

Abstract
N-Demethylation of trialkylamines is a useful transformation, but typically requires harsh reaction conditions and stepwise procedures, as well as judicious protection of labile functional groups. Herein we report a mild, catalytic approach for the demethylation of trialkylamines by utilizing photoinduced nickel catalysis wherein C(sp2)–bromides serve as hydrogen-atom transfer (HAT) reagents. This method achieves direct demethylation of trialkylamines with wide functional group compatibility making it highly suitable for late-stage derivatization of complex molecules. Mechanistic investigations provide evidence that C(sp2) radicals generated via photoinduced Ni─C(sp2) bond homolysis are involved in hydrogen atom abstraction from trialkylamines. Utilizing steric control of the C(sp2)–bromides, our HAT approach achieves demethylation with excellent site selectivity in the presence of benzylsubstituted amines, which is complementary to the selectivity of classical approaches that afford debenzylation product instead.
Graphical Abstract
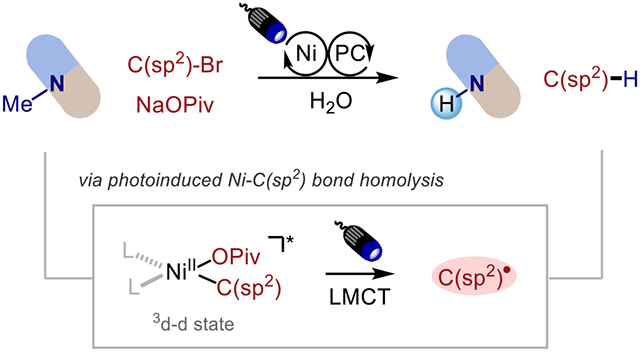
Trialkylamines are important motifs in alkaloid natural products and pharmaceutical compounds.1 The removal or replacement of N-methyl substituents is a useful transformation that has been employed in drug synthesis and in the development of tools for pharmacological and drug metabolic studies.2,3 The classical and frequently employed approaches for N-demethylation of tertiary amines include the von Braun Reaction, the use of chloroformate reagents (Scheme 1A), and the Polonovski reaction, which are two-step protocols involving a cyanamide, carbamate, or N-oxide intermediate respectively, and require harsh reaction conditions (high temperature, employment of toxic reagent such as cyanogen bromide, or a strong oxidant), with limited functional group tolerance and poor site-selectivity.4,5 Other approaches such as biochemical,6,7 photochemical,8-11 electrochemical,12,13 and transition-metal catalyzed14,15 methods have also been reported, albeit are restricted to specific structures and lack broad applicability.
Scheme 1.

Reaction Design
Over the past decade, the merger of photoredox and nickel catalysis has enabled novel disconnection approaches and new reaction pathways, especially broadening cross-coupling reactions.16 Previously, our group developed a nickel/photoredox dual catalytic platform for selective C(sp2)─C(sp3) N─Me arylation of trialkylamines under mild conditions, including in the context of late-stage functionalization of pharmaceutical compounds.17 In an effort to expand the scope of late-stage derivatization of trialkylamine scaffolds, we questioned whether it would be possible to employ C(sp2)–bromides as hydrogen-atom transfer (HAT) reagents instead of coupling partners. In recent years, photoelimination (photoinduced ligand dissociation) of a Ni─Cl or Ni─Br bond has been applied in C─H cross-coupling reactions,18,19 where C(sp2)–chlorides20-23 or C(sp2)–bromides24-26 serve as both coupling partners and the chlorine/bromine radical source for hydrogen atom abstraction. Inspired by Doyle’s27 and Hadt’s28 recent mechanistic investigations on photoinduced Ni─aryl bond homolysis, we envisioned the use of aryl and vinyl bromides as a C(sp2) radical source for hydrogen abstraction from the α-nitrogen position followed by an oxidation event with nickel to achieve N-demethylation of trialkylamines (Scheme 1B). This HAT approach for demethylation would allow complementary site selectivity compared to the classical chloroformate approach, where substrates such as benzyl amines are not tolerated since chloride attacks the most electrophilic benzylic position (Scheme 1A, bottom). In contrast to Ready’s recent functionalization of alkyl/benzyl amines via photoredox chemistry governed by differences in acidity and bond dissociation energy (BDE),29 in our catalytic system, the relative ambiphilicity of aryl radicals30,31 would allow abstraction of hydrogen from the most accessible methyl position instead, and the selectivity for demethylation could be further improved via steric control by tuning the sterics of C(sp2)-bromides.
