

Abstract
Glucosylceramide synthase (GCS) is an enzyme that catalyzes the first reaction of ceramide glycosylation in sphingolipid metabolism. It represents a primary target in the pharmacological treatment of some lysosomal storage diseases (LSDs), such as Gaucher and Niemann-Pick syndromes. In this study, starting from the model reported in the AlphaFold Protein Structure Database, the location and conformations of GCS substrates and cofactors have been provided by a step-by-step in silico procedure, by which the functional manganese ion and the substrates have been inserted in the GCS structure through combined molecular docking and full-atomistic molecular dynamics approaches, including metadynamics. A detailed analysis by structural dynamics of the complete model system, i.e., the enzyme anchored to the plasma membrane, containing the manganese ion and the two substrates, has been carried out to identify its complex conformational landscape by means of well-tempered metadynamics. A final structure was selected, in which both substrates were present in the active site of the enzyme at minimum distance, thus giving support to a SNi-type reaction mechanism for catalysis. Asp236, Glu235, and Asp144 are found to interact with the metal cofactor, which is able to trap the phosphates of UDP-glucose, while Gly210, Trp276, and Val208 cooperate to provide its correct orientation. Phe205, Cys207, Tyr237, and Leu284 form a pocket for the polar head of the ceramide, which is transiently placed in position to determine the catalytic event, when His193 interacts with the head of the ceramide, thus anchoring the substrate to the active site.
Introduction
Lysosomal storage diseases (LSDs) are a group of over 70 diseases, mainly autosomal recessive disorders, that are characterized by lysosomal impairment. These disorders are individually rare, but taken together, they affect 1 in 5000 live births.1 LSDs are characterized by shortage in normal lysosomal function and by lysosomal accumulation of nondegraded substrates. Therapeutic approaches may include strategies to increase the residual activity of a missing enzyme through enzyme replacement therapy, as well as hematopoietic stem cell transplantation, pharmacological chaperone therapy, gene therapy, and approaches based on reducing the number of substrates to lysosomes.2 Niemann-Pick disease type C (NPC) is an autosomal recessive lipid storage disorder characterized by visceral symptoms, progressive deterioration of the central nervous system, and premature death in most patients.3 The frequency of the disease is about 1 case in every 120,000 live births. NPC is caused by mutations of the NPC1 (95% of cases) or NPC2 (5% of cases) genes that decrease intracellular cholesterol trafficking, resulting in accumulation of unesterified cholesterol. Despite the fact that cholesterol is largely recognized as the principal storage lipid in NPC, many species of sphingolipids also accumulate. This accumulation leads to the initial assumption that a deficiency in sphingolipid catabolism contributes to the biology and pathology observed in the NPC disease.4
To study the contribution of glycosphingolipid accumulation to the pathogenesis of NPC, animal models were treated with the iminosugar Miglustat (N-butyldeoxynojirimycin), an inhibitor of glucosylceramide synthase (GCS). In these animals, a reduced accumulation of glycosphingolipids was observed together with a delay in the onset of symptoms and an increase in lifespan;5 the same behavior was also confirmed in human patients,6 therefore indicating this enzyme as a possible hotspot for a therapeutic approach.
Thus, we have focused our study on the GCS enzyme (EC 2.4.1.80), a membrane-anchored protein with its C-terminal catalytic domain located in the cytoplasm, which is able to catalyze the first reaction of ceramide glycosylation in sphingolipid metabolism7,8 (UniProt code: Q16739)9 (see Figure 1).
Figure 1.
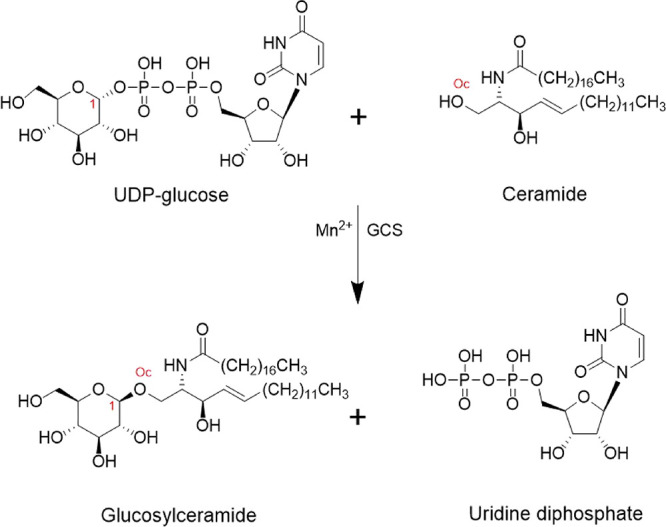
Reaction catalyzed by GCS. Involving C1 of UDP-glucose and OH (Oc) of the polar head of the ceramide.
