting therapeutic targets to pTau and Aβ accumulations has already been demonstrated to be too restrictive and largely ineffective for providing significant and sustained disease remediation in AD.
Neuronal Loss and Degeneration
Neurodegeneration in AD is associated with loss of neurons, nerve terminals and fibers beginning in the hippocampus, parahippocampal gyrus (entorhinal cortex), and medial temporal structures (Mizutani et al., 1990). However, what characteristically distinguishes AD from other disease processes is that neuronal loss is accompanied by progressive accumulations of neurofibrillary tangles, dystrophic neurites, neuritic plaques, and neuropil threads. These lesions mark loss of inter-neuronal connections, compromised neuronal plasticity, and cognitive impairment.
Neurofibrillary tangles are composed of aggregated twisted insoluble fibrillar proteins (paired-helical filaments; PHF) whose main constituent is the hyper-phosphorylated tau. Tau is a microtubule-associated protein. Microtubules provide structure and intracellular nutrient transport functions. Neuropil threads (Braak et al., 1986, Perry et al., 1991) and dystrophic (irregular, swollen) axonal neurites distributed in the neuropil and around plaques also contain PHF (Su et al., 1993). Over time (years), neurons continue to degenerate, disconnect, and die while PHF-associated neuronal, axonal, and dendritic pathologies increase, progressively extending from medial temporal to cortical-limbic, followed by parietal and frontal regions and beyond (Serrano-Pozo et al., 2011). Neuronal death is ultimately mediated by several factors including oxidative (Gotz et al., 1994, Mancuso et al., 2007, Mangialasche et al., 2009, de la Monte and Wands, 2006), endoplasmic reticulum (de la Monte, 2012c) and nitrosative (Mangialasche et al., 2009, Swomley and Butterfield, 2015, de la Monte et al., 2007) stress, apoptosis, mitochondrial dysfunction with energy failure (Kidd, 2005, Gonzalez-Lima et al., 2014, Daulatzai, 2017, de la Monte and Wands, 2006), brain hypo-perfusion (Aliev et al., 2003, Daulatzai, 2017), neuroinflammation (McGeer and McGeer, 2013), neurotoxic effects of Aβ1–42, and impaired signaling through insulin and insulin-like growth factor (IGF) pathways that promote cell survival and energy metabolism (Steen et al., 2005, de la Monte and Wands, 2008).
Synaptic terminal degeneration
Loss of cortical pre-synaptic terminals and dendritic spines, and dystrophic deformation of axonal and dendritic spines correlate with severity of dementia and are better markers of disease severity than senile plaques (Masliah et al., 1989, Masliah et al., 1991). Synaptic disconnection due to degeneration and loss of nerve terminals is associated with reduced synaptophysin immunoreactivity (Masliah et al., 1989, Masliah et al., 1991), and accompanied by loss of neuronal and neuritic sprouting in AD (Masliah, 1995, Brun et al., 1995, Liu et al., 1996). Irregularly swollen, dystrophic cortical neurites are detectable by silver impregnation histochemical techniques and Immunohistochemical staining for synaptophysin (Su et al., 1993) and ubiquitin (Wilson et al., 2001, Whatley et al., 2008). The histological correlate of neuritic dystrophy resulting from synaptic disconnection is cortical spongiosis manifested by vacuolation of the neuropil, especially in superficial layers of cerebral cortex.
Dystrophic neurites are often distributed along the periphery of Aβ1–42 plaques (Wippold et al., 2008), a phenomenon that could be linked to the presence of trophic factors such as basic fibroblast growth factor (bag) within plaques, potentially drawing in disconnected sprouting neurites (Cummings et al., 1993). Furthermore, the finding in a genetic mouse model that anti-Aβ1–42 therapy prevented synaptic degeneration (Buttini et al., 2005) suggested that the neurotoxic effects of Aβ1–42 have causal roles in the impairment of synaptic plasticity. Subsequently, the finding that mitoxantrone inhibition of Aβ1–42 oligomer fibril growth was neuroprotective in reducing the loss of cortical synapses in an experimental model of AD (Eleuteri et al., 2015) provided evidence that synaptic loss in AD is mediated by the neurotoxic effects of Aβ1–42 oligomers. However, it should also be noted that cortical dystrophic neurites are often distributed independent of Aβ1–42 deposits (Masliah et al., 1991), and in brains of ApoE-ε4 allele carriers, the presence of abundant cortical senile plaques in AD does not contribute to synaptic pathology or loss of cholinergic function (Corey-Bloom et al., 2000). Therefore, alternative mechanisms of synaptic disconnection should be evaluated.
Gliosis
Progressive loss of neurons, nerve terminals and fibers in AD is associated with gliosis, characterized by proliferation and activation of astrocytic glia (Brun et al., 1995, de la Monte, 1989) and deposition of extracellular matrix composed of glial fibrillary acidic protein (GFAP) in both gray and white matter structures (Chalmers et al., 2005). Combined neurodegenerative and reactive gliotic changes cause the underlying parenchymal architecture to become distorted. For example, instead of their normal laminar arrangements, cortical neurons become irregularly distributed and mis-oriented among hypertrophic astrocytes, plaques, microglia, and metabolic astrocytes. In addition, loss of nerve terminals and fibers combined with the proliferation of dystrophic neurites lead to collapse (shrinkage) and vacuolation of the neuropil. As neurodegeneration advances, hypertrophic, reactive astrocytes increase in abundance throughout the cortex, but most prominently in Layers III, V and at cortical-white matter boundaries. Gliosis is also evident throughout white matter and medial temporal structures, particularly the ventromedial amygdala.
The distribution and severity of gliosis are best revealed by immunohistochemical staining for GFAP (Figures 1A–1C), which typically shows that glial activation in AD is substantially more pronounced than pTau and Aβ pathologies. Astrocytic gliosis is not simply a scarring response to neurodegeneration since activated astrocytes have documented roles in mediating inflammatory and stress associated tissue injury (Birch, 2014, Garwood et al., 2016, Verkhratsky et al., 2014). Pro-inflammatory cytokines, chemokines, and reactive oxygen and nitrogen species can all damage synapses and disrupt neuronal plasticity (Agostinho et al., 2010). Therefore, once activated, gliosis can exacerbate neurodegeneration.
Figure 1.
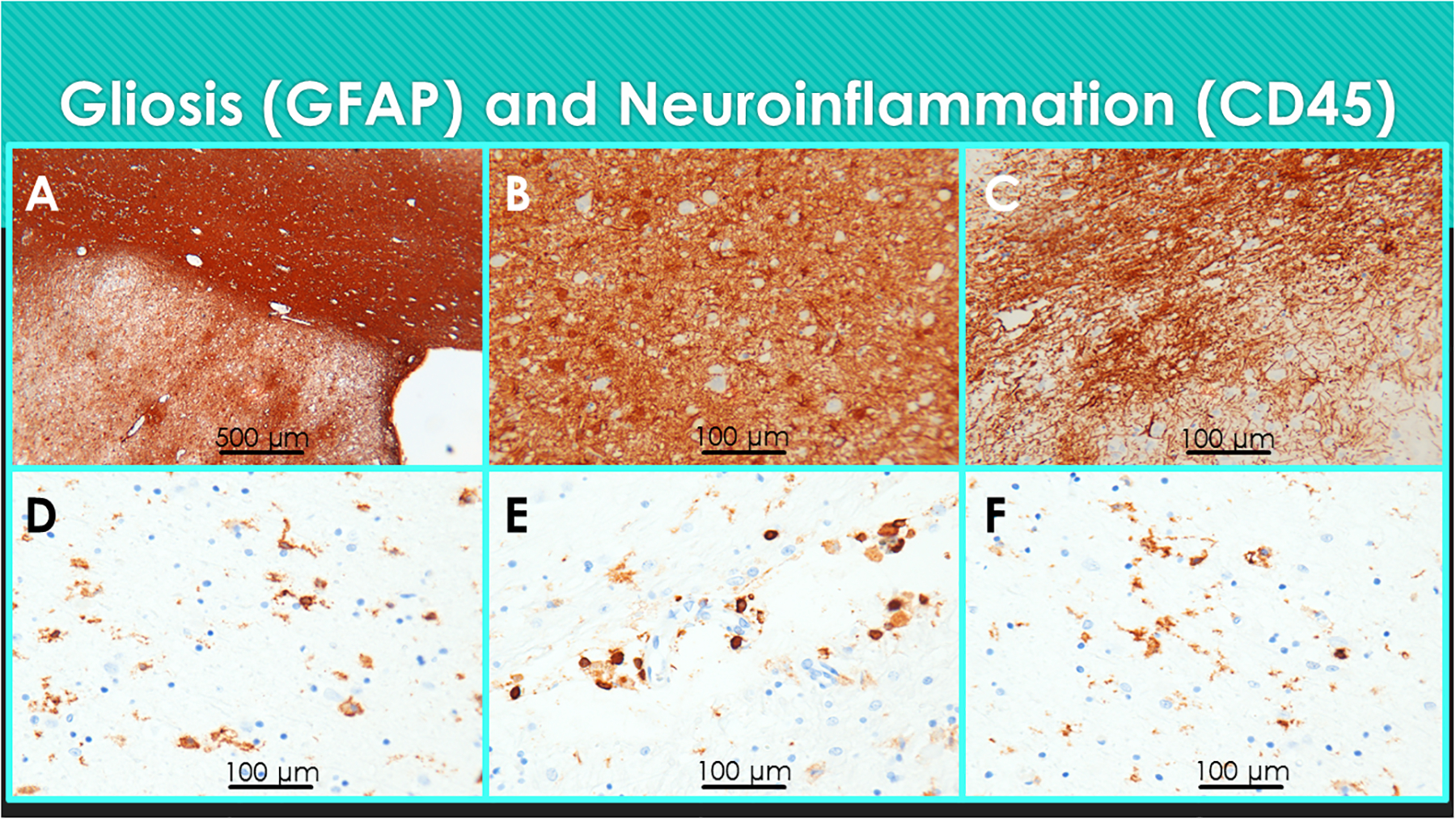
Gliosis and neuroinflammation in AD. Formalin fixed, paraffin-embedded (FFPE) sections of medial temporal structures including the amygdala from a postmortem human brain with AD were immunostained for (A-C) GFAP or (D-F) CD45 to detect reactive gliosis or microglia, respectively. Immunoreactivity was detected with HRP-polymer conjugated secondary antibodies and diaminobenzidine (DAB) as the chromogen (brown precipitate). The sections were counterstained with hematoxylin. Note (A) dense band of GFAP immunoreactivity in white matter (upper right) and diffuse labeling of the amygdala (lower left). Higher magnification images show (B) abundant hypertrophic astrocytes and (C) glial fibrils distributed throughout the amygdala. (D-F) Activated microglia (D) distributed throughout the neuropil, (E) surrounding small blood vessels, and (F) in white matter. (D-F)
Neuroinflammation
Neuroinflammation in AD is largely mediated by microglia and astrocytes. In Hematoxylin and Eosin stained histological sections, activated microglia exhibit irregularly twisted rod-shaped cell bodies distributed in the neuropil and closely associated with senile plaques. Immunohistochemical staining with CD45 or Iba1 antibodies demonstrates microglia abundantly distributed in the cortical neuropil, subcortical nuclei, and white matter. Microglia infiltrate white matter diffusely, but also prominently surrounding vessels and within the central, periventricular, and sub-cortical U-fiber zones (Figures 1D–1F), along with reactive astrocytes (gliosis). In addition, overlapping microgliosis and astrogliosis are abundantly distributed in corticolimbic and more distant structures, indicating that the distributions extend well beyond structures marred by abundant neurofibrillary tangle and plaques pathologies.
Neuroinflammatory injury occurs via pro-inflammatory cytokine activation, chemokine and complement release, and increased generation of membrane fatty acids, eicosanoids, lipid peroxidation products, reactive oxygen species, and reactive nitrogen species (Piro et al., 2012, Agostinho et al., 2010, Singhal et al., 2014). Consequences of neuroinflammation include damage nerve terminals, ultimately resulting in synaptic dysfunction followed by cognitive impairment (Agostinho et al., 2010). Neuronal injury and death linked to increased generation of fatty acids may be due to phospholipase activation and attendant hydrolysis of membrane phospholipids (Stephenson et al., 1999).
The sources of neuroinflammation are not well understood. One hypothesis that stemmed from the observation that microglial and reactive astrocyte pro-inflammatory cytokines such as IL-1β, IL-6, interferon-gamma, and macrophage migration inhibitory factor, are increased around plaques, was that Aβ promotes neuroinflammation (Mehlhorn et al., 2000, Dandrea et al., 2001). In addition, Aβ-activated microglia and reactive astrocytes could potentially cause neuronal injury and cholinergic dysfunction by increasing inducible cyclooxygenase, inducible nitric oxide synthase, and p38 MAPK activities (Giovannini et al., 2002). Alternatively, brain metabolic dysfunction linked to impaired insulin signaling may be the driving force toward neuroinflammation, vasculopathy, and oxidative stress (Misiak et al., 2012, Samaras and Sachdev, 2012, Gaspar et al., 2016).
Peroxisome proliferator activator receptors (PPARs) are nuclear hormone receptors that have both insulin sensitizing and anti-inflammatory effects in response to agonist binding (Collino et al., 2008, Dunn et al., 2010, Kalinin et al., 2009). Delta is the most abundantly expressed PPAR isoform in the brain (de la Monte and Wands, 2006) and its down-regulation in AD brains (de la Monte and Wands, 2006) has been implicated in the pathogenesis of neuroinflammation and oxidative stress. This concept is supported by the findings that experimental depletion of PPAR-delta increases neuroinflammation, gliosis, oxidative stress, Aβ deposition, and PHF tau (Barroso et al., 2013), while PPAR-delta agonist treatments reduce neuroinflammation and Aβ deposition in AD models (Kalinin et al., 2009, de la Monte et al., 2017b).
Oxidative stress
Oxidative stress is an important underlying mediator of neurodegeneration in AD (Agostinho et al., 2010, de la Monte, 2012b, de la Monte et al., 2017a) due to its relationships to neuroinflammation, insulin resistance, lipid peroxidation, and cell death. Potential sources of stress and inflammation include increased levels of advanced glycation end-products (AGE) and expression of the receptor for advanced glycation end products (RAGE) (Deane et al., 2003, Donahue et al., 2006, Lovestone and Smith, 2014, Yamagishi et al., 2015), impaired insulin/IGF signaling through Akt pathways, lipid peroxidation linked to myelin breakdown, and ceramide accumulation. Neuroinflammation in AD is associated with increased pro-inflammatory cytokine expression in astrocytes and microglia (Agostinho et al., 2010, Piro et al., 2012, Singhal et al., 2014). Correspondingly, postmortem brain levels of AGE and 4-hydroxy-2-noneal (4-HNE), a marker of lipid peroxidation, increase with severity of AD progression (Figure 9). However, at earlier stages of disease, 4-HNE was found selectively increased in AD CSF, along with elevated levels of pTau and Aβ (Figure 10), suggesting that stress-mediated white matter myelin degeneration marks the early stages of neurodegeneration.
Figure 9.
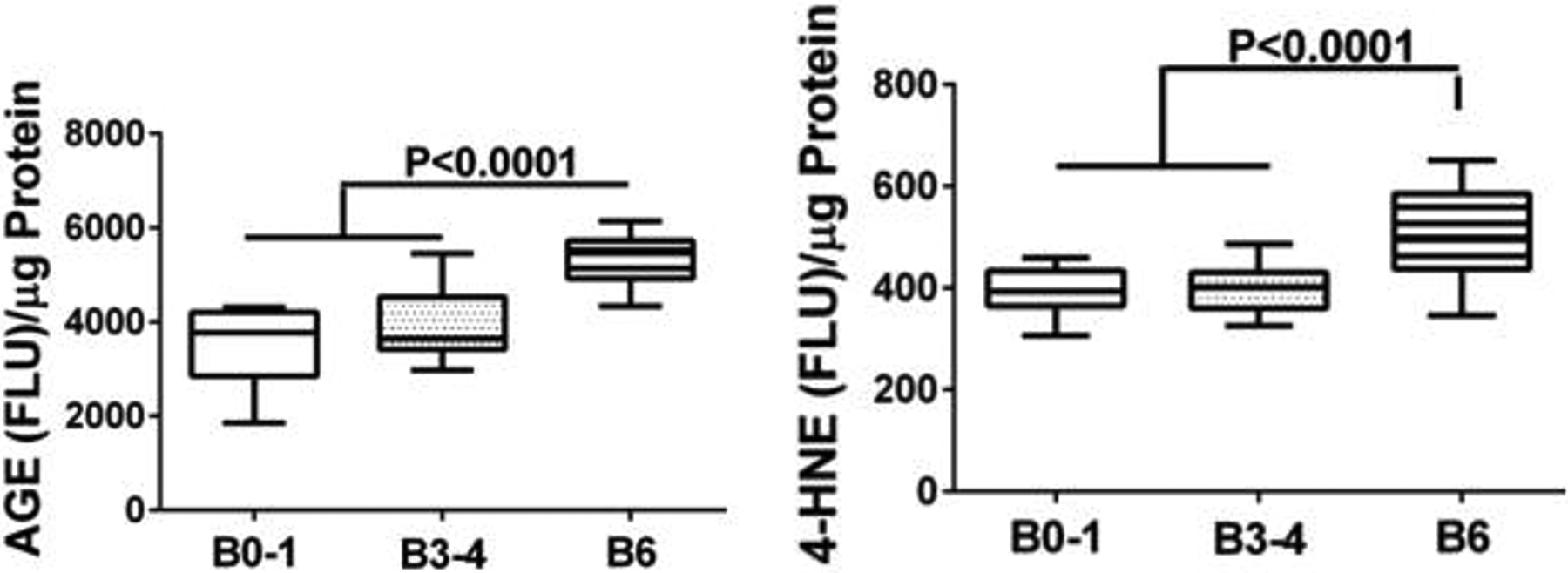
Increased levels of cerebral oxidative stress with AD progression marked by Braak stage (B). Levels of (Left) advanced glycation end-product (AGE) and (Right) 4-hydroxynonenal (4-HNE) lipid peroxidation markers were measured in postmortem frontal lobe homogenates by direct binding ELISA (Lee et al., 2013). B1–2 corresponds to control. B3–4 represents early- to intermediate-stage AD. B5–6 represents severe AD. Immunoreactivity was detected with horseradish peroxidase-conjugated secondary antibody and Amplex Red. Fluorescence light units (FLU) were measured (Ex 579 nm/Em 595 nm) in a Spectromax M5, and results were normalized to protein content. Boxplots depict calculated group means (horizontal bars), 95% confidence limits (upper and lower borders of the boxes), and ranges (stems). Intergroup statistical comparisons were made by repeated measures one-way ANOVA with the post hoc Tukey test. Significant P-values are indicated over the bars.
