

Abstract
Objective:
We sought to define how sensory neurotransmitters substance P and calcitonin gene-related peptide (CGRP) affect membrane potential of vascular smooth muscle and endothelium.
Methods:
Microelectrodes recorded membrane potential of smooth muscle from pressurized mouse mesenteric arteries (diameter, ~150 μm) and in endothelial tubes.
Results:
Resting potential was similar (~ −45 mV) for each cell layer. Substance P hyperpolarized smooth muscle and endothelium ~ −15 mV; smooth muscle hyperpolarization was abolished by endothelial disruption or NO synthase inhibition. Blocking KCa channels (apamin + charybdotoxin) attenuated hyperpolarization in both cell types. CGRP hyperpolarized endothelium and smooth muscle ~ −30 mV; smooth muscle hyperpolarization was independent of endothelium. Blocking KCa channels prevented hyperpolarization to CGRP in endothelium but not smooth muscle. Inhibiting KATP channels with glibenclamide or genetic deletion of KIR6.1 attenuated hyperpolarization in smooth muscle but not endothelium. Pinacidil (KATP channel agonist) hyperpolarized smooth muscle more than endothelium (~ −35 vs. ~ −20 mV).
Conclusions:
Calcitonin gene-related peptide elicits greater hyperpolarization than substance P. Substance P hyperpolarizes both cell layers through KCa channels and involves endothelium-derived NO in smooth muscle. Endothelial hyperpolarization to CGRP requires KCa channels, while KATP channels mediate hyperpolarization in smooth muscle. Differential K+ channel activation in smooth muscle and endothelium through sensory neurotransmission may selectively tune mesenteric blood flow.
Keywords: membrane potential, neurotransmitters, nitric oxide, sensory nerves
1 |. INTRODUCTION
Perivascular nerves are integral to blood flow regulation. Activation of sensory nerves releases calcitonin gene-related peptide (CGRP) and substance P (SP).1,2 Of these neurotransmitters, CGRP has been most widely studied and elicits vasodilation via binding to its receptors on vascular smooth muscle cells (SMCs) and endothelial cells (ECs).3,4 Functional CGRP receptors are composed of three subunits: calcitonin receptor-like receptor (CRLR), receptor activity modifying protein 1 (RAMP1), and receptor component protein.5 In SMCs from mesenteric,6 renal,7 and coronary8 arteries, CGRP elicits vasorelaxation via protein kinase A-mediated activation of K+ channels, with a primary contribution of ATP-sensitive K+ (KATP) channels.6,8–11 The ensuing efflux of K+ leads to hyperpolarization, which reduces constitutive voltage-gated calcium channel (VGCC) activity and thereby attenuates SMC [Ca2+]i to promote vasodilation.12 CGRP can also elicit endothelium-dependent vasodilation through production of NO, which further hyperpolarizes SMCs.13 While these effects of CGRP on membrane potential (Vm) of SMCs have been resolved, little is known of how CGRP may affect Vm of ECs.
How SP affects Vm of SMCs and ECs has received little attention. Nevertheless, endothelial disruption abolishes dilation of mesenteric arteries (MAs) to SP.14,15 Binding to neurokinin receptors on ECs,16 SP increases [Ca2+]i to activate endothelial NO synthase,15 with NO able to trigger SMC membrane hyperpolarization through Ca2+-activated K+ (KCa) and KATP channels.17 While KCa channels have been implicated in hyperpolarization of cultured porcine coronary ECs to SP,18,19 how SP may affect Vm in SMCs of pressurized resistance arteries or their native ECs has not been determined.
The objective of this study was to define how CGRP and SP effect Vm of SMCs and ECs in MAs freshly isolated from the mouse. Vessels were cannulated and pressurized with intact or denuded endothelium to evaluate EC-dependent and intrinsic SMC responses, with ECs studied in endothelial tubes following dissociation of SMCs. With both vasodilatory neurotransmitters released from perivascular sensory nerves, we hypothesized that CGRP and SP have similar actions on KCa and KATP channels in respective cell layers. Our findings illustrate that EC hyperpolarization to CGRP is mediated primarily by KCa channels, as is hyperpolarization of both ECs and SMCs to SP. In contrast, SMC hyperpolarization to CGRP is mediated by KATP channels. Such differences in how sensory neurotransmission affects Vm in respective cell layers of resistance arteries may be integral to precisely regulating local blood flow.
2 |. MATERIALS AND METHODS
2.1 |. Animal care and use
All protocols and experimental procedures were reviewed and approved by the Animal Care and Use Committee of the University of Missouri (Columbia, MO; Protocol #9220), were performed in compliance with all local, state, and federal guidelines for the humane care and use of animals, and in accord with the animal ethics guidelines for this journal. Experiments were performed on C57BL/6J male “wildtype” mice (n = 75, 27–32 g; 4–6 months old) purchased from Jackson Laboratorie (Bar Harbor, ME, USA). Complementary experiments were performed on MAs from KATP/KIR6.1 knockout mice (KATP−/−, mice lacking KCNJ8 gene on C57BL/6 background; n = 6) obtained from Dr. Michael Davis (University of Missouri; Columbia, MO).20
Mouse MAs provide an animal model which recapitulates aspects of human vascular behavior.21 Mice were housed on a 12:12 h light-dark cycle at ~23°C with fresh water and food available ad libitum. Male mice were utilized to avoid sex differences as a confounding variable. Each mouse was anesthetized with ketamine and xylazine (100 and 10 mg kg−1, respectively; intraperitoneal injection) for tissue harvest and killed by exsanguination.
2.2 |. Materials
2.2.1 |. Reagents
Acetylcholine (ACh, Cat. #A6625), glibenclamide (Cat. #G2539), NG-nitro-l-arginine methyl ester (L-NAME; Cat. #N5751), norepinephrine (NE, Cat. #A7256), and pinacidil (Cat. #P154) were acquired from Sigma-Aldrich (St. Louis, MO, USA). Apamin (Cat. #60772), CGRP (Cat. #20682), charybdotoxin (Cat. #28244), and SP (Cat. #24279) were acquired from AnaSpec (Fremont, CA, USA).
2.2.2 |. Solutions
Isolated arteries and endothelial tubes were superfused with physiological salt solution (PSS; pH 7.4) containing (in mM): 140 NaCl (Fisher Scientific, Pittsburgh, PA, USA), 5 KCl (Fisher), 2 CaCl2 (Fisher), 1 MgCl2 (Sigma), 10 HEPES (Sigma), and 10 glucose (Fisher). During vessel dissection and isolation of endothelial tubes, the PSS contained 0.1% bovine serum albumin (#10856, USB Corp., Cleveland, OH, USA). During dissection, CaCl2 was absent and 10−5 M sodium nitroprusside was added to relax SMCs. For dissociation of SMCs, 10−4 M CaCl2 (Sigma) was included (required for enzyme activity) and sodium nitroprusside was omitted.
2.3 |. Vascular preparations
The intestines were accessed via a laparotomy, excised, and pinned on to transparent silicone rubber (Sylgard 184; Dow Corning, Midland, MI, USA) then immersed in PSS within a dissection chamber maintained at 4°C. Individual MAs [inner diameter (ID), ~150 μm] supplying the small intestine were dissected and transferred to a tissue chamber (RC-27N, volume ~1 mL; Warner Instruments, Hamden, CT, USA) secured on an aluminum platform (length: 24 cm; width: 14.5 cm; thickness: 0.4 cm). Cannulation pipettes were pulled (P-97; Sutter Instruments, Novato, CA, USA) from glass capillaries (Cat. #64–1781, Warner; 0.94 mm ID, 1.2 mm outer diameter) and heat-polished (outer diameter, 100 μm). Each pipette was secured in a custom holder (Warner) held in a three-axis micromanipulator (DT3-100; Siskiyou Corp., Grants Pass, OR, USA) secured to the aluminum platform at each end of the tissue chamber. Pipettes were positioned in the chamber; individual MAs were cannulated and secured at each end with a strand of silk suture.22,23
2.3.1 |. Endothelial disruption
To evaluate SMC responses independent of EC influences, the endothelium was disrupted by rubbing a small tungsten wire (diameter, 50 μm; Goodfellow, Huntington, UK) through the lumen of the vessel three times prior to cannulation.22 Once secured and pressurized (as above), selective EC damage was confirmed by ~50% constriction to NE (l.7 × 10−7 M; EC50) with loss of dilation to ACh (10−5 M).22 Dilation to ACh was calculated as: [(IDAch−IDNE)/(ID Ca2+ free−IDNE)] × 100%, where IDCa2+ free is maximal internal diameter in Ca2+-free PSS, IDNE is the internal diameter during constriction to NE, and IDACh is internal diameter during the addition of ACh. All endothelium-disrupted vessels studied in these experiments responded to NE but not ACh (vasodilation: intact = 76 ± 5%, endothelium-disrupted = 3 ± 6%).
2.3.2 |. Endothelial tube isolation
Isolated MAs were placed into PSS containing 0.62 mg·mL−1 papain (P4762, Sigma), 1.0 mg·mL−1 dithioerythritol (D8255, Sigma), and 1.5 mg·mL−1 collagenase (C8051, Sigma) then incubated for 25 min at 33°C. Following incubation, the enzyme solution was replaced with dissociation PSS and arterial segments were transferred into the tissue chamber. Trituration pipettes were pulled from borosilicate glass capillary tubes [Cat. #1B100-4, World Precision Instruments (WPI), Sarasota, FL, USA], heat polished to a tip ID of ~120 μm and connected to a Nanoliter Injector (WPI) to control fluid movement. During visual observation at 200× magnification, arterial segments were gently triturated to remove SMCs.24 Once isolated, each end of the endothelial tube was gently pressed against the bottom of the tissue chamber (a 24 × 54 mm coverslip) with a heat-blunted pipette (tip, diameter ~80 μm) and extended to approximate in situ length.23,25
Completed preparations were placed on an inverted microscope (Eclipse TS100; Nikon, Tokyo, Japan) mounted on a vibration-isolated table (Technical Manufacturing Corp., Peabody, MA, USA) and superfused at 4 mL·min−1 with PSS. An inline heater (SH-27B, Warner) and heating platform (PH6, Warner) coupled to a controller (TC-344B, Warner) maintained the superfusion solution temperature. Cannulated MAs were pressurized to 100 cm H2O while being warmed to 37°C22 for experiments; any vessels with leaks were discarded. Endothelial tubes were studied at 32°C to maintain viability.25 Pharmacological agents were added to the superfusion solution to achieve final concentrations in the vessel chamber.
2.4 |. Intracellular recording
The Vm of SMCs of intact and endothelium-disrupted MAs and of ECs in endothelial tubes was recorded with an Axoclamp 2B amplifier (Molecular Devices, Sunnyvale, CA, USA) using microelectrodes pulled (P-97, Sutter) from glass capillary tubes (GC100F-10, Warner) and backfilled with 2 M KCl (tip resistance: ~150 MΩ). A Ag/AgCl pellet in the effluent PSS served as a reference electrode. The output of the amplifier was connected to a data acquisition system (Digidata 1322A, Molecular Devices) and an audible baseline monitor (ABM-3, WPI). Successful impalements were indicated by sharp negative deflection of Vm, shift in tone of the ABM, stable Vm for >1 min, depolarization (≥20 mV) to 0.1 M KCl (SMCs) or hyperpolarization (≥20 mV) to 10−5 M ACh (ECs),23,26 recovery to resting Vm after KCl or ACh washout, and prompt return to ~0 mV upon electrode withdrawal. For intact vessels, SMCs are the first cell to be penetrated when approached from the bath; ECs comprise endothelial tubes. Data were acquired at 1 kHz on a personal computer using Axoscope 10.1 software (Molecular Devices). Following cell impalement, baseline Vm was recorded for 10 min to ensure stable electrode placement prior to pharmacological treatments. Experimental protocols were completed within 90 min. Each protocol was performed on a separate vessel preparation with 1 SMC or 1 EC continuously recorded (Figure 1). One neurotransmitter was studied in each preparation.