The three-dimensional structure of this monomeric enzyme is not experimentally known, probably due to intrinsic low propensity to crystallize (a common characteristic of the membrane-bound proteins), and is not present in the Protein Data Bank, but its predicted conformation has been provided via the recently reported state-of-the-art AI (artificial intelligence) AlphaFold system, which is able to computationally predict protein structures with unprecedented accuracy.10 Notably, the structure of the enzyme is freely available in the AlphaFold Protein Structure Database.11 The 3D structure (Figure 2 and Figure S1) includes an inlet channel on the solvent-exposed surface of the enzyme (most likely for nucleotide sugar), extending to an inlet channel located at the membrane–solvent interface (most likely for the ceramide), in an approximately linear geometry. In the present study, we provide the site location of a divalent manganese ion (Mn2+), which is reported as a key cofactor for its catalytic activity,12,13 together with the conformation of the endogenous substrates UDP-glucose and ceramide. A wide in silico characterization of the enzyme–substrate complex has been performed by means of molecular dynamics and well-tempered metadynamics. Our results may be used as a starting point for a future characterization of this pharmacological hotspot in the presence of a lead compound that is able to mimic the structure of the active state extracted by in silico characterization.
Figure 2.

Starting GCS structure in a model membrane. The enzyme–membrane complex was built according to protein domain topological localization as reported on UniProt and using the CHARMM-GUI server.14 (A) The GCS structure is shown in surface representation in blue color. (B) Different and close-up view of the GCS structure, where the access to the active site of the enzyme is shown.
Materials and Methods
Model System Setup and Characterization
The computational model confidence was estimated using different parameters. The first one was the local distance difference test (pLDDT) that exhibited a very high score (>90) for most of the residues, as shown in Figure S1A. The second one was the predicted aligned error plot, which is useful for evaluating the domain packing and has been revealed to be satisfying for the entire structure (Figure S1B) as demonstrated by a quite uniform dark green intensity for the relative position of all residues.
Moreover, the retrieved structure underwent further analysis and characterization; specifically, the minimized structure via Maestro tools was submitted to the ProSA-web server,15 obtaining the Z-score and local model quality plot. The Z-score value is displayed as a black dot in Figure S1C, where all the experimentally determined values in the current PDB, both X-ray and NMR structures, were represented. As shown in the selected panel, the Z-score of the GCS model is perfectly within the score range corresponding to experimentally known structures of similar size proteins. Furthermore, the local model quality, reported in Figure S1D, was achieved by plotting the energy score as a function of amino acid sequence. In general, positive values correspond to problematic or erroneous sections of the input structure, while negative values reflect the good quality of the model system. A plot of single-residue energies usually contains large fluctuations and is of limited value for model evaluations. Therefore, the plot is smoothed by calculating the average energy over a selected range of residues; accordingly, the system was challenged using two different window sizes: a first one was done considering a fragment of 40 residues, s(i,i + 39), which is then assigned to the “central” residue of the fragment at position i + 19 (thick line in the plot); a second one was prepared using a smaller, 10-residue fragment window size, and the result is shown in the background of the plot (thin line). In both cases, overall, the energy score obtained is largely in the negative side of the plot with both window sizes, thus confirming the good quality of the model. The 40-residue energies are weakly positive in the sequence of amino acids around positions 175–201, 203–209, 291–314, and 321–332. These amino acids belong to the membrane spanning domain or are close to the membrane, which thus are expected to have larger values, due to the lower capacity of ProSA-web to predict their conformation.15 On the contrary, these amino acids are well predicted by the AlphaFold algorithm due to the different metric used to analyze the confidence.16
Metadynamics Simulations of Mn2+ Uptake
We used all-atom metadynamics to simulate the process of uptake of divalent ions within the active site of the GCS enzyme, i.e., Mg2+ and Mn2+. The same binding site was found by both simulations; thus, we report here only the Mn2+ case. The ion was placed outside the binding pocket, and its motion was driven by a metadynamics bias potential,17 combined with a cylinder-shaped restraint potential as a special case of the more general funnel shape, as shown in Figure S2. The PLUMED 2.8 package,18,19 which includes the funnel metadynamics (FM) code,20 has been patched to the GROMACS 2021.421 engine to carry out the metadynamics runs. The cylinder shape of the potential has been customized on the target structure according to the following parameters: Zcc = 0.2 nm as the distance between the switching point from the cone to the cylinder section, Alpha = 0.01 rad as the angle defining the amplitude of the cone section, and Rcyl = 1.1 nm as the radius of the cylinder section.20 The cylinder axis lies approximately along the axis of the substrate entry channels, and the position of the ion is restrained inside the cylinder. Two biased collective variables (CVs) were chosen according to the FM protocol, i.e., the position along the axis of the cylinder and its distance from the rotation axis. Gaussian functions, describing the bias potential added in time at the position defined by CVs, had a height of 1 kJ/mol and a sigma value of 0.05 and were saved every 1 ps. At the end of the simulation, we determined the position of the ion binding site; thereafter, the distance between the ion and the binding site and the ion hydration degree were recalculated, and their free energy-like diagrams were obtained. Although classical molecular dynamics and metadynamics based on point-charge atomistic force fields are inadequate to provide the accurate binding geometry of the coordination metal Mn2+, we are fully confident that its approximate location within the enzyme (the same as Mg2+), mainly determined by its charge, is correctly defined.
Molecular Dynamics Simulations of GCS Protein
Further computational analysis of GCS protein and in combination with its endogenous ligands was performed using MD simulation by the GROMACS 2021.4 package using the CHARMM36m force field22 with the WYF parameter for π–cation interactions23 at the full atomistic level using a TIP3P water solvent.22 All the systems were prepared via the CHARMM-GUI server14 using Membrane Builder to create a phospholipidic double-layer membrane so that the conformation of the protein was inserted into a POPC double layer (134 numbers of lipids) surrounded by water (30,477 atoms). The total charge of the system was neutralized with K+ ions and Cl– ions to obtain neutrality with 0.15 M salt concentration, and all runs were performed at 303.15 K.
