

Abstract
The N-end rule relates the in vivo half-life of a protein to the identity of its N-terminal residue. In the yeast Saccharomyces cerevisiae, the UBR1-encoded ubiquitin ligase (E3) of the N-end rule pathway mediates the targeting of substrate proteins in part through binding to their destabilizing N-terminal residues. The functions of the yeast N-end rule pathway include fidelity of chromosome segregation and the regulation of peptide import. Our previous work described the cloning of cDNA and a gene encoding the 200-kDa mouse UBR1 (E3α). Here we show that mouse UBR1, in the presence of a cognate mouse ubiquitin-conjugating (E2) enzyme, can rescue the N-end rule pathway in ubr1Δ S. cerevisiae. We also constructed UBR1−/− mouse strains that lacked the UBR1 protein. UBR1−/− mice were viable and fertile but weighed significantly less than congenic +/+ mice. The decreased mass of UBR1−/− mice stemmed at least in part from smaller amounts of the skeletal muscle and adipose tissues. The skeletal muscle of UBR1−/− mice apparently lacked the N-end rule pathway and exhibited abnormal regulation of fatty acid synthase upon starvation. By contrast, and despite the absence of the UBR1 protein, UBR1−/− fibroblasts contained the N-end rule pathway. Thus, UBR1−/− mice are mosaics in regard to the activity of this pathway, owing to differential expression of proteins that can substitute for the ubiquitin ligase UBR1 (E3α). We consider these UBR1-like proteins and discuss the functions of the mammalian N-end rule pathway.
Many biological processes are regulated by circuits that involve conditionally or constitutively short-lived proteins. Features of proteins that confer metabolic instability are called degradation signals, or degrons (16, 36, 72). The essential component of one degradation signal, termed the N-degron, is a destabilizing N-terminal residue of a protein (3, 71). A set of amino acid residues that are destabilizing in a given cell yields a rule, called the N-end rule, which relates the in vivo half-life of a protein to the identity of its N-terminal residue. Variants of the underlying proteolytic system, called the N-end rule pathway, are present in all organisms examined, from mammals and plants to fungi and prokaryotes (51, 71).
In eukaryotes, an N-degron consists of two determinants: a destabilizing N-terminal residue and an internal lysine of a substrate protein (4, 29, 66). The recognition of N-degron by the targeting machinery involves stochastic selection of second-determinant Lys residues from among the substrate's sterically suitable lysines (4, 29, 66). This Lys residue is the site of formation of a substrate-linked multiubiquitin (multi-Ub) chain (10, 48, 78). The N-end rule pathway is, thus, one pathway of the Ub system (15, 20, 24–26, 57). Ub is a 76-residue protein whose covalent conjugation to other proteins plays a role in a vast range of biological processes, including cell growth, division, differentiation, and responses to stress (24, 26, 49, 74). In most of these processes, Ub acts through routes that involve processive degradation of Ub-protein conjugates by the 26S proteasome, an ATP-dependent protease (14, 52, 75).
The N-end rule has a hierarchic structure. In the yeast Saccharomyces cerevisiae, Asn and Gln are tertiary destabilizing N-terminal residues in that they function through their deamidation, by the NTA1-encoded N-terminal amidase (Nt-amidase), to yield the secondary destabilizing N-terminal residues Asp and Glu (6, 63). The destabilizing activity of N-terminal Asp and Glu requires their conjugation, by the ATE1-encoded Arg-tRNA-protein transferase (R-transferase), to Arg, one of the primary destabilizing residues (7, 40, 71) (Fig. 1). These latter residues are bound directly by UBR1 (N-recognin), the E3 (recognition) component of the N-end rule pathway. S. cerevisiae UBR1 is a 225-kDa E3 which binds to potential N-end rule substrates through its type 1 and type 2 substrate-binding sites. The type 1 site binds the basic N-terminal residues Arg, Lys, and His. The type 2 site binds the bulky hydrophobic N-terminal residues Phe, Leu, Trp, Tyr, and Ile (34, 71) (Fig. 1). S. cerevisiae UBR1 also contains a third substrate-binding site which targets proteins such as CUP9 and GPA1 through their internal (non-N-terminal) degrons (9, 56, 68). The UBR1-encoded E3, in a complex with the RAD6-encoded E2 (Ub-conjugating) enzyme, catalyzes the synthesis of a substrate-linked multi-Ub chain (17, 71) and may also mediate the delivery of substrates to the 26S proteasome (80). UBR1 contains a functionally essential RING-H2 domain (79), a feature of many otherwise distinct E3s (28, 70, 78).
FIG. 1.

(A) The N-end rule pathway in mammals (12, 32, 33). N-terminal residues are indicated by single-letter abbreviations for amino acids. The ovals denote the rest of a protein substrate. The Asn-specific N-terminal amidase (NtN-amidase) NTAN1 converts N-terminal Asn into Asp (19, 32). N-terminal Gln is deamidated by a distinct NtQ-amidase, NTAQ1, which remains to be characterized. In mammals, the secondary destabilizing N-terminal residues Asp, Glu, and Cys are arginylated by the Arg-tRNA-protein transferases (R-transferases) encoded by ATE1 (33 and Y. T. Kwon, A. Kashina, and A. Varshavsky, unpublished data). The set of primary destabilizing N-terminal residues—Arg, Lys, His, Phe, Leu, Trp, Tyr, and Ile—is recognized in mammals by at least three distinct E3 enzymes of similar binding specificities, including the UBR1-encoded E3α and UBR2 (see Discussion). N-terminal Ala, Ser, and Thr are primary destabilizing residues in mammals but are stabilizing residues in S. cerevisiae (18, 22). An E3 that recognizes these N-terminal residues remains to be characterized. In mammals, either of the two highly similar Ub-conjugating (E2) enzymes, HR6A and HR6B (E214K), can be a component of E2-E3 complexes (Ub ligases) that mediate ubiquitylation of N-end rule substrates. The term “Ub ligase” is used to denote either an E2-E3 complex or its specific E3 component. A targeted, multi-Ub chain-bearing substrate is degraded by the 26S proteasome. (B) The N-end rule pathway in the yeast S. cerevisiae differs from its mammalian counterpart by the presence of a single Nt-amidase, NTA1, which mediates deamidation of N-terminal Asn or Gln (6) by Cys being a stabilizing residue; by the absence of E3 that recognizes N-terminal Ala, Ser, and Thr; and by the presence of a single E3, UBR1, that recognizes other primary destabilizing N-terminal residues (71).
The term Ub ligase denotes either an E2-E3 complex or its E3 component. The numerous proteolytic pathways of the Ub system have in common their dependence on Ub conjugation and the proteasome and differ largely through their utilization of distinct E2-E3 complexes. The RAD6-UBR1 (E2-E3) Ub ligase of the N-end rule pathway is one example of such a complex. Specific E3s recognize (bind to) specific degrons of their protein substrates. The diversity of E3s underlies the enormous range of substrates that are recognized and destroyed by the Ub system in ways that are regulated both temporally and spatially. There are dozens of E3s in S. cerevisiae and possibly hundreds of distinct E3s in mammals (78).
In contrast to yeast, where N-terminal Asn and Gln are deamidated by a single Nt-amidase, in mammals there are two enzymes, NtN-amidase and NtQ-amidase, which are specific for N-terminal Asn and Gln, respectively (19, 32, 64) (Fig. 1). In vertebrates, the set of secondary destabilizing residues contains not only Asp and Glu but also Cys, the latter being a stabilizing residue in the yeast N-end rule (11, 18). The two known species of mammalian R-transferase, ATE1-1 and ATE1-2, are produced through alternative splicing of ATE1 pre-mRNA (33). The substrate specificities of ATE1-1 and ATE1-2 are similar to those of the ATE1-encoded R-transferase of S. cerevisiae in that they can arginylate N-terminal Asp and Glu but cannot arginylate N-terminal Cys (33). However, recent work revealed that mouse ATE1−/− cells are incapable of arginylating any of the three secondary destabilizing N-terminal residues—Asp, Glu, and Cys (Y. T. Kwon, A. Kashina, I. Davydov, and A. Varshavsky, unpublished data).
The known functions of the N-end rule pathway include the control of peptide import in S. cerevisiae through the conditional degradation of CUP9, a transcriptional repressor of the peptide transporter PTR2 (1, 9, 68). It remains to be determined whether the N-end rule pathway has similar import-regulating functions in prokaryotes and multicellular eukaryotes. The S. cerevisiae N-end rule pathway is also essential for chromosome stability, through degradation, at the metaphase-anaphase transition, of a fragment of cohesin complexes that hold together sister chromatids (51). Given the evolutionary conservation of separase and cohesin (76), this function of the yeast N-end rule pathway may be relevant to other eukaryotes as well.
Besides CUP9 and SCC1, several other proteins were also found to be substrates of the N-end rule pathway. These proteins include Sindbis virus RNA polymerase (and homologous polymerases of other alphaviruses) (13), HIV integrase (45), a bacterial protein, p60, which is secreted by Listeria monocytogenes into the cytosol of infected mammalian cells (59), the mammalian GTPase-accelerating (GAP) proteins RGS4 and RGS16 (12), the S. cerevisiae GPA1-encoded Gα protein (43, 56), and the encephalomyocarditis (EMC) virus 3C protease (37). Physiological functions, if any, of the degradation of these proteins by the N-end rule pathway are either unknown or have not been established definitively.
In yeast only one E3, encoded by UBR1, mediates the recognition of substrates by the N-end rule pathway (8, 71). Studies of the Ub-dependent proteolysis in rabbit reticulocyte extracts suggested that the same may be true in mammalian cells, since only one E3 of the N-end rule pathway, called E3α, was apparent in these extracts (23). However, the cloning of cDNA and genes encoding mouse UBR1 (E3α) indicated the existence of at least two UBR1 homologs in the mouse (and human) genome, termed UBR2 and UBR3 (35). The sequences of UBR2 and UBR3 cDNAs and genes (Y. T. Kwon, T. Tasaki, and A. Varshavsky, unpublished data) suggested that at least mouse UBR2 may functionally overlap with UBR1. To address this and related questions, we initiated genetic and biochemical dissection of the UBR protein family in the mouse. In the present study, the first in a projected series, we constructed and analyzed mouse strains lacking UBR1.
MATERIALS AND METHODS
Strains and plasmids.
The S. cerevisiae strains used were JD52 (MATa ura3-52 his3-Δ200 leu2-3,112 trp1-Δ63 lys2-801) (30) and the ubr1Δ strain AVY107 (MATa ura3-52 his3-Δ200 leu2-3,112 trp1-Δ63 lys2-801 ubr1Δ::myc3) (H. Rao and A. Varshavsky, unpublished data). Cells were grown in rich medium (yeast-peptone-dextrose) or in synthetic glucose-containing medium (standard-dextrose [SD]) (58). The pUB23-X plasmids (X = Arg, Leu, or Met) (3) were used for expressing Ub–X–β-galactosidase (βgal) proteins from the galactose-inducible PGAL promoter. Transformation of S. cerevisiae was performed using the lithium acetate method (2).
Construction of mouse strains lacking UBR1.
BAC3, a BAC clone containing the mouse UBR1 gene (35), was the source of homology arms. The 7.0-kb ClaI-NsiI fragment of BAC3 (encompassing exons 3 and 4) and the 1,670-bp PvuII-NsiI fragment (encompassing exons 6 to 8) were used as the long and short homology arms of the targeting vector, respectively. These fragments were inserted into pPGK-SA containing a PGK/neo cassette and a PGK/TK cassette (32), yielding pUBR1-KO (Fig. 2A). The ClaI-linearized targeting vector (Fig. 2A) was electroporated into CJ7 embryonic stem (ES) cells (derived from the mouse strain 129/SvJ) (2). Selection with G418 (at 0.4 mg/ml) and 1-(2′-deoxy, 2′fluoro-β-d-arabinofuranosyl)-5-iodouracil (FIAU; at 0.4 μM) was started 24 h after electroporation. Correctly targeted ES cells were identified by PCR and Southern hybridization by using the 5′ and 3′ probes (Fig. 2A to C). Cells of the 10 independent ES cell clones were injected into 3.5-days postcoitum C57BL/6J blastocysts. The resulting male chimeras were bred with C57BL/6J females to test for germline transmission of the mutated UBR1 gene. The UBR1+/− mice resulting from this cross (5 of 10 independent ES clones were found to populate the germline in these tests) were intercrossed to produce UBR1−/− mice in the mixed 129/C57 strain background. Alternatively, the initial male chimeras were mated with 129/SvEv females, yielding, through the analogous procedures, UBR1−/− mice in the strain 129 background. For genotyping the tail-derived DNA was analyzed by PCR or digested with either SphI or BamHI and analyzed by Southern hybridization. The 0.8- and 1.1-kb PCR-produced fragments (indicated in Fig. 2A) were used as the 5′ and 3′ Southern hybridization probes, respectively.
FIG. 2.

