

Abstract
We have determined in mice the minimum composition required for forming a vaccine adjuvant that stimulates a Treg response to immunization, and named the adjuvant “complete tolerogenic adjuvant.” This new kind of adjuvant may let us use the well-proven “antigen with adjuvant” form of immunization for inducing Treg cell-mediated antigen-specific immunosuppression. The minimum composition consists of dexamethasone, rapamycin, and monophosphoryl lipid A at a mass ratio of 8:20:3. By dissecting the respective role of each of these components during immunization, we have further shown why immunosuppressive and immunogenic agents are both needed for forming true adjuvants for Treg cells. This finding may guide the design of additional, and potentially more potent, complete tolerogenic adjuvants with which we may form numerous novel vaccines for treating immune diseases.
Introduction
Autoimmunity, allergy, and graft rejection are all perpetuated by an adaptive immune response that is both antigen (“Ag”)-specific and already active. If we were able to induce Ag-specific immunosuppression against this kind of response, we might have a cure for all these diseases.
One approach to this goal is to raise endogenous immunosuppressive cells, particularly the CD4+CD25+Foxp3+ regulatory T (“Treg”) cells (1) specific for the Ag that is causing this kind of response (“recall Ag”). To that end, we have conceptualized a different kind of vaccine adjuvant called “tolerogenic adjuvant” that permits raising the Treg cells via immunization (2). Immunization is arguably the simplest way of raising T cells. But to raise the Treg cells selectively, the novel adjuvant is needed to prevent other T cells, many of which are pathogenic and already active during disease, from reacting to immunization.
Because of that need, tolerogenic adjuvants have been sought mostly from “biased” immunosuppressants that inhibit the conventional types of CD4+ T (“Teff”) cells while sparing Treg cells. The synthetic glucocorticoid dexamethasone was the first tolerogenic adjuvant (2), chosen for this bias (3, 4). In mouse models, immunization with a recall Ag and dexamethasone led to preferential expansion of recall Ag-specific Treg cells (over Teff cells) that correlated with suppression of established allo- and autoimmunity (2). The term “suppressed immunization” was coined for this form of immunization that combines Ag and immunosuppressant (2). Subsequently, various other immunosuppressants, including immunomodulators, were explored (5, 6). Collectively, these studies showed the generality of using biased immunosuppressants as tolerogenic adjuvants. Today, the combination of Ag and immunosuppressant is a common part of many forms of Ag-specific immunotherapy besides suppressed immunization.
Mechanistic understanding of how biased immunosuppressants work as tolerogenic adjuvants has come mostly from studies of dexamethasone (7). As an analog of the body’s naturally produced immunosuppressant, dexamethasone preferentially inhibits not only Teff cells, but also other cell types that are essential for a Teff response, in a pattern that shows coordination. In mice, dexamethasone kills Teff cells, B-2 cells, and dendritic cells (“DCs”) while sparing Treg cells, B-1 cells, and the CD11cloCD40lo monocyte-derived macrophages (“MMΦs”) (3, 4, 8, 9). Dexamethasone kills cells by engaging glucocorticoid receptor-α (“GR-α”), whereas cells can avoid being killed by expressing glucocorticoid receptor-β (“GR-β”), a dominant-negative isoform (10, 11). Therefore, we have proposed the GR isoform hypothesis that the pattern of bias seen among the aforementioned cell types is set intrinsically via the expression of the GR isoforms (9). Influenced by this pattern, Treg cells, B-1 cells, and MMΦs are enriched at the expense of Teff cells, B-2 cells, and DCs during suppressed immunization (8, 9). Among the enriched cells, MMΦs differentiate into tolerogenic Ag presenting cells that activate Treg cells but not memory Teff cells (9). These studies thus point to MMΦs as a mediator for tolerogenic adjuvants.
Despite these advances, there is a pressing need to improve tolerogenic adjuvants. Biased immunosuppressants are not true adjuvants by the conventional standard because they lack the ability to expand, via immunization, Ag-specific Treg cells to a greater absolute number than that via immunization with Ag alone. Rather, by relying on inhibiting Teff cells, they expand Treg cells only relatively (versus the Teff cells), which limits efficacy. In retrospect, preventing Teff cells from reacting to immunization was not the only condition a tolerogenic adjuvant must meet. To be more efficacious, the adjuvant should also have the ability to expand Treg cells in absolute numbers, thereby positively stimulating a Treg response. Not meeting this second condition, most of the biased immunosuppressants are actually “incomplete” adjuvants.
Guided by this notion, we have composed a “complete” adjuvant that meets both of the said conditions. Our study shows that a conventional (immunogenic) adjuvant that actively attracts MMΦs is required to complete the incomplete adjuvants.
Materials and Methods
Mice
Use of mice was approved by the Biologic Resource Committee of University of Illinois College of Medicine at Rockford. BALB/c Foxp3-eGFP and C57BL/6 Foxp3-eGFP mice were originally purchased from the Jackson Laboratory. BALB/c DO11.10 TCR-transgenic Foxp3-eGFP (“DO11.10 Foxp3-eGFP”) mice were generated in our facility by crossing BALB/c Foxp3-eGFP mice with BALB/c DO11.10 TCR-transgenic mice (from the Jackson Laboratory). All mice were used at age of 8 weeks unless indicated otherwise. Female and male mice were used in equal numbers in separate groups. Although sex difference was noted in the baseline counts of CD4+ T cells and CD11cloCD40lo monocyte-derived macrophages (“MMΦs”) in lymph nodes and the spleen, the difference was modest and did not change the responses of the mice to the adjuvants tested. Consequently, all the data reported were pooled from both female and male groups; and the word “mice” is used to indicate the inclusion of both sexes.
Reagents
Dexamethasone sodium phosphate was from ValleyVet.com. Rapamycin (dissolved in DMSO first) and FTY720 (fingolimod HCL; dissolved in DMSO first) were from Selleckchem.com. DQ ovalbumin, sodium heparin, ACK lysing buffer, Ficoll-Paque Plus, Freund’s complete adjuvant (“FCA”), and Freund’s incomplete adjuvant (“FIA”) were from ThermoFisher. Hen ovalbumin (“OVA”), hen egg lysozyme (“HEL”), and monophosphoryl lipid A (“MPLA”; dissolved in DMSO first) were from Sigma-Aldrich. Protease- and Ig-free BSA was from SouthernBiotech. Antibodies for flow cytometry were from Biolegend, eBioscience, and Jackson ImmunoResearch Laboratories. Cell counting beads for flow cytometry were from Bangs Laboratories. Mouse Th1/Th2/Th17 CBA kit was from BD Biosciences. Clodronate liposomes and control liposomes were a gift from Roche Diagnostics GmbH. Mouse CD4+ T cell isolation kit (negative selection), mouse CD4+CD25+ regulatory T cell isolation kit, and mouse CD3ε MicroBead kit were from Miltenyi Biotec. eFluor 670 was from eBioscience (dissolved in DMSO first). The peptide OVA323–339 was from AnaSpec.
Footpad injection
“A footpad” means the footpad of the right hind leg. Adjuvant components, Ags, or their combinations were injected subcutaneously (s.c.) between the 3rd and 4th toes in a volume of ~20 μl in PBS with a BD insulin syringe.
Dose-response analysis
For dose-response analysis of MPLA, the reagent was serially diluted in DMSO; 5 μl of the diluted reagent were mixed with 100 μl of PBS and the resulting mixture was injected at 20 μl/footpad. For dose 0, 5 μl of DMSO were mixed with 100 μl of PBS and the resulting mixture was injected. Dose-response analysis of rapamycin was performed similarly, except that PBS containing 3 μg of MPLA was used in place of PBS.
Preparation of single cell suspension from lymphoid organs
Lymph nodes or the spleen from studied mice were placed in a 1.5 ml microtube containing blood buffer (PBS/0.5% protease- and Ig-free BSA/2 mM EDTA); in the tube, the organs were cut with 4-inch fine scissors for 200 times and then homogenized gently with a microtube sample pestle. Released cells were filtered through an SP Bel-Art Flowmi 70 μM cell strainer.
Analysis of adjuvant-induced potentiation in lymphoid organs
A cell suspension was prepared from the draining lymph node (“DLN,” popliteal), non-draining lymph nodes (“NLNs,” contralateral brachial and axillary nodes), or the spleen, as described above. For counting MMΦs and dendritic cells (“DCs”), a portion of the cell suspension was stained with biotin anti-CD3, biotin anti-CD19, PE/Cy7 anti-CD11c, APC anti-CD40, and PE streptavidin in the presence of mIgG as a blocker. Cells were analyzed by flow cytometry. PE+ cells were gated out. The total counts of MMΦs (CD11loCD40lo) (9) and DCs (all the CD11+CD40+ cells after excluding the CD11loCD40lo cells) (9) in the cell suspension were determined by the method of Schlenke et al. (12). The MMΦ count in percent was calculated as % of the total MMΦ and DC counts combined.
For counting Treg and Teff cells, a portion of the cell suspension was stained with PE/Cy7 anti-CD3 and APC anti-CD4 and analyzed by flow cytometry. The total counts of Treg (CD3+CD4+Foxp3-eGFP+) and Teff (CD3+CD4+Foxp3-eGFP−) cells in the suspension were determined by the method of Schlenke et al. (12). The Treg count in percent was calculated as % of the total Treg and Teff counts combined.
Preparation of 8203
Composition 8203 was prepared in the amount of ten doses as follows: 27 μl of dexamethasone sodium phosphate (3 μg dexamethasone/μl) were added to 165 μl of PBS in a microtube. Next, 3 μl of rapamycin (60 μg/μl in DMSO) were added, and the tube was vortexed at the top speed for 13 seconds, which produced a cloudy solution. Lastly, 5 μl of MPLA (6 μg/μl in DMSO) were added and mixed. The final solution remained cloudy and was used at 20 μl/dose.
Mouse model for testing tolerogenic adjuvants (“the model”)
The model was generated as follows: On day 0, whole blood was collected with heparin as the anticoagulant from a DO11.10 Foxp3-eGFP mouse. The blood was diluted 1:50 in PBS and transfused intravenously (i.v.) into sex-matched syngeneic BALB/c Foxp3-eGFP mice at ~100 μl (containing ~4,000 CD4+KJ1-26+ cells) per mouse. On day 1, the recipients were injected at a footpad with 100 μg of OVA in 20 μl of PBS (see “Footpad injection”) and let rest. On day 5, they were used as the model.
To determine the origin of OVA-in-8203-expanded Treg cells, an alternative model was generated. Briefly, blood was collected by cardiac puncture from DO11.10 Foxp3-eGFP mice. Leukocytes were isolated via Ficoll-Paque density gradient centrifugation, from which CD4+ Teff cells were enriched untouched using the Miltenyi mouse CD4+CD25+ regulatory T cell isolation kit. The enriched cells were flow sorted twice with a FACS Melody sorter, excluding any Treg (Foxp3-eGFP+) cells. The purified Teff cells were transfused (i.v.) into BALB/c Foxp3-eGFP mice at 4,000 cells/mouse (day 0). The recipient mice were primed on day 1 and used on day 5, as described above.
Determination of CD4+ T cell counts in the model
A cell suspension was prepared from the DLN, NLNs, or the spleen as described earlier. For counting CD4+ T cells in the DLN, a portion of the DLN cell suspension was stained with PE/Cy7 anti-CD3 and APC anti-CD4 and analyzed by flow cytometry. The total CD4+ T cell count (CD3+CD4+) in the suspension was determined by the method of Schlenke et al. (12). Another portion was stained with biotin anti-F4/80, biotin anti-CD8, biotin anti-CD19, PE KJ1-26 (anti-DO11.10 TCR), APC anti-CD3, and PE/Cy7 streptavidin in the presence of mIgG as a blocker. Cells were analyzed by flow cytometry. The total counts in the suspension for KJ1-26+ Treg, KJ1-26+ Teff, host Treg, and host Teff cells were calculated as portions of the total CD4+ T cell count. The count in percent for Treg cells (“% Treg”) was calculated as % of the total Treg and Teff counts combined.
