

Abstract
Both infection and autoimmune disease can disrupt pre-existing antibody titers leading to diminished serological memory, yet the underlying mechanisms are not well understood. Here, we report that tumor necrosis factor-alpha (TNFa), an inflammatory cytokine, is a master regulator of the plasma cell (PC) niche in the bone marrow. Acute recombinant TNFa treatment depletes previously existing antibody titers following vaccination by limiting PC occupancy or retention in the bone marrow. Consistent with this phenomenon, mice lacking TNFa signaling have elevated PC capacity in the bone marrow and higher antibody titers. Using bone marrow chimeric mice, we found that PC egress from the bone marrow is regulated in a cell-extrinsic manner, by radiation-resistant cells via TNFa receptor 1 signaling, leading to increased vascular permeability and CD138 downregulation on PCs. PC motility and egress in the bone marrow are triggered within six hours of recombinant TNFa treatment. In addition to promoting egress, TNFa signaling also prevented re-engraftment into the bone marrow, leading to reduced PC survival. While other inflammatory stimuli can promote PC egress, TNFa signaling is necessary for limiting the PC capacity in the bone marrow. Collectively, these data characterize how TNFa-mediated inflammation attenuates the durability of serological memory and shapes the overall size and composition of the antibody secreting cell (ASC) pool in the bone marrow.
Introduction
The longevity of prophylaxis against infection is directly linked to the long-term survival of antibody-secreting cells (ASCs) (1–3). The bone marrow is a major niche for maintaining long-lived plasma cells (PCs), a critical subset of ASCs in the body, particularly in humans (4). Hematopoietic and non-hematopoietic cells that reside in the bone marrow produce survival factors, like APRIL (5) and IL-6 (6), and retention factors such as CXCL12 chemokine (7–10) that are critical for PC survival.
Various infections have been shown to reduce pre-existing antigen-specific antibody titers, suggesting long-lived PCs have been disrupted. For salmonella infection, SiiE protein directly targets integrin β1 expressed on ASCs and inhibits ASC interaction with bone marrow stromal cells (11). Unvaccinated children exposed to measles have reported 40–70% loss in antibody titer repertoire (12, 13), potentially by depletion of ASCs through engagement of the signaling lymphocyte activation molecule, SLAMF1 (14). Malaria infection leads to reductions in both malaria-specific and bystander ASCs (15–17), but the mechanism of depletion has not been identified. In addition to these reported pathways targeting ASCs, these infections are highly inflammatory, and linked with reduced antibody titers. For example, reports have shown that children who developed higher temperatures during malaria infection were found to have reduced tetanus, measles, and hepatitis B titers (15).
Tumor necrosis factor alpha (TNFa) is highly-potent inflammatory cytokine found to be elevated in patients infected with malaria (18, 19), salmonella (20), and measles (21). Direct injection of human recombinant TNFa in patients can induce responses such as fever, increased plasma cortisol and C-reactive protein, increased blood pressure, and hypotension (22, 23). TNFa is highly secreted by macrophages and T cells, and it has two known receptors, TNFa receptor 1 (TNFR1) and TNFa receptor 2 (TNFR2) (24, 25). TNFR1 is ubiquitously-expressed, whereas TNFR2 expression is limited to immune cells (26), endothelial cells (27), cardiac myocytes (28), mesenchymal stem cells (29), neurons (30), and oligodendrocytes (31). Signaling through TNFR1 can lead to activation of cell survival and pro-inflammatory gene expression via complex 1 signaling (TRADD-RIP1-TRAF2) or apoptosis via complex 2 signaling (TRADD-RIP-1-TRAF-2-FADD-procaspase 8/10) (32, 33). TNFR2 has been reported to promote cell activation, migration, proliferation, tissue repair, and enhancement of TNFR1 signaling via crosstalk (25, 34).
Previous work by Slocombe et al., (35) has demonstrated that salmonella infection, alum adjuvant, or repeated injections with recombinant TNFa (rTNFa) can decrease the number of ASCs in the bone marrow. It is unclear however, what effect TNFa signaling has on humoral immunity and LLPCs as well as which mechanisms are involved. In this study, we aim to determine the effects of TNFa-mediated inflammation on the longevity of serological memory and characterize the role of TNFa on retention and survival of bone marrow PCs.
Methods
Mice
TNF receptor double knock-out mice (DKO) (B6.129S-Tnfrsf1atm1lmx Tnfrs1btm1lmx/J) (Jackson Laboratories) were crossed with C57BL/6J (Jackson Laboratories) to generate single TNF receptor knock-out mice (R1KO and R2KO). TNF-alpha knock-out mice (TNFaKO) (B6.129-TNFtm1Gkl) (Jackson Laboratories), C57BL/6 CD45.1 and C57BL/6 CD45.2 (B6) (Charles River) were used for steady state analysis, immunizations, and as recipients for chimera experiments. B1–8high, Blimp-1YFP, and Sdc1−/− mice were described previously and bred in-house (36, 37).
Bone marrow chimeras
For chimeric animals, two million donor bone marrow cells were intravenously (i.v) injected into lethally irradiated (950 RAD) recipient mice. Ten weeks later, recipient mice were treated with 100 PBS or 1 of recombinant tumor necrosis factor alpha (rTNFa) (Biolegend; 575206) for three consecutive days and then sacrificed. Mice were age-matched within experiments, and all ranged between 10 and 16 weeks old. The Institutional Animal Care and Use Committee approved the animal work conducted in this study.
Enzyme-linked immunosorbent assay (ELISA)
IgG-specific antibodies against nitrophenyl (NP) were detected by coating plates [CORNING, REF#3361] with 2 of NP25-Ovalbumin (OVA) (Biosearch Technologies) overnight at 4℃. Plates were washed four times with 0.05% Tween 20 in PBS and blocked with 100 1% BSA in PBS at room temperature for two hours. Serial dilutions of 100 serum samples were plated at 1:1000 starting dilutions for NP IgG antibody, followed by four-fold serial dilutions. Samples were incubated for two hours at room temperature. Purified NP-specific antibody, 9T13 (100 ), was used as a standard to quantify NP-IgG antibody levels and plated at a 1:100 dilution as an initial titration. Plates were then washed and loaded with 50 IgG horseradish peroxidase (Jackson Immunoresearch Laboratories; 115-035-146) capture antibody at a 1:5000 dilution. Plates were incubated for one hour and then washed. After the last wash, 50 tetramethylbenzidine substrate (TMB) (Fisher Scientific) was used for detection for 5–10 minutes at room temperature in the dark, and reactions were stopped by adding 25 sulfuric acid before the measurement of absorbance at 450nm by a plate reader (Wallac 1420).
To detect total IgG antibody titers, plates were coated with 4 of AffiniPure Donkey Anti-Mouse IgG(H+L) (Jackson Immunoresearch Laboratories, 715-005-150) and incubated overnight as previously described. Plates were then washed and blocked as described above. Serum samples for systemic knock-out experiments (Figure 1) were plated at a starting dilution of 1:1000 and followed by 5-fold serial dilutions. Plates were incubated for two hours at room temperature. Plates were then washed, and IgG horseradish peroxidase (115-035-146 Jackson Immunoresearch Laboratories) was used as detection antibody and loaded at 50 at a 1:5000 dilution. Plates were treated and quantified as previously described.
Figure 1: TNFa signaling restricts the ASC pool in the bone marrow and spleen.
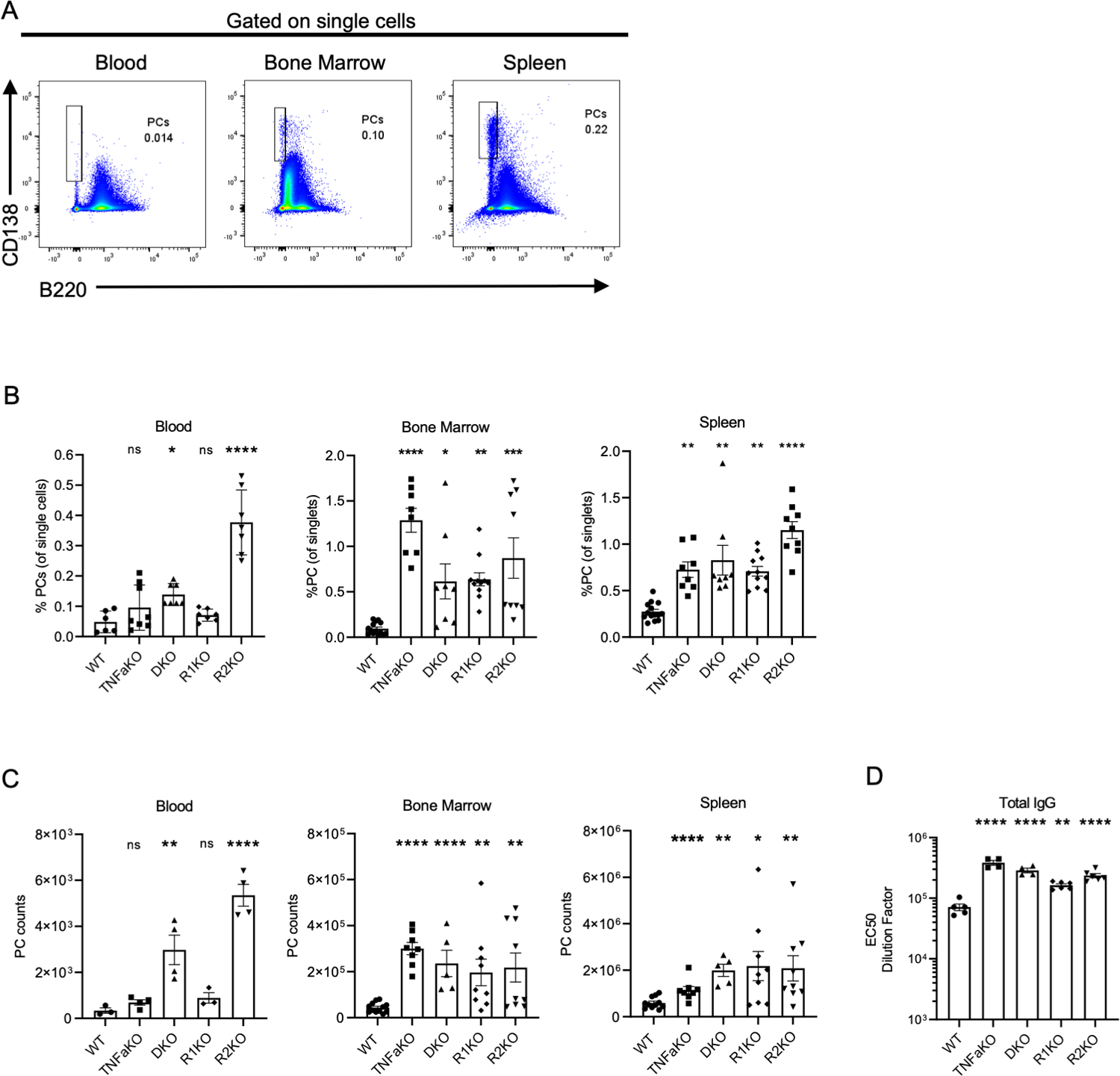
Gating used for PCs in blood, bone marrow, and spleen in WT mice. (B) Frequencies of PCs in the blood, bone marrow, and spleen in various mice at steady-state . (C) Total counts of PCs in the blood (per 0.2 mL), bone marrow (per leg), and spleen. (D) EC50 dilution factor of the total IgG titer in WT, TNFaKO, DKO, R1KO, and R2KO mice. Each experiment is combined data of 2–3 experiments. Groups contained 3–5 mice per group. Each dot represents a mouse. Analysis of PC frequency and numbers were done using one-way ANOVA *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. For total IgG titers (D), non-parametric analysis was done using a Kruskal-Wallis test, **p<0.01, ****p<0.0001.