FIGURE 1.
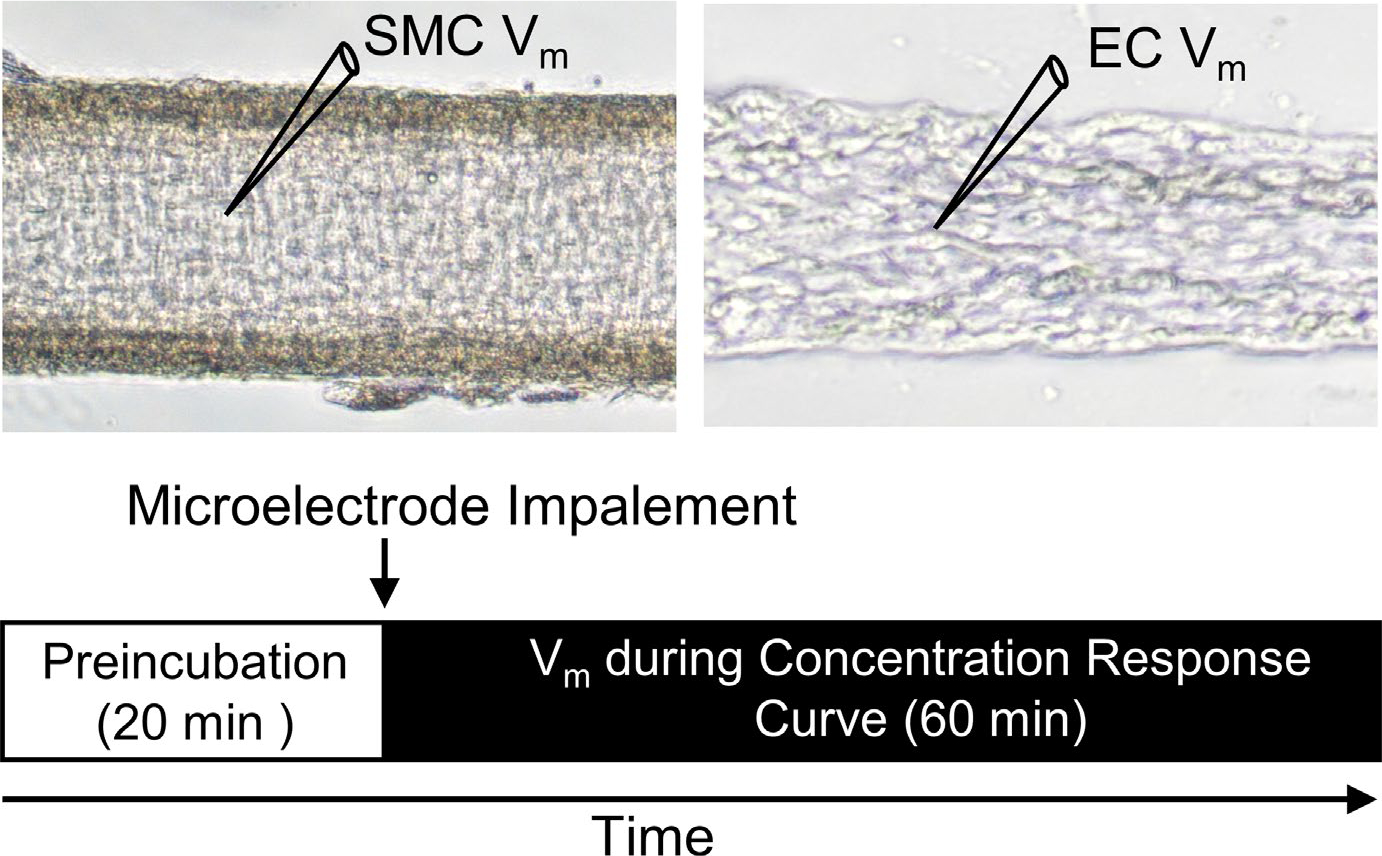
Experimental design. Diagram illustrating experimental setup illustrating sharp microelectrode impalement for measurement of Vm in a SMC of an intact artery (left) or an EC in an endothelial tube (right). Following 20 min preincubation with a pharmacological agent or vehicle control, Vm was recorded during cumulative addition of SP or CGRP. Responses to each neurotransmitter and pharmacological agent were tested in separate preparations
2.5 |. Pharmacology
Increasing concentrations of CGRP (10−10–10−6 M) or SP (10−10–10−6 M) were added in half log molar increments, each for 5 min, with continuous recording of Vm. Responses to SP and CGRP were then evaluated in the presence of L-NAME (10−4 M) in separate preparations. Responses were also evaluated in the presence the KATP channel antagonist, glibenclamide (10−6 M), which is selective for KATP over other K+ channels including KV, KCa, and KIR.12 We have previously shown that 10−6 M glibenclamide prevents hyperpolarization to the KATP agonist pinacidil (10−5 M).27 We also evaluated responses in the presence of charybdotoxin (10−7 M; inhibits BK and IK) + apamin (3×10−7 M; inhibits SK) to inhibit KCa channels in SMCs and ECs.28–30 The KCa channels were inhibited simultaneously as they are each activated through a similar Ca2+-dependent mechanism, and their inhibitors can overlap among channel subtypes.30,31 All antagonists were equilibrated for 20 min prior to evaluating their effect on electrophysiological responses to SP or CGRP. For each reagent requiring a solvent, controls refer to experiments performed with its stated vehicle added to control PSS; the control solution for water-soluble compounds was PSS. Concentration-response curves to pinacidil (10−9–10−5 M) were evaluated in the same manner as responses to CGRP. For analyses, data were sampled for 30 s once a stable Vm response was achieved at each concentration of agonist.
2.6 |. Data and statistical analysis
Electrophysiological analyses included: resting Vm (mV) under control conditions; peak Vm (mV) = Vm during maximal response to SP, CGRP, or pinacidil; and change in Vm (ΔVm) = peak response Vm – preceding baseline Vm. The order of experiments was randomized between control and treatment groups. Blinding of the investigator was not feasible as protocols required the operator to be aware of which agent(s) were added to the PSS and recordings were analyzed immediately after completion of the protocol. Data were analyzed using analysis of variance (Prism 5, GraphPad Software, La Jolla, Ca, USA). For each experimental condition, a repeated measures ANOVA was performed to determine whether the intervention had a significant effect on Vm compared to baseline. For comparisons between groups, two-way ANOVA was performed to compare effects of cell types and pharmacological interventions. When significant main effects were detected with ANOVA, post hoc comparisons were performed using Bonferroni tests. p ≤ 0.05 was considered statistically significant. Summary data are presented as means ± SE. Results were obtained from n = 5 vessels in each group. Data supporting the findings of this study are available from the corresponding author upon reasonable request.
3 |. RESULTS
3.1 |. Mesenteric arteries are hyperpolarized by both sensory neurotransmitters
The baseline Vm was similar (~ −45 mV) between cell types in respective preparations (Table 1). During continuous recording of Vm, SP (10−10–10−6 M) hyperpolarized SMCs of intact MAs in a concentration-dependent manner (Figure 2A,B). Following EC disruption, SMCs were no longer hyperpolarized by SP (Figure 2C). SP also hyperpolarized ECs in a concentration-dependent manner but the change in Vm from baseline was less than observed for SMCs with intact endothelium (Figure 2D). The EC50 values for hyperpolarization to SP were not significantly different between SMCs of intact MAs vs. ECs of endothelial tubes (Table 1).
TABLE 1.
Membrane potential of ECs and SMCs at rest and in response to SP and CGRP
| Substance P |
CGRP |
|||||||
|---|---|---|---|---|---|---|---|---|
| Rest | Max | ΔVm | EC50 | Rest | Max | ΔVm | EC50 | |
| SMC (+EC) | −46 ± 2 | −66 ± 2 | −20 ± 2 | 12.6 | −42 ± 1 | −73 ± 2 | −30 ± 1† | 10.0 |
| SMC (−EC) | −48 ± 2 | −50 ± 2* | −2 ± 1* | NA | −43 ± 3 | −69 ± 2 | −27 ± 2† | 10.3 |
| EC | −42 ± 1 | −56 ± 2* | −15 ± 3 | 7.4 | −44 ± 2 | −75 ± 1 | −31 ± 1† | 7.8 |
Note: Summary data for resting Vm (Rest), Vm at maximum response (Max), ΔVm (Max-Rest) and EC50 values (×10−9 M) for hyperpolarization (mV) to SP and CGRP in ECs of endothelial tubes, SMCs of MAs with disrupted endothelium (−EC) and SMCs of endothelium-intact (+EC) MAs. Values are means ± SEM for n = 5 vessels per group.
p < 0.05 vs. SMC (+EC).
p < 0.05 vs. maximal response to SP.
FIGURE 2.