Cell suspensions prepared from NLNs and the spleen contained far more cells. To shorten the time needed for flow cytometry, irrelevant cells were removed from the suspension with the Miltenyi CD4+ T cell isolation kit after the total CD3+CD4+ cell count in the suspension was determined. The rest of the steps were the same as described before.
Analysis of CD4+ T cell proliferation in the model
A cell suspension was prepared from the DLN, NLNs, or the spleen as described earlier and stained with biotin anti-F4/80, biotin anti-CD8, biotin anti-CD19, PE/Cy7 anti-CD3, and APC KJ1-26 in the presence of mIgG as a blocker. Biotin+ cells were depleted with anti-biotin MACS beads (from the mouse CD4+ T cells isolation kit). The remaining cells were fixed in 4% paraformaldehyde, permeabilized in 0.5% Tween-20, stained with PE anti-Ki67 (clone SolA15 from eBioscience) in the presence of mIgG, and analyzed by flow cytometry. The count in percent for Ki67+ cells (“% Ki67+”) was calculated as % of Ki67+ and Ki67− cells combined.
Preparation of OVA-in-8203
OVA-in-8203 was prepared in the amount of ten doses by mixing 10 μl of OVA (100 μg/μl in PBS) with ten doses of 8203 (200 μl). The final solution was used at 21 μl/dose. As controls, ten doses of OVA-in-Dex were prepared by mixing 10 μl of OVA, 27 μl of dexamethasone sodium phosphate (3 μg dexamethasone/μl), and 173 μl of PBS; and ten doses of OVA-in-PBS by mixing 10 μl of OVA with 200 μl of PBS.
Clodronate liposome treatment
The model was injected (i.v.) with clodronate liposomes or plain liposomes (control) on day 4 (150 μl/mouse), day 5 (100 μl/mouse), and day 6 (100 μl/mouse) (defined in Mouse model for testing tolerogenic adjuvants). The mice were also injected at the primed footpad with OVA-in-8203 on day 5. MMΦs in the DLN were counted (see “Analysis of adjuvant-induced potentiation in lymphoid organs”) on days 6–8. KJ1-26+ Treg cells in the same DLN were counted (see “Analysis of CD4+ T cell counts in the model”) on day 8.
Inhibition of T cell egress with FTY720
FTY720 was dissolved in DMSO at 20 μg/μl. On day 5 in the model, 1 μl of FTY720 was mixed with one dose of OVA-in-8203, and the mixture was injected into the primed footpad. As a control, 1 μl of DMSO was used in place of FTY720, and the mixture was injected into a control group. On days 6 and 7, the mice were given additional injections (i.p.) of FTY720 (or DMSO) at 1 μl (diluted in 100 μl PBS)/mouse/day. On day 8, KJ1-26+ Treg and Teff cells in the DLN, NLNs, and the spleen were counted, as described earlier.
Analysis of Treg markers
A cell suspension was prepared from pooled DLNs from the model that had been given an optimal regime of OVA-in-8203. The cells were stained with biotin anti-F4/80, biotin anti-CD8, biotin anti-CD19, PE/Cy7 anti-CD3, and APC KJ1-26 in the presence of mIgG as a blocker. Biotin+ cells were depleted with anti-biotin MACS beads. The remaining cells were stained with one of the following PE-labeled mAbs: anti-CD25, anti-CTLA4, anti-PD1, anti-TNFR2, and anti-4-1BB. Some of the cells were fixed/permeabilized and intracellularly stained with PE anti-IL-10. The results were analyzed by flow cytometry. KJ1-26+ Treg cells (CD3+KJ1-26+Foxp3-eGFP+) were gated. The same analysis was performed on naïve KJ1-26+ Treg cells (from pooled popliteal lymph nodes of naïve DO11.10 Foxp3-eGFP mice) as a reference.
Suppression assay
Splenocytes were isolated from BALB/c mice, depleted of T cells with the anti-CD3ε MicroBead kit, and used as stimulators. Untouched Teff cells were isolated from pooled lymph nodes of naïve DO11.10 Foxp3-eGFP mice with the mouse CD4+CD25+ regulatory T cells isolation kit, stained with eFluor 670 (9), and used as responders. Naïve “source Treg cells” (“Treg1”) were isolated along with the Teff cells, from which the Foxp3-eGFP+ fraction was purified using a FACS Melody sorter. Expanded source Treg cells (“Treg2”) were isolated by the same method for Treg1, except that they were from pooled DLNs from mice receiving the optimal regimen of OVA-in-8203. The responders (5 × 104 cells/well) were cultured in RPMI 1640/10% FCS in multiple U-bottom wells with the stimulators (2 × 105 cells/well) and the OVA323–339 peptide Ag (0.1 μg/ml) for 3 days in the presence of Treg1 (5 × 104 cells/well), Treg2 (5 × 104 cells/well), or neither. The cultured cells were analyzed by flow cytometry, gating on the responders (CD4+KJ1-26+ Foxp3-eGFP−). Proliferation of the responders was measured by the count in percent for eFLuor 670lo Teff cells (“% eFLuor 670lo Teff”), calculated as % of total eFLuor 670+ Teff cells.
Analysis of Ag-specific immune suppression
The model was given the optimal regimen of OVA-in-8203 or a similar regimen of OVA-in-PBS (control). Four months later, both groups were challenged by an injection (s.c.) of 100 μg of OVA/100 μg of HEL in FCA, followed by a boost injection (s.c.) three weeks later of the same dose of Ags in FIA. KJ1-26+ Treg and Teff cells in the spleen were counted immediately before the start of the challenge, and five days after the end of the challenge. Delayed-type hypersensitivity (“DTH”) to HEL was analyzed three weeks after the end of the challenge by injecting the left footpad with 20 μl of PBS (control) and the right footpad with 20 μl of PBS containing 20 μg of heated HEL, which was prepared by heating HEL (1 mg/ml in PBS) at 99°C for 20 min (in a PCR thermal cycler). Footpad swelling was measured at 24 h with a Mitutoyo digital micrometer. Swelling = thickness of the right footpad – that of the left footpad. DTH to OVA was similarly analyzed after another three weeks with OVA at 20 μg/footpad. KJ1-26+ Teff cells in the DLN were counted five days after the OVA DTH test.
Statistics
Unpaired t test was used for comparisons between two independent groups. When multiple t tests were performed in parallel, the resulting p-values were adjusted by Bonferroni correction. One-way ANOVA was used for comparisons involving three or more independent groups, with post hoc Tukey’s honest significant difference test for multiple-to-multiple comparisons, or with Bonferroni correction or Holm’s method for multiple-to-one comparisons. Statistical significance was set at p ≤ 0.05. The GraphPad Prism software was used for curve fitting.
Results
Higher GRβ:GRα ratio attributes to lower sensitivity to dexamethasone
We began by testing the GR isoform hypothesis in order to understand why dexamethasone has limited adjuvanticity. Our studies, and the studies by others, had shown that Treg cells, B-1 cells, and MMΦs all have a lower sensitivity to dexamethasone than their respective immunogenic counterparts, i.e., the Teff cells, B-2 cells, and DCs (3, 4, 8, 9). We therefore asked whether a similar pattern could be seen in their GR isoform ratio. This was indeed the case: Treg cells, B-1 cells, and MMΦs all had 3−6-fold higher GR-β/GR-α ratios in mRNA than their counterparts (Fig. 1A). In B-1 cells, knockdown of GR-α and GR-β led to decrease and increase in the cells’ sensitivity to dexamethasone, respectively (Fig. 1B). Of note, we could not perform the same study in the other cell types because they could not, in our hand, survive the knockdown procedure. Collectively, these results supported the GR isoform hypothesis. It was therefore understood that dexamethasone acts essentially as a differential inhibitor for the T, B, and Ag presenting cell types. As a result, it does not stimulate any of the cell types actively.
FIGURE 1.
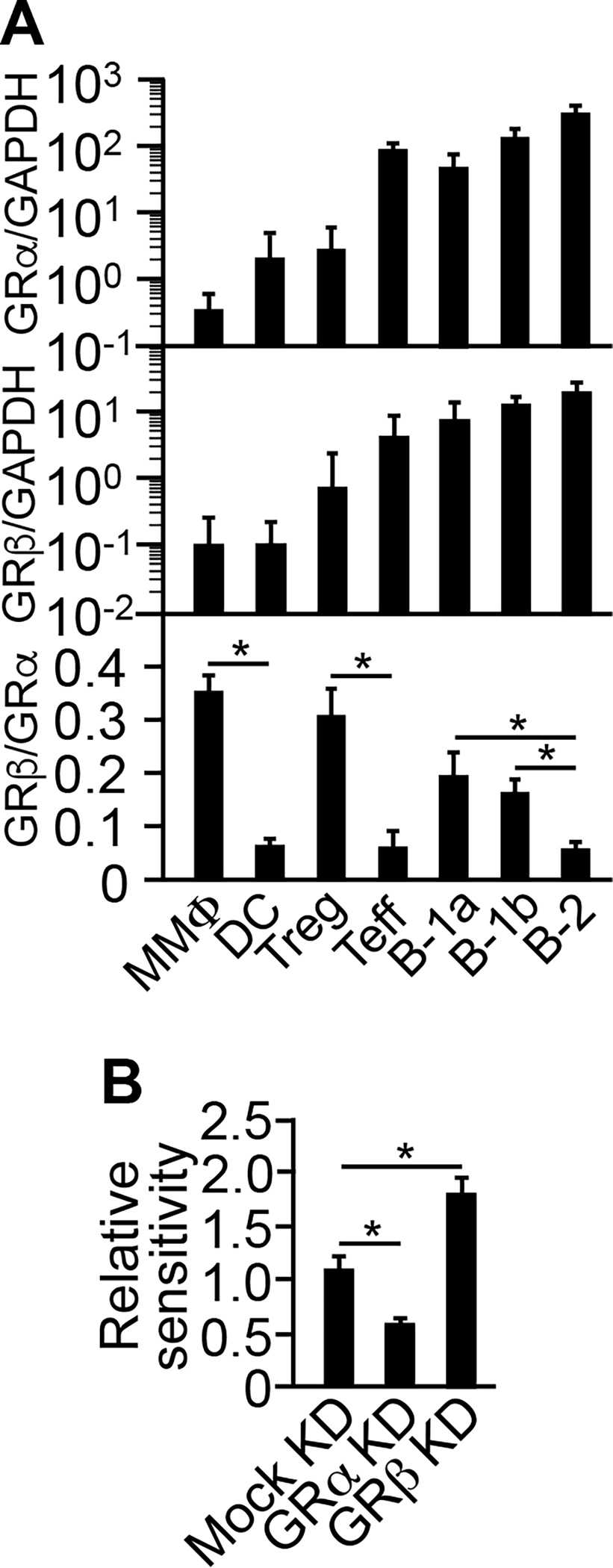
Higher GRβ:GRα ratio attributes to lower sensitivity to Dex. (A) Treg cells, B-1 cells, and MMΦs all have a higher GR-β/GR-α ratio in mRNA than their immunogenic counterparts. CD3+CD4+CD25+Foxp3-eGFP+ Treg cells, CD3+CD4+CD25− Foxp3-eGFP− Teff cells, CD5+CD19+CD23−FSClarge B-1a cells, CD5−CD19+CD23−FSClarge B-1b cells, CD19+CD23+ FSCsmall B-2 cells, CD11cloCD40lo MMΦs, and CD11chi DCs were flow sorted from BALB/c Foxp3-eGFP mice. Total RNA was isolated and reverse-transcribed, and qRT-PCR was performed using the GRα- or GRβ-specific primer pair and GAPDH as an internal standard (10). Data are mean ± SD of five experiments. *p ≤ 0.001 (one-way ANOVA). (B) Knockdown (“KD”) of GRα decreases, whereas KD of GRβ increases, Dex-induced depletion of B-1 cells in vivo. B-1 cells were treated ex vivo with siRNAs (10). KD efficiencies were within 58–73% (not shown). The treated cells were adoptively transferred into the peritoneal cavity, followed by an intramuscular injection of dexamethasone (8). Cell depletion from the peritoneal cavity was measured as relative to that of untreated B-1 cells (“Relative sensitivity”). Data are mean ± SD of three experiments. *p ≤ 0.003 (one-way ANOVA).