Docking of Different Ligands to the GCS Enzyme
The GCS starting structure for the in silico induced fit docking procedure24 was obtained as the minimum found with metadynamics to define the position of Mn2+ in the active site. The protein was prepared using Maestro Protein Preparation Wizard with default parameters. Induced fit docking accounts for receptor flexibility in ligand-receptor docking by iteratively combining rigid receptor docking, using Glide,25,26 with protein structure prediction and refinement, using Prime.27,28 Both GCS substrates were obtained from PubChem, a substance and compound database.29 Each ligand (substrate) was subjected to a preparation using LigPrep with the OPLS3e30 force field. Mn2+ was chosen for the center of the box, and the size of the box was 20 Å. While performing induced fit docking, receptor and ligand van der Waals scaling was maintained to 0.50 and generated the maximum of 20 poses of the protein with ligands. The calculations were performed by considering the standard precision protocol. The best pose of the ligand–protein complex was selected for performing the MD simulation.
HOLE Analysis for the GCS Binding Site
To determine the shape and dimension of the extended channel, which represents the GCS binding site, we used the HOLE program.31 In the resulting image (see Figure 7), the surface is colored according to the water-accessible parts, i.e., the surface is red where the pore radius is too tight for a water molecule, green where there is room for a single water, and blue where the radius is double the minimum for a single water. The starting point for this analysis corresponds to the coordinates of the Mn2+ ion, while the values used for the van der Waals atomic radii were taken from the AMBER ff19SB force field;32 the solvent and ions found in the channel were excluded from the HOLE analysis.
Figure 7.

GCS pore: the surface is colored according to the water-accessible parts; therefore, the surface is green where there is room for a single water and blue where the radius is double. The restriction is generated by amino acids Phe205, Cys207, Tyr237, and Leu284.
Well-Tempered Funnel Metadynamics (FM) Simulations for the Ceramide
To analyze the location and conformation of the GCS substrates, an FM protocol was used.20 A starting ceramide conformation inside the enzyme was chosen between those found after preliminary MD runs to be compatible with glycosylation, i.e., when the distance dC1-Oc between the terminal hydroxyl oxygen of the ceramide’s polar head and the carbon C1 of glucose in UDP-glucose (atoms involved in the glycosylation reaction were minimal). The starting system is shown in Figure S3. According to FM, biased CVs chosen were the position along the rotation axis of the funnel (CV1), its distance from the axis (CV2), and the distance dC1-Oc (CV3). The funnel parameters that were used are Zcc = 3.0 nm, Alpha = 0.5 rad, and Rcyl = 0.6 nm. Gaussian functions with a height of 2 kJ/mol and a sigma value of 0.05 were deposited every 1 ps.
Results and Discussion
The computational model confidence of the GCS enzyme was estimated according to a set of different parameters to check the model quality. The first two are the pLDDT and predicted aligned error plot from AlphaFold, while the Z-score and local model quality plot have been obtained using the ProSA-web server as reported in Materials and Methods. All the parameters were satisfactory.
Identification of the Metal Site through Classical Metadynamics
A divalent ion binding site has been reported to be present inside the GCS binding pocket; to provide its correct position, metadynamics simulations were performed with Mn2+, as this is the supposed key cofactor.8 Its initial position was set outside the binding site, and then two biased CVs were selected: the position along the rotation axis and the distance from the rotation axis of the above-mentioned potential-restraint cylinder, which includes the active site channel (see Materials and Methods). A position restraint, with a constant force of 200 kJ/mol nm2 applied to each spatial dimension, was further applied to the protein backbone alpha carbons (C-alpha atoms), and a 500 ns-long metadynamics simulation was performed.
At the end of simulation, since we detected the binding site close to Glu235, we recalculated the ion-binding site distance between the amino acid Glu235 and the manganese ion (d_Mn) and the manganese hydration (Mn_hydr) all along the simulation. Accordingly, we have been able to obtain a 2D free energy-like histogram and its projection on the distance parameter using the PLUMED plugin.33 As shown in Figure 3, during the 500 ns of metadynamics, the ion crossed the binding site several times (panel A), thus allowing one to consider the result obtained with a successful convergency procedure. The 2D histogram (panel B) allows one to identify an absolute minimum in the position of the ion in the binding site of the protein. The Mn2+ ion at the starting point (S) of the simulation outside the binding site of GCS is fully hydrated, while in its final position, it is close to the amino acid Glu235 and no longer hydrated. The free energy-like profile as a function of the distance between Mn2+ and the amino acid Glu235 (panel C) confirms that the energy minimum lies at a binding distance of about 2.0 Å to this residue.
Figure 3.

(A) Plot of d_Mn along the metadynamics simulation. (B) Free energy-like 2D histogram as a function of the manganese hydration (Mn_hydr) and distance (d_Mn, in nanometers) from the amino acid Glu235. (C) Integrated free energy-like histogram as a function of d_Mn. (D) Mn2+ binding site corresponding to the lowest energy.
