

Abstract

There is a continuing demand of new inhibitors of HIV-1 Integrase (HIV-1 IN) due to mutations of HIV-1. This study aims to develop the synthesis of 3,6-diaryl 7-azaindoles and introspect the role of aryl groups on the strand transfer (ST) inhibition of HIV-1 IN. An efficient and chemo-selective one-pot method is established for the synthesis of the unexplored diverse C3 → C6 diaryl 7-azaindoles starting from 6-chloro-3-iodo-N-protected 7-azaindoles. Here we report Pd2dba3/SPhos catalyzed synthesis of eight selective C3 monoaryl 7-azaindoles (10a–h) and eight C3,C6-diaryl 7-azaindoles (11a–f, 12a,b) with yields in the ranges of 67–93% and 43–88% respectively. The synthesized derivatives inhibit the strand transfer (ST) activity of HIV-1 IN enzyme at 10 μM dose with 11d and 11f exhibiting %ST inhibitions of 72% and 71%, respectively. SAR studies indicate the para-substitution on the C3 aryl ring and C6 aryl is essential for enhanced %ST inhibition. 11b,c, 11e–f, and 12b showed lower cytotoxicity (IC50 > 200 μM) against TZM-bl cells. Molecular docking of the diaryl 7-azaindoles and Raltegravir (RAL), to the PFV-integrase revealed favorable binding interactions.
Introduction
Integrase strand transfer inhibitors (INSTI) are widely used in antiretroviral therapy (ART) to efficiently manage HIV-1 infection.1 The integrase viral enzyme (IN) catalyzes the nucleophilic attack of 3′-OH of dAs (deoxy adenine) on the host genome, followed by insertion of the 3′-ends of the viral DNA into the host DNA.2 This process is known as strand transfer (ST), and its inhibition is an essential prospect in targeting HIV-1 viral infections. INSTIs interact with the DNA strands, Mg2+ ions, and key amino acid residues (Asp64, Asp116, and Glu142)3 leading to ST inhibition, and thus IN is highly targeted in anti-HIV-1 therapy.4 The crystal structure of FDA-approved INSTI, Raltegravir, with PFV-IN (Prototype Foamy Virus-INtegrase) shows that inhibition of the strand transfer (ST) phenomena by binding of diketo groups to the two Mg2+ ions.5 In addition, the π–π interaction of the Tyr212 with the oxadiazole ring and the hydrophobic interactions of the ligand with Pro214 are important for the inhibitory activity. HIV-1 has shown resistance to all FDA-approved integrase inhibitors.6 The resistance offered by the enzymes is caused by specific mutations such as Gly155His, Gly140Ser, Gln148His, Tyr143Arg, and Gln148Arg which are known to decrease the efficacy of Raltegravir.7 Dolutagravir is also an FDA-approved drug used in ART and is known to have a high resistance barrier; however, there are reports on the emergence of resistance to this drug as well.8 Thus, there is an evolving need for the development of new inhibitors against HIV-1 to counter drug resistance. Azaindole derivatives are known for their antiviral properties and various reports have been published in the last two decades (Figure 1A).9 An extensive study on the hydroxamic acids of 6-azaindole exhibited favorable structural modifications for good potency in IN inhibition10 (Figure 1B). The hydroxamic acids present on the C5 position bind to the cofactors (Mg2+ ions) in the binding pocket of HIV-IN. The C3 substitution in 6-azaindole with the aliphatic chain, was an important modification to render stability of the molecule toward glucorinidation reaction, thus decreasing the clearance rate in human hepatocytes.11
Figure 1.

A) Biologically relevant azaindole derivatives. B) Design of HIV-1 IN inhibitors.
The benzyl group at the N1 position occupies a hydrophobic pocket which is important for the activity of the drug.12 Therefore, both metal binding moiety and a hydrophobic region are responsible for the activity of these molecules. The metabolic instability of the hydroxamic acids13 has led to the development of alternatives but could not be pursued due to their detrimental pharmacokinetic profile.14 The presence of metal binding heteroatoms and hydrophobicity are perquisites to the development of HIV-1 IN inhibitors and we wished to understand the role of 7-azaindole core and the electrostatic interaction borne out of a 3,6- diaryl pattern in the inhibition of IN.
7-Azaindoles being privileged structures in biological processes15 have been explored from the synthetic point of view to generate mono-/di-/triarylated 7-azaindoles.16 Diverse aryl groups can be introduced in a site-selective manner using Suzuki–Miyaura C-arylations on halogenated precursors.17 However, achieving substrate selectivity still requires extensive optimizations, which we previously encountered while generating tetra-aryl 7-azaindoles.18 The sequential Suzuki–Miyaura cross-coupling reaction is established on C3 and C5 position of indole; however, it was done in a sequential stepwise manner.19 The only known report on the synthesis of a 3,6-diaryl 7-azaindole in our knowledge, is by Schirock et al. (Scheme 1A), where in one step, an epoxide ring at third position of 2,6-dichloro pyridine, 1, was opened by an amine in microwave conditions, followed by nucleophilic aromatic substitution and dehydration.20 This reaction procured C3-Me/-Ph incorporated 7-azaindoles and further C6 Suzuki–Miyaura arylation of 2 gave diaryl 7-azaindoles, 3. The limits posed by the aforementioned synthesis are the tedious synthetic steps to prepare starting materials and the nonavailability of starting substrates.
Scheme 1. A) Previous Work on the Synthesis of 3,6-Diaryl 7-Azaindoles and B) Present Work on One-Pot Synthesis of C3,C6 Diarylation of 7-Azaindoles20.

C3,C6-Diaryl 7-azaindoles have not been explored for their medicinal impact, and therefore we endeavored to derive a procedure for incorporating diverse aryl groups with varying electronic natures by selective C3 → C6 Suzuki–Miyaura cross-coupling reactions (Scheme 1B). Herein we report an efficient method for a site-selective sequential C3,C6 C-arylation of 7-azaindole in a one-pot manner, generating diversified diaryl 7-azaindoles as potential anti-HIV inhibitors and analyzing their in silico binding interactions. This work is part of our continuing efforts to develop novel inhibitors of HIV-1 IN in our lab.21
Results and Discussions
6-Chloro-3-iodo-1-methyl-1H-pyrrolo[2,3-b]pyridine (9) and 6-chloro-3-iodo-1-(4-methoxybenzyl)-1H-pyrrolo[2,3-b]pyridine (9′) were synthesized bearing in mind the different rates of oxidative additions of C–I and C–Cl bond in SMC reaction (Scheme 2).17 Synthesis of 9 has been established in our lab in previous literature reports.22 The synthesis is as follows: electrophilic C3 iodination of commercially available 7-azaindole, 4 using N-iodosuccinimide (1 equiv) and a base KOH (0.5 equiv) in DCM furnished C3-iodinated 1H-7-azaindole, 5, in 95% yield. mCPBA (2.5 equiv) in Et2O oxidizes 5, giving 57% of N-oxide, 6. The N-oxide facilitates electrophilic C6 chlorination of 6 to furnish 7 in 46% yield. The carboxylate group was removed in basic conditions, i.e., 1 N NaOH in MeOH, stirred at r.t. for 24 h to give 8 in 98% yield. Methylation using sodium hydride as a base and methyl iodide as a methylating agent, produced 9 in 73% yield and the desired precursor in 29% overall yield from 5 steps. Similarly, a p-methoxy benzyl group can be used for N-protection using K2CO3 (3 equiv) and p-methoxy benzyl chloride (1.2 equiv) to give 9′ in 88% yield and 35% overall yield over 5 steps.
Scheme 2. Synthesis of Starting Materials 6-Chloro-3-iodo-1-methyl-1H-pyrrolo[2,3-b]pyridine (9) and 6-Chloro-3-iodo-1-(4-methoxybenzyl)-1H-pyrrolo[2,3-b]pyridine (9′).

Reagents and conditions: (i) KOH (0.5 equiv), NIS (1 equiv), DCM, r.t., 15 h; (ii) mCPBA (2.5 equiv), Et2O, r.t., 1 h followed by basification of 6-int with aq. K2CO3 at pH 9–10 (iii) HMDS (1 equiv), CH3OCOCl (2.5 equiv), dry THF, r.t., N2 atm., 1 h; (iv) 1N NaOH, MeOH, r.t., 24 h; (v) NaH, MeI, DMF, 0–3 h, r.t. or K2CO3 (3 equiv), p-methoxy benzylchloride (1.2 equiv), DMF, 10 h, r.t., aPMB = p-methoxybenzyl.
The following reaction condition was chosen as the best parameter for C3 selective arylation of N-protected 7-azaindole after extensive optimization (Table S1): 3-iodo-6-chloro-7-azaindole (1 equiv), phenyl boronic acid (1.2 equiv) at 60 °C, Pd2dba3 (5 mol %), SPhos (5 mol %), Cs2CO3 (2 equiv), toluene/ethanol (1:1). Additionally, the reaction did not require an inert atmosphere and can be easily done in an open flask setup. The unsubstituted phenyl was incorporated with 85% yield (10a) (Scheme 3). The aryl boronic acids bearing electron-donating −Me at the para position gave product 10b in an excellent yield of 89%. Change in the position of the −Me group on C3-aryl did not affect the yield greatly as 10c was formed in 93% yield. The +R effect rendered by the p-methoxy substituent led to a higher yield of 93% for 10d. The electron-withdrawing p-fluoro substituent on aryl boronic acids reacted with the 7-azaindole precursor to furnish a good yield of 79% of 10e. The 3,5-bis(trifluormethyl) substituted aryl boronic acid reacts with the 7-azaindole precursor to give a good yield of 67% for 10f. This shows this reaction condition tolerates both electron-donating and withdrawing groups. The reaction condition is also suitable for bulky bicyclic groups such as naphthyl (10g) and benzo[d][1,3]dioxole (10h) groups to procure the desired compounds in 92% and 76% yield, respectively.