Figure 10:
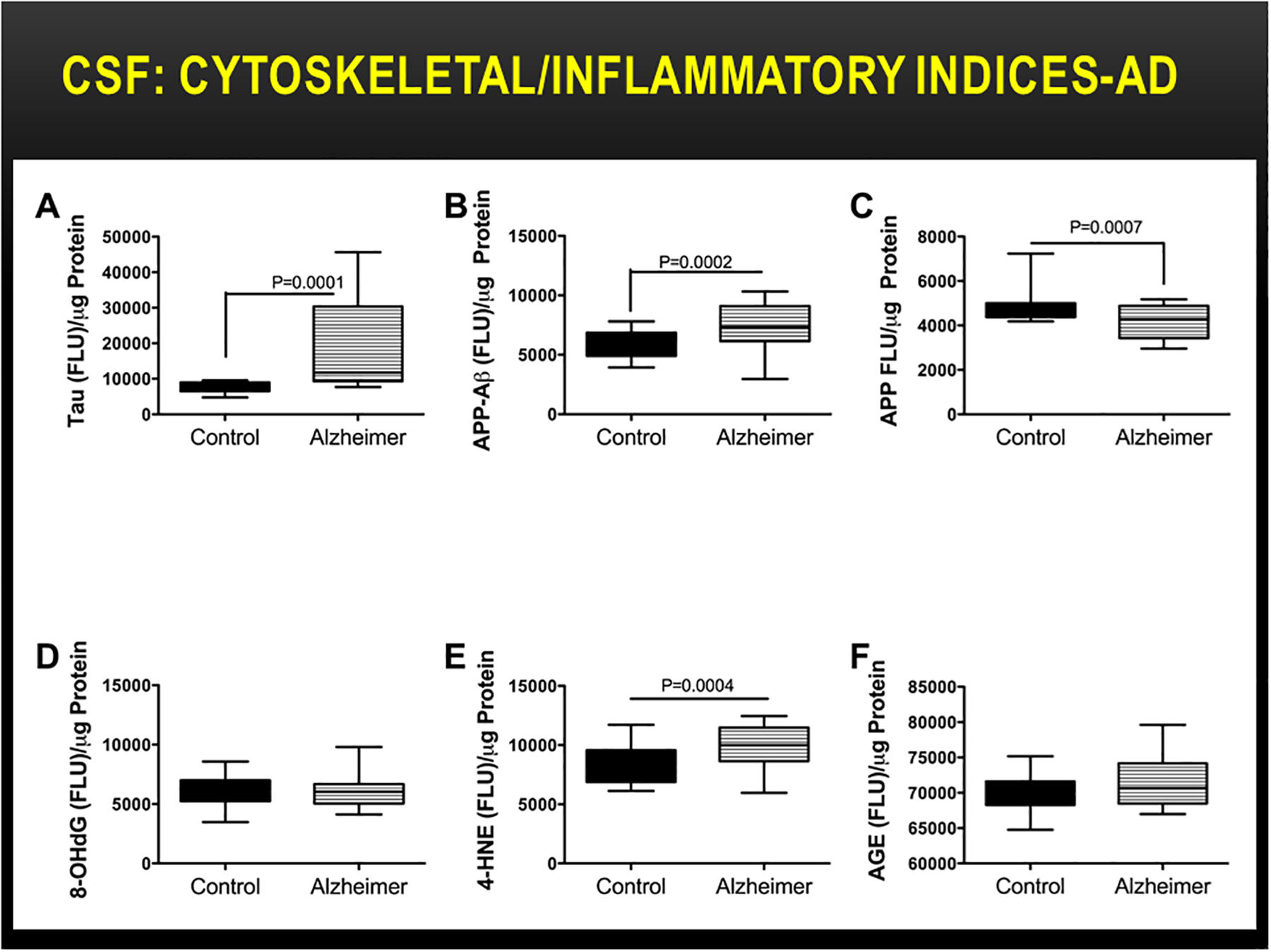
Increased CSF indices of oxidative stress in early AD. Lumbar puncture CSF from controls (N=12) and patients with early AD (N=16) were used to measure (A) phosphorylated tau (Tau), (B) the Aβ fragment of amyloid precursor protein (APP-Aβ), (C) APP, (D) 8-hydroxy-2’-deoxyguanosine (8-OHdG), (E) 4-HNE, and (F) AGE by direct binding ELISA (Lee et al., 2013). Immunoreactivity was detected with horseradish peroxidase-conjugated secondary antibody and Amplex Red. Fluorescence light units (FLU) were measured (Ex 579 nm/Em 595 nm) in a Spectromax M5, and results were normalized to protein content. Boxplots depict calculated group means (horizontal bars), 95% confidence limits (upper and lower borders of the boxes), and ranges (stems). Intergroup statistical comparisons were made with Student T-tests.
White matter degeneration
Nature and Distribution:
White matter mainly consists of myelinated axons together with glial cells, including oligodendrocytes and astrocytes, and vascular elements ranging from penetrating arteries and veins to capillaries and venules. Axons are long cytoplasmic extensions of neuronal cell bodies that serve to connect neurons in different regions of the CNS. Axonal structure is maintained by an elaborate cytoskeleton that is rich in neurofilament proteins and microtubule-associated proteins such as Tau. CNS myelin is composed of lipid-rich oligodendrocyte membranes that wrap around axons to provide insulation and ensure efficient conductivity. Loss of myelin or axons compromises neurotransmission and plasticity.
White matter degeneration is a major and consistent abnormality in AD. Its occurrence was initially characterized in 1986 by Brun and Englund (Brun and Englund, 1986). Subsequently, it was demonstrated that white matter atrophy emerges early and in the pre-clinical stages of AD (de la Monte, 1989). White matter atrophy can be quantified in relation to overall brain atrophy and cortical or subcortical nuclear atrophy using morphometric analysis (de la Monte, 1989). With that approach, white matter atrophy in postmortem AD brains was shown to be most pronounced in the parietal and temporal lobes, followed by frontal lobes, whereas the occipital lobes were relatively spared (de la Monte, 1989). In addition, the anterior commissure, fornix, and corpus callosum undergo atrophy while the internal, external, and extreme capsules remain intact. The histopathological correlates of white matter atrophy in AD have been characterized mainly via histochemical staining with Luxol fast blue dye. Luxol fast blue reacts with phospholipids or lipoproteins and detects myelin loss. Silver-impregnation, such by Bielschowsky staining which binds to neuronal cytoskeletal proteins, is used to characterize axonal pathology.
Associated Critical Pathologies:
In AD, white matter atrophy is associated with loss of myelin and axons. Myelin loss, manifested by pallor of Luxol fast blue staining (Figure 2), is associated with reduced populations of oligodendrocytes which are needed to maintain myelin, increased reactive gliosis (scarring) (Figures 1 and 3), and degenerative vasculopathy (Brun and Englund, 1986, de la Monte, 1989, Brickman et al., 2008, Burns et al., 2005, Brilliant et al., 1995, Englund, 1998, Sjobeck et al., 2003, Sjobeck et al., 2005) (Figure 4). White matter degeneration is most pronounced in periventricular and central regions, corresponding with the distribution of leukoaraiosis (Figure 2) and white matter hyperintensities seen by MRI (Scheltens et al., 1995). White matter degenerative vasculopathy mainly impacts micro-vessels which exhibit mural thickening and fibrosis with narrowing of the lumens, damage to endothelial cells, and perivascular tissue attrition and fibrosis. Although the consequence of cerebral micro-vasculopathy, including micro-infarcts and atrophy can be detected by MRI (Scheltens et al., 1995), the vasculopathy/vascular degeneration cannot be assessed directly using non-invasive neuroimaging tests.
Figure 2:
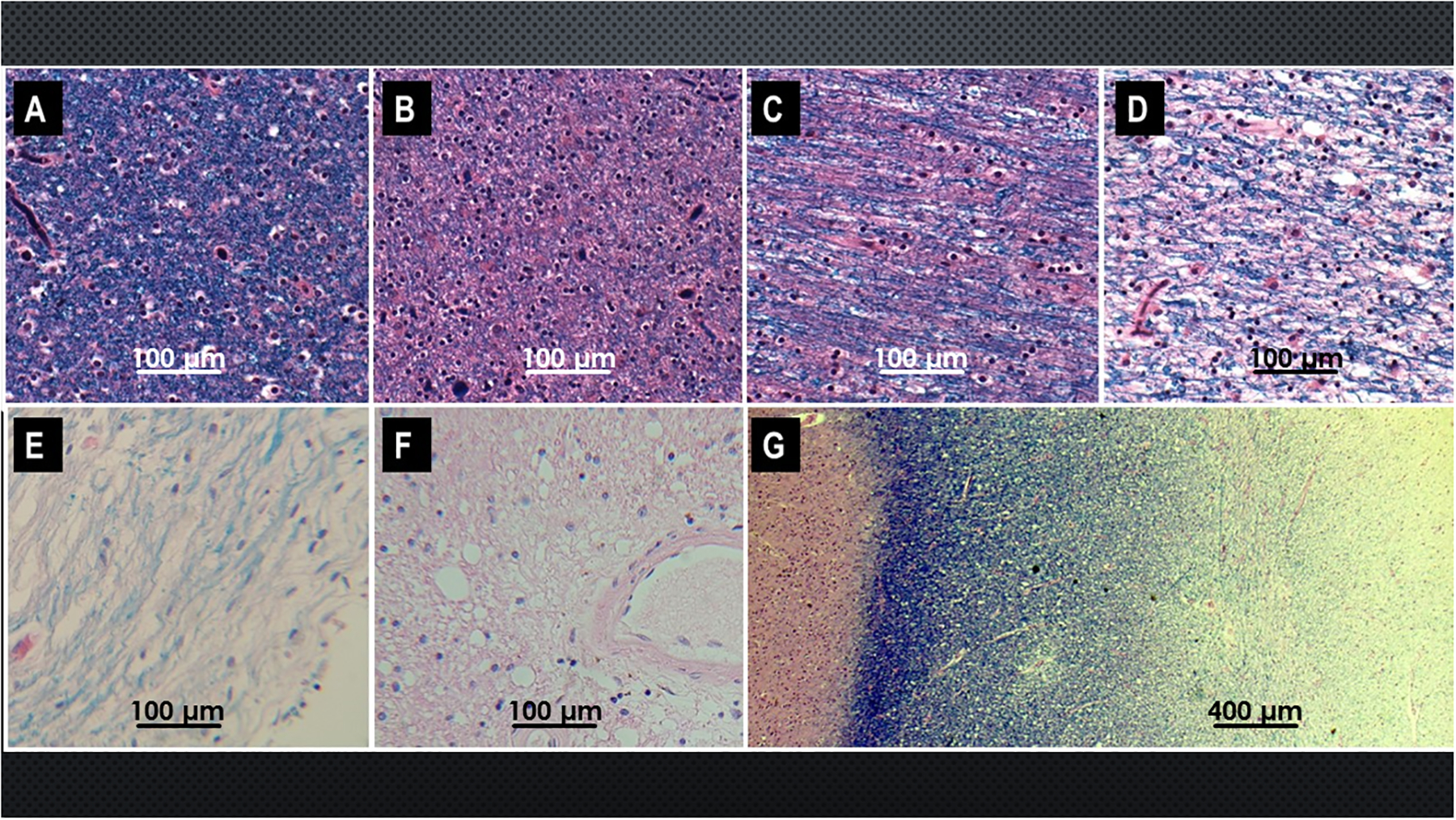
Spectrum of white matter (WM) degeneration in AD. Sections of FFPE posterior cingulate gyrus with underlying WM from human postmortem brains with different Braak stage severities of AD were stained with Luxol Fast Blue (LFB) to show myelin integrity, and counterstained with hematoxylin and eosin (LHE). High-intensity blue staining reflects intact myelin and low intensity staining indicates myelin loss. (A) Normal aged control WM with dense blue myelin staining. (B-F) Progressive reductions in WM LFB staining with increasing severity of AD: (B) Braak 2; (C) Braak 3; (D) Braak 4; (E) Braak 5; (F) Braak 6. (G) Leukoaraiosis manifested by graded WM degeneration from subcortical U-fiber region (left dense blue band) to central WM (right) where LFB staining is minimal LFB staining.
Figure 3.
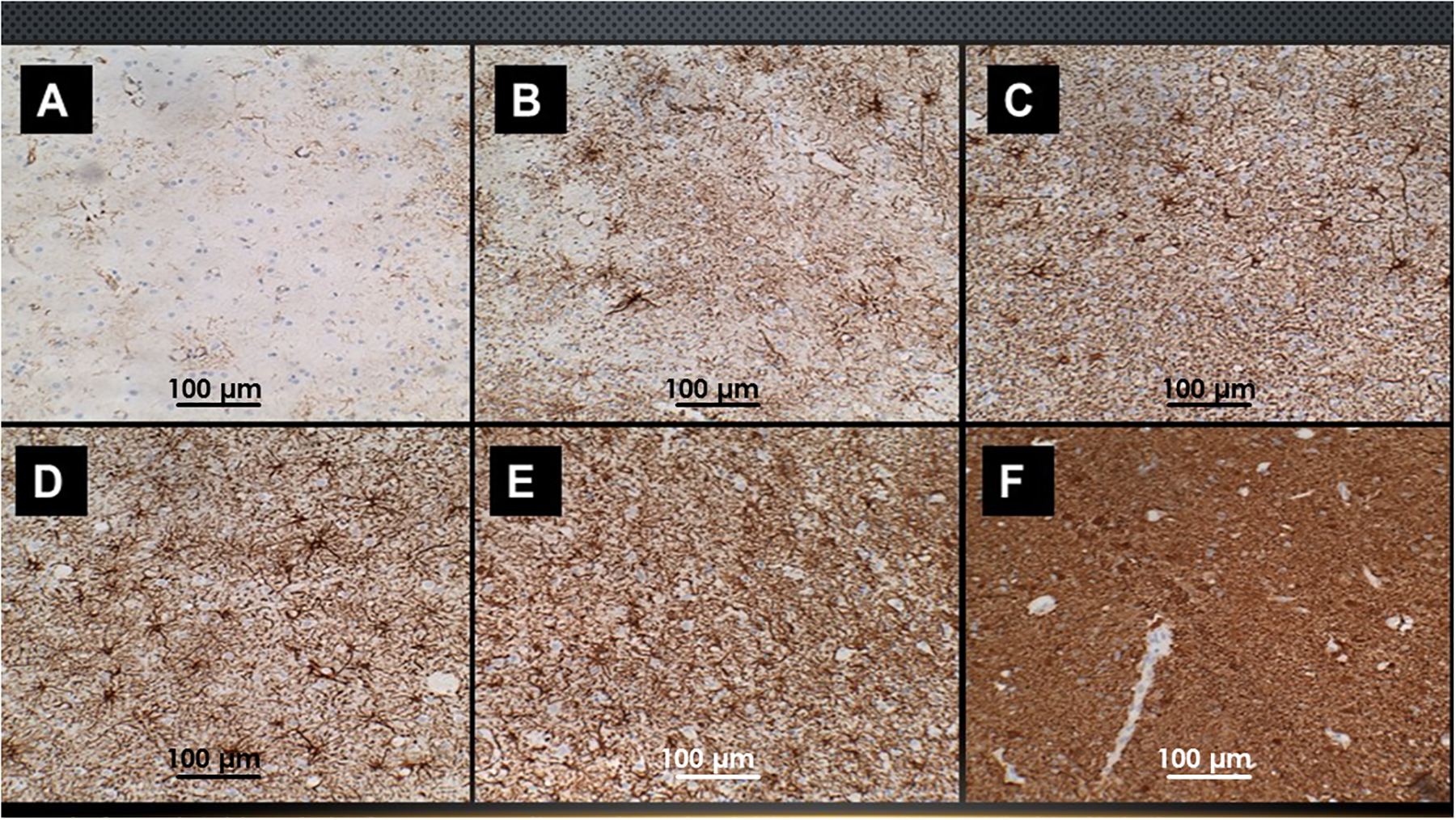
WM gliosis increases with severity of AD. FFPE sections of posterior cingulate from brains with (A) normal aging or different Braak stage severities of AD: (B) Braak 2; (C) Braak 3; (D) Braak 4; (E) Braak 5; (F) Braak 6 were immunostained for GFAP (See Legend to Figure 1). Higher intensities of GFAP immunoreactivity correspond to greater degrees of gliosis.
Figure 4.
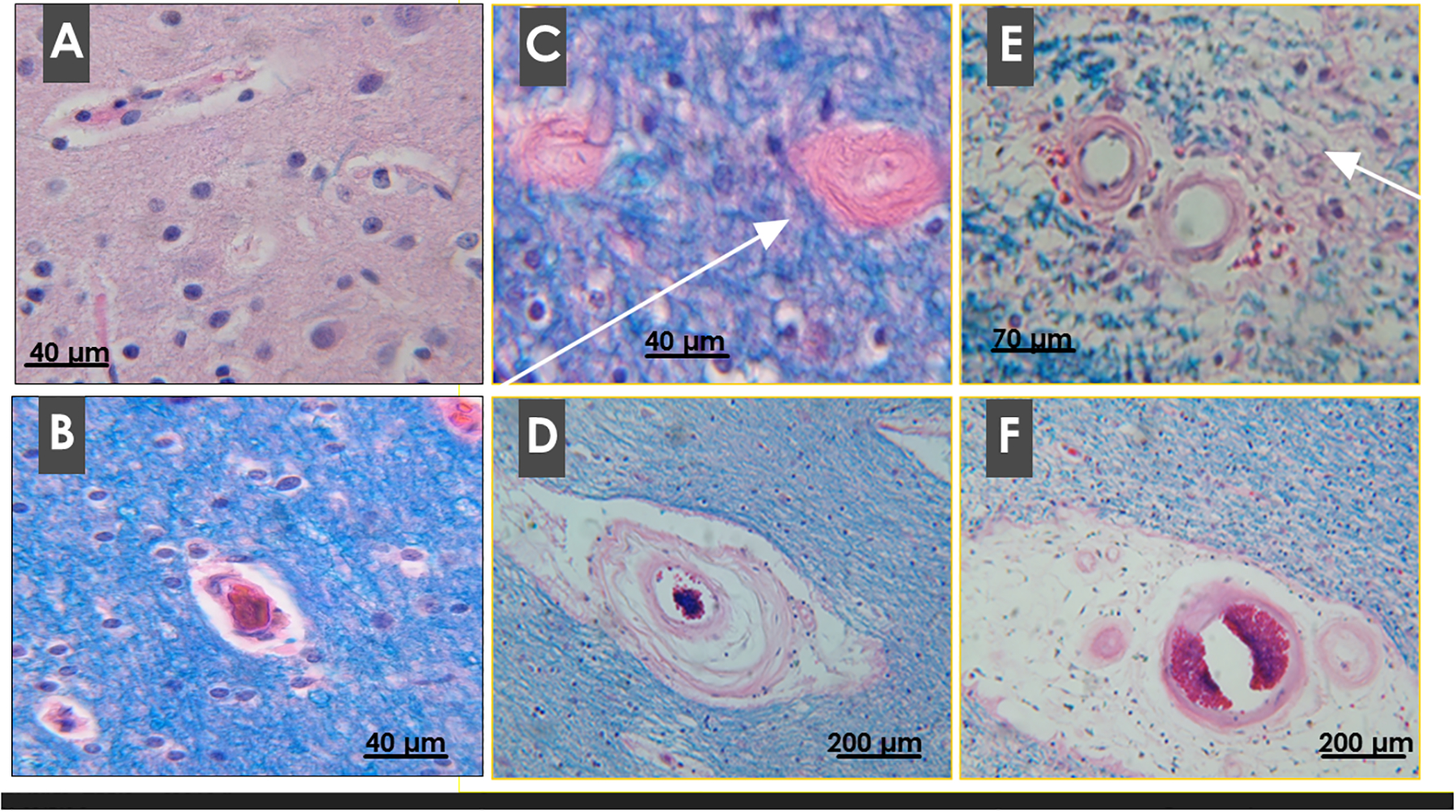
Microvascular pathology in AD. FFPE sections of human postmortem parietal (A) control cortical, (B) control WM, and (C-F) AD WM were stained with LHE. Note delicate walls and plump, regularly spaced endothelial cells in control micro-vessels. In AD, WM vascular disease affects small arteries, arterioles and capillaries and results in: (C) severe fibrotic thickening of the walls with extreme narrowing of lumens (arrow); (D, E) degeneration of vascular smooth muscle and loss of perivascular tissue and increased perivascular inflammatory cells (arrow in E); and (F) extensive attrition of perivascular WM tissue resulting in loss of contact between vessels and WM parenchyma.