We began our investigation of N-demethylation of trialkylamines with the introduction of water as a nucleophile to the photoredox/nickel catalytic system. Gratifyingly, after optimization we observed exclusive N-demethylation product 2, alongside reduced arene 3, the product that would form after aryl radical formation and HAT. Further screening revealed the optimized conditions to include 4-bromobenzonitrile Br1 (1.1 equiv), NiCl2(H2O)6 (5 mol%), organic photocatalyst 5CzBN (1 mol%),32,33 NaOPiv (1.0 equiv) as base, and H2O (36 μL, 10 equiv) in acetonitrile (0.1 M) under blue LEDs irradiation (427 nm), providing 2 in 66% yield (Table 1, entry 1). Using excess of aryl bromide (2 equiv) decreases the yield, with homocoupling of ArBr observed as side pathway (Table 1, entry 2). Changing aryl bromide into vinyl bromide Br2 affords comparable yield, generating propene as a low-molecular weight byproduct, thus increasing atom efficiency (Table 1, entry 3). Subsequent one-pot Boc protection was performed for facile isolation of the secondary amine, affording a 61% isolated yield of the product (Table 1, entry 3). Base was shown to be unnecessary but promotes the reaction (Table 1, entry 4). The reaction also works well with Ni(cod)2 in the absence of chloride sources (Table 1, entry 5), eliminating the necessity of Cl radical as HAT reagent in our system.18 Control studies showed that nickel, light, photocatalyst, and aryl bromide are all necessary, as their removal results in no desired product (Table 1, entries 6-9).
Table 1.
Optimization and Control Studiesa

| |||
|---|---|---|---|
| Entry | Deviation | Yield 2 (%)b |
Ar-H 3 (%)b |
| 1 | none | 66 | 92 |
| 2 | 2 equiv Br1 | 58 | 99 |
| 3 | Br2 instead of Br1 | 63 (61)c | / |
| 4 | no base | 27 | 48 |
| 5 | Ni(cod)2 instead of NiCl2(H2O)6 | 59 | 86 |
| 6 | no Ni/dtbbpy | 0 | 21 |
| 7 | in the dark | 0 | 0 |
| 8 | no photocatalyst | 0 | 2 |
| 9 | no aryl bromide | 0 | / |

| |||
Optimizations were performed on a 0.2 mmol scale.
GC-MS yield with 1,3,5-trimethoxybenzene as external standard.
Yield in bracket refers to isolated amine products after subsequent one-pot Boc protection with NEt3 (2 equiv) and Boc2O (1 equiv) followed by purification using flash chromatography.