Construction of UBR1−/− mice. (A) Top diagram, a restriction map of the ∼24-kb 5′-proximal region of the ∼120-kb mouse UBR1 gene. Middle diagram, the targeting vector. Bottom diagram, the deletion and/or disruption UBR1− allele. Exons are denoted by solid vertical rectangles. The directions of transcription of the neomycin (neo) and the thymidine kinase (tk) genes are indicated. Homologous recombination resulted in the replacement of the UBR1 exons 4 to 6 with the neo cassette. Exon 4, marked by an asterisk, contains Gly147 and Asp150, the residues that are essential, in S. cerevisiae UBR1, for binding type 1 destabilizing N-terminal residues (see Results). Exon 6, also marked by an asterisk, contains Asp233 and His236, the residues that are essential, in S. cerevisiae UBR1, for binding type 2 destabilizing N-terminal residues (see Results). Probes used for Southern hybridization are indicated by solid rectangles. Restriction sites: Sp, SphI; Cl, ClaI; BI, BamHI, Ns, NsiI, PII, PvuII. (B) PCR analysis of mouse tail DNA. The primers were 5′-GCCACTTGTGTAGCGCCAAGTGCCAG-3′ (for neo; forward), 5′-GAGATAGGAAACTGCATGCGCTGC-3′ (for UBR1; forward), and 5′-CAAGAGTGCAACAGTTACCACATG-3′ (for UBR1; reverse). DNA bands corresponding to the wild-type (wt) and mutant (mut) UBR1 alleles are indicated on the right. (C) Southern analysis of BamHI-cut (3′ probe) and SphI-cut (5′ probe) tail DNA from +/+, UBR1+/−, and UBR1−/− mice. The 3′ probe detected 13- and 5.5-kb UBR1 fragments in BamHI-cut DNA corresponding to the wild-type and mutant UBR1 alleles, respectively. The 5′ probe detected 16- and 10-kb fragments in the same SphI-cut alleles. The depicted organization of the deletion and/or disruption UBR1−/− allele was additionally verified using Southern analysis with other restriction endonucleases (data not shown). (D) Northern analysis. Total electrophoretically fractionated RNA from brain, testis, or liver of +/+ and UBR1−/− mice was probed either with the UBR1 cDNA fragment (nucleotides 555 to 888) which was deleted in the UBR1−/− allele (gel a) or with the ∼2-kb cDNA fragment (nucleotides 116 to 2124) that contained both the deleted region (nucleotides 532 to 885) and its flanking sequences (gel b) or with the human β-actin cDNA fragment (gel c). (E) Immunoblot analysis of total extracts from liver, skeletal muscle, and EFs with affinity-purified antibody (38) against the N-terminal ∼35-kDa fragment of mouse UBR1 (gels a to c) or with affinity-purified antibody specific for the 2-1 UBR1 peptide (see Materials and Methods) encoded by the deleted region of UBR1 in the UBR1− allele (gel d).
Cloning of the mouse HR6A cDNA.
We searched GenBank's expressed sequence tag (EST) database for a close mouse homolog of the mouse E214K Ub-conjugating enzyme (m HR6B, accession no. U57690). A putative amino acid sequence deduced from EST clones was 95% identical to that of mouse HR6B (E214K). The corresponding full-length cDNA was amplified by reverse transcription (RT)-PCR using total RNA isolated from skeletal muscle (33). First-strand cDNA was synthesized using Superscript II polymerase (GIBCO, Frederick, Md.), and PCR was carried out using primers specific for the 5′ end (GGCGGATCCTGAGCCCGCTAAAGCCATGTCGAC, forward primer; single and double underlinings denote the BamHI restriction site and start codon, respectively) and the 3′ end (CTGGCGCGACTGTTGACTCAGGGTCTCGAGCGG, reverse primer; single and double underlinings denote the XhoI restriction site and stop codon, respectively) of the mouse HR6A open reading frame (ORF). For the expression of mouse HR6A in S. cerevisiae, the amplified mHR6A cDNA was cloned into BamHI-XhoI-cut p413-MET25 and p415-MET25 (46), yielding p413-MET25-mHR6A and p415-MET25-mHR6A, respectively. For the expression of mouse HR6B (E214K) in S. cerevisiae, its ORF was amplified by PCR from the plasmid 44.83 encoding mHR6B (E214K) (a gift from H. P. Roest, Erasmus University, Rotterdam, The Netherlands) and was then subcloned into BamHI-XhoI-cut p413-MET25 and p415-MET25 (46), yielding p413-MET25-E214K and p415-MET25-E214K, respectively.
Subcloning and expression of mouse UBR1 cDNA in S. cerevisiae.
Because the mouse UBR1 cDNA was toxic to all of the tested Escherichia coli strains (data not shown), we subcloned it directly in S. cerevisiae. An AnfII (blunt-ended)/ClaI-produced cDNA fragment containing the 5.3-kb mouse UBR1 ORF in the MR26 plasmid (35) was ligated to SmaI/ClaI-cut p414-MET25. The ligation mixture was used to transform the ubr1Δ S. cerevisiae strain AVY107 on SD plates lacking Trp, followed by incubation at 30°C for 3 to 4 days. Selected transformants were grown in SD (lacking Trp) liquid medium, followed by isolation of the plasmid DNA and by PCR screening for the presence of full-length UBR1 ORF. The resulting plasmid, pMET414-mUBR1, expressed the 200-kDa mouse UBR1 in S. cerevisiae.
Assay of βgal activity and pulse-chase analysis in S. cerevisiae.
For the assay of βgal activity, S. cerevisiae cells in a 5-ml culture (A600 of ∼1) were pelleted by centrifugation and resuspended in 5 ml of buffer Z (10 mM KCl, 1 mM MgSO4, 50 mM β-mercaptoethanol, 60 mM Na2HPO4, 40 mM NaH2PO4 [pH 7.0]). After determining the A600 of the suspension, 50- or 100-μl samples were diluted to 1 ml with buffer Z, and 0.1% SDS (20 μl) and CHCl3 (50 μl) were then added; the suspension was vortexed for 10 to 15 s and incubated for 15 min at 30°C, followed by the addition of 0.2 ml of o-nitrophenyl-β-d-galactopyranoside (ONPG) (4 mg/ml in buffer Z) and incubation at 30°C, until a medium-yellow color had developed, at which point the reaction was stopped by the addition of 1 M Na2CO3 (0.4 ml). The mixture was centrifuged for 5 min at 1,100 × g, and A420 and A500 of the samples were measured. The ONPG units (UONPG) of βgal activity were calculated as follows: UONPG = 1,000 × [(A420) − (1.75 × A500)]/(t) × (v) × (A600), where (t) and (v) were the time of incubation (min) and the sample volume (ml), respectively.
For pulse-chase analysis, cells were grown at 30°C in SD (lacking Ura, Trp, and Leu) medium, harvested by centrifugation, washed twice with sterile water, and inoculated into SG (lacking Ura, Trp, Leu, and Met) medium to a final A600 of 0.2. When the A600 reached 1.0, cells from a 10-ml culture were harvested by centrifugation, washed with 0.8 ml SG (lacking Ura, Trp, Leu, and Met) medium, resuspended in 0.4 ml of the same medium, and labeled for 5 min at 30°C with 35S-methionine–cysteine (0.16 mCi of 35S-EXPRESS; New England Nuclear, Boston, Mass.). Cells were pelleted and resuspended in 0.4 ml of SG (lacking Ura, Trp, Leu, and Met) medium containing 10 mM l-methionine and 5 mM l-cysteine. Samples (0.1 ml) were taken at the indicated time points, followed by preparation of extracts and immunoprecipitation (34) by using a monoclonal anti-βgal antibody (Promega, Madison, Wis.).
Overexpression, labeling, and purification of Ub-X-eK-DHFR-His6 proteins.
A pT7-UbXeKDHFRhis plasmid (X = Met, Arg, or Phe) (11) was transformed into E. coli BL21(DE3) (2). Following the addition of isopropyl-1-thio-β-d-galactopyranoside (IPTG), the cells were labeled with 35S-methionine–cysteine (35S-EXPRESS; New England Nuclear) as described previously (32). The cells were collected by centrifugation and disrupted by sonication, and 35S-labeled Ub-eK-DHFR-His6 (Ub-X-DHFR) test proteins were purified by affinity chromatography under nondenaturing conditions by using the Ni-NTA Spin Kit (Qiagen, Chatsworth, Calif.). The eluted proteins were dialyzed against a solution containing 1 mM MgCl2, 1 mM dithiothreitol (DTT), 0.1 M Tris-HCl (pH 7.7) and were rapidly frozen and stored at −80°C in samples that were to be thawed just once. The specific radioactivity of [35S]Ub-X-DHFR proteins was 5 × 105 to 10 × 105 cpm/μg.
Extracts of skeletal muscle and embryonic fibroblast (EF) cells.
To prepare muscle extracts, leg muscles from three to six +/+ male mice or their littermate (also male) UBR1−/− counterparts (strain 129 background) were combined and homogenized in 20 mM Tris (pH 7.6) containing 1 mM β-mercaptoethanol, 1% glycerol, 1 mM EDTA, 1 mM EGTA, 50 μM chymostatin, and 50 μM E64 (Sigma, St. Louis, Mo.) using a rotor-stator homogenizer (Biospec Products, Bartlesville, Okla.).
Homogenates were centrifuged at 30,000 × g for 30 min to remove myofibrils, followed by centrifugation of the supernatants at 100,000 × g for 1 h. The resulting extracts were either assayed directly or fractionated on DEAE-cellulose (23, 61) to produce fraction II (which contained the proteasome and most of the Ub-system enzymes) and the flowthrough fraction I (which contained Ub and ∼70% of proteins in the initial extract). Both whole-cell extracts and fraction II were dialyzed against a solution containing 20% glycerol, 1 mM DTT, 5 mM MgCl2, 50 mM Tris (pH 7.6) and were stored at −80°C in samples that were to be thawed and used only once. The same procedures were employed to prepare extracts from +/+ and UBR1−/− EF cells, except that a Dounce homogenizer was used to disrupt the cells.
In vitro proteolysis and Ub conjugation assays.
Degradation assays were carried out in samples of 0.1 ml containing the following components: 20 mM Tris-HCl (pH 7.6), 5 mM MgCl2, 2 mM DTT, ATP-regenerating system (10 μg of creatine phosphokinase and 10 mM creatine phosphate), 1 mM ATP, 25 μg of purified Ub, 50 μM bestatin, a dipeptide at 2 mM (when present), ∼0.5 mg of dialyzed whole-cell extract, and a 35S-labeled X-DHFR (X-eK-DHFR-His6) test substrate, produced from Ub-X-eK-DHFR-His6 through the cleavage by deubiquitylating enzymes (DUBs) in the extract. The following dipeptides were used: Lys-Ala, Ala-Lys, Phe-Ala, and Ala-Phe (Sigma, St. Louis, Mo.). Dipeptides were stored at −20°C at 0.5 M in 10 mM K-HEPES, pH 7.5. Bestatin (Sigma) was added to decrease degradation of dipeptides in the extract (12, 53). Control experiments (not shown) showed that the addition of bestatin alone did not significantly inhibit the degradation of test proteins by the N-end rule pathway. In some experiments the reaction mixtures were incubated for 10 min without ATP, followed by the addition of ATP and an ATP-regenerating system. Incubations were carried out at 37°C. Samples of 10 μl were withdrawn in the course of incubation, and the degradation of [35S]Ub-X-DHFR proteins was assessed by measuring 5% trichloroacetic acid (TCA)-soluble 35S as follows:
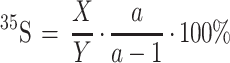 |
where X was the amount of TCA-soluble 35S (cpm); Y was the total amount of 35S (cpm) in the same sample; and a was the number of Met residues in a Ub-X-DHFR test protein (a = 10 for Ub-Met-DHFR; a = 9 for the other Ub-X-DHFRs). The a-1 term corrected for the presence of one Met residue in Ub. To determine TCA-soluble 35S, a 10-μl sample was added to a tube containing 90 μl of ice-cold 5.6% TCA. The tube was briefly vortexed, incubated on ice for 10 min, and centrifuged in a microcentrifuge for 10 min at 4°C. 35S in a fraction of supernatant was then determined using scintillation counter.
To assay Ub conjugation to a test protein, fraction II (35 μg of protein, 20 μl total volume) was incubated with 125I-labeled α-lactalbumin (∼150,000 cpm, ∼1 μM) and unlabeled Ub (50 μM) in buffer A (20 mM Tris-HCl [pH 7.6], 20 mM KCl, 5 mM MgCl2, 2 mM AMP-PNP, 1 mM DTT, 30 μM MG132 [Sigma], and 10% glycerol). Samples (20 μl) were incubated at 37°C for 1 h. The reactions were terminated by the addition of 5× Laemmli sample buffer (6 μl) (2) followed by SDS–13% polyacrylamide gel electrophoresis (PAGE) and quantitation by using PhosphorImager (Molecular Dynamics, Sunnyvale, Calif.). To assay Ub conjugation to endogenous muscle proteins, fraction II (35 μg of protein) was incubated with 125I-Ub (∼150,000 cpm, 5 to 10 μM) in buffer A for 1 h at 37°C, followed by SDS-PAGE analysis as described above. Purified E214K(C88S), a dominant-negative inhibitor of UBR1 (E3α) (65), or Lys-Ala, a type 1 dipeptide inhibitor of UBR1 (18), was added to some of the assays. Human α-lactalbumin was radioiodinated by using the chloramine T method (61).
Antibodies to mouse UBR1 and immunoblotting
Rabbit polyclonal antibodies to mouse UBR1 were raised against the synthetic peptides EMDPDLEKQEESVQ (UBR1 residues 54 to 67; antibody UBR1 [1-1]), HEPGRAGTTKESLH (UBR1 residues 66 to 179; antibody UBR1 [2-1]), and EYLDRNNKFNFQGYSQDK (UBR1 residues 451 to 468; antibody UBR1 [3-1]). The antibodies were affinity-purified using immobilized peptides. The sequence of the second peptide (antibody UBR1 [2-1]) was in the region of UBR1 that was absent from the UBR1− allele.
For immunoblotting, mouse tissues were homogenized in 50 mM Tris-HCl (pH 7.2) containing 0.5% SDS, 5 mM EDTA, 0.25 mM phenylmethylsulfonyl fluoride (PMSF; freshly prepared as a 125 mM solution in isopropanol), and protease inhibitor tablets containing inhibitors of several proteases (Boehringer Mannheim, Indianapolis, Ind.). The samples were centrifuged at 12,000 × g for 1 h, and proteins in the supernatants (50 to 70 μg per lane) were subjected to SDS–7.5% PAGE, followed by electrophoretic transfer to Immobilon-P membranes (Millipore, Bedford, Mass.). The membranes were blocked with TBS-T buffer (20 mM Tris-HCl [pH 7.6], 137 mM NaCl, 0.1% Tween 20) containing 5% nonfat dry milk (Carnation) and thereafter incubated with one of the above antibodies to mouse UBR1 (diluted 1:1,000), washed with TBS-T buffer, incubated with horseradish peroxidase (HRP)-coupled goat anti-rabbit immunoglobulin G (Bio-Rad, Hercules, Calif.), and developed using a ECL kit (Amersham, Piscataway, N.J.) (21). Another antibody to mouse UBR1 employed in the present work was previously raised against the 35-kDa N-terminal fragment of UBR1 (38). This affinity-purified polyclonal rabbit antibody was used at a 1:1,500 dilution.