Cell Types Affected:
Despite their widespread use, histochemical stains including Luxol fast blue, are relatively non-specific and offer few prospects for uncovering disease mechanisms. On the other hand, immunohistochemical staining provides an effective means of characterizing pathological processes. For example, GFAP immunoreactivity marks increased abundance and activation of reactive astrocytes, astrocytic fibrils, and glial fibrillary matrix (white matter scarring). Antibodies to pTau or neurofilament are used to demonstrate altered patterns and abundance of neuronal cytoskeletal proteins in relation to axonal loss and degeneration. Myelin basic protein antibodies help assess the integrity of mature myelin. Oligodendrocytes express Olig transcription factors; therefore, immunohistochemical staining for Olig1, Olig2, and Olig3 could potentially be used to quantify loss the of oligodendrocytes that accompanies white matter atrophy and degeneration AD.
Gliosis:
In AD, white matter gliosis is associated with markedly increased GFAP fibrillar and astrocytic cytoplasm immunoreactivity (Figures 1 and 3). Dense fibrillary gliosis is typically observed in periventricular zones. Central white matter exhibits prominently increased populations of reactive astrocytes in a background of variably increased GFAP-positive fibrils. Next is severity of gliosis are the sub-cortical U-fiber zones and white matter cores within gyri. Gliosis is also evident in white matter fibers that are intrinsic to nuclei that exhibit neuronal degeneration, but least prominent in major sensory, motor and cerebellar tracts systems.
Neuroinflammation:
Although the histopathology of white matter atrophy and degeneration in AD has been well characterized, the mechanisms remain unknown. Empirical studies showing that increased GFAP immunoreactivity overlaps with the distribution of microgliosis in white matter (Figures 1D–1F) suggest that gliosis and neuroinflammation may be inter-related. Increased generation of pro-inflammatory cytokines by microglia and astrocytes could potentially lead to oxidative injury and degeneration of myelin as well as axons. Although attractive, the underlying cause of neuroinflammation has not yet been determined. However, one potential consideration is that white matter degeneration is mediated by increased Aβ1–40 and Aβ1–42 accumulation and attendant neuroinflammation (Roher et al., 2002). Against this argument is that Aβ1–40 and Aβ1–42 do not accumulate in white matter except at cortical-white matter boundaries, and their increased immunoreactivity is not present in regions of white matter that exhibit the most severe myelin loss, fiber rarefaction, gliosis, and neuroinflammation.
Role of Ischemic Injury:
One compelling hypothesis is that white matter degeneration is mediated by chronic ischemic injury secondary to micro-vascular disease (Brun and Englund, 1986, Verny et al., 1991, Englund, 1998, de la Monte et al., 2000, van der Vlies et al., 2012, Love and Miners, 2015, Poliakova et al., 2016). Correspondingly, white matter fiber attrition frequently occurs in perivascular distributions and is associated with vascular fibrosis and luminal narrowing (Jellinger, 2002, Suter et al., 2002, Thal et al., 2003, Ervin et al., 2004). In addition, large areas of leukoaraiosis, characterized by ill-defined regions of myelin loss and incomplete infarction (Prencipe and Marini, 1989) are nearly always associated with white matter vasculopathy in AD, but is regarded as unspecific since elderly controls and patients with vascular dementia have similar abnormalities by neuroimaging (Verny et al., 1991, Meyer et al., 1992). Furthermore, it is now well-recognized that white matter ischemic injury contributes to cognitive impairment in AD, and that if this aspect of AD were prevented or effectively treated, incident rates and severity of AD would decline (Etiene et al., 1998). Correspondingly, the recent decline in AD prevalence has been attributed to increased use of cerebrovascular and cardiovascular preventive and intervention measures in older individuals (Langa, 2015).
Oligodendrocyte dysfunction:
Another potential mediator of white matter degeneration in AD is loss of impaired function of oligodendrocytes. Maintenance of oligodendrocytes is required for myelin homeostasis, and loss of myelin impairs neuronal conductivity and compromises the insulation of axons from the extracellular environment. Correspondingly, elevated levels of neurofilament light chain and myelin sulfatides were detected in CSF of patients with subcortical vascular dementia, even prior to significant onset of symptoms (Wallin et al., 2016a). Increased neurofilament and myelin sulfatides in CSF mark degeneration of white matter axons and myelin, reinforcing the concept that white matter degeneration occurs early in AD. Furthermore, myelin lipid breakdown can lead to oxidative damage via lipid peroxidation with attendant neuroinflammation and reactive gliosis. Previous studies have demonstrated major abnormalities in oligodendrocyte myelin-associated gene expression in both humans and experimental models of AD (Love and Miners, 2015, Tong et al., 2016, de la Monte, 2017). Mechanistically, oligodendrocyte survival and function and myelin maintenance are supported through stimulation of insulin and insulin-like growth factor (IGF) signaling pathways (Barres et al., 1993, Carson et al., 1993, Chesik et al., 2008). Correspondingly, in experimental models of impaired insulin and IGF signaling in the brain, oligodendrocyte dysfunction with white matter atrophy accompanies AD-type pathology and cognitive impairment (de la Monte et al., 2006, Lester-Coll et al., 2006).
Current Limitations in our Assessments of White Matter Degeneration in AD:
The major barriers to incorporating indices of white matter degeneration into the diagnostic scheme for AD are that: 1) practical tools and strategies for systematically characterizing white matter degeneration and quantifying changes over time or responses to treatment remain limited; 2) the full spectrum of abnormalities including alterations in lipid composition has not been characterized; and 3) the mechanistic framework remains uncertain and under-studied. Another limitation is that neuroimaging approaches are not sufficiently refined to enable distinctions among the many causes of white matter degeneration. Finally, surrogate CSF or serum biomarkers of white matter degeneration have not been discovered. Fortunately, progress in the application of mass spectrometry and lipidomics (Han et al., 2002, Lam et al., 2014, Mendis et al., 2016, Tong et al., 2017) may help provide new insights into the nature and pathogenesis of white matter degeneration in AD.
Microvascular Degeneration
Role of vascular degeneration in dementia:
Progressive declines in cerebral blood flow, glucose metabolism, and oxygen utilization in AD have been recognized for decades and suggest that impairments in brain perfusion contribute to neurodegeneration (de la Torre and Mussivand, 1993, de la Torre and Stefano, 2000). However, the structural pathology that mediates deficits in cerebral blood flow and metabolism remain somewhat controversial. For example, in one postmortem study vascular pathology was observed in over 80% of AD cases (Toledo et al., 2013), yet in other clinical and postmortem studies, dementia was linked to cases in which cerebrovascular disease overlapped with mild or moderate severity AD, and not when it occurred in isolation (Nolan et al., 1998, Etiene et al., 1998). Correspondingly, although substantial losses in brain volume due to large or multiple small infarcts can lead to depression and cognitive impairment, they do not cause AD (Ballard et al., 2000). Perhaps the most compelling data concerning the interactive roles of cerebrovascular disease and AD was provided by the 14-year Gothenburg study in which 63% of the cases had dementia due to AD or mixed AD+vascular disease, another 23% had other forms of dementia, and only14% had pure vascular dementia (Wallin et al., 2016b). Furthermore, the recent decline in AD incidence rates has been attributed to improved vascular care rather than recovery from AD (Scheltens et al., 2016), and supports the earlier hypothesis that cerebrovascular diseases lowers the threshold for AD to become clinically manifested (Etiene et al., 1998).
Nature of microvascular disease in AD:
Two types of vascular pathology are characteristically present in AD: Aβ-associated and Aβ-unassociated. Amyloid angiopathy affects cortical and leptomeningeal but not white matter vessels (Vinters et al., 1990, Joachim and Selkoe, 1992). In the cortex, microvessels, including capillaries contain Aβ deposits in their walls and in the adjacent perivascular parenchyma (Vinters et al., 1996, Vinters, 2015). The most clinically significant consequence of amyloid angiopathy is hemorrhage.
Non-Aβ vascular degeneration occurs in the cerebral cortex, white matter and subcortical nuclei. The earliest descriptions of AD vasculopathy noted that the cortical microvessels, including capillaries, arterioles and venules, exhibit thickened basement membranes and attrition of perivascular tissue (Figure 4) (Scheibel et al., 1989). Pathophysiologically, non-Aβ vascular degeneration most frequently occurs in the settings of diabetes mellitus (Luchsinger, 2012) and systemic arterial hypertension (Naderali et al., 2009, Middleton and Yaffe, 2009). Blood vessels, particularly in white matter, become fibrotic with thickened walls and severely narrowed lumens, which together reduce vessel wall compliance, restrict brain perfusion, and impair responsiveness to metabolic demands (Farkas et al., 2000).
Consequences of Brain Microvascular Disease:
Degeneration of vessel walls also causes them to weaken and become leaky, enabling circulating toxins and inflammatory mediators to enter the brain (Brun and Englund, 1986, Englund, 1998, Perlmutter and Chui, 1990). Consequently, white matter tissue surrounding damaged, leaky vessels is vulnerable to injury and degeneration (de la Torre and Stefano, 2000, Neltner et al., 2014, Kalaria et al., 2012, Chalmers et al., 2005, Jellinger, 2002, Verny et al., 1991, Ferrer et al., 1990, Thal et al., 2003) with attendant ischemic atrophy, incomplete infarction, and leukoaraiosis, (Brun and Englund, 1986, Englund, 1998). White matter vascular dysfunction and pathology is variably associated with dementia, but consistently correlated with slowness in mental processing, decline in executive function, and compromise of blood-brain barrier (BBB) integrity (Wallin et al., 2016a).
Blood-Brain Barrier Disruption in AD
The blood brain barrier is critical for regulating brain exposures to substances in the peripheral circulation, including toxins. Metalloprotein-9 (MMP-9) regulates opening and activity of the BBB, while tissue inhibitor of metalloproteinase 1 (TIMP-1) counteracts this effect (Wallin et al., 2016a). Plasminogen activator inhibitor-1 (PAI-1), which is produced by endothelial cells, inhibits fibrinolysis as well as MMP activity (Wallin et al., 2016a). In AD and other dementias, white matter microvascular disease is associated with elevated CSF levels of MMP-9 (Wallin et al., 2016a), BBB dysfunction marked by increased permeability to albumin, and altered pro-inflammatory and pro-coagulation events that enhance microvascular occlusions and thereby promote ischemic injury (Grammas, 2011).
In AD, BBB disruption could account for: 1) neurotoxic injury leading to neurodegeneration (Mann, 1985, Hoyer, 1982); 2) dysregulation of the brain pool of Aβ and its equilibrium status between cerebrospinal fluid and plasma (Johanson et al., 2018); and 3) neuroinflammation (Ott et al., 2018). Aβ homeostasis is regulated by influx of soluble Aβ across the BBB via interactions with and efflux via the low-density lipoprotein (LDL) receptor on brain endothelial cells. AD is associated with increased RAGE influx or decreased LDL receptor efflux, preventing Aβ clearance (Grammas, 2011).
MMP-9
What Comes First?
As AD progresses from its pre-symptomatic stages to dementia different aspects of disease emerge and evolve (Daviglus et al., 2010). Brain accumulations of Aβ due to reduced clearance (Zafari et al., 2015) and neuroinflammation increase during the presymptomatic period and peak early in the course of neurodegeneration (Serrano-Pozo et al., 2011). pTau accumulation begins later than Aβ and the progressively increased levels associated with neurofibrillary tangle and dystrophic neurite pathologies correlate with cognitive decline, memory impairment, and hippocampal atrophy (Daviglus et al., 2010). However, the two abnormalities that correlate best with cognitive impairment and progressive dementia are hippocampal atrophy and impairment in glucose utilization. In addition, evidence suggests that, in contrast to senile plaque burden which does not correlate with dementia, Aβ-derived diffusible ligands (ADDLs) also increase in association with brain metabolic dysfunction and cognitive impairment through the late stages of AD (Viola and Klein, 2015). Altogether, the data suggest that most of the bio-indices studied in relation to AD may be informative and carry diagnostic potential only at limited stages of disease. In contrast, brain metabolic dysfunction linked to deficits in glucose utilization reliably marks the course of progressive cognitive decline and neurodegeneration. ADDL accumulation may provide a supplementary index of neurotoxicity-mediated neurodegeneration in AD (Schuster and Funke, 2016).
Brain Metabolic Dysfunction in AD
Impairments in brain glucose utilization
The predictably consistent clinical, pathophysiological, and neuropathological abnormalities that characterize the progressive course of AD suggest the existence of a fundamental underlying process that drives neurodegeneration. In our re-conceptualization of its etiopathic basis, it will be critical to link seemingly unrelated pathologies utilizing evidence-based approaches to determine how they are shared. Deficits in brain energy metabolism, particularly with respect to glucose utilization in AD have been recognized for years, but more recent approaches such as PET imaging with (18)F-fluoro-deoxyglucose (FDG) have standardized its detection in the early stages of neurodegeneration (Daulatzai, 2012, Schaffer et al., 2015). Correspondingly, one of the most significant findings across multiple studies is that AD is associated with global reductions in brain glucose metabolism relative to normal healthy control brains (de Leon et al., 1983, Faulstich, 1991, Waldron et al., 2015, Wurtman, 2015). The importance of these observations is that glucose metabolism is critical for most brain functions, but particularly those related to cognition, behavior and plasticity, which are most significantly impacted in AD. Metabolic derangements pertaining to glucose and oxygen utilization in the brain worsen with severity of AD. Since glucose and oxygen are the main fuels, deficits in their utilization effectively cause brain starvation (Figure 5). A key mediator of brain metabolic dysfunction in AD is the progressive compromise of insulin’s actions, starting in the earliest stages of disease (de la Monte, 2012b, de la Monte, 2012a, de la Monte, 2012c, de la Monte et al., 2009a, de la Monte et al., 2011b).
Figure 5:

The AD neuropathological spectrum includes an array of abnormalities linked to neurodegeneration and goes well beyond progressive accumulations of pTau and Aβ1–42-associated pathologies. Most other major aspects of AD neurodegeneration stem from metabolic derangements linked to insulin deficiency and insulin resistance, which essentially produce states of brain starvation due to impaired glucose uptake and utilization.
Insulin and Insulin-Like Growth Factor (IGF) Functions in the Brain
Insulin and its cousin IGF-1, and their receptors are abundantly expressed in brain regions that are most vulnerable to AD neurodegeneration (de la Monte and Wands, 2005, see Chapter 1 in this book). Insulin and IGF-1 signaling networks regulate neuronal survival, plasticity, growth, metabolism, and repair, and mediate memory, cognition, motor functions, and behavior (de la Monte, 2012b, de la Monte and Wands, 2005). The findings that brain and cerebrospinal fluid (CSF) insulin levels are reduced in the early and intermediate stages of AD (Lee et al., 2013) and that insulin administration improves working memory and cognition (Benedict et al., 2011, de la Monte, 2013, Kidd, 2008, Reger et al., 2008) highlight the etiopathic role of insulin-linked metabolic dysfunction in AD. In addition, counter-regulatory roles of insulin and Aβ have been suggested by a large number of experimental reports that are supported in part by human studies demonstrating co-occurrences of brain insulin and IGF-1 deficiencies with higher brain levels of Aβ and AGE levels (Lee et al., 2013, de la Monte and Tong, 2014, Shuvaev et al., 2001), and enhancement of Aβ clearance with insulin administration (Reger et al., 2008). Since insulin and IGF-1 regulate neuronal and oligodendroglial cell functions including survival (de la Monte, 2012b, de la Monte and Wands, 2005), it is evident that deficiencies in these trophic factors or their receptor responsiveness would lead to pathology in both gray and white matter structures. Although there has been relatively little research linking impairments in insulin and IGF signaling to white matter and oligodendrocyte pathology in human cases of AD, there is ample evidence that both gray and white matter structures undergo atrophy and neurodegeneration with disease progression.
Lessons from Experimental Models of Brain Insulin Deficiency and Resistance
Experimental models have been instrumental in demonstrating that brain insulin deficiency and resistance impair learning and memory and mediate neurodegeneration (de la Monte et al., 2011a). The models included: 1) silencing of brain insulin or IGF polypeptide or receptor expression; 2) intracerebral (i.c.) treatment with streptozotocin (STZ); and 3) diet-induced obesity-associated systemic insulin resistance. Selective silencing of brain insulin, IGF-1 or IGF-2 polypeptide or receptor genes by i.c. administration of targeted short interfering RNA molecules caused significant atrophy of the hippocampus and temporal lobes with neuronal loss and impairments in spatial learning and memory (de la Monte et al., 2011a). Although STZ is a well-established pro-diabetes toxin, i.c. administration causes impairments in spatial learning and memory (Lester-Coll et al., 2006, Tong et al., 2016) and AD-type neurodegeneration characterized by temporal lobe and hippocampal atrophy with neuronal loss, gliosis, white matter degeneration, increased oxidative stress, and elevated levels of pTau, Aβ42, and ubiquitin (de la Monte et al., 2017b, Tong et al., 2016). Further analysis revealed that i.c. STZ disrupts brain insulin and IGF-1 signaling through their receptors and at multiple downstream points in the cascades, corresponding with increased activation of stress and pro-apoptosis mechanisms, and inhibition of cell survival and metabolism pathways (de la Monte et al., 2017b, Tong et al., 2016). Therefore, the i.c. STZ model replicates most of the characteristic neuropathological findings in human AD, warranting the conclusion that AD is a brain metabolic disorder with diabetes-like abnormalities.
The finding that experimental high fat diet-induced obesity was associated with brain insulin resistance, cognitive impairment (Figure 6), and increased brain levels of pTau, Aβ and oxidative stress (Moroz et al., 2008, Lyn-Cook et al., 2009, Tong et al., 2010) was initially surprising. However, further investigations revealed that those models also had visceral obesity, steatohepatitis with hepatic insulin resistance, type 2 diabetes mellitus, and systemic inflammation (Lyn-Cook et al., 2009, Jiao et al., 2009). Two key findings were that steatohepatitis progressed in parallel with brain insulin resistance and was associated with increased liver and serum levels of ceramides due to dysregulated sphingolipid metabolism (Lyn-Cook et al., 2009). Since ceramides can be pro-inflammatory and inhibit insulin signaling through PI3K-Akt (Czubowicz and Strosznajder, 2014, Zinda et al., 2001, de la Monte and Tong, 2014), additional experiments determined whether ceramides, due to their lipid soluble nature, could cross the BBB and cause neurodegeneration. Those studies showed that ceramides can cross the BBB and exert neurotoxic/neurodegenerative effects manifested by increased levels of oxidative stress, Aβ, and pTau, pro-apoptosis pathway activation, and impairments in insulin signaling (Tong and de la Monte, 2009). Therefore, these investigations provided a potential mechanism by which peripheral insulin resistance diseases could be linked to cognitive impairment, brain insulin resistance, and AD-type neurodegeneration.