Composition 8203 potentiates draining lymph node (DLN) for Treg response
Because no other biased immunosuppressants were known to have the stimulatory capacity either (5), it would be difficult to replace dexamethasone. Therefore, we sought to complement dexamethasone with a biased immune stimulator in order to improve its adjuvanticity. From many candidates (cytokines, costimulators, proinflammatory mediators, and small molecule agonists), we found an unexpected combination that consists of monophosphoryl lipid A (“MPLA”), a lipopolysaccharide-derived conventional adjuvant that activates TLR4 (13); and rapamycin (sirolimus), a macrolide immunosuppressant that inhibits mTOR (14). This pair stood out largely because of their unique interaction that attracted MMΦs preferentially. When MPLA was injected alone into a footpad of a BALB/c mouse, it induced an influx of MMΦs and Teff cells preferentially over that of DCs and Treg cells, respectively, to the draining lymph node (“DLN,” popliteal) (Fig. 2, A&B). However, if MPLA was co-injected with rapamycin, the Teff cell influx was blocked, resulting in more selective accumulation of MMΦs (Fig. 2B). In BALB/c mice, the combination of 3 μg MPLA and 20 μg rapamycin was sufficient to produce the MMΦ-accumulating effect (Fig. 2B).
FIGURE 2.
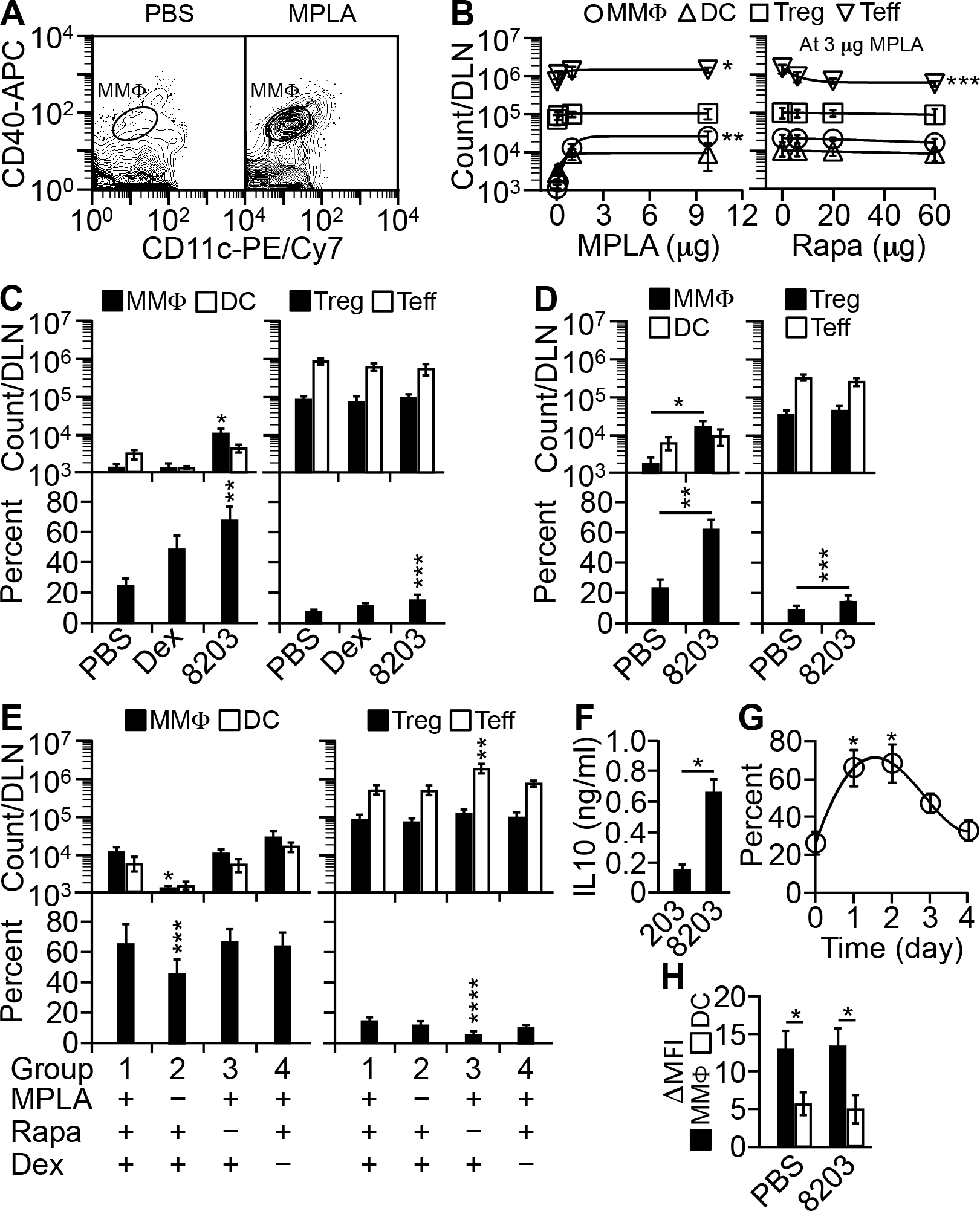
Composition 8203 potentiates DLN for Treg response. (A) MPLA preferentially attracts MMΦs to DLN. Shown is flow cytometry of DLN CD11c+CD40+ cells one day after a single injection of 10 μg of MPLA, or a PBS control, into a footpad. (B) Rapamycin (“Rapa”) blocks MPLA-induced influx of Teff cells to DLN. Shown are dose-response curves generated by nonlinear regression. Left: absolute cell counts for MMΦs, DCs, Treg cells, and Teff cells one day after a single injection into a footpad of a graded dose of MPLA. Right: the same set of cells after injection of 3 μg (based on the left panel) of MPLA mixed with a graded dose of Rapa. Each dose point is mean ± SD of four experiments. *p ≤ 0.03 and **p ≤ 0.01, for Teff and MMΦ counts, respectively, at MPLA doses of ≥1 μg versus 0; ***p ≤ 0.03, Teff count at Rapa doses of ≥6 μg versus 0 (one-way ANOVA). (C) Composition 8203 preferentially attracts MMΦs and Treg cells to DLN. DLN cells were counted one day after a single injection into a footpad of PBS, Dex (8 μg), or 8203. Data are mean ± SD of four experiments. *p ≤ 0.001 and **p ≤ 0.02, for absolute and relative MMΦ counts, respectively, of 8203 injected versus any other; ***p ≤ 0.01, relative Treg count of 8203 injected versus PBS injected (one-way ANOVA). (D) Composition 8203 shows similar effects in C57BL/6 Foxp3-eGFP mice. DLN cells were counted one day after a single injection into a footpad of PBS or 8203. Data are mean ± SD of four experiments. *p = 0.005, **p = 0.00009, and ***p = 0.04 (two-sided t test). (E) Role of each component of 8203. DLN cells were counted one day after a single injection into a footpad of 8203 or a composition missing one of the three components. Data are mean ± SD of four experiments. *p ≤ 0.02 and ***p ≤ 0.03, for absolute and relative MMΦ counts, respectively, in group 2 versus any other group; **p ≤ 0.0002 and ****p ≤ 0.0009, absolute Teff count and relative Treg count, respectively, in group 3 versus any other group (one-way ANOVA). (F) Dexamethasone is required for converting MMΦs (IL-10lo) into Dex-MΦs (IL-10hi) (9). MMΦs were isolated from the DLN by flow sorting one day after injection at a footpad with either 8203 or “203” (20 μg rapamycin/3 μg MPLA, as a negative control for dexamethasone), and cultured at 250,000 cells/ml in U-bottom wells in the presence of 0.1 μg/ml of LPS. At 20 h, the medium was analyzed for IL-10 by a CBA assay. Data are mean ± SD of four experiments. *p ≤ 0.00002 (two-sided t test). (G) Effect of 8203 on MMΦ influx lasts for three days after a single injection. Shown is a nonlinearly fitted decay curve for the relative count of MMΦs (% of total CD11c+CD40+ cells) in the DLN. Each point is mean ± SD of four experiments. *p ≤ 0.02, for days 1 and 2 versus any other day (one-way ANOVA). (H) Composition 8203 allows normal uptake and processing of whole protein Ag. Mice were injected at a footpad with 5 μg of DQ OVA in either PBS or 8203; after 2 days, MMΦs and DCs in the DLN were analyzed for mean fluorescence intensity (MFI) resulted from intracellular cleavage of DQ OVA. ΔMFI = MFI from DQ OVA injected − MFI from PBS (control) injected. Data are mean ± SD of four experiments. *p ≤ 0.004 (two-sided t test).
MMΦs are precursor cells that differentiate into Treg cell-stimulating, IL-10+ tolerogenic Ag presenting cells called dexamethasone-enriched macrophages (“Dex-MΦs”) (9). The phenotype and function of both MMΦs and Dex-MΦs have been described in detail (9). We therefore asked whether the dose above may work well with dexamethasone in producing a large number of Dex-MΦs. Combining the dose with 8 μg dexamethasone, an effective dose used previously for suppressed immunization (2), we had in hand a new composition called “8203” (8 μg dexamethasone:20 μg rapamycin:3 μg MPLA). When injected into a footpad of a BALB/c mouse, 8203 increased the abundance of MMΦs in the DLN among total Ag presenting cells by increasing the absolute number of MMΦs preferentially over that of DCs (Fig. 2C). It also increased the abundance of Treg cells in the DLN among total CD4+ T cells (albeit less drastically) by increasing the absolute number of Treg cells and decreasing that of Teff cells (Fig. 2C). Lymphoid organs not connected directly from the injection site, such as the contralateral brachial and axillary lymph nodes (“NLNs”) and the spleen, were less affected (data not shown). In comparison, injection of dexamethasone alone was less effective compared to 8203 because dexamethasone did not increase the absolute number of MMΦs or Treg cells in the DLN (Fig. 2C). We therefore concluded that 8203 is a stronger potentiating agent than is dexamethasone.
It was possible that the effect of 8203 is peculiar to the BALB/c strain. We thus tested 8203 in C57BL/6 mice as well. The possibility was excluded because 8203 showed a similar potentiating effect in the latter mice (Fig. 2D).
We also determined the respective role of each component within 8203. MPLA was required for attracting MMΦs (Fig. 2E). Although dexamethasone could enrich MMΦs relatively by killing DCs preferentially (9), as confirmed in Fig. 2C, this effect was masked by the much stronger effect of MPLA on the MMΦ influx (Fig. 2E). Rather, dexamethasone was required for converting MMΦs into Dex-MΦs (9), as confirmed in Fig. 2F. Rapamycin was required for blocking the influx of Teff cells (Fig. 2E).