After the development of N-demethylation conditions, we then examined the site selectivity of our method with benzylamines as substrates. The classical approach for demethylation has poor tolerance of substrates such as benzylsubstituted amines, affording benzyl chloride as the debenzylation product exclusively in 83% yield instead of the demethylation product when methyl chloroformate is employed (Table 2A).34 In contrast, our method provides demethylation of N,N-dimethylbenzylamine 4a as the major product, achieving selectivity of 4:1 to the debenzylation product (benzaldehyde) when using bromobenzonitrile Br1, or 2:1 when using the smaller vinyl bromide Br2 (Table 2B). The change in selectivity depending on size of the C(sp2)–bromide used inspired us to utilize steric control35 to further improve selectivity for N-demethylation. Gratifyingly, bulky TRIP-Br (2,4,6-triisopropylbromobenzene) Br3 furnishes the demethylated amine with 7:1 selectivity. Considering the slow oxidative addition with sterically-hindered aryl bromides, we premixed Br3 with Ni(0) source Ni(cod)2 prior to irradiation (with a noticeable color change to orange-brown and a characteristic peak at δ 9.33 (d, J = 6.0 Hz, 1H) in 1H NMR as evidence for NiBr(TRIP)(dtbbpy) formation),36 which increases the yield to 51%, and interestingly, also improves selectivity to >20:1. Next, we demonstrated the generality of our approach with a representative panel of benzylamines. In most cases the bulky TRIP-Br Br3 leads to better site selectivity of demethylation compared to the smaller vinyl bromide Br2. In general, electron-deficient benzylamines (7-9) show higher selectivity than electron-rich benzylamines (5, 6, and 10), which we postulate is due to more favorable abstraction of weaker and more hydridic benzylic C(sp3)─H bonds when benzylic positions are more electron-rich.18 In addition, benzylamine 13 possessing a cyclohexyl group also provides selective demethylation even in the presence of a weaker tertiary C(sp3)─H bond. Furthermore, Br3 combined with Ni(cod)2 affords excellent selectivity of >20:1 and good yield for all the examples, except benzylamines 11 and 12 bearing sterically-hindered benzylic positions which give higher or comparable yield with less sterically encumbered Br2, with no loss of enantiopurity at the benzylic center.
Table 2.

|
Reactions were run on a 0.2 mmol scale at ambient temperature in MeCN (0.1 M) using 5CzBN (1 mol%), Ni (5 mol%), dtbbpy (8 mol%), C(sp2)-Br (1.1-1.5 equiv), NaOPiv (1 equiv) and H2O (36 μL) under 8-30 h of blue LEDs irradiation (427 nm) until complete conversion of the amine substrate. Yields of secondary amines and ratios were determined by GC-MS with 1,3,5-trimethoxybenzene as standard. Yields in parentheses refer to isolated amine products after subsequent one-pot Boc protection with NEt3 and Boc2O.
Br2 and Ni(cod)2 was used.
Trace side products such as primary amine and imine were observed as a result of over-oxidation, along with unreacted substrate. Driving the reaction to completion also results in overoxidation by-products.
Having identified conditions that enable high selectivity for N-demethylation, we then sought to study the reaction mechanism. Direct excitation with 390 nm UV light affords the demethylation product without the participation of photocatalyst, which demonstrates that an oxidation/deprotonation pathway17 to the α-amino radical is not dominant (Scheme 2B). On the other hand, we noted that the reaction features a strong dependence on sodium pivalate as the base to achieve efficient demethylation. Indeed, a minor byproduct observed is C─O coupling between the aryl bromide and pivalate, suggesting that pivalate displacement of bromide is operative in the reaction. This led us to employ sodium adamantane-1-carboxylate instead of sodium pivalate to track the possible formation of a tertiary carbon-centered radical that could mediate HAT. However, no admantane is detected, rendering the possibility of carboxyl radical generation via Ni–carboxyl bond homolysis unlikely (otherwise adamantane should be observed as the rate of decarboxylation (k ≈ 109 s−1)37 is orders of magnitude faster than HAT (k ≈ 107M−1s−1)38). Furthermore, an aryl radical is trapped by N-methylpyrrole with nickel catalyst under 390 nm light, and control studies showed that light is necessary for the generation of aryl radical (Scheme 2C).
Scheme 2.