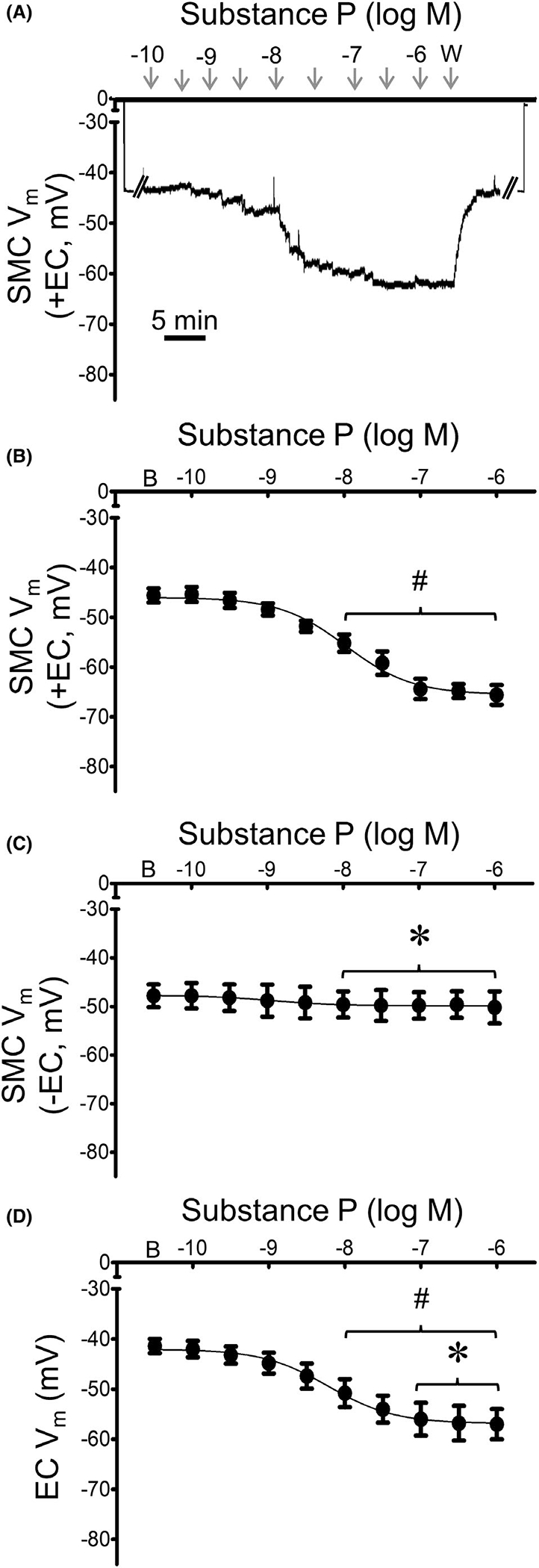
Mesenteric artery SMCs and ECs are hyperpolarized by SP. (A) Representative recording of membrane potential (Vm) illustrates concentration-dependent hyperpolarization of a SMC by SP in an intact MA (+EC). From zero baseline, note abrupt change in Vm upon entry and exit from cell. (B) Summary data for Vm of SMCs in intact MAs (+EC) studied at 37°C to increasing [SP] (10−10–10−6 M). (C) Summary data for Vm of SMCs of endothelium-disrupted MAs (−EC) studied at 37°C to SP as in B. (D) Summary data for Vm of ECs within endothelial tubes studied at 32°C to SP as in B. Values are means ± SEM for n = 5 per group. B = baseline Vm. W = washout in control PSS. #p < 0.05 vs. baseline (prior to application of SP) Vm. *p < 0.05 vs. SMC (+EC)
During continuous recording of Vm, CGRP (10−10–10−6 M) hyperpolarized SMCs in a concentration-dependent manner and to a similar level irrespective of the endothelium (Figure 3). Hyperpolarization of ECs with CGRP was not different from its effect on SMCs. Further, the magnitude of hyperpolarization with CGRP was greater than SP in both cell layers (Table 1). Respective EC50 values for CGRP hyperpolarization were not significantly different between preparations (Table 1).
FIGURE 3.
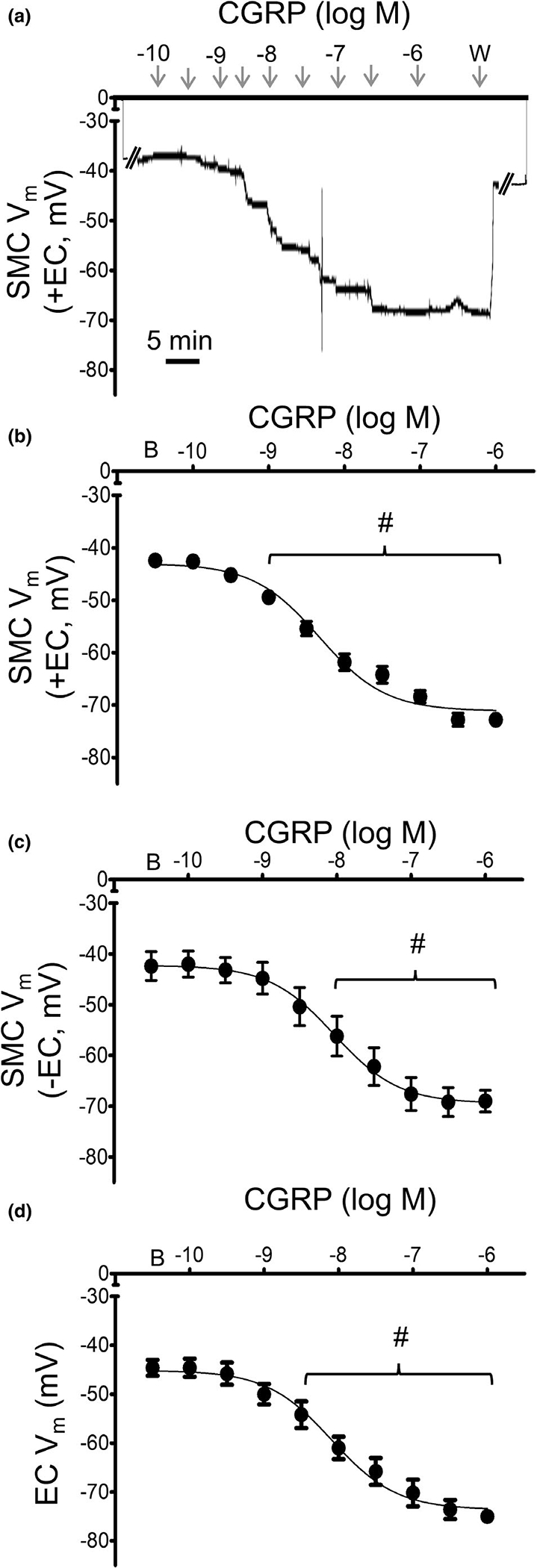
Mesenteric artery SMCs and ECs are hyperpolarized by CGRP. (A) Representative recording of Vm illustrates hyperpolarization of a SMC by CGRP in an intact MA (+EC). (B) Summary data for Vm in SMCs of intact MAs (+EC) studied at 37°C to increasing concentrations of CGRP (10−10–10−6 M). C) Summary data for Vm of SMCs of endothelium-disrupted MAs (−EC) studied at 37°C. (D) Summary data for Vm of ECs within endothelial tubes studied at 32°C. Values are means ± SEM for n = 5 per group. B = baseline Vm. W = washout in control PSS. #p < 0.05 vs. baseline (prior to application of CGRP) Vm
3.2 |. Effects of NO on SMC hyperpolarization in response to CGRP
Substance P and CGRP can signal SMCs through NO.1 To test for the contribution of NO to hyperpolarization, concentration-response curves were repeated for SMCs of intact MAs in the presence of the NO synthase inhibitor, L-NAME (10−4 M). Consistent with the effect of endothelial disruption (Figure 2), EC hyperpolarization to SP was eliminated during NO synthase inhibition (Figure 4A,B). To confirm the viability of SMCs in these preparations, addition of 0.1 M KCl following each L-NAME protocol consistently elicited depolarization (ΔVm = 34 ± 5 mV; Figure 4A). In contrast to inhibiting SMC hyperpolarization to SP, L-NAME had no effect on hyperpolarization to CGRP (Figure 4C), nor did it alter EC50 values from those in Table 1.
FIGURE 4.
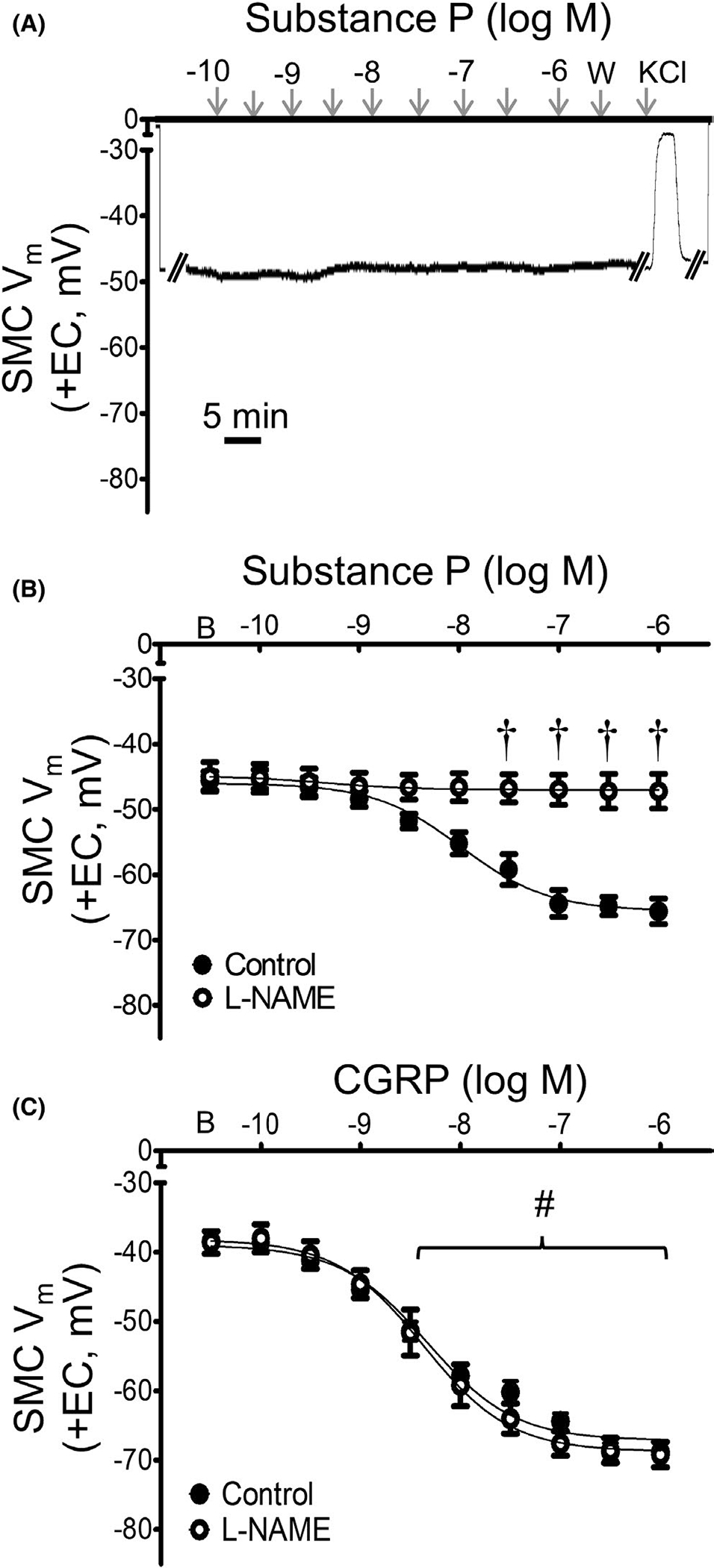
SP hyperpolarizes SMCs through NO signaling. A) Representative recording of Vm demonstrates lack of response to SP of a SMC in an intact MA (+EC) in the presence of L-NAME (10−4 M); response to acute administration of KCl (5 min) confirms preparation viability. (B) Summary data for Vm responses to SP in SMCs from intact MAs studied at 37°C under control conditions (from Figure 1B) or in the presence of L-NAME. (C) Summary data for Vm responses to CGRP in SMCs from intact MAs in the absence (from Figure 2B) or presence of L-NAME (10−4 M). Values are means ± SEM for n = 5 per group. B = baseline Vm. W = washout in control PSS. #p < 0.05 vs. baseline (prior to application of SP or CGRP) Vm during L-NAME. †p < 0.05 vs. control
3.3 |. KATP channels do not contribute to hyperpolarization elicited by SP
To test whether KATP channels contribute to hyperpolarization by SP, the channel inhibitor glibenclamide was added at a concentration (10−6 M) that blocks hyperpolarization to the KATP channel opener pinacidil (10−5 M).27 Inhibition of KATP channels had no significant effect on hyperpolarization to SP in either SMCs or ECs (Figure 5), nor did glibenclamide alter EC50 values from those in Table 1. Responses to SP with its vehicle (DMSO) in PSS did not differ significantly from PSS alone.
FIGURE 5.