Kinetically, the effect of 8203 on MMΦs after a single injection peaked in 2 days in the DLN and was gone after 4 days (Fig. 2G). At the peak time, the MMΦs picked up and proteolytically processed DQ ovalbumin (“DQ OVA”), a model whole-protein Ag, in the DLN as actively as those in mock (PBS) injected mice (Fig. 2H).
In summary, these results show that 8203 has a stronger potentiating effect on the Treg response than does dexamethasone. This effect is local and transient and does not interfere with Ag uptake or processing. The latter property further indicates that 8203 is compatible with whole-protein Ags, the preferred form of immunogen for overcoming the HLA diversity in humans.
Model for testing tolerogenic adjuvants
To determine whether 8203’s potentiating effect actually helps stimulate a Treg response to immunization, we constructed a mouse model as follows. BALB/c Foxp3-eGFP mice were transfused with a small number of hen ovalbumin (“OVA”)-specific TCR-transgenic CD4+ T cells bearing the KJ1-26+ marker (~4,000 cells from DO11.10 Foxp3-eGFP mice). By a previous estimate, ~6% of the donor T cells (240 cells) entered the lymph nodes in the recipients (15). This number was small enough to be physiological (16), yet big enough for tracking the cells (data not shown).
As a potential tolerogenic adjuvant, 8203 must be able to work against already active Teff cells. To test that, we activated the donor T cells (KJ1-26+) in the recipients by injecting 100 μg of OVA into a footpad. While having a far limited effect in NLNs and the spleen, the injection expanded KJ1-26+ Teff cells >100-fold in the DLN five days after transfusion (Fig. 3, A&B). Few KJ1-26+ Teff or Treg cells were found in other sites such as blood and bone marrow (data not shown); therefore, these sites were not further studied. In the DLN, nearly all the KJ1-26+ Teff cells (Foxp3-) were activated by day 5, with ~94% and ~89% of the cells expressing the memory-stage marker CD44hi (Fig. 3C) and proliferation marker Ki67 (Fig. 3D), respectively. In contrast, most host Teff cells (KJ1-26−Foxp3−) remained inactive, as assessed by the same markers (Fig. 3, C&D). These results indicated that the recipients were ready to be used on day 5 as “the model.”
FIGURE 3.
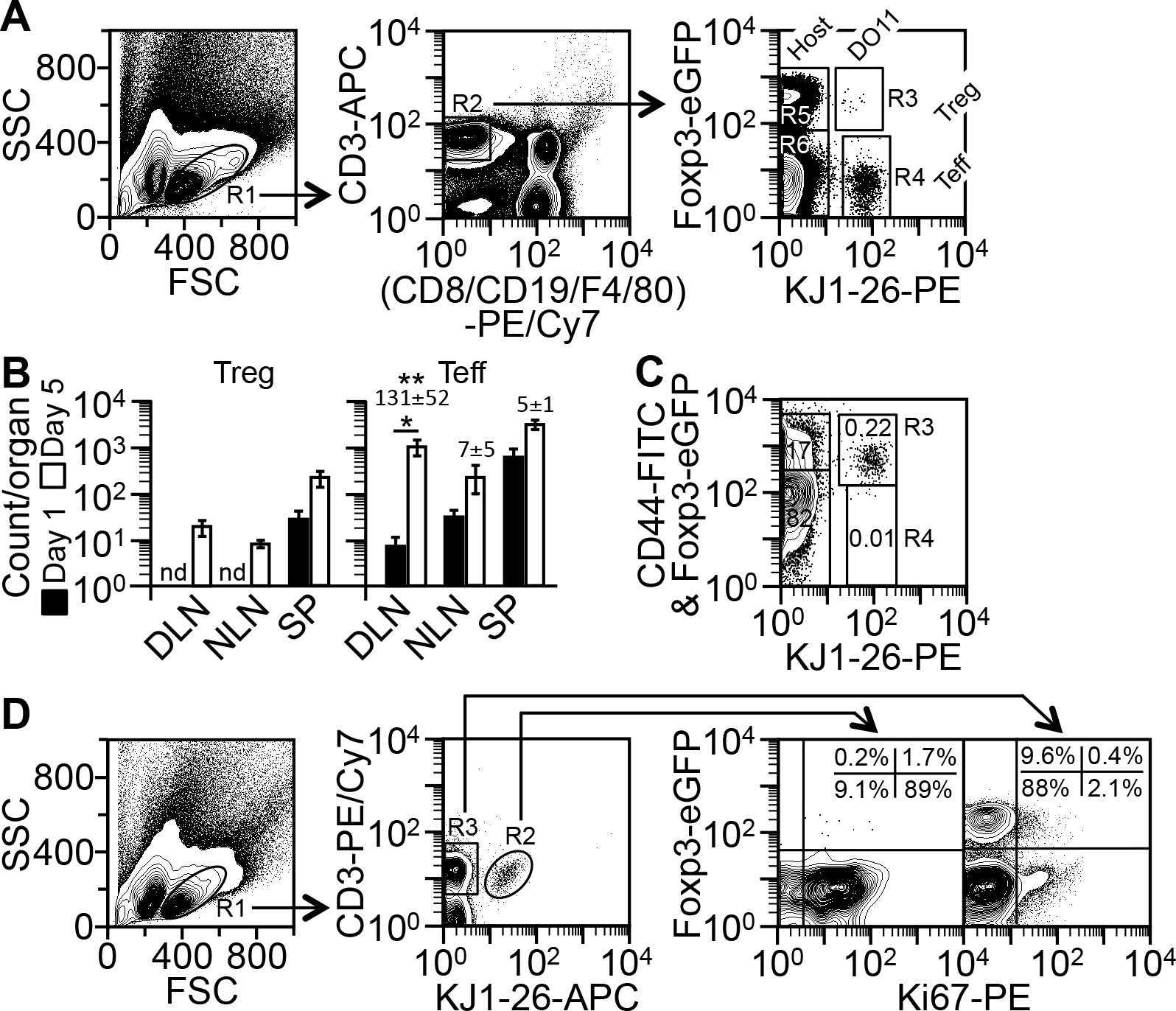
Model for testing tolerogenic adjuvants. (A) Method for detecting TCR-transgenic (“DO11”) Treg (R1*R2*R3), TCR-transgenic Teff (R1*R2*R4), host Treg (R1*R2*R5), and host Teff (R1*R2*R6) cells in the model (see “Analysis of CD4+ T cell counts in the model”). Depicted are the cells from the DLN on day 5. (B) Counts of KJ1-26+ Treg and Teff cells in DLN, non-draining lymph nodes (“NLN”), and the spleen (“SP”) on day 5 versus day 1, as determined by the method in A. Data are mean ± SD of four experiments. Numbers on the top of the bars indicate the fold change in Teff counts since day 1. *p = 0.004 (two-sided t test); **p ≤ 0.0002, for the fold change in DLN versus NLN and SP (one-way ANOVA); nd, not determined due to low counts. (C) Method for CD44 analysis. Cells shown in A were additionally stained with FITC anti-CD44 and analyzed by the method in A. Numbers in the gates indicate percentages of the total CD4+ T cells. The R3 gate contains both KJ1-26+CD44+ Teff cells (FITC+; 0.22%) and KJ1-26+ Treg cells (Foxp3-eGFP+; 0.004%), as determined with an isotype-matched FITC control (not shown). Shown is one of two similar results. (D) Method for detecting Ki67+ Treg and Teff cells (see “Analysis of CD4+ T cell proliferation in the model”). Depicted are the cells from the DLN on day 5. Quadrant plots on the right are drawn based on the locations of the Ki67− subpopulation. Shown is one of two similar results.
Composition 8203 is a complete tolerogenic adjuvant for Treg cells
Because in the model KJ1-26+ Teff cells were activated in the DLN, an injection at the primed footpad was the most direct route to the cells. Indeed, a single footpad injection of a mixed solution containing 8203 and OVA (“OVA-in-8203”) was sufficient to raise the absolute count of KJ1-26+ Treg cells in the DLN beyond that by an injection of the Ag alone (“OVA-in-PBS”) (Fig. 4A). Such effect was not seen if dexamethasone was used in place of 8203 (“OVA-in-Dex”) (Fig. 4A). Furthermore, the OVA-in-8203-expanded Treg cells also contained a larger fraction of proliferating cells identified by the proliferation marker Ki67 (Fig. 4A). Thus, 8203 augmented absolute expansion of the Treg cells by increasing their proliferation. It was also noted that 8203 did so even when KJ1-26+ Treg (and Teff) cells underwent spontaneous reduction in proliferation (Fig. 4A, “Start” versus “OVA-in-PBS”), which further revealed the stimulating effect of this adjuvant on Treg cell proliferation. This effect was OVA-specific because host Treg cells, which were mostly non-OVA-specific, were not significantly expanded and remained mostly Ki67− (Fig. 4B). This effect was also OVA-dependent (Fig. 4C, group 6 versus group 2), which confirmed that 8203 acted only as an adjuvant. Thus, by all accounts 8203 acted as a “true” Treg-stimulating adjuvant.
FIGURE 4.
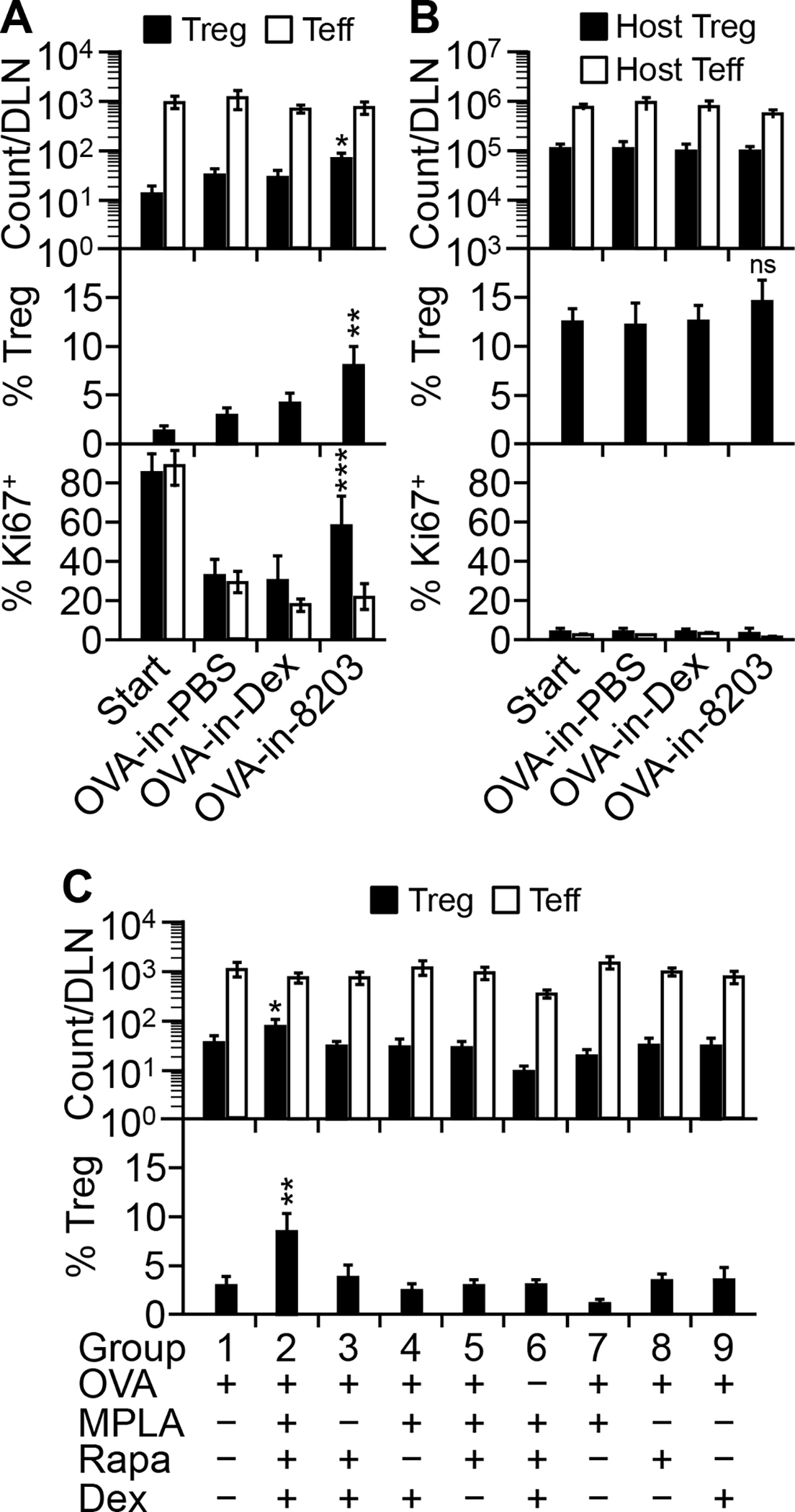
Composition 8203 is a complete tolerogenic adjuvant for Treg cells. (A) Composition 8203 augments absolute expansion of KJ1-26+ Treg cells in DLN. The model was injected on day 5 at the primed footpad with OVA-in-PBS, OVA-in-Dex, or OVA-in-8203. DLN T cells were analyzed on day 8. The baseline of the model (on day 5) is shown as “Start.” Data are mean ± SD of six (for cell count analysis) or four (for Ki67 analysis) experiments. *p ≤ 0.001 and **p ≤ 0.001, for absolute and relative Treg counts, respectively, in the OVA-in-8203 injected versus any other group (one-way ANOVA). ***p ≤ 0.04, for % Ki67+ Treg cells in the OVA-in-8203 injected versus any other group (one-way ANOVA). (B) Composition 8203 has little effect on host Treg or Teff cells in the same DLN shown in A. Data are mean ± SD of six (count analysis) or four (Ki67 analysis) experiments. “ns,” not significant. (C) All the OVA-in-8203 components are required for absolute KJ1-26+ Treg cell expansion. The model was injected with indicated compositions and analyzed as in A. Data are mean ± SD of four experiments. *p ≤ 0.01 and **p ≤ 0.001, for absolute and relative counts, respectively, of KJ1-26+ Treg cells in the OVA-in-8203 treated (group 2) versus any other group (one-way ANOVA).