Mechanistic Investigations
To eliminate the potential involvement of bromine radicals generated via photoelimination of Ni(II)─Br bond as HAT reagents,24,25 we perfomed a control study without base using benzylamine 5, and achieved debenzylation instead of demethylation providing benzaldehyde 5b as the major product (Scheme 2D), which is a result of Br radical (BDE (H─Br) = 87.5 kcal/mol) abstracting the weak benzylic C─H bond (BDE = 83 kcal/mol). For comparison, in the presence of sodium pivalate, demethylation is provided as the major product with site-selectivity of 14:1. It is possible that pivalate displacement of a halide ligand on nickel is important for blocking halogen radical photoelimination pathways, instead liberating aryl radicals selectively, and that the involvement of aryl radical is responsible for the excellent site-selectivity of demethylation. Therefore, we propose a mechanistic pathway where an α-amino radical is generated upon hydrogen atom abstraction from the trialkylamine involving aryl radical (Scheme 2A), which is formed via photoelimination of Ni(II)─Ar bond of Ni(II)ArOPiv complex (Ni-3).27,28 Interception of the α-amino radical by Ni(I)-OPiv intermediate (Ni-4) affords a Ni(II) intermediate (Ni-5), which is in equilibrium with the iminium ion (Ni-6),17,39 providing N-demethylation product and formaldehyde upon hydrolysis.
Next, we investigated the role of photocatalyst under 427 nm visible light. Employment of photocatalysts that are less oxidizing than 5CzBN (*Ered = 1.31 V vs SCE; ET = 65.3 kcal/mol),32 including Ir(ppy)3 (*Ered = 0.31 V vs SCE; ET = 55.2 kcal/mol) and Ir(dFppy)3 (*Ered = 0.77 V vs SCE; ET = 63.5 kcal/mol) also improves the yield compared to using no photocatalysts under 427 nm light (Scheme 2E), which lends additional evidence to an energy transfer (EnT) pathway proceeding via triplet sensitization of Ni(II)ArOPiv to a 3d-d excited state (Ni-3*), instead of single electron transfer (SET), although we cannot rigorously exclude the latter.
The improved selectivity of demethylation vs. debenzylation observed with Ni(cod)2 compared to NiCl2 also captured our interest. Introducing chloride into the Ni(cod)2 system through the addition of exogenous tetrabutylammonium chloride (TBACl, 10 mol%) results in excellent selectivity of demethylation similar to the system with Ni(cod)2, rather than the NiCl2(H2O)6 system (Scheme 2F). We reasoned that an off-cycle pathway could be operative where Cl radical acts as a competitive HAT reagent via a Ni(I)/Ni(III) cycle when a Ni(II) source NiCl2(H2O)6 is employed (Scheme 2G). This pathway can be induced by the reduction of NiCl2 with the reduced state of the photocatalyst while trialkylamine serves as a sacrificial reductant, generating Ni(I)-Cl (Ni-7). Oxidative addition of ArBr leads to a Ni(III) intermediate (Ni-8), followed by photoelimination to afford Cl radical. In addition to selective abstraction of methyl C(sp3)─H bond enabled by the C(sp2) radical (Scheme 2A), the smaller and much more electrophilic Cl radical abstracts hydrogen from benzylic positions competitively, thus lowering the selectivity for demethylation when NiCl2(H2O)6 is employed. The high bond strength of HCl (BDE = 102 kcal/mol), in addition to the stoichiometry of abstractable α-amino C─H bonds (2 benzylic: 6 methyl) could also be consistent with the involvement of chlorine radicals in HAT.22