Scheme 3. Substrate Scope for Suzuki–Miyaura C3-Arylation of 6-Chloro-3-iodo-1-methyl-1H-pyrrolo[2,3-b]pyridine (9).

Isolated yields
We commenced the study of one-pot sequential synthesis of 7-azaindoles (Table 1). Temperature and sequential loading of the catalytic system were important parameters for procuring good yields of 11a. Diarylation was attempted with 5 mol % Pd2dba3 and 5 mol % SPhos at 60 and 80 °C but was unsuccessful in getting desired isolable product. A second addition of SPhos (10 mol %) in one-pot synthesis yielded 45% of 11a at 110 °C (entry 1), whereas a second addition of 20 mol % SPhos yielded 67% of 11a at 110 °C (entry 2). The subsequent addition of Pd2dba3 (10 mol %) and SPhos (20 mol %) enhanced the yield to 88% (entry 3). Changing the ligand to electron-deficient 1,1′-bis(diphenylphosphino)ferrocene (dppf) in subsequent addition gave a low yield of 48% (entry 4), whereas when Pd2dba3 (10 mol %) and SPhos (20 mol %) were used without any second addition, the yield of the diarylated product was 59% (entry 5). We observed the following conditions as best to synthesize diverse C3,C6 diaryl 7-azaindoles in one-pot synthesis: 5 mol % Pd2dba3, 5 mol % SPhos, 1.1 equiv Ar1–B(OH)2, Cs2CO3 (2 equiv) in toluene/ethanol (1:1) at 60 °C followed by addition of 10 mol % Pd2dba3, 20 mol % SPhos, 110 °C. The single-crystal X-ray of 11a corroborates with the desired structure and was also deposited to Cambridge Crystallographic Data Center with CCDC no. 2211709 (Figure S1 and Table S2).
Table 1. Optimization for C3 → C6 One-Pot Diarylation of 3-Iodo-6-chloro-N-methyl-7-azaindolea.

| entry | cond. | Pd2dba3 | ligand | yield (%)b |
|---|---|---|---|---|
| 1 | (i) | 5 mol % | 5 mol % | 45% |
| (ii) | SPhos (10 mol %) | |||
| 2 | (i) | 5 mol % | SPhos (5 mol %) | 67% |
| (ii) | SPhos (20 mol %) | |||
| 3 | (i) | 5 mol % | SPhos (5 mol %) | 88% |
| (ii) | 10 mol % | SPhos (20 mol %) | ||
| 4 | (i) | 5 mol % | SPhos (5 mol %) | 48% |
| (ii) | 10 mol % | dppf (5 mol %) | ||
| 5 | (i) | 10 mol % | SPhos (20 mol %) | 59% |
| (ii) |
Reaction conditions: (i) 9 (1 equiv), Ph-B(OH)2 (1.2 equiv), Pd2dba3 (mol %), ligand (mol %), Cs2CO3 (2 equiv) in toluene/ethanol (1:1, 5 mL), 60 °C, 0.5 h; (ii) 4OMe-Ph-B(OH)2, Pd2dba3 (mol %), ligand (mol %), 110 °C.
Isolated yields
Exploring the substrate scope of this one-pot procedure, we observed phenyl and 4-methoxy phenyl group can be installed at C3 → C6 sequence in 88% yield (11a) (Scheme 4). 4-Methyl phenyl can be installed at the C6 position to give 11b in 87% yield. Incorporating phenyl at C3 and electron-withdrawing 3,5-bis(trifluoromethyl) phenyl group at C6 lowers the yield as exemplified by 11c (53%). One pot coupling of the electron-withdrawing group at the C3 position and 4-methoxy phenyl at the C6 position procured the diaryl derivative (11d) in a good yield of 66%. Heteroarenes are responsible for a majority of bonding interactions with the receptor-binding site when used as potential inhibitors. The C3 phenyl and 3-thienyl group at C6 was incorporated in a high yield of 81% (11e). Benzo[d][1,3]dioxole at the C3 position and 4-pyridyl group at C6 position gave 11f in 43% yield. N-Protected 7-azaindoles were also found to show a moderate to good yield as shown by 12a and 12b where examples are shown of introducing two heteroaryl rings and two bicyclic rings on 7-azaindole substrate (12a–b).
Scheme 4. Substrate Scope for Suzuki–Miyaura C3 → C6 Diarylation of 6-Chloro-3-iodo-1-methyl-1H-pyrrolo[2,3-b]pyridine (9) or 6-Chloro-3-iodo-1-(4-methoxybenzyl)-1H-pyrrolo[2,3-b]pyridine (9′).
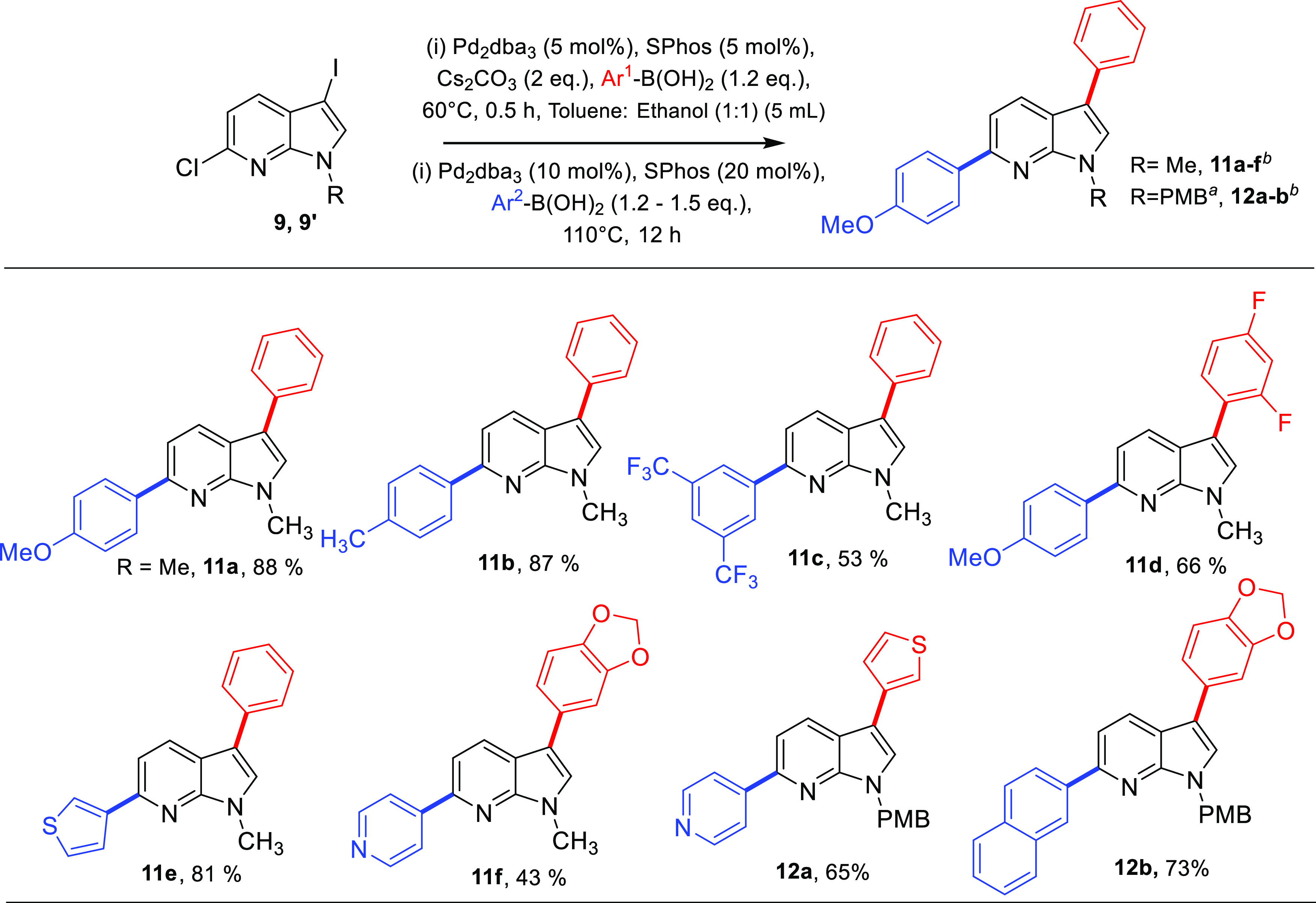
PMB = p-methoxybenzyl.
Isolated yields.