The validity of this result was compared with previous experimental data. Indeed, as reported by Marks et al.,34 there are important amino acids involved in the enzymatic function. Their role in the GCS function was studied with site-directed mutagenesis, and specifically, substitution of Asp144, Asp236, Arg275, or Trp-276 with alanine was always considered to perturb a significantly enzymatic function. The manganese ion, as we can see in Figure 3D, coordinates directly Glu235 and Asp236, thus confirming the positioning of this ion in the active site of the enzyme as a prerequisite to favor the formation of the active complex with the endogenous substrates.
Molecular Dynamics of GCS-apo and GCS-Mn2+
The two models obtained, GCS-apo and GCS-Mn2+, were subjected to an MD protocol to verify their stability. The C-alpha root mean square deviation (RMSD) plot of two consecutive 2 μs MD simulations relative to both GCS-apo and GCS-Mn2+ (Figure 4A,B) shows the stability of the backbone throughout the simulations; it is interesting to note that the difference appearing between the two RMSD plots corresponds to a conformational change occurring in the first 250 ns that is triggered by a small repositioning of Mn2+. This conformational change is represented in Figure 4C, where there is a backward shift of amino acids 213–235 and a downward shift of amino acids 236–250. Furthermore, the root mean square fluctuation (RMSF), relative to the average structures in the last 500 ns, reported in Figure 4D, shows the difference of residue flexibility between GCS-apo and GCS-Mn2+. In particular, Thr209, Lys124, Glu235, and Asp236 are affected by the presence of Mn2+.
Figure 4.

(A) RMSD trajectory (2 μs) of CGS-apo. (B) RMSD trajectory (2 μs) of CGS-Mn2+. (C) Residue-based C-alpha RMSF of CGS-apo (blue line) and CGS-Mn2+ (orange line). (D) Shift representation of amino acid 213–250 CGS-apo (blue) and CGS-Mn2+ (orange).
Docking and Molecular Dynamics of the Complex UDP-Glucose and GCS-Mn2+
According to the protocol described in Materials and Methods, induced fit docking of UDP-glucose to the GCS-Mn2+ structure was obtained by the Maestro Schrodinger procedure software suite. At the end of this procedure, nine different docking poses of UDP-glucose were obtained and the best one (docking score, 7.559) was selected (gray structure in Figure 5A). After induced fit docking adjustment, MD simulations were used to characterize the binding site of the enzyme and two 1 μs-long simulations were also performed. A clustering procedure was then applied to the last 500 ns of each simulation after concatenating them together. A blow-up of the result is displayed in Figure 5A, where the final pose of UDP-glucose is reported in blue, and it is compared to the initial position in gray.
Figure 5.

(A) In blue, the most representative structure of UDP-glucose binding mode obtained after a clustering of the MD runs is shown, while in gray, the starting configuration obtained after the induced fit docking procedure is reported. (B) Interaction histogram of protein and UDP-glucose. Results include hydrophobic contacts, hydrogen bonds, salt bridges, π–cation contacts, π-stacking contacts, water bridges, and metal coordination and are obtained by analyzing the last 500 ns of two 1 μs-long molecular dynamics. (C) Scheme of the Mn2+ coordination cluster obtained by the MD analysis.
Moreover, in Figure 5B, a histogram reporting the residues mainly involved in the binding with UDP-glucose along the final part of the trajectories analyzed is shown. The Asp144, Asp236, Arg275, and Trp276 residues, previously reported as key amino acids for the enzymatic activity,34 are all found to be involved in the formation of the UDP-glucose-GCS complex. Glu235 and Asp236 establish a water bridge and hydrogen bonds to the glucose moiety. Arg275 establishes a salt bridge with the phosphate group, and Trp276 establishes a hydrogen bond and hydrophobic interaction with the glucose and phosphate group, while the ribose moiety of UDP-glucose is strengthened by Asp144 and Lys124. Moreover, we found that in the formation of the complex with the UDP-glucose substrate, Pro58 and Arg275 are also important for correct positioning of the uracil ring of UDP via hydrophobic and π–cation interactions, respectively.
To further examine the coordination of manganese during MD simulations, we calculated the percentage of bonds as shown in Figure 5C. The phosphate groups of UDP-glucose are directly involved in the coordination of Mn2+ together with Asp236 and Glu235, which directly bind to the metal ion, whereas Asp144 coordinates Mn2+ via a water molecule with a percentage of 140%. A percentage greater than 100% means that in agreement with what was reported by Belyi and Aktories35 for the structure of the glucosyltransferase A-domain of toxin B, there is another water molecule that intervenes in the coordination of manganese (Figure 5C).
Docking and Molecular Dynamics of the Full Complex UDP-Glucose and Ceramide with GCS-Mn2+
Induced fit docking of UDP-glucose and ceramide to the GCS-Mn2+ structure was attempted by the Maestro Schrodinger procedure. At the end of this procedure, 10 docking poses were obtained and the best pose (docking score, 6.848) was chosen. Starting from the selected docking pose, two 1 μs MD simulations were performed, trying to find conditions as close as possible to those compatible to trigger the enzyme activation. These simulations were analyzed considering only the first 500 ns time course; as for longer times, the ceramide returns into the phospholipid milieu of the plasma membrane (which is expected in the absence of any conformational rearrangement, leading to an activated complex transition state). The analysis of MD simulations allowed one to identify a specific pose of the ceramide with a distance dC1-Oc of 2.90 Å, which is compatible for triggering ceramide glycosylation (Figure 6).