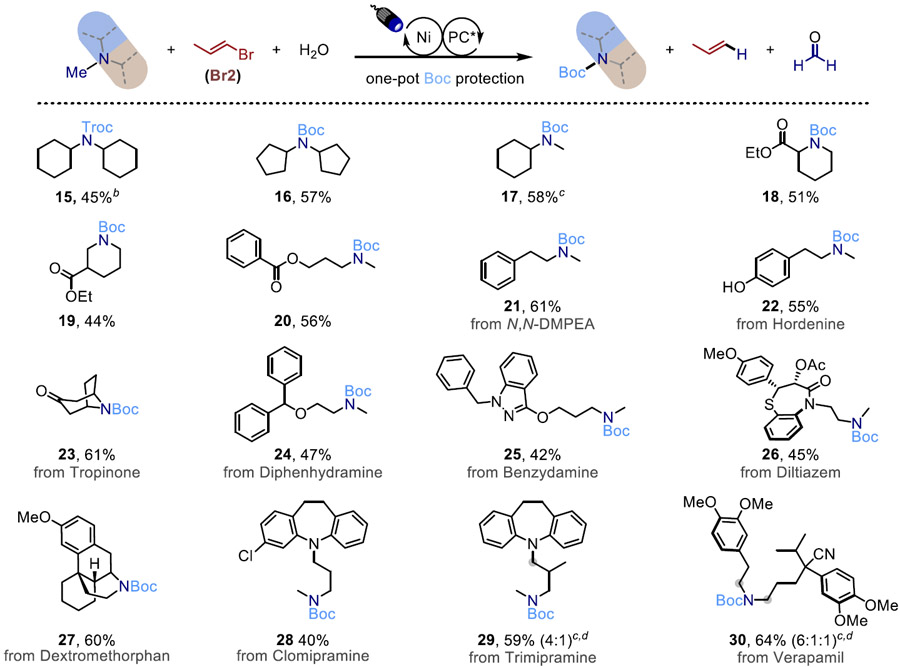
With rationalizations of the reactivity and selectivity in hand, we anticipated that our method would not only be applicable to derivitization of simple benzylamines but also to a broader scope of substrates. Vinyl bromide Br2 was employed for higher atom economy, and in the same pot the amine was Boc-protected at the conclusion of the reaction (Table 3). Trialkylamines bearing α-cyclohexyl and α-cyclopentyl substituent(s) provide selective demethylation (15-17). Cyclic amines such as piperidine decorated with an ethyl ester undergo the reaction in moderate to good yield (18-19), and benzoate is also well tolerated (20).
Table 3.
Scope of Selective N-Demethylation and Late-Stage Applicationsa
|
Reactions were performed with 0.2 mmol of substrate at ambient temperature in MeCN (0.1 M) using 1 mol% PC (5CzBN), NiCl2(H2O)6 (5 mol%), dtbbpy (8 mol%), Br2 (1.5 equiv), NaOPiv (1 equiv) and H2O (36 μL) under 12-30 h of blue LED light irradiation (427 nm), followed by adding NEt3 (2 equiv) and Boc2O (1 equiv). Yields refer to isolated products after purification by flash chromatography. Regioselectivities were determined by isolated yield.
Troc-Cl (1 equiv) was used in place of Boc2O.
TRIP-Br Br3 (1.1 equiv) and Ni(cod)2 was used in place of Br2 and NiCl2(H2O)6 for improved site selectivity.
The ratios in parentheses refer to site selectivity between the methyl position and position(s) highlighted in grey determined by crude 1H NMR.
Additionally, we performed late-stage functionalization on commercially available drugs and natural products containing trialkylamine scaffolds and other potential reactive sites (21-30), providing moderate to good yields with excellent selectivity.40 Functional groups such as alcohol (22) are tolerated with our method, while the traditional chloroformate approach requires extra protection and deprotection steps,41 enabling novel approaches to protecting group strategies in complex target synthesis. Furthermore, our approach opens up previously inaccessible secondary amine functionalities on biologically active scaffolds for various late-stage transformations such as N-arylation, amide coupling, or reductive amination, as well as provides new exit vectors for the synthesis of various novel therapeutic modalities such as protein degraders, drug-conjugates, and fluorescence labeling.42
In conclusion, we have developed a photoinduced Ni-catalyzed approach to the N-demethylation of trialkylamines using C(sp2)–bromides as hydrogen-atom transfer (HAT) reagents. The reaction displays excellent selectivity for N-methyl C(sp3)─H bonds, and mechanistic investigations indicate a C(sp2) (vinyl or aryl) radical, rather than a halogen radical, mediated HAT to provide the excellent selectivity observed. As such, our approach offers an opportunity to complement traditional protocols for demethylation under milder conditions and with better functional group compatibility, and we anticipate that this strategy can be used to rapidly diversify amine analogs in library synthesis.
Supplementary Material
ACKNOWLEDGMENT
We thank NIGMS for support (GM125206). Research reported in this publication was supported by the Office Of The Director, National Institutes Of Health of the National Institutes of Health under Award Number S10OD026749. We thank Fereshteh Zandkarimi for assistance in attaining HRMS data.