The HIV-1 IN inhibitory activity was evaluated for the 16 synthesized compounds, through in vitro biochemical strand transfer (ST) assay at 10 μM dose, as described by literature method (Tables 2 and S3).2310a which contains an unsubstituted phenyl ring exhibits as %ST = 14% (entry 2). A ∼3-fold increase in %ST inhibition is observed for para-substituted methyl on C3 aryl (10b) (entry 3). This could indicate the importance of para-substitution in the C3 monoaryl derivatives. Meta substitution on phenyl at C3 was detrimental to the biological activity (entry 4). Changing the para substituent to π -donor methoxy group (10d), we observed retention of %ST inhibition (%ST = 56%), which justifies the need of adding π-donor groups at the para-position. Fluoro derivatives 10e and 10f containing both para and meta substitution on C3 aryl shed light on the moderate to good %ST inhibition. 10g containing a bicyclic naphthyl on the C3 position showed a higher %ST inhibition. This higher activity cannot be fully explained as the naphthyl group is unsubstituted. Corroborating with our good results for p-substitution on C3 aryl, 10h containing a benzo[d][1,3]dioxole at the C3 position of 7-azaindoles showed good strand transfer inhibition of 54%. Among the monoaryl 7-azaindoles, the presence of π-donating para-substituent is observed to be beneficial for good %ST inhibition. The addition of aryl at C6 position (11a) was seen to give a ∼2-fold %ST inhibition when compared to 10a. The 3,6-diaryl 7-azaindoles in general proved to be better inhibitors than the C3 monoaryl derivatives as can be seen for 11b–c and 11e. 2,4-Difluoro phenyl at C3 and 4-methoxy phenyl at C6 (11d) presents a good %ST inhibition of 72%. The presence of 3-thienyl group at C6 decreased the inhibition of strand transfer in HIV-1 IN. Comparing 10h and 11f we notice a ∼1.3-fold increase in %ST inhibition when appending a 4-pyridyl ring at the C6 position. PMB-protected diaryl compounds (12a,b) were not effective against HIV-1 IN. The SAR study provides insight into the importance of the C6 aryl group to increase HIV-1 IN inhibition. In addition presence of π-donor groups on para-position and bicyclic nature of C3 aryl is incremental to %ST inhibition (Figure 2).
Table 2. Strand Transfer Inhibition Assay of HIV-1 IN and Cytotoxicity Study on TZM-bl Cell Lines of Synthesized 7-Azaindole Derivatives.
| entry | compound | IC50 in TZM-bl (μM)a | %ST inhibition (%)b |
|---|---|---|---|
| 1 | RALc | >200 | 82 |
| 2 | 10a | 17.47 | 14.41 |
| 3 | 10b | 25.26 | 41.03 |
| 4 | 10c | N.D.d | 19.42 |
| 5 | 10d | 79.52 | 55.96 |
| 6 | 10e | 5.53 | 38.34 |
| 7 | 10f | 88.54 | 48.28 |
| 8 | 10g | 170 | 55.82 |
| 9 | 10h | 44.87 | 53.97 |
| 10 | 11a | 50.46 | 39.31 |
| 11 | 11b | >200 | 51.72 |
| 12 | 11c | >200 | 36.41 |
| 13 | 11d | 49.32 | 71.61 |
| 14 | 11e | >200 | 41.65 |
| 15 | 11f | >200 | 71.27 |
| 16 | 12a | 17.29 | 15.87 |
| 17 | 12b | 58.12 | 24.14 |
IC50 values at 48 h.
ST%: percentage inhibition of strand transfer activity of integrase enzyme at 10 μM dose in WT (wild-type).
RAL = Raltegravir.
N.D. = not determined.
Figure 2.

Structure–activity relationship of synthesized derivatives; %ST = percentage strand transfer inhibition in HIV-1 IN, Th = thienyl.
The cell-based cytotoxicity was determined at 3.125 to 200 μM concentrations for all the synthesized derivatives (Table 2 and Figure S2). The C3 monoaryl 7-azaindoles (10a-h) were found to be toxic at IC50 values ranging from 5.53–170 μM. Among the eight diaryl derivatives, 11b, 11c, 11e, 11f, and 12b showed low cell toxicity, IC50 > 200 μM. Among the two molecules showing good inhibition of HIV-1 IN enzyme, 11d was found to have IC50 = 49.32 μM and 11f was found to have low cytotoxicity (IC50 > 200 μM) (Figure 3). Thus, results suggest, 11f could be a potential lead compound for further development as HIV-1 IN inhibitors.
Figure 3.

Cyto-toxicity of diaryl compounds 11d and 11f determined by MTT assay on TZM-bl cell line.
A crystal structure of a complex having HIV-1 IN bound with Raltegravir and associated DNA has not been determined yet. Prototype Foamy virus (PFV) and HIV-1 belong to the family Retroviridae, and both contain a highly conserved binding site of integrase.24 O’Hare reported the X-ray crystal structure of PFV-IN enzyme (PDB ID: 3OYA) complexed with DNA and cocrystallized with Raltegravir and Elvitegravir,5 and has been useful in guiding the design of new HIV-1 IN inhibitors.6b,25 The 16 synthesized derivatives of 7-azaindole scaffolds were subjected to molecular docking studies on PFV-IN (Table 3). The Glu221, Asp128, and Asp185 are bound to the two Mg2+ ions and form the catalytic site in PFV-IN. The cocrystallized Raltegravir is bound to the two Mg2+ ions through its diketo group and the Tyr212 forms a face-to-face π–π interaction with the oxadiazole group. The extra-precision molecular docking of all compounds proved 11f to have the best docking score = −7.29 kcal/mol, among other compounds. The docking results suggest hydrophobic interactions of arylated 7-azaindoles with Pro214 and Tyr212 in all synthesized molecules except 10c. Electrostatic interactions between Mg2+ and the N atom of pyridine were observed for 10a–h (Figure 4). Azine moiety of 10c showed π–π interaction with the aryl group of Tyr212, pi-cation interaction with the Arg329, and halogen bond with the Ser209, whereas no such interactions were observed in monoaryl 7-azaindoles. Among the diaryl 7-azaindoles, 11f exhibits electrostatic interactions between the oxygen atoms on the benzo[d][1,3]dioxole and two Mg2+ ions. In addition, a 4.2 Å π–π interaction between the azine group of 7-azaindole and Tyr212 was observed. The C6 4-pyridyl group formed aromatic H bonds with Gln186. These molecular recognitions within the PFV-IN binding pocket corroborate with the in vitro studies proving 11f to be a good starting point for further SAR study.
Table 3. Docking Study of Compounds on PFV IN Enzyme (PDB ID: 3OYA)a.
| compb | SP (glide score)c | XP (glide score)d | interaction with Mg2+e | H bondf | π–πg | hydropho-bic interaction | halogen bond |
|---|---|---|---|---|---|---|---|
| RAL | –11.03 | –9.34 | C=O···Mg2+ (2.08) | Tyr212 | Tyr212 | Tyr212 | |
| C–OH···Mg2+ (2.09 Å) | Pro211 | ||||||
| C–OH···Mg2+ (2.29 Å) | Pro214 | ||||||
| C=O···Mg2+ (1.83 Å) | |||||||
| 10a | –7.08 | –5.09 | Pyr(N)···Mg2+ (2.10 Å) | Pro214 | Tyr129 | ||
| 10b | –7.01 | –5.152 | Pyr(N)···Mg2+ (2.10 Å) | Tyr212 | Tyr129 | ||
| Pro214 | |||||||
| 10c | –7.08 | –4.51 | Tyr212 | Tyr212 | Ser209 | ||
| Arg329h | Pro214 | ||||||
| 10d | –7.08 | –6.25 | O···Mg2+ (1.95 Å) | Tyr212 | |||
| Pro214 | |||||||
| 10e | –6.91 | –5.05 | Pyr(N)···Mg2+ (2.10 Å) | Pro214 | Tyr129 | ||
| 10f | –6.37 | –5.07 | Pyr(N)···Mg2+ (2.16 Å) | Tyr212 | Tyr129 | ||
| Pro214 | |||||||
| 10g | –7.31 | –5.69 | Pyr(N)···Mg2+ (2.14 Å) | Tyr212 | Tyr129 | ||
| Pro214 | |||||||
| 10h | –6.98 | –7.1 | Pyr(N)···Mg2+ (2.17 Å) | Asp185 | Tyr212 | Tyr212 | |
| Pro214 | |||||||
| 11a | –6.45 | –5.51 | O···Mg2+ (2.02 Å) | Asp185 | Tyr212 | Tyr212 | |
| Pro211, Pro214 | |||||||
| 11b | –4.84 | –4.73 | Asp185Gln186 | Tyr212 | Tyr212 Pro214 | ||
| 11c | –3.88 | –2.89 | Tyr212 | Tyr212 Pro214 | |||
| 11d | –6.37 | –6.24 | Glu221, Asp185 | Tyr212, Arg329g | Tyr212 Pro211 | ||
| Pro214 | |||||||
| 11e | –4.55 | –4.98 | S···Mg2+ (2.41 Å) | Tyr212 Pro211 | |||
| Pro214 | |||||||
| 11f | –7.18 | –7.29 | O···Mg2+ (2.02 Å) | Gln186, Asp185 | Tyr212 | Tyr212 | |
| Pro211 | |||||||
| Pro214 | |||||||
| 12a | –7.36 | –7.05 | 4Pyr(N)···Mg2+ (2.01 Å) | Ala328 | Tyr212 | ||
| Pro214 | |||||||
| 12b | –7.75 | –6.97 | O···Mg2+ (2.15 Å) | Pro214 |
Docking scores of compounds: RAL, 10a–h, 11a–f, 12a–b.
Compound.
SP = Standard precision docking,
XP = Extra precision docking,
Electrostatic interaction with Mg2+ions and hydrogen bond distance in parentheses,
Hydrogen bonds,
π–π interactions,
π-cation interaction.
Figure 4.

Binding interactions of A) Raltegravir and B) 11f with the Mg2+ ions (lavender color) and Tyr212. (H-bonds: black; π–π interaction: purple; aromatic H bonds: blue).