Immortalization of embryonic fibroblasts, transfection, and pulse-chase analysis.
Primary EFs were isolated from 13.5-day old (E13.5) +/+ and littermate UBR1−/− embryos of the mixed (129/C57) genetic background as described previously (32, 54). EFs were grown in Dulbecco's modified Eagle medium-F12 medium (GIBCO) supplemented with 15% fetal bovine serum, antibiotics, and 2 mM l-glutamine. Permanent cell lines were established from primary EFs through crisis-mediated immortalization over 2 months by replating, every 3 days, ∼1.5 × 106 cells onto a 10-cm-diameter plate. DNA transfection efficiency was considerably higher with immortalized EFs than with primary EFs (data not shown).
Immortalized +/+ and UBR1−/− EF cell lines were transiently transfected, using Lipofectamine (GIBCO), with pRC/dhaUbXnsP4βgal (X = Met, Arg, or Phe) expressing the fusion proteins DHFRh-UbR48-X-nsP4βgal (superscript “h” denotes the ha epitope [2]) from the PCMV promoter. These and analogous Ub/protein/reference (UPR)-based fusions (39, 66) are cotranslationally cleaved in vivo at the Ub-protein junction, yielding, in the present case, the reference protein DHFRh-UbR48 (DHFR-Ub) and a test protein X-nsP4βgal (see Results). Similarly, the transfected plasmids pcDNA3-flag-DHFR-ha-Ub-X-nsP4-flag (X = Met, Arg, or Tyr) expressed fDHFRh-UbR48 (DHFR-Ub) and X-nsP4f (X-nsP4) (full-length nsP4 of the Sindbis virus bearing N-terminal Met, Arg, or Tyr) (superscript f denotes the flag epitope) (2). The latter plasmids were produced by subcloning PCR-produced SmaI-XbaI fragments encoding X-nsP4-flag into EheI-XbaI-cut UPR vector pcDNA3(dEheI)FDHUMCM (J. Sheng and A. Varshavsky, unpublished data) encoding fDHFRh-UbR48. About 24 h after transfection cells were labeled with 35S-methionine–cysteine (35S-EXPRESS; New England Nuclear), followed by chases for 0, 1, and 2 h in the presence of cycloheximide, preparation of extracts, immunoprecipitation, SDS–10% PAGE, autoradiography, and quantitation using a PhosphorImager, essentially as described previously (32, 39, 66). Cells expressing X–β-gal(X-nsP4βgal) tests were labeled for 1 h without a chase and then processed as above.
Blood plasma measurements.
UBR1−/− mice (in the inbred strain 129 background) and their congenic +/+ littermates (produced through matings of UBR1+/− mice) were used. The experiments were performed twice (experiments 1 and 2), with 28 pairs of animals total. Blood from 12 pairs of mice, with water and chow diet ad libitum, was collected before fasting. Blood from 8 pairs of mice was collected after 24 h of food deprivation (from 10 a.m. to 10 a.m., with free access to water). Blood from 8 pairs of mice was collected after 24 h of fasting followed by 24 h of refeeding, with chow diet ad libitum. Body weights were determined before and after fasting and refeeding. Blood was withdrawn by cardiac puncture (∼0.6 ml per animal) and transferred into heparin-coated tubes. The plasma fraction was prepared by centrifugation immediately thereafter, quickly frozen, and stored at −70°C before the measurements. The levels of glucose, triglycerides, and cholesterol, together with sodium, potassium, chloride, calcium, phosphorus, blood urea nitrogen, creatine, total protein, albumin, total bilirubin, aspartate aminotransferase (AST), alanine aminotransferase (ALT), alkaline phosphatase, and γ-glutamyltransferase (GGT) were determined by the Biomedical Testing Services (San Diego, Calif.) by using a Vitros 950 chemistry analyzer (Johnson and Johnson, Rochester, N.Y.). Two independent experiments, 1 and 2, yielded average values that differed by, at most, 1% of the measured values for sodium, potassium, and chloride, 6.4% for calcium and phosphorus, 3.2% for glucose, 5.3% for triglycerides and cholesterol, and 13% for albumin, urea nitrogen, total protein, GGT, AST, and ALT. The level of glucose was independently determined by the hexokinase/G6PD method, using a commercial kit (Sigma).
Double-mutant NTAN1−/− UBR1−/− mouse strains.
NTAN1−/− mice (32) of the 129Sv background (129SvJ/129SvEv) and UBR1−/− mice of the mixed (129SvJ and C57BL/6) background were mated to produce F1 progeny heterozygous for both genes. Siblings were then intercrossed to produce, in particular, the NTAN1−/− UBR1−/− progeny, as determined by using PCR and tail-derived DNA, with primers specific for NTAN1 (32) and UBR1 (Fig. 2A and B).
Other methods.
For Northern hybridization, total RNA was isolated from the brain, testis, skeletal muscle, and EF cells of +/+ and UBR1−/− mice as described previously (32). RNA was fractionated by electrophoresis in formaldehyde–1% agarose gels, blotted onto Hybond N+ membranes (Amersham), and hybridized with 32P-labeled cDNA probes (2). The fasting and refeeding protocols for Northern analysis were identical to those used for plasma chemistry measurements, except that fasting and refeeding were carried out for 48 and 24 h, respectively. For histological examination tissues and organs were fixed in Bouin's fixative or 10% buffered formalin, paraffin embedded, sectioned, and either stained with hematoxylin and eosin or assayed for apoptosis by using the terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling (TUNEL) technique and an in situ cell death detection kit (Boehringer Mannheim), with fluorescein-dUTP. For behavioral tests, the strain 129 UBR1−/− mice and their congenic +/+ littermates (produced through matings of UBR1+/− mice) were used. The rotarod, weight retention, and coat-hanger tests were performed as described previously (32).
Nucleotide sequence accession number.
The nucleotide sequence of mouse HR6A cDNA was submitted to GenBank with accession no. AF383148.
RESULTS
Mouse UBR1, in the presence of cognate mouse E2, rescues the N-end rule pathway in ubr1Δ S. cerevisiae
The amino acid sequence encoded by the mouse UBR1 ORF contained all 14 sequences of peptide-size UBR1 fragments isolated from two independently produced preparations of rabbit E3α (35), indicating that mouse UBR1 was, in fact, E3α, the N-recognin of the N-end rule pathway. To verify directly whether mouse UBR1 could function as N-recognin in vivo, we asked whether its expression in ubr1Δ S. cerevisiae, which lacked the N-end rule pathway, could rescue the pathway in these cells. Since the full-length mouse UBR1 cDNA was found to be toxic to E. coli, we constructed a mouse UBR1-expessing plasmid directly in S. cerevisiae (see Materials and Methods). ubr1Δ S. cerevisiae, which lacked S. cerevisiae UBR1 but retained RAD6, the UBR1-interacting E2 enzyme of the N-end rule pathway (71), were cotransformed with a pair of plasmids that expressed mouse UBR1 from the PMET25 promoter and one of three test proteins, expressed as Ub-X-βgal fusions (X = Arg, Leu, or Met). In eukaryotes, Ub fusions are cleaved cotranslationally by DUBs after the last residue of the Ub moiety (69, 73). With Ub-X-βgals, this cleavage yielded X-βgal proteins bearing either Arg, Leu, or Met at their N termini. Arg and Leu are, respectively, type 1 and type 2 primary destabilizing residues in the N-end rule. Met is a stabilizing residue (71).
Previous work showed that the steady-state level of an X-βgal protein is a sensitive measure of its metabolic stability (17, 33, 42). The relative levels of X-βgals in S. cerevisiae were determined by measuring the enzymatic activity of βgal in yeast extracts. We found that the activity of Arg-βgal and Leu–β-gal in extracts from ubr1Δ cells expressing mouse UBR1 was reproducibly ∼20% lower than that in control ubr1Δ extracts, whereas the activity of Met-βgal was not affected by the presence of mouse UBR1 (Fig. 3A and data not shown). In addition, a ∼90-kDa, long-lived βgal cleavage product that is specific for short-lived X-βgal test proteins (3) was observed in pulse-chase assays with S. cerevisiae that coexpressed Arg-βgal and mouse UBR1 but not with Arg-βgal in the absence of mouse UBR1 (data not shown). The detectable but low N-recognin activity of mouse UBR1 in S. cerevisiae may be caused by a poor fit between the yeast RAD6-encoded E2 enzyme and the mouse counterpart of yeast UBR1. Therefore we asked whether coexpression of mouse HR6B (E214K), a homolog of S. cerevisiae RAD6, could increase the activity of mouse UBR1 in yeast without decreasing its specificity. Indeed, coexpression of mHR6B and mouse UBR1 increased the activity of the N-end rule pathway in ubr1Δ S. cerevisiae (Fig. 3A). Koken et al. (31) described a human E2 enzyme, termed HR6A, whose deduced sequence was 95% identical to that of human HR6B (E214K). We used RT-PCR to isolate a cDNA encoding the mouse counterpart (mHR6A) of human HR6A. The deduced sequence of mHR6A was 95% identical to that of the mouse HR6B (E214K) enzyme and 99% identical to the sequence of human HR6A (data not shown). The mouse HR6A E2 enzyme was found to be indistinguishable from mHR6B (E214K) in its ability to cooperate with mouse UBR1 in mediating the activity of the N-end rule pathway in ubr1Δ S. cerevisiae (Fig. 3A). No additional enhancement of the N-end rule pathway's activity was observed upon coexpression of mouse UBR1, mHR6A, and mHR6B (Fig. 3A). Thus, both mHR6A and mHR6B (E214K) are likely to be the cognate E2 components of the UBR1-containing Ub ligase.
FIG. 3.

Mouse UBR1, in the presence of mouse E214K or HR6A, can rescue the N-end pathway in ubr1Δ S. cerevisiae. (A) Relative enzymatic activities of βgal in ubr1Δ S. cerevisiae expressing different combinations of the following components. (i) Type 1 (Arg-βgal) or type 2 (Leu-βgal) N-end rule substrates; (ii) mouse UBR1 or the p414-MET25 vector alone (designated 414); and (iii) mouse E214K, mouse HR6A (homolog of E214K), both of them together, or vector(s) alone (p415-MET25, designated 415, and p413-MET25, designated 413). The activities of X-βgal test proteins in wild-type (UBR1) S. cerevisiae are shown in the rightmost column. 100%, the activity of X-βgal in ubr1Δ cells transformed with the p414-MET25 vector alone. (B) Same as for panel A but with type 3 N-end rule substrates, Ala-βgal (shaded bars), Ser-βgal (hatched bars), or Thr–β-gal (black bars). (C) Pulse-chase analysis of Arg-βgal (produced from Ub-Arg-βgal) in ubr1Δ S. cerevisiae coexpressing either mouse UBR1 and the p415-MET25 vector (denoted 415), mouse HR6A E2 enzyme and the p414-MET25 vector (denoted 414), mouse UBR1 and HR6A, or vectors alone. Time zero refers to the end of 5-min pulse. The asterisk indicates the ∼90-kDa, long-lived βgal cleavage product specific for short-lived X-βgal test proteins (3) in the pulse-chase with cells coexpressing mouse UBR1 and mouse HR6A. (Much smaller amounts of the 90-kDa species could also be detected in the pulse-chase with cells expressing mouse UBR1 alone.) (D) Quantitation of Arg-βgal degradation depicted in panel C (the decay curves shown are averages from two independent experiments). 100%, the initial amount of Arg-βgal in cells transformed with vectors alone (p414-MET25 and p415-MET25); □, vectors alone; ▵, UBR1 and p415-MET25 (vector); ○, HR6A and p414-MET25 (vector); ●, UBR1 and HR6A.
The above results, based on the measurements of X-βgal concentrations (Fig. 3A), were in agreement with direct (pulse-chase) assays under the same conditions: significant degradation of Arg-βgal in ubr1Δ S. cerevisiae was observed only upon coexpression of mouse UBR1 and a cognate mouse E2 enzyme (Fig. 3C and D). The N-end rule pathway's activity conferred on ubr1Δ S. cerevisiae through a combination of mouse UBR1 and a cognate E2 enzyme (mHR6A or mHR6B) was significant but much lower than the activity of this pathway in wild-type (UBR1) S. cerevisiae (Fig. 3A). This is not surprising, given the evolutionary distance between fungi and mammals and the likely presence of substrate targeting steps in the yeast N-end rule pathway that can be compromised through interactions with a mammalian version of Ub ligase.
In contrast to yeast, where primary destabilizing residues are either of type 1 or of type 2 (see the introduction), there are also type 3 N-end rule substrates in mammals. The substrates of this latter class bear N-terminal Ala, Ser, or Thr (18) and are recognized by a distinct E3, termed E3β (22). The molecular identity of E3β is unknown. We asked whether mouse UBR1, in the presence of either mHR6A or mHR6B (E214K), could mediate degradation of the type 3 N-end rule substrates Ala-βgal, Ser-βgal, or Thr-βgal in ubr1Δ S. cerevisiae. (These proteins are long-lived in either wild-type or ubr1Δ yeast [71].) The results (Fig. 3B) indicated that mouse UBR1 lacked the activity of E3β Ub ligase.
Construction of UBR1−/− mice.