Figure 6.

Selected state of the GCS-ceramide-UDP-glucose complex. (A) Full enzyme representation with the substrates reported as van der Waals spheres. (B) Close-up view of the active site positioning of the UDP-glucose head, in green, and the ceramide molecule, in blue, with a distance dC1-Oc = 2.90 Å.
Analysis of the Active Site of the GCS-Substrate Complex via the HOLE Program
To further characterize the GCS enzyme in the last identified pose, when the distance dC1-Oc is minimal, we analyzed the surface of the pore containing the active site using the HOLE program as described in Materials and Methods, and the results are visualized in Figure 7. The entire pore surface, colored according to water accessibility and represented using the VMD graphical interface,36 shows one restriction, colored green, closer to the membrane edge (see Figure 7). The shrinkage is generated by amino acids Phe205, Cys207, Tyr237, and Leu284. On the cytosolic side of this restriction, a blue pocket is visible, containing the Mn2+ site and the UDP-glucose substrate, correctly oriented to trigger the catalytic event. The pore allows the entering from the other side of the polar head of the ceramide while reducing the opening toward the membrane milieu and transiently trapping the lipidic substrate in a properly oriented configuration. On the lower side of the green knot, where the protein is anchored to the membrane, a larger blue area opens up to allow the exchange of the substrate product with the phospholipidic bilayer.
Funnel Metadynamics Analysis of the Active Site of the GCS-Substrate Complex
A deeper study of the active site of the GCS-substrate complex was attempted using a well-tempered metadynamics (WTM) protocol as reported in Materials and Methods. As a starting point for WTM, the best pose obtained from previous MD analysis was selected. In this configuration, both substrates are placed at a distance such as to favor the ceramide glycosylation, corresponding to a dC1-Oc of 2.90 Å. In the following WTM procedure, three CVs were selected: CV1 represents the position along the rotation axis of the funnel (see Figure S2), CV2 represents the distance from the rotation axis of the above-mentioned funnel, and CV3 represents the distance dC1-Oc. A positional restraint to the protein alpha-carbons of the protein backbone (C-alpha atoms) and a positional restraint to UDP-glucose were also applied, and a 1800 ns-long WTM simulation was performed. The Gaussian heights deposited during the time tend to be zero, indicating that WTM has converged after 1500 ns as reported in Figure S4.
In Figure 8A, the distance dC1-Oc is reported during the WMT simulation starting from its 0 value corresponding to the starting position previously determined. As evident from the figure, the lipidic substrate was able to exit and re-enter its inlet channel from the membrane milieu, reaching three times in all the cases a position potentially allowing its glycosylation, with a favorable orientation of the polar head. At t = 1148 ns, the minimum distance dC1-Oc of 2.87 Å was found, and the corresponding pose is represented in Figure 8B.
Figure 8.

(A) Time evolution of the CV1 value during the WTM simulation. The ceramide reaches three times a binding distance with UDP-glucose, and at 1148 ns, we find the minimum distance between the oxygen of the ceramide and the carbon of UDP-glucose obtained during the simulation. (B) Close-up representation of the minimum distance between the two endogenous substrates. In purple, the Mn2+ ion is visible.
Therefore, the entire computational procedure provides clues on the possible reaction mechanism and on the specific roles played by the Mn2+ ion and by specific enzyme amino acids. As reported by Mestrom et al.37 for Leloir glycosyltransferases, there are different possible reaction mechanisms utilizing divalent metals, which are available to this class of enzymes and may include inversion or retention of the anomeric glycosidic bond. As in our study, we cannot identify an amino acid that can be used for general base catalysis; according to the publication mentioned, we propose that a SNi-type retaining mechanism is responsible for the activity of human GCS, as shown in Figure 9.
Figure 9.

Proposed SNi-type mechanism for the reaction catalyzed by GCS.
The first part of the figure shows the amino acids involved in the correct positioning of the two substrates as a result of the docking procedure coupled with MD and metadynamics simulations. Indeed, Asp236, Glu235, and Asp144 have a structural role, being the residues required to position the metal cofactor that is able to trap the phosphate groups of UDP-glucose, while Gly210, Trp276, and Val208 participate in its oriention, in agreement with a site-directed mutagenesis study.34 A specific role is also emerging for His193, which during MD trajectories directly binds the second hydroxyl group of the ceramide polar head, transiently anchoring the lipid substrate to the active site in the right orientation. This result is in perfect agreement with Wu et al.,38 who demonstrated that as in the homologous GCS enzyme of rats, the replacement of the conserved His193 residue eliminates sensitivity to the GCS inhibitor d-threo-1-phenyl-2-decanoylamino-3-morpholinopropan-1-ol (PDMP), which is an analogue of the ceramide substrate. This interaction is also favored by the restriction imposed by Phe205, Cys207, Tyr237, and Leu284 that considerably decrease the space available in the active site for the ceramide polar head, allowing it to be correctly positioned to determine the catalytic event. Furthermore, the best approximation of the transition state, defined like the trapped pose at the shortest distance dC1Oc, is highlighted by a red circle in Figure 8A, and it is represented in Figure 8B. This protein conformation is able to trigger the catalytic event, as shown inside the cyan frame of Figure 9. Indeed, in this conformation, the position of glucose is accompanied by the Val208 movement that changes its interaction partner from carbon C3 to carbon C2, causing the displacement of Gly210. This displacement allows the shortening of the binding distance dC1Oc from the initial 2.93 Å to the final 2.87 Å. The reaction mechanism continues as hypothesized from Moremen and Haltiwanger39 and Breton et al.40 for the SNi-type retaining mechanism: the acceptor hydroxyl nucleophile of the ceramide is deprotonated by the donor phosphate oxygen on UDP-glucose and attacks the carbon C1 of the sugar head, with the transient formation of an oxocarbenium ion and the final production of glucosylceramide and UDP, which are ready to be released in the corresponding milieu.