Figure 6:
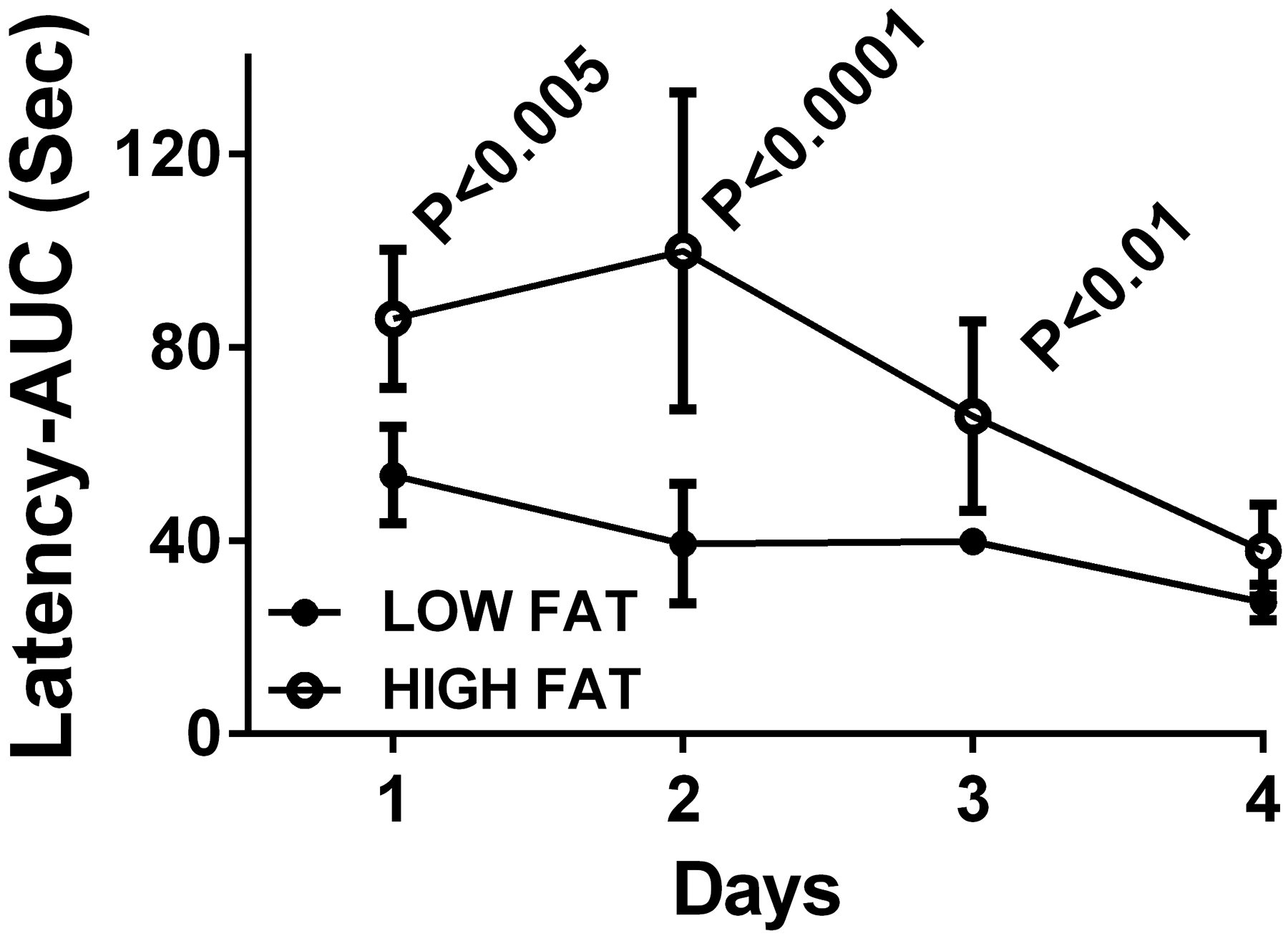
Chronic high fat diet-induced obesity with insulin resistance impairs spatial learning and memory in an experimental rat model. Adult male Long Evans rats were fed with low (LFD) or high (HFD) fat diets for 6 weeks, which resulted in steatohepatitis, peripheral insulin resistance, visceral obesity and cognitive impairment (de la Monte et al., 2009c). The Morris Water Maze test was performed over a 4-day period with 3 trials per day. Performance on the first two days reflects learning and memory acquisition while the platform is visible but entry points into the maze are varied, and performance on Days 3 and 4 reflects memory consolidation when the maze platform is hidden, and entry points are varied. Area-under-the-curve (AUC) calculations for the latencies (seconds) required to locate and land on the platform were made for each of the 3 daily trials. Intergroup statistical comparisons were made with paired T-tests.
Brain Diabetes and the Pathogenesis of AD
Primary Brain Insulin Deficiency and Resistance in AD—Type 3 Diabetes
To validate the roles of brain insulin and IGF deficiency and resistance, insulin, IGF, and corresponding receptor expression were measured in human postmortem brains that were diagnosed with different Braak stage severities of AD (de la Monte and Wands, 2008, Steen et al., 2005, Rivera et al., 2005) (Figure 7). Those investigations demonstrated significant and progressive disease-stage declines in insulin and IGF-1 signaling mechanisms, including through PI3K-Akt pathways that regulate energy metabolism, cell survival, and neuronal plasticity and inhibit cellular stress and apoptosis (de la Monte and Wands, 2008, Steen et al., 2005, Rivera et al., 2005, Talbot et al., 2012). In addition, sustained deficits in brain insulin/IGF signaling were linked to reduced expression of choline acetyltransferase, which is needed to generate acetylcholine (de la Monte and Wands, 2008, Steen et al., 2005, Rivera et al., 2005).
Figure 7:
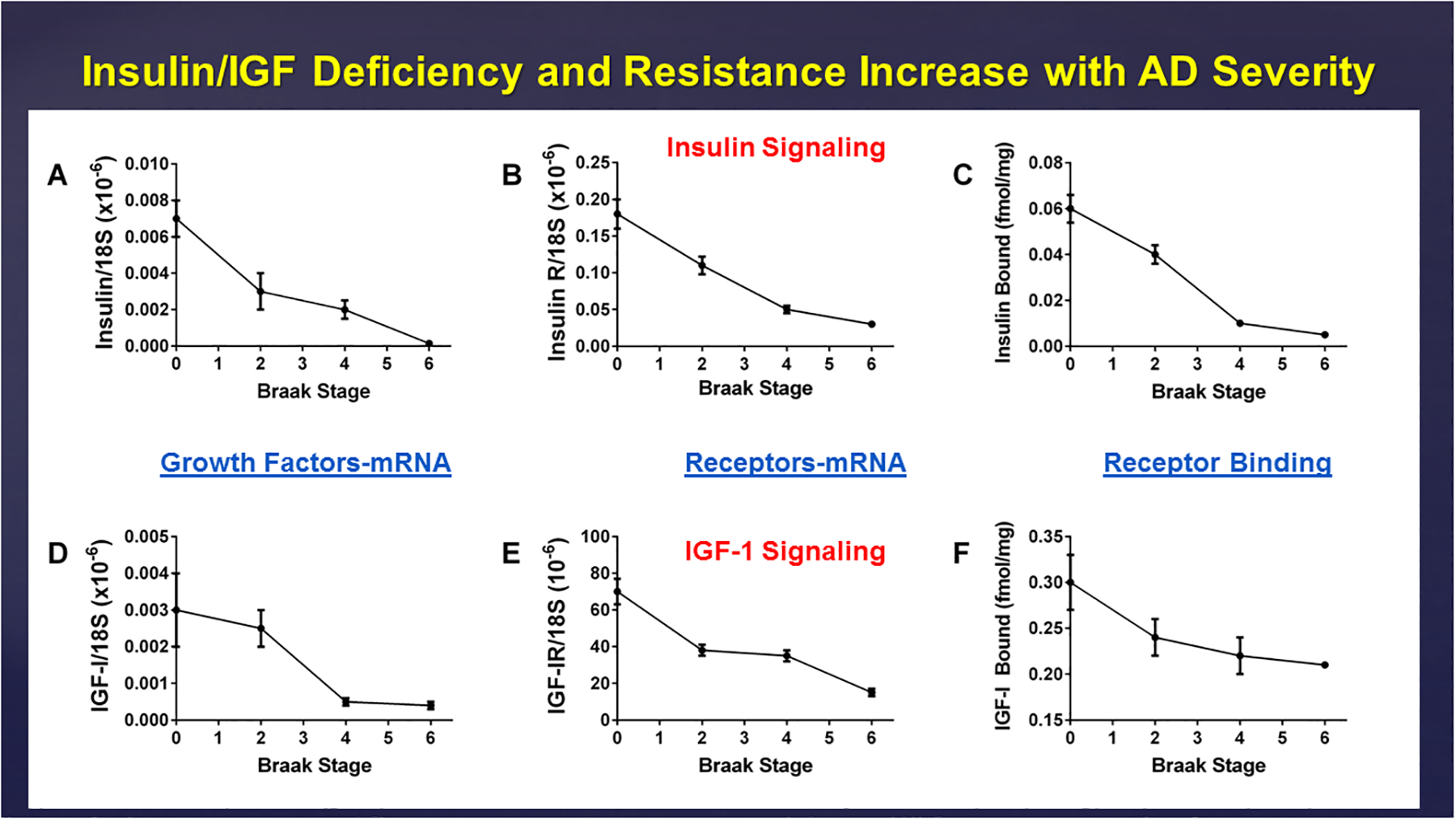
Progressive declines in frontal lobe expression of (A) insulin and (D) IGF-1 polypeptide genes, (B) insulin and (E) IGF-1 receptor genes, and (C) insulin and (F) IGF-1 receptor binding with increasing Braak Stage severity of AD. The mRNA levels were measured by qRT-PCR analysis, and ligand-receptor interactions were measured using competitive equilibrium binding assays (Rivera et al., 2005).
In accord with the experimental data showing causal roles of brain insulin and IGF-1 deficiency and resistance in the pathogenesis of AD-type neurodegeneration, it was important to determine whether related abnormalities occurred early during disease and could be detected in cerebrospinal fluid (CSF). The availability of paired clinical and postmortem CSF samples from confirmed cases of AD enabled us to demonstrate that insulin and IGF-1 levels in CSF are significantly decreased in the mild cognitive impairment stages of AD (Lee et al., 2013) (Figure 8). In addition, CSF levels of nerve growth factor and glial-derived neurotrophic factor were also shown to be reduced in AD (Lee et al., 2013). Therefore, besides pTau and Aβ, indices of brain metabolic and neurotrophic dysfunction should be included in CSF screening panels to aid in earlier diagnosis of AD.
Figure 8:
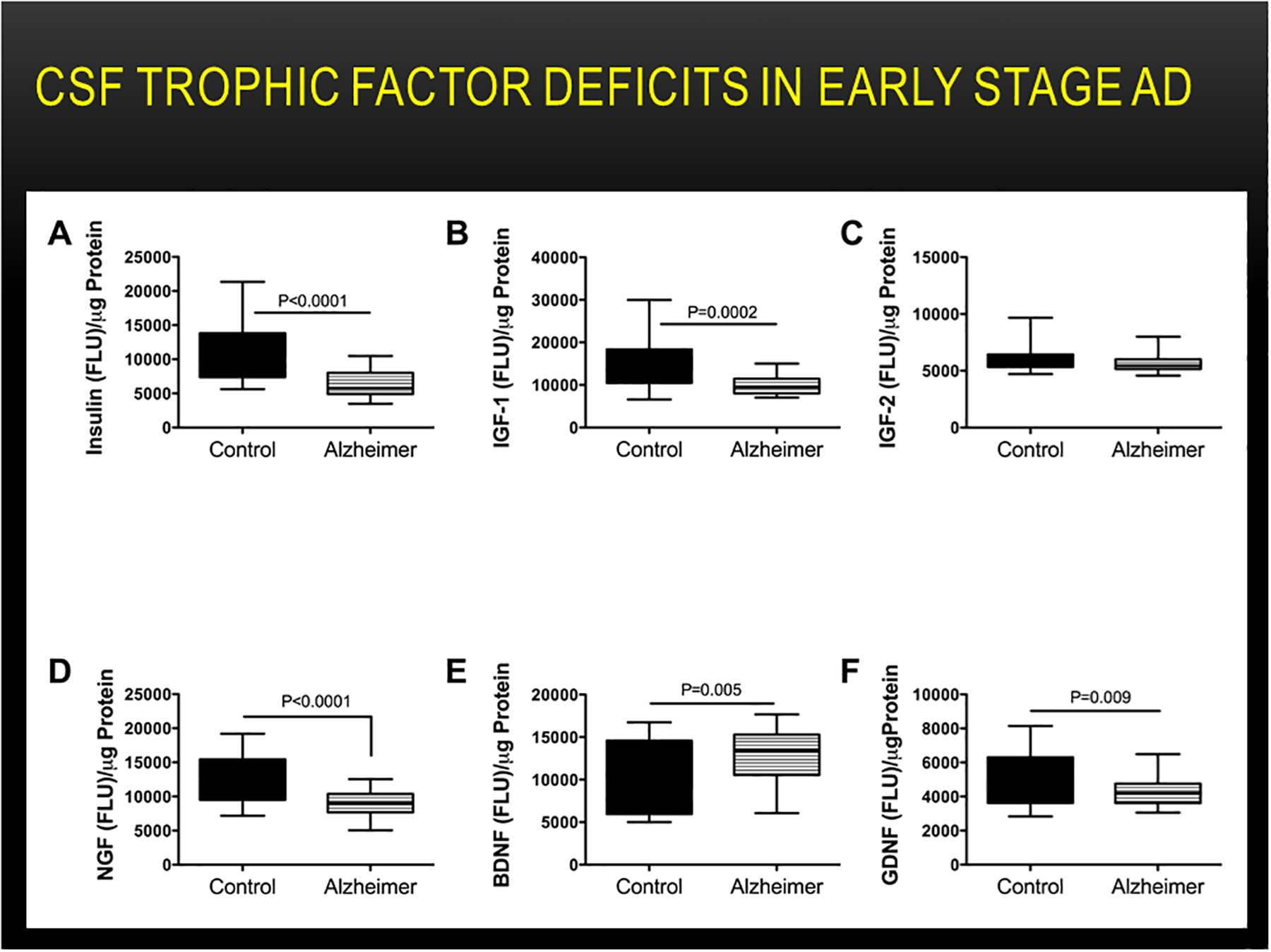
Altered CSF levels of insulin, IGFs, and neurotrophins in early AD. Lumbar puncture CSF samples from controls (N=12) and patients with early probable AD (N=16) were used to measure (A) insulin, (B) IGF-1, (C) IGF-2 (D) nerve growth factor-NGF, (E) brain-derived neurotrophic factor-BDNF, and (F) glial-derived neurotrophic factor-GDNF immunoreactivity by ELISA (Lee et al., 2013). Immunoreactivity is expressed in fluorescence light units (FLU) normalized to protein content. Data were analyzed with Student T tests.
Secondary Systemic Insulin Resistance Diseases and Cognitive Impairment
Epidemiological trend analyses showed that the rates of AD had increased sharply across all age groups, 50 years and above, over a period of several decades (de la Monte et al., 2009b). Importantly, those trends paralleled shifting rates of diabetes mellitus (de la Monte et al., 2009b), suggesting that the underlying cause factors might be related. Correspondingly, several human studies demonstrated that the risks of developing cognitive impairment and dementia were significantly elevated in overweight or obese people and diabetics relative to lean controls (Pedditizi et al., 2016, Alosco and Gunstad, 2014, Luchsinger et al., 2007, Noble et al., 2012, Naderali et al., 2009, see Cahpter 2 in this book). Moreover, inadequate medical control of Type 1 and Type 2 diabetes also increased risk for cognitive impairment (Drab, 2009, Roriz-Filho et al., 2009, de la Monte, 2012c, Fotuhi et al., 2012, Sridhar et al., 2015, de la Monte, 2014) and AD (Li et al., 2015). Still others linked various forms of peripheral insulin resistance (Kim and Feldman, 2015, Cholerton et al., 2011) including pre-diabetes (Roriz-Filho et al., 2009), metabolic syndrome (Frisardi et al., 2010), and non-alcoholic fatty liver disease (de la Monte et al., 2009a, Nagoshi, 2014) to cognitive decline. Finally, multivariate analysis of a late onset AD international, multicenter cohort identified gene clusters associated with inflammation, diabetes, and obesity as pathologic processes linked to neurodegeneration (Meda et al., 2012). Altogether, evidence that peripheral insulin resistance diseases contribute to or cause cognitive impairment and neurodegeneration in humans is strong and corresponds well with data generated from experimental models.
Type 3 versus Type 2 Diabetes
Because the human AD associated abnormalities in insulin and IGF-1 signaling are highly reminiscent of the pathophysiological findings in both Type 1 and Type 2 diabetes mellitus, yet they selectively involve the brain, we coined the term ‘Type 3 diabetes’ (Table 1) (Steen et al., 2005, de la Monte and Wands, 2008). Type 3 diabetes concisely conveys the concept that AD is a brain form of diabetes in which both ligand (insulin and IGF-1) deficiencies and receptor resistances coexist and mediate functional impairments in signaling pathways that mediate glucose utilization, metabolism, neuronal plasticity, cell survival and a range of functions needed to maintain the integrity of the CNS. In ‘Type 3 diabetes’, we hypothesize that the brain is the main target of metabolic dysfunction since most people with typical AD have pathology limited to the brain and lack clinical evidence of peripheral insulin resistance. On the other hand, it is impossible to ignore both human and experimental evidence that peripheral insulin resistance diseases in general are often associated with cognitive impairment and AD-type neurodegeneration. This phenomenon could account for the rapid increases in the incidence and prevalence rates of mild cognitive impairment (MCI) and AD. The good news is that the excess MCI/AD case burdens are potentially preventable and treatable using strategies like those employed to manage diabetes mellitus, metabolic syndrome and non-alcoholic fatty liver disease.