In the deletion-2 disruption allele of mouse UBR1, exons 4 to 6 were replaced by a neo cassette (Fig. 2A). Exons 4 to 6 encompassed a region of high sequence conservation between the S. cerevisiae and mouse UBR1 proteins (35). Moreover, these exons encompassed amino acid positions that were previously found to be essential for the integrity of the type 1 substrate-binding site (Gly147 and Asp150) and the type 2 site (Asp233 and His236) in S. cerevisiae UBR1. Specifically, mutations of these residues greatly impaired the activity of yeast UBR1 in mediating degradation of either type 1 or type 2 N-end rule substrates (A. Webster, M. Ghislain, and A. Varshavsky, unpublished data). Of the ∼1,000 ES cell clones resistant to both G418 and FIAU, 33 clones contained the expected deletion or disruption (Fig. 2A), as verified by PCR and Southern analyses (data not shown). Ten of these correctly targeted ES cell clones were used to generate male chimeras, and in five of them the UBR1− allele was transmitted through the germ line. Male chimeras were mated with either 129/SvEv or C57BL/6 females, yielding UBR1+/− heterozygotes. Intercrosses of UBR1+/− mice produced UBR1−/− progeny (Fig. 4B) at the expected Mendelian frequency of ∼25%. In addition, examination of embryos from heterozygous matings did not suggest a significantly increased embryonic lethality of UBR1−/− mice (data not shown). This, and the evidence below that UBR1−/− mice lacked the UBR1 protein, indicated that UBR1 was not required for either mouse embryonic development or postnatal viability.
FIG. 4.

Growth retardation and altered fat metabolism in UBR1−/− mice. (A) Growth retardation in UBR1−/− mice of the 129/C57 strain background. Body masses of the offspring (total of 141 mice) from UBR1+/− heterozygous matings were determined from 1 day after birth until adulthood. To keep track of newborn pups before they could be distinguished using ear punching (at 3 weeks of age), the pups were marked by applying spots of paint every day. At week 3 the genotypes of pups were determined by using PCR with tail-derived DNA. (A, graph a) Male masses as a function of age: 18 +/+ (□), 34 UBR1+/− (▴), and 20 UBR1−/− mice (●) were used. (A, graph b) Same as for panel A, graph a, but with females: 18 +/+ (□), 35 UBR1+/− (▴), and 16 UBR1−/− mice (●) were used. (A, graph c) The mass ratios of UBR1−/− males to +/+ males (▪) and of UBR1−/− females to +/+ females (○) as a function of age. The arrows in graphs a to c denote the time of weaning (day 20). (B) A pair of 6-week-old +/+ and UBR1−/− male littermates. The mass of the UBR1−/− mouse shown here was 15% lower than that of its +/+ littermate. (C) Relative masses of organs and tissues of UBR1−/− mice, expressed as percentages of the weights of age-matched +/+ counterparts. Standard deviations are indicated. For determining the masses of hind legs and hind leg fat pads, 94 pairs of 2- to 4-month-old mice (69 male pairs and 25 female pairs) were used (63 +/+, 31 UBR1+/−, and 94 UBR1−/− mice, produced through matings of UBR1+/− mice). For other measurements in this panel, 24 pairs of 2- to 4-month-old mice (17 +/+, 7 UBR1+/−, and 24 UBR1−/− mice) were used. (D) Altered regulation of the FAS mRNA in skeletal muscle of UBR1−/− mice. Lanes a to f, Northern analysis of FAS mRNA from skeletal muscle of +/+ and UBR1−/− mice that were either fed ad libitum, fasted for 48 h, or refed for 24 h after the fast. Lanes g and h, FAS mRNA from growing EF cells (+/+ and UBR1−/−) in culture. Lanes i and j, the same as lanes c and d but with a longer autoradiographic exposure. A 32P-labeled 1.1-kb FAS cDNA fragment (nucleotides 535 to 1642; accession no. AAG02285) was used as a probe.
The relevant genotype of UBR1−/− mice was verified by using both PCR and Southern analysis (Fig. 2B and C). Northern analysis with a UBR1 cDNA-derived probe containing exclusively the region deleted in the UBR1− allele did not detect UBR1-specific transcripts in the brain, testis, and liver of UBR1−/− mice (Fig. 2D, gel a). By contrast, a UBR1 probe that encompassed both the deleted region and the 3′-flanking (undeleted) region of UBR1 cDNA detected UBR1-specific transcripts in both +/+ and UBR1−/− tissues (Fig. 2D, gel b). The latter transcripts were smaller than +/+ ones (Fig. 2D, gel b) and were presumably the aberrantly spliced RNAs transcribed from the PUBR1 promoter. To determine whether a version of UBR1 protein was present in UBR1−/− tissues, we carried out immunoblot analyses with different antibodies to mouse UBR1 (see Materials and Methods). With SDS-PAGE-fractionated extracts from the liver, skeletal muscle, and EF cells, these antibodies detected the band of expected (∼200 kDa) size in +/+ tissues but not in their UBR1−/− counterparts (Fig. 2E and Fig. 5B and data not shown), indicating that UBR1−/− mice (Fig. 4B) lacked the UBR1 protein.
FIG. 5.

Expression analysis of mouse UBR1 and functionally related genes in +/+ and UBR1−/− mice. (A) Northern analysis of skeletal muscle mRNAs encoding UBR1, other components of the N-end rule pathway, and the UBR1 homologs UBR2 and UBR3. RNA was isolated from skeletal muscle of control, 48-h fasted, or 24-h refed +/+ and UBR1−/− mice. See Materials and Methods and the legend to Fig. 4D for the fasting and refeeding protocols. Northern blots were hybridized with 32P-labeled cDNA probes specific for the following genes: UBR1 (nucleotides 555 to 888, accession no. AF061555; this probe was specific for the deleted region in the UBR1− allele); UBR2 (a 0.3-kb probe encompassing the region homologous to the one deleted in the UBR1−/− allele; Y. T. Kwon and A. Varshavsky, unpublished data); UBR3 (a 0.4-kb probe; Y. T. Kwon and A. Varshavsky, unpublished data); ATE1 (nucleotides 638 to 1734, accession no. AF079098); NTAN1 (nucleotides 34 to 900, accession no. U57692); mHR6B (E214K) (nucleotides 115 to 569, accession no. U57690); and β-actin. (B) The level of UBR1 protein in skeletal muscle is not significantly altered upon fasting. Total extracts (∼70 μg per lane) from skeletal muscles of normally fed or 48-h fasted +/+ and UBR1−/− mice were analyzed by immunoblotting, using affinity-purified antibody (38) against the N-terminal ∼35-kDa fragment of mouse UBR1. (C) Northern analysis of ATE1, NTAN1, and mHR6B (E214K) expression in the brain, liver, and testis of +/+ and UBR1−/− mice by using DNA probes described in the legend to panel A.
Expression of mRNAs encoding components of the N-end rule pathway and related proteins in UBR1−/− and +/+ mice.
Northern analysis was used to assess the levels of mouse mRNAs encoding NTAN1, ATE1, mHR6B, UBR1 (E3α), and its homologs UBR2 and UBR3 (Fig. 5A). After a 48-h fast, the level of UBR1 mRNA in the +/+ muscle increased by ∼2-fold, returning to approximately basal levels 24 h after refeeding (Fig. 5A). The levels of UBR2 and UBR3 mRNAs also increased upon fasting. Expression of UBR2 mRNA returned to the basal (or slightly higher) level 24 h after refeeding, but the level of UBR3 mRNA remained high (Fig. 5A). The level of UBR2 mRNA was reproducibly higher in the UBR1−/− muscle under both normal and fasting conditions (Fig. 5A), suggesting compensatory overexpression of UBR2 in UBR1−/− mice. The level of mRNA encoding mHR6B (E214K), one of UBR1-interacting E2s (see above), changed in parallel with that of UBR1. As to the N-end rule pathway's components upstream of Ub ligase, the levels of ATE1 mRNA also increased upon fasting, in contrast to that of NTAN1 mRNA. A Northern survey of other tissues showed that the levels of NTAN1, ATE1, and mHR6B mRNAs in the UBR1−/− brain, testis, and liver did not change significantly, except that the testis-specific NTAN1 transcript (1.1 kb) and ATE1 transcript (2 kb) were decreased in UBR1−/− mice (Fig. 5C).
We also asked whether an increased level of UBR1 mRNA during fasting of +/+ mice (Fig. 5A) was accompanied by an increased level of the UBR1 protein. We observed no increase (Fig. 5B), in agreement with data of Lecker et al. (38), who assessed the levels of UBR1 mRNA and protein in the muscle of normal versus diabetic rats. One possibility is that the UBR1 expression circuit is designed to maintain a constant level of the UBR1 protein. Thus, the observed induction of UBR1 mRNA upon fasting or diabetes may be caused by enhanced degradation of UBR1 under these conditions.
Extracts of UBR1−/− skeletal muscle lack the N-end rule pathway, in contrast to +/+ extracts.
Previous studies with ATP-supplemented extracts from the skeletal muscle of rats and rabbits demonstrated the presence of the N-end rule pathway in the muscle and also showed that this pathway was further induced during muscle atrophy caused by tumors, sepsis, or diabetes (38, 60, 62). To assess the activity of the N-end rule pathway in skeletal muscles of +/+ and UBR1−/− mice, we measured the ATP-dependent degradation of model N-end rule substrates in muscle extracts (Fig. 6). The substrates used were 35S-labeled, purified Ub-X-DHFR proteins (X = Met, Arg, or Phe) (see Materials and Methods). Ub-X-DHFRs were rapidly deubiquitylated in a muscle extract by ATP-independent DUBs, yielding X-DHFR test proteins, similarly to the results with extracts from other eukaryotic cells (12, 18). Arg-DHFR and Phe-DHFR are, respectively, a type 1 and type 2 N-end rule substrates (see the introduction), whereas Met-DHFR is not a substrate of the N-end rule pathway. Degradation of X-DHFRs in ATP-supplemented +/+ and UBR1−/− extracts was measured by determining 5% TCA-soluble 35S. Although the N-end rule-specific degradation of Arg-DHFR and Phe-DHFR was only ∼2-fold above the background of nonspecific degradation (i.e., the degradation of Met-DHFR), a much lower ratio than the one observed with rabbit reticulocyte extracts and similar substrates (18, 23, 29), the specific degradation was reproducible in independent experiments (data not shown). Moreover, the N-end rule specificity of degradation could be independently verified by adding to an extract dipeptides bearing either type 1 or type 2 destabilizing N-terminal residues. These dipeptides act as specific inhibitors of either type 1 or type 2 substrate-binding sites of UBR1 (12, 18, 53).
FIG. 6.

Extracts from skeletal muscle of UBR1−/− mice lack the N-end rule pathway. Degradation of 35S-labeled, purified Ub-X-DHFR test proteins in extracts from skeletal muscle of +/+ and UBR1−/− littermates (strain 129 background) was monitored by measuring TCA-soluble 35S (see Materials and Methods). Open and closed symbols denote, respectively, the data with +/+ and UBR1−/− extracts. (A) Degradation of Phe-DHFR, a type 2 N-end rule substrate. □ and ▪, no dipeptide inhibitor; ○ and ●, Lys-Ala; ▵ and ▴, Phe-Ala; ▿ and ▾, Ala-Lys (control dipeptide). (B) Degradation of Arg-DHFR, a type 1 substrate. □ and ▪, no dipeptide inhibitor; ○ and ●, Lys-Ala; ▵ and ▴, Phe-Ala; ▿ and ▾, Ala-Lys. (C and D) The N-end rule-specific degradation of Phe-DHFR is ATP-dependent. Reaction mixtures were incubated at 37°C for 10 min without ATP followed by assays either in the absence or the presence of added ATP. (Time zero corresponds to the end of 10-min preincubation in the absence of ATP to allow deubiquitylation of Ub-X-DHFRs.) (C) Degradation of Phe-DHFR in the presence (□ and ▪) or absence (○ and ●) of added ATP, and of Met-DHFR (not an N-end substrate) in the presence (▵ and ▴) or absence (⋄ and ♦) of ATP. (D) Effect of Lys-Ala, a type 1 dipeptide inhibitor, on the ATP-dependent degradation of Phe-DHFR, a type 2 N-end rule substrate. □ and ▪, no inhibitor; ○ and ●, Lys-Ala; ▵ and ▴, Phe-Ala; ⋄ and ♦, Ala-Lys; ▿ and ▾, Ala-Phe. (E) Conjugation of unlabeled Ub to 125I-α-lactalbumin (a type 1 N-end rule substrate) is decreased in the extract from UBR1−/− muscle. Some of the samples contained either the dominant-negative E214K(C88S) mutant protein (at 2 μM), 2 mM Lys-Ala, or 2 mM Ala-Lys. Double arrowheads on the left indicate the excess of unconjugated 125I-α-lactalbumin. Lane 1, 125I-α-lactalbumin alone. (F) Conjugation of 125I-Ub to endogenous proteins is marginally decreased in a UBR1−/− muscle extract compared to that in the wild-type extract. Fraction II (see Materials and Methods) of muscle extracts from +/+ and UBR1−/− mice was supplemented with AMP-PNP, and the formation of 125I-Ub-protein con- jugates was assayed by SDS-PAGE in the ab- sence (lanes 2 and 3) or presence (lanes 4 and 5) of 2 μM E214K(C88S). Arrowhead on the right indicates the position of 125I-Ub-E214K(C88S) conjugate. Lane 1, 125I-Ub alone. The results shown in panels A through D are typical of those obtained in at least five independent experiments, using three independently produced muscle extract preparations, and two independent EF cell extracts (see Fig. 7), in combination with two independent preparations of [35S]Ub-X-DHFR.
The degradation of Phe-DHFR (a type 2 substrate) in extract from +/+ muscle was reproducibly higher than in the extract from UBR1−/− muscle (Fig. 6A). Crucially, the extent of degradation of Phe-DHFR in UBR1−/− extract was essentially indistinguishable from the level of background degradation observed with Met-DHFR (not an N-end rule substrate) in either +/+ or UBR1−/− extracts (Fig. 6A and data not shown). Moreover, the addition of Phe-Ala dipeptide to +/+ extract decreased the degradation of Phe-DHFR to background levels, whereas the same peptide had no effect on the (already background-level) degradation of Phe-DHFR in UBR1−/− extract (Fig. 6A). Ala-Phe, a dipeptide of the same composition but bearing a type 3 destabilizing N-terminal residue, had no effect in either +/+ or UBR1−/− extracts (Fig. 6D). Finally, the addition of Lys-Ala dipeptide, bearing a type 1 destabilizing N-terminal residue, to +/+ extract enhanced the degradation of Phe-DHFR (Fig. 6A), whereas the same dipeptide inhibited the degradation of Arg-DHFR (Fig. 6B). These findings (Fig. 6A, B, and D) reproduced, in a muscle extract, the previously observed phenomenon of type 1 dipeptides enhancing the degradation of type 2 N-end rule substrates either in vivo (5) or in reticulocyte extract (18). Note that the addition of type 1 dipeptide to UBR1−/− extract had no effect on the (background) levels of degradation of either Arg-DHFR or Phe-DHFR in this extract, in contrast to the effect of type 1 and type 2 dipeptides on the same substrates in +/+ extract (Fig. 6A to D and data not shown).