Within 8203, all three components were required for stimulating the absolute expansion of KJ1-26+ Treg cells (Fig. 4C). All three were also required for the best relative expansion of the Treg cells within total KJ1-26+ T cells (Fig. 4C). In aggregate, these results define 8203 as the minimum composition for a true adjuvant for Treg cells. We therefore call 8203 “complete tolerogenic adjuvant” to distinguish it from incomplete adjuvants such as dexamethasone.
Composition 8203 augments absolute expansion of Treg cells via MMΦs
The dependence of 8203 on MPLA (Fig. 4C) presumably reflects the involvement of MMΦs. To confirm that, we reduced the number of MMΦs in the DLN. Intravenous injection of clodronate liposome is known to prevent influx of monocytes or macrophages into lymph nodes by selectively depleting the cells from the blood circulation (17). Thus, we repeated the experiment in Fig. 4A while giving the model clodronate liposome. As shown in Fig. 5A, the clodronate liposome treatment (CL) reduced the influx of MMΦs in the DLN by ~73% and kept the MMΦ count low during the period of Treg cell expansion (days 6–8). As a result, the adjuvant effect of 8203 in the OVA-in-8203 injected (compared to the OVA-in-PBS injected) mice was completely abolished (Fig. 5B). Thus, MMΦs are an essential cell mediator for the effect of 8203.
FIGURE 5.
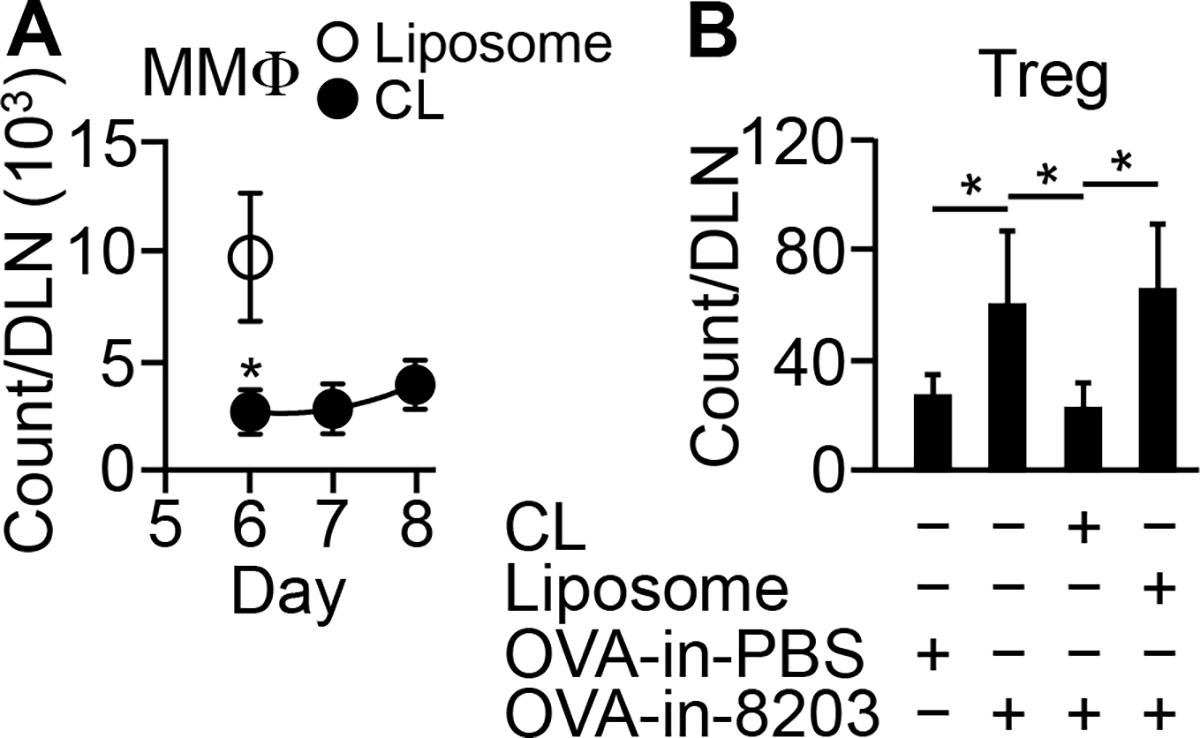
Composition 8203 augments absolute expansion of Treg cells via MMΦs. (A) Clodronate liposomes (“CL”) reduce OVA-in-8203-induced influx of MMΦs into DLN. CL was injected i.v. on days 4–6, and OVA-in-8203 was injected at the primed footpad on day 5. MMΦs in the DLN were counted on the indicated days. Data are mean ± SD of four experiments. *p ≤ 0.002 versus plain liposomes (“Liposome”) (one-way ANOVA). (B) CL diminishes the effect of 8203 on absolute expansion of Treg cells in DLN (analyzed on day 8). Mice in A were analyzed for KJ1-26+ Treg cells in the same DLN on day 8. Data are mean ± SD of six experiments. *p ≤ 0.02 (one-way ANOVA).
Composition 8203 promotes systemic dissemination of expanded Treg cells
Composition 8203 also outperformed dexamethasone in augmenting the absolute KJ1-26+ Treg cell expansion in distant lymphoid organs (not directly draining the injection site) such as the NLNs and the spleen (Fig. 6A). But it did so mainly by dissemination because the KJ1-26+ Treg cells in the NLNs and spleen, unlike their counterpart in the DLN, did not have a large fraction of proliferating (i.e., Ki67+) cells (Fig. 6B). Moreover, the gain of KJ1-26+ Treg (but not Teff) counts there could be blocked with FTY720 (Fig. 6C), an inhibitor for T cell egress from lymph nodes (18). In fact, the gain in KJ1-26+ Treg counts in the distant organs correlated with a loss of the same cells in the DLN (Fig. 6C). It was estimated that ~55% and ~71% of the KJ1-26+ Treg cells in NLNs and the spleen, respectively, came from other lymphoid organs (Fig. 6D); whereas <4% of the KJ1-26+ Teff cells came that way (Fig. 6D). The lack of KJ1-26+ Teff cell ingress in these organs correlated with the lack of KJ1-26+ Teff cell expansion in the DLN (Fig. 4A). The latter might cause the former by reducing the number of cells available for egress. Collectively, these results suggest that the cells’ systemic dissemination depends on their local expansion; and that 8203 promotes the systemic dissemination of Ag-specific Treg cells by promoting their expansion in the DLN.
FIGURE 6.
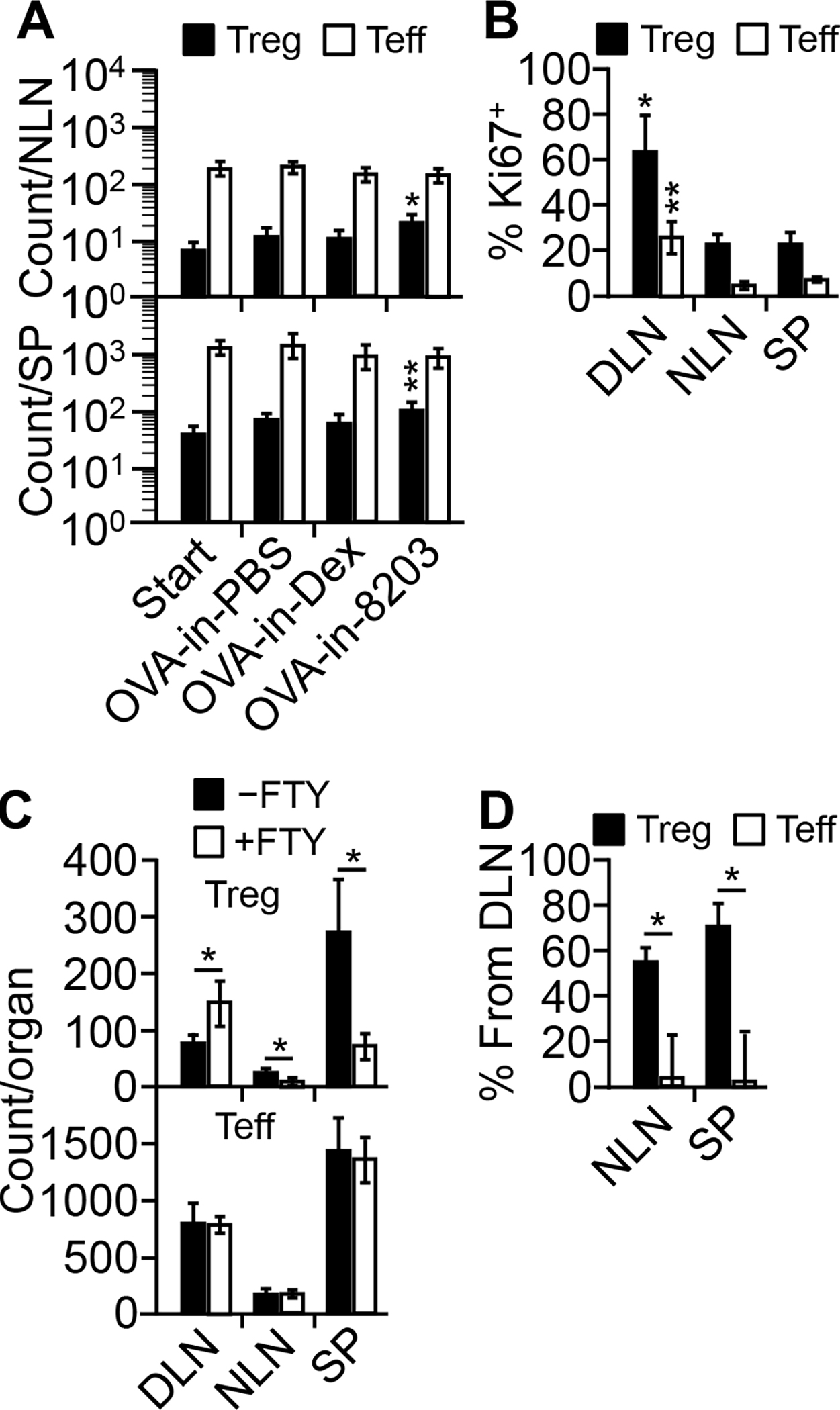
Composition 8203 promotes systemic dissemination of the Treg cells expanded in DLN. (A) OVA-in-8203 expands KJ1-26+ Treg cells in nondraining lymph nodes (“NLN”) and the spleen (“SP”). Data are mean ± SD of six (NLN) or eight (SP) experiments. *p ≤ 0.01 and **p ≤ 0.04, for absolute KJ1-26+ Treg counts in NLN and SP, respectively, in the OVA-in-8203 treated versus any other group (one-way ANOVA). (B) NLN and SP have a smaller fraction of proliferating KJ1-26+ T cells than does DLN. Data are mean ± SD of four experiments. *p ≤ 0.0002 and **p ≤ 0.0004, for % Ki67+ KJ1-26+ Treg and Teff cells, respectively, in DLN versus in NLN and SP (one-way ANOVA). (C) FTY720 (“FTY”) blocks dissemination of OVA-in-8203-expanded Treg cells. The model was injected with OVA-in-8203 either alone (“−FTY”) or with FTY (“+FTY”) on day 5, and cell counts were analyzed on day 8. Data are mean ± SD of four experiments. *p ≤ 0.012 (two-sided t test). (D) Percentages of DLN-derived KJ1-26+ Treg and Teff cells in NLN and SP (“% from DLN“) were calculated from data in C as follows: % from DLN = (count obtained without FTY − average count obtained with FTY) / count obtained without FTY × 100%. *p ≤ 0.001 (two-sided t test).