To detect spike-specific IgG antibodies, plates were coated with 0.2 of spike protein (SARS-CoV-2 Spike, expiCHO Lot: LHD8.1) and incubated overnight as previously described. Plates were then washed and blocked as described above. Serum samples from spike-expressing vaccinia infected mice were plated at a starting dilution of 1:50 and followed by a 3-fold serial dilution. Plates were incubated at room temperature for two hours and then washed. IgG-horseradish peroxidase detection antibody was used to quantify spike-specific IgG antibody titers as previously described. Recombinant spike protein was produced in house by Dr. Kartik Chandran lab.
Enzyme-linked immunosorbent spot
To quantify NP-specific IgG plasma cells, plates were coated with 20 of NP32-BSA (bovine serum albumin) and incubated overnight at 37 C. Plates were then washed four times with 200 of PBS and then blocked with 1% BSA/PBS. Plates were then washed with sterile PBS and blocked with 1% BSA/PBS for two hours. Bone marrow (two femurs + tibia) or spleen cells from NP-KLH (emulsified in Alum) immunized mice were processed into single cell suspension and resuspended in RMPI 1600 media (10% FCS, Glutamine, Penicillin/Streptomycin). Cells were then plated in triplicate (MAHAS4510, Millipore) at a starting dilution of 500,000 cells/100 followed by a two-fold dilution and incubated at 37 C overnight. Plates were then washed with PBS and PBS-Tween (0.05%). A 1:5000 dilution of IgG-horseradish peroxidase detection antibody was added to quantify NP-specific IgG for two hours at room temperature. Plates were then washed with PBS-Tween (0.05%) and PBS. TMB substrate (3,3’,5,5’-Tetramethylbenzidine, 3651-10, MABTECH INC) was added until cells were detected and then washed vigorously with water. Plates were analyzed using an AID Elispot reader (Version 3.2.3).
To quantify spike-specific IgG plasma cells, plates were coated with 4 of spike protein (SARS-CoV-2 Spike, expiCHO Lot: LHD8.1) and incubated overnight at 37 C. Plates were then washed as previously described. Bone marrow and splenic cells were processed and resuspended in media as stated above. Cells were then plated in triplicate at a starting dilution of 4,000,000 cells/100ul followed by a two-fold dilution and incubated at 37 C overnight. Plates were then washed with PBS and PBS-Tween (0.05%). A 1:5000 dilution of IgG-horseradish peroxidase detection antibody was added to detect spike-specific IgG for two hours at room temperature. Plates were then washed with PBS-Tween (0.05%) and PBS. TMB substrate (3,3’,5,5’-Tetramethylbenzidine, 3651-10, MABTECH INC) was added until cells were detected and then washed vigorously with water. Plates were analyzed using AID Elispot reader (Version 3.2.3).
Recombinant vaccinia virus expressing SARS-CoV-2 spike (VacV-S)
A recombinant vaccinia virus expressing the SARS-CoV-2 2P (S2P) stabilized spike protein ectodomain fused to a transmembrane targeting sequence from the vaccinia A56R protein (VacV-S) was generated as described below. The parental strain used was Western Reserve VacV expressing eGFP from the tk locus and bearing the K151E “super-secretor” mutation in the A34R gene (PMID 8497053) (38). The S2P insert, from a construct described in ref (PMID: 32075877; gift from J.S. McLellan), was subcloned into a transfer plasmid containing the VacV A56R gene and 500-bp arms homologous to the tk locus. Recombinant VacV-S was then generated by the trimolecular recombination approach (PMID 15114008). Briefly, purified parental vaccinia DNA was cut with ApaI and NotI-HF at pre-engineered unique restriction sites in the tk gene. The spike-containing transfer plasmid and the digested parental DNA were then co-transfected into BSC-40 cells infected with fowl pox virus (VR-250 from ATCC). Virus production was allowed to occur for one week before individual plaques were grown under 0.5% methylcellulose and sequenced.
Immunizations and infections
For NP-IgG titer experiments, C57Bl/6 mice were immunized intraperitoneally (i.p) with 50 nitrophenyl (NP)-conjugated keyhole limpet hemocyanin (NP-KLH) (NP[24]; Biosearch Technologies) emulsified in 50 of alum. For spike-IgG titer experiments, mice were infected with 2×106 pfu vaccinia expressing spike from SARS-CoV-2 (VacV-S) in 200 of 1X PBS intraperitoneally (i.p). Mice were also infected with a parental vaccinia strain that did not express spike protein (VacV) as a negative control for spike-IgG titers. At six weeks post-immunization, mice in both models were treated with 100 of PBS or 1 of rTNFa for three consecutive days. Serum was collected by retro-orbital bleeding on the day before the first treatment and on days 7, 14, and 30 after the last PBS or rTNFa treatment (Figure 6).
Figure 6. TNFa treatment limits Antigen-specific Antibody titer duration.
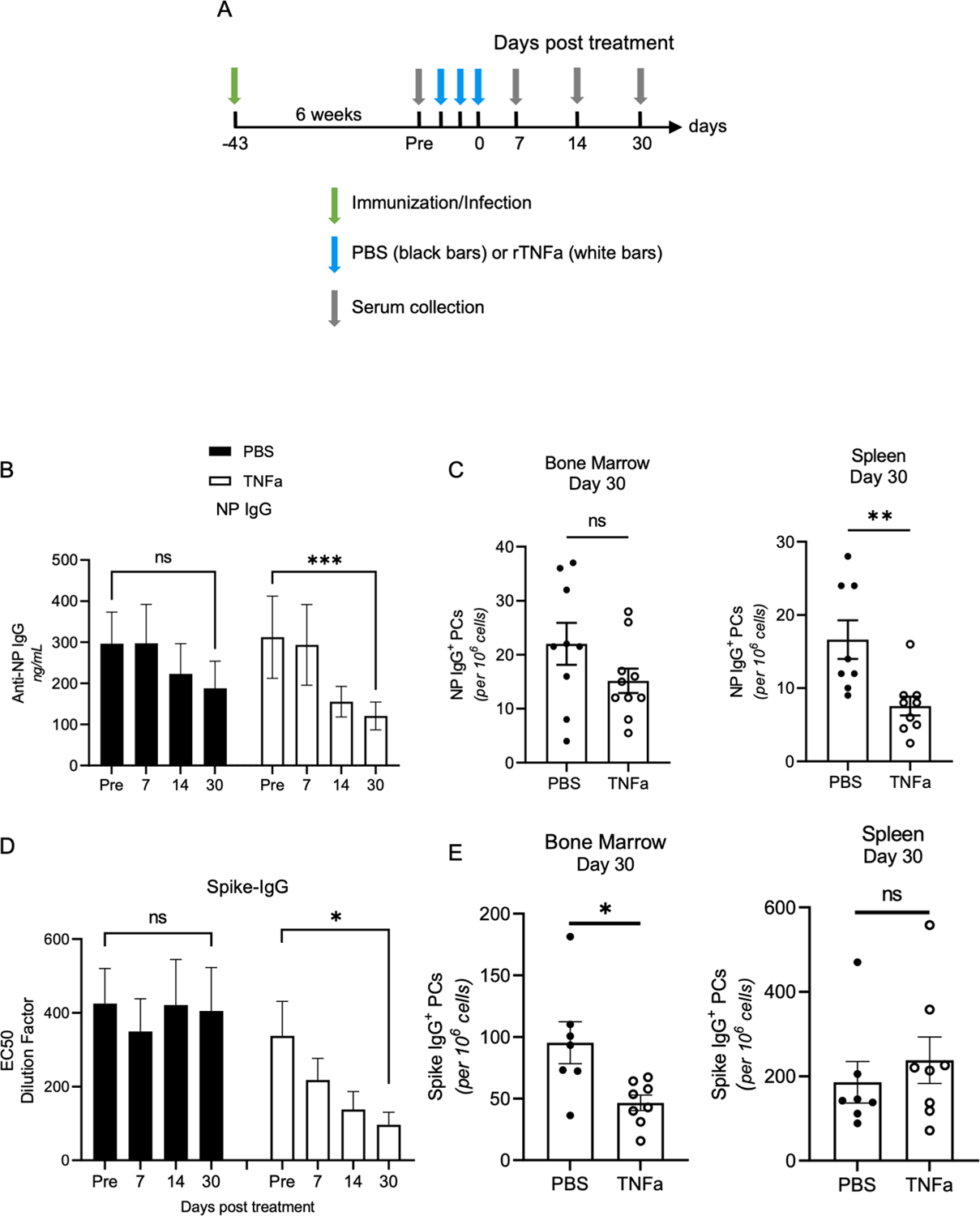
(A) Diagram of immunization and infection experiments. PBS (black-filled bars) and TNFa (white-filled bars) treated groups are indicated in each experiment. (B) NP-IgG titers of NP-KLH in alum immunized WT mice were measured by ELISA. Data is representative of two experiments with 5 mice per group. (C) Bone marrow and spleen NP-specific plasma cell quantification was done using ELISPOT. Data is representative of two experiments. (D) Spike IgG titers in vaccinia-infected mice were quantified using ELISA with 8 mice per group. Data is representative of two experiments. (E) Bone marrow and spleen spike-specific plasma cell were quantified using ELISPOT. Data is representative of two experiments. To analyze anti-NP-IgG and anti-spike-IgG titer decay in PBS and TNFa treated groups (B and D), one-way ANOVA and one-phase decay analysis were used. *p<0.05, **p<0.01 and ***p<0.001. (B) PBS T1/2 = 47.52 days and TNFa T1/2 = 26.54 days. PBS T1/2 = not established and TNFa T1/2 = 7.33 days.
Adoptive Transfer
B1-8high and Blimp-1YFP mice were crossed to generate B1-8high, Blimp-1 YFP mice (BHY). C57Bl/6 (WT) or R1KO mice were treated with PBS, 1ug rTNFa, monoclonal anti-TNFa (BioXCell, BE0058), or isotype control anti-horseradish peroxidase (BioXCell, BE0088) antibodies every four days for the duration of the experiments. BHY donor mice were primed with 50 NP-KLH in alum and boosted with 50 NP-KLH. Donor mice were sacrificed and 2×107 splenocytes (≈ 2×105 ASCs) were adoptively transferred i.v into WT or R1KO mice 24 hours after the second treatment (Figure 7). All groups were sacrificed or bled 14 days after the adoptive transfer. Transferred ASCs were quantified in the bone marrow and spleen and NP-IgG titers were analyzed.
Figure 7. TNFa reduces PC survival and antigen-specific titers by limiting bone marrow retention and engraftment.