Table 1:
Characteristic Features of Type 1, Type 2 and Type 3 Diabetes
| Target Effects | Type 1 Diabetes | Type 2 Diabetes | Type 3 Diabetes |
|---|---|---|---|
| Insulin Ligand | Reduced | Increased | Reduced |
| Insulin Receptor | Unaffected or Increased | Reduced Activation | Reduced activation and expression |
| Glucose Utilization | Decreased | Decreased | Decreased |
| Primary Tissue/Organs | Pancreas (Brain) | Skeletal muscle, adipose tissue, blood vessels | Brain neurons, white matter, micro-vessels |
| Secondary Tissue/Organs | Brain, retina, blood vessels, kidneys, peripheral and autonomic nerves | Brain, retina, blood vessels, kidneys, peripheral and autonomic nerves | Brain satiety centers with increased proneness to obesity |
Major consequences of diabetes include impaired glucose utilization, energy metabolism, alterations in the lipidome, inflammation, and oxidative stress. At the core of this complex dysregulated metabolic disease state is the inability of cells, tissues and organs to utilize insulin signaling networks. In Type 1 diabetes, the primary problem stems from reduced insulin production. In Type 2 diabetes, insulin production is abundant, but the receptors have reduced responsiveness (insulin resistance). In Type 3 diabetes, the brain undergoes neurodegeneration due to combined effects of insulin deficiency and insulin resistance. The structures targeted by Types 1 and 2 diabetes are nearly identical. However, in Type 3 diabetes, the brain, including neurons, white matter and micro-vessels are the main targets, but Type 3 diabetes is driven by most factors that lead to or result from Types 1 and 2 diabetes.
Going forward, it is important to recognize that the severities of cognitive impairment and neurodegeneration which accompany peripheral insulin resistance diseases including Type 2 diabetes tend to correspond to MCI or early AD, whereas in Type 3 diabetes neurodegeneration progresses to end-stage dementia. However, another way to conceptualize the problem is to regard insulin resistance diseases as a single pathophysiological process that can afflict one or multiple organs and tissues in the same way that atherosclerosis can target one or more vessels and produce distinct manifestations of disease. Atherosclerotic damage to carotid vessels produces CNS deficits, whereas coronary atherosclerosis correlates with myocardial ischemic injury and infarction and renal artery atherosclerosis leads to kidney disease, yet we regard the underlying pathologies as the same and largely mediated by hypertension and diabetes mellitus. Therefore, it would not be exceptional to suggest that insulin resistance diseases can preferentially attack different organ systems, either singularly or in combination, and with dominant or minor effects on function. Type 3 diabetes is the case in which insulin resistance disease dominantly attacks the brain and has mainly minor effects on other organs, whereas Type 2 diabetes, metabolic syndrome, and non-alcoholic steatohepatitis represent peripheral insulin resistance diseases that have variable to modest adverse effects on CNS function.
Broadening the Therapeutic Options for AD
Nearly all pathologies in AD, including Aβ1-42 deposits, phospho-tau containing, paired helical filament-associated pathologies, and the broader array of disease processes linked to metabolic dysfunction, neuroinflammation, cellular stress, synaptic disconnection, aberrant proliferation of dystrophic neurites (reflecting loss of neuronal plasticity), cell death, white matter atrophy and degeneration, and microvascular disease, could be attributed to impairments in brain insulin and IGF signaling. Therefore, going forward, therapeutic interventions should include measures that increase insulin supply, insulin/IGF receptor responsiveness, downstream signaling through IRS, and appropriate modulation of target gene expression. Therefore, therapeutic interventions for AD should be multi-pronged to address all components of neurodegeneration at various stages of AD rather than focused only on reducing burdens of Aβ1–42 and pTau. Potential strategies for therapeutic targeting of brain metabolic dysfunction and associated or cofactor pathologies are summarized below.
Lifestyle modifications:
Although proven effective for restoring and preserving insulin sensitivity, implementing healthful lifestyle changes through diet and physical activity remains challenging because they are difficult to maintain. In addition, it has been difficult to assess the full benefits of caloric restriction and physical activity on long-term cognitive function. A third concern is that lifestyle measures may be ineffective once neurodegeneration advances to intermediate stages or if systemic insulin resistance diseases cannot be controlled. Most likely, lifestyle interventions will be coupled with medical interventions to more effectively halt progression of neurodegeneration.
Anti-inflammatory/anti-oxidant treatments:
Neuroinflammation and cellular stress (oxidative, nitrosative, and endoplasmic reticulum) are associated with broad activation of cytokines and chemokines (de la Monte et al., 2017a, Ott et al., 2018) that are largely generated in microglia and astrocytes (Dickson et al., 1993, Mrak et al., 1995, Mrak, 2009, Agostinho et al., 2010), or from systemic sources (de la Monte et al., 2017a, Ott et al., 2018). Furthermore, neuroinflammation and cellular stress are linked to major AD-associated neuropathological lesions, including Aβ1–42 deposits and PHF-associated neurofibrillary tangles, dystrophic neurites, tau hyper-phosphorylation, cell death, and loss of synaptic terminals (Du Yan et al., 1997, Agostinho et al., 2010, de la Monte and Tong, 2014). However, clinical trials have failed to demonstrate that anti-inflammatory and anti-oxidant agents alone can remediate cognitive impairment and neurodegeneration, suggesting that neuroinflammation has a cofactor rather than causal role in AD progression.
Insulins, Incretins, and Beyond:
Insulin has positive effects on neurocognitive function in people with MCI or early-stage AD (de la Monte, 2017). Mechanistically, insulin stimulates glucose utilization in the brain (Jauch-Chara et al., 2012, Reger et al., 2008, Stockhorst et al., 2004, Talbot et al., 2012). Nasal administration of long-acting or ultralong-acting insulins has been shown to benefit subsets of MCI or AD patients (de la Monte, 2017). Nasal delivery of insulin penetrates the blood-brain barrier enabling CNS targeting while minimizing risk of hypoglycemia (de la Monte, 2012b). Incretins, particularly long-acting synthetic analogues, are attractive alternatives to insulin because they stimulate insulin secretion(Yamamoto et al., 2003, Freeman, 2009, Irwin et al., 2010) and can be administered 1 or 2 times per week and stability of long-acting forms ensures predictable levels of insulin release (de la Monte, 2017). Furthermore, using experimental models of AD, investigators have shown that incretins can clear a broad range of AD pathologies, including neuroinflammation, Aβ accumulations and dysregulated lipid metabolism (Duffy and Holscher, 2013, Holscher, 2014, McClean and Holscher, 2014). However, the insulinotropic effects of incretins can decline over time, possibly due to exhaustion or resistance of insulin producing cells (Meier and Nauck, 2010).
Insulin Sensitizers:
Peroxisome Proliferator-Activated Receptor (PPAR) agonists offer potentially excellent opportunities for treating insulin resistance diseases in general, and brain diabetes specifically, because insulin resistance worsens as disease progresses. Insulin resistance begins at the cell surface receptor which is problematic because eventually insulin therapy will have reduced effectiveness. However, PPAR agonists target nuclear hormone receptors to broadly activate target genes. Moreover, PPAR agonists can activate both insulin and IGF-1 regulated pathways and suppress neuroinflammation (de la Monte et al., 2017b). Although several studies using PPAR-γ agonists provided mixed results, future treatment strategies should consider the need to target PPAR-δ, which in the CNS is far more abundantly expressed than PPAR-γ (de la Monte and Wands, 2005). Experimental data generated in AD models treated with PPAR-δ or hybrid PPAR-δ/γ agonists strongly support the conclusion that disease remediation can be achieved by proper targeting of PPAR subtypes in the brain (de la Monte et al., 2006, de la Monte et al., 2017b).
Conclusions
AD should be regarded as a form of brain metabolic dysfunction in which insulin resistance and deficiency develop primarily in the brain (Type 3 Diabetes). However, similar but generally less severe forms of AD-type neurodegeneration often accompany systemic insulin resistance diseases such as Type 2 diabetes mellitus, metabolic syndrome, and non-alcoholic fatty liver disease. Given the recent striking increases in rates of AD, Type 2 diabetes, metabolic syndrome, obesity, and non-alcoholic fatty liver disease and growing evidence that people with systemic insulin resistance diseases have significantly higher risks of developing cognitive impairment, it is likely -alcoholic fatty liver disease that the underlying factors causing these diseases are either identical or closely related (Figure 11). These concepts suggest that effective treatment and preventive strategies for insulin resistance diseases, including AD, could substantially overlap with one another, although tissue/organ-specific targeting of pharmaceuticals may be required to optimize outcomes.
Figure 11:
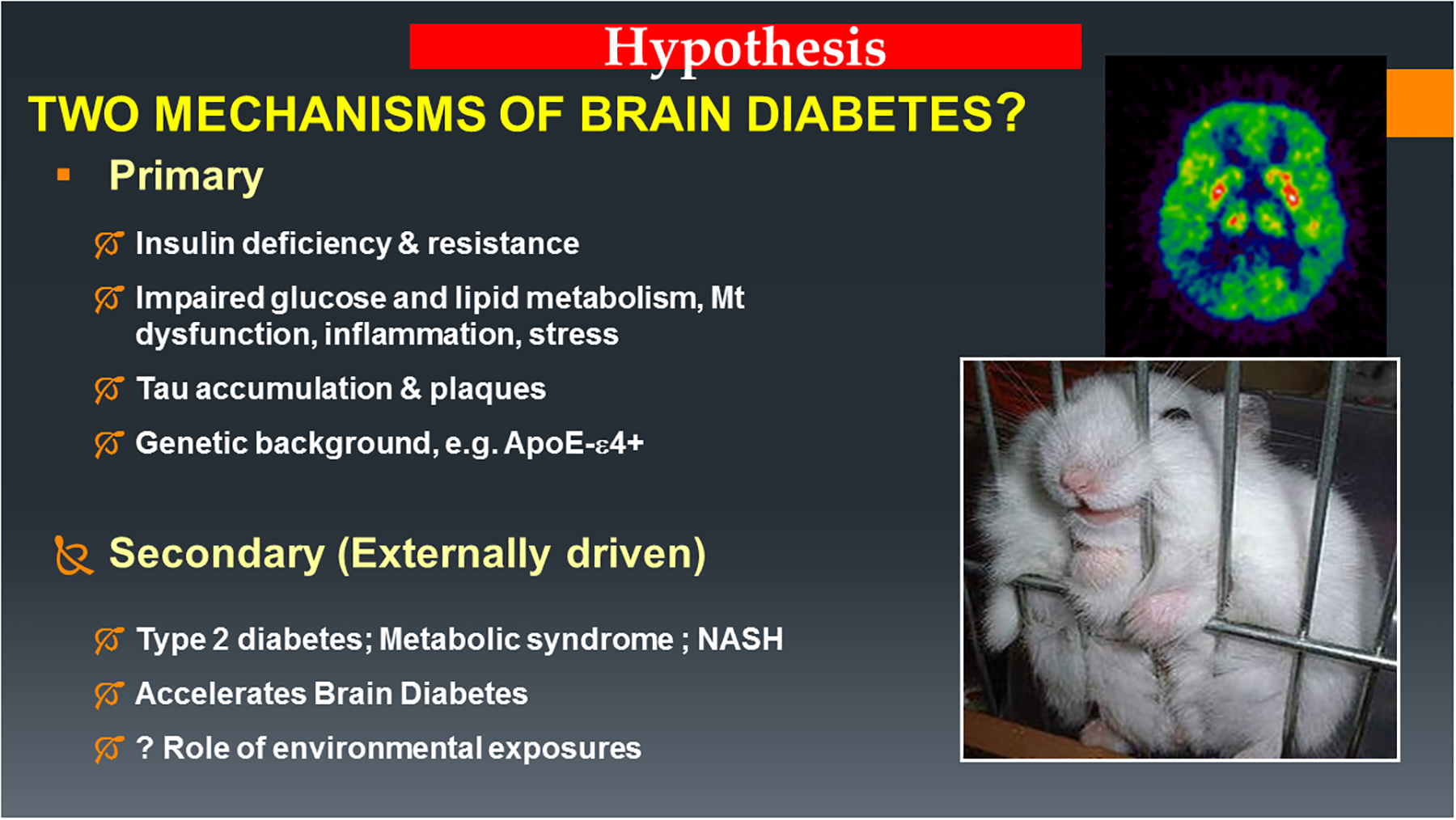
Two proposed mechanisms of brain insulin resistance and cognitive impairment. In the Primary Disease Model, brain insulin resistance is an early dominant and progressive metabolic abnormality that leads to end-stage AD dementia and usually occurs independent of clinically manifested systemic insulin resistance diseases. In the Secondary (extrinsic factor) Disease Model, insulin resistance mainly targets peripheral organ systems and has modest impact on the brain compared with primary brain insulin resistance, and results in mild cognitive impairment or early to moderate-stage AD. However, secondary brain insulin resistance can exacerbate underlying primary brain insulin resistance and accelerate the course of AD. Growing evidence indicates systemic/secondary brain insulin resistance diseases are linked to obesogenic diets and sedentary lifestyles. In addition, experimental data highlight the role of chronic low-dose nitrosamine exposures as potential mediators of the array of insulin resistance diseases (de la Monte et al., 2009c, Tong et al., 2010, de la Monte et al., 2011b, Yalcin et al., 2015, Lester-Coll et al., 2006).
Acknowledgments
Supported by AA-11431 from the National Institutes of Health
REFERENCES
- AGOSTINHO P, CUNHA RA & OLIVEIRA C 2010. Neuroinflammation, oxidative stress and the pathogenesis of Alzheimer’s disease. Curr Pharm Des, 16, 2766–78. [DOI] [PubMed] [Google Scholar]
- ALIEV G, SMITH MA, OBRENOVICH ME, DE LA TORRE JC & PERRY G 2003. Role of vascular hypoperfusion-induced oxidative stress and mitochondria failure in the pathogenesis of Azheimer disease. Neurotox Res, 5, 491–504. [DOI] [PubMed] [Google Scholar]
- ALOSCO ML & GUNSTAD J 2014. The negative effects of obesity and poor glycemic control on cognitive function: a proposed model for possible mechanisms. Curr Diab Rep, 14, 495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- AMLIEN IK & FJELL AM 2014. Diffusion tensor imaging of white matter degeneration in Alzheimer’s disease and mild cognitive impairment. Neuroscience, 276, 206–15. [DOI] [PubMed] [Google Scholar]
- BALLARD C, MCKEITH I, O’BRIEN J, KALARIA R, JAROS E, INCE P & PERRY R 2000. Neuropathological substrates of dementia and depression in vascular dementia, with a particular focus on cases with small infarct volumes. Dement Geriatr Cogn Disord, 11, 59–65. [DOI] [PubMed] [Google Scholar]
- BARRES BA, JACOBSON MD, SCHMID R, SENDTNER M & RAFF MC 1993. Does oligodendrocyte survival depend on axons? Curr Biol, 3, 489–97. [DOI] [PubMed] [Google Scholar]
- BARRIO JR, KEPE V, SATYAMURTHY N, HUANG SC & SMALL G 2008. Amyloid and tau imaging, neuronal losses and function in mild cognitive impairment. J Nutr Health Aging, 12, 61S–5S. [DOI] [PubMed] [Google Scholar]
- BARRIO JR, SMALL GW, WONG KP, HUANG SC, LIU J, MERRILL DA, GIZA CC, FITZSIMMONS RP, OMALU B, BAILES J & KEPE V 2015. In vivo characterization of chronic traumatic encephalopathy using [F-18]FDDNP PET brain imaging. Proc Natl Acad Sci U S A, 112, E2039–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BARROSO E, DEL VALLE J, PORQUET D, VIEIRA SANTOS AM, SALVADO L, RODRIGUEZ-RODRIGUEZ R, GUTIERREZ P, ANGLADA-HUGUET M, ALBERCH J, CAMINS A, PALOMER X, PALLAS M, MICHALIK L, WAHLI W & VAZQUEZ-CARRERA M 2013. Tau hyperphosphorylation and increased BACE1 and RAGE levels in the cortex of PPARbeta/delta-null mice. Biochim Biophys Acta, 1832, 1241–8. [DOI] [PubMed] [Google Scholar]
- BENEDICT C, FREY WH 2ND, SCHIOTH HB, SCHULTES B, BORN J & HALLSCHMID M 2011. Intranasal insulin as a therapeutic option in the treatment of cognitive impairments. Exp Gerontol, 46, 112–5. [DOI] [PubMed] [Google Scholar]
- BERTI V, PUPI A & MOSCONI L 2011. PET/CT in diagnosis of dementia. Ann N Y Acad Sci, 1228, 81–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BEYDOUN MA, BEYDOUN HA, GAMALDO AA, TEEL A, ZONDERMAN AB & WANG Y 2014. Epidemiologic studies of modifiable factors associated with cognition and dementia: systematic review and meta-analysis. BMC Public Health, 14, 643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BIRCH AM 2014. The contribution of astrocytes to Alzheimer’s disease. Biochem Soc Trans, 42, 1316–20. [DOI] [PubMed] [Google Scholar]
- BLENNOW K, DUBOIS B, FAGAN AM, LEWCZUK P, DE LEON MJ & HAMPEL H 2015a. Clinical utility of cerebrospinal fluid biomarkers in the diagnosis of early Alzheimer’s disease. Alzheimers Dement, 11, 58–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BLENNOW K, MATTSSON N, SCHOLL M, HANSSON O & ZETTERBERG H 2015b. Amyloid biomarkers in Alzheimer’s disease. Trends Pharmacol Sci, 36, 297–309. [DOI] [PubMed] [Google Scholar]
- BRAAK H, BRAAK E, GRUNDKE-IQBAL I & IQBAL K 1986. Occurrence of neuropil threads in the senile human brain and in Alzheimer’s disease: a third location of paired helical filaments outside of neurofibrillary tangles and neuritic plaques. Neurosci Lett, 65, 351–5. [DOI] [PubMed] [Google Scholar]
- BRASKIE MN, KLUNDER AD, HAYASHI KM, PROTAS H, KEPE V, MILLER KJ, HUANG SC, BARRIO JR, ERCOLI LM, SIDDARTH P, SATYAMURTHY N, LIU J, TOGA AW, BOOKHEIMER SY, SMALL GW & THOMPSON PM 2010. Plaque and tangle imaging and cognition in normal aging and Alzheimer’s disease. Neurobiol Aging, 31, 1669–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BRICKMAN AM, HONIG LS, SCARMEAS N, TATARINA O, SANDERS L, ALBERT MS, BRANDT J, BLACKER D & STERN Y 2008. Measuring cerebral atrophy and white matter hyperintensity burden to predict the rate of cognitive decline in Alzheimer disease. Arch Neurol, 65, 1202–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BRIDGES LR, ANDOH J, LAWRENCE AJ, KHOONG CH, POON WW, ESIRI MM, MARKUS HS & HAINSWORTH AH 2014. Blood-brain barrier dysfunction and cerebral small vessel disease (arteriolosclerosis) in brains of older people. J Neuropathol Exp Neurol, 73, 1026–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BRILLIANT M, HUGHES L, ANDERSON D, GHOBRIAL M & ELBLE R 1995. Rarefied white matter in patients with Alzheimer disease. Alzheimer Dis Assoc Disord, 9, 39–46. [DOI] [PubMed] [Google Scholar]
- BRUN A & ENGLUND E 1986. A white matter disorder in dementia of the Alzheimer type: a pathoanatomical study. Ann Neurol, 19, 253–62. [DOI] [PubMed] [Google Scholar]