Footnotes
Supporting Information.
Reaction optimization, detailed experimental procedures and compound characterization. This material is available free of charge on the ACS Publications website.
The authors declare no competing financial interest.
REFERENCES
- (1).Blakemore DC; Castro L; Churcher I; Rees DC; Thomas AW; Wilson DM; Wood A Organic Synthesis Provides Opportunities to Transform Drug Discovery. Nat Chem. 2018, 10 (4), 383–394. [DOI] [PubMed] [Google Scholar]
- (2).Thavaneswaran S; McCamley K; Scammells PJ N-Demethylation of Alkaloids. Nat. Prod. Commun 2006, 1 (10), 1934578X0600101008. [Google Scholar]
- (3).Najmi AA; Bischoff R; Permentier HP N-Dealkylation of Amines. Molecules 2022, 27 (10), 3293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Cooley JH; Evain EJ Amine Dealkylations with Acyl Chlorides. Synthesis 1989, 1989 (1), 1–7. [Google Scholar]
- (5).Kapnang H; Charles G Reaction Des Chloroformiates Sur Les Amines Tertiaires: Competition Entre n-Demethylation, Desamination, n-Debenzylation et n-Desallylation. Tetrahedron Lett. 1983, 24 (31), 3233–3236. [Google Scholar]
- (6).Ren X; Yorke JA; Taylor E; Zhang T; Zhou W; Wong LL Drug Oxidation by Cytochrome P450BM3: Metabolite Synthesis and Discovering New P450 Reaction Types. Chem. – Eur. J 2015, 21 (42), 15039–15047. [DOI] [PubMed] [Google Scholar]
- (7).Augustin MM; Augustin JM; Brock JR; Kutchan TM Enzyme Morphinan N-Demethylase for More Sustainable Opiate Processing. Nat. Sustain 2019, 2 (6), 465–474. [Google Scholar]
- (8).Pandey G; Sudha Rani K; Bhalerao UT Photooxidative Set Initiated N-Demethylation of N,N’-Dimethylanlines: Mimicking the Cytochrome P-450 Type Oxygenations. Tetrahedron Lett. 1990, 31 (8), 1199–1202. [Google Scholar]
- (9).Chen Y; Glotz G; Cantillo D; Kappe CO Organophotocatalytic N-Demethylation of Oxycodone Using Molecular Oxygen. Chem. – Eur. J 2020, 26 (13), 2973–2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Firoozi S; Hosseini-Sarvari M Nanosized CdS as a Reusable Photocatalyst: The Study of Different Reaction Pathways between Tertiary Amines and Aryl Sulfonyl Chlorides through Visible-Light-Induced N-Dealkylation and C─H Activation Processes. J. Org. Chem 2021, 86 (3), 2117–2134. [DOI] [PubMed] [Google Scholar]
- (11).Zhao H; Leonori D Minimization of Back-Electron Transfer Enables the Elusive Sp3 C─H Functionalization of Secondary Anilines. Angew. Chem. Int. Ed 2021, 60 (14), 7669–7674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Portis LC; Bhat VV; Mann CK Electrochemical Dealkylation of Aliphatic Tertiary and Secondary Amines. J. Org. Chem 1970, 35 (7), 2175–2178. [Google Scholar]
- (13).Sommer F; Gerber Aeschbacher R; Thurnheer U; Kappe CO; Cantillo D Sustainable Synthesis of Noroxymorphone via a Key Electrochemical N-Demethylation Step. ACS Sustain. Chem. Eng 2022, 10 (27), 8988–8996. [Google Scholar]
- (14).Carroll RJ; Leisch H; Scocchera E; Hudlicky T; Cox DP Palladium-Catalyzed N-Demethylation/N-Acylation of Some Morphine and Tropane Alkaloids. Adv. Synth. Catal 2008, 350 (18), 2984–2992. [Google Scholar]
- (15).Murahashi S; Naota T; Yonemura K Ruthenium-Catalyzed Cytochrome P-450 Type Oxidation of Tertiary Amines with Alkyl Hydroperoxides. J. Am. Chem. Soc 1988, 110 (24), 8256–8258. [Google Scholar]