Conclusion
This manuscript presents the synthesis of novel 3,6-diaryl 7-azaindoles in an efficient one-pot manner via a sequential Suzuki–Miyaura cross-coupling reaction on a 3-iodo-6-chloro 7-azaindole precursor. The C3 selectivity was established first on 3-iodo-6-chloro-N-methyl 7-azaindole (9) followed by optimization of a one-pot diarylation method. To show the substrate scope of this methodology, different electron-withdrawing and electron-donating arenes were introduced on 9/9′. The structure–activity relationship between synthesized derivatives and strand transfer inhibition of HIV-IN agree with the importance of para-substituent having a π-donor character on the C3-aryl and the addition of the arenes at C6 position enhances the %ST inhibition in HIV-1. Out of 16 derivatives of 7-azaindoles, 11f showed 71% inhibition of strand transfer activity of HIV-1 IN with minimal cytotoxicity in TZM-bl cells. Among the 16 compounds, benzo[d][1,3]dioxole substitution at the C3 position of 7-azaindole contributes toward the strong electrostatic interactions between Mg2+ ions of enzyme and the 7-azaindole derivatives. Thus, we surmise that the presence of π-donor atoms (O, N, F) at the para position of the C3 aryl of 7-azaindoles may be beneficial in interacting with the catalytic domain of the HIV-1 IN. These results prove that the diaryl 7-azaindoles can be developed to be potential candidates as HIV-1 IN inhibitors.
Experimental Section
All commercial reagents and solvents were obtained from the commercial provider and used without further purification. Thin-layer chromatography (TLC) was performed using precoated silica gel 60 F254 Merck. TLC plates were visualized by exposing UV light or by iodine vapors. Organic solutions were concentrated by rotary evaporation on BUCHI-Switzerland; R-210 rotary evaporator and vacuum pump V-700. Melting points of solid compounds were determined on BUCHI-B-540-Switzerland melting point apparatus and are uncorrected. 1H and 13C NMR spectra were recorded with BRUKER 500 and Jeol 400 MHz NMR instrument. Proton and carbon magnetic resonance spectra (1H NMR and 13C NMR) were recorded using tetramethylsilane (TMS) in the solvent of CDCl3 and DMSO-d6 as the internal standard. All the NMR spectra were processed in MestReNova. Mass spectra were recorded with Waters SYNAPT G2 with 2D nano ACQUITY System at USIC, Delhi University, India, and Agilent LCMS with Quadropole time-of-flight at AIRF, JNU, India.
Synthesis of 3-Iodo-1H-pyrrolo[2,3-b]pyridine (5)
This compound was synthesized using literature methods.16g,22 In a round-bottomed flask equipped with a magnetic bead was added 7-azaindole, 4 (10 g, 1 equiv, 84.65 mmol), along with KOH (0.5 equiv) and stirred for 30 min in DCM at room temperature. N-iodosuccinimide (1 equiv) was then added to it and stirred for 15 h. The reaction was monitored using TLC and on completion, it was washed with a saturated solution of sodium thiosulfate (3 × 100 mL) and brine (1 × 100 mL). The organic layer was dried over anhydrous sodium sulfate and evaporated using a rotary evaporator. The white solid obtained, 5 (19.62 g) was used further without purification. The NMR and the melting point matched with the literature report. Yield: 95%, mp = 189.5–190.3 °C. 1H NMR (400 MHz, DMSO-d6) δ 12.14 (brs, 1H), 8.27 (dd, J = 4.7 Hz, 1.5 Hz, 1H), 7.74 (s, 1H), 7.70 (dd, J = 7.9 Hz, 1.6 Hz, 1H), 7.17 (dd, J = 7.9 Hz, 4.7 Hz, 1H). 13C NMR (101 MHz, DMSO-d6) δ 148.5, 144.3, 131.0, 128.6, 122.5, 117.0, 54.9. HRMS (ESI) m/z calculated for C7H6IN2 [M + H]+ 244.9576, found 244.9575.
Synthesis of 3-Iodo-1H-pyrrolo[2,3-b]pyridine 7-oxide (6)
6 was synthesized using literature methods.26 In a round-bottomed flask containing a solution of 3-iodo-1H-pyrrolo[2,3-b]pyridine, 5 (18 g, 1 equiv, 73.75 mmol) in diethyl ether (100 mL) was added 77% m-chloroperbenzoic acid (2.5 equiv) portionwise. The mixture was stirred at room temperature for 1 h as white precipitation was observed. The reaction was checked for completion using TLC and filtered to form intermediate 6-int. 1H NMR (400 MHz, CDCl3) δ 13.00 (brs, 1H), 8.25 (dd, J = 6.1 Hz, 0.6 Hz, 1H), 7.93–7.90 (m, 2H), 7.73–7.70 (m, 2H), 7.56 (t, J = 8.1 Hz, 1H), 7.39 (dd, J = 8.1 Hz, 0.8 Hz, 1H), 7.19 (dd, J = 8.1 Hz, 6.1 Hz, 1H). 13C NMR (101 MHz, DMSO-d6) δ 166.6, 138.6, 133.8, 133.5, 133.2, 132.5, 131.3, 131.2, 129.4, 128.4, 126.7, 120.0, 117.4, 56.9.
The white solid (6-int) obtained was then dried and added to 200 mL of distilled water and to it saturated solution of potassium carbonate was added until the solution attained a pH of 9–10. The mixture was stirred overnight. The solid was filtered, washed with cold distilled water (50 mL), and dried to give 6 (17.65 g) as white solid, which was used further without purification. Yield = 92%.
Synthesis of Methyl 6-Chloro-3-iodo-1H-pyrrolo[2,3-b]pyridine-1-carboxylate (7)
7 was synthesized using available methods in literature.27 In a dry round-bottomed flask was added 3-iodo-1H-pyrrolo[2,3-b]pyridine 7-oxide, 6 (16 g, 1 equiv, 61.53 mmol) in dry THF. The mixture was purged with N2 gas and an inert atmosphere was maintained. HMDS (1 equiv) was added along with slow addition of orthochloroformate with constant stirring on an ice bath. The reaction was then stirred at room temperature for 1 h. On completion of the reaction, THF was evaporated and the mixture was extracted with ethyl acetate which was washed with a saturated solution of sodium bicarbonate (3 × 100 mL) followed by washing with brine solution (100 mL). The combined organic layer was dried using anhydrous sodium sulfate and purified using column chromatography (ethyl acetate/hexane (2:98)) to give 7 (9.5 g). Yield = 46%, white solid, mp = 101.4–102.3 °C. 1H NMR (400 MHz, CDCl3) δ 7.89 (s, 1H), 7.68 (d, J = 8.1 Hz, 1H), 7.32 (d, J = 8.3 Hz, 1H), 4.11 (s, 3H). 13C NMR (101 MHz, CDCl3)δ 149.4, 148.0, 145.4, 132.4, 131.0, 124.3, 120.2, 62.3, 54.9. HRMS (ESI) m/z calculated for C9H7ClIN2O2 [M + H]+ 336.9240, found 336.9253.
Synthesis of 6-Chloro-3-iodo-1H-pyrrolo[2,3-b]pyridine (8)
Methyl 6-chloro-3-iodo-1H-pyrrolo[2,3-b]pyridine-1-carboxylate, 7 (8 g, 1 equiv, 23.77 mmol), was added to methanol (200 mL) in a round-bottomed flask equipped with magnetic bead. 1 N NaOH (100 mL) was added to it and stirred overnight at room temperature. The solvent was evaporated and the product was extracted with ethyl acetate. The organic layer was washed with water and brine and dried over sodium sulfate. The organic layer was evaporated and the residue was purified using column chromatography on silica gel with ethyl acetate/hexanes (30:70) as eluent to give a white product, 8 (6.4 g). Yield = 98%, white solid, Decomposes >200 °C, 1H NMR (400 MHz, DMSO-d6) δ 12.29 (s, 1H), 7.72–7.69 (m, 2H), 7.16 (d, J = 7.7 Hz, 1H). 13C NMR (101 MHz, DMSO-d6) δ 147.2, 144.7, 131.8, 131.8, 121.6, 116.8, 55.3. HRMS (ESI) m/z calculated for C7H5ClIN2 [M + H]+ 278.9186, found 278.9201.
Synthesis of 6-Chloro-3-iodo-1-methyl-1H-pyrrolo[2,3-b]pyridine (9)
6-Chloro-3-iodo-1H-pyrrolo[2,3-b]pyridine, 8 (6 g, 1 equiv, 21.55 mmol), was taken in a round-bottomed flask, and DMF was added. NaH (1 equiv) was added to it portionwise at 0 °C-5 °C and stirred for 30 min in an N2 atmosphere. Methyl iodide (1.1 equiv) was then added to it and stirred for 3 h. On completion of the reaction as observed in TLC, ethyl acetate was added to the reaction mixture. The organic layer was washed with water and brine followed by drying using anhydrous sodium sulfate. The organic layer was evaporated and purification was done using column chromatography (ethyl acetate/hexanes (2:98)) to give white product, 9 (4.6 g). Yield = 73%, white solid, mp = 112.2–114.8 °C. 1H NMR (CDCl3, 500 MHz) δ 7.63 (d, J = 8.3 Hz, 1H), 7.25 (s, 1H), 7.12 (d, J = 8.2 Hz, 1H), 3.86 (s, 3H). 13C NMR (CDCl3, 126 MHz) δ 146.5, 145.9, 133.4, 131.6, 121.9, 116.7, 53.1, 31.8. HRMS (HRMS-ESI) m/z calculated for C8H7IClN2 [M + H]+ 292.9342, found: 292.9345.