Composition 8203 helps prolong Treg cell expansion
In the model, measuring the effect of a single injection of OVA-in-8203 after three days was adequate because by a kinetics study, the Treg cell expansion in the DLN peaked in three days (Fig. 7A). Led by that finding, we further found that repeated injections of OVA-in-8203 once every 3 days expanded KJ1-26+ Treg cells progressively over a 9-day period, causing between 3-fold (in the spleen) to 13-fold (in the DLN) increases in the absolute cell number (Fig. 7B). In contrast, KJ1-26+ Teff cells declined progressively during the same period. The combination of the two opposite events resulted in a marked shift of cell balance, with the KJ1-26+ Treg cells representing between ~34% (in the DLN) to ~60% (in the spleen) of the total KJ1-26+ T cells in the end (Fig. 7B). This was not the case if OVA was injected without 8203 (i.e., OVA-in-PBS), which resulted in a shortened Treg cell expansion and a slightly prolonged Teff cell expansion (Fig. 7C). Focusing on these processes in the DLN, we deteremined that the combination of rapamycin and OVA was both necessary and sufficient for prolonging the Treg cell expansion (Fig. 7D), and this action was Treg cell-selective (Fig. 7D). On the othe hand, the combination of rapamycin and dexamethasone was necessary for shortening the Teff cell expansion (Fig. 7D), and this action was Teff cell-selective (Fig. 7D). MPLA was not required here for either effect (Fig. 7D). These results suggest that in mice, the optimal regimen of 8203-adjuvanted immunization is one subcutaneous injetion every three days for a total of three injections.
FIGURE 7.
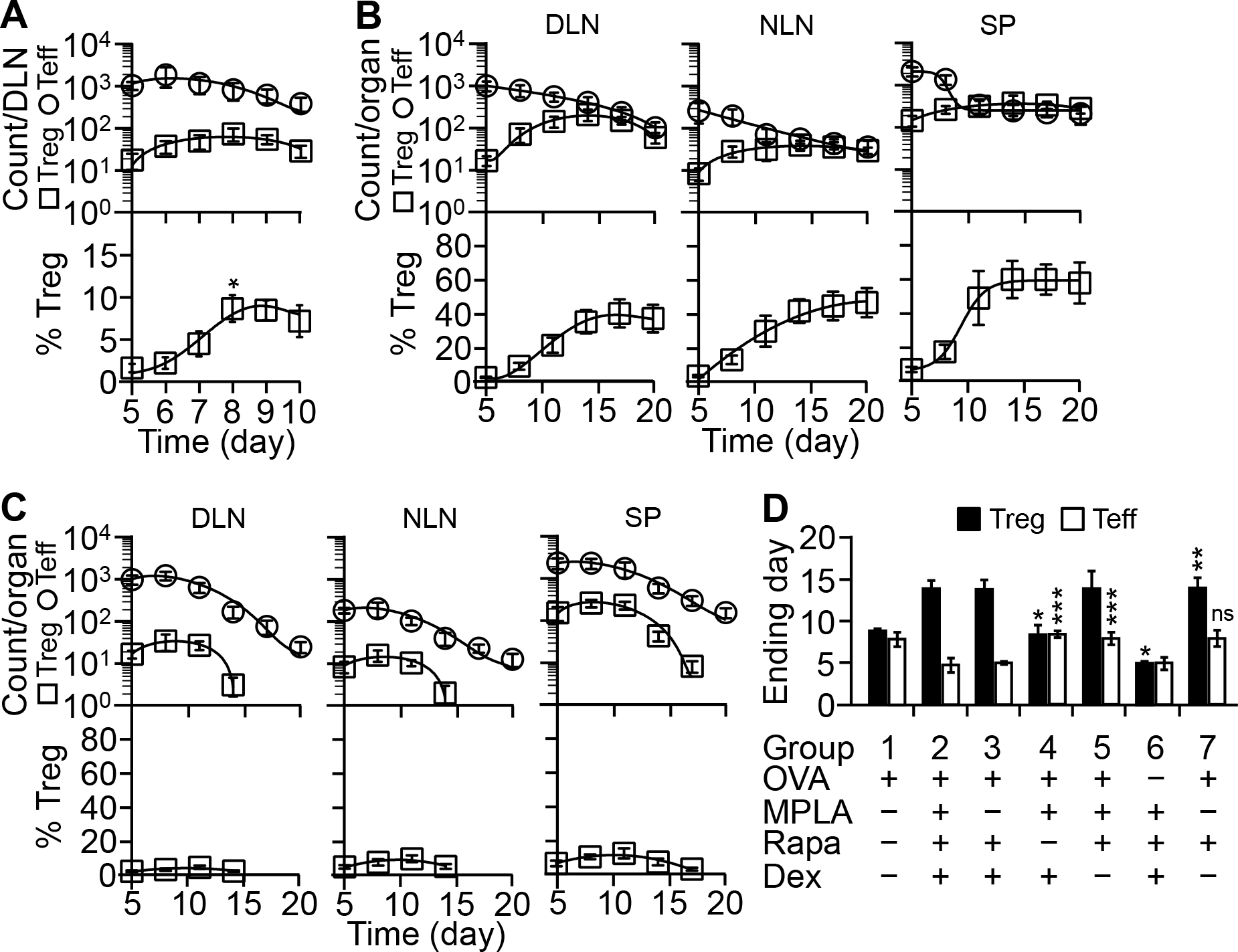
Composition 8203 helps prolong Treg cell expansion. (A) Kinetics after a single injection. The model was injected once on day 5 at the primed footpad with OVA-in-PBS or OVA-in-8203. KJ1-26+ Treg and Teff cells in the DLN were counted daily thereafter. Shown are daily count curves generated by nonlinear regression. Each time point is mean ± SD of four experiments. *p ≤ 0.002, for % Treg on day 8 versus days 5, 6, and 7 (one-way ANOVA). (B&C) Kinetics during repeated injections. The model was injected on day 5 with OVA-in-8203 (B) or OVA-in-PBS (C) once every 3 days. Expansion of KJ1-26+ Treg and Teff cells in indicated organs was followed in the same interval, and cell count curves generated as described in A. Each time point is mean ± SD of four experiments. (D) Role of Ag and each component of 8203 in T cell expansion. The model was injected and analyzed as in B and C. The day when KJ1-26+ T cells in the DLN reached the maximal number is defined as the “Ending day” for cell expansion. Data are mean ± SD of four experiments. *p ≤ 0.001, for Treg cells in groups 4 and 6 versus group 2 (OVA-in-8203); **p ≤ 0.001, for Treg cells in group 7 versus group 1 (OVA-in-PBS); ***p ≤ 0.001, for Teff cells in groups 4 and 5 versus group 2; “ns” (not significant), for Teff cells in group 7 versus group 1 (one-way ANOVA).
Composition 8203 helps expand nTreg cells
To identify the type of Treg cells that 8203 helps expand, we traced the cells’ origin. These cells originated from donor CD4+Foxp3+ Treg cells (“the source Treg cells”) because non-recipient mice or mice that received only donor CD4+Foxp3− Teff cells could not produce the Treg cells in question despite being given the optimal regimen of OVA-in-8203 (Fig. 8A). The source Treg cells were nTreg cells because they existed naturally in donor mice, constituting ~3.8% of the CD4+ T cells in blood at age of 8 weeks (data not shown). They also expressed a set of markers typical of nTreg cells. In comparison, the Treg cells in question expressed the same set of markers at the same relative levels, with the exception of PD1, which was at a lowered level (Fig. 8B). Collectively, these findings indicate that 8203 helps expand nTreg cells.
FIGURE 8.