- BRUN A, LIU X & ERIKSON C 1995. Synapse loss and gliosis in the molecular layer of the cerebral cortex in Alzheimer’s disease and in frontal lobe degeneration. Neurodegeneration, 4, 171–7. [DOI] [PubMed] [Google Scholar]
- BURNS JM, CHURCH JA, JOHNSON DK, XIONG C, MARCUS D, FOTENOS AF, SNYDER AZ, MORRIS JC & BUCKNER RL 2005. White matter lesions are prevalent but differentially related with cognition in aging and early Alzheimer disease. Arch Neurol, 62, 1870–6. [DOI] [PubMed] [Google Scholar]
- BUTTINI M, MASLIAH E, BARBOUR R, GRAJEDA H, MOTTER R, JOHNSON-WOOD K, KHAN K, SEUBERT P, FREEDMAN S, SCHENK D & GAMES D 2005. Beta-amyloid immunotherapy prevents synaptic degeneration in a mouse model of Alzheimer’s disease. J Neurosci, 25, 9096–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CARSON MJ, BEHRINGER RR, BRINSTER RL & MCMORRIS FA 1993. Insulin-like growth factor I increases brain growth and central nervous system myelination in transgenic mice. Neuron, 10, 729–40. [DOI] [PubMed] [Google Scholar]
- CHALMERS K, WILCOCK G & LOVE S 2005. Contributors to white matter damage in the frontal lobe in Alzheimer’s disease. Neuropathol Appl Neurobiol, 31, 623–31. [DOI] [PubMed] [Google Scholar]
- CHANDRA A 2015. Role of amyloid from a multiple sclerosis perspective: a literature review. Neuroimmunomodulation, 22, 343–6. [DOI] [PubMed] [Google Scholar]
- CHESIK D, DE KEYSER J & WILCZAK N 2008. Insulin-like growth factor system regulates oligodendroglial cell behavior: therapeutic potential in CNS. J Mol Neurosci, 35, 81–90. [DOI] [PubMed] [Google Scholar]
- CHETELAT G, LA JOIE R, VILLAIN N, PERROTIN A, DE LA SAYETTE V, EUSTACHE F & VANDENBERGHE R 2013. Amyloid imaging in cognitively normal individuals, at-risk populations and preclinical Alzheimer’s disease. Neuroimage Clin, 2, 356–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHOLERTON B, BAKER LD & CRAFT S 2011. Insulin resistance and pathological brain ageing. Diabet Med, 28, 1463–75. [DOI] [PubMed] [Google Scholar]
- COLLINO M, PATEL NS & THIEMERMANN C 2008. PPARs as new therapeutic targets for the treatment of cerebral ischemia/reperfusion injury. Ther Adv Cardiovasc Dis, 2, 179–97. [DOI] [PubMed] [Google Scholar]
- COREY-BLOOM J, TIRABOSCHI P, HANSEN LA, ALFORD M, SCHOOS B, SABBAGH MN, MASLIAH E & THAL LJ 2000. E4 allele dosage does not predict cholinergic activity or synapse loss in Alzheimer’s disease. Neurology, 54, 403–6. [DOI] [PubMed] [Google Scholar]
- CSELENYI Z, JONHAGEN ME, FORSBERG A, HALLDIN C, JULIN P, SCHOU M, JOHNSTROM P, VARNAS K, SVENSSON S & FARDE L 2012. Clinical validation of 18F-AZD4694, an amyloid-beta-specific PET radioligand. J Nucl Med, 53, 415–24. [DOI] [PubMed] [Google Scholar]
- CUMMINGS BJ, SU JH & COTMAN CW 1993. Neuritic involvement within bFGF immunopositive plaques of Alzheimer’s disease. Exp Neurol, 124, 315–25. [DOI] [PubMed] [Google Scholar]
- CZUBOWICZ K & STROSZNAJDER R 2014. Ceramide in the molecular mechanisms of neuronal cell death. The role of sphingosine-1-phosphate. Mol Neurobiol, 50, 26–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DALY RM, GIANOUDIS J, PROSSER M, KIDGELL D, ELLIS KA, O’CONNELL S & NOWSON CA 2015. The effects of a protein enriched diet with lean red meat combined with a multi-modal exercise program on muscle and cognitive health and function in older adults: study protocol for a randomised controlled trial. Trials, 16, 339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DANDREA MR, REISER PA, GUMULA NA, HERTZOG BM & ANDRADE-GORDON P 2001. Application of triple immunohistochemistry to characterize amyloid plaque-associated inflammation in brains with Alzheimer’s disease. Biotech Histochem, 76, 97–106. [PubMed] [Google Scholar]
- DAULATZAI MA 2012. Quintessential risk factors: their role in promoting cognitive dysfunction and Alzheimer’s disease. Neurochem Res, 37, 2627–58. [DOI] [PubMed] [Google Scholar]
- DAULATZAI MA 2017. Cerebral hypoperfusion and glucose hypometabolism: Key pathophysiological modulators promote neurodegeneration, cognitive impairment, and Alzheimer’s disease. J Neurosci Res, 95, 943–972. [DOI] [PubMed] [Google Scholar]
- DAVIGLUS ML, BELL CC, BERRETTINI W, BOWEN PE, CONNOLLY ES JR., COX NJ, DUNBAR-JACOB JM, GRANIERI EC, HUNT G, MCGARRY K, PATEL D, POTOSKY AL, SANDERS-BUSH E, SILBERBERG D & TREVISAN M 2010. NIH state-of-the-science conference statement: Preventing Alzheimer’s disease and cognitive decline. NIH Consens State Sci Statements, 27, 1–30. [PubMed] [Google Scholar]
- DE LA MONTE SM 1989. Quantitation of cerebral atrophy in preclinical and end-stage Alzheimer’s disease. Ann Neurol, 25, 450–9. [DOI] [PubMed] [Google Scholar]
- DE LA MONTE SM 2012a. Brain insulin resistance and deficiency as therapeutic targets in Alzheimer’s disease. Current Alzheimer Research, 9, 35–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DE LA MONTE SM 2012b. Contributions of brain insulin resistance and deficiency in amyloid-related neurodegeneration in Alzheimer’s disease. Drugs, 72, 49–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DE LA MONTE SM 2012c. Metabolic derangements mediate cognitive impairment and Alzheimer’s disease: role of peripheral insulin-resistance diseases. Panminerva Med, 54, 171–8. [PMC free article] [PubMed] [Google Scholar]
- DE LA MONTE SM 2013. Intranasal insulin therapy for cognitive impairment and neurodegeneration: current state of the art. Expert Opin Drug Deliv, 10, 1699–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DE LA MONTE SM 2014. Relationships between diabetes and cognitive impairment. Endocrinol Metab Clin North Am, 43, 245–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DE LA MONTE SM 2017. Insulin Resistance and Neurodegeneration: Progress Towards the Development of New Therapeutics for Alzheimer’s Disease. Drugs, 77, 47–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DE LA MONTE SM, DAIELLO LA, HAPEL AJ, TONG M & OTT BR 2017a. Altered Serum and Cerebrospinal Fluid Inflammatory Cascades in Mild Cognitive Impairment and Alzheimer’s Disease. Article ID: 100004. J Neuroinflam Neurodegen, 1, 1–24. [Google Scholar]
- DE LA MONTE SM, JHAVERI A, MARON BA & WANDS JR 2007. Nitric oxide synthase 3-mediated neurodegeneration after intracerebral gene delivery. J Neuropathol Exp Neurol, 66, 272–83. [DOI] [PubMed] [Google Scholar]
- DE LA MONTE SM, LONGATO L, TONG M & WANDS JR 2009a. Insulin resistance and neurodegeneration: roles of obesity, type 2 diabetes mellitus and non-alcoholic steatohepatitis. Curr Opin Investig Drugs, 10, 1049–60. [PMC free article] [PubMed] [Google Scholar]
- DE LA MONTE SM, NEUSNER A, CHU J & LAWTON M 2009b. Epidemiological Trends Strongly Suggest Exposures as Etiologic Agents in the Pathogenesis of Sporadic Alzheimer’s Disease, Diabetes Mellitus, and Non-Alcoholic Steatohepatitis. J Alzheimers Dis, 17, 519–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DE LA MONTE SM, SOHN YK, ETIENNE D, KRAFT J & WANDS JR 2000. Role of aberrant nitric oxide synthase-3 expression in cerebrovascular degeneration and vascular-mediated injury in Alzheimer’s disease. Ann N Y Acad Sci, 903, 61–71. [DOI] [PubMed] [Google Scholar]
- DE LA MONTE SM & TONG M 2014. Brain metabolic dysfunction at the core of Alzheimer’s disease. Biochem Pharmacol, 88, 548–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DE LA MONTE SM, TONG M, BOWLING N & MOSKAL P 2011a. si-RNA inhibition of brain insulin or insulin-like growth factor receptors causes developmental cerebellar abnormalities: relevance to fetal alcohol spectrum disorder. Molecular Brain, 4, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DE LA MONTE SM, TONG M, LAWTON M & LONGATO L 2009c. Nitrosamine exposure exacerbates high fat diet-mediated type 2 diabetes mellitus, non-alcoholic steatohepatitis, and neurodegeneration with cognitive impairment. Mol Neurodegener, 4, 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DE LA MONTE SM, TONG M, LESTER-COLL N, PLATER M JR. & WANDS JR 2006. Therapeutic rescue of neurodegeneration in experimental type 3 diabetes: relevance to Alzheimer’s disease. J Alzheimers Dis, 10, 89–109. [DOI] [PubMed] [Google Scholar]
- DE LA MONTE SM, TONG M, SCHIANO I & DIDSBURY J 2017b. Improved Brain Insulin/IGF Signaling and Reduced Neuroinflammation with T3D-959 in an Experimental Model of Sporadic Alzheimer’s Disease. J Alzheimers Dis, 55, 849–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DE LA MONTE SM, TONG M & WANDS JR 2011b. Insulin resistance, cognitive impairment, and neurodegeneration: roles of nitrosamine exposure, diet, and lifestyles. InTech, 20, 1–39. [Google Scholar]
- DE LA MONTE SM & WANDS JR 2005. Review of insulin and insulin-like growth factor expression, signaling, and malfunction in the central nervous system: relevance to Alzheimer’s disease. J Alzheimers Dis, 7, 45–61. [DOI] [PubMed] [Google Scholar]
- DE LA MONTE SM & WANDS JR 2006. Molecular indices of oxidative stress and mitochondrial dysfunction occur early and often progress with severity of Alzheimer’s disease. J Alzheimers Dis, 9, 167–81. [DOI] [PubMed] [Google Scholar]
- DE LA MONTE SM & WANDS JR 2008. Alzheimer’s disease is type 3 diabetes: Evidence reviewed. J Diabetes Sci Technol, 2, 1101–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DE LA TORRE JC & MUSSIVAND T 1993. Can disturbed brain microcirculation cause Alzheimer’s disease? Neurol Res, 15, 146–53. [DOI] [PubMed] [Google Scholar]
- DE LA TORRE JC & STEFANO GB 2000. Evidence that Alzheimer’s disease is a microvascular disorder: the role of constitutive nitric oxide. Brain Res Brain Res Rev, 34, 119–36. [DOI] [PubMed] [Google Scholar]
- DE LEON MJ, GEORGE AE, FERRIS SH, ROSENBLOOM S, CHRISTMAN DR, GENTES CI, REISBERG B, KRICHEFF II & WOLF AP 1983. Regional correlation of PET and CT in senile dementia of the Alzheimer type. AJNR Am J Neuroradiol, 4, 553–6. [PMC free article] [PubMed] [Google Scholar]
- DEANE R, DU YAN S, SUBMAMARYAN RK, LARUE B, JOVANOVIC S, HOGG E, WELCH D, MANNESS L, LIN C, YU J, ZHU H, GHISO J, FRANGIONE B, STERN A, SCHMIDT AM, ARMSTRONG DL, ARNOLD B, LILIENSIEK B, NAWROTH P, HOFMAN F, KINDY M, STERN D & ZLOKOVIC B 2003. RAGE mediates amyloid-beta peptide transport across the blood-brain barrier and accumulation in brain. Nat Med, 9, 907–13. [DOI] [PubMed] [Google Scholar]
- DECLERCQ L, CELEN S, LECINA J, AHAMED M, TOUSSEYN T, MOECHARS D, ALCAZAR J, ARIZA M, FIERENS K, BOTTELBERGS A, MARIEN J, VANDENBERGHE R, ANDRES IJ, VAN LAERE K, VERBRUGGEN A & BORMANS G 2016. Comparison of New Tau PET-Tracer Candidates With [18F]T808 and [18F]T807. Mol Imaging, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DICKSON DW, LEE SC, MATTIACE LA, YEN SH & BROSNAN C 1993. Microglia and cytokines in neurological disease, with special reference to AIDS and Alzheimer’s disease. Glia, 7, 75–83. [DOI] [PubMed] [Google Scholar]
- DONAHUE JE, FLAHERTY SL, JOHANSON CE, DUNCAN JA 3RD, SILVERBERG GD, MILLER MC, TAVARES R, YANG W, WU Q, SABO E, HOVANESIAN V & STOPA EG 2006. RAGE, LRP-1, and amyloid-beta protein in Alzheimer’s disease. Acta Neuropathol, 112, 405–15. [DOI] [PubMed] [Google Scholar]
- DOUBLE KL, HALLIDAY GM, KRIL JJ, HARASTY JA, CULLEN K, BROOKS WS, CREASEY H & BROE GA 1996. Topography of brain atrophy during normal aging and Alzheimer’s disease. Neurobiol Aging, 17, 513–21. [DOI] [PubMed] [Google Scholar]
- DRAB SR 2009. Recognizing the rising impact of diabetes in seniors and implications for its management. Consult Pharm, 24 Suppl B, 5–10. [PubMed] [Google Scholar]
- DU YAN S, ZHU H, FU J, YAN SF, ROHER A, TOURTELLOTTE WW, RAJAVASHISTH T, CHEN X, GODMAN GC, STERN D & SCHMIDT AM 1997. Amyloid-beta peptide-receptor for advanced glycation endproduct interaction elicits neuronal expression of macrophage-colony stimulating factor: a proinflammatory pathway in Alzheimer disease. Proc Natl Acad Sci U S A, 94, 5296–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DUFFY AM & HOLSCHER C 2013. The incretin analogue D-Ala2GIP reduces plaque load, astrogliosis and oxidative stress in an APP/PS1 mouse model of Alzheimer’s disease. Neuroscience, 228, 294–300. [DOI] [PubMed] [Google Scholar]
- DUNCAN GW, FIRBANK MJ, O’BRIEN JT & BURN DJ 2013. Magnetic resonance imaging: a biomarker for cognitive impairment in Parkinson’s disease? Mov Disord, 28, 425–38. [DOI] [PubMed] [Google Scholar]
- DUNN SE, BHAT R, STRAUS DS, SOBEL RA, AXTELL R, JOHNSON A, NGUYEN K, MUKUNDAN L, MOSHKOVA M, DUGAS JC, CHAWLA A & STEINMAN L 2010. Peroxisome proliferator-activated receptor delta limits the expansion of pathogenic Th cells during central nervous system autoimmunity. J Exp Med, 207, 1599–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ELEUTERI S, DI GIOVANNI S, ROCKENSTEIN E, MANTE M, ADAME A, TREJO M, WRASIDLO W, WU F, FRAERING PC, MASLIAH E & LASHUEL HA 2015. Novel therapeutic strategy for neurodegeneration by blocking Abeta seeding mediated aggregation in models of Alzheimer’s disease. Neurobiol Dis, 74, 144–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ENGLER H, SANTILLO AF, WANG SX, LINDAU M, SAVITCHEVA I, NORDBERG A, LANNFELT L, LANGSTROM B & KILANDER L 2008. In vivo amyloid imaging with PET in frontotemporal dementia. Eur J Nucl Med Mol Imaging, 35, 100–6. [DOI] [PubMed] [Google Scholar]
- ENGLUND E 1998. Neuropathology of white matter changes in Alzheimer’s disease and vascular dementia. Dement Geriatr Cogn Disord, 9 Suppl 1, 6–12. [DOI] [PubMed] [Google Scholar]
- ERVIN JF, PANNELL C, SZYMANSKI M, WELSH-BOHMER K, SCHMECHEL DE & HULETTE CM 2004. Vascular smooth muscle actin is reduced in Alzheimer disease brain: a quantitative analysis. J Neuropathol Exp Neurol, 63, 735–41. [DOI] [PubMed] [Google Scholar]
- ETIENE D, KRAFT J, GANJU N, GOMEZ-ISLA T, GEMELLI B, HYMAN BT, HEDLEY-WHYTE ET, WANDS JR & DE LA MONTE SM 1998. Cerebrovascular pathology contributes to the heterogeneity of Alzheimer’s Disease. J Alzheimers Dis, 1, 119–134. [DOI] [PubMed] [Google Scholar]
- EWERS M, CHENG X, ZHONG Z, NURAL HF, WALSH C, MEINDL T, TEIPEL SJ, BUERGER K, HE P, SHEN Y & HAMPEL H 2011. Increased CSF-BACE1 activity associated with decreased hippocampus volume in Alzheimer’s disease. J Alzheimers Dis, 25, 373–81. [DOI] [PubMed] [Google Scholar]
- FARKAS E, DE JONG GI, APRO E, DE VOS RA, STEUR EN & LUITEN PG 2000. Similar ultrastructural breakdown of cerebrocortical capillaries in Alzheimer’s disease, Parkinson’s disease, and experimental hypertension. What is the functional link? Ann N Y Acad Sci, 903, 72–82. [DOI] [PubMed] [Google Scholar]
- FAULSTICH ME 1991. Brain imaging in dementia of the Alzheimer type. Int J Neurosci, 57, 39–49. [DOI] [PubMed] [Google Scholar]
- FERRER I, BELLA R, SERRANO MT, MARTI E & GUIONNET N 1990. Arteriolosclerotic leucoencephalopathy in the elderly and its relation to white matter lesions in Binswanger’s disease, multi-infarct encephalopathy and Alzheimer’s disease. J Neurol Sci, 98, 37–50. [DOI] [PubMed] [Google Scholar]
- FLEISHER AS, CHEN K, QUIROZ YT, JAKIMOVICH LJ, GOMEZ MG, LANGOIS CM, LANGBAUM JB, AYUTYANONT N, ROONTIVA A, THIYYAGURA P, LEE W, MO H, LOPEZ L, MORENO S, ACOSTA-BAENA N, GIRALDO M, GARCIA G, REIMAN RA, HUENTELMAN MJ, KOSIK KS, TARIOT PN, LOPERA F & REIMAN EM 2012. Florbetapir PET analysis of amyloid-beta deposition in the presenilin 1 E280A autosomal dominant Alzheimer’s disease kindred: a cross-sectional study. Lancet Neurol, 11, 1057–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FOTUHI M, DO D & JACK C 2012. Modifiable factors that alter the size of the hippocampus with ageing. Nat Rev Neurol, 8, 189–202. [DOI] [PubMed] [Google Scholar]
- FREEMAN JS 2009. Role of the incretin pathway in the pathogenesis of type 2 diabetes mellitus. Cleve Clin J Med, 76 Suppl 5, S12–9. [DOI] [PubMed] [Google Scholar]