Synthesis of 6-Chloro-3-iodo-1-(4-methoxybenzyl)-1H-pyrrolo[2,3-b]pyridine (9′)
6-Chloro-3-iodo-1H-pyrrolo[2,3-b]pyridine, 8 (6 g, 1 equiv, 21.55 mmol), was taken in a round-bottomed flask and DMF was added. K2CO3 (3 equiv) was added at room temperature followed by stirring for 10 min at N2 atmosphere. P-methoxy benzyl chloride (1.2 equiv) was then added to it and stirred for 10 h. On completion of the reaction as observed in TLC, ethyl acetate was added to the reaction mixture. The organic layer was washed with water and brine followed by drying using anhydrous sodium sulfate. The organic layer was evaporated and purification was done using column chromatography (ethyl acetate/hexanes (2:98–5:95)) to give a white product, 9′ (7.56 g). Yield = 88%, white solid, mp = 85.0–86.0 °C, 1H NMR (CDCl3, 500 MHz) δ 7.59 (d, J = 8.2 Hz, 1H), 7.18 (d, 2H), 7.16 (s, 1H), 7.11 (d, J = 8.2 Hz, 1H), 6.82 (d, J = 8.5 Hz, 2H), 5.32 (s, 2H), 3.75 (s, 3H). 13C NMR (CDCl3, 126 MHz) δ 159.4, 146.2, 145.8, 131.9, 131.6, 129.5, 128.5, 121.8, 116.9, 114.3, 55.3, 54.0, 47.8.
General Procedure for the Synthesis of C3-Arylated 7-Azaindoles
9 (0.2 g, 1 equiv, 0.684 mmol) was taken in a sealed tube and to it was added Ar1–B(OH)2 (1.2 equiv), Pd2dba3 (5 mol %), SPhos (5 mol %), and Cs2CO3 (2 equiv) in toluene/ethanol (1:1, 5 mL). The reaction was stirred at 60 °C for 15 min. On completion of the reaction monitored by TLC, the reaction mixture was diluted with ethyl acetate and filtered through a bed of Celite. The organic mixture was dried over sodium sulfate, concentrated, and purified through column chromatography (ethyl acetate/hexane (2:98 to 20:98)).
6-Chloro-1-methyl-3-phenyl-1H-pyrrolo[2,3-b]pyridine (10a)
85%, 0.141 g; white solid, mp = 116.9–118.5 °C. 1H NMR (400 MHz, CDCl3) δ 8.12 (d, J = 8.2 Hz, 1H, H8), 7.58 (d, J = 7.96 Hz, 2H, H4 and H4′), 7.44 (m, 2H, H5 and H5′), 7.34 (s, 1H, H2), 7.32–7.28 (m, 1H, H6), 7.12 (d, J = 8.2 Hz, 1H, H9), 3.91 (s, 3H, N–CH3). 13C NMR (101 MHz, CDCl3) δ 147.5 (C11), 144.9 (C10), 134.5 (C8), 130.5 (C3), 129.1 (C4, C4′), 127.0 (C5, C5′), 126.5(C6), 126.3 (C2), 117.2 (C7), 116.1 (C9), 115.6 (C3), 31.6 (N–CH3). HRMS (ESI) m/z calculated for C14H12ClN2 [M + H]+ 243.0689, found 243.0684.
6-Chloro-1-methyl-3-(p-tolyl)-1H-pyrrolo[2,3-b]pyridine (10b)
89%, 0.156 g; white solid, mp = 110.1–110.9 °C, 1H NMR (400 MHz, CDCl3) δ 8.09 (d, J = 8.24 Hz, 1H), 7.46 (d, J = 8.0 Hz, 2H), 7.29 (s, 1H), 7.25 (d, J = 7.8 Hz, 2H), 7.10 (d, J = 8.2 Hz, 1H), 3.89 (s, 3H), 2.39 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 147.5, 144.8, 136.2, 131.5, 130.5, 129.8, 126.9, 126.0, 117.3, 116.0, 115.6, 31.5, 21.2. HRMS (ESI) m/z calculated for C14H14ClN2 [M + H]+ 257.0846, found 257.0822.
6-Chloro-1-methyl-3-(m-tolyl)-1H-pyrrolo[2,3-b]pyridine (10c)
93%, 0.163 g; white solid, mp = 228.5–229.2 °C. 1H NMR (400 MHz, CDCl3) δ 8.12 (d, J = 8.2 Hz, 1H), 7.39–7.37 (m, 2H), 7.35–7.31 (m, 2H), 7.12–7.10 (m, 2H), 3.89 (s, 3H), 2.42 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 147.5, 144.8, 138.7, 134.4, 130.6, 129.0, 127.7, 127.3, 126.3, 124.1, 117.3, 116.1, 115.6, 31.6, 21.7. HRMS (ESI) m/z calculated for C14H14ClN2 [M + H]+ 257.0845, found 257.0851.
6-Chloro-3-(4-methoxyphenyl)-1-methyl-1H-pyrrolo[2,3-b]pyridine (10d)
93%, 0.173 g; white solid, mp = 111.0–112.1 °C. 1H NMR (500 MHz, CDCl3) δ 8.05 (d, J = 8.2 Hz, 1H), 7.48 (d, J = 8.5 Hz, 2H), 7.24 (s, 1H), 7.09 (d, J = 8.2 Hz, 1H), 6.98 (d, J = 8.7 Hz, 2H), 3.87 (s, 3H), 3.85 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 158.4, 147.3, 144.7, 130.4, 128.1, 126.9, 125.6, 117.2, 115.8, 115.2, 114.5, 55.4, 31.5. HRMS (ESI) m/z calculated for C14H14ClN2O [M + H]+ 273.0795, found 273.0802.
6-Chloro-3-(4-fluorophenyl)-1-methyl-1H-pyrrolo[2,3-b]pyridine (10e)
79%, 0.141 g; white solid, mp = 112.8–114.1 °C. 1H NMR (400 MHz, CDCl3) δ 8.05 (d, J = 8.2 Hz, 1H), 7.53–7.50 (m, 2H), 7.29 (s, 1H), 7.16–7.11 (m, 3H), 3.90 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 161.7 (d, J = 245.7 Hz), 147.4, 145.0, 130.5 (d, J = 3.1 Hz), 130.3, 128.5 (d, J = 7.8 Hz), 126.2, 117.1, 116.2, 116.1, 115.9, 114.6, 31.6. HRMS (ESI) m/z calculated for C14H11ClFN2 [M + H]+ 261.0595, found 261.0585.
3-(3,5-Bis(trifluoromethyl)phenyl)-6-chloro-1-methyl-1H-pyrrolo[2,3-b]pyridine (10f)
67%, 0.087 g starting from 0.1 g, 9; white solid, mp = 132.0–132.8 °C. 1H NMR (400 MHz, CDCl3) δ 8.08 (s, J = 8.3 Hz, 1H), 7.99 (s, 1H), 7.78 (s, 1H), 7.22 (d, J = 8.4 Hz, 1H), 3.96 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 147.5, 144.7, 136.7, 132.4 (q, J = 33.0 Hz), 129.9, 127.5, 126.4 (d, J = 3.3 Hz), 123.5 (d, J = 272.7 Hz), 119.8 (m), 117.1, 116.5, 112.8, 31.8. HRMS (ESI) m/z calculated for C16H10ClF6N2 [M + H]+ 379.0436, found 379.0416.
0.6-Chloro-1-methyl-3-(naphthalen-2-yl)-1H-pyrrolo[2,3-b]pyridine (10g)
92%, 0.092 g starting from 0.1 g 9; white solid, mp = 126.0–127.0 °C. 1H NMR (500 MHz, CDCl3) 8.20 (d, J = 8.2 Hz, 1H), 7.99 (s, 1H), 7.89–7.83 (m, 3H), 7.68 (d, J = 8.5 Hz, 1H), 7.52–7.43 (m, 2H), 7.41 (s, 1H), 7.14 (d, J = 8.2 Hz, 1H), 3.90 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 147.5, 144.9, 133.9, 132.2, 131.9, 130.6, 128.6, 127.8, 127.8, 126.7, 126.4, 125.6, 125.6, 124.8, 117.3, 116.2, 115.4, 31.6. HRMS (ESI) m/z calculated for C18 H14ClN2 [M + H]+ 293.0845, found 293.0821.
3-(Benzo[d][1,3]dioxol-5-yl)-6-chloro-1-methyl-1H-pyrrolo[2,3-b]pyridine (10h)
76%, 0.075 g starting from 0.1 g 9; white solid, mp = 136.0–137.2 °C. 1H NMR (500 MHz, CDCl3) δ 8.06 (d, J = 7.9 Hz, 1H), 7.24 (s, 1H), 7.11 (d, J = 8.0 Hz, 1H), 7.03 (s, 2H), 6.89 (d, J = 7.4 Hz, 1H), 6.00 (s, 2H), 3.89 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 148.2, 147.3, 144.8, 130.3, 128.4, 125.8, 120.3, 117.1, 115.9, 115.4, 108.9, 107.6, 101.1, 31.4. HRMS (ESI) m/z calculated for C15H11ClN2O2 [M + H]+ 287.0587, found 287.0597.
General One-Pot Synthesis of C3,C6-Diaryl 7-Azaindoles
9 (0.2 g, 0.1 equiv, 0.684 mmol) or 9′ (0.2 g, 1 equiv, 0.502 mmol) was added to the 15 mL sealed tube and to it was added Ar1–B(OH)2 (1.1 equiv) followed by Pd2dba3 (5 mol %), SPhos (5 mol %), and Cs2CO3 (2 equiv) in toluene/ethanol (1:1, 5 mL). The reaction was stirred at 60 °C for 0.25 h. The completion was noted using TLC analysis and Ar2–B(OH)2 (1.2–1.5 equiv) was added followed by the addition of Pd2dba3 (10 mol %) and SPhos (20 mol %) and stirred at 110 °C for 6–12 h. On completion of the reaction, the reaction mixture was diluted with ethyl acetate and filtered through a bed of Celite. The organic solution was dried over sodium sulfate, dried, and evaporated using the rotary evaporator. The crude was purified using ethyl acetate/hexane mixture (2:98–20:80) to obtain a pure product.