We also measured, in +/+ and UBR1−/− extracts, the conjugation of Ub to 125I-labeled human α-lactalbumin, a type 1 N-end rule substrate bearing N-terminal Lys (23, 38). For these experiments, +/+ and UBR1−/− muscle extracts were fractionated by DEAE-cellulose chromatography to yield fraction II preparations, which contained most of the Ub system, including components of the N-end rule pathway, but lacked free Ub (23, 61). AMP-PNP was used as the energy source in these assays because it supported the activation of Ub by the E1 enzyme but could not be utilized by the 26S proteasome (61). The conjugation of added Ub to 125I-labeled α-lactalbumin in fraction II from +/+ muscle resulted, upon SDS-PAGE and autoradiography, in a smear of multiubiquitylated α-lactalbumin derivatives (Fig. 5E, lane 2; compare with lane 1). Previous work (65) has shown that E214K(C88S), an active-site mutant of the E214K (HR6B) Ub-conjugating enzyme, traps Ub in a stable ester-bond complex (through a reaction mediated by E1 enzyme) and therefore acts as a dominant-negative inhibitor of the E3α-containing Ub ligase. Indeed, the addition of either the purified E214K(C88S) protein or Lys-Ala dipeptide (a type 1 N-end rule inhibitor) to +/+ fraction II abolished the bulk of α-lactalbumin ubiquitylation in this system (Fig. 6E, lanes 4 and 6; compare with lane 2). By contrast, the addition of Ala-Lys, bearing a type 3 destabilizing N-terminal residue, had no effect on ubiquitylation of α-lactalbumin (Fig. 6E, lane 8; compare with lane 2). Strikingly, the extent of ubiquitylation of α-lactalbumin in fraction II from UBR1−/− muscle (Fig. 6E, lane 3; compare with lane 2) was essentially indistinguishable from background ubiquitylation in the inhibited +/+ fraction II (Fig. 6E, lane 3; compare with lanes 4 and 6). Moreover, the background-level ubiquitylation in UBR1−/− fraction II remained essentially unchanged upon the addition of either E214K(C88S) or Lys-Ala, in contrast to the results with +/+ fraction II (Fig. 6E, lane 3; compare with lanes 5 and 7).
Previous work, which used dipeptide inhibitors and dominant-negative E214K(C88S) in an attempt to distinguish between protein ubiquitylation due to the N-end rule pathway and the total ubiquitylation of endogenous proteins in a fraction II preparation from a +/+ muscle extract suggested that 60 to 80% of the ATP-dependent degradation of soluble muscle proteins was mediated by the N-end rule pathway (62). We assayed the conjugation of 125I-Ub to endogenous proteins in fraction II preparations from +/+ and UBR1−/− muscle. This conjugation was found to be 25 to 30% lower in UBR1−/− fraction II than in +/+ fraction II (Fig. 6F, lanes 2 and 3; compare with lane 1). Ubiquitylation of endogenous proteins in both +/+ and UBR1−/− fraction II preparations was strongly inhibited by the addition of dominant-negative E214K(C88S) protein (Fig. 6F, lanes 4 and 5; compare with lanes 2 and 3).
Thus, both degradation assays with purified N-end rule substrates in +/+ versus UBR1−/− muscle extracts (Fig. 6A to D) and ubiquitylation assays with 125I-labeled α-lactalbumin in +/+ versus UBR1−/− fraction II (Fig. 6E) indicated that the N-end rule pathway was virtually absent from skeletal muscle extracts of UBR1−/− mice, in contrast to extracts from +/+ muscle. It remains to be determined whether the N-end rule pathway is absent from the intact UBR1−/− muscle.
The N-end rule pathway is active in UBR1−/− fibroblasts.
We assayed the degradation of purified, 35S-labeled Phe-DHFR, a type 2 N-end rule substrate, in ATP-supplemented extracts from +/+ and UBR1−/− EF cell lines. Met-DHFR, bearing a stabilizing N-terminal residue, was used as a negative control. In striking contrast to the findings with UBR1−/− muscle extracts (Fig. 6), the N-end-rule-specific degradation of Phe-DHFR not only was retained in UBR1−/− EF extract but was actually significantly higher in that extract than in the otherwise identical extract from +/+ EF cells (Fig. 7A, graph a). A large fraction of Phe-DHFR degradation in both +/+ and UBR1−/− EF extracts was mediated by the N-end rule pathway (Fig. 7A, graph a; compare the decay curves of Phe-DHFR with those of Met-DHFR, both in the presence and absence of added ATP). As expected, Phe-Ala, a type 2 dipeptide inhibitor, reduced the degradation of Phe-DHFR in both +/+ and UBR1−/− EF extracts (Fig. 7A, graph c). Ala-Phe (Fig. 7A, graph c) and Ala-Lys (not shown), both of them type 3 dipeptide inhibitors, did not have any effect on either +/+ or UBR1−/− EF extracts, again as expected. Strikingly, however, Lys-Ala, a type 1 dipeptide inhibitor, enhanced the degradation of Phe-DHFR only in +/+ EF extract (see above for a description of this previously discovered crossover enhancing effect of type 1 dipeptides on degradation of type 2 N-end rule substrates [18]). Specifically, whereas the degradation of Phe-DHFR in +/+ EF extract was enhanced by Lys-Ala (Fig. 7A, graph b), which is similar to the results with +/+ muscle extracts described above (Fig. 6A and D), the same Lys-Ala dipeptide not only did not enhance degradation of the same substrate, Phe-DHFR, in UBR1−/− EF extract but slightly (and reproducibly) decreased it (Fig. 7A, graph b, and data not shown).
FIG. 7.

The N-end rule pathway is retained in UBR1−/− EF cells. (A) Degradation of 35S-labeled Ub-X-DHFR proteins in extracts from +/+ and UBR1−/− EF cell lines was monitored by measuring TCA-soluble 35S (see Materials and Methods). Open and closed symbols denote the data with +/+ and UBR1−/− extracts, respectively. Reaction mixtures were incubated at 37°C for 10 min without ATP, and then ATP-dependent degradation of X-DHFR was initiated by the addition of ATP (time zero). No ATP was added to control samples. (A, graph a) ATP-dependent degradation of Phe-DHFR, a type 2 N-end rule substrate, is faster in extracts from UBR1−/− EF cells. Squares and circles, Phe-DHFR with or without ATP, respectively; diamonds and triangles, Met-DHFR (not an N-end rule substrate) with or without ATP, respectively. (A, graph b) Lys-Ala, a type 1 dipeptide inhibitor, does not enhance degradation of the type 2 N-end rule substrate Phe-DHFR in UBR1−/− EF extracts. □ and ▪, no inhibitor; ○ and ●, Lys-Ala. (A, graph c) Effect of Phe-Ala, a type 2 dipeptide inhibitor, on the ATP-dependent degradation of Phe-DHFR. □ and ▪, no inhibitor; ○ and ●, Phe-Ala; ▿ and ▾, Ala-Phe. (B) Immortalized +/+ and UBR1−/− EF cells were transiently transfected with pRC/dhaUbXnsP4βgal (X = Met, Arg, or Phe) expressing X–β-gal test proteins as parts of UPR-based DHFRh-UbR48-X-nsP4βgal fusions (39, 66, 68, 69). Cells were labeled for 1 h with 35S-methionine–cysteine followed by immunoprecipitation and SDS-PAGE analysis of an X–β-gal test protein and the DHFR-based reference protein. (C) Quantitation of the patterns shown in panel B using PhosphorImager. Note a small but significant additional destabilization of Phe-DHFR in UBR1−/− EF cells. (D) Pulse-chase analysis of the N-end rule pathway in +/+ and UBR1−/− EF cells. Immortalized +/+ and UBR1−/− EF cells were transiently transfected with pcDNA3flagDHFRhaUbXnsP4flag (X = Met, Arg, or Tyr) expressing fDHFRh-UbR48-X-nsP4f, a UPR-based fusion yielding, cotranslationally, the fDHFRh-UbR48 reference protein (denoted DHFR on the right) and X-nsP4f (X-nsP4-flag) test protein, denoted X-nsP4 on the right. Cells were labeled for 10 min with 35S-methionine–cysteine followed by a chase for 0, 1, and 2 h in the presence of cycloheximide, preparation of extracts, immunoprecipitation, SDS-PAGE, autoradiography, and quantitation, essentially as described previously (39). (E) Quantitation of the patterns shown in panel D using a PhosphorImager. The amounts of 35S in an X-nsP4f relative to 35S in the fDHFRh-UbR48 reference protein at the same time points were plotted as percentages of this ratio for Met-nsP4f (bearing a stabilizing N-terminal residue) at time zero. □ and ▪, Met-nsP4; ○ and ●, Arg-nsP4; ▵ and ▴, Tyr-nsP4.
Thus, the N-end rule pathway is active in UBR1−/− EF extracts (Fig. 7A, graph a) despite the absence of the UBR1 protein from UBR1−/− EF cells (Fig. 2E, gel c). The selective absence of enhancing effect of a type 1 dipeptide on degradation of type 2 N-end rule substrate in UBR1−/− EF extract (Fig. 7A, graph b) suggests that non-UBR1 N-recognins that mediate the N-end rule pathway in UBR1−/− EF cells lack a structural feature that underlies the above allosteric effect in the UBR1 N-recognin (E3α).
To examine the in vivo degradation of N-end rule substrates in +/+ and UBR1−/− EF cells, they were transiently transfected with plasmids that expressed an X-nsP4βgal test protein (X = Met, Arg, or Tyr) as part of the DHFRh-UbR48-X-nsP4βgal fusion, a UPR construct (39, 66, 68, 69). In this Ub fusion, the reference moiety DHFRh-UbR48 contained the ha epitope-tagged mouse dihydrofolate reductase (DHFRh). DHFRh-UbR48-X-nsP4βgal is cotranslationally cleaved by DUBs at the UbR48-X junction, yielding the long-lived DHFRh-UbR48 reference protein and a test protein, X-nsP4βgal (X = Met, Arg, or Tyr). In the UPR technique, the reference protein serves as an internal control for the levels of expression, immunoprecipitation yields, sample volumes, and other sources of sample-to-sample variation, thereby increasing the accuracy of pulse-chase and related assays (39, 51, 66, 68, 69). The nsP4βgal moiety of X-nsP4βgal comprised the first 165 residues of nsP4 (Sindbis virus polymerase) (13, 69), followed by the E. coli βgal moiety that lacked the first 5 residues of wild-type βgal. Due to a built-in reference protein in DHFRh-UbR48-X-nsP4βgals, it was possible to compare metabolic stabilities of X-nsP4βgals not only in pulse-chase assays but also in pulse-only assays where we determined, after a 60-min pulse, the ratio of an X-nsP4βgal protein to the reference protein DHFRh-UbR48 (Fig. 7B and C). Arg-nsP4βgal was metabolically unstable in both +/+ and UBR1−/− EF cells. Specifically, 44 and 42% of Arg-nsP4βgal (bearing a type 1 destabilizing N-terminal residue) remained after the 60-min pulse in, respectively, the +/+ and UBR1−/− EF cells relative to the long-lived Met-nsP4βgal, whose amount at the end of a 60-min pulse was taken as 100% (Fig. 7B and C). Remarkably, Phe-nsP4βgal, bearing a type 2 destabilizing N-terminal residue, was reproducibly more unstable in UBR1−/− EF cells (42%) than in +/+ EF cells (64%) (Fig. 7B and C). This in vivo result was in agreement with the independent observation of enhanced Phe-DHFR degradation in UBR1−/− versus +/+ EF extracts (Fig. 7A, graph a).
We carried out pulse-chase assays of N-end rule substrates in +/+ and UBR1−/− EF cells by using UPR-type fusions of fDHFRh-UbR48-X-nsP4f (X = Met, Arg, or Tyr). In these fusions, the reporter moiety was X-nsP4f, the full-length 69-kDa X-nsP4 protein (X = Met, Arg, or Tyr) bearing the C-terminal flag epitope (2). UPR-based assays with X-nsP4 test proteins in UBR1−/− and +/+ EF cells employed 10-min pulse and chase times of 0, 1, and 2 h. The results (Fig. 7D and E) confirmed that Arg-nsP4 (a type 1 substrate) and Tyr-nsP4 (a type 2 substrate) were short-lived in both UBR1−/− and +/+ EF cells. Moreover, the degradation of Arg-nsP4 and Tyr-nsP4 was slightly but reproducibly faster in UBR1−/− cells (Fig. 7D and E), in agreement with the findings using 1-h in vivo pulse (Fig. 7B and C) as well as proteolysis in EF extracts (Fig. 7A). Met-nsP4, bearing a stabilizing N-terminal residue, was long-lived in both UBR1−/− and +/+ EF cells (Fig. 7D and E). In UBR1−/− EF cells, 23% of Arg-nsP4 remained at the end of the 1-h chase, in contrast to 32% Arg-nsP4 in +/+ EF cells under the same conditions (Fig. 7D and E). Similarly, in UBR1−/− EF cells 25% of Tyr-nsP4 remained at the end of the 1-h chase, in contrast to 35% of Tyr-nsP4 in +/+ EF cells under the same conditions (Fig. 7D and E). We conclude that the N-end rule activity was present and, moreover, hyperactive in UBR1−/− EF cells, which lacked the UBR1 protein (Fig. 7).