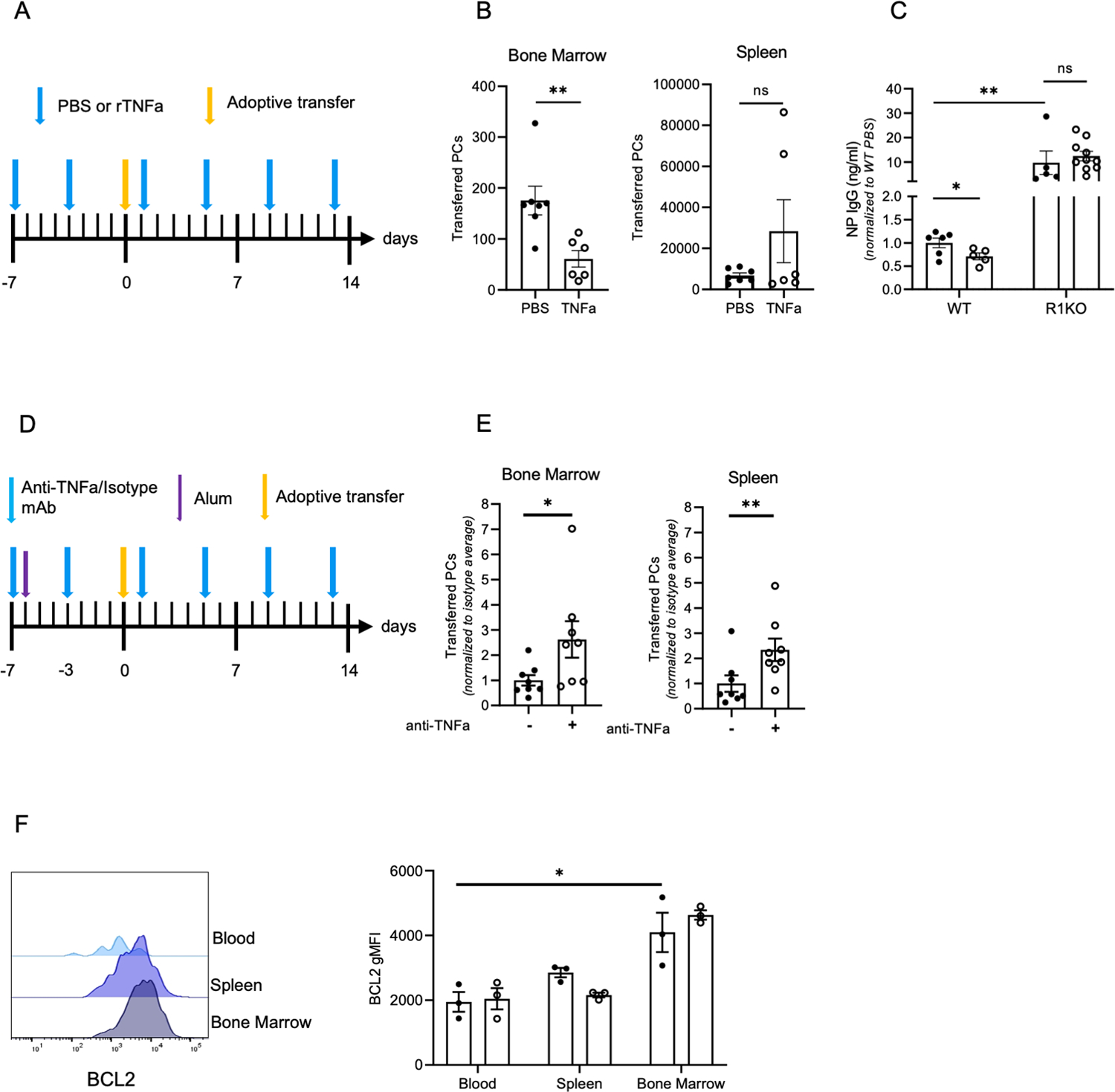
(A) Diagram of experimental design for (B-C) using B18-high Blimp-YFP splenocytes for adoptive transfer into PBS and rTNFa treated WT recipients (B) Quantification of transferred ASCs in the bone marrow and spleen of WT recipient mice from (A). Data are from two pooled experiments, with 3–4 mice per group. (C) NP IgG titers of WT and R1KO recipient mice from adoptive transfer experiment depicted in (A). Titers were normalized to WT PBS treated control average. Data are from two pooled experiments with 3–5 mice per group. (D) Diagram of ASC adoptive transfer into anti-TNFa or isotype control treated, alum treated WT recipient mice. (E) Quantification of transferred ASCs in the bone marrow and spleen of recipient WT mice. Transferred ASCs were normalized to the average of the isotype control treated group. Data is representative of two pooled experiments with 3–4 mice per group. (F) BCL2 expression in (CD138high B220low CD3neg YFPhigh) ASCs in the bone marrow, blood, and spleen of PBS and rTNFa treated Blimp-1 YFP mice. Each dot is representative of one mouse. Significance was calculated for adoptive transfer experiments using a student’s T-test. For non-parametric data, a Mann Whitney’s test was performed. *p<0.05, **p<0.01. To quantify the effect of environment and TNFa treatment on YFP ASCs (K), a two-way ANOVA was performed. *p<0.05
Dissemination and kinetic assays
To track ASCs that egressed from the bone marrow, mice were injected intra-tibially (i.t) with 10 500 carboxyfluorescein succinimidyl ester (CFSE) (Sigma Aldrich), approximately 15–20% of the PCs in the injected bone marrow are labelled (Figure 2F). 24hrs later, mice were treated with 100 PBS or 1 rTNFa and sacrificed the following day (Figure 2E). To quantify PC egress, the total number of CFSE-positive PCs in the spleen or contralateral leg (femur+tibia) was divided by the total number of CFSE-positive PCs in the injected tibia. We have defined this number as the PC dissemination index.
Figure 2. rTNFa treatment induces ASC egress from the bone marrow.
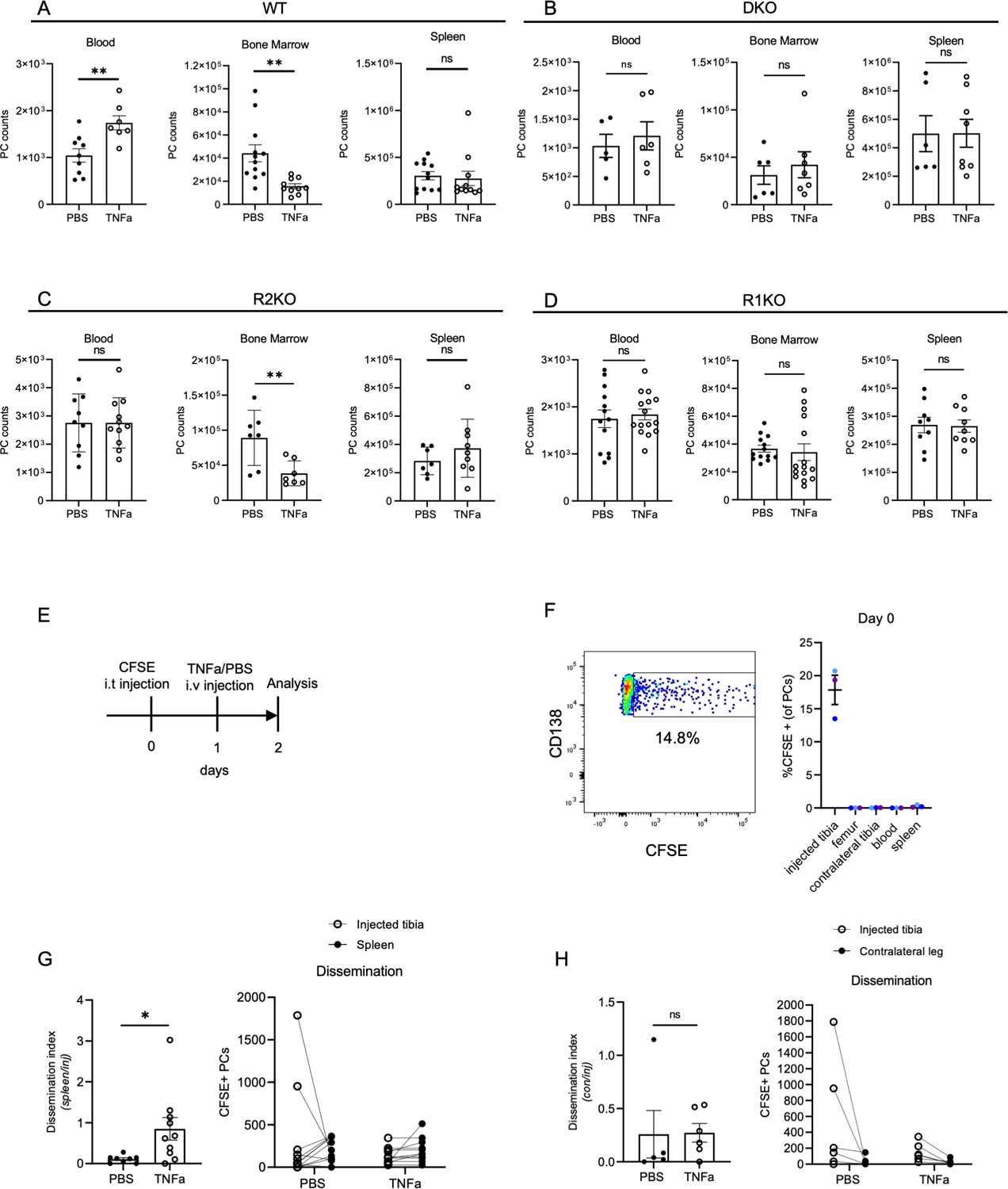
(A-D) Quantification of PC total counts in the bone marrow, blood, and spleen of PBS- and rTNFa-treated mice, one day after treatment. Analysis of (A) WT, (B) DKO, (C) R2KO, and (D) R1KO mice. Each dot represents a mouse, combined from 2–3 experiments (E) Scheme for CFSE i.t. experiment. (F) CFSE representative plot of PCs in the injected tibia bone marrow and quantification of CFSE+ PCs in various organs and bones, immediately after i.t. injection n=3 mice, with color-coded tissues from matching mouse. (G-H) Quantification of CFSE+ ASCs in the injected tibia, contralateral tibia, and spleen. Dissemination is quantified by dividing the number of CFSE+ PCs in the spleen (G) or contralateral leg bones (femur+tibia) (H), ratioed to the number of CFSE+ PCs in the injected tibia. n=2 for (G) and n=2 (H). Each dot represents a mouse, pooled from multiple experiments. Comparisons of PC numbers, dissemination, and survival means were analyzed using student T-test. *p<0.05, *P<0.01 (A-D, G-H).
Permeability, and survival assays
To quantify changes in vascular permeability, WT and TNFa receptor-deficient mice were treated with rTNFa. Six hours later, mice were injected with 100µg 70kD Texas Red dextran (D1380, Life Technologies Corp) and sacrificed fifteen minutes later. CD45+ Dextran+ cells were quantified in the bone marrow of rTNFa-treated mice and compared to non-treated controls.