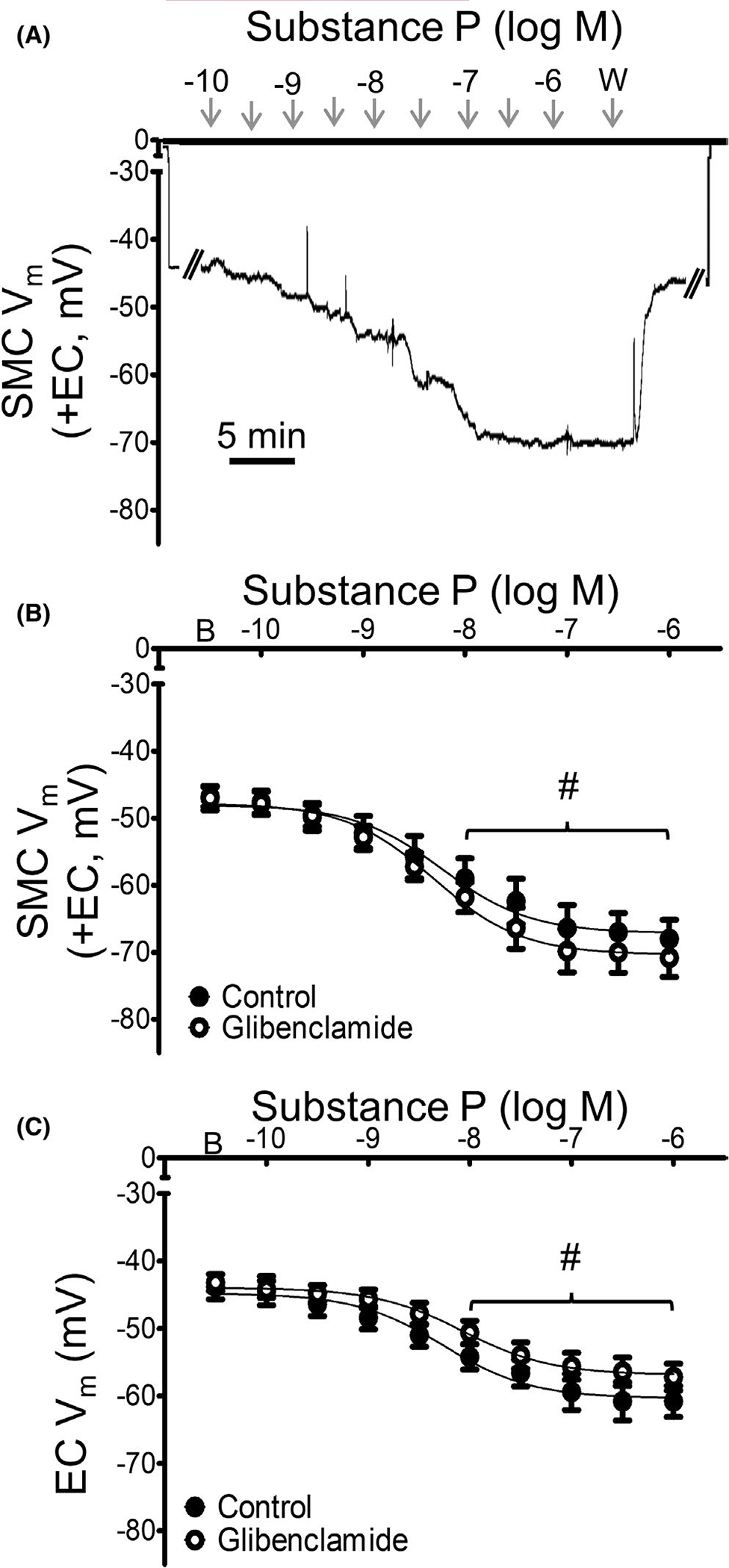
Hyperpolarization to SP does not occur through KATP channels. (A) Representative recording of Vm in an intact MA (+EC) illustrates maintained SMC hyperpolarization to SP in the presence of glibenclamide (10−6 M). (B) Summary data for Vm responses to SP in SMCs from intact MAs (+EC) studied at 37°C under control (vehicle = 0.1% DMSO) conditions or during exposure to the KATP channel inhibitor glibenclamide. (C) Summary data for Vm responses to SP for ECs in endothelial tubes studied at 32°C in the absence or presence of glibenclamide. Values are means ± SEM for n = 5 per group. B = baseline Vm. W = washout in control PSS. #p < 0.05 vs. baseline (prior to application of SP) Vm in preparations treated with glibenclamide
3.4 |. KCa channels contribute to SMC and EC hyperpolarization in response to SP
The role of KCa channels in mediating hyperpolarization to SP was tested with the antagonists charybdotoxin (10−7 M) + apamin (3 × 10−7 M).28,29 For SMCs, KCa channel inhibition attenuated hyperpolarization to SP (Figure 6A,B), although some hyperpolarization remained at concentrations of SP ≥10−7 M. In ECs, KCa channel inhibition nearly eliminated hyperpolarization (peak ΔVm = −4 ± 1 mV) and significantly increased EC50 values (control = 7.4 × 10−9 M; charybdotoxin + apamin = 13.6 × 10−9 M; p < 0.05). KCa channel inhibition did not alter EC50 values in SMCs from those in Table 1.
FIGURE 6.
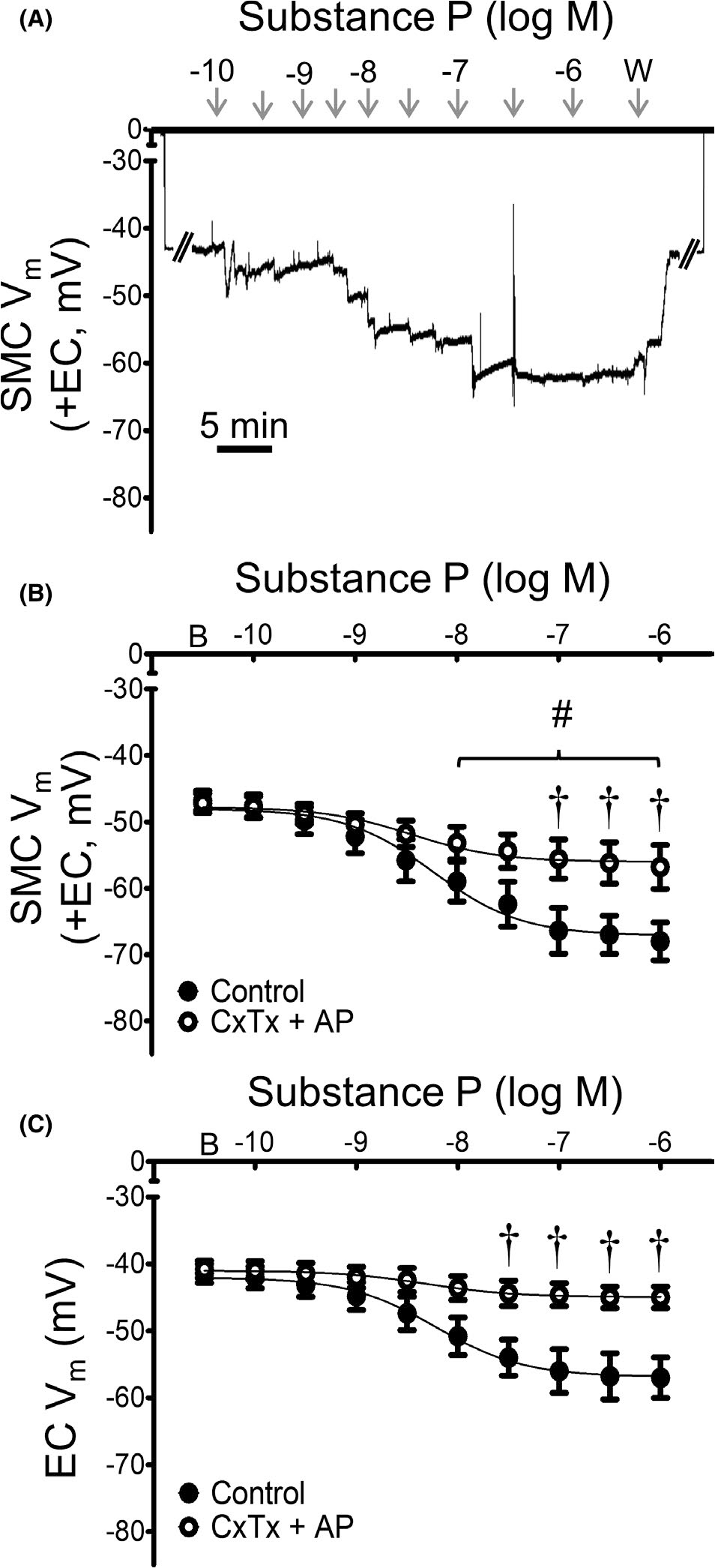
Hyperpolarization to SP occurs through KCa channels. (A) Representative recording of Vm in an intact MA (+EC) demonstrates attenuation of SMC hyperpolarization to SP in the presence of KCa channel inhibitors charybdotoxin (ChTx; 10−7 M) + apamin (AP; 3 × 10−7 M). B) Summary data for Vm responses to SP in SMCs from intact MAs (+EC) studied at 37°C under control (from Figure 1) conditions and during KCa channel inhibition. (C) Summary data for Vm responses to SP for ECs within endothelial tubes studied at 32°C during the absence or presence of KCa channel inhibitors. Values are means ± SEM for n = 5 per group. B = baseline Vm. W = washout in control PSS. #p < 0.05 vs. baseline (prior to application of SP) Vm in vessels treated with ChTx + AP. †p < 0.05 vs. control
3.5 |. KATP channels contribute to SMC but not EC hyperpolarization elicited by CGRP
In the presence of glibenclamide (10−6 M), SMC hyperpolarization to CGRP was significantly attenuated (Figure 7), with a maximal ΔVm of ~ −10 mV vs. ~ −30 mV under control conditions. Contrary its effect on SMCs (and to our hypothesis), EC hyperpolarization to CGRP was maintained in the presence of glibenclamide. These findings were verified in KATP−/− mice (global deletion), where EC responses to CGRP were unaffected while SMC responses were significantly attenuated compare to those of wildtype mice (Figure 8). In SMCs from mice lacking KATP channels, there was a significant increase in the EC50 value to CGRP (wildtype = 10 × 10−9 M; KATP−/− = 20 × 10−9 M; p < 0.05). The absence of KATP function in KATP−/− mice was confirmed by testing with the KATP channel activator pinacidil (10−5 M); from a baseline Vm of −45 mV, the ΔVm was <5 mV.
FIGURE 7.