Reduced mass and other phenotypes of UBR1−/− mice.
Most UBR1−/− mice weighed significantly less than their +/+ and UBR1+/− littermates of the same gender (Fig. 4A). The ∼20% difference in mass observed at birth transiently increased to ∼32% for males and ∼26% for females at the time of weaning (Fig. 4A, graph c, arrow), when the pups' nutrition changes from mother's milk to solid food. By 4 months the mean difference in mass was ∼20% (Fig. 7A, graph c) and decreased to ∼12% at 1 year (data not shown). Despite the lower mass of UBR1−/− mice, their mean body length (excluding their tails) was nearly indistinguishable from that of age-matched, same-gender +/+ mice (Fig. 4B and data not shown). The lengths and weights of bones from UBR1−/− mice were also similar to those of their littermate +/+ controls (data not shown). The weights of brain, liver, spleen, kidney, and testis of UBR1−/− mice were ∼90% or more of the corresponding organ weights of either UBR1+/− or +/+ littermates (Fig. 4C). However, the average weights of hind leg muscle and the hind leg fat pad of UBR1−/− mice were, respectively, ∼82 and ∼61% of their UBR1+/− and +/+ counterparts (Fig. 7C), suggesting that the lower total mass of UBR1−/− mice stemmed from a disproportionate decrease in the mass of skeletal muscle and adipose tissues.
No gross morphological differences were observed between UBR1−/− and +/+ embryos at any stage of development (data not shown). The body masses of eight E17.5 +/+ male embryos, fifteen UBR1+/− male embryos, and six UBR1−/− male embryos were, respectively, 714 ± 41, 716 ± 39, and 616 ± 58 mg. Histological examinations of adult UBR1−/− tissues (small intestine, liver, pancreas, adrenal gland, thyroid gland, kidney, ovary, heart, spleen, thymus, skeletal muscle, brain, and sciatic nerve) did not detect significant abnormalities (data not shown). UBR1−/− mice appeared to be healthy; their limb movements and overall behavior were apparently normal; they oriented to sound and cared for offspring. Several behavioral and motor coordination tests were also carried out. We used 24 adult UBR1−/− mice (2 to 4 months old) whose weights were ∼15% lower than those of their control littermates (8 UBR1+/− and 16 +/+ mice). Motor coordination of UBR1−/− mice was assessed using rotarod (32). UBR1−/− mice exhibited a slightly lower ability to stay on a rotating horizontal rod than their UBR1+/− and +/+ littermates (data not shown). The cause of this phenotype is likely to be a reduced motor coordination of UBR1−/− mice rather than defects in learning and memory (data not shown). No significant differences between UBR1−/− mice and their UBR1+/− or +/+ littermates were observed in several previously described tests (32), including the weight retention test (assessment of physical strength), the coat hanger test (assessment of both physical strength and coordination), and the hind paw footprint test (assessment of walking patterns). Thus, despite the 15 to 20% lower mass of UBR1−/− mice, the above tests detected no overt behavioral or locomotor impairments in these mice.
Mouse UBR1 (E3α) functions in association with either E214K (mHR6B) or mHR6A, two highly similar Ub-conjugating enzymes (Fig. 3). Previous work has shown that mice lacking E214K (HR6B) are defective in spermatogenesis (55). We examined whether UBR1−/− mice exhibited defects in spermatogenesis. Comparison of various mating combinations [(+/+ × +/+), (+/+ × −/−), (−/− × +/+), and (−/− × −/−)] revealed no significant fertility defects in either male or female UBR1−/− mice, in that UBR1−/− mice were apparently normal in copulatory behavior, the size of testes, the number of litters, and the average litter size (data not shown). Histological examination of UBR1−/− seminiferous tubules showed no obvious abnormalities in either the structure of tubules, differentiation of germ cells (mitotic, meiotic, and postmeiotic), or morphology of supporting cells, such as Sertoli and Leydig cells (data not shown). No differences in testicular cell apoptosis was detected between UBR1−/− and +/+ testes by using the TUNEL assay (data not shown). Thus, UBR1, in an otherwise wild-type genetic background, is not required for fertility-related functions.
Abnormal fat metabolism in UBR1−/− mice.
To address the cause of adipose tissue reduction in UBR1−/− mice, we used Northern analysis of RNA from +/+ and UBR1−/− skeletal muscles, comparing the levels of mRNAs encoding some of the proteins involved in fat metabolism, specifically gamma interferon, interleukin 1β (IL-1β), IL-6, tumor necrosis factor alpha, glycerol-3-phosphate transferase, triacyl glycerol lipase, and fatty acid synthase (FAS). No significant differences between +/+ and UBR1−/− mice were observed in the levels of the above mRNAs under normal, fasting, and refeeding conditions (data not shown), except for the levels of FAS mRNA (Fig. 4D). FAS is a multicatalytic enzyme containing domains for acyl-carrier peptide and seven enzymatic modules required for the conversion of acetyl-coenzyme A (CoA) and malonyl-CoA to palmitate. The expression of FAS is tightly regulated through nutritional, hormonal, and developmental inputs (41, 77)
Under normal conditions the level of FAS mRNA in the UBR1−/− muscle was ∼56% of that in +/+ muscle (Fig. 4D, lanes a and b), suggesting that reduction of adipose tissue in UBR1−/− mice was caused at least in part by a decrease in FAS expression. In contrast to skeletal muscle, the levels of FAS mRNA in growing UBR1−/− and +/+ EF cell lines were nearly identical (Fig. 4D). After 48 h of fasting, the level of FAS mRNA in +/+ skeletal muscle was reduced to less than 3% of its level under normal conditions (Fig. 4D, lanes c and i). In striking contrast, the level of FAS mRNA in the muscle of fasting UBR1−/− mice remained nearly unchanged (Fig. 7D, lanes d and j; compare with lanes c and i). Thus, whereas under normal conditions the UBR1−/− muscle contained half as much FAS mRNA as the +/+ muscle, after 48 h of fasting the level of FAS mRNA became ∼14-fold higher in the UBR1−/− muscle than in the congenic +/+ muscle (Fig. 4D). After 24 h of refeeding, the levels of FAS mRNA in the UBR1−/− and +/+ muscles became nearly equal (Fig. 4D, lanes e and f).
Given the above results, several blood plasma parameters were determined in multiple pairs of 2- to 4-month old +/+ and UBR1−/− littermates (Table 1). Under normal conditions the plasma glucose level of UBR1−/− mice was 12% lower than that of +/+ mice. This difference in glucose levels between UBR1−/− and +/+ mice increased to 21% after a 24-h fast and remained nearly unchanged (20%) 24 h after refeeding (Table 1). These differences were reproducible in independent measurements (data not shown). Thus, UBR1−/− mice were mildly hypoglycemic under normal conditions. The levels of plasma triglycerides, urea nitrogen, total protein, and albumin were also lower in UBR1−/− mice, suggesting a state of mild malnutrition (Table 1). There were no significant differences between UBR1−/− mice and +/+ littermates in the levels of plasma cholesterol, as well as sodium, potassium, calcium, chloride, and phosphorus (Table 1). UBR1−/− mice were also normal in their levels of several markers of the liver function, including GGT, AST, and ALT (data not shown).
TABLE 1.
Plasma parameters of UBR1−/− and congenic +/+ micea
| Parameter (unit of measurement) | Results for the
following mice groups:
|
|||||
|---|---|---|---|---|---|---|
| Control
|
Fasted
|
Refed
|
||||
| Wild-type | UBR1−/− | Wild-type | UBR1−/− | Wild-type | UBR1−/− | |
| Glucose (mg/dl) | 172 ± 9.6 | 152 ± 10.3c | 162 ± 10.4 | 128 ± 8.5c | 186 ± 12.5 | 149 ± 15.1c |
| Triglycerides (mg/dl) | 56.8 ± 6.7 | 44.5 ± 5.1c | 44.7 ± 5.5 | 35.2 ± 4.7d | 61.0 ± 6.9 | 46.2 ± 5.8d |
| Cholesterol (mg/dl) | 57.3 ± 4.5 | 53.1 ± 5.1 | 68.5 ± 5.2 | 70.5 ± 4.6 | 54.4 ± 4.3 | 54.2 ± 4.8 |
| Albumin (g/dl) | 1.40 ± 0.17 | 1.27 ± 0.12e | 2.13 ± 0.25 | 1.44 ± 0.31c | NDb | ND |
| Urea nitrogen (mg/dl) | 21.3 ± 4.3 | 16.3 ± 3.2d | 27.3 ± 5.1 | 20.5 ± 4.7e | ND | ND |
| Total protein (g/dl) | 3.23 ± 0.41 | 3.02 ± 0.34 | 4.33 ± 0.57 | 3.34 ± 0.52 | ND | ND |
| Sodium (mmol/liter) | 147 ± 1.3 | 148 ± 1.6 | 153 ± 1.5 | 153 ± 1.5 | 148 ± 1.7 | 149 ± 1.9 |
| Potassium (mmol/liter) | 6.43 ± 0.12 | 6.57 ± 0.15 | 5.25 ± 0.12 | 5.45 ± 0.13 | 6.52 ± 0.15 | 6.48 ± 0.13 |
| Chloride (mmol/liter) | 116 ± 1.6 | 116 ± 1.5 | 122 ± 1.4 | 124 ± 0.6 | 116 ± 1.7 | 117 ± 1.5 |
| Calcium (mg/dl) | 7.47 ± 0.21 | 7.43 ± 0.23 | 8.15 ± 0.27 | 7.35 ± 0.21 | 7.18 ± 0.36 | 7.42 ± 0.26 |
| Phosphorus (mg/dl) | 11.5 ± 0.46 | 12.4 ± 0.34c | 12.7 ± 0.48 | 13.4 ± 0.32d | 11.7 ± 0.51 | 12.1 ± 0.69 |
The experiments were performed twice (experiments 1 and 2), with 28 pairs of animals total. The blood was collected from 12 pairs before fasting (control), 8 pairs after 24 h of fasting (fasted), and 8 pairs after 24 h of fasting and 24 h of refeeding (refed).
ND, not determined.
P < 0.001.
P < 0.01.
P < 0.05.
NTAN1−/−UBR1−/− double knockout mice.
Our previous work (32) described NTAN1−/− mice which lacked the Asn-specific NtN-amidase, a component of the N-end rule pathway upstream of UBR1 (see the introduction). NTAN1−/− mice were viable, fertile, and of normal size, physical strength, and motor coordination, but they exhibited altered behaviors, including a socially conditioned exploratory phenotype (32). NtN-amidase mediates the destabilizing activity of Asn, 1 of 16 destabilizing residues that are recognized directly or indirectly by UBR1 (19, 35). In situ hybridization indicated similar spatial patterns of UBR1 and NTAN1 expression in embryogenesis (32, 35). Whereas NTAN1−/− mice completely lacked NtN-amidase activity (32), UBR1−/− mice contained proteins that at least partially complemented the activity of missing UBR1 (present work). It is unknown whether the function of NtN-amidase is confined to the N-end rule pathway. To address the possibility of unexpected interactions between NTAN1- and UBR1-dependent functions, we produced double mutant NTAN1−/−UBR1−/− mice through double heterozygous matings (see Materials and Methods). Adult NTAN1−/− UBR1−/− mice were fertile and apparently healthy. Intercrosses between NTAN1+/− UBR1−/− mice or between NTAN1−/− UBR1+/− mice yielded expected Mendelian frequencies of NTAN1−/−UBR1−/− mice, indicating that the absence of both NTAN1 and UBR1 did not significantly increase embryonic lethality. NTAN1−/−UBR1−/− mice resembled UBR1−/− mice in being of lower weight (by ∼20%) than their NTAN1+/+UBR1+/+, NTAN1+/−UBR1+/+, and NTAN1+/+ UBR1+/− littermates (data not shown), in contrast to the normal weight of NTAN1−/− mice (32). General behavior, physical strength, and motor coordination of NTAN1−/−UBR1−/− mice were not overtly abnormal, as assayed with the previously described (32) rotarod test, weight retention test, and coat hanger test (data not shown).
DISCUSSION
This study is the first in a projected series that aims to decipher, in functional and mechanistic detail, the mammalian N-end rule pathway at the level of the pathway's E3 Ub ligases. It was found that more than one E3 (N-recognin) mediates the mouse N-end rule pathway, in contrast to S. cerevisiae. We report the following results.
(i) The mouse UBR1-encoded 200-kDa N-recognin (E3α) (35) was shown to rescue the N-end rule pathway in ubr1Δ S. cerevisiae that lacked the 225-kDa yeast homolog of mouse UBR1. The rescue's efficiency was strongly increased in the presence of a cognate mouse E2 enzyme, either mHR6A or mHR6B (E214K) (Fig. 3).
(ii) UBR1−/− mouse strains lacking the UBR1 protein (Fig. 2) were viable, of normal fertility, and outwardly healthy, but their mass, from birth through adulthood, was significantly lower than that of their congenic +/+ littermates (on average, 20% lower at 2 months of age) (Fig. 4). The lower mass of UBR1−/− mice stemmed at least in part from reduced mass of skeletal muscle and adipose tissues. FAS mRNA, encoding FAS, was underexpressed in UBR1−/− muscle from normally fed UBR1−/− mice and was strikingly misregulated after a 48-h fast (Fig. 7C). The growth retardation and decreased fat content in UBR1−/− mice were consistent with a lower level of glucose and triglycerides in the blood plasma of these mice (Table 1).
(ii) Extracts of UBR1−/− skeletal muscle lacked the N-end rule pathway, in contrast to otherwise identical extracts from +/+ muscle. This and other evidence (Fig. 6) suggested that the N-end rule pathway was absent from cells of the UBR1−/− skeletal muscle. By contrast, the N-end rule pathway was found to be active and even slightly enhanced both in UBR1−/− EF cell lines in vivo and in EF extracts (Fig. 7).
(iv) Double-mutant NTAN1−/−UBR1−/− mice, which lacked both NtN-amidase (see the introduction) and the UBR1-encoded E3α Ub ligase, were viable and did not exhibit phenotypes other than the expected ones.