6-(4-Methoxyphenyl)-1-methyl-3-phenyl-1H-pyrrolo[2,3-b]pyridine, (11a)
88%, 0.189 g; white solid, mp = 135.5–136.8 °C. 1H NMR (500 MHz, CDCl3) δ 8.23 (d, J = 8.3 Hz, 1H, H8), 8.09 (d, J = 8.8 Hz, 2H, H14 and H14′)), 7.65 (d, J = 7.1 Hz, 2H, H4 and H4′), 7.55 (d, J = 8.3 Hz, 1H, H9), 7.45 (t, J = 7.7 Hz, 2H, H5 and H5′), 7.37 (s, 1H, H1), 7.28 (t, J = 7.4 Hz, 1H, H6), 7.02 (d, J = 8.8 Hz, 2H, H13 and H13′), 3.98 (s, 3H, O–CH3), 3.89 (s, 3H, N–CH3). 13C NMR (126 MHz, CDCl3) δ 160.0 (C15), 151.0 (C10), 148.5 (C11), 135.2 (C12), 133.0 (C8), 128.9 (C4 and C4′), 128.6, 128.3 (C5 and C5′), 126.8 (C13 and C13′), 126.2 (C1), 126.0 (C3), 116.8 (C7), 115.0 (C2), 114.1 (C14 and C14′), 112.9 (C9), 55.4 (O–CH3), 31.2 (N–CH3). HRMS (ESI) calculated for C21H18N2O [M + H]+ 315.1497, found 315.1510.
1-Methyl-3-phenyl-6-(p-tolyl)-1H-pyrrolo[2,3-b]pyridine (11b)
87%, 0.177 g; white solid, mp = 137.6–138.8 °C. 1H NMR (500 MHz, CDCl3) 8.20 (d, J = 8.3 Hz, 1H), 8.03 (d, J = 7.9 Hz, 2H), 7.62 (d, J = 7.4 Hz, 2H), 7.56 (d, J = 8.2 Hz, 1H), 7.42 (t, J = 7.5 Hz, 2H), 7.33 (s, 1H), 7.28- 7.27 (m, 3H), 3.93 (s, 3H), 2.40 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 151.3, 148.6, 138.2, 137.6, 135.2, 129.5, 129.0, 128.6, 126.9, 126.8, 126.4, 126.0, 117.1, 114.9, 113.2, 31.1, 21.4. HRMS (ESI) calculated for C21H18N2 [M + H]+ 299.1548, found 299.1562.
6-(3,5-Bis(trifluoromethyl)phenyl)-1-methyl-3-phenyl-1H-pyrrolo[2,3-b]pyridine (11c)
53%, 0.076 g from 0.1 g 9; light yellow solid, mp = 136.3–136.7 °C. 1H NMR (500 MHz, CDCl3) δ 8.61 (s, 2H), 8.32 (d, J = 8.2 Hz, 1H), 7.88 (s, 1H), 7.68–7.65 (m, 3H), 7.49–7.47 (m, 3H), 7.33 (t, J = 7.4 Hz, 1H), 4.04 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 148.5, 147.50, 142.3, 134.6, 131.9 (q, J = 33.2 Hz), 129.0, 129.0, 127.8, 126.9, 126.8 (d, J = 2.6 Hz), 126.4, 123.6 (d, J = 272.6 Hz), 121.6–121.5 (m), 118.5, 115.3, 113.2, 31.3. HRMS (ESI) calculated for C22H14F6N2 [M + H]+ 421.1139, found 421.1127.
3-(2,4-Difluorophenyl)-6-(4-methoxyphenyl)-1-methyl-1H-pyrrolo[2,3-b]pyridine (11d)
66%, 0.158 g; white solid, mp = 120.2–121.5 °C. 1H NMR (500 MHz, CDCl3) δ 8.08 (d, J = 8.7 Hz, 2H), 8.06 (d, J = 8.9 Hz, 1H), 7.62–7.58 (m, 1H), 7.54 (d, J = 8.3 Hz, 1H), 7.42 (s, 1H), 7.01 (d, J = 8.6 Hz, 2H), 6.98–6.93 (m, 2H), 3.97 (s, 3H), 3.86 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 161.2 (dd, J = 246.9 Hz, 11.8 Hz), 160.0, 159.6 (dd, J = 249.5 Hz, 11.8 Hz), 151.2, 148.1, 132.9, 130.1 (dd, J = 9.3, 5.7 Hz), 128.3, 128.3, 128.2, 119.0 (dd, J = 14.8, 3.9 Hz), 116.9, 114.1, 113.0, 111.5 (dd, J = 21.3, 3.6 Hz), 107.2, 104.5 (t, 26.3 Hz), 55.4, 31.2. HRMS (ESI) m/z calculated for C21H16FN2O [M + H]+ 351.1309, found 351.1295.
1-Methyl-3-phenyl-6-(thiophen-3-yl)-1H-pyrrolo[2,3-b]pyridine (11e)
81%, 0.081 g from 0.1 g 9; white solid, mp = 112.2–114.8 °C. 1H NMR (500 MHz, CDCl3) δ 8.20 (d, J = 8.2 Hz, 1H), 7.94–7.91 (m, 1H), 7.80 (d, J = 5.0 Hz, 1H), 7.63 (d, J = 7.9 Hz, 2H), 7.49 (d, J = 8.2 Hz, 1H), 7.44 (t, J = 7.7 Hz, 2H), 7.39 (m, 1H), 7.37 (s, 1H), 7.28 (t, J = 7.4 Hz, 1H), 3.96 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 148.4, 147.6, 143.3, 135.1, 128.9, 128.5, 126.8, 126.7, 126.3, 126.0, 125.9, 122.3, 117.1, 115.1, 113.4, 31.1. HRMS (ESI) m/z calculated for C18H14N2S [M + H]+ 291.0956, found 291.0968.
3-(Benzo[d][1,3]dioxol-5-yl)-1-methyl-6-(pyridin-4-yl)-1H-pyrrolo[2,3-b]pyridine (11f)
43%, 0.049 g; yellow solid, mp = 167.8–168.9 °C. 1H NMR (500 MHz, CDCl3) δ 8.71 (d, J = 5.3 Hz, 2H), 8.21 (d, J = 8.2 Hz, 1H), 8.03 (d, J = 5.1 Hz, 2H), 7.64 (d, J = 8.2 Hz, 1H), 7.35 (s, 1H), 7.09–7.07 (m, 2H), 6.91 (d, J = 8.4 Hz, 1H), 6.00 (s, 2H), 3.97 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 150.2, 148.4, 148.2, 147.8, 147.3, 146.2, 128.7, 128.5, 127.3, 121.1, 120.2, 118.7, 115.1, 113.3, 108.9, 107.6, 101.1, 31.1. HRMS (ESI) m/z calculated for C20H15N3O2 [M + H]+ 330.1242, found 330.1221.
1-(4-Methoxybenzyl)-6-(pyridin-4-yl)-3-(thiophen-3-yl)-1H-pyrrolo[2,3-b]pyridine (12a)
65%, 0.065 g; yellow solid, mp = 154.0–155.0 °C. 1H NMR (500 MHz, CDCl3) δ 8.72 (s, 1H), 8.26 (d, J = 8.2 Hz, 1H), 8.06 (d, J = 4.3 Hz, 1H), 7.71 (d, J = 8.2 Hz, 1H), 7.43–7.40 (m, 2H), 7.35 (d, J = 2.7 Hz, 1H), 7.30 (d, J = 8.2 Hz, 1H), 6.86 (d, J = 8.3 Hz, 1H), 5.52 (s, 2H), 3.78 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 159.3, 150.3, 148.1, 147.9, 147.2, 135.0, 129.5, 129.3, 128.8, 126.5, 126.4, 126.0, 121.1, 118.8, 118.6, 114.2, 113.6, 111.2, 55.3, 47.4. HRMS (ESI) m/z calculated for C24H19N3OS [M + H]+ 398.1327, found 398.1328.
3-(Benzo[d][1,3]dioxol-5-yl)-1-(4-methoxybenzyl)-6-(naphthalen-2-yl)-1H-pyrrolo[2,3-b]pyridine (12b)
73%, 0.177 g, white solid, mp = 152.4–153.4 °C. 1H NMR (500 MHz, CDCl3) δ 8.59 (s, 1H), 8.37 (d, J = 8.5 Hz, 1H), 8.24 (d, J = 8.2 Hz, 1H), 7.96–7.95 (m, 2H), 7.88 (d, J = 7.4 Hz, 1H), 7.79 (d, J = 8.2 Hz, 1H), 7.54–7.46 (m, 2H), 7.34 (d, J = 8.1 Hz, 2H), 7.29 (s, 1H), 7.10–7.08 (m, 2H), 6.92–6.85 (m, 3H), 5.99 (s, 2H), 5.56 (s, 2H), 3.78 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 159.2, 150.9, 148.3, 148.1, 146.1, 137.6, 133.7, 133.4, 129.9, 129.5, 129.1, 128.6, 128.5, 128.3, 127.7, 126.2, 126.0, 125.1, 124.9, 120.1, 117.4, 115.5, 114.2, 113.8, 108.8, 107.6, 101.0, 55.3, 47.4. HRMS (ESI) m/z calculated for C32H24N2O3 [M + H]+ 485.1865, found 485.1762.