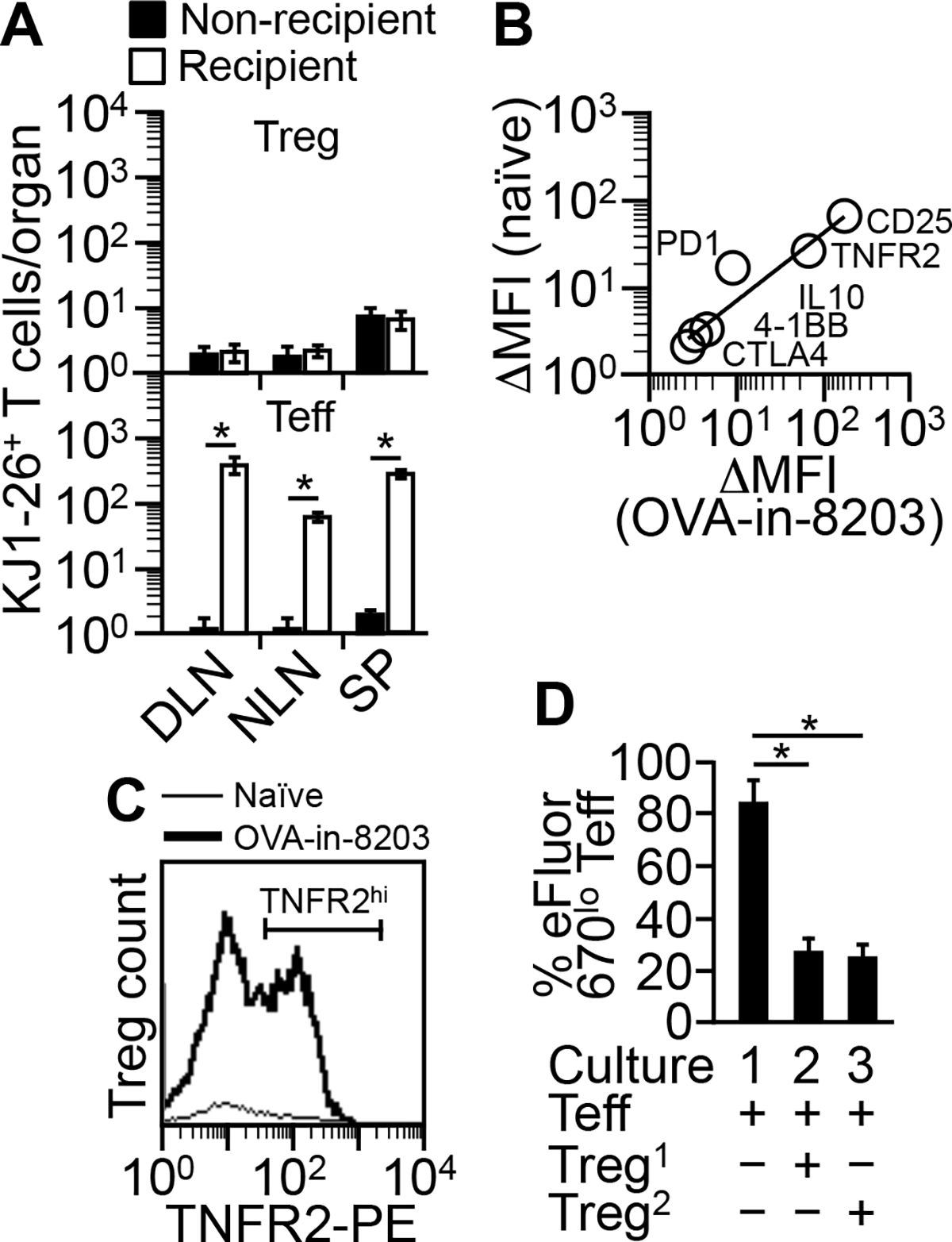
Composition 8203 helps expand nTreg cells. (A) Expansion of KJ1-26+ Treg cells by OVA-in-8203 requires “source Treg cells.” Mice transfused with pure KJ1-26+ Teff cells (see “Mouse model for testing tolerogenic adjuvants”) (“Recipient”) or with PBS (“Non-recipient) were given the optimal regimen of OVA-in-8203. KJ1-26+ Teff and Treg cells were counted three days after the end of the regimen. Data are mean ± SD of two experiments. *p ≤ 0.05 (one-sided t test). (B) Spearman correlation analysis for expression of nTreg markers. KJ1-26+ Treg cells expanded in the model by the optimal regimen of OVA-in-8203 (“OVA-in-8203”) were compared with the source Treg cells in naïve DO11.10 Foxp3-eGFP mice (“naïve”). ΔMFI = MFI of marker-specific staining − MFI of control Ab staining. Data points are average of two experiments. There was a positive correlation between the OVA-in-8203-expanded and the source Treg cells (Rho = 1, p = 0.0028). Of note, the TNFR2 expression depicted is for the TNFR2+ fraction. (C) Expansion of source Treg cells by OVA-in-8203. DO11.10 Foxp3-eGFP mice were primed and given the optimal regimen of OVA-in-8203 in the same manner as described for the model. Three days after the end of the regimen, Treg cells in the DLN (“OVA-in-8203”) were counted and analyzed for TNFR2 expression. The popliteal lymph nodes from naïve mice (PBS injected; “Naïve”) were used as a control. Shown is one of two similar results. (D) Suppression assay. eFluor 670-stained KJ1-26+ Teff responder cells were co-cultured with naïve KJ1-26+ nTreg cells (“Treg1”) or OVA-in-8203-expanded KJ1-26+ Treg cells (“Treg2”). Data are mean ± SD of two experiments. *p ≤ 0.008 (one-way ANOVA).
It was possible that 8203 helped expand nTreg cells without preserving their function. We could not study the cells directly because they were too scarce to isolate. Hence, we studied the source Treg cells as a surrogate. To that end, we gave donor mice the optimal regimen of OVA-in-8203. The source Treg cells responded to the regimen by expanding their number ~14-fold in the DLN while upregulating the TNFR2+ Treg activation marker (19, 20) in nearly half of their population (Fig. 8C). Consistent with what was seen in the model, this response was also Treg cell-biased because Teff cells in the same DLN expanded only ~6-fold and remained mostly TNFR2− (not shown). Taking advantage of their abundance, we analyzed the expanded source Treg cells for their suppressive activity using an in vitro suppression assay. They were equivalent to naïve source Treg cells from untreated donor mice (Fig. 8D). Hence, nTreg cells remain functional after being expanded by 8203-adjuvanted immunization.
Composition 8203-adjuvanted immunization induces Ag-specific immune suppression
To determine whether 8203-adjuvanted immunization induces Ag-specific immunosuppression, we gave the model the optimal regimen of OVA-in-8203 and then challenged it with OVA in the most stringent way: an injection of OVA emulsified in Freund’s complete adjuvant (“FCA”), followed by a boost injection of OVA in Freund’s incomplete adjuvant (“FIA”). We reasoned that if the regimen induced OVA-specific immunosuppression, the mice would respond poorly to the challenge. The results showed that the challenge failed to lower the level of KJ1-26+ Treg cells in the OVA-in-8203 immunized mice to that in OVA-in-PBS immunized control mice (Fig. 9, A versus B). The former mice also developed a weaker delayed-type sensitivity (“DTH”) to OVA than did the latter (Fig. 9C, lower panel), which correlated with fewer KJ1-26+ Teff cells in the DLN (Fig. 9C, upper panel). However, the former mice developed a normal DTH to hen egg lysozyme (“HEL”), a control Ag co-injected with OVA during the challenge (Fig. 9C, lower panel). In aggregate, these results support the notion that in mice, 8203-adjuvanted immunization induces Ag-specific immunosuppression.
FIGURE 9.
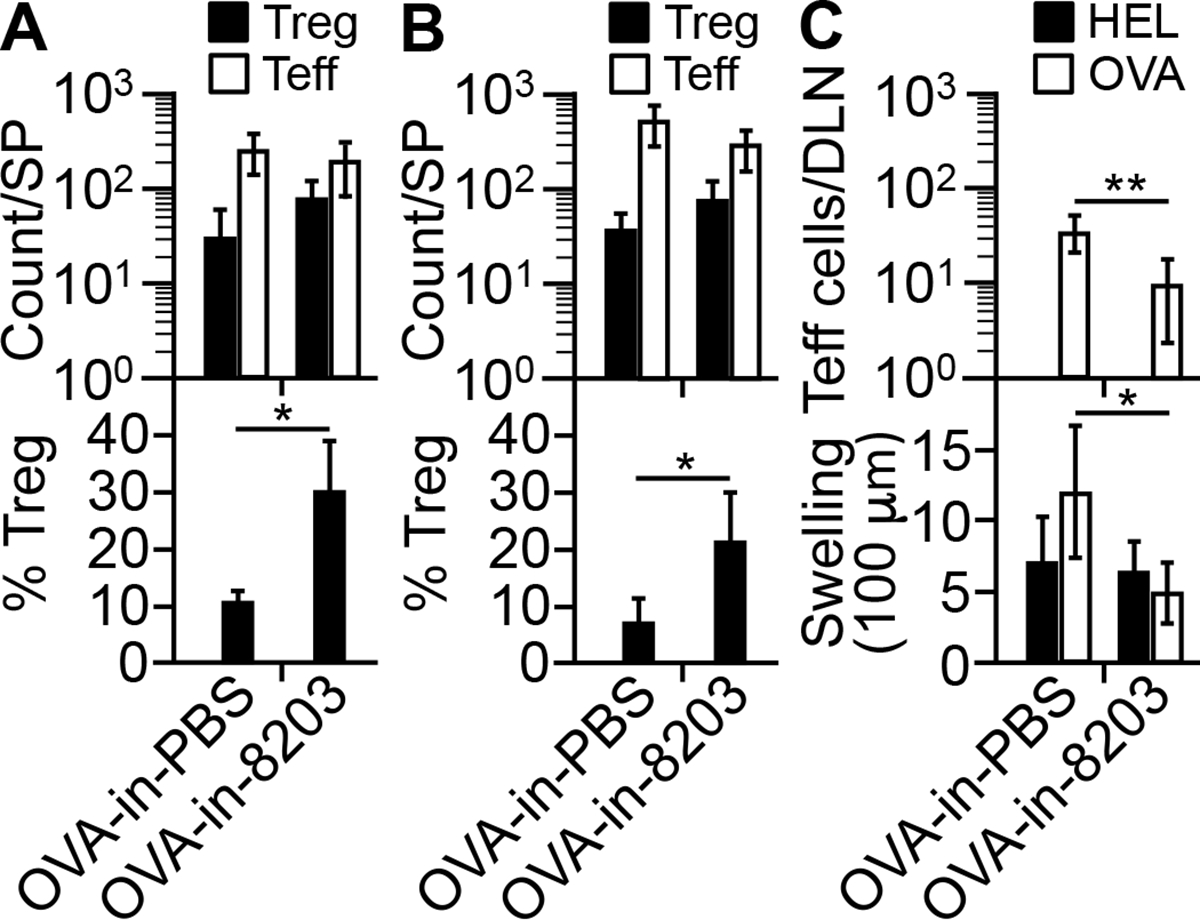
Composition 8203-adjuvanted immunization induces Ag-specific immune suppression. (A) Counts of KJ1-26+ Treg and Teff cells in the spleen (“SP”) of immunized (“OVA-in-8203”) or control immunized (“OVA-in-PBS”) mice immediately before the challenge. Data are mean ± SD of four experiments. *p = 0.009 (two-sided t test). (B) Counts of KJ1-26+ Treg and Teff cells in the SP five days after the challenge. Data are mean ± SD of four experiments. *p = 0.03 (two-sided t test). (C) Teff response to the challenge. Lower: DTH reactions. Data are mean ± SD of 8 mice/group. *p = 0.001 (two-sided t test). Upper: count of KJ1-26+ Teff cells in pooled DLNs five days after the DTH test for OVA. Of note, KJ1-26+ Treg cells were not analyzed due to low counts. Data are mean ± SD of four experiments. **p = 0.001 (two-sided t test).
Discussion
We have shown that 8203 is the minimum composition for a true tolerogenic adjuvant that augments the proliferation and, thus, absolute expansion of nTreg cells. Each of the three components plays at least one critical role in this process: MPLA increases the influx of MMΦs into the DLN, providing ample Ag presenting cells in the microenvironment. Dexamethasone converts the MMΦs into Dex-MΦs, the tolerogenic Ag presenting cells that stimulate Treg cell proliferation (9). Rapamycin blocks the influx of Teff cells into the DLN, preventing MPLA from inducing a Teff cell-dominated response. It also prolongs Treg cell expansion together with an Ag, permitting the growth of more Treg cells via repeated immunization. Dexamethasone and rapamycin together inhibit Teff cell proliferation. Their inhibitory effects may also explain why there was no conversion of Teff cells to Treg cells during the 8203-adjuvanted immunization. These effects might be additive because rapamycin, like dexamethasone, is a biased immunosuppressant that preferentially inhibits Teff cells (21, 22). Alternatively, rapamycin might potentiate the effect of dexamethasone, as reported previously (23, 24). In comparison, the original tolerogenic adjuvant dexamethasone cannot fulfill most of the mentioned roles, which explains why it is less effective. Hence, by the definition given earlier, 8203 is a complete tolerogenic adjuvant (whereas dexamethasone is not).