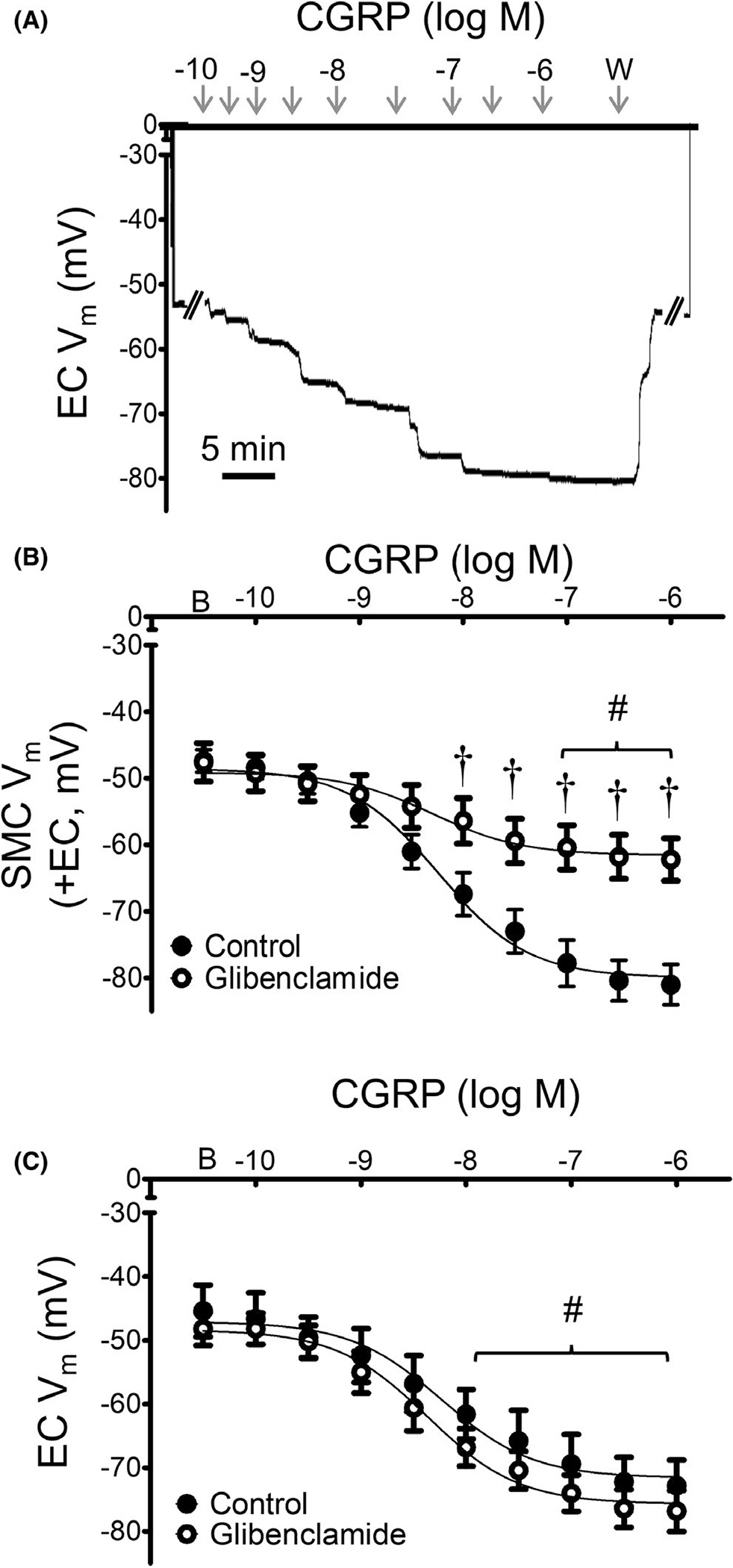
Hyperpolarization to CGRP in SMCs, but not ECs, requires KATP channels. (A) Representative recording of Vm from a MA endothelial tube in the presence of glibenclamide (10−6 M) illustrates normal EC response to CGRP (10−10–10−6 M). (B) Summary data for SMCs of intact MAs (+EC) studied at 37°C to CGRP in the absence or presence of glibenclamide or its vehicle. (C) Summary data for ECs within endothelial tubes studied at 32°C in response to CGRP in the absence or presence glibenclamide. Values are means ± SEM for n = 5 per group. B = baseline Vm. W = washout in control PSS. #p < 0.05 vs. baseline (prior to application of CGRP) Vm in vessels treated with glibenclamide. †p < 0.05 vs. control
FIGURE 8.
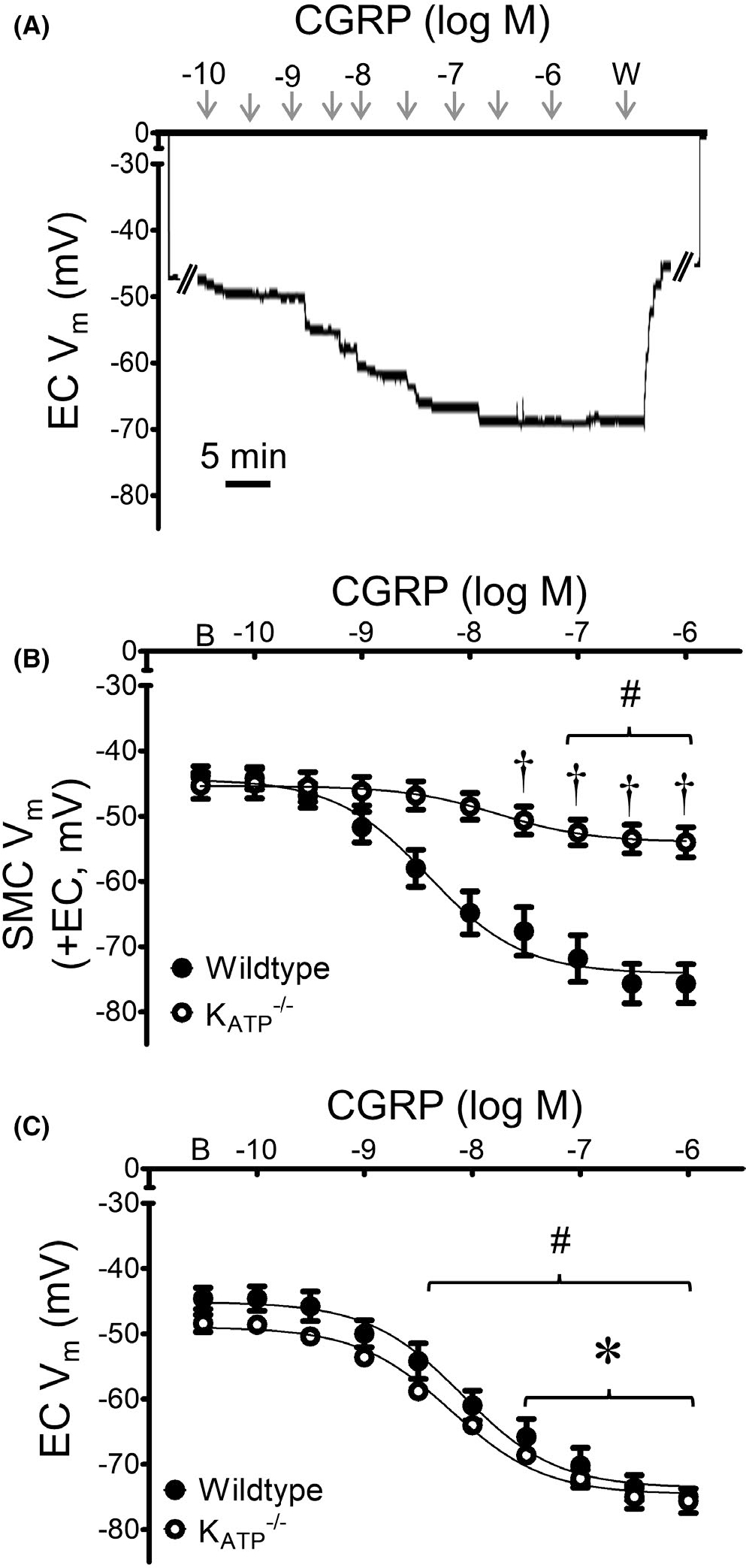
Genetic deletion of KATP channels inhibits hyperpolarization to CGRP. (A) Representative recording of Vm from an EC within a MA endothelial tube of a mouse lacking KATP channels illustrates normal (wildtype) response to CGRP (10−10–10−6 M). (B) Summary data for responses to CGRP in SMCs of intact MAs (+EC) studied at 37°C from KATP−/− vs. wildtype mice. (C) Summary data for responses to CGRP in endothelial tubes studied at 32°C from KATP−/− vs. wildtype mice. Values are means ± SEM for n = 5 per group. B = baseline Vm. W = washout in control PSS. #p < 0.05 vs. baseline (prior to application of CGRP) Vm in KATP−/− mice. *p < 0.05 EC vs. SMC Vm in KATP−/− mice. †p < 0.05 vs. wildtype
To determine whether KATP channel activation can hyperpolarize SMCs and ECs, we stimulated intact MAs and endothelial tubes of wildtype mice with the KATP agonist pinacidil (10−9–10−5 M). Pinacidil evoked concentration-dependent hyperpolarization of SMCs within intact MAs and of ECs within endothelial tubes (Figure 9). From a similar resting Vm (~ −45 mV), maximal hyperpolarization was nearly twofold greater in SMCs (ΔVm = −33 ± 2 mV) vs. ECs (ΔVm = −18 ± 1 mV). Furthermore, EC50 values for pinacidil were significantly greater in ECs (1.2×10−7 M) vs. SMCs (4.1×10−8 M; p < 0.05).
FIGURE 9.
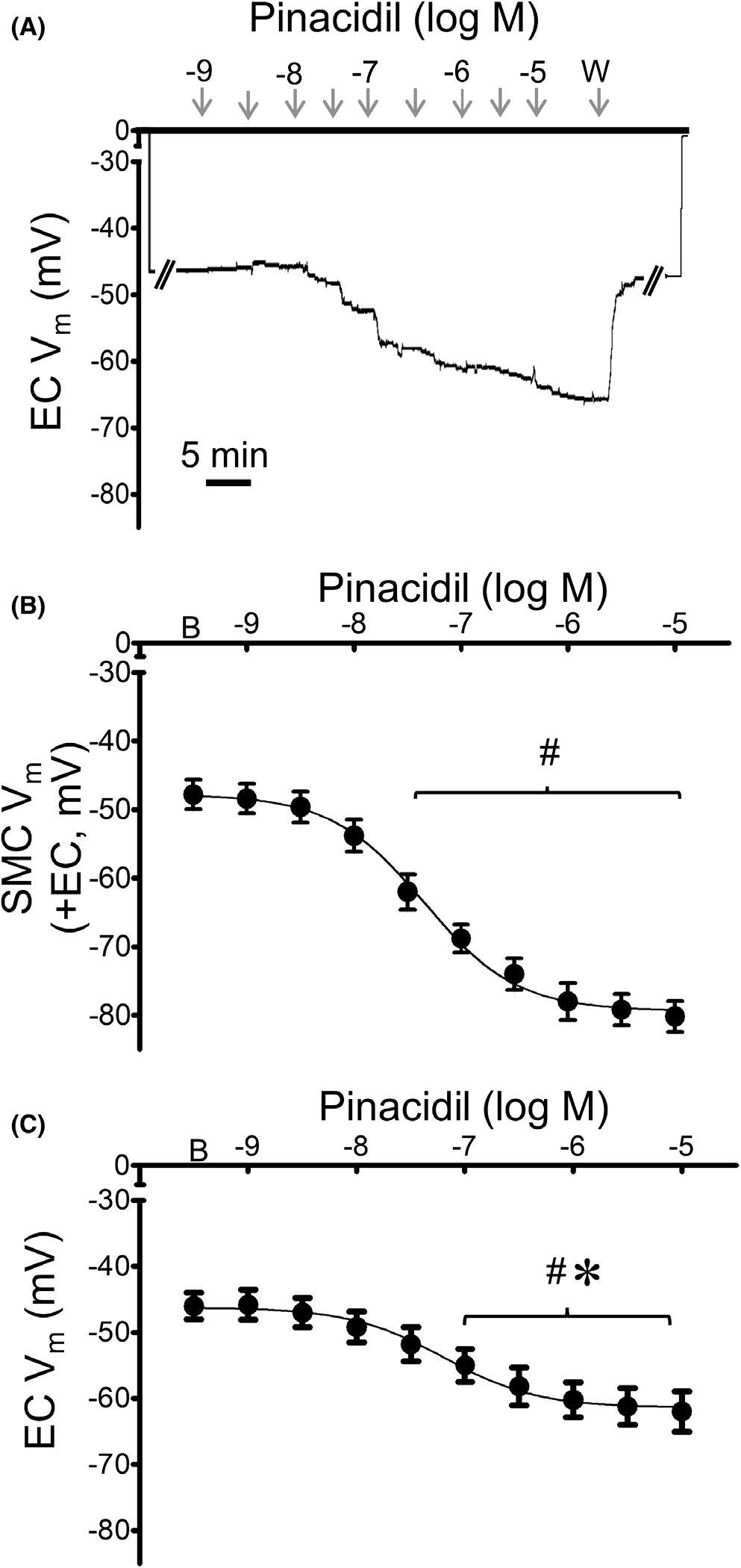
KATP channel activation hyperpolarizes SMCs more than ECs. (A) Representative recording from an EC within a MA endothelial tube illustrates concentration-dependent hyperpolarization to pinacidil (10−9–10−5 M). (B) Summary data from SMCs of intact MAs (+EC) studied at 37°C in response to increasing concentrations of pinacidil. (C) Summary data for ECs within endothelial tubes studied at 32°C in response to increasing concentrations of pinacidil. Values are means ± SEM for n = 5 per group. B = baseline Vm. W = washout in control PSS. # p < 0.05 vs. baseline (prior to application of pinacidil) Vm. *p < 0.05 EC vs. SMC Vm
We next tested the role of KCa channels to mediate hyperpolarization to CGRP. Addition of charybdotoxin + apamin attenuated SMC hyperpolarization to CGRP by ~30% (Figure 10). KCa channel inhibition had a greater effect in ECs, nearly abolishing hyperpolarization to CGRP (peak ΔVm = −3 ± 1 mV). To ensure that the observed lack of hyperpolarization was not due to nonviable EC tube preparations, responses to ACh (10−5 M) were verified at the end of each protocol following washout of the channel blockers (ΔVm = −31 ± 3 mV; Figure 10A). Inhibition of KCa channels did not significantly alter EC50 values to CGRP in SMCs compared to those in Table 1.