- FRISARDI V, SOLFRIZZI V, SERIPA D, CAPURSO C, SANTAMATO A, SANCARLO D, VENDEMIALE G, PILOTTO A & PANZA F 2010. Metabolic-cognitive syndrome: a cross-talk between metabolic syndrome and Alzheimer’s disease. Ageing Res Rev, 9, 399–417. [DOI] [PubMed] [Google Scholar]
- GARWOOD CJ, RATCLIFFE LE, SIMPSON JE, HEATH PR, INCE PG & WHARTON SB 2016. Review: Astrocytes in Alzheimer’s disease and other age-associated dementias; a supporting player with a central role. Neuropathol Appl Neurobiol. [DOI] [PubMed] [Google Scholar]
- GASPAR JM, BAPTISTA FI, MACEDO MP & AMBROSIO AF 2016. Inside the Diabetic Brain: Role of Different Players Involved in Cognitive Decline. ACS Chem Neurosci, 7, 131–42. [DOI] [PubMed] [Google Scholar]
- GIOVANNINI MG, SCALI C, PROSPERI C, BELLUCCI A, VANNUCCHI MG, ROSI S, PEPEU G & CASAMENTI F 2002. Beta-amyloid-induced inflammation and cholinergic hypofunction in the rat brain in vivo: involvement of the p38MAPK pathway. Neurobiol Dis, 11, 257–74. [DOI] [PubMed] [Google Scholar]
- GONZALEZ-LIMA F, BARKSDALE BR & ROJAS JC 2014. Mitochondrial respiration as a target for neuroprotection and cognitive enhancement. Biochem Pharmacol, 88, 584–93. [DOI] [PubMed] [Google Scholar]
- GOTZ ME, KUNIG G, RIEDERER P & YOUDIM MB 1994. Oxidative stress: free radical production in neural degeneration. Pharmacol Ther, 63, 37–122. [DOI] [PubMed] [Google Scholar]
- GRAMMAS P 2011. Neurovascular dysfunction, inflammation and endothelial activation: implications for the pathogenesis of Alzheimer’s disease. J Neuroinflammation, 8, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GRAMMAS P, MARTINEZ J & MILLER B 2011. Cerebral microvascular endothelium and the pathogenesis of neurodegenerative diseases. Expert Rev Mol Med, 13, e19. [DOI] [PubMed] [Google Scholar]
- HAN X, D MH, MCKEEL DW JR., KELLEY J & MORRIS JC 2002. Substantial sulfatide deficiency and ceramide elevation in very early Alzheimer’s disease: potential role in disease pathogenesis. J Neurochem, 82, 809–18. [DOI] [PubMed] [Google Scholar]
- HOLSCHER C 2014. The incretin hormones glucagonlike peptide 1 and glucose-dependent insulinotropic polypeptide are neuroprotective in mouse models of Alzheimer’s disease. Alzheimers Dement, 10, S47–54. [DOI] [PubMed] [Google Scholar]
- HONG YT, VEENITH T, DEWAR D, OUTTRIM JG, MANI V, WILLIAMS C, PIMLOTT S, HUTCHINSON PJ, TAVARES A, CANALES R, MATHIS CA, KLUNK WE, AIGBIRHIO FI, COLES JP, BARON JC, PICKARD JD, FRYER TD, STEWART W & MENON DK 2014. Amyloid imaging with carbon 11-labeled Pittsburgh compound B for traumatic brain injury. JAMA Neurol, 71, 23–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HOYER S 1982. The abnormally aged brain. Its blood flow and oxidative metabolism. A review - part II. Arch Gerontol Geriatr, 1, 195–207. [DOI] [PubMed] [Google Scholar]
- HULETTE CM, ERVIN JF, EDMONDS Y, ANTOINE S, STEWART N, SZYMANSKI MH, HAYDEN KM, PIEPER CF, BURKE JR & WELSH-BOHMER KA 2009. Cerebrovascular smooth muscle actin is increased in nondemented subjects with frequent senile plaques at autopsy: implications for the pathogenesis of Alzheimer disease. J Neuropathol Exp Neurol, 68, 417–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HYMAN BT, PHELPS CH, BEACH TG, BIGIO EH, CAIRNS NJ, CARRILLO MC, DICKSON DW, DUYCKAERTS C, FROSCH MP, MASLIAH E, MIRRA SS, NELSON PT, SCHNEIDER JA, THAL DR, THIES B, TROJANOWSKI JQ, VINTERS HV & MONTINE TJ 2012. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimers Dement, 8, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IRWIN N, GAULT V & FLATT PR 2010. Therapeutic potential of the original incretin hormone glucose-dependent insulinotropic polypeptide: diabetes, obesity, osteoporosis and Alzheimer’s disease? Expert Opin Investig Drugs, 19, 1039–48. [DOI] [PubMed] [Google Scholar]
- JAUCH-CHARA K, FRIEDRICH A, REZMER M, MELCHERT UH, H GS-E, HALLSCHMID M & OLTMANNS KM 2012. Intranasal insulin suppresses food intake via enhancement of brain energy levels in humans. Diabetes, 61, 2261–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JELLINGER KA 2002. The pathology of ischemic-vascular dementia: an update. J Neurol Sci, 203–204, 153–7. [DOI] [PubMed] [Google Scholar]
- JELLINGER KA 2003. Neuropathological spectrum of synucleinopathies. Mov Disord, 18 Suppl 6, S2–12. [DOI] [PubMed] [Google Scholar]
- JIAO P, CHEN Q, SHAH S, DU J, TAO B, TZAMELI I, YAN W & XU H 2009. Obesity-related upregulation of monocyte chemotactic factors in adipocytes: involvement of nuclear factor-kappaB and c-Jun NH2-terminal kinase pathways. Diabetes, 58, 104–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JOACHIM CL & SELKOE DJ 1992. The seminal role of beta-amyloid in the pathogenesis of Alzheimer disease. Alzheimer Dis Assoc Disord, 6, 7–34. [DOI] [PubMed] [Google Scholar]
- JOHANSON CE, STOPA EG, DAIELLO LA, DE LA MONTE SM, KEANE M & OTT BR 2018. Disrupted Blood-CSF Barrier to Urea and Creatinine in Mild Cognitive Impairment and Alzheimer’s Disease. Journal of Alzheimer’s Disease & Parkinsonism, 08, 435. [Google Scholar]
- JONES RS & WALDMAN AD 2004. 1H-MRS evaluation of metabolism in Alzheimer’s disease and vascular dementia. Neurol Res, 26, 488–95. [DOI] [PubMed] [Google Scholar]
- KALARIA RN, AKINYEMI R & IHARA M 2012. Does vascular pathology contribute to Alzheimer changes? J Neurol Sci, 322, 141–7. [DOI] [PubMed] [Google Scholar]
- KALARIA RN & BALLARD C 1999. Overlap between pathology of Alzheimer disease and vascular dementia. Alzheimer Dis Assoc Disord, 13 Suppl 3, S115–23. [DOI] [PubMed] [Google Scholar]
- KALININ S, RICHARDSON JC & FEINSTEIN DL 2009. A PPARdelta agonist reduces amyloid burden and brain inflammation in a transgenic mouse model of Alzheimer’s disease. Curr Alzheimer Res, 6, 431–7. [DOI] [PubMed] [Google Scholar]
- KIDD PM 2005. Neurodegeneration from mitochondrial insufficiency: nutrients, stem cells, growth factors, and prospects for brain rebuilding using integrative management. Altern Med Rev, 10, 268–93. [PubMed] [Google Scholar]
- KIDD PM 2008. Alzheimer’s disease, amnestic mild cognitive impairment, and age-associated memory impairment: current understanding and progress toward integrative prevention. Altern Med Rev, 13, 85–115. [PubMed] [Google Scholar]
- KIM B & FELDMAN EL 2015. Insulin resistance as a key link for the increased risk of cognitive impairment in the metabolic syndrome. Exp Mol Med, 47, e149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LAM SM, WANG Y, DUAN X, WENK MR, KALARIA RN, CHEN CP, LAI MK & SHUI G 2014. Brain lipidomes of subcortical ischemic vascular dementia and mixed dementia. Neurobiol Aging, 35, 2369–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LANGA KM 2015. Is the risk of Alzheimer’s disease and dementia declining? Alzheimers Res Ther, 7, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEE S, TONG M, HANG S, DEOCHAND C & DE LA MONTE S 2013. CSF and Brain Indices of Insulin Resistance, Oxidative Stress and Neuro-Inflammation in Early versus Late Alzheimer’s Disease. J Alzheimers Dis Parkinsonism, 3, 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LESTER-COLL N, RIVERA EJ, SOSCIA SJ, DOIRON K, WANDS JR & DE LA MONTE SM 2006. Intracerebral streptozotocin model of type 3 diabetes: relevance to sporadic Alzheimer’s disease. J Alzheimers Dis, 9, 13–33. [DOI] [PubMed] [Google Scholar]
- LI X, SONG D & LENG SX 2015. Link between type 2 diabetes and Alzheimer’s disease: from epidemiology to mechanism and treatment. Clin Interv Aging, 10, 549–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIU X, ERIKSON C & BRUN A 1996. Cortical synaptic changes and gliosis in normal aging, Alzheimer’s disease and frontal lobe degeneration. Dementia, 7, 128–34. [DOI] [PubMed] [Google Scholar]
- LOVE S & MINERS JS 2015. White matter hypoperfusion and damage in dementia: post-mortem assessment. Brain Pathol, 25, 99–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LOVESTONE S & SMITH U 2014. Advanced glycation end products, dementia, and diabetes. Proc Natl Acad Sci U S A, 111, 4743–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LUCHSINGER JA 2012. Type 2 diabetes and cognitive impairment: linking mechanisms. J Alzheimers Dis, 30 Suppl 2, S185–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LUCHSINGER JA, REITZ C, PATEL B, TANG MX, MANLY JJ & MAYEUX R 2007. Relation of diabetes to mild cognitive impairment. Arch Neurol, 64, 570–5. [DOI] [PubMed] [Google Scholar]
- LYN-COOK LE JR., LAWTON M, TONG M, SILBERMANN E, LONGATO L, JIAO P, MARK P, WANDS JR, XU H & DE LA MONTE SM 2009. Hepatic ceramide may mediate brain insulin resistance and neurodegeneration in type 2 diabetes and non-alcoholic steatohepatitis. J Alzheimers Dis, 16, 715–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MADSEN SK, RAJAGOPALAN P, JOSHI SH, TOGA AW, THOMPSON PM & ALZHEIMER’S DISEASE NEUROIMAGING I 2015. Higher homocysteine associated with thinner cortical gray matter in 803 participants from the Alzheimer’s Disease Neuroimaging Initiative. Neurobiol Aging, 36 Suppl 1, S203–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MANCUSO C, BATES TE, BUTTERFIELD DA, CALAFATO S, CORNELIUS C, DE LORENZO A, DINKOVA KOSTOVA AT & CALABRESE V 2007. Natural antioxidants in Alzheimer’s disease. Expert Opin Investig Drugs, 16, 1921–31. [DOI] [PubMed] [Google Scholar]
- MANGIALASCHE F, POLIDORI MC, MONASTERO R, ERCOLANI S, CAMARDA C, CECCHETTI R & MECOCCI P 2009. Biomarkers of oxidative and nitrosative damage in Alzheimer’s disease and mild cognitive impairment. Ageing Res Rev, 8, 285–305. [DOI] [PubMed] [Google Scholar]
- MANN DM 1985. The neuropathology of Alzheimer’s disease: a review with pathogenetic, aetiological and therapeutic considerations. Mech Ageing Dev, 31, 213–55. [DOI] [PubMed] [Google Scholar]
- MASLIAH E 1995. Mechanisms of synaptic dysfunction in Alzheimer’s disease. Histol Histopathol, 10, 509–19. [PubMed] [Google Scholar]
- MASLIAH E, HANSEN L, ALBRIGHT T, MALLORY M & TERRY RD 1991. Immunoelectron microscopic study of synaptic pathology in Alzheimer’s disease. Acta Neuropathol, 81, 428–33. [DOI] [PubMed] [Google Scholar]
- MASLIAH E, TERRY RD, DETERESA RM & HANSEN LA 1989. Immunohistochemical quantification of the synapse-related protein synaptophysin in Alzheimer disease. Neurosci Lett, 103, 234–9. [DOI] [PubMed] [Google Scholar]
- MCCLEAN PL & HOLSCHER C 2014. Lixisenatide, a drug developed to treat type 2 diabetes, shows neuroprotective effects in a mouse model of Alzheimer’s disease. Neuropharmacology, 86, 241–58. [DOI] [PubMed] [Google Scholar]
- MCGEER PL & MCGEER EG 2013. The amyloid cascade-inflammatory hypothesis of Alzheimer disease: implications for therapy. Acta Neuropathol, 126, 479–97. [DOI] [PubMed] [Google Scholar]
- MCKHANN GM, KNOPMAN DS, CHERTKOW H, HYMAN BT, JACK CR JR., KAWAS CH, KLUNK WE, KOROSHETZ WJ, MANLY JJ, MAYEUX R, MOHS RC, MORRIS JC, ROSSOR MN, SCHELTENS P, CARRILLO MC, THIES B, WEINTRAUB S & PHELPS CH 2011. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement, 7, 263–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MEDA SA, NARAYANAN B, LIU J, PERRONE-BIZZOZERO NI, STEVENS MC, CALHOUN VD, GLAHN DC, SHEN L, RISACHER SL, SAYKIN AJ & PEARLSON GD 2012. A large scale multivariate parallel ICA method reveals novel imaging-genetic relationships for Alzheimer’s disease in the ADNI cohort. Neuroimage, 60, 1608–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MEHLHORN G, HOLLBORN M & SCHLIEBS R 2000. Induction of cytokines in glial cells surrounding cortical beta-amyloid plaques in transgenic Tg2576 mice with Alzheimer pathology. Int J Dev Neurosci, 18, 423–31. [DOI] [PubMed] [Google Scholar]
- MEIER JJ & NAUCK MA 2010. Is the diminished incretin effect in type 2 diabetes just an epi-phenomenon of impaired beta-cell function? Diabetes, 59, 1117–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MENDIS LH, GREY AC, FAULL RL & CURTIS MA 2016. Hippocampal lipid differences in Alzheimer’s disease: a human brain study using matrix-assisted laser desorption/ionization-imaging mass spectrometry. Brain Behav, 6, e00517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MEYER JS, KAWAMURA J & TERAYAMA Y 1992. White matter lesions in the elderly. J Neurol Sci, 110, 1–7. [DOI] [PubMed] [Google Scholar]
- MIDDLETON LE & YAFFE K 2009. Promising strategies for the prevention of dementia. Arch Neurol, 66, 1210–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MISIAK B, LESZEK J & KIEJNA A 2012. Metabolic syndrome, mild cognitive impairment and Alzheimer’s disease--the emerging role of systemic low-grade inflammation and adiposity. Brain Res Bull, 89, 144–9. [DOI] [PubMed] [Google Scholar]
- MIZUTANI T, AMANO N, SASAKI H, MORIMATSU Y, MORI H, YOSHIMURA M, YAMANOUCHI H, HAYAKAWA K & SHIMADA H 1990. Senile dementia of Alzheimer type characterized by laminar neuronal loss exclusively in the hippocampus, parahippocampus and medial occipitotemporal cortex. Acta Neuropathol, 80, 575–80. [DOI] [PubMed] [Google Scholar]
- MONTINE TJ, PHELPS CH, BEACH TG, BIGIO EH, CAIRNS NJ, DICKSON DW, DUYCKAERTS C, FROSCH MP, MASLIAH E, MIRRA SS, NELSON PT, SCHNEIDER JA, THAL DR, TROJANOWSKI JQ, VINTERS HV, HYMAN BT, NATIONAL INSTITUTE ON A & ALZHEIMER’S A 2012. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: a practical approach. Acta Neuropathol, 123, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MOREIRA PI 2013. High-sugar diets, type 2 diabetes and Alzheimer’s disease. Curr Opin Clin Nutr Metab Care, 16, 440–5. [DOI] [PubMed] [Google Scholar]
- MOROZ N, TONG M, LONGATO L, XU H & DE LA MONTE SM 2008. Limited Alzheimer-type neurodegeneration in experimental obesity and type 2 diabetes mellitus. J Alzheimers Dis, 15, 29–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MRAK RE 2009. Neuropathology and the neuroinflammation idea. J Alzheimers Dis, 18, 473–81. [DOI] [PubMed] [Google Scholar]
- MRAK RE, SHENG JG & GRIFFIN WS 1995. Glial cytokines in Alzheimer’s disease: review and pathogenic implications. Hum Pathol, 26, 816–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NAASAN G, RABINOVICI GD, GHOSH P, ELOFSON JD, MILLER BL, COPPOLA G, KARYDAS A, FONG J, PERRY D, LEE SE, YOKOYAMA JS, SEELEY WW, KRAMER JH, WEINER MW, SCHUFF N, JAGUST WJ, GRINBERG LT, PRIBADI M, YANG Z, SEARS R, KLEIN E, WOJTA K & ROSEN HJ 2016. Amyloid in dementia associated with familial FTLD: not an innocent bystander. Neurocase, 22, 76–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NADERALI EK, RATCLIFFE SH & DALE MC 2009. Obesity and Alzheimer’s disease: a link between body weight and cognitive function in old age. Am J Alzheimers Dis Other Demen, 24, 445–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NAGOSHI S 2014. [Liver diseases]. Nihon Rinsho, 72, 726–9. [PubMed] [Google Scholar]
- NELSON PT, ALAFUZOFF I, BIGIO EH, BOURAS C, BRAAK H, CAIRNS NJ, CASTELLANI RJ, CRAIN BJ, DAVIES P, DEL TREDICI K, DUYCKAERTS C, FROSCH MP, HAROUTUNIAN V, HOF PR, HULETTE CM, HYMAN BT, IWATSUBO T, JELLINGER KA, JICHA GA, KOVARI E, KUKULL WA, LEVERENZ JB, LOVE S, MACKENZIE IR, MANN DM, MASLIAH E, MCKEE AC, MONTINE TJ, MORRIS JC, SCHNEIDER JA, SONNEN JA, THAL DR, TROJANOWSKI JQ, TRONCOSO JC, WISNIEWSKI T, WOLTJER RL & BEACH TG 2012. Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature. J Neuropathol Exp Neurol, 71, 362–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NELTNER JH, ABNER EL, BAKER S, SCHMITT FA, KRYSCIO RJ, JICHA GA, SMITH CD, HAMMACK E, KUKULL WA, BRENOWITZ WD, VAN ELDIK LJ & NELSON PT 2014. Arteriolosclerosis that affects multiple brain regions is linked to hippocampal sclerosis of ageing. Brain, 137, 255–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NOBLE JM, MANLY JJ, SCHUPF N, TANG MX & LUCHSINGER JA 2012. Type 2 diabetes and ethnic disparities in cognitive impairment. Ethn Dis, 22, 38–44. [PMC free article] [PubMed] [Google Scholar]