To assess changes in PC survival, age-matched Blimp-YFP mice were treated with 1µg of rTNFa and sacrificed 6 hours post treatment and compared to non-treated controls. Blimp-1 expression was used to compare PCs of similar maturation in the blood, spleen, and bone marrow. BCL-2 expression (geometric mean) in CD138high B220low CD3negBLIMP-1high ASCs was quantified to determine the microenvironment’s effect on PC survival.
Intravital Imaging Analysis
Bone marrow intra-tibial imaging has been previously described (39). Briefly, mice were anesthetized using isoflurane gas and hydrated with lactated ringers via subcutaneous injection. Imaging was conducted with an Olympus FVE-1200 upright microscope (40). Time lapses were conducted every 90 seconds. All image analysis was conducted using Imaris software 9.2 to track cells and drift correct images. For drift correction, sessile tomato-positive cells were tracked using manual methods to correct XYZ registrations over time. Subtracted channels were used to isolate antibody secreting cell fluorescence from background and bleed-through channel noise. The distance between ASCs and vessels were calculations from vessels obtained by creating surfaces of vessels and then performing a distance transformation. Distances were normalized by dividing the distance by the average distance at all time points in pre-treated movies. All imaging and conditions were independently repeated by two authors to confirm reproducibility.
Antibodies and flow cytometry
All flow cytometric analysis was conducted on BD FACS Aria III or LSR II. The data were analyzed using Flowjo software. To stain for ASCs, CD138 APC or PerCP-Cy5.5 (clone 281-2, BD Biosciences or Biolegend), B220 APC-Cy7 (clone RA3-6B2, Biolegend), CD98 PerCP-Cy5.5 or PE (clone 4F2, Biolegend), CD93 APC (clone AA4.1, Biolegend), CD3 Efluor 450 (17A2, eBioscience), and CD184 BV421 or PE (clone 2B11/CXCR4; BD Biosciences) were used. For B-cell lymphoma 2 (BCL-2 PeCy7; clone BCL/10C4; Biolegend) and mantle cell leukemia (MCL-1 APC; clone D2W9E; Cell Signaling Technologies) staining, cytofix/cytoperm kit (BD) was used. CD45.1 PE (clone A20; Invitrogen) and CD45.2 BV450 (clone 104; BD Biosciences) antibodies were used to distinguish donor and recipient cells in chimera studies. Fluorescent counting beads (Spherotech) were used to quantify cell numbers routinely.
Quantification and statistical analysis
Statistical analyses were performed using Graphpad Prism (version 8 and 9). Specific tests used are listed in the figure legend with p values. As imaging data can produce highly significant data due to large sample sizes, we compared groups from multiple experiments independently and compared averages of averages from multiple experiments to ensure that differences were not artifacts of large sample size. To calculate the EC50 value (the reciprocal of the dilution that caused a 50% reduction of total IgG titer), the non-linear model (Y = Bottom + (XHillslope)*(Top − Bottom) / (XHillSlope + EC50HillSlope)) was used to calculate the EC50 value using Graphpad Prism 9. The reciprocal of the EC50 was then calculated to quantify the dilution factor at which the total IgG concentration is half of the total concentration at that timepoint.
Results
TNFa signaling in radiation-resistant cells limits PC numbers in the bone marrow.
Since acute treatment with recombinant TNF-alpha (rTNFa) can rapidly reduce the number of ASCs in the bone marrow (35), we hypothesized that naïve mice that lack TNFa signaling might retain more PCs in the bone marrow under steady-state conditions. We analyzed PCs (CD138highb220low) in the blood, bone marrow (BM) and spleen of age matched C57BL/6 (WT), mice deficient for TNFa (TNFaKO), TNFa receptor 1 (R1KO), TNFa receptor 2 (R2KO), and mice lacking both TNFa receptors (DKO) (Figure 1A). Using Blimp-1-YFP mice, in which YFPhigh cells are predominantly PCs (Figure S1A), subsets within the PC gate (CD138highb220low), plasmablast gate (CD138high B220+) or B cell gate (B220high, CD138-) were phenotyped (Figure S1B). Indeed cells in the PC gate were found to be mature PCs (YFPhigh CD93high CD98high MHC class IIlow ) particularly in the BM and spleen, and less so in the blood, but still distinct from plasmablasts (CD138high B220+) or B cells (B220high, CD138-) (Figure S1C). At steady-state, in comparison to WT mice, there was a significant increase in the frequency and number of PCs found in the BM, blood, and spleen in all groups with TNFa deficiencies, with the exception of blood PC from TNFaKO and R1KO mice (Figure 1B–C).
Furthermore, all mice deficient for TNFa cytokine or receptor expression all had elevated antibody titers, in line with increased PC numbers (Figure 1D). Differences in total cellularity of the bone marrow were not altered in TNFa and TNF receptor deficient mice (Figure S1D), thus basal or homeostatic TNFa signaling in naïve mice limits PC numbers and ultimately antibody titers.
rTNFa treatment induces PC egress from the bone marrow and limits ASC bone marrow re-engraftment
A previous study had shown that chronic rTNFa treatment could reduce the number of PCs in the bone marrow specifically (35), but the mechanisms involved were not fully resolved. To determine what TNF signaling pathways were involved, WT, DKO, R1KO, and R2KO mice were treated with rTNFa (1μg/day) and analyzed the next day. In WT mice treated with rTNFa, we observed a 50% decrease in the number of BM PCs, with a concomitant 2-fold increase in PCs in peripheral blood, as compared to PBS-treated controls (Figure 2A). No changes in total PC number were detected in the spleen. As expected, we did not see a significant in PC number in the BM, blood, or spleen of DKO mice upon treatment (Figure 2B). R2KO mice treated with rTNFa showed a decrease in BM PCs, as in WT mice, however, this was not accompanied by a comparable increase of PCs in the peripheral blood (Figure 2C) beyond their hyper-elevated steady-state levels (Figure 1B). In contrast, rTNFa treatment of R1KO mice did not result any significant changes in PC number in the blood, bone marrow, and spleen (Figure 2D). These results indicate that TNFR1 signaling is required in TNFa-mediated reduction of bone marrow PCs, while TNFR2 is not.
Although rTNFa treatment reduced PCs numbers in the BM while increasing them in the blood, it was unclear if these were changes were causally linked, or other possibilities might be at play. Thus, to track the fate of BM PCs after rTNFa treatment, we developed a method to directly labeled BM cells in situ by intra-tibial (i.t.) injection of fluorescent cell tracking dye,, carboxyfluorescein succinimidyl ester (CFSE) (Figure 2E). CFSE-labeled PCs were only detectable in the injected tibia on the day of injection (Figure 2F). This dye is rapidly internalized in cells, cleaved, and becomes cell restricted, in a test tube or in vivo. However, while labeling in cell suspension (e.g. in a test tube) is highly uniform, labeling here was done within an intact bone, and thus, the amount of dye incorporation was heterogenous, presumably based on their proximity to the injection site at the knee joint (Figure 2F). Immediately after CFSE injection, we quantified frequency of CFSE+ PCs (CD138+ B220−) in various organs, and found labeled PCs were only detectable in the injected tibia, indicating the labeling approach was highly specific to the injected bone (Figure 2F). To quantify dissemination and normalize for varying labeling efficiencies, the number of CFSE+ PCs in the distal sites, spleen or contralateral leg bones (femur+tibia) were normalized to the number of CFSE+ PCs in the injected tibia, to generate a dissemination index. Following TNFa treatment, PC dissemination index to the spleen was significantly higher as compared to PBS-treated controls (Figure 2G). However, there was no increase in dissemination to the contralateral leg bones, indicating that re-engraftment of mobilized CFSE+ PCs into a new bone might also be disrupted by TNFa signaling (Figure 2H). Thus, using in situ labeling BM PCs, we were able to confirm that rTNFa treatment increases PC mobilization from the BM, over steady-state recirculation levels (40).
TNFR1 signaling in radiation-resistant cells regulates bone marrow retention of PCs in a cell extrinsic manner
TNFa receptors are differentially-expressed in cells of both hematopoietic and non-hematopoietic origin, and signaling could be occurring in a cell autonomous or non-autonomous fashion to trigger mobilization. Therefore, we generated bone marrow chimeras to determine which TNFa receptors expressed by which cell populations regulates PC retention in the context of inflammation. Chimeras were generated using lethally-irradiated recipients and donor cells from congenically-distinct WT, R1KO, R2KO and DKO mice (Figure 3A, S2A). We confirmed donor-derived B cells constituted 90% or higher of the total B cell pool in the spleen (Figure S2B). At ten weeks post reconstitution, all chimera groups, generated using TNFa-deficient mice as donor or recipients, showed significantly higher number of PCs in the spleen than the control group of WT recipients reconstituted with WT donor cells (WT → WT) (Figure 3B), which mirrored results in non-chimeric animals (Figure 1B). In contrast, PC numbers in the BM and blood from chimeric groups using TNF deficient mice were all comparable to control chimera (WT→ WT) group, in contrast to what was observed in non-chimeric TNF deficient mice at steady state (Figure 1B), suggesting some steady-state changes in the chimeric setting. Nevertheless, acute rTNFa could still mobilize BM PCs into the blood in WT-> WT control chimeras (Figure 3C). Based on chimeras using DKO mice as donor cells or as recipient mice, rTNFa treatment did not mobilize PCs from the BM into blood in mice lacking TNF receptors in radiation-resistant cells (WT→ DKO chimeras). In contrast, chimeras with DKO donor cells (DKO→ WT), treatment with rTNFa was capable of mobilizing PCs (Figure 3C). Similarly, chimeras lacking TNFR1 expression only in radiation-resistant cells (R1KO→ WT) were also unable to mobilize PCs required TNFa-mediated egress of bone marrow PCs (Figure 3D). However, in chimeras generated with R2KO mice, rTNFa treatment decreased bone marrow PCs in all groups, indicating that TNFR2 expression was dispensable for PC mobilization (Figure 3E). rTNFa treatment had no effect on overall PC numbers in the spleen of chimeric animals (Figure S2C), similar to what was found in non-chimeric animals (Figure 2A–D). Next, we confirmed that the decline in bone marrow PC and increase in blood PC numbers seen in R1KO chimeras were linked, as seen in non-chimeric mice (Figure 2F). Tibial PCs were in situ CFSE-labeled in chimeric mice as before (Figure 2E), and tracked after rTNFa treatment. Results showed that dissemination of CFSE+ PCs to the spleen after rTNFa required TNFR1 expression in radiation-resistant cells (Figure 3F). Taken together, these data show that TNFR1 signaling in radiation-resistant cells regulate PC retention in the bone marrow.
Figure 3. TNFR1 signaling in radiation-resistant cells regulates ASC retention in the bone marrow.