FIGURE 10.
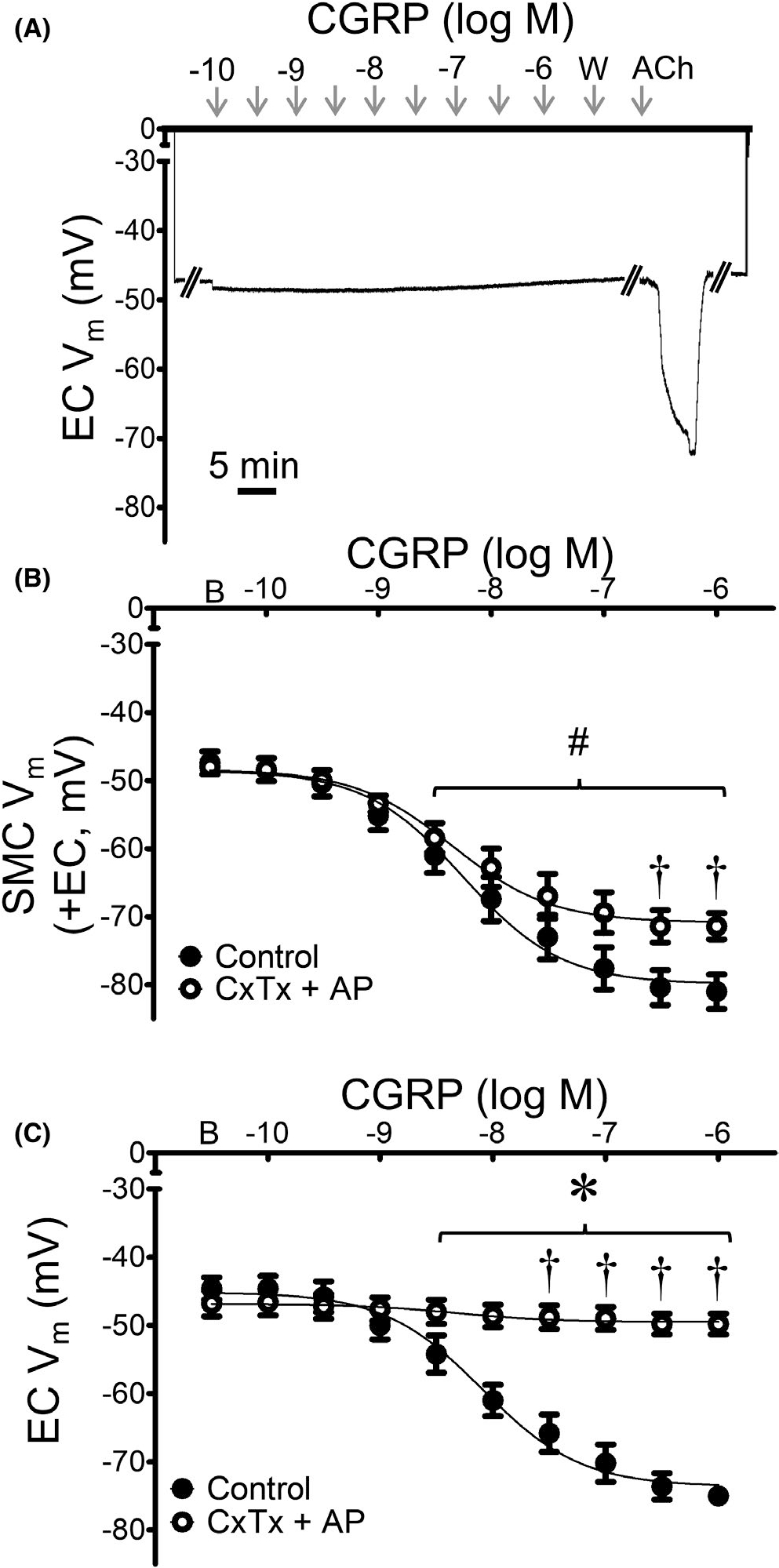
KCa channels contribute to hyperpolarization of ECs more than SMCs in response to CGRP. (A) Representative recording of Vm from an EC within a MA endothelial tube demonstrates lack of hyperpolarization to CGRP in the presence of charybdotoxin (ChTx; 10−7 M) + apamin (AP; 3 × 10−7 M); note hyperpolarization to ACh (5 min) following washout, confirming EC viability. (B) Summary data for Vm responses to CGRP in SMCs from intact MAs (+EC) studied at 37°C under control conditions (from Figure 2) or during KCa channel inhibition. (C) Summary data for Vm responses to CGRP in ECs within endothelial tubes studied at 32°C in the presence or absence of KCa channel inhibitors. Values are means ± SEM for n = 5 per group. B = baseline Vm. W = washout in control PSS. #p < 0.05 vs. baseline (prior to application of CGRP) Vm in vessels treated with ChTx + AP. †p < 0.05 vs. control. *p < 0.05 EC vs. SMC Vm
4 |. DISCUSSION
The present experiments define the electrophysiological responses of SMCs and ECs comprising mouse MAs during controlled exposure to CGRP and SP. The stability of our Vm recordings facilitated continuous, uninterrupted evaluation of complete concentration-response relationships of SMCs within intact MAs, endothelium-disrupted MAs, and of ECs within intact endothelial tubes following dissociation of SMCs. Our major findings (Figure 11) show that: 1) both sensory neurotransmitters hyperpolarize ECs and SMCs, with CGRP having ~twofold greater effect than SP; 2) SMC hyperpolarization to CGRP can occur independent of ECs while SMC hyperpolarization to SP is mediated through endothelium-derived NO; 3) for ECs, KCa channels are primary mediators of hyperpolarization to both SP and CGRP; and 4) KATP channels are primary mediators of SMC hyperpolarization to CGRP but have a negligible role in hyperpolarization of either cell layer to SP.
FIGURE 11.
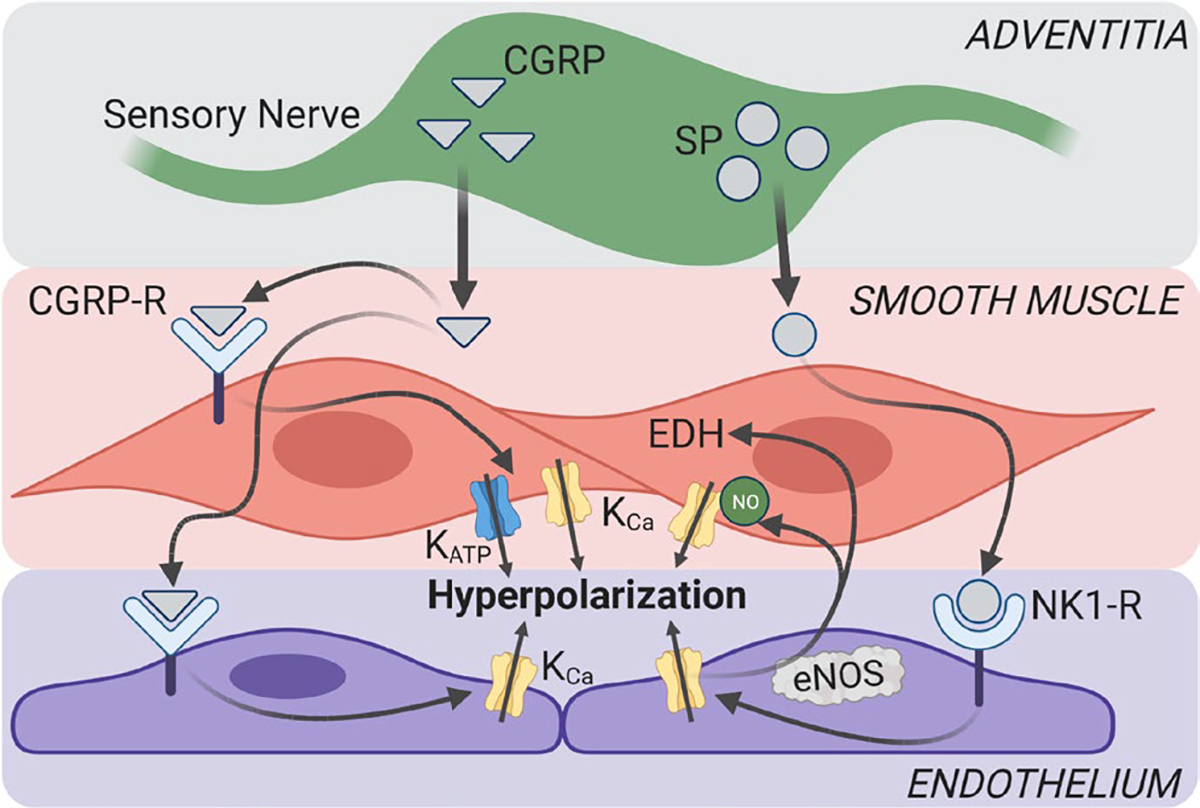
Summary of findings. Schematic depicts perivascular neurotransmitter signaling in mouse mesenteric arteries. NO signaling mediates endothelium-dependent hyperpolarization (EDH) of SMCs to SP but not CGRP. KCa channels mediate hyperpolarization to SP in both SMCs and ECs. KCa channels are required for EC hyperpolarization to CGRP, while KATP channels mediate most SMC hyperpolarization to CGRP. CGRP-R, CGRP receptor; eNOS, endothelial nitric oxide synthase; NK1-R, neurokinin 1 receptor. Figure created using BioRender
4.1 |. Sensory neurotransmitters hyperpolarize vascular smooth muscle and endothelial cells
Substance P hyperpolarized SMCs within intact vessels and ECs within endothelial tubes; however, endothelial disruption prevented SMC hyperpolarization (Figure 2). These findings are consistent with endothelial denudation preventing hyperpolarization of SMCs to SP in unpressurized porcine coronary artery segments.32 Substance P exerts its actions in the vasculature through neurokinin receptors,16 and our previous findings confirm that neurokinin 1 receptors are expressed on ECs, but not SMCs, in mouse MAs.23,33 The present data are also consistent with neurokinin receptor expression on human aortic ECs.16 The magnitude of EC hyperpolarization in endothelial tubes (~ −15 mV from a resting Vm of ~ −45 mV) was less than observed in cultured porcine coronary artery ECs (ΔVm ~ −25 mV from resting Vm of ~ −30 mV).18 Such differences may reflect a difference between vascular beds, animal species, and/or cultured EC monolayers vs. native EC preparations. Differences between our experimental conditions also encompass endothelial tubes being studied at 32°C to maintain viability25 as compared to SMCs in intact MAs studied at 37°C. Finding that the inhibition of NO synthase prevented SMC hyperpolarization to SP in intact MAs (Figure 4) indicates that endothelium-derived NO mediates SMC hyperpolarization rather than SP acting directly on SMCs to elicit hyperpolarization. Although SP released from sensory nerves does not act directly on SMCs, electrical field stimulation of MAs releases SP that acts upon the endothelium.23 Furthermore, the neurokinin receptors on ECs can also be activated by circulating SP.34 Because we have not been able to consistently evaluate Vm of ECs within the intact MA, the present data cannot address the contribution of myoendothelial coupling between respective cell layers of the vessel wall.