Conclusions
The availability of the GCS structure with unprecedented accuracy, predicted by the AlphaFold AI system, allowed us a computational characterization of the enzyme, which we studied in the presence of the manganese cofactor ion and both endogenous substrates, UDP-glucose and ceramide. The manganese ion uptake within the apo-enzyme was simulated by means of a metadynamics approach, and we have further determined how it coordinates the phosphate groups of UDP-glucose, trapping and properly orienting the soluble substrate. The most important amino acids involved in coordination of manganese and in the interaction with UDP-glucose are in perfect agreement with previous experimental work of site-directed mutagenesis.34 Finally, WTM allowed us to determine a structure of the fully active enzyme in the presence of both substrates at a distance favorable to trigger catalysis, thus justifying a possible SNi-type reaction mechanism, as shown in Figure 9. Accordingly, Asp236, Glu235, and Asp144 directly interact with the metal cofactor, which traps the phosphates of UDP-glucose, while Gly210, Trp276, and Val208 cooperate to its orientation. His193 directly binds the head of the ceramide, transiently anchoring the substrate to the active site. Phe205, Cys207, Tyr237, and Leu284 form a pocket for the polar head of the ceramide, which is placed in the correct position to determine the catalytic event.
These results are the prerequisite for further development of a computational platform, in which, starting from a subset of specific configurations of GCS in complex with UDP-glucose and ceramide at a catalytic distance, a possible drug repurposing strategy may be attempted, specifically focusing on lead compounds that are able to simulate the chemical property of this activation state.
Acknowledgments
Financial support by the Italian Ministry of University and Research (Linea D1 Università Cattolica del Sacro Cuore) is gratefully acknowledged. This work was partially supported by the CINECA supercomputing centers through the project IsC97 OriginID HP10CKTQ6H.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.2c08219.
Figure S1: AlphaFold model confidence, predicted aligned error, model quality according to ProSA-web, and local model quality according to ProSA-web; Figure S2: representation of the cylinder used to sample the position of divalent ions in the active site with classical metadynamics; Figure S3: representation of the funnel metadynamics system used to analyze the location and conformation of the GCS substrates; Figure S4: plot of evolution of Hills heights as a function of time in FM simulation with GCS substrates (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Platt F. M.; d’Azzo A.; Davidson B. L.; Neufeld E. F.; Tifft C. J. Lysosomal storage diseases. Nat. Rev. Dis. Primers. 2018, 4, 27. 10.1038/s41572-018-0025-4. [DOI] [PubMed] [Google Scholar]
- Parenti G.; Andria G.; Ballabio A. Lysosomal storage diseases: from pathophysiology to therapy. Annu. Rev. Med. 2015, 66, 471–486. 10.1146/annurev-med-122313-085916. [DOI] [PubMed] [Google Scholar]
- Naureckiene S.; Sleat D. E.; Lackland H.; Fensom A.; Vanier M. T.; Wattiaux R.; Jadot M.; Lobel P. Identification of HE1 as the second gene of Niemann-Pick C disease. Science 2000, 290, 2298–2301. 10.1126/science.290.5500.2298. [DOI] [PubMed] [Google Scholar]
- Newton J.; Milstien S.; Spiegel S. Niemann-Pick type C disease: The atypical sphingolipidosis. Adv. Biol. Regul. 2018, 70, 82–88. 10.1016/j.jbior.2018.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zervas M.; Somers K. L.; Thrall M. A.; Walkley S. U. Critical role for glycosphingolipids in Niemann-Pick disease type C. Curr. Biol. 2001, 11, 1283–1287. 10.1016/s0960-9822(01)00396-7. [DOI] [PubMed] [Google Scholar]
- Lachmann R. H.; te Vruchte D.; Lloyd-Evans E.; Reinkensmeier G.; Sillence D. J.; Fernandez-Guillen L.; Dwek R. A.; Butters T. D.; Cox T. M.; Platt F. M. Treatment with miglustat reverses the lipid-trafficking defect in Niemann-Pick disease type C. Neurobiol Dis. 2004, 16, 654–658. 10.1016/j.nbd.2004.05.002. [DOI] [PubMed] [Google Scholar]