- (16).Chan AY; Perry IB; Bissonnette NB; Buksh BF; Edwards GA; Frye LI; Garry OL; Lavagnino MN; Li BX; Liang Y; Mao E; Millet A; Oakley JV; Reed NL; Sakai HA; Seath CP; MacMillan DWC Metallaphotoredox: The Merger of Photoredox and Transition Metal Catalysis. Chem. Rev 2022, 122 (2), 1485–1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Shen Y; Rovis T Late-Stage N-Me Selective Arylation of Trialkylamines Enabled by Ni/Photoredox Dual Catalysis. J. Am. Chem. Soc 2021, 143 (40), 16364–16369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Kariofillis SK; Doyle AG Synthetic and Mechanistic Implications of Chlorine Photoelimination in Nickel/Photoredox C(Sp3)─H Cross-Coupling. Acc. Chem. Res 2021, 54 (4), 988–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Cao H; Tang X; Tang H; Yuan Y; Wu J Photoinduced Intermolecular Hydrogen Atom Transfer Reactions in Organic Synthesis. Chem Catal. 2021, 1 (3), 523–598. [Google Scholar]
- (20).Shields BJ; Doyle AG Direct C(Sp3)─H Cross Coupling Enabled by Catalytic Generation of Chlorine Radicals. J. Am. Chem. Soc 2016, 138 (39), 12719–12722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Ackerman LKG; Martinez Alvarado JI; Doyle AG Direct C─C Bond Formation from Alkanes Using Ni-Photoredox Catalysis. J. Am. Chem. Soc 2018, 140 (43), 14059–14063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Kariofillis SK; Shields BJ; Tekle-Smith MA; Zacuto MJ; Doyle AG Nickel/Photoredox-Catalyzed Methylation of (Hetero)Aryl Chlorides Using Trimethyl Orthoformate as a Methyl Radical Source. J. Am. Chem. Soc 2020, 142 (16), 7683–7689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Maiti S; Roy S; Ghosh P; Kasera A; Maiti D Photo-Excited Nickel-Catalyzed Silyl-Radical-Mediated Direct Activation of Carbamoyl Chlorides To Access (Hetero)Aryl Carbamides. Angew. Chem. Int. Ed 2022, 61 (44), e202207472. [DOI] [PubMed] [Google Scholar]
- (24).Heitz DR; Tellis JC; Molander GA Photochemical Nickel-Catalyzed C─H Arylation: Synthetic Scope and Mechanistic Investigations. J. Am. Chem. Soc 2016, 138 (39), 12715–12718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Huang L; Rueping M Direct Cross-Coupling of Allylic C(sp3)─H Bonds with Aryl- and Vinylbromides by Combined Nickel and Visible-Light Catalysis. Angew. Chem. Int. Ed 2018, 57 (32), 10333–10337. [DOI] [PubMed] [Google Scholar]
- (26).Cheng X; Lu H; Lu Z Enantioselective Benzylic C─H Arylation via Photoredox and Nickel Dual Catalysis. Nat. Commun 2019, 10 (1), 3549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Ting SI; Garakyaraghi S; Taliaferro CM; Shields BJ; Scholes GD; Castellano FN; Doyle AG 3d-d Excited States of Ni(II) Complexes Relevant to Photoredox Catalysis: Spectroscopic Identification and Mechanistic Implications. J. Am. Chem. Soc 2020, 142 (12), 5800–5810. [DOI] [PubMed] [Google Scholar]