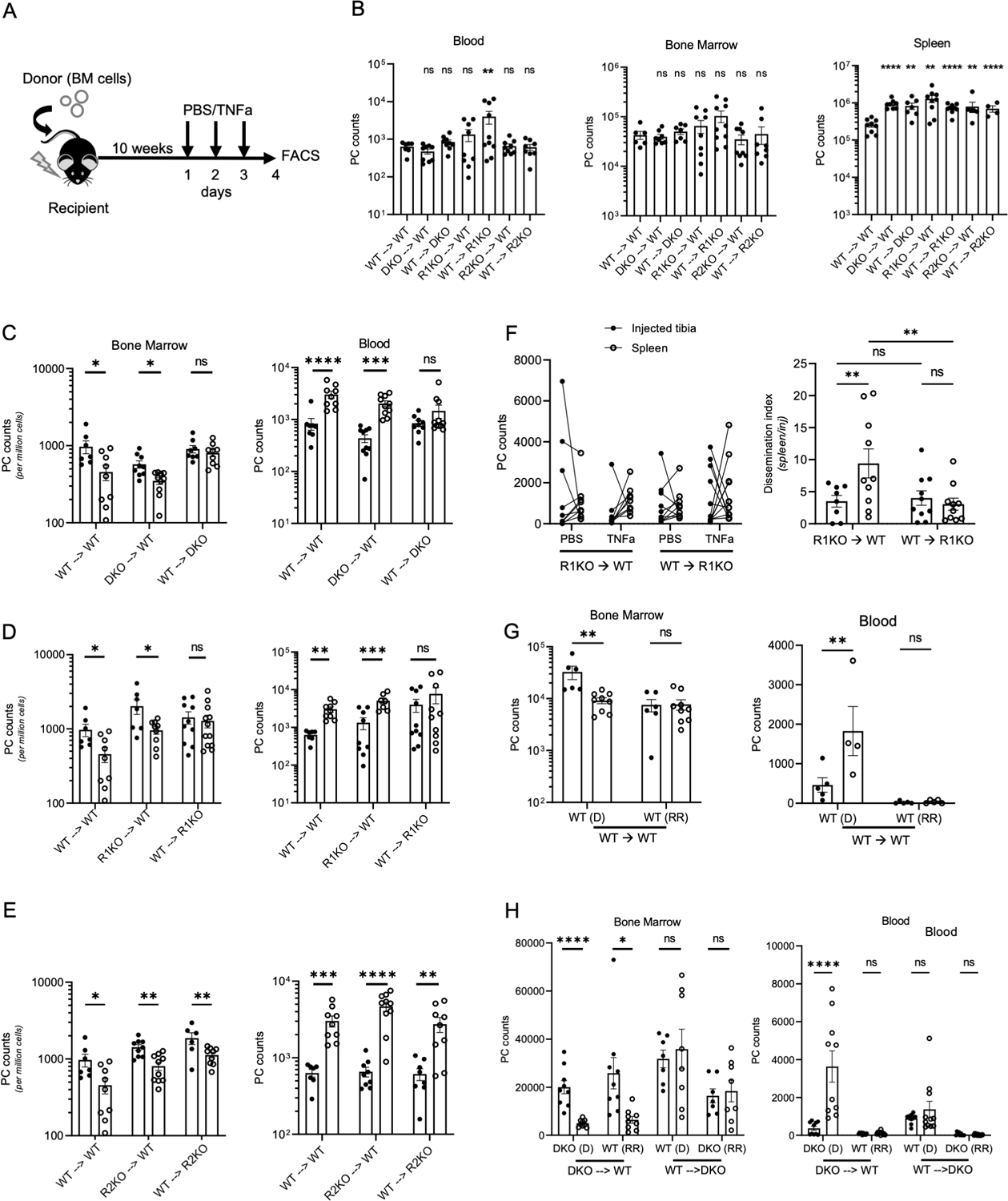
(A) Scheme for chimera generation and TNFa treatment schedule. (B) Quantification of blood, bone marrow, and splenic PCs in PBS treated chimeras, labeled using donor → recipient format. (C-E) Analysis of PCs counts in the bone marrow (per million live mononuclear cells) and in blood (per mL) of PBS-treated (black-filled circles) and rTNFa-treated (white-filled circles). (C) Analysis of WT, and DKO chimeras. (D) Analysis of R1KO and WT chimeras. (E) Analysis of R2KO and WT chimeras. (F) Matching counts of i.t.-labeled CFSE+ PCs in the injected tibia and spleen in PBS- treated (black-filled) or TNFa-treated R1KO chimeras, and calculated dissemination index, as described in Figure 2. (G-H) Quantification of donor-derived (D) and radiation-resistant (RR) PCs in the bone marrow and blood of WT (G) and DKO (H) chimeras. Chimera analysis was done using a student T-test or Mann Whitney test for parametric and non-parametric data, respectively. N=2–3 experiments per chimera and each group contained between 3–5 mice per group. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
Potentially different subsets of ASCs could be mobilized by TNFa in various chimeric groups, either based on TNFa receptor expression, ASC maturity, or both. In lethally-irradiated chimeras, while new PCs are continuously-generated from donor-derived hematopoietic stem cells, some recipient ASCs persist following irradiation as they do not proliferate and are highly differentiated, and this is consistent with a long-lived plasma cell (LLPC) subset (41). These LLPCs, are distinguishable donor-derived PCs on the basis of CD45 isotype (Figure S2A). To determine if different ASC subsets were more sensitive to rTNFa-induced egress, we compared the change in number of PCs in both donor-derived and radiation-resistant host populations after TNFa treatment in WT control (Figure 3G). rTNFa treatment decreased donor-derived PCs in WT control chimeras, which also correlated with an increase in donor-derived PCs in the blood. However, radiation-resistant LLPCs were not affected by TNFa treatment. In DKO chimeras, TNFa treatment reduced both donor and radiation-resistant PC populations in the DKO → WT chimera, indicating that mobilization was cell non-autonomous process and not dependent on PC expression of TNFa receptors (Figure 3H). TNFa treatment induced an increase in donor-derived (DKO) PCs in the blood, whereas long-lived radiation-resistant (WT) PCs remained unchanged. As expected, TNFa treatment had no effect either donor-derived nor radiation-resistant PC populations in the bone marrow or blood of WT → DKO chimeras. Collectively, these data indicate that TNFa-mediated inflammation preferentially mobilizes less mature bone marrow PCs in a cell non-autonomous manner.
TNFa signaling rapidly induces PC motility and vascular permeability in the bone marrow
To identify the mechanisms of TNFa-mediated cell-extrinsic egress, we conducted a kinetic time course to determine how early bone marrow PC dissemination could be captured. PC numbers were significantly decreased in the bone marrow and significantly increased in the blood as early as six hours post initial rTNFa i.v. treatment when compared to non-treated control mice (Figure 4A). At this six-hour timepoint, there was no change in splenic PCs. Furthermore, this timepoint did not show the induction of TNFa-mediated apoptosis (Figure 4B–D). Because egress occurs rapidly after rTNFa, we used intra-vital two-photon imaging of tibial bone marrow to visualize how treatment triggered PC mobilization. To visualize PC motility and egress in situ, in real-time, we bred the PC reporter, Blimp1-YFP, with cdh5-cre (VeCad) Rosa26LSL-tdTomato alleles, in which bone marrow endothelial cells are tdTomato+. Previous work in our laboratory established that bone marrow PCs are motile in steady state conditions, at track and displacements velocities averaging 1 micron/min and 0.2 micron/min, respectively (40). Treatment with rTNFa increased YFPhigh ASC average track speed by 20%, displacement velocity by 60%, and mean squared displacement (MSD) within 2–6 hours after treatment (Figure 4E–G, Video S1). Furthermore, the distance between ASCs and vessels was significantly reduced in response to rTNFa treatment, consistent with egress (Figure 4H).
Figure 4. rTNFa treatment enhances ASC motility and vascular permeability in the bone marrow.
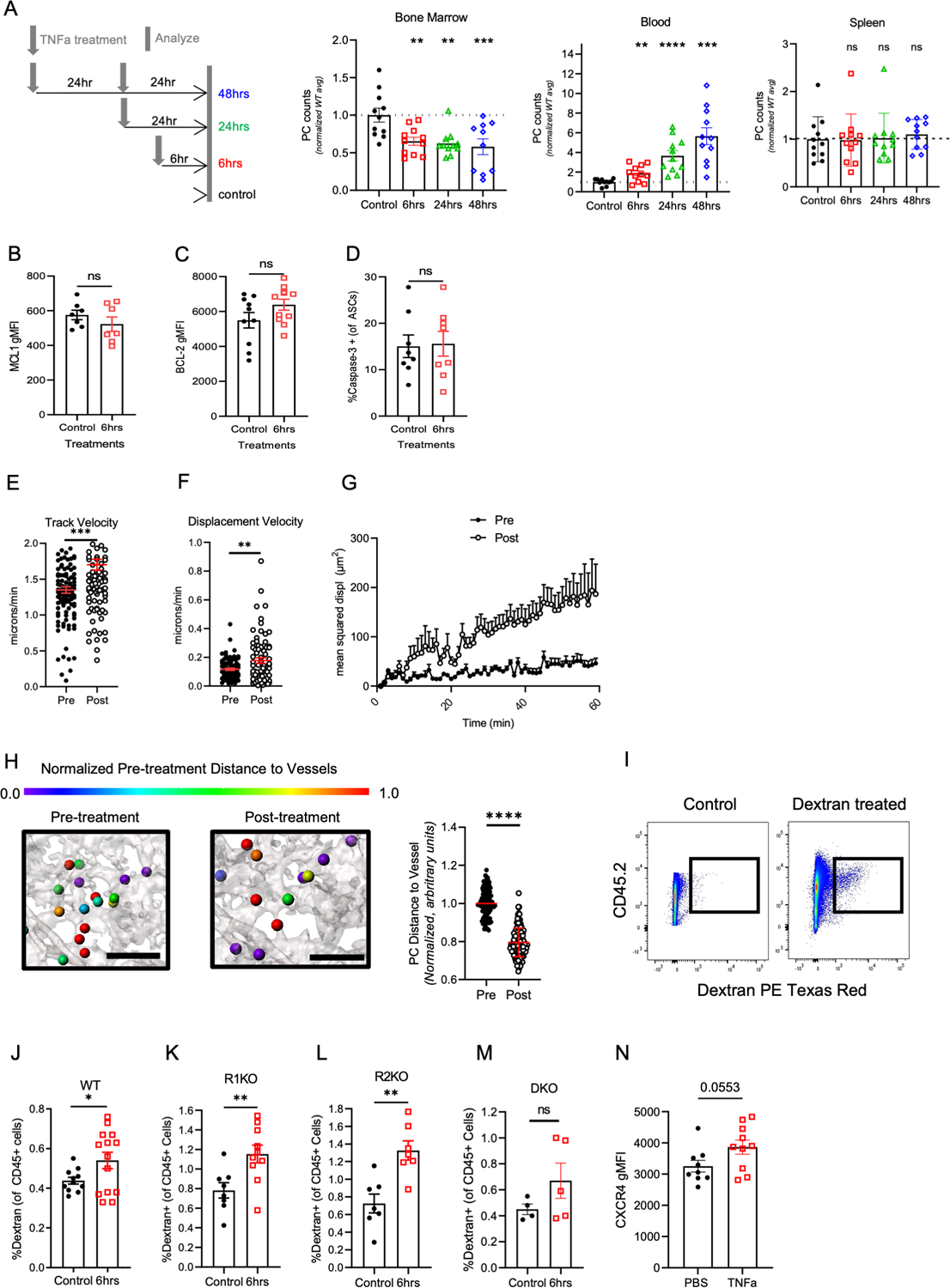
(A) Conditions for time course of assessing BM PC egress following TNFa treatment. Mice were treated once with rTNFa and sacrificed 6 hours (red), 24 hours (green) or treated twice with rTNFa and sacrificed 24 hours after the last treatment (blue), analyzing BM, spleen, and blood, normalized to control (untreated) mice. Data are pooled from 2–3 experiments and groups contained 3–4 mice per group. Each experiment was normalized to the mean of the non-treated control (black). (B-D) Comparison of in MCL-1 (B), BCL-2 (C), and active caspase-3 (D) expression in control (black) and 6 hours post TNFa treatment WT mice (red). Comparison of ASC dynamics pre and post rTNFa treatment based on (E) track velocity, (F) displacement velocity, and (G) mean squared displacement analysis. Data is pooled from 2 Blimp1-YFP, cdh5-cre (VeCad) Rosa26LSL-tdTomato mice. (H) Diagram and quantification of changes in distance between ASCs (spheres) and vasculature (grey). The distance from the ASC edge to the nearest VeCad-Cre+ vessel was quantified pre- and post-rTNFa treatment and normalized to the average ASC-vessel distance in the pre-treatment segment of the movie. Color represents normalized 3D distance to the vessel (0 = closest to vessel, 1 no change or farther than pre-treatment distance). Scale bar (black line in diagram), 100um. (I) Representative dot plot for gating dextran+ of CD45+ mononuclear BM cells. (J-M) Analysis of dextran uptake in control (untreated) mice (black) or in mice 6 hours after TNFa treatment using WT (J), R1KO (K), R2KO (L), and DKO (M) mice. N=3 for WT data and N=2 for R1KO, R2KO, and DKO data, with 3–4 mice per group. (N) Quantification of CXCR4 expression in PBS and TNFa-treated WT mice. N=2, with 4–5 mice per group. For kinetic experiments, significance was calculated using one-way ANOVA. For changes in vascular permeability, track velocity, displacement velocity, and CXCR4 expression, significance was calculated using a student T-test. *p<0.05, **p<0.01, and ***p<0.001, *** p<0.0001.