- Yamashita T.; Wada R.; Sasaki T.; Deng C.; Bierfreund U.; Sandhoff K.; Proia R. L. A vital role for glycosphingolipid synthesis during development and differentiation. Proc. Natl. Acad. Sci. U. S. A. 1999, 96, 9142–9147. 10.1073/pnas.96.16.9142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y. Y.; Hill R. A.; Li Y. T. Ceramide glycosylation catalyzed by glucosylceramide synthase and cancer drug resistance. Adv. Cancer Res. 2013, 117, 59–89. 10.1016/B978-0-12-394274-6.00003-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uni Prot: the Universal Protein Knowledgebase in 2023. Nucleic Acids Res. 2023, 51, D523–D531. 10.1093/nar/gkac1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jumper J.; Evans R.; Pritzel A.; Green T.; Figurnov M.; Ronneberger O.; Tunyasuvunakool K.; Bates R.; Žídek A.; Potapenko A.; Bridgland A.; Meyer C.; Kohl S. A. A.; Ballard A. J.; Cowie A.; Romera-Paredes B.; Nikolov S.; Jain R.; Adler J.; Back T.; Petersen S.; Reiman D.; Clancy E.; Zielinski M.; Steinegger M.; Pacholska M.; Berghammer T.; Bodenstein S.; Silver D.; Vinyals O.; Senior A. W.; Kavukcuoglu K.; Kohli P.; Hassabis D. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varadi M.; Anyango S.; Deshpande M.; Nair S.; Natassia C.; Yordanova G.; Yuan D.; Stroe O.; Wood G.; Laydon A.; Žídek A.; Green T.; Tunyasuvunakool K.; Petersen S.; Jumper J.; Clancy E.; Green R.; Vora A.; Lutfi M.; Figurnov M.; Cowie A.; Hobbs N.; Kohli P.; Kleywegt G.; Birney E.; Hassabis D.; Velankar S. Alpha Fold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50, D439–D444. 10.1093/nar/gkab1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu S.; Kaufman B.; Roseman S. Enzymatic synthesis of ceramide-glucose and ceramide-lactose by glycosyltransferases from embryonic chicken brain. J. Biol. Chem. 1968, 243, 5802–5804. 10.1016/S0021-9258(18)91935-6. [DOI] [PubMed] [Google Scholar]
- Shukla G. S.; Radin N. S. Glucosyceramide synthase of mouse kidney: further characterization with an improved assay method. Arch. Biochem. Biophys. 1990, 283, 372–378. 10.1016/0003-9861(90)90657-k. [DOI] [PubMed] [Google Scholar]
- Brooks B. R.; Brooks C. L. III; Mac Kerell A. D.; Nilsson L.; Petrella R. J.; Roux B.; Won Y.; Archontis G.; Bartels C.; Boresch S.; Caflisch A.; Caves L.; Cui Q.; Dinner A. R.; Feig M.; Fischer S.; Gao J.; Hodoscek M.; Im W.; Kuczera K.; Lazaridis T.; Ma J.; Ovchinnikov V.; Paci E.; Pastor R. W.; Post C. B.; Pu J. Z.; Schaefer M.; Tidor B.; Venable R. M.; Woodcock H. L.; Wu X.; Yang W.; York D. M.; Karplus M. CHARMM: The Biomolecular Simulation Program. J. Comput. Chem. 2009, 30, 1545–1614. 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiederstein M.; Sippl M. J. ProSA-web: interactive web service for the recognition of errors in three- dimensional structures of proteins. Nucleic Acids Res. 2007, 35, W407–W410. 10.1093/nar/gkm290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariani V.; Biasini M.; Barbato A.; Schwede T. lDDT: a local superposition-free score for comparing protein structures and models using distance difference tests. Bioinformatics 2013, 29, 2722–2728. 10.1093/bioinformatics/btt473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laio A.; Parrinello M. Escaping free-energy minima. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 12562–12566. 10.1073/pnas.202427399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Promoting transparency and reproducibility in enhanced molecular simulations. Nat. Methods 2019, 16, 670–673. 10.1038/s41592-019-0506-8. [DOI] [PubMed] [Google Scholar]
- Tribello G. A.; Bonomi M.; Branduardi D.; Camilloni C.; Bussi G. PLUMED2: New feathers for an old bird. Comp. Phys. Commun. 2014, 185, 604–613. 10.1016/j.cpc.2013.09.018. [DOI] [Google Scholar]
- Raniolo S.; Limongelli V. Ligand binding free-energy calculations with funnel metadynamics. Nat. Protoc. 2020, 15, 2837–2866. 10.1038/s41596-020-0342-4. [DOI] [PubMed] [Google Scholar]
- Abraham M. J.; Murtola T.; Schulz R.; Páll S.; Smith J. C.; Hess B.; Lindahl E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. Software X 2015, 1-2, 19–25. 10.1016/j.softx.2015.06.001. [DOI] [Google Scholar]
- Huang J.; Rauscher S.; Nawrocki G.; Ran T.; Feig M.; de Groot B. L.; Grubmuller H.; Mac Kerell A. D. Jr. CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat. Methods 2017, 14, 71–73. 10.1038/nmeth.4067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan H. M.; Mac Kerell A. D.; Reuter Jr. Cation-pi interactions between methylated ammonium groups and tryptophan in the CHARMM36 additive force field. J. Chem. Theory Comput. 2019, 15, 7–12. 10.1021/acs.jctc.8b00839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman W.; Day T.; Jacobson M. P.; Friesner R. A.; Farid R. Novel Procedure for Modeling Ligand/Receptor Induced Fit Effects. J. Med. Chem. 2006, 49, 534–553. 10.1021/jm050540c. [DOI] [PubMed] [Google Scholar]