The main finding of this work is that the recognition of type 1 and type 2 N-end rule substrates by the mammalian N-end rule pathway is mediated by more than one E3 Ub ligase (N-recognin), in contrast to the yeast S. cerevisiae, where UBR1 is the only E3 of the N-end rule pathway. Functionally overlapping, differentially expressed N-recognins render UBR1−/− mice mosaic in regard to activity of the N-end rule pathway. The previously identified mouse UBR2 and UBR3 genes (35) encode proteins that are, respectively, 47 and 25% identical and 67 and 51% similar to mouse UBR1 (Y. T. Kwon, T. Tasaki, and A. Varshavsky, unpublished data). S. cerevisiae UBR1 is equally similar (22% identity, 42% similarity) to mouse UBR1 and UBR2. Although UBR3 is clearly an E3 Ub ligase of the same RING-H2 class as UBR1 and is a member of the UBR sequence family, it lacks the N-terminus-proximal residues that have previously been found to be essential for the integrity of the type 1 and type 2 substrate-binding sites of S. cerevisiae UBR1 (A. Webster, M. Ghislain, and A. Varshavsky, unpublished data). UBR3 might be the still-unidentified E3 Ub ligase that recognizes N-terminal Ala, Ser, or Thr, the type 3 destabilizing residues of the mammalian N-end rule (18, 22).
In contrast to UBR3, mouse UBR2 is identical in size to mouse UBR1 (200 kDa) and contains the residues identified as essential for the recognition of type 1 or 2 substrates in S. cerevisiae UBR1 (see above). In addition, the mouse UBR1 and UBR2 genes have identical exon and intron junctions in the first 12 exons that have been examined in detail (Y. T. Kwon and A. Varshavsky, unpublished data). Recent in vitro binding assays with model N-end rule substrates identified mouse UBR2 as a type 1 and 2 N-recognin (Z. Xia, Y. T. Kwon, F. Du, and A. Varshavsky, unpublished data), making it likely that UBR2 is an E3 that at least partially rescues the N-end rule pathway in UBR1−/− mice. If so, it remains to be explained why extracts from the UBR1−/− skeletal muscle, where UBR2 is apparently expressed, lack the N-end rule pathway, in contrast to identically prepared extracts from +/+ muscle and also in contrast to extracts from either UBR1−/− or +/+ EF cells (see Results). Remarkably, recent results indicated that UBR1 and UBR2 cannot be the only type 1 or 2 N-recognins in the mouse. The UBR1−/−UBR2−/− double mutant mice are embryonic lethals, in contrast to UBR1−/− and UBR2−/− mice; nevertheless, EF cells rescued from arrested UBR1−/−UBR2−/− embryos do contain the N-end rule pathway, albeit of significantly lower activity (Y. T. Kwon, I. Davydov, and A. Varshavsky, unpublished data). The identity of a third mouse type 1 and 2 N-recognin (E3) is unknown. This N-recognin might be closer in sequence to that of the plant (Arabidopsis thaliana) protein PRT1, which is not a member of the UBR sequence family but shares with it the presence of a RING finger and the ability to recognize destabilizing N-terminal residues in model substrates (50). In contrast to UBR1, which binds to both type 1 (basic) and type 2 (bulky hydrophobic) destabilizing N-terminal residues, plant PRT1 binds to a subset of type 2 N-terminal residues (A. Bachmair, personal communication), raising the possibility that a third mouse N-recognin is PRT1-like in that it may be specific for subsets of N-terminal residues that are recognized by N-recognins such as UBR1 and UBR2.
The recently constructed UBR2−/− mice are viable as males but not as females, most of which die as embryos (Y. T. Kwon and A. Varshavsky, unpublished data). UBR2−/− EF cells contain the N-end rule pathway, as would be expected given the presence of UBR1 (E3α) in UBR2−/− mice. UBR2−/− males are of normal weight and appearance but are infertile owing to death, through apoptosis, of meiotic spermatocytes in UBR2−/− testes (Y. T. Kwon and A. Varshavsky, unpublished data). The death of meiotic spermatocytes in UBR2−/− mice may be caused by the lack of UBR1 (E3α) expression in these cells, a conjecture to be verified.
Several studies utilized dipeptides as inhibitors of the N-end rule pathway in metazoan cells. The reported results include the inhibition of mammalian cell differentiation (27, 47), the inhibition of apoptosis in human lymphocytes (44), and the inhibition of limb regeneration in newts (67) by dipeptides bearing destabilizing N-terminal residues. In contrast to S. cerevisiae, where dipeptides added to the growth medium are strong in vivo inhibitors of the N-end rule pathway (5), the same dipeptides are, at most, weak inhibitors of the N-end rule pathway in mammalian cells (F. Lévy and A. Varshavsky, unpublished data). Moreover, if the design of S. cerevisiae UBR1 is relevant to mammalian N-recognins, it is clear that dipeptides can also activate rather than inhibit a substrate-binding site of N-recognin that recognizes internal degrons in a subset of its substrates (68). Given this issue alone, the discovery of circuits controlled by the N-end rule pathway in multicellular eukaryotes will require genetic analyses as well as identification of the pathway's physiological substrates.
Four functions of the N-end rule pathway have been identified so far: its essential roles in the control of peptide import (68) and chromosome stability (51) in S. cerevisiae (see the introduction), its essential role in mammalian spermatogenesis (the phenotype of UBR2−/− mice described above [Y. T. Kwon and A. Varshavsky, unpublished data]), and its requirement for cardiogenesis and angiogenic remodeling during mouse embryogenesis (Y. T. Kwon, A. Kashina, and A. Varshavsky, unpublished data). The latter function was discovered through the construction of ATE1−/− mouse strains that lacked R-transferase (see the introduction). In contrast to the two roles of the N-end rule pathway in yeast, where both the relevant circuits and physiological substrates of UBR1 are partially understood (51, 68), we do not know, as yet, the identity of proteins whose metabolic stabilization underlies the observed phenotypes of UBR1−/−, UBR2−/−, and ATE1−/− mouse strains. The UBR1−/− mice and cells of the present work will be essential for further advances in the understanding of the N-end rule pathway.
ACKNOWLEDGMENTS
We are grateful to members of the Caltech Transgenic and Knockout Core Facility, especially to S. Pease, B. Kennedy, and A. Granados for their care of mice and expert technical help. We thank B. Kennedy for his assistance with mouse weighing, W. Rivas for help with the cardiac puncture procedure, and Greg Cope for assistance with the Northern analysis. We are grateful to H. P. Roest (Erasmus University, Rotterdam, The Netherlands) for a gift of plasmid 44.83 and to members of the Varshavsky laboratory for helpful discussions and support. We also thank T. Tasaki and F. Du for their comments on the manuscript.
A.V. gratefully acknowledges support by the Fellows Program of the International Institute for Advanced Studies (Kyoto, Japan). This work was supported by grants GM31530 and DK39520 from the National Institutes of Health to A.V.
REFERENCES
- 1.Alagramam K, Naider F, Becker J M. A. Recognition component of the ubiquitin system is required for peptide transport in Saccharomyces cerevisiae. Mol Microbiol. 1995;15:225–234. doi: 10.1111/j.1365-2958.1995.tb02237.x. [DOI] [PubMed] [Google Scholar]
- 2.Ausubel F M, Brent R, Kingston R E, Moore D D, Smith J A, Seidman J G, Struhl K, editors. Current protocols in molecular biology. New York, N.Y: Wiley-Interscience; 2000. [Google Scholar]
- 3.Bachmair A, Finley D, Varshavsky A. In vivo half-life of a protein is a function of its amino-terminal residue. Science. 1986;234:179–186. doi: 10.1126/science.3018930. [DOI] [PubMed] [Google Scholar]
- 4.Bachmair A, Varshavsky A. The degradation signal in a short-lived protein. Cell. 1989;56:1019–1032. doi: 10.1016/0092-8674(89)90635-1. [DOI] [PubMed] [Google Scholar]
- 5.Baker R T, Varshavsky A. Inhibition of the N-end rule pathway in living cells. Proc Natl Acad Sci USA. 1991;87:2374–2378. doi: 10.1073/pnas.88.4.1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baker R T, Varshavsky A. Yeast N-terminal amidase. A new enzyme and component of the N-end rule pathway. J Biol Chem. 1995;270:12065–12074. doi: 10.1074/jbc.270.20.12065. [DOI] [PubMed] [Google Scholar]
- 7.Balzi E, Choder M, Chen W, Varshavsky A, Goffeau A. Cloning and functional analysis of the arginyl-tRNA-protein transferase gene ATE1of Saccharomyces cerevisiae. J Biol Chem. 1990;265:7464–7471. [PubMed] [Google Scholar]
- 8.Bartel B, Wünning I, Varshavsky A. The recognition component of the N-end rule pathway. EMBO J. 1990;9:3179–3189. doi: 10.1002/j.1460-2075.1990.tb07516.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Byrd C, Turner G C, Varshavsky A. The N-end rule pathway controls the import of peptides through degradation of a transcriptional repressor. EMBO J. 1998;17:269–277. doi: 10.1093/emboj/17.1.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chau V, Tobias J W, Bachmair A, Marriott D, Ecker D J, Gonda D K, Varshavsky A. A multiubiquitin chain is confined to specific lysine in a targeted short-lived protein. Science. 1989;243:1576–1583. doi: 10.1126/science.2538923. [DOI] [PubMed] [Google Scholar]
- 11.Davydov I V, Patra D, Varshavsky A. The N-end rule pathway in Xenopus egg extracts. Arch Biochem Biophys. 1998;357:317–325. doi: 10.1006/abbi.1998.0829. [DOI] [PubMed] [Google Scholar]
- 12.Davydov I V, Varshavsky A. RGS4 is arginylated and degraded by the N-end rule pathway in vitro. J Biol Chem. 2000;275:22931–22941. doi: 10.1074/jbc.M001605200. [DOI] [PubMed] [Google Scholar]
- 13.deGroot R J, Rümenapf T, Kuhn R J, Strauss J H. Sindbis virus RNA polymerase is degraded by the N-end rule pathway. Proc Natl Acad Sci USA. 1991;88:8967–8971. doi: 10.1073/pnas.88.20.8967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.DeMartino G N, Slaughter C A. The proteasome, a novel protease regulated by multiple mechanisms. J Biol Chem. 1999;274:22123–22126. doi: 10.1074/jbc.274.32.22123. [DOI] [PubMed] [Google Scholar]
- 15.Deshaies R J. SCF and cullin/RING-H2-based ubiquitin ligases. Annu Rev Cell Dev Biol. 1999;15:435–467. doi: 10.1146/annurev.cellbio.15.1.435. [DOI] [PubMed] [Google Scholar]
- 16.Dohmen R J. Primary destruction signals. In: Hilt W, Wolf D, editors. Proteasomes: the world of regulatory proteolysis. Georgetown, Tex: R. G. Landes Biosciences; 2000. pp. 188–205. [Google Scholar]
- 17.Dohmen R J, Madura K, Bartel B, Varshavsky A. The N-end rule is mediated by the UBC2(RAD6) ubiquitin-conjugating enzyme. Proc Natl Acad Sci USA. 1991;88:7351–7355. doi: 10.1073/pnas.88.16.7351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gonda D K, Bachmair A, Wünning I, Tobias J W, Lane W S, Varshavsky A. Universality and structure of the N-end rule. J Biol Chem. 1989;264:16700–16712. [PubMed] [Google Scholar]
- 19.Grigoryev S, Stewart A E, Kwon Y T, Arfin S M, Bradshaw R A, Jenkins N A, Copeland N G, Varshavsky A. A mouse amidase specific for N-terminal asparagine. The gene, the enzyme, and their function in the N-end rule pathway. J Biol Chem. 1996;271:28521–28532. doi: 10.1074/jbc.271.45.28521. [DOI] [PubMed] [Google Scholar]
- 20.Haas A J, Siepman T J. Pathways of ubiquitin conjugation. FASEB J. 1997;11:1257–1268. doi: 10.1096/fasebj.11.14.9409544. [DOI] [PubMed] [Google Scholar]
- 21.Harlow E, Lane D, editors. Using antibodies: a laboratory manual. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 1999. [Google Scholar]
- 22.Heller H, Hershko A. A ubiquitin-protein ligase specific for type III protein substrates. J Biol Chem. 1990;265:6532–6535. [PubMed] [Google Scholar]
- 23.Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem. 1998;76:425–479. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- 24.Hershko A, Ciechanover A, Varshavsky A. The ubiquitin system. Nat Med. 2000;10:1073–1081. doi: 10.1038/80384. [DOI] [PubMed] [Google Scholar]
- 25.Hicke L. Protein regulation by monoubiquitin. Nat Rev Mol Cell Biol. 2001;2:195–201. doi: 10.1038/35056583. [DOI] [PubMed] [Google Scholar]
- 26.Hochstrasser M. Ubiquitin-dependent protein degradation. Annu Rev Genet. 1996;30:405–439. doi: 10.1146/annurev.genet.30.1.405. [DOI] [PubMed] [Google Scholar]