Next, we tested if TNFa altered vascular permeability in the bone marrow by the six-hour timepoint, thus promoting PC egress. Previous studies have shown that TNFR1 stimulation increased vascular permeability in vitro in HUVEC cells and in vivo in liver vessels (42). However, in the absence of TNFR1 signaling, TNFR2 signaling in vivo could slightly increase vascular permeability (42). WT, DKO, R1KO, and R2KO mice were treated with rTNFa and at six hours, i.v. injected with Texas-red conjugated dextran and assessed vascular leakage based on uptake of dextran by bone marrow mononuclear cells (Figure 4I). In WT mice, rTNFa increased dextran uptake by bone marrow mononuclear cells (Figure 4J). To see which TNFa receptor mediated this effect, we compared dextran uptake in R1KO and R2KO mice (Figure 4K and L). Expression of either TNFa receptor was sufficient to increase dextran uptake in the bone marrow. However, TNFa treatment of DKO mice did not change dextran uptake in the bone marrow (Figure 4M).
Previously, we have shown that there is an inverse correlation between CXCR4 receptor expression on bone marrow PCs and CXCL12 concentration in the bone marrow serum (40). Additionally, inhibition of the CXCR4-CXCL12 axis can alter ASC motility and retention within the bone marrow. Furthermore, studies have shown that TNFa treatment can inhibit CXCL12 secretion from stromal cells (43–45). Here, we observed a significant increase in CXCR4 expression with rTNFa treatment in bone marrow PCs (Figure 4N), indicating a disruption in the CXCR4-CXCL12 axis. Collectively, these data show that rTNFa enhances PC motility, association with vessels, increases vascular permeability and disrupts CXCR4-CXCL12-mediated retention, which could potentiate bone marrow egress.
TNFa signaling promotes egress by downregulating CD138 expression in bone marrow PCs
We analyzed the phenotype of in bone marrow PCs at six hours post rTNFa treatment. Syndecan-1, or CD138, is a transmembrane proteoglycan that is highly expressed on PCs and has been shown to engage with hundreds of proteins to enhance cell adhesion, endocytosis, and wound healing (37, 46). CD138 expression has been shown to enhance PC survival (37), and a decrease in its surface expression on myeloma cells has been associated with apoptosis and egress from the bone marrow (47). Treatment with rTNFa caused CD138 expression to decrease in PCs in the BM of WT mice but not in the blood or spleen (Figure 5A). Furthermore, in both PBS and TNFa treated groups, CD138 expression on PCs in the bone marrow and spleen were significantly higher when compared to the blood. As in WT mice, BM PCs in R2KO mice showed a decrease in CD138 expression with TNFa treatment, however, not in R1KO mice (Figure 5B). To assess if TNFa downregulation of CD138 expression on bone marrow PCs was mediated by cell-intrinsic or cell-extrinsic TNFa receptor signaling, we analyzed CD138 expression in donor and radiation-resistant PCs populations in our chimeric groups (Figure 3). In chimeras using DKO mice, CD138 expression was down-regulated on BM PCs when radiation-resistant compartment expressed TNFa receptors (DKO→ WT) but not when it was absent (WT→ DKO) (Figure 5C). TNFR2 expression by radiation-resistant cells was dispensable for CD138 downregulation (Figure 5D), however, in the absence of TNFR1, CD138 expression downregulation is disrupted (Figure 5E). These results indicate that TNFR1 signaling down-regulates CD138 expression on PCs in a cell non-autonomous fashion. To determine if CD138 modulation is necessary for TNFa-mediated egress of bone marrow PCs, we treated CD138−/− (CD138KO) and WT mice with TNFa and quantified mature PC numbers at 24 hours post treatment. In CD138KO mice and controls, PCs were identified as B220low CD3neg CD93high CD98high cells (Figure 5F). PC survival factor CD93 and amino acid transporter CD98 are highly expressed on bone marrow PCs (48). TNFa treatment did not induce significant egress of CD138−/− PCs from the bone marrow (Figure 5F), indicating CD138 expression may support PC retention in the bone marrow. These results suggest that TNFR1 signaling may trigger down-regulation of CD138 on bone marrow PCs, thereby contributing to egress.
Figure 5. rTNFa promotes bone marrow ASC egress by downregulating CD138 expression.
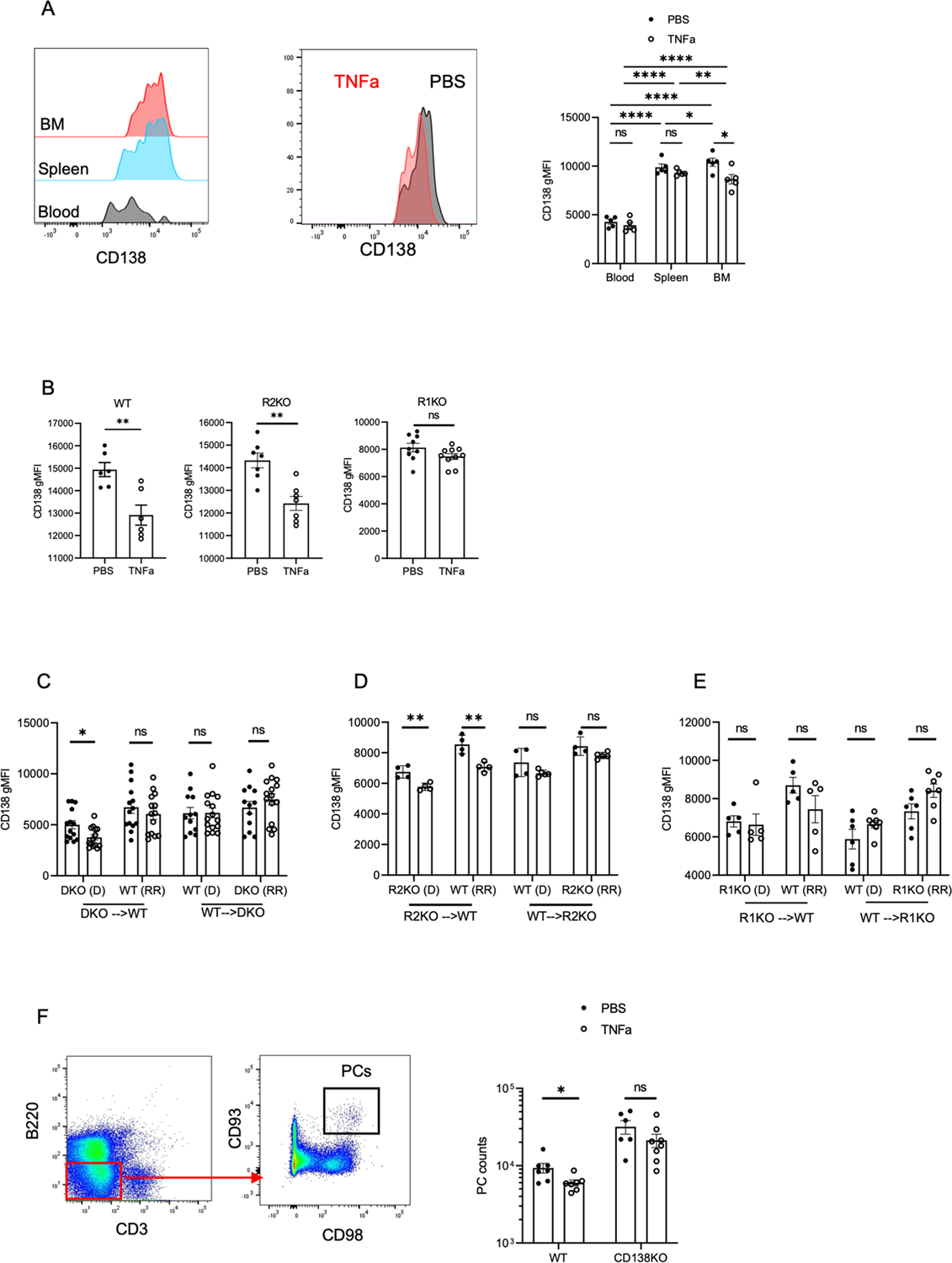
(A) Analysis of CD138 expression on PCs at 6 hours after PBS and rTNFa treatment in WT BM, spleen and blood. (B) Analyses of BM PCs for WT, R1KO and R2KO mice in PBS and rTNFa treated mice. Data for WT, R1KO, and R2KO mice are two pooled experiments with 3–5 mice per group. (C-E) CD138 expression in donor (D) and radiation-resistant (RR) ASCs in PBS- and rTNFa-treated DKO, R1KO, and R2KO chimeras. Data are from two pooled experiments, expect for R2KO chimera with a representative experiment in (D). (F) Gating strategy and quantification of bone marrow PCs in PBS and rTNFa-treated WT and CD138KO (B220low CD3neg CD93high CD98high) mice. Data are from two pooled experiments, with 3 mice per group. For all figures, analysis of CD138 in PBS and rTNFa-treated groups were quantified using a student T-test or Mann Whitney test for parametric and non-parametric data, respectively. *p<0.05, **p<0.01.