- Friesner R. A.; Banks J. L.; Murphy R. B.; Halgren T. A.; Klicic J. J.; Mainz D. T.; Repasky M. P.; Knoll E. H.; Shelley M.; Perry J. K.; Shaw D. E.; Francis P.; Shenkin P. S. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- Halgren T. A.; Murphy R. B.; Friesner R. A.; Beard H. S.; Frye L. L.; Pollard W. T.; Banks J. L. Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750. 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
- Jacobson M. P.; Kaminski G. A.; Friesner R. A.; Rapp C. S. Force field validation using protein side chain prediction. J. Phys. Chem. B 2002, 106, 11673–11680. 10.1021/jp021564n. [DOI] [Google Scholar]
- Jacobson M. P.; Friesner R. A.; Xiang Z.; Honig B. On the role of the crystal environment in determining protein side-chain conformations. J. Mol. Biol. 2002, 320, 597–608. 10.1016/s0022-2836(02)00470-9. [DOI] [PubMed] [Google Scholar]
- Kim S.; Thiessen P. A.; Bolton E. E.; Chen J.; Fu G.; Gindulyte A.; Han L.; He J.; He S.; Shoemaker B. A.; Wang J.; Yu B.; Zhang J.; Bryant S. H. PubChem Substance and Compound databases. Nucleic Acids Res. 2016, 44, D1202–D1213. 10.1093/nar/gkv951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harder E.; Damm W.; Maple J.; Wu C.; Reboul M.; Xiang J. Y.; Wang L.; Lupyan D.; Dahlgren M. K.; Knight J. L.; Kaus J. W.; Cerutti D. S.; Krilov G.; Jorgensen W. L.; Abel R.; Friesner R. A. OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J. Chem. Theory Comput. 2016, 12, 281–296. 10.1021/acs.jctc.5b00864. [DOI] [PubMed] [Google Scholar]
- Smart O. S.; Neduvelil J. G.; Wang X.; Wallace B. A.; Sansom M. S. P. HOLE: a program for the analysis of the pore dimensions of ion channel structural models. J. Mol. Graph. 1996, 14, 354–360. 10.1016/s0263-7855(97)00009-x. [DOI] [PubMed] [Google Scholar]
- Weiner S. J.; Kollman P. A.; Case D. A.; Singh U. C.; Ghio C.; Alagona G.; Profeta S.; Weiner P. A new force field for molecular mechanical simulation of nucleic acids and proteins. J. Am. Chem. Soc. 1984, 106, 765–784. 10.1021/ja00315a051. [DOI] [Google Scholar]
- Bonomi M.; Branduardi D.; Bussi G.; Camilloni C.; Provasi D.; Raiteri P.; Donadio D.; Marinelli F.; Pietrucci F.; Broglia R.; Parrinello M. PLUMED: A Portable Plugin for Free-Energy Calculations with Molecular Dynamics. Comput. Phys. Commun. 2009, 180, 1961–1972. 10.1016/j.cpc.2009.05.011. [DOI] [Google Scholar]
- Marks D. L.; Dominguez M.; Wu K.; Pagano R. E. Identification of active site residues in glucosylceramide synthase. A nucleotide-binding catalytic motif conserved with processive beta-glycosyltransferases. J. Biol. Chem. 2001, 276, 26492–26498. 10.1074/jbc.M102612200. [DOI] [PubMed] [Google Scholar]
- Belyi Y.; Aktories K. Bacterial toxin and effector glycosyltransferases. Biochim. Biophys. Acta 2010, 1800, 134–143. 10.1016/j.bbagen.2009.07.022. [DOI] [PubMed] [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. VMD-Visual Molecular Dynamics. J. Molec. Graphics 1996, 14, 33–38. 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Mestrom L.; Przypis M.; Kowalczykiewicz D.; Pollender A.; Kumpf A.; Marsden S. R.; Bento I.; Jarzębski A. B.; Szymańska K.; Chruściel A.; Tischler D.; Schoevaart R.; Hanefeld U.; Hagedoorn P. L. Leloir Glycosyltransferases in Applied Biocatalysis: A Multidisciplinary Approach. Int. J. Mol. Sci. 2019, 20, 5263. 10.3390/ijms20215263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu K.; Marks D. L.; Watanabe R.; Paul P.; Rajan N.; Pagano R. E. Histidine-193 of rat glucosylceramide synthase resides in a UDP-glucose- and inhibitor (D-threo-1-phenyl-2-decanoylamino-3-morpholinopropan-1-ol)-binding region: a biochemical and mutational study. Biochem. J. 1999, 341, 395–400. 10.1042/bj3410395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moremen K. W.; Haltiwanger R. S. Emerging structural insights into glycosyltransferase-mediated synthesis of glycans. Nat. Chem. Biol. 2019, 15, 853–864. 10.1038/s41589-019-0350-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breton C.; Fournel-Gigleux S.; Palcic M. M. Recent structures, evolution and mechanisms of glycosyltransferases. Curr. Opin. Struct. Biol. 2012, 22, 540–549. 10.1016/j.sbi.2012.06.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.