- NOLAN KA, LINO MM, SELIGMANN AW & BLASS JP 1998. Absence of vascular dementia in an autopsy series from a dementia clinic. J Am Geriatr Soc, 46, 597–604. [DOI] [PubMed] [Google Scholar]
- OLSSON B, LAUTNER R, ANDREASSON U, OHRFELT A, PORTELIUS E, BJERKE M, HOLTTA M, ROSEN C, OLSSON C, STROBEL G, WU E, DAKIN K, PETZOLD M, BLENNOW K & ZETTERBERG H 2016. CSF and blood biomarkers for the diagnosis of Alzheimer’s disease: a systematic review and meta-analysis. Lancet Neurol, 15, 673–84. [DOI] [PubMed] [Google Scholar]
- OTT BR, JONES R, DAIELLO LA, DE LA MONTE SM, STOPA EG, JOHANSON CE, DENBY C & GRAMMAS P 2018. Blood-Cerebrospinal Fluid Barrier Gradients in Mild Cognitive Impairment and Alzheimer’s Disease: Relationship to Inflammatory Cytokines and Chemokines. Front Aging Neurosci, (In Press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- PANTANO P, CARAMIA F & PIERALLINI A 1999. The role of MRI in dementia. Ital J Neurol Sci, 20, S250–3. [DOI] [PubMed] [Google Scholar]
- PEDDITIZI E, PETERS R & BECKETT N 2016. The risk of overweight/obesity in mid-life and late life for the development of dementia: a systematic review and meta-analysis of longitudinal studies. Age Ageing, 45, 14–21. [DOI] [PubMed] [Google Scholar]
- PERLMUTTER LS & CHUI HC 1990. Microangiopathy, the vascular basement membrane and Alzheimer’s disease: a review. Brain Res Bull, 24, 677–86. [DOI] [PubMed] [Google Scholar]
- PERRY G, KAWAI M, TABATON M, ONORATO M, MULVIHILL P, RICHEY P, MORANDI A, CONNOLLY JA & GAMBETTI P 1991. Neuropil threads of Alzheimer’s disease show a marked alteration of the normal cytoskeleton. J Neurosci, 11, 1748–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PIRO JR, BENJAMIN DI, DUERR JM, PI Y, GONZALES C, WOOD KM, SCHWARTZ JW, NOMURA DK & SAMAD TA 2012. A dysregulated endocannabinoid-eicosanoid network supports pathogenesis in a mouse model of Alzheimer’s disease. Cell Rep, 1, 617–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- POLIAKOVA T, LEVIN O, ARABLINSKIY A, VASENINA E & ZERR I 2016. Cerebral microbleeds in early Alzheimer’s disease. J Neurol. [DOI] [PubMed] [Google Scholar]
- PRENCIPE M & MARINI C 1989. Leuko-araiosis: definition and clinical correlates--an overview. Eur Neurol, 29 Suppl 2, 27–9. [DOI] [PubMed] [Google Scholar]
- REGER MA, WATSON GS, GREEN PS, WILKINSON CW, BAKER LD, CHOLERTON B, FISHEL MA, PLYMATE SR, BREITNER JC, DEGROODT W, MEHTA P & CRAFT S 2008. Intranasal insulin improves cognition and modulates beta-amyloid in early AD. Neurology, 70, 440–8. [DOI] [PubMed] [Google Scholar]
- RIVERA EJ, GOLDIN A, FULMER N, TAVARES R, WANDS JR & DE LA MONTE SM 2005. Insulin and insulin-like growth factor expression and function deteriorate with progression of Alzheimer’s disease: link to brain reductions in acetylcholine. J Alzheimers Dis, 8, 247–68. [DOI] [PubMed] [Google Scholar]
- ROHER AE, WEISS N, KOKJOHN TA, KUO YM, KALBACK W, ANTHONY J, WATSON D, LUEHRS DC, SUE L, WALKER D, EMMERLING M, GOUX W & BEACH T 2002. Increased A beta peptides and reduced cholesterol and myelin proteins characterize white matter degeneration in Alzheimer’s disease. Biochemistry, 41, 11080–90. [DOI] [PubMed] [Google Scholar]
- ROLANDI E, FRISONI GB & CAVEDO E 2016. Efficacy of lifestyle interventions on clinical and neuroimaging outcomes in elderly. Ageing Res Rev, 25, 1–12. [DOI] [PubMed] [Google Scholar]
- RORIZ-FILHO JS, SA-RORIZ TM, ROSSET I, CAMOZZATO AL, SANTOS AC, CHAVES ML, MORIGUTI JC & RORIZ-CRUZ M 2009. (Pre)diabetes, brain aging, and cognition. Biochim Biophys Acta, 1792, 432–43. [DOI] [PubMed] [Google Scholar]
- ROWE CC, BOURGEAT P, ELLIS KA, BROWN B, LIM YY, MULLIGAN R, JONES G, MARUFF P, WOODWARD M, PRICE R, ROBINS P, TOCHON-DANGUY H, O’KEEFE G, PIKE KE, YATES P, SZOEKE C, SALVADO O, MACAULAY SL, O’MEARA T, HEAD R, COBIAC L, SAVAGE G, MARTINS R, MASTERS CL, AMES D & VILLEMAGNE VL 2013a. Predicting Alzheimer disease with beta-amyloid imaging: results from the Australian imaging, biomarkers, and lifestyle study of ageing. Ann Neurol, 74, 905–13. [DOI] [PubMed] [Google Scholar]
- ROWE CC, PEJOSKA S, MULLIGAN RS, JONES G, CHAN JG, SVENSSON S, CSELENYI Z, MASTERS CL & VILLEMAGNE VL 2013b. Head-to-head comparison of 11C-PiB and 18F-AZD4694 (NAV4694) for beta-amyloid imaging in aging and dementia. J Nucl Med, 54, 880–6. [DOI] [PubMed] [Google Scholar]
- SAMARAS K & SACHDEV PS 2012. Diabetes and the elderly brain: sweet memories? Ther Adv Endocrinol Metab, 3, 189–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHAFFER C, SARAD N, DECRUMPE A, GOSWAMI D, HERRMANN S, MORALES J, PATEL P & OSBORNE J 2015. Biomarkers in the Diagnosis and Prognosis of Alzheimer’s Disease. J Lab Autom, 20, 589–600. [DOI] [PubMed] [Google Scholar]
- SCHEIBEL AB, DUONG TH & JACOBS R 1989. Alzheimer’s disease as a capillary dementia. Ann Med, 21, 103–7. [DOI] [PubMed] [Google Scholar]
- SCHELTENS P, BARKHOF F, LEYS D, WOLTERS EC, RAVID R & KAMPHORST W 1995. Histopathologic correlates of white matter changes on MRI in Alzheimer’s disease and normal aging. Neurology, 45, 883–8. [DOI] [PubMed] [Google Scholar]
- SCHELTENS P, BLENNOW K, BRETELER MM, DE STROOPER B, FRISONI GB, SALLOWAY S & VAN DER FLIER WM 2016. Alzheimer’s disease. Lancet, 388, 505–17. [DOI] [PubMed] [Google Scholar]
- SCHUSTER J & FUNKE SA 2016. Methods for the Specific Detection and Quantitation of Amyloid-beta Oligomers in Cerebrospinal Fluid. J Alzheimers Dis, 53, 53–67. [DOI] [PubMed] [Google Scholar]
- SERRANO-POZO A, FROSCH MP, MASLIAH E & HYMAN BT 2011. Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect Med, 1, a006189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHIN J, KEPE V, SMALL GW, PHELPS ME & BARRIO JR 2011. Multimodal Imaging of Alzheimer Pathophysiology in the Brain’s Default Mode Network. Int J Alzheimers Dis, 2011, 687945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHUVAEV VV, LAFFONT I, SEROT JM, FUJII J, TANIGUCHI N & SIEST G 2001. Increased protein glycation in cerebrospinal fluid of Alzheimer’s disease. Neurobiol Aging, 22, 397–402. [DOI] [PubMed] [Google Scholar]
- SINGHAL G, JAEHNE EJ, CORRIGAN F, TOBEN C & BAUNE BT 2014. Inflammasomes in neuroinflammation and changes in brain function: a focused review. Front Neurosci, 8, 315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SJOBECK M, HAGLUND M & ENGLUND E 2005. Decreasing myelin density reflected increasing white matter pathology in Alzheimer’s disease--a neuropathological study. Int J Geriatr Psychiatry, 20, 919–26. [DOI] [PubMed] [Google Scholar]
- SJOBECK M, HAGLUND M, PERSSON A, STURESSON K & ENGLUND E 2003. Brain tissue microarrays in dementia research: white matter microvascular pathology in Alzheimer’s disease. Neuropathology, 23, 290–5. [DOI] [PubMed] [Google Scholar]
- SMALL GW, KEPE V, ERCOLI LM, SIDDARTH P, BOOKHEIMER SY, MILLER KJ, LAVRETSKY H, BURGGREN AC, COLE GM, VINTERS HV, THOMPSON PM, HUANG SC, SATYAMURTHY N, PHELPS ME & BARRIO JR 2006. PET of brain amyloid and tau in mild cognitive impairment. N Engl J Med, 355, 2652–63. [DOI] [PubMed] [Google Scholar]
- SRIDHAR GR, LAKSHMI G & NAGAMANI G 2015. Emerging links between type 2 diabetes and Alzheimer’s disease. World J Diabetes, 6, 744–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STEEN E, TERRY BM, RIVERA EJ, CANNON JL, NEELY TR, TAVARES R, XU XJ, WANDS JR & DE LA MONTE SM 2005. Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease--is this type 3 diabetes? J Alzheimers Dis, 7, 63–80. [DOI] [PubMed] [Google Scholar]
- STEPHENSON D, RASH K, SMALSTIG B, ROBERTS E, JOHNSTONE E, SHARP J, PANETTA J, LITTLE S, KRAMER R & CLEMENS J 1999. Cytosolic phospholipase A2 is induced in reactive glia following different forms of neurodegeneration. Glia, 27, 110–28. [DOI] [PubMed] [Google Scholar]
- STOCKHORST U, DE FRIES D, STEINGRUEBER HJ & SCHERBAUM WA 2004. Insulin and the CNS: effects on food intake, memory, and endocrine parameters and the role of intranasal insulin administration in humans. Physiol Behav, 83, 47–54. [DOI] [PubMed] [Google Scholar]
- SU JH, CUMMINGS BJ & COTMAN CW 1993. Identification and distribution of axonal dystrophic neurites in Alzheimer’s disease. Brain Res, 625, 228–37. [DOI] [PubMed] [Google Scholar]
- SUTER OC, SUNTHORN T, KRAFTSIK R, STRAUBEL J, DAREKAR P, KHALILI K & MIKLOSSY J 2002. Cerebral hypoperfusion generates cortical watershed microinfarcts in Alzheimer disease. Stroke, 33, 1986–92. [DOI] [PubMed] [Google Scholar]
- SWOMLEY AM & BUTTERFIELD DA 2015. Oxidative stress in Alzheimer disease and mild cognitive impairment: evidence from human data provided by redox proteomics. Arch Toxicol, 89, 1669–80. [DOI] [PubMed] [Google Scholar]
- TALBOT K, WANG HY, KAZI H, HAN LY, BAKSHI KP, STUCKY A, FUINO RL, KAWAGUCHI KR, SAMOYEDNY AJ, WILSON RS, ARVANITAKIS Z, SCHNEIDER JA, WOLF BA, BENNETT DA, TROJANOWSKI JQ & ARNOLD SE 2012. Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J Clin Invest, 122, 1316–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- THAL DR, GHEBREMEDHIN E, ORANTES M & WIESTLER OD 2003. Vascular pathology in Alzheimer disease: correlation of cerebral amyloid angiopathy and arteriosclerosis/lipohyalinosis with cognitive decline. J Neuropathol Exp Neurol, 62, 1287–301. [DOI] [PubMed] [Google Scholar]
- TOLEDO JB, CAIRNS NJ, DA X, CHEN K, CARTER D, FLEISHER A, HOUSEHOLDER E, AYUTYANONT N, ROONTIVA A, BAUER RJ, EISEN P, SHAW LM, DAVATZIKOS C, WEINER MW, REIMAN EM, MORRIS JC, TROJANOWSKI JQ & ALZHEIMER’S DISEASE NEUROIMAGING I 2013. Clinical and multimodal biomarker correlates of ADNI neuropathological findings. Acta Neuropathol Commun, 1, 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TONG M & DE LA MONTE SM 2009. Mechanisms of ceramide-mediated neurodegeneration. J Alzheimers Dis, 16, 705–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TONG M, DEOCHAND C, DIDSBURY J & DE LA MONTE SM 2016. T3D-959: A Multi-Faceted Disease Remedial Drug Candidate for the Treatment of Alzheimer’s Disease. J Alzheimers Dis, 51, 123–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TONG M, LEAO R, VIMBELA GV, YALCIN EB, KAY J, KROTOW A & DE LA MONTE SM 2017. Altered temporal lobe white matter lipid ion profiles in an experimental model of sporadic Alzheimer’s disease. Mol Cell Neurosci, 82, 23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TONG M, LONGATO L & DE LA MONTE SM 2010. Early limited nitrosamine exposures exacerbate high fat diet-mediated type 2 diabetes and neurodegeneration. BMC Endocr Disord, 10, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- UENO M, CHIBA Y, MATSUMOTO K, NAKAGAWA T & MIYANAKA H 2014. Clearance of beta-amyloid in the brain. Curr Med Chem, 21, 4085–90. [DOI] [PubMed] [Google Scholar]
- VAN DER VLIES AE, GOOS JD, BARKHOF F, SCHELTENS P & VAN DER FLIER WM 2012. Microbleeds do not affect rate of cognitive decline in Alzheimer disease. Neurology, 79, 763–9. [DOI] [PubMed] [Google Scholar]
- VERKHRATSKY A, PARPURA V, PEKNA M, PEKNY M & SOFRONIEW M 2014. Glia in the pathogenesis of neurodegenerative diseases. Biochem Soc Trans, 42, 1291–301. [DOI] [PubMed] [Google Scholar]
- VERNY M, DUYCKAERTS C, PIEROT L & HAUW JJ 1991. Leuko-araiosis. Dev Neurosci, 13, 245–50. [DOI] [PubMed] [Google Scholar]
- VINTERS HV 2015. Emerging concepts in Alzheimer’s disease. Annu Rev Pathol, 10, 291–319. [DOI] [PubMed] [Google Scholar]
- VINTERS HV, SECOR DL, PARDRIDGE WM & GRAY F 1990. Immunohistochemical study of cerebral amyloid angiopathy. III. Widespread Alzheimer A4 peptide in cerebral microvessel walls colocalizes with gamma trace in patients with leukoencephalopathy. Ann Neurol, 28, 34–42. [DOI] [PubMed] [Google Scholar]
- VINTERS HV, WANG ZZ & SECOR DL 1996. Brain parenchymal and microvascular amyloid in Alzheimer’s disease. Brain Pathol, 6, 179–95. [DOI] [PubMed] [Google Scholar]
- VIOLA KL & KLEIN WL 2015. Amyloid beta oligomers in Alzheimer’s disease pathogenesis, treatment, and diagnosis. Acta Neuropathol, 129, 183–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WALDRON AM, WINTMOLDERS C, BOTTELBERGS A, KELLEY JB, SCHMIDT ME, STROOBANTS S, LANGLOIS X & STAELENS S 2015. In vivo molecular neuroimaging of glucose utilization and its association with fibrillar amyloid-beta load in aged APPPS1–21 mice. Alzheimers Res Ther, 7, 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WALLIN A, NORDLUND A, JONSSON M, BLENNOW K, ZETTERBERG H, OHRFELT A, STALHAMMAR J, ECKERSTROM M, CARLSSON M, OLSSON E, GOTHLIN M, SVENSSON J, ROLSTAD S, ECKERSTROM C & BJERKE M 2016a. Alzheimer’s disease--subcortical vascular disease spectrum in a hospital-based setting: Overview of results from the Gothenburg MCI and dementia studies. J Cereb Blood Flow Metab, 36, 95–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WALLIN A, NORDLUND A, JONSSON M, LIND K, EDMAN A, GOTHLIN M, STALHAMMAR J, ECKERSTROM M, KERN S, BORJESSON-HANSON A, CARLSSON M, OLSSON E, ZETTERBERG H, BLENNOW K, SVENSSON J, OHRFELT A, BJERKE M, ROLSTAD S & ECKERSTROM C 2016b. The Gothenburg MCI study: Design and distribution of Alzheimer’s disease and subcortical vascular disease diagnoses from baseline to 6-year follow-up. J Cereb Blood Flow Metab, 36, 114–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WASHINGTON PM, VILLAPOL S & BURNS MP 2016. Polypathology and dementia after brain trauma: Does brain injury trigger distinct neurodegenerative diseases, or should they be classified together as traumatic encephalopathy? Exp Neurol, 275 Pt 3, 381–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHATLEY BR, LI L & CHIN LS 2008. The ubiquitin-proteasome system in spongiform degenerative disorders. Biochim Biophys Acta, 1782, 700–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WILSON CM, GRACE GM, MUNOZ DG, HE BP & STRONG MJ 2001. Cognitive impairment in sporadic ALS: a pathologic continuum underlying a multisystem disorder. Neurology, 57, 651–7. [DOI] [PubMed] [Google Scholar]
- WIPPOLD FJ 2ND, CAIRNS N, VO K, HOLTZMAN DM & MORRIS JC 2008. Neuropathology for the neuroradiologist: plaques and tangles. AJNR Am J Neuroradiol, 29, 18–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WURTMAN R 2015. Biomarkers in the diagnosis and management of Alzheimer’s disease. Metabolism, 64, S47–50. [DOI] [PubMed] [Google Scholar]
- YALCIN EB, NUNEZ K, TONG M & DE LA MONTE SM 2015. Differential Sphingolipid and Phospholipid Profiles in Alcohol and Nicotine-Derived Nitrosamine Ketone-Associated White Matter Degeneration. Alcohol Clin Exp Res, 39, 2324–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YAMAGISHI S, NAKAMURA N, SUEMATSU M, KASEDA K & MATSUI T 2015. Advanced Glycation End Products: A Molecular Target for Vascular Complications in Diabetes. Mol Med, 21 Suppl 1, S32–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YAMAMOTO H, KISHI T, LEE CE, CHOI BJ, FANG H, HOLLENBERG AN, DRUCKER DJ & ELMQUIST JK 2003. Glucagon-like peptide-1-responsive catecholamine neurons in the area postrema link peripheral glucagon-like peptide-1 with central autonomic control sites. J Neurosci, 23, 2939–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YANG L, RIEVES D & GANLEY C 2012. Brain amyloid imaging--FDA approval of florbetapir F18 injection. N Engl J Med, 367, 885–7. [DOI] [PubMed] [Google Scholar]
- ZAFARI S, BACKES C, MEESE E & KELLER A 2015. Circulating Biomarker Panels in Alzheimer’s Disease. Gerontology, 61, 497–503. [DOI] [PubMed] [Google Scholar]
- ZHANG W, ARTEAGA J, CASHION DK, CHEN G, GANGADHARMATH U, GOMEZ LF, KASI D, LAM C, LIANG Q, LIU C, MOCHARLA VP, MU F, SINHA A, SZARDENINGS AK, WANG E, WALSH JC, XIA C, YU C, ZHAO T & KOLB HC 2012. A highly selective and specific PET tracer for imaging of tau pathologies. J Alzheimers Dis, 31, 601–12. [DOI] [PubMed] [Google Scholar]
- ZINDA MJ, VLAHOS CJ & LAI MT 2001. Ceramide induces the dephosphorylation and inhibition of constitutively activated Akt in PTEN negative U87mg cells. Biochem Biophys Res Commun, 280, 1107–15. [DOI] [PubMed] [Google Scholar]