TNFa-mediated inflammation diminishes humoral longevity
The effects of TNFa-mediated PC egress from the bone marrow on antibody titer longevity remains unknown. To assess the effect of TNFa on the longevity of antigen-specific titers following vaccination, we employed several models. C57BL/6 mice were immunized with NP conjugated to keyhole limpet hemocyanin (NP-KLH) with alum adjuvant (37, 49). At 6-weeks post immunization, mice were treated with PBS or rTNFa and NP-IgG titers were tracked over time (Figure 6A). rTNFa treatment significantly reduced anti-NP IgG titers by days 14 post treatment (Figure 6B). The half-life of NP-IgG titers was reduced to 26.54 days compared to the 47.52 days in PBS treated mice. High variance in BM NP-specific PC counts between mice, particularly in PBS control group, made any decreases in PCs due to TNFa undiscernible (Figure 6C). However, NP-specific PC numbers were significantly decreased in the spleen of TNFa treated mice when compared to PBS control mice. Next, to determine if TNFa could deplete antibody titers produced by a viral infection, mice were infected with 2×106 pfu vaccinia expressing SARS CoV-2 spike. Results showed that rTNFa treatment caused a significant decline in spike-binding IgG titers (EC50) by day 30 post treatment (Figure 6D). Here, TNFa treatment in vaccinia-infected mice caused a significant decline in spike-specific PCs in the bone marrow (Figure 6E). Collectively, these results show that TNFa-mediated inflammation can significantly reduce the longevity of serological memory by reducing numbers of antigen-specific plasma cells in survival niches.
PC access to the bone marrow survival niche is critical for long-term humoral memory. Previous results (Figure 2G) showed that rTNFa treatment mobilized BM PCs to the spleen but they did not reengraft into the contralateral tibia. To determine if TNFa inhibited PC engraftment into the bone marrow, WT recipient mice were chronically treated with PBS and rTNFa (2x per week for three weeks). After the first week of treatment, splenocytes were transferred into mice from transgenic mice, which express the NP-specific BCR heavy chain, B18-high, and the PC reporter, Blimp-1 YFP. Fourteen days after transfer, mice were sacrificed and transferred (YFP+) PC numbers and NP-specific IgG titers were quantified (Figure 7A, S3A). rTNFa treatment inhibited transferred PC engraftment into the bone marrow but not the spleen (Figure 7B). Furthermore, reduction in transferred PCs in the bone marrow correlated with a reduction in NP-specific IgG titers in serum (Figure 7C). When R1KO were used as recipients in these experiments, NP-specific IgG titers were 10-fold higher than in WT mice, and rTNFa treatment did not dampen NP-specific IgG titers. Therefore, TNFR1 signaling inhibits antigen-specific PC engraftment into the bone marrow and concomitantly reduces corresponding circulating antibody titers.
While TNFa was sufficient to inhibit bone marrow engraftment, it may not be necessary or uniquely capable of disrupting bone marrow PCs. Thus, to test the specific requirement of TNFa for inflammation-induced PC egress from the bone marrow, recipient WT mice were immunized intraperitoneally (i.p.) with alum, to induce Th2-specific general inflammation and inflammasome activation (50). WT recipient mice were chronically treated with either anti-TNFa blocking antibody (BE0058) or isotype-matched control (BE0088) and adoptively transferred with splenocytes containing transferred PCs. At fourteen days post adoptive transfer (Figure 7D), anti-TNFa treated mice had increased transferred PC numbers in the bone marrow and the spleen compared to isotype-treated mice (Figure 7E), indicating that TNFa was necessary for limiting PC engraftment.
Under non-inflammatory steady-state conditions, the blood lacks any appreciable number of mature PCs, suggesting cells rapidly engraft into survival niches. To determine if TNA-induced egress of BM PCs affects their survival, Blimp-1 YFP mice were treated with rTNFa and the expression of anti-apoptotic marker BCL-2 in highly mature PCs (CD138high B220low CD3neg YFPhigh) (Figure S3B–C) was assessed in the bone marrow, spleen, and blood was assessed. Bcl-2 expression in mature PCs was significantly lower in the blood when compared to PCs in the bone marrow (Figure 7F), illustrating that the bone marrow microenvironment is critical to long-term PC survival. Thus, by driving PC egress from the bone marrow and keeping PCs mobilized, TNFa-mediated inflammation may ultimately lead to reduced PC survival and faster decay of antigen-specific antibody titers.
Discussion
In this study, we report that TNFa-mediated inflammation is a key inhibitor of humoral immune memory, depleting circulating antibody titers and PC survival through direct disruption of the bone marrow niche. These findings are in line with initial studies (35) indicating that TNFa and other inflammatory factors such as alum or infection, can disrupt bone marrow PCs. Here, we find that TNFa is both necessary and sufficient for regulating PC egress from and homing to the bone marrow, and that this process occurs within hours after treatment. While alum can disrupt PC engraftment, anti-TNFa was sufficient to block this effect, indicating that TNFa signaling is uniquely inhibiting engraftment. Within hours of rTNFa treatment, PC motility and vascular permeability are increased within the bone marrow leading to rapid PC mobilization into circulation. Both plasmablasts and plasma cells could be mobilized, but radiation-resistant long-lived plasma cells were less sensitive, suggesting they are protected from inflammation-induced egress, although the mechanisms are unclear. Once in the blood and detached from their niche, PC survival in the blood is likely compromised. Although we were not able to detect cell death in vivo, this decrease in BCL-2 is consistent with previous work showing human ASCs in the blood have lower expression of BCL-2 survival molecule expression than ASCs in the bone marrow as well as increased expression of death receptor CD95 (51). Consequently, we conclude that the decreases in PC survival and antibody titer is largely due to the mobilization of PCs into the blood and further inhibition of re-engraftment.
We also report that TNFa-mediated PC mobilization is a cell-extrinsic cell non-autonomous process, mediated by TNFa receptor 1 signaling by radiation-resistant cells. While PCs do not require TNFa receptors for mobilization, it is unclear which radiation-resistant stromal subsets are involved. It is tempting to speculate that bone marrow stromal cells play a direct and local role in this process. Both human and murine endothelial cells are known to respond to TNFa signaling through TNFR1 leading to increased expression of integrin ligands, ICAM-1 and VCAM-1 (52–55), and they are likely responsible for inducing increased vascular permeability and intravasation. These changes may promote egress of PCs and other cells into circulation. In addition, bone marrow perivascular stromal cells may also be responding locally to TNFa leading to PC egress. Previous work has shown that three days following TNFa treatment, CXCL12 concentrations are reduced in the bone marrow (45). As CXCL12 is a key regulator of ASC retention in the bone marrow (40), it is likely this chemokine could be involved. While we could not detect decreases in CXCL12 within the first 24 hours post treatment (data not shown), CXCR4 levels were elevated by PCs. However, we do not yet know if changes in surface CXCR4 reflect upregulating of expression, reductions in CXCL12 ligand, or selective depletion of PCs with lower CXCR4. While all three mechanisms may be at play, the last model is consistent with the limited mobilization of radiation resistant LLPCs, which have the highest CXCR4 expression.
In addition to potential disruption of known retention mechanisms, TNFa treatment also led to the down-regulation of syndecan-1 (CD138) expression on bone marrow PCs. Again, this was mediated in a cell-extrinsic manner by TNFR1 signaling in radiation-resistant cells. Previous work from our laboratory has shown that CD138 downregulation on malignant myeloma increases their motility, recirculation and dissemination throughout the body (47). Since PCs do not require TNFa receptors to downregulate CD138 directly, this may involve additional cytokines or signals, such as IL-6, which can trigger CD138 downregulation on normal PC (56) or induction of CD138 shedding (57).
Previous studies have proposed that the PC niche is limited and there is competition for access to the bone marrow (58, 59). Our finding that mature PCs and antibody titers are increased two- to five-fold in naïve mice lacking TNFa or its receptors indicates that steady-state tonic TNFa signaling may reduce the PC pool and thereby limit the duration of antibody titer, particularly those from less mature ASCs. While this may seem detrimental to overall humoral immunity, production of TNFa in the immune response may help recruit effector PCs to sites of inflammation and infection (60, 61), and it may also be providing new vacancies in the bone marrow niche for newly generated ASCs to engraft. This trade-off may come at the cost of pre-existing PCs generated from previous vaccinations or infections, as we have shown. While it is tempting to conclude that increases in PCs in the spleen and BM in the TNFa-deficient mice at steady-state is solely due to changes in retention in the BM, as TNFa can play important roles in organizing splenic architecture, and TNFa plays pleiotropic roles throughout immune responses. Indeed, in chimeric mice lacking TNFa signaling in either donor or recipient compartments, increases in splenic PCs were maintained while BM niche was no longer enriched. Thus, multiple mechanisms are likely at play and some of them are disrupted following lethal irradiation and BM transplantation.
Given that TNFa can disrupt PC-bone marrow niche interactions, it serves as a potential therapeutic target in multiple contexts. Designing vaccines that either block or limit the production of TNFa specifically may have pleiotropic effects of protecting pre-existing PCs and their antibody titers as well as minimizing TNFa-induced blockade of bone marrow engraftment. Indeed, while alum can induce multiple pathways of inflammation (62, 63), blocking TNFa was sufficient to increase ASC engraftment. On the contrary, in diseases such as multiple myeloma, the bone marrow protects pathological ASCs and reduces efficacy of chemotherapy (64–66). TNFa treatments can mobilize myeloma cells, increasing their susceptibility to treatments with proteasomal inhibitors like bortezomib class drugs (47). In patients with rheumatoid arthritis, treatments with anti-TNFa drugs, which may reduce T cell-mediated inflammation in joints, may inadvertently retain anti-self ASCs in the bone marrow, leading to additional effector damage (67). Previous studies have shown that targeting a single TNF receptor can have beneficial effects in the context of autoimmune diseases (68–70). Identification of the non-hematopoietic cell that regulates PC retention in the bone marrow with TNF receptor 1 signaling may also provide potential therapeutic targets to disrupt the retention of pathological PCs in the bone marrow and pave the way for more successful therapies. Thus, harnessing TNFa signaling pathways to target the PC niche may unveil new treatments in plasma cell dyscrasias.
Supplementary Material
Key Points:
TNFa depletes antigen specific BM PCs and shortens serological memory.
TNFaR1 in radiation-resistant cells is required to mobilize BM PCs.
Acknowledgements
We thank Dr. Gregoire Lauvau of Albert Einstein College of Medicine for gifting the spike protein and Dr. Zhixin Jing and Rosa Park for assistance with editing this manuscript.
This work was supported by R01HL141491 (DRF), F32HL149155 (ZB), R01AI132633 (KC) and the Eric J. Heyer, M.D., Ph.D. Translational Research Pilot Project Grant Award with support from the Albert Einstein NCI Cancer Center grant P30CA013330.