- (28).Cagan DA; Bím D; Silva B; Kazmierczak NP; McNicholas BJ; Hadt RG Elucidating the Mechanism of Excited-State Bond Homolysis in Nickel–Bipyridine Photoredox Catalysts. J. Am. Chem. Soc 2022, 144, 6516–6531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Leng L; Fu Y; Liu P; Ready JM Regioselective, Photocatalytic α-Functionalization of Amines. J. Am. Chem. Soc 2020, 142 (28), 11972–11977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Kvasovs N; Gevorgyan V Contemporary Methods for Generation of Aryl Radicals. Chem. Soc. Rev 2021, 50 (4), 2244–2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Parsaee F; Senarathna MC; Kannangara PB; Alexander SN; Arche PDE; Welin ER Radical Philicity and Its Role in Selective Organic Transformations. Nat. Rev. Chem 2021, 5 (7), 486–499. [DOI] [PubMed] [Google Scholar]
- (32).Speckmeier E; Fischer TG; Zeitler K A Toolbox Approach To Construct Broadly Applicable Metal-Free Catalysts for Photoredox Chemistry: Deliberate Tuning of Redox Potentials and Importance of Halogens in Donor–Acceptor Cyanoarenes. J. Am. Chem. Soc 2018, 140 (45), 15353–15365. [DOI] [PubMed] [Google Scholar]
- (33).Ou W; Zou R; Han M; Yu L; Su C Tailorable Carbazolyl Cyanobenzene-Based Photocatalysts for Visible Light-Induced Reduction of Aryl Halides. Chin. Chem. Lett 2020, 31 (7), 1899–1902. [Google Scholar]
- (34).Brine GA; Boldt KG; Hart CK; Carroll FI The N-Demethylation of Morphine and Codeine Using Methyl Chloroformate. Org. Prep. Proced. Int 1976, 8 (3), 103–106. [Google Scholar]
- (35).Czaplyski WL; Na CG; Alexanian EJ C─H Xanthylation: A Synthetic Platform for Alkane Functionalization. J. Am. Chem. Soc 2016, 138 (42), 13854–13857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Yakhvarov DG; Kvashennikova SV; Sinyashin OG Reactions of Activated Organonickel σ-Complexes with Elemental (White) Phosphorus. Russ. Chem. Bull 2013, 62 (11), 2472–2476. [Google Scholar]
- (37).Acetoxy radical decarboxylates with a rate of 2.2 x 109 s−1 at 70 °C. Tertiary carboxyls should be much faster. See: Giese B, in Houben-Weyl, Methoden der Organischen Chemie, Band E 19a, C-Radikale, ed. Regitz M and Giese B, Georg Thieme Verlag, Stuttgart, New York, 1989, p. 140. [Google Scholar]
- (38).Hu X-Q; Liu Z-K; Hou Y-X; Gao Y Single Electron Activation of Aryl Carboxylic Acids. iScience 2020, 23 (7), 101266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Sepelak DJ; Pierpont CG; Barefield EK; Budz JT; Poffenberger CA Organometallic Chemistry of the Carbon-Nitrogen Double Bond. 1. Nickel Complexes Prepared from Iminium Cations and the x-Ray Structure of {[(C6H5)3P]Ni[CH2N(CH3)2]Cl}. J. Am. Chem. Soc 1976, 98 (20), 6178–6185. [Google Scholar]
- (40).Trimipramine leads to demethylation (29) in 59% yield and 4:1 selectivity over dealkylation at the diarylamine fragment rather than the aliphatic portion of the dimethylamine. Subjection of diphenylme-thylamine to these reaction conditions leads to demethylation in 33% yield (see SI).
- (41).Olofson RA; Schnur RC; Bunes L; Pepe JP Selective N-Dealkylation of Tertiary Amines with Vinyl Chloroformate: An Improved Synthesis of Naloxone. Tetrahedron Lett. 1977, 18 (18), 1567–1570. [Google Scholar]
- (42).Müller CE; Hansen FK; Gütschow M; Lindsley CW; Liotta D New Drug Modalities in Medicinal Chemistry, Pharmacology, and Translational Science: Joint Virtual Special Issue by Journal of Medicinal Chemistry, ACS Medicinal Chemistry Letters, and ACS Pharmacology & Translational Science. ACS Med. Chem. Lett 2021, 12 (10), 1508–1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.