HIV-1 Integrase Strand Transfer Inhibition Assays
The assay for the detection of HIV-1 integrase strand transfer (ST) inhibitors was adapted from previously described methods.21 A double-stranded biotinylated donor DNA, corresponding to the HIV U5 viral DNA end, was added to the wells of Streptavidin-coated 96-well microtiter plates (R&D systems). Following a 1 h incubation at r.t. and three wash steps with PBS, purified recombinant integrase (1 μM) was assembled using DTT (dithiothreitol, 1 M) onto the preprocessed donor DNA through incubation for 30 min at 22 °C. After the wash step, the test compounds and a positive control inhibitor (Raltegravir, Merck) were added into individual wells at a final concentration of 10 μM. The microtiter plates were incubated for 30 min at 37 °C and washed. The strand transfer reaction was initiated through the addition of FITC-labeled target dsDNA (5′-TGACCAAGGGCTAATTCACT-FITC-3′ and 5′-AGTGAATTAGCCCTTGGTCA-FITC-3′). The plates were incubated for a period of 1 h at 37 °C followed by washing as before. An AP (Alkaline S2 phosphatase)-conjugated anti-FITC secondary antibody (Sigma) was added and the plates were washed. The chromogenic substrate (BluePhos, KPL) was added to allow for photometric measurement at 620 nm using a microplate reader (xMark, Biorad). Percentage inhibition was calculated utilizing the formula:
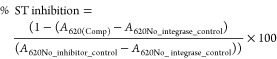 |
where A620(Comp) = Absorbance at 620 nm with compounds, A620_No_integrase_control = Absorbance at 620 nm containing no enzyme, and A620_No_inhibitor_control = Absorbance at 620 nm containing no inhibitor. All inhibition values are the average of triplicate experiments.
Cytotoxicity Assay of Compounds on Mammalian Cells
The in vitro cytotoxicity assay of various test compounds was performed on TZM-BL cells and incubated at 37 °C and 5% CO2 in an incubator for 24 h. The chemical compounds were tested at 24, 48, and 72 h with various concentrations ranging from 3.125 to 200 μM. Following the incubation period, test compounds were removed from each well of the culture plate, and 50 μL of MTT reagent (3-(4, 5-dimethylthiazolyl-2)-2,5-diphenyltetrazolium bromide) (5 mg/mL) was added and incubated for 4 h. The MTT solution was then discarded, and 150 μL of DMSO was added to each well. The plates were placed on a shaker to solubilize the purple formazan crystals. The absorbance was measured using a microplate reader (TECAN) at a wavelength of 570 nm with reference wavelength of 630 nm. The results were plotted as cell viability percentage against compound concentrations.
Acknowledgments
S.C. thanks CSIR, P.Y. thanks DST, and A.A. thanks DBT for financial support. The work was supported by the DST and DBT grant at V.T.’s lab, SCMM, JNU. The authors are grateful for facilities provided by AIRF-JNU and USIC-DU for compound characterization.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.2c07372.
Optimization for C3-arylation of 3-iodo-6-chloro-N-methyl-7-azaindole, X-ray Crystal data of 11a, HIV-1 integrase inhibition assay at 10 μM drug concentration, Plot of % viability of cells and concentration of compounds 10a–h, 11a–f, 12a,b (MTT assay), 1H and 13C spectra (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- a Brooks K. M.; Sherman E. M.; Egelund E. F.; Brotherton A.; Durham S.; Badowski M. E.; Cluck D. B. Integrase Inhibitors: After 10 Years of Experience, Is the Best Yet to Come?. Pharmacotherapy 2019, 39 (5), 576–598. 10.1002/phar.2246. [DOI] [PubMed] [Google Scholar]; b Smith S. J.; Zhao X. Z.; Passos D. O.; Lyumkis D.; Burke T. R.; Hughes S. H. Integrase Strand Transfer Inhibitors Are Effective Anti-HIV Drugs. Viruses 2021, 13 (2), 205. 10.3390/v13020205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu T. K.; Davies D. R. Structure and function of HIV-1 integrase. Curr. Top. Med. Chem. 2004, 4 (9), 965–977. 10.2174/1568026043388547. [DOI] [PubMed] [Google Scholar]
- Craigie R. The molecular biology of HIV integrase. Future Virol. 2012, 7 (7), 679–686. 10.2217/fvl.12.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Naidu B. N.; Patel M.; McAuliffe B.; Ding B.; Cianci C.; Simmermacher J.; Jenkins S.; Parker D. D.; Sivaprakasam P.; Khan J. A.; Kish K.; Lewis H.; Hanumegowda U.; Krystal M.; Meanwell N. A.; Kadow J. F. Design, Synthesis, and Preclinical Profiling of GSK3739936 (BMS-986180), an Allosteric Inhibitor of HIV-1 Integrase with Broad-Spectrum Activity toward 124/125 Polymorphs. J. Med. Chem. 2022, 65 (6), 4949–4971. 10.1021/acs.jmedchem.1c02169. [DOI] [PubMed] [Google Scholar]; b Cook N. J.; Li W.; Berta D.; Badaoui M.; Ballandras-Colas A.; Nans A.; Kotecha A.; Rosta E.; Engelman A. N.; Cherepanov P. Structural basis of second-generation HIV integrase inhibitor action and viral resistance. Science 2020, 367 (6479), 806–810. 10.1126/science.aay4919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hare S.; Vos A. M.; Clayton R. F.; Thuring J. W.; Cummings M. D.; Cherepanov P. Molecular mechanisms of retroviral integrase inhibition and the evolution of viral resistance. Proc. Natl. Acad. Sci. 2010, 107 (46), 20057–20062. 10.1073/pnas.1010246107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a You J.; Wang H.; Huang X.; Qin Z.; Deng Z.; Luo J.; Wang B.; Li M. Therapy-Emergent Drug Resistance to Integrase Strand Transfer Inhibitors in HIV-1 Patients: A Subgroup Meta-Analysis of Clinical Trials. PLoS One 2016, 11 (8), e0160087 10.1371/journal.pone.0160087. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Smith S. J.; Zhao X. Z.; Passos D. O.; Pye V. E.; Cherepanov P.; Lyumkis D.; Burke T. R. Jr.; Hughes S. H. HIV-1 Integrase Inhibitors with Modifications That Affect Their Potencies against Drug Resistant Integrase Mutants. ACS Infect. Dis. 2021, 7 (6), 1469–1482. 10.1021/acsinfecdis.0c00819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco J.; Varghese V.; Rhee S.; Gatell J. M.; Shafer R. W. HIV-1 Integrase Inhibitor Resistance and Its Clinical Implications. J. Infect. Dis. 2011, 203 (9), 1204–1214. 10.1093/infdis/jir025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Underwood M.; Horton J.; Nangle K.; Hopking J.; Smith K.; Aboud M.; Wynne B.; Sievers J.; Stewart E. L.; Wang R. Integrase Inhibitor Resistance Mechanisms and Structural Characteristics in Antiretroviral Therapy-Experienced, Integrase Inhibitor-Naive Adults with HIV-1 Infection Treated with Dolutegravir plus Two Nucleoside Reverse Transcriptase Inhibitors in the DAWNING Study. Antimicrob. Agents Chemother. 2022, 66 (1), e0164321 10.1128/AAC.01643-21. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Revollo B.; Viñuela L.; de la Mora L.; García F.; Noguera-Julián M.; Parera M.; Paredes R.; Llibre J. M. Integrase resistance emergence with dolutegravir/lamivudine with prior HIV-1 suppression. J. Antimicrob. Chemother. 2022, 77 (6), 1738–1740. 10.1093/jac/dkac082. [DOI] [PubMed] [Google Scholar]
- a Stanton R. A.; Lu X.; Detorio M.; Montero C.; Hammond E. T.; Ehteshami M.; Domaoal R. A.; Nettles J. H.; Feraud M.; Schinazi R. F. Discovery, characterization, and lead optimization of 7-azaindole non-nucleoside HIV-1 reverse transcriptase inhibitors. Bioorg. Med. Chem. Lett. 2016, 26 (16), 4101–5. 10.1016/j.bmcl.2016.06.065. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Urvashi; Senthil Kumar J. B.; Das P.; Tandon V. Development of Azaindole-Based Frameworks as Potential Antiviral Agents and Their Future Perspectives. J. Med. Chem. 2022, 65 (9), 6454–6495. 10.1021/acs.jmedchem.2c00444. [DOI] [PubMed] [Google Scholar]; c Bandarage U. K.; Clark M. P.; Perola E.; Gao H.; Jacobs M. D.; Tsai A.; Gillespie J.; Kennedy J. M.; Maltais F.; Ledeboer M. W.; Davies I.; Gu W.; Byrn R. A.; Nti Addae K.; Bennett H.; Leeman J. R.; Jones S. M.; O’Brien C.; Memmott C.; Bennani Y.; Charifson P. S. Novel 2-Substituted 7-Azaindole and 7-Azaindazole Analogues as Potential Antiviral Agents for the Treatment of Influenza. ACS Med. Chem. Lett. 2017, 8 (2), 261–265. 10.1021/acsmedchemlett.6b00487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plewe M. B.; Butler S. L. R.; Dress K.; Hu Q.; Johnson T. W.; Kuehler J. E.; Kuki A.; Lam H.; Liu W.; Nowlin D.; et al. Azaindole hydroxamic acids are potent HIV-1 integrase inhibitors. J. Med. Chem. 2009, 52 (22), 7211–7219. 10.1021/jm900862n. [DOI] [PubMed] [Google Scholar]