- 27.Hondermarck H, Sy J, Bradshaw R A, Arfin S M. Dipeptide inhibitors of ubiquitin-mediated protein turnover prevent growth factor-induced neurite outgrowth in rat pheochromocytoma PC12 cells. Biochem Biophys Res Commun. 1992;30:280–288. doi: 10.1016/0006-291x(92)91555-5. [DOI] [PubMed] [Google Scholar]
- 28.Joazeiro C A P, Hunter T. Ubiquitination: more than two to tango. Science. 2000;289:2061–2062. doi: 10.1126/science.289.5487.2061. [DOI] [PubMed] [Google Scholar]
- 29.Johnson E S, Gonda D K, Varshavsky A. Cis-trans recognition and subunit-specific degradation of short-lived proteins. Nature. 1990;346:287–291. doi: 10.1038/346287a0. [DOI] [PubMed] [Google Scholar]
- 30.Johnson E S, Ma P C, Ota I M, Varshavsky A. A proteolytic pathway that recognizes ubiquitin as a degradation signal. J Biol Chem. 1995;270:17442–17456. doi: 10.1074/jbc.270.29.17442. [DOI] [PubMed] [Google Scholar]
- 31.Koken M H, Reynolds P, Jaspers-Dekker I, Prakash L, Prakash S, Bootsma D, Hoeijmakers J H. Structural and functional conservation of two human homologs of the yeast DNA repair gene RAD6. Proc Natl Acad Sci USA. 1991;88:8865–8869. doi: 10.1073/pnas.88.20.8865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kwon Y T, Balogh S A, Davydov I V, Kashina A S, Yoon J K, Xie Y, Gaur A, Hyde L, Denenberg V H, Varshavsky A. Altered activity, social behavior, and spatial memory in mice lacking the NTAN1p amidase and the asparagine branch of the N-end rule pathway. Mol Cell Biol. 2000;20:4135–4148. doi: 10.1128/mcb.20.11.4135-4148.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kwon Y T, Kashina A S, Varshavsky A. Alternative splicing results in differential expression, activity, and localization of the two forms of arginyl-tRNA-protein transferase, a component of the N-end rule pathway. Mol Cell Biol. 1999;19:182–193. doi: 10.1128/mcb.19.1.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kwon Y T, Levy F, Varshavsky A. Bivalent inhibitor of the N-end rule pathway. J Biol Chem. 1999;274:18135–18139. doi: 10.1074/jbc.274.25.18135. [DOI] [PubMed] [Google Scholar]
- 35.Kwon Y T, Reiss Y, Fried V A, Hershko A, Yoon J K, Gonda D K, Sangan P, Copeland N G, Jenkins N A, Varshavsky A. The mouse and human genes encoding the recognition component of the N-end rule pathway. Proc Natl Acad Sci USA. 1998;95:7898–7903. doi: 10.1073/pnas.95.14.7898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Laney J D, Hochstrasser M. Substrate targeting in the ubiquitin system. Cell. 1999;97:427–430. doi: 10.1016/s0092-8674(00)80752-7. [DOI] [PubMed] [Google Scholar]
- 37.Lawson T G, Gronros D L, Evans P E, Bastien M C, Michalewich K M, Clark J K, Edmonds J H, Graber K H, Werner J A, Lurvey B A, Cate J M. Identification and characterization of a protein destruction signal in the encephalomyocarditis virus 3C protease. J Biol Chem. 1999;274:9871–9880. [PubMed] [Google Scholar]
- 38.Lecker S H, Solomon V, Price S R, Kwon Y T, Mitch W E, Goldberg A L. Ubiquitin conjugation by the N-end rule pathway and mRNAs for its components increase in muscles of diabetic rats. J Clin Investig. 1999;104:1411–1420. doi: 10.1172/JCI7300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lévy F, Johnsson N, Rumenapf T, Varshavsky A. Using ubiquitin to follow the metabolic fate of a protein. Proc Natl Acad Sci USA. 1996;93:4907–4912. doi: 10.1073/pnas.93.10.4907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li J, Pickart C M. Binding of phenylarsenoxide to Arg-tRNA-protein transferase is independent of vicinal thiols. Biochemistry. 1995;34:15829–15837. doi: 10.1021/bi00048a028. [DOI] [PubMed] [Google Scholar]
- 41.Loftus T M, Jaworsky D E, Frehywot G L, Townsend C A, Ronnett G V, Lane M D, Kuhajda F P. Reduced food intake and body weight in mice treated with fatty acid synthase inhibitors. Science. 2000;288:2379–2381. doi: 10.1126/science.288.5475.2379. [DOI] [PubMed] [Google Scholar]
- 42.Madura K, Dohmen R J, Varshavsky A. 1993. N-recognin/Ubc2 interactions in the N-end rule pathway. J Biol Chem. 2054;268:12046–12051. [PubMed] [Google Scholar]
- 43.Madura K, Varshavsky A. Degradation of Gα by the N-end rule pathway. Science. 1994;265:1454–1458. doi: 10.1126/science.8073290. [DOI] [PubMed] [Google Scholar]
- 44.Masdehors P, Glaisner S, Maciorowski Z, Magdelenat H, Delic J. Ubiquitin-dependent protein processing controls radiation-induced apoptosis through the N-end rule pathway. Exp Cell Res. 2000;257:48–57. doi: 10.1006/excr.2000.4870. [DOI] [PubMed] [Google Scholar]
- 45.Mulder L C F, Muesing M A. Degradation of HIV-1 integrase by the N-end rule pathway. J Biol Chem. 2000;275:29749–29753. doi: 10.1074/jbc.M004670200. [DOI] [PubMed] [Google Scholar]
- 46.Mumberg D, Muller R, Funk M. Regulatable promoters of Saccharomyces cerevisiae—comparison of transcriptional activity and their use for heterologous expression. Nucleic Acids Res. 1994;22:5767–5768. doi: 10.1093/nar/22.25.5767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Obin M, Mesco E, Gong X, Haas A L, Joseph J, Taylor A. Neurite outgrowth in PC12 cells. Distinguishing the roles of ubiquitylation and ubiquitin-dependent proteolysis. J Biol Chem. 1999;274:11789–11795. doi: 10.1074/jbc.274.17.11789. [DOI] [PubMed] [Google Scholar]
- 48.Pickart C M. Targeting of substrates to the 26S proteasome. FASEB J. 1997;11:1055–1066. doi: 10.1096/fasebj.11.13.9367341. [DOI] [PubMed] [Google Scholar]
- 49.Plemper R K, Wolf D H. Retrograde protein translocation: ERADication of secretory proteins in health and disease. Trends Biochem Sci. 1999;24:266–270. doi: 10.1016/s0968-0004(99)01420-6. [DOI] [PubMed] [Google Scholar]
- 50.Potuschak T, Stary S, Schlogelhofer P, Becker F, Nejinskaia V, Bachmair A. PRT1 of Arabidopsis thaliana encodes a component of the plant N-end rule pathway. Proc Natl Acad Sci USA. 1998;95:7904–7908. doi: 10.1073/pnas.95.14.7904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rao H, Uhlmann F, Nasmyth K, Varshavsky A. Degradation of a cohesin subunit by the N-end rule pathway is essential for chromosome stability. Nature. 2001;410:955–960. doi: 10.1038/35073627. [DOI] [PubMed] [Google Scholar]
- 52.Rechsteiner M. The 26S proteasome. In: Peters J M, Harris J R, Finley D, editors. Ubiquitin and the biology of the cell. New York, N.Y: Plenum Press; 1998. pp. 147–189. [Google Scholar]
- 53.Reiss Y, Kaim D, Hershko A. Specificity of binding of N-terminal residues of proteins to ubiquitin-protein ligase. Use of amino acid derivatives to characterize specific binding sites. J Biol Chem. 1988;263:2693–2698. [PubMed] [Google Scholar]
- 54.Robertson E J. Embryo-derived stem cell lines. In: Robertson E J, editor. Teratocarcinomas and embryonic stem cells: a practical approach. Oxford, United Kingdom: IRL Press; 1987. pp. 71–112. [Google Scholar]
- 55.Roest H P, van Klaveren J, de Wit J, van Gurp C G, Koken M H, Vermey M, van Roijen J H, Hoogerbrugge J W, Vreeburg J T, Baarends W M, Bootsma D, Grootegoed J A, Hoeijmakers J H. Inactivation of the HR6B ubiquitin-conjugating DNA repair enzyme in mice causes male sterility associated with chromatin modification. Cell. 1996;86:799–810. doi: 10.1016/s0092-8674(00)80154-3. [DOI] [PubMed] [Google Scholar]
- 56.Schauber C, Chen L, Tongaonkar P, Vega I, Madura K. Sequence elements that contribute to the degradation of yeast G-alpha. Genes Cells. 1998;3:307–319. doi: 10.1046/j.1365-2443.1998.00192.x. [DOI] [PubMed] [Google Scholar]
- 57.Scheffner M, Smith S, Jentsch S. The ubiquitin conjugation system. In: Peters J-M, Harris J R, Finley D, editors. Ubiquitin and the biology of the cell. New York, N.Y: Plenum Press; 1998. pp. 65–98. [Google Scholar]
- 58.Sherman F. Getting started with yeast. Methods Enzymol. 1991;194:3–21. doi: 10.1016/0076-6879(91)94004-v. [DOI] [PubMed] [Google Scholar]
- 59.Sijts A J, Pilip I, Pamer E G. The Listeria monocytogenes-secreted p60 protein is an N-end rule substrate in the cytosol of infected cells. Implications for major histocompatibility complex class I antigen processing of bacterial proteins. J Biol Chem. 1997;272:19261–19268. doi: 10.1074/jbc.272.31.19261. [DOI] [PubMed] [Google Scholar]
- 60.Solomon V, Baracos V, Sarraf P, Goldberg A. Rates of ubiquitin conjugation increase when muscles atrophy, largely through activation of the N-end rule pathway. Proc Natl Acad Sci USA. 1998;95:12602–12607. doi: 10.1073/pnas.95.21.12602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Solomon V, Goldberg A L. Importance of the ATP-ubiquitin-proteasome pathway in the degradation of soluble and myofibrillar proteins in rabbit muscle extracts. J Biol Chem. 1996;271:26690–26697. doi: 10.1074/jbc.271.43.26690. [DOI] [PubMed] [Google Scholar]
- 62.Solomon V, Lecker S H, Goldberg A L. The N-end rule pathway catalyzes a major fraction of the protein degradation in skeletal muscle. J Biol Chem. 1998;273:25216–25222. doi: 10.1074/jbc.273.39.25216. [DOI] [PubMed] [Google Scholar]
- 63.Stewart A. Trends in genetics nomenclature guide. Cambridge, United Kingdom: Elsevier Science, Ltd.; 1995. [Google Scholar]
- 64.Stewart A E, Arfin S M, Bradshaw R A. The sequence of porcine protein NH2-terminal asparagine amidohydrolase. A new component of the N-end rule pathway. J Biol Chem. 1995;270:25–28. doi: 10.1074/jbc.270.1.25. [DOI] [PubMed] [Google Scholar]
- 65.Sung P, Prakash S, Prakash L. Stable ester conjugate between the S. cerevisiae RAD6 protein and ubiquitin has no biological activity. J Mol Biol. 1991;221:745–749. doi: 10.1016/0022-2836(91)80169-u. [DOI] [PubMed] [Google Scholar]
- 66.Suzuki T, Varshavsky A. Degradation signals in the lysine-asparagine sequence space. EMBO J. 1999;18:6017–6026. doi: 10.1093/emboj/18.21.6017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Taban C H, Hondermarck H, Bradshaw R A, Boilly B. Effect of a dipeptide inhibiting ubiquitin-mediated protein degradation on nerve-dependent limb regeneration in the newt. Experientia. 1996;52:865–870. doi: 10.1007/BF01938871. [DOI] [PubMed] [Google Scholar]
- 68.Turner G C, Du F, Varshavsky A. Peptides accelerate their uptake by activating a ubiquitin-dependent proteolytic pathway. Nature. 2000;405:579–583. doi: 10.1038/35014629. [DOI] [PubMed] [Google Scholar]
- 69.Turner G C, Varshavsky A. Detecting and measuring cotranslational protein degradation in vivo. Science. 2000;289:2117–2120. doi: 10.1126/science.289.5487.2117. [DOI] [PubMed] [Google Scholar]
- 70.Tyers M, Jorgensen P. Proteolysis and the cell cycle: with this RING I do thee destroy. Curr Opin Genet Dev. 2000;10:54–64. doi: 10.1016/s0959-437x(99)00049-0. [DOI] [PubMed] [Google Scholar]
- 71.Varshavsky A. The N-end rule: functions, mysteries, uses. Proc Natl Acad Sci USA. 1996;93:12142–12149. doi: 10.1073/pnas.93.22.12142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Varshavsky A. Naming a targeting signal. Cell. 1991;64:13–15. doi: 10.1016/0092-8674(91)90202-a. [DOI] [PubMed] [Google Scholar]
- 73.Varshavsky A. Ubiquitin fusion technique and its descendants. Methods Enzymol. 2000;327:578–593. doi: 10.1016/s0076-6879(00)27303-5. [DOI] [PubMed] [Google Scholar]
- 74.Varshavsky A. The ubiquitin system. Trends Biochem Sci. 1997;22:383–387. doi: 10.1016/s0968-0004(97)01122-5. [DOI] [PubMed] [Google Scholar]
- 75.Voges D, Zwickl P, Baumeister W. The 26S proteasome: a molecular machine designed for controlled proteolysis. Annu Rev Biochem. 1999;68:1015–1068. doi: 10.1146/annurev.biochem.68.1.1015. [DOI] [PubMed] [Google Scholar]
- 76.Waizenegger I C, Hauf S, Meinke A, Peters J-M. Two distinct pathways remove mammalian cohesin from chromosome arms in prophase and from centromeres in anaphase. Cell. 2000;103:399–410. doi: 10.1016/s0092-8674(00)00132-x. [DOI] [PubMed] [Google Scholar]
- 77.Wakil S J. Fatty acid synthase, a proficient multifunctional enzyme. Biochemistry. 1989;28:4523–4530. doi: 10.1021/bi00437a001. [DOI] [PubMed] [Google Scholar]
- 78.Weissman A M. Themes and variations on ubiquitylation. Nat Rev Mol Cell Biol. 2001;2:169–178. doi: 10.1038/35056563. [DOI] [PubMed] [Google Scholar]
- 79.Xie Y, Varshavsky A. The E2-E3 interaction in the N-end rule pathway: the RING-H2 finger of E3 is required for the synthesis of multiubiquitin chain. EMBO J. 1999;18:6832–6844. doi: 10.1093/emboj/18.23.6832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Xie Y, Varshavsky A. Physical association of ubiquitin ligases and the 26S proteasome. Proc Natl Acad Sci USA. 2000;97:2497–2502. doi: 10.1073/pnas.060025497. [DOI] [PMC free article] [PubMed] [Google Scholar]