In contrast to SP effecting SMC hyperpolarization indirectly via the endothelium, we show here that CGRP acts directly on each cell layer of the MA. Thus, neither endothelium-derived NO nor myoendothelial coupling through gap junctions26 are required for SMC hyperpolarization to CGRP. These data are consistent with hyperpolarization observed in SMCs from rabbit MAs6,11 and illustrate the similarity in response of ECs to SMCs to this sensory neurotransmitter. The ability of CGRP to hyperpolarize SMCs and ECs individually is also consistent with expression of CGRP receptor components RAMP1 and CRLR on both cell types.22,23 Nevertheless, our findings raise the question of why ECs hyperpolarize to CGRP if the endothelium is unnecessary for SMC hyperpolarization. Cell-cell coupling through gap junctions along and between ECs and SMCs may coordinate the electrical and vasomotor response along the vascular wall to ensure that CGRP elicits SMC hyperpolarization and vasodilation irrespective of whether it is presented abluminally from perivascular nerves or intraluminally via the circulation. Additionally, diffusion of endothelium-derived NO can relax SMCs by reducing sensitivity to [Ca2+]i independent of affecting Vm.35
4.2 |. KCa channels mediate hyperpolarization of SMCs and ECs to SP
KCa channels are integral mediators of endothelium-dependent dilation.12,36,37 They are tetrameric proteins formed of four α and β subunits and are unique among K+ channels in their ability couple intracellular Ca2+ signals to membrane hyperpolarization. Based on their single-channel conductance, KCa channels are classified into SK, IK, and BK channels. Whereas SMCs preferentially contain BK channels, SK and IK channels are primarily located in ECs.36 While KCa channels are established mediators of EC hyperpolarization to SP,18,19,38 the channels mediating SMC hyperpolarization to SP had remained undefined. The present data show that, while inhibiting KATP channels had little effect on EC or SMC hyperpolarization to SP (Figure 5), blocking KCa channels attenuated hyperpolarization in both cell types (Figure 6). While KCa inhibition nearly eliminated hyperpolarization to SP in ECs, the remaining endothelium-dependent hyperpolarization in SMCs (from Figure 2C) suggests that the endothelium activates additional ion channels on SMCs that contribute to hyperpolarization to SP. Because L-NAME prevented hyperpolarization to SP in SMCs (Figure 4), we suggest that KCa channels (likely BK) on SMCs are activated by endothelium-derived NO to mediate hyperpolarization, which can occur through cGMP- and PKG-dependent phosphorylation.37,39 Alternatively, direct activation of BK channels by NO has been reported in vascular smooth muscle independent of guanylate cyclase signaling.40 Because SK and IK channels can regulate NO synthesis in ECs,30,41 it is possible KCa inhibition in ECs prevents SMC hyperpolarization by limiting NO production. Furthermore, because SK and IK channels contribute to endothelium-dependent hyperpolarization,30,42 myoendothelial coupling may also contribute to SMC hyperpolarization elicited by SP.
4.3 |. KATP channels mediate CGRP-dependent hyperpolarization of SMCs but not ECs
KATP channels are integral to the regulation of vasomotor tone.43,44 While KATP channels in SMCs of systemic arteries are well documented,6,8–11,45 ECs of the aorta, mesenteric, and coronary arteries are also reported to express KATP channels.46–49 KATP channels are formed from octameric protein complexes composed of four inward rectifying subunits (KIR 6.1 and 6.2), each associated with a larger sulphonylurea receptor (SUR).50,51 The KIR6.1 and SUR2B isoform is the primary form of the channel in arterial myocytes.50,52 In mice lacking the KIR6.1 subunit, SMC hyperpolarization to CGRP was greatly reduced, validating the role of KATP channels as mediators of CGRP-dependent hyperpolarization in MAs utilizing a genetic approach (Figure 8). However, KATP channels contribute only a portion of SMC hyperpolarization to CGRP in MAs (~10 mV remaining following inhibition or knockout). Because KCa channels have also been implicated in SMC hyperpolarization to CGRP,10,53,54 we tested their contribution to SMC hyperpolarization in the intact mouse MA. As shown in Figure 10, inhibition of KCa channels reduced hyperpolarization to CGRP by ~10 mV, which approximates the portion of this response not mediated by KATP channels.
Despite the expression of KATP channels in ECs,46–49 hyperpolarization to CGRP remained unaltered in endothelial tubes treated with glibenclamide and in ECs from KATP−/− mice (Figures 7 and 8). Therefore, we tested whether activation of KATP channels could elicit hyperpolarization in ECs. Stimulation of KATP channels with pinacidil evoked hyperpolarization of ECs (Figure 9). These findings indicate that while KATP channels are present in ECs of the mouse MA, they are not functionally coupled to CGRP receptors. In contrast, the combination of charybdotoxin and apamin nearly abolished the response to CGRP in ECs, pointing to KCa channels as the primary mediator of hyperpolarization to this neurotransmitter (Figure 10). Therefore, while CGRP elicits hyperpolarization in both cell layers, its receptors are functionally coupled to different classes of K+ channels in SMCs vs. ECs.
5 |. SUMMARY AND CONCLUSION
We have investigated the effect of CGRP and SP on the membrane potential of ECs and SMCs of MAs from C57BL/6J male mice. While both sensory neurotransmitters elicit hyperpolarization of respective cell layers, their signaling pathways differ. Whereas SP hyperpolarizes the endothelium through KCa channels, these channels in smooth muscle are activated via NO. CGRP also hyperpolarizes both cell layers but does so via KCa channels in endothelium and KATP channels in smooth muscle.
Impaired sensory nerve function contributes to impaired vasomotor function in aging, diabetes, and hypertension,22,55,56 under-scoring the importance of perivascular sensory nerves in both health and disease. Recent findings have revealed a novel role for sensory neurotransmitter dysfunction in MAs during inflammatory bowel disease, whereby aberrant SP signaling impedes CGRP-mediated vasodilation.23 Based on the integral role of membrane potential in regulating arterial diameter and organ blood flow, resolving the determinants of electrophysiological responses in smooth muscle and EC layers of the vessel wall identifies selective targets for treating vascular disease associated with sensory neurotransmitter dysfunction.
6 |. PERSPECTIVES
In mouse MAs, vascular smooth muscle is hyperpolarized by substance P via endothelium-derived NO activation of KCa channels and by CGRP via activation of KATP channels independent of the endothelium. The endothelium is hyperpolarized by both SP and CGRP through activation of KCa channels. New insight into differential signaling underlying hyperpolarization of the arterial wall advances the ability to selectively target conditions of sensory neurotransmitter dysfunction.
ACKNOWLEDGMENTS
We have no acknowledgments. The authors are entirely responsible for the content of this article.