- Tanis S. P.; Plewe M. B.; Johnson T. W.; Butler S. L.; Dalvie D.; DeLisle D.; Dress K. R.; Hu Q.; Huang B.; Kuehler J. E.; et al. Azaindole N-methyl hydroxamic acids as HIV-1 integrase inhibitors-II. The impact of physicochemical properties on ADME and PK. Bioorg. Med. Chem. Lett. 2010, 20 (24), 7429–7434. 10.1016/j.bmcl.2010.10.022. [DOI] [PubMed] [Google Scholar]
- Goldgur Y.; Craigie R.; Cohen G. H.; Fujiwara T.; Yoshinaga T.; Fujishita T.; Sugimoto H.; Endo T.; Murai H.; Davies D. R. Structure of the HIV-1 integrase catalytic domain complexed with an inhibitor: a platform for antiviral drug design. Proc. Natl. Acad. Sci. 1999, 96 (23), 13040–13043. 10.1073/pnas.96.23.13040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Lipczynska-Kochany E.; Iwamura H.; Takahashi K.; Hakura A.; Kawazoe Y. Mutagenicity of pyridine-and quinoline-carbohydroxamic acid derivatives. Mutation Research/Genetic Toxicology 1984, 135 (3), 139–148. 10.1016/0165-1218(84)90114-9. [DOI] [PubMed] [Google Scholar]; b Skipper P. L.; Tannenbaum S. R.; Thilly W. G.; Furth E. E.; Bishop W. W. Mutagenicity of hydroxamic acids and the probable involvement of carbamoylation. Cancer Res. 1980, 40 (12), 4704–4708. [PubMed] [Google Scholar]
- Pryde D. C.; Webster R.; Butler S. L.; Murray E. J.; Whitby K.; Pickford C.; Westby M.; Palmer M. J.; Bull D. J.; Vuong H.; et al. Discovery of an HIV integrase inhibitor with an excellent resistance profile. Med. Chem. Comm. 2013, 4 (4), 709–719. 10.1039/c3md00014a. [DOI] [Google Scholar]
- a Walker S. R.; Carter E. J.; Huff B. C.; Morris J. C. Variolins and related alkaloids. Chem. Rev. 2009, 109 (7), 3080–3098. 10.1021/cr900032s. [DOI] [PubMed] [Google Scholar]; b Welsch M. E.; Snyder S. A.; Stockwell B. R. Privileged scaffolds for library design and drug discovery. Curr. Opin. Chem. Biol. 2010, 14 (3), 347–361. 10.1016/j.cbpa.2010.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Xu X.; Ou M.; Wang Y.-E.; Lin T.; Xiong D.; Xue F.; Walsh P. J.; Mao J. Alkali-amide controlled selective synthesis of 7-azaindole and 7-azaindoline through domino reactions of 2-fluoro-3-methylpyridine and aldehydes. Org. Chem. Front. 2022, 9 (9), 2541–2548. 10.1039/D2QO00339B. [DOI] [Google Scholar]; b Pires M. J.; Poeira D. L.; Purificacao S. I.; Marques M. M. B. Synthesis of substituted 4-, 5-, 6-, and 7-azaindoles from aminopyridines via a cascade C–N cross-coupling/heck reaction. Org. Lett. 2016, 18 (13), 3250–3253. 10.1021/acs.orglett.6b01500. [DOI] [PubMed] [Google Scholar]; c Kim Y.; Hong S. Rh (iii)-catalyzed 7-azaindole synthesis via C–H activation/annulative coupling of aminopyridines with alkynes. Chem. Commun. 2015, 51 (56), 11202–11205. 10.1039/C5CC03497C. [DOI] [PubMed] [Google Scholar]; d Santos A. S.; Mortinho A. C.; Marques M. M. B. Metal-catalyzed cross-coupling reactions on azaindole synthesis and functionalization. Molecules 2018, 23 (10), 2673. 10.3390/molecules23102673. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Kim Y.; Hong S. Rh(iii)-catalyzed 7-azaindole synthesis via C–H activation/annulative coupling of aminopyridines with alkynes. Chem. Commun. 2015, 51 (56), 11202–11205. 10.1039/C5CC03497C. [DOI] [PubMed] [Google Scholar]; f Price D. J.; Drewry D. H.; Schaller L. T.; Thompson B. D.; Reid P. R.; Maloney P. R.; Liang X.; Banker P.; Buckholz R. G.; Selley P. K.; McDonald O. B.; Smith J. L.; Shearer T. W.; Cox R. F.; Williams S. P.; Reid R. A.; Tacconi S.; Faggioni F.; Piubelli C.; Sartori I.; Tessari M.; Wang T. Y. An orally available, brain-penetrant CAMKK2 inhibitor reduces food intake in rodent model. Bioorg. Med. Chem. Lett. 2018, 28 (10), 1958–1963. 10.1016/j.bmcl.2018.03.034. [DOI] [PubMed] [Google Scholar]; g Gourdain S.; Dairou J.; Denhez C.; Bui L. C.; Rodrigues-Lima F.; Janel N.; Delabar J. M.; Cariou K.; Dodd R. H. Development of DANDYs, new 3, 5-diaryl-7-azaindoles demonstrating potent DYRK1A kinase inhibitory activity. J. Med. Chem. 2013, 56 (23), 9569–9585. 10.1021/jm401049v. [DOI] [PubMed] [Google Scholar]
- Almond-Thynne J.; Blakemore D. C.; Pryde D. C.; Spivey A. C. Site-selective Suzuki–Miyaura coupling of heteroaryl halides – understanding the trends for pharmaceutically important classes. Chem. Sci. 2017, 8 (1), 40–62. 10.1039/C6SC02118B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardoza S.; Das P.; Tandon V. Pd-Catalyzed Sequential Arylation of 7-Azaindoles: Aggregate-Induced Emission of Tetra-Aryl 7-Azaindoles. J. Org. Chem. 2019, 84 (21), 14015–14029. 10.1021/acs.joc.9b02187. [DOI] [PubMed] [Google Scholar]
- Nishiguchi G. A.; Atallah G.; Bellamacina C.; Burger M. T.; Ding Y.; Feucht P. H.; Garcia P. D.; Han W.; Klivansky L.; Lindvall M. Discovery of novel 3, 5-disubstituted indole derivatives as potent inhibitors of Pim-1, Pim-2, and Pim-3 protein kinases. Bioorg. Med. Chem. Lett. 2011, 21 (21), 6366–6369. 10.1016/j.bmcl.2011.08.105. [DOI] [PubMed] [Google Scholar]
- Schirok H. Microwave-Assisted Flexible Synthesis of 7-Azaindoles. J. Org. Chem. 2006, 71 (15), 5538–5545. 10.1021/jo060512h. [DOI] [PubMed] [Google Scholar]
- a Yadav P.; Sur S.; Desai D.; Kulkarni S.; Sharma V.; Tandon V. Interaction of HIV-1 integrase with polypyrimidine tract binding protein and associated splicing factor (PSF) and its impact on HIV-1 replication. Retrovirology 2019, 16 (1), 12. 10.1186/s12977-019-0474-1. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Singh R.; Yadav P.; Urvashi; Tandon V. Novel Dioxolan Derivatives of Indole as HIV-1 Integrase Strand Transfer Inhibitors Active Against RAL Resistant Mutant Virus. ChemistrySelect 2016, 1 (17), 5471–5478. 10.1002/slct.201601024. [DOI] [Google Scholar]; c Tandon V.; Urvashi; Yadav P.; Sur S.; Abbat S.; Tiwari V.; Hewer R.; Papathanasopoulos M. A.; Raja R.; Banerjea A. C.; et al. Design, Synthesis, and Biological Evaluation of 1,2-Dihydroisoquinolines as HIV-1 Integrase Inhibitors. ACS Med. Chem. Lett. 2015, 6 (10), 1065–1070. 10.1021/acsmedchemlett.5b00230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urvashi; Dar M. O.; Bharatam P. V.; Das P.; Kukreti S.; Tandon V. Cu(II)-catalyzed sulfonylation of 7-azaindoles using DABSO as SO2-Source and its mechanistic study. Tetrahedron 2020, 76 (28-29), 131337. 10.1016/j.tet.2020.131337. [DOI] [Google Scholar]
- David C. A.; Middleton T.; Montgomery D.; Lim H. B.; Kati W.; Molla A.; Xuei X.; Warrior U.; Kofron J. L.; Burns D. J. Microarray Compound Screening (μARCS) to Identify Inhibitors of HIV Integrase. J. Biomol. Screen. 2002, 7 (3), 259–266. 10.1089/108705702760047754. [DOI] [PubMed] [Google Scholar]
- a Kulkosky J.; Jones K. S.; Katz R. A.; Mack J. P.; Skalka A. M. Residues critical for retroviral integrative recombination in a region that is highly conserved among retroviral/retrotransposon integrases and bacterial insertion sequence transposases. Mol. Cell. Biol. 1992, 12 (5), 2331–2338. 10.1128/MCB.12.5.2331. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Engelman A.; Craigie R. Identification of conserved amino acid residues critical for human immunodeficiency virus type 1 integrase function in vitro. J.Virol. 1992, 66 (11), 6361–6369. 10.1128/jvi.66.11.6361-6369.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha C. H. X.; Lee N. K.; Rahman T.; Hwang S. S.; Yam W. K.; Chee X. W. Repurposing FDA-approved drugs as HIV-1 integrase inhibitors: an in silico investigation. J. Biomol. Struct. Dyn. 2022, 1–14. 10.1080/07391102.2022.2028677. [DOI] [PubMed] [Google Scholar]
- Huestis M. P.; Fagnou K. Site-selective azaindole arylation at the azine and azole rings via N-oxide activation. Org. Lett. 2009, 11 (6), 1357–1360. 10.1021/ol900150u. [DOI] [PubMed] [Google Scholar]
- Minakata S.; Itoh S.; Komatsu M.; Ohshiro Y. Functionalization of 1 H-Pyrrolo [2, 3-b] pyridine. Bull. Chem. Soc. Jpn. 1992, 65 (11), 2992–2997. 10.1246/bcsj.65.2992. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.