As the first of its kind, 8203 opens the door to additional, and potentially stronger, complete tolerogenic adjuvants including those tailored to humans. Combining these adjuvants with various known autoantigens, allergens, and transplantation Ags, we may create numerous novel “Ag-with-complete tolerogenic adjuvant” vaccines for treating specific auto- and alloimmune diseases in the same straightforward manner as giving a subunit vaccine. The core concept of combining an Ag and 8203, or its equivalent, can also be adapted to other forms of Ag-specific immunotherapy besides immunization.
For future design of tolerogenic adjuvants, the need to combine immunosuppressive and immunogenic agents may emerge as a principle for obtaining high adjuvanticity. Previously, García-González et al. showed that a combination of dexamethasone and MPLA converted human peripheral blood monocytes into tolerogenic DCs in culture (25). Maggi et al. showed in culture that the dexamethasone/MPLA-induced tolerogenic DCs rendered naïve and memory human CD4+ T cells hyporesponsive to Ag stimulation and conferred suppressive features to the naïve T cells (26). Campos-Acuña et al. showed in a mouse skin-allograft model that a combination of rapamycin and MPLA produced tolerogenic DCs in culture that prolonged skin-allograft survival following adoptive transfer (27). Lastly, Northrup et al. showed in a mouse experimental autoimmune encephalomyelitis model that co-delivery of autoantigen and dexamethasone in FIA, which effectively combined dexamethasone with an immunogenic adjuvant, ameliorated the disease (28). These prior studies showed that the two groups of “opposing” agents, i.e., biased immunosuppressants and conventional adjuvants, can work together to alter the Ag presentation pathways. Our present study has moved further by showing that their combination is also required for attracting MMΦs to the site of immunization before converting them into tolerogenic Ag presenting cells (i.e. the Dex-MΦs), and that the influx of the MMΦs is in fact required for augmenting absolute expansion of Treg cells. Hence, a specific prediction from this discussion is that highly active tolerogenic adjuvants besides 8203 may be found from other combinations that strongly attract MMΦs.
Interestingly, most subunit vaccines in use today contain an immunogenic adjuvant that strongly attracts MMΦs. With an appropriate immunosuppressant, such as the dexamethasone-rapamycin combination used in this study, these vaccines might be converted into their own Ag-specific “antidotes” when their immunogenic adjuvant forms a complete tolerogenic adjuvant with the immunosuppressant. Thus, what differentiates conventional subunit vaccines and vaccines for Ag-specific immunosuppression might just be the exclusion or inclusion of an immunosuppressant. This notion may help unify the design of both types of vaccines.
In conclusion, we have formed a vaccine adjuvant that stimulates a Treg response to immunization by promoting Treg cell proliferation. With this new kind of adjuvant, we may be able to induce Treg cell-mediated Ag-specific immunosuppression simply by standard Ag-with-adjuvant immunization. This means that for the first time since the inception of immunization, we may cross the boundary between induction of immunity and induction of immunosuppression by choosing an appropriate adjuvant or, more precisely, an appropriate immunosuppressant.
Key Points:
We have composed the first true adjuvant for Treg cells.
It requires immunosuppressive and immunogenic agents as two essential ingredients.
Acknowledgments
This work was supported by the National Institutes of Health/National Heart, Lung, and Blood Institute Grant R21 HL106340 (to A.C. and G.Z.), a grant from the American Diabetes Association (to G.Z.), and in part by the Master of Science in Medical Biotechnology Program at the University of Illinois College of Medicine Rockford.
Footnotes
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Sakaguchi S, Mikami N, Wing JB, Tanaka A, Ichiyama K, and Ohkura N. 2020. Regulatory T Cells and Human Disease. Annu Rev Immunol 38: 541–566. [DOI] [PubMed] [Google Scholar]
- 2.Kang Y, Xu L, Wang B, Chen A, and Zheng G. 2008. Cutting edge: Immunosuppressant as adjuvant for tolerogenic immunization. J Immunol 180: 5172–5176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen X, Murakami T, Oppenheim JJ, and Howard OM. 2004. Differential response of murine CD4+CD25+ and CD4+CD25− T cells to dexamethasone-induced cell death. Eur J Immunol 34: 859–869. [DOI] [PubMed] [Google Scholar]
- 4.Chen X, Oppenheim JJ, Winkler-Pickett RT, Ortaldo JR, and Howard OM. 2006. Glucocorticoid amplifies IL-2-dependent expansion of functional FoxP3(+)CD4(+)CD25(+) T regulatory cells in vivo and enhances their capacity to suppress EAE. Eur J Immunol 36: 2139–2149. [DOI] [PubMed] [Google Scholar]
- 5.Northrup L, Sullivan BP, Hartwell BL, Garza A, and Berkland C. 2017. Screening Immunomodulators To Skew the Antigen-Specific Autoimmune Response. Mol Pharm 14: 66–80. [DOI] [PubMed] [Google Scholar]
- 6.Northrup L, Christopher MA, Sullivan BP, and Berkland C. 2016. Combining antigen and immunomodulators: Emerging trends in antigen-specific immunotherapy for autoimmunity. Adv Drug Deliv Rev 98: 86–98. [DOI] [PubMed] [Google Scholar]
- 7.Stagliano KE, and Oppenheim JJ. 2013. DEXterity of tolerogenic APCs. Eur J Immunol 43: 38–41. [DOI] [PubMed] [Google Scholar]
- 8.Chen A, Geng Y, Ke H, Constant L, Yan Z, Pan Y, Lee P, Tan I, Williams K, George S, Munirathinam G, Reardon CA, Getz GS, Wang B, and Zheng G. 2014. Cutting edge: Dexamethasone potentiates the responses of both regulatory T cells and B-1 cells to antigen immunization in the ApoE(−/−) mouse model of atherosclerosis. J Immunol 193: 35–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zheng G, Zhong S, Geng Y, Munirathinam G, Cha I, Reardon C, Getz GS, van Rooijen N, Kang Y, Wang B, and Chen A. 2013. Dexamethasone promotes tolerance in vivo by enriching CD11c(lo) CD40(lo) tolerogenic macrophages. Eur J Immunol 43: 219–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hinds TD Jr., Ramakrishnan S, Cash HA, Stechschulte LA, Heinrich G, Najjar SM, and Sanchez ER. 2010. Discovery of glucocorticoid receptor-beta in mice with a role in metabolism. Mol Endocrinol 24: 1715–1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Oakley RH, and Cidlowski JA. 2011. Cellular processing of the glucocorticoid receptor gene and protein: new mechanisms for generating tissue-specific actions of glucocorticoids. J Biol Chem 286: 3177–3184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schlenke P, Frohn C, Kluter H, Saballus M, Hammers HJ, Zajac SR, and Kirchner H. 1998. Evaluation of a flow cytometric method for simultaneous leukocyte phenotyping and quantification by fluorescent microspheres. Cytometry 33: 310–317. [DOI] [PubMed] [Google Scholar]
- 13.Casella CR, and Mitchell TC. 2008. Putting endotoxin to work for us: monophosphoryl lipid A as a safe and effective vaccine adjuvant. Cell Mol Life Sci 65: 3231–3240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Abraham RT, and Wiederrecht GJ. 1996. Immunopharmacology of rapamycin. Annu Rev Immunol 14: 483–510. [DOI] [PubMed] [Google Scholar]
- 15.Quiel J, Caucheteux S, Laurence A, Singh NJ, Bocharov G, Ben-Sasson SZ, Grossman Z, and Paul WE. 2011. Antigen-stimulated CD4 T-cell expansion is inversely and log-linearly related to precursor number. Proc Natl Acad Sci U S A 108: 3312–3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moon JJ, Chu HH, Pepper M, McSorley SJ, Jameson SC, Kedl RM, and Jenkins MK. 2007. Naive CD4(+) T cell frequency varies for different epitopes and predicts repertoire diversity and response magnitude. Immunity 27: 203–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fink K, Ng C, Nkenfou C, Vasudevan SG, van Rooijen N, and Schul W. 2009. Depletion of macrophages in mice results in higher dengue virus titers and highlights the role of macrophages for virus control. Eur J Immunol 39: 2809–2821. [DOI] [PubMed] [Google Scholar]
- 18.Cyster JG, and Schwab SR. 2012. Sphingosine-1-phosphate and lymphocyte egress from lymphoid organs. Annu Rev Immunol 30: 69–94. [DOI] [PubMed] [Google Scholar]
- 19.Chen X, and Oppenheim JJ. 2011. The phenotypic and functional consequences of tumour necrosis factor receptor type 2 expression on CD4(+) FoxP3(+) regulatory T cells. Immunology 133: 426–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen X, Subleski JJ, Kopf H, Howard OM, Mannel DN, and Oppenheim JJ. 2008. Cutting edge: expression of TNFR2 defines a maximally suppressive subset of mouse CD4+CD25+FoxP3+ T regulatory cells: applicability to tumor-infiltrating T regulatory cells. J Immunol 180: 6467–6471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Battaglia M, Stabilini A, and Roncarolo MG. 2005. Rapamycin selectively expands CD4+CD25+FoxP3+ regulatory T cells. Blood 105: 4743–4748. [DOI] [PubMed] [Google Scholar]
- 22.Strauss L, Whiteside TL, Knights A, Bergmann C, Knuth A, and Zippelius A. 2007. Selective survival of naturally occurring human CD4+CD25+Foxp3+ regulatory T cells cultured with rapamycin. J Immunol 178: 320–329. [DOI] [PubMed] [Google Scholar]
- 23.Ishizuka T, Sakata N, Johnson GL, Gelfand EW, and Terada N. 1997. Rapamycin potentiates dexamethasone-induced apoptosis and inhibits JNK activity in lymphoblastoid cells. Biochem Biophys Res Commun 230: 386–391. [DOI] [PubMed] [Google Scholar]
- 24.Stromberg T, Dimberg A, Hammarberg A, Carlson K, Osterborg A, Nilsson K, and Jernberg-Wiklund H. 2004. Rapamycin sensitizes multiple myeloma cells to apoptosis induced by dexamethasone. Blood 103: 3138–3147. [DOI] [PubMed] [Google Scholar]
- 25.Garcia-Gonzalez P, Morales R, Hoyos L, Maggi J, Campos J, Pesce B, Garate D, Larrondo M, Gonzalez R, Soto L, Ramos V, Tobar P, Molina MC, Pino-Lagos K, Catalan D, and Aguillon JC. 2013. A short protocol using dexamethasone and monophosphoryl lipid A generates tolerogenic dendritic cells that display a potent migratory capacity to lymphoid chemokines. J Transl Med 11: 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maggi J, Schinnerling K, Pesce B, Hilkens CM, Catalan D, and Aguillon JC. 2016. Dexamethasone and Monophosphoryl Lipid A-Modulated Dendritic Cells Promote Antigen-Specific Tolerogenic Properties on Naive and Memory CD4(+) T Cells. Front Immunol 7: 359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Campos-Acuna J, Perez F, Narvaez E, Campos-Mora M, Gajardo T, Catalan D, Aguillon JC, and Pino-Lagos K. 2015. Rapamycin-conditioned dendritic cells activated with monophosphoryl lipid-A promote allograft acceptance in vivo. Immunotherapy 7: 101–110. [DOI] [PubMed] [Google Scholar]
- 28.Northrup L, Griffin JD, Christopher MA, Antunez LR, Hartwell BL, Pickens CJ, and Berkland C. 2017. Co-delivery of autoantigen and dexamethasone in incomplete Freund’s adjuvant ameliorates experimental autoimmune encephalomyelitis. J Control Release 266: 156–165. [DOI] [PMC free article] [PubMed] [Google Scholar]