Funding information
This research was supported by National Institutes of Health grants R37-HL041026 (S.S.S.) and R00-HL129196 (E.M.B)
Abbreviations:
- ACh
acetylcholine
- BK
large conductance KCa channel
- CGRP
calcitonin gene-related peptide
- DMSO
dimethylsulfoxide
- EC
endothelial cell
- ID
inner diameter
- IK
intermediate conductance KCa channel
- KATP
ATP-sensitive potassium channels
- KCa
Ca2+-activated potassium channels
- L-NAME
NG-nitro-l-arginine methyl ester
- MA
mesenteric artery
- NE
norepinephrine
- NO
nitric oxide
- PGP
protein gene product
- PSS
physiological saline solution
- SK
small conductance KCa channel
- SMC
smooth muscle cell
- SP
substance P
- SUR
sulphonylurea receptor
- VGCC
voltage-gated Ca2+ channel
- Vm
membrane potential
Footnotes
CONFLICT OF INTEREST
The authors have no competing interests. The content of this article is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
REFERENCES
- 1.Westcott EB, Segal SS. Perivascular innervation: a multiplicity of roles in vasomotor control and myoendothelial signaling. Microcirculation. 2013;20:217–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Burnstock G, Ralevic V. New insights into the local regulation of blood flow by perivascular nerves and endothelium. Br J Plast Surg. 1994;47:527–543. [DOI] [PubMed] [Google Scholar]
- 3.Brain SD, Williams TJ, Tippins JR, Morris HR, MacIntyre I. Calcitonin gene-related peptide is a potent vasodilator. Nature. 1985;313:54–56. [DOI] [PubMed] [Google Scholar]
- 4.Kawasaki H Regulation of vascular function by perivascular calcitonin gene-related peptide-containing nerves. Jpn J Pharmacol. 2002;88:39–43. [DOI] [PubMed] [Google Scholar]
- 5.McLatchie LM, Fraser NJ, Main MJ, et al. RAMPs regulate the transport and ligand specificity of the calcitonin-receptor-like receptor. Nature. 1998;393:333–339. [DOI] [PubMed] [Google Scholar]
- 6.Nelson MT, Huang Y, Brayden JE, Herscheler J, Standen NB. Arterial dilations in response to calcitonin gene-related peptide involve activation of K+ channels. Nature. 1990;344:770–773. [DOI] [PubMed] [Google Scholar]
- 7.Reslerova M, Loutzenhiser R. Renal microvascular actions of calcitonin gene-related peptide. Am J Physiol Renal Physiol. 1998;274:1078–1085. [DOI] [PubMed] [Google Scholar]
- 8.Wellman GC, Quayle JM, Standen NB. ATP-sensitive K+ channel activation by calcitonin gene related peptide and protein kinase A in pig coronary arterial smooth muscle. J Physiol. 1998;507:117–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Blanco-Rivero J, Marquez-Rodas I, Sastre E, et al. Cirrhosis decreases vasoconstrictor response to electrical field stimulation in rat mesenteric artery: role of calcitonin gene-related peptide. Exp Physiol. 2011;96:275–286. [DOI] [PubMed] [Google Scholar]
- 10.Miyoshi H, Nakaya Y. Calcitonin gene-related peptide activates the K+ channels of vascular smooth muscle cells via adenylate cyclase. Basic Res Cardiol. 1995;90:332–336. [DOI] [PubMed] [Google Scholar]
- 11.Quayle JM, Bonev AD, Brayden JE, Nelson MT. Calcitonin gene-related peptide activated ATP-sensitive K+ currents in rabbit arterial smooth muscle via protein kinase A. J Physiol. 1994;475:9–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nelson MT, Quayle JM. Physiological roles and properties of potassium channels in arterial smooth muscle. Am J Physiol Cell Physiol. 1995;268:C799–C822. [DOI] [PubMed] [Google Scholar]
- 13.Si J, Zhao H, Yang Y, Jian Z, Nuttall AL. Nitric oxide induces hyperpolarization by opening ATP-sensitive K+ channels in guinea pig spiral modiolar artery. Hear Res. 2002;171:167–176. [DOI] [PubMed] [Google Scholar]
- 14.Brain SD, Cox HM. Neuropeptides and their receptors: innovative science providing novel therapeutic targets. Br J Pharmacol. 2006;147:S202–S211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bolton TB, Clapp LH. Endothelial-dependent relaxant actions of carbachol and substance P in arterial smooth muscle. Br J Pharmacol. 1986;87:713–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Greeno EW, Mantyh P, Vercellotti GM, Moldow CF. Functional neurokinin 1 receptors for substance P are expressed by human vascular endothelium. J Exp Med. 1993;177:1269–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cohen RA, Vanhoutte PM. Endothelium-dependent hyperpolarization: beyond nitric oxide and cyclic GMP. Circulation. 1995;92:3337–3349. [DOI] [PubMed] [Google Scholar]
- 18.Frieden M, Sollini M, Beny JL. Substance P and bradykinin activate different types of KCa currents to hyperpolarize cultured procine coronary artery endothelial cells. J Physiol. 1999;519:361–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sollini M, Frieden M, Beny JL. Charybdotoxin-sensitive small conductance KCa channel activation by bradykinin and substance P in endothelial cells. Br J Pharmacol. 2002;136:1201–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Davis MJ, Kim HJ, Zawieja SD, et al. Kir6.1-dependent KATP channels in lymphatic smooth muscle and vessel dysfunction in mice with Kir6.1 gain-of-function. J Physiol. 2020;598:3107–3127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Outzen EM, Zaki M, Abdolalizadeh B, Sams A, Boonen HCM, Sheykhzade M. Translational value of mechanical and vasomotor properties of mouse isolated mesenteric resistance-sized arteries. Pharmacol Res Perspect. 2015;3:e00200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Boerman EM, Segal SS. Depressed perivascular sensory innervation of mouse mesenteric arteries with advanced age. J Physiol. 2016;594:2323–2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Norton CE, Grunz-Borgmann EA, Hart ML, Jones BW, Franklin CL, Boerman EM. Role of perivascular nerve and sensory neurotransmitter dysfunction in inflammatory bowel disease. Am J Physiol Heart Circ Physiol. 2021;320:H1887–H1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Socha MJ, Segal SS. Isolation of microvascular endothelial tubes from mouse resistance arteries. J vis Exp. 2013;81:50759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Socha MJ, Hakim CH, Jackson WF, Segal SS. Temperature effects on morphological integrity and Ca2+ signaling in freshly isolated murine feed artery endothelial cell tubes. Am J Physiol Heart Circ Physiol. 2011;301:H773–H783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Behringer EJ, Socha MJ, Polo-Parada L, Segal SS. Electrical conduction along endothelial cell tubes from mouse feed arteries: confounding actions of glycyrrhetinic acid derivatives. Br J Pharmacol. 2012;166:774–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Norton CE, Segal SS. Calcitonin gene-related peptide hyperpolarizes mouse pulmonary artery endothelial tubes through KATP activation. Am J Physiol Lung Cell Mol Physiol. 2018;315:L212–L226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Behringer EJ, Segal SS. Tuning electrical conduction along endothelial tubes of resistance arteries through Ca2+-activated K+ channels. Circ Res. 2012;110:1311–1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Behringer EJ, Shaw RL, Westcott EB, Socha MJ, Segal SS. Aging impairs electrical conduction along endothelium of resistance arteries through enhanced Ca2+-activated K+ channel activation. Arterioscler Thromb Vasc Biol. 2013;33:1892–1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ledoux J, Werner ME, Brayden JE, Nelson MT. Calcium-activated potassium channels and the regulation of vascular tone. Physiology. 2006;21:69–78. [DOI] [PubMed] [Google Scholar]
- 31.Leonard RJ, Garcia ML, Slaughter RS, Reuben JP. Selective blockers of voltage-gated K+ channels depolarize human T lymphocytes: mechanism of the antiproliferative effect of charybdotoxin. Proc Natl Acad Sci USA. 1992;89:10094–10098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Edwards G, Thollon C, Gardener MJ, et al. Role of gap junctions and EETs in endothelium-dependent hyperpolarization of porcine coronary artery. Br J Pharmacol. 2000;129:1145–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Boerman EM, Sen S, Shaw RL, Joshi T, Segal SS. Gene expression profiles of ion channels and receptors in mouse resistance arteries: effects of cell type, vascular bed, and age. Microcirculation. 2018;25:e12425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dehlin HM, Levick SP. Substance P in heart failure: the good and the bad. Int J Cardiol. 2014;170:270–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bolz SS, Vogel L, Sollinger D, et al. Nitric oxide-i nduced decrease in calcium sensitivity of resistance arteries is attributable to activation of the myosin light chain phosphatase and antagonized by the RhoA/Rho kinase pathway. Circulation. 2003;107:3081–3087. [DOI] [PubMed] [Google Scholar]
- 36.Feletou M Calcium-activated potassium channels and endothelial dysfunction: therapeutic options? Br J Pharmacol. 2009;156:545–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Archer SL, Huang JM, Hampl V, Nelson CP, Shultz PJ, Weir EK. Nitric oxide and cGMP cause vasorelaxation by activation of a charybdotoxin-sensitive K channel by cGMP-dependent protein kinase. Proc Natl Acad Sci USA. 1994;91:7583–7587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Blychkov R, Burnham MP, Richards GR, et al. Characterization of a charybdotoxin-sensitive intermediate conductance Ca2+-activated K+ channel in porcine coronary endothelium: relevance to EDHF. Br J Pharmacol. 2002;137:1346–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Robertson BE, Schubert R, Hescheler J, Nelson MT. cGMP-dependent protein kinase activates Ca-activated K channels in cerebral artery smooth muscle cells. Am J Physiol. 1993;265:C299–C303. [DOI] [PubMed] [Google Scholar]
- 40.Bolotina VM, Najibi S, Palacino JJ, Pagano PJ, Cohen RA. Nitric oxide directly activates calcium-dependent potassium channels in vascular smooth muscle. Nature. 1994;368:850–853. [DOI] [PubMed] [Google Scholar]
- 41.Sheng JZ, Braun AP. Small-and intermediate-conductance Ca2+-activated K+ channels directly control agonist-evoked nitric oxide synthesis in human vascular endothelial cells. Am J Physiol Cell Physiol. 2007;293:C458–467. [DOI] [PubMed] [Google Scholar]
- 42.Crane GJ, Gallagher N, Dora KA, Garland CJ. Small- and intermediate-conductance calcium-activated K+ channels provide different facets of endothelium-dependent hyperpolarization in rat mesenteric artery. J Physiol. 2003;553:183–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Quayle JM, Nelson MT, Standen NB. ATP-sensitive and inwardly rectifying potassium channels in smooth muscle. Physiol Rev. 1997;77:1165–1232. [DOI] [PubMed] [Google Scholar]
- 44.Jackson WF. Potassium channels in the peripheral microcirculation. Microcirculation. 2005;12:113–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Park WS, Hong DH, Son YS, et al. Alteration of ATP-sensitive K+ channels in rabbit aortic smooth muscle during left ventricular hypertrophy. Am J Physiol Cell Physiol. 2012;303:C170–C178. [DOI] [PubMed] [Google Scholar]
- 46.Katnik C, Adams DJ. An ATP-sensitive potassium conductance in rabbit arterial endothelial cells. J Physiol. 1995;485:595–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kuo L, Chancellor JD. Adenosine potentiates flow-induced dilation of coronary arterioles by activating KATP channels in endothelium. Am J Physiol. 1995;269:H857–H869. [DOI] [PubMed] [Google Scholar]
- 48.White R, Hiley CR. Hyperpolarisation of rat mesenteric endothelial cells by ATP-sensitive K(+) channel openers. Eur J Pharmacol. 2000;397:279–290. [DOI] [PubMed] [Google Scholar]
- 49.Yoshida H, Feig JE, Morrissey A, Ghiu IA, Artman M, Coetzee WA. KATP channels of primary human coronary artery endothelial cells consist of a heteromultimeric complex of Kir6.1, Kir6.2, and SUR2B subunits. J Mol Cell Cardiol. 2004;37:857–869. [DOI] [PubMed] [Google Scholar]
- 50.Li L, Wu J, Jiang C. Differential expression of Kir6.1 and SUR2B mRNAs in the vasculature of various tissues in rats. J Membr Biol. 2003;196:61–69. [DOI] [PubMed] [Google Scholar]
- 51.Babenko AP, Aguilar-Bryan L, Bryan J. A view of sur/KIR6.X, KATP channels. Annu Rev Physiol. 1998;60:667–687. [DOI] [PubMed] [Google Scholar]
- 52.Morrissey A, Rosner E, Lanning J, et al. Immunolocalization of KATP channel subunits in mouse and rat cardiac myocytes and the coronary vasculature. BMC Physiol. 2005;5:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sheykhzade M, Berg Nyborg NC. Mechanism of CGRP-i nduced relaxation in rat intramural coronary arteries. Br J Pharmacol. 2001;132:1235–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vedernikoy YP, Fulep EE, Saade GR, Garfield RE. Calcitonin gene-related peptide dilates the pregnant rat uterine vascular bed via guanylate cyclase, ATP- and Ca-sensitive potassium channels and gap junctions. Curr Med Res Opin. 2002;18:465–470. [DOI] [PubMed] [Google Scholar]
- 55.Ralevic V, Belai A, Burnstock G. Impaired sensory-motor nerve function in the isolated mesenteric arterial bed of streptozotoc-indiabetic and ganglioside-treated streptozotocin-diabetic rats. Br J Pharmacol. 1993;110:1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Boerman EM, Segal SS. Aging alters spontaneous and neurotransmitter-mediated Ca2+ signaling in smooth muscle cells of mouse mesenteric arteries. Microcirculation. 2020;27:e12607. [DOI] [PMC free article] [PubMed] [Google Scholar]