References
- 1.Slifka MK, Antia R, Whitmire JK, and Ahmed R. 1998. Humoral immunity due to long-lived plasma cells. Immunity 8: 363–372. [DOI] [PubMed] [Google Scholar]
- 2.Amanna IJ, Carlson NE, and Slifka MK. 2007. Duration of humoral immunity to common viral and vaccine antigens. The New England journal of medicine 357: 1903–1915. [DOI] [PubMed] [Google Scholar]
- 3.Ahuja A, Anderson SM, Khalil A, and Shlomchik MJ. 2008. Maintenance of the plasma cell pool is independent of memory B cells. Proc Natl Acad Sci U S A 105: 4802–4807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wols HAM, Underhill GH, Kansas GS, and Witte PL. 2002. The Role of Bone Marrow-Derived Stromal Cells in the Maintenance of Plasma Cell Longevity. The Journal of Immunology 169: 4213–4221. [DOI] [PubMed] [Google Scholar]
- 5.Belnoue E, Pihlgren M, McGaha TL, Tougne C, Rochat AF, Bossen C, Schneider P, Huard B, Lambert PH, and Siegrist CA. 2008. APRIL is critical for plasmablast survival in the bone marrow and poorly expressed by early-life bone marrow stromal cells. Blood 111: 2755–2764. [DOI] [PubMed] [Google Scholar]
- 6.Jourdan M, Cren M, Robert N, Bollore K, Fest T, Duperray C, Guilloton F, Hose D, Tarte K, and Klein B. 2014. IL-6 supports the generation of human long-lived plasma cells in combination with either APRIL or stromal cell-soluble factors. Leukemia 28: 1647–1656. [DOI] [PubMed] [Google Scholar]
- 7.Belnoue E, Tougne C, Rochat AF, Lambert PH, Pinschewer DD, and Siegrist CA. 2012. Homing and adhesion patterns determine the cellular composition of the bone marrow plasma cell niche. J Immunol 188: 1283–1291. [DOI] [PubMed] [Google Scholar]
- 8.Biajoux V, Natt J, Freitas C, Alouche N, Sacquin A, Hemon P, Gaudin F, Fazilleau N, Espeli M, and Balabanian K. 2016. Efficient Plasma Cell Differentiation and Trafficking Require Cxcr4 Desensitization. Cell reports 17: 193–205. [DOI] [PubMed] [Google Scholar]
- 9.Hauser AE, Debes GF, Arce S, Cassese G, Hamann A, Radbruch A, and Manz RA. 2002. Chemotactic responsiveness toward ligands for CXCR3 and CXCR4 is regulated on plasma blasts during the time course of a memory immune response. J Immunol 169: 1277–1282. [DOI] [PubMed] [Google Scholar]
- 10.Tokoyoda K, Egawa T, Sugiyama T, Choi BI, and Nagasawa T. 2004. Cellular niches controlling B lymphocyte behavior within bone marrow during development. Immunity 20: 707–718. [DOI] [PubMed] [Google Scholar]
- 11.Manne C, Takaya A, Yamasaki Y, Mursell M, Hojyo S, Wu TY, Sarkander J, McGrath MA, Cornelis R, Hahne S, Cheng Q, Kawamoto T, Hiepe F, Kaufmann SHE, Yamamoto T, Radbruch A, and Tokoyoda K. 2019. Salmonella SiiE prevents an efficient humoral immune memory by interfering with IgG(+) plasma cell persistence in the bone marrow. Proc Natl Acad Sci U S A 116: 7425–7430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Behrens L, Cherry JD, Heininger U, and Swiss G Measles Immune Amnesia Study. 2020. The Susceptibility to Other Infectious Diseases Following Measles During a Three Year Observation Period in Switzerland. Pediatr Infect Dis J 39: 478–482. [DOI] [PubMed] [Google Scholar]
- 13.Mina MJ, Kula T, Leng Y, Li M, de Vries RD, Knip M, Siljander H, Rewers M, Choy DF, Wilson MS, Larman HB, Nelson AN, Griffin DE, de Swart RL, and Elledge SJ. 2019. Measles virus infection diminishes preexisting antibodies that offer protection from other pathogens. Science 366: 599–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goncalves-Carneiro D, McKeating JA, and Bailey D. 2017. The Measles Virus Receptor SLAMF1 Can Mediate Particle Endocytosis. J Virol 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Banga S, Coursen JD, Portugal S, Tran TM, Hancox L, Ongoiba A, Traore B, Doumbo OK, Huang CY, Harty JT, and Crompton PD. 2015. Impact of acute malaria on pre-existing antibodies to viral and vaccine antigens in mice and humans. PLoS One 10: e0125090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ng DH, Skehel JJ, Kassiotis G, and Langhorne J. 2014. Recovery of an antiviral antibody response following attrition caused by unrelated infection. PLoS Pathog 10: e1003843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wykes MN, Zhou YH, Liu XQ, and Good MF. 2005. Plasmodium yoelii can ablate vaccine-induced long-term protection in mice. J Immunol 175: 2510–2516. [DOI] [PubMed] [Google Scholar]
- 18.Kwiatkowski D, Hill AV, Sambou I, Twumasi P, Castracane J, Manogue KR, Cerami A, Brewster DR, and Greenwood BM. 1990. TNF concentration in fatal cerebral, non-fatal cerebral, and uncomplicated Plasmodium falciparum malaria. Lancet 336: 1201–1204. [DOI] [PubMed] [Google Scholar]
- 19.Kwiatkowski D, Molyneux ME, Stephens S, Curtis N, Klein N, Pointaire P, Smit M, Allan R, Brewster DR, Grau GE, and et al. 1993. Anti-TNF therapy inhibits fever in cerebral malaria. Q J Med 86: 91–98. [PubMed] [Google Scholar]
- 20.de Jong HK, Parry CM, van der Poll T, and Wiersinga WJ. 2012. Host-pathogen interaction in invasive Salmonellosis. PLoS pathogens 8: e1002933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lin WW, Nelson AN, Ryon JJ, Moss WJ, and Griffin DE. 2017. Plasma Cytokines and Chemokines in Zambian Children With Measles: Innate Responses and Association With HIV-1 Coinfection and In-Hospital Mortality. J Infect Dis 215: 830–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chapman PB, Lester TJ, Casper ES, Gabrilove JL, Wong GY, Kempin SJ, Gold PJ, Welt S, Warren RS, Starnes HF, and et al. 1987. Clinical pharmacology of recombinant human tumor necrosis factor in patients with advanced cancer. J Clin Oncol 5: 1942–1951. [DOI] [PubMed] [Google Scholar]
- 23.Michie HR, Manogue KR, Spriggs DR, Revhaug A, O’Dwyer S, Dinarello CA, Cerami A, Wolff SM, and Wilmore DW. 1988. Detection of circulating tumor necrosis factor after endotoxin administration. N Engl J Med 318: 1481–1486. [DOI] [PubMed] [Google Scholar]
- 24.Engelmann H, Aderka D, Rubinstein M, Rotman D, and Wallach D. 1989. A tumor necrosis factor-binding protein purified to homogeneity from human urine protects cells from tumor necrosis factor toxicity. J Biol Chem 264: 11974–11980. [PubMed] [Google Scholar]
- 25.Bradley JR 2008. TNF-mediated inflammatory disease. J Pathol 214: 149–160. [DOI] [PubMed] [Google Scholar]
- 26.Chen X, Wu X, Zhou Q, Howard OM, Netea MG, and Oppenheim JJ. 2013. TNFR2 is critical for the stabilization of the CD4+Foxp3+ regulatory T. cell phenotype in the inflammatory environment. J Immunol 190: 1076–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zelova H, and Hosek J. 2013. TNF-alpha signalling and inflammation: interactions between old acquaintances. Inflamm Res 62: 641–651. [DOI] [PubMed] [Google Scholar]
- 28.Irwin MW, Mak S, Mann DL, Qu R, Penninger JM, Yan A, Dawood F, Wen WH, Shou Z, and Liu P. 1999. Tissue expression and immunolocalization of tumor necrosis factor-alpha in postinfarction dysfunctional myocardium. Circulation 99: 1492–1498. [DOI] [PubMed] [Google Scholar]
- 29.Bocker W, Docheva D, Prall WC, Egea V, Pappou E, Rossmann O, Popov C, Mutschler W, Ries C, and Schieker M. 2008. IKK-2 is required for TNF-alpha-induced invasion and proliferation of human mesenchymal stem cells. J Mol Med (Berl) 86: 1183–1192. [DOI] [PubMed] [Google Scholar]
- 30.Yang L, Lindholm K, Konishi Y, Li R, and Shen Y. 2002. Target depletion of distinct tumor necrosis factor receptor subtypes reveals hippocampal neuron death and survival through different signal transduction pathways. J Neurosci 22: 3025–3032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Arnett HA, Mason J, Marino M, Suzuki K, Matsushima GK, and Ting JP. 2001. TNF alpha promotes proliferation of oligodendrocyte progenitors and remyelination. Nat Neurosci 4: 1116–1122. [DOI] [PubMed] [Google Scholar]
- 32.Hsu H, Shu HB, Pan MG, and Goeddel DV. 1996. TRADD-TRAF2 and TRADD-FADD interactions define two distinct TNF receptor 1 signal transduction pathways. Cell 84: 299–308. [DOI] [PubMed] [Google Scholar]
- 33.Liu ZG, Hsu H, Goeddel DV, and Karin M. 1996. Dissection of TNF receptor 1 effector functions: JNK activation is not linked to apoptosis while NF-kappaB activation prevents cell death. Cell 87: 565–576. [DOI] [PubMed] [Google Scholar]
- 34.Naude PJ, den Boer JA, Luiten PG, and Eisel UL. 2011. Tumor necrosis factor receptor cross-talk. FEBS J 278: 888–898. [DOI] [PubMed] [Google Scholar]
- 35.Slocombe T, Brown S, Miles K, Gray M, Barr TA, and Gray D. 2013. Plasma cell homeostasis: the effects of chronic antigen stimulation and inflammation. J Immunol 191: 3128–3138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fooksman DR, Schwickert TA, Victora GD, Dustin ML, Nussenzweig MC, and Skokos D. 2010. Development and migration of plasma cells in the mouse lymph node. Immunity 33: 118–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McCarron MJ, Park PW, and Fooksman DR. 2017. CD138 mediates selection of mature plasma cells by regulating their survival. Blood 129: 2749–2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Laudermilch E, and Chandran K. 2021. MAVERICC: Marker-free Vaccinia Virus Engineering of Recombinants through in vitro CRISPR/Cas9 Cleavage. J Mol Biol 433: 166896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pitt LA, Tikhonova AN, Hu H, Trimarchi T, King B, Gong Y, Sanchez-Martin M, Tsirigos A, Littman DR, Ferrando AA, Morrison SJ, Fooksman DR, Aifantis I, and Schwab SR. 2015. CXCL12-Producing Vascular Endothelial Niches Control Acute T Cell Leukemia Maintenance. Cancer cell 27: 755–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Benet Z, Jing Z, and Fooksman DR. 2021. Plasma cell dynamics in the bone marrow niche. Cell reports 34: 108733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Slifka MK, Antia R, Whitmire JK, and Ahmed R. 1998. Humoral immunity due to long-lived plasma cells. Immunity 8: 363–372. [DOI] [PubMed] [Google Scholar]
- 42.Mackay F, Loetscher H, Stueber D, Gehr G, and Lesslauer W. 1993. Tumor necrosis factor alpha (TNF-alpha)-induced cell adhesion to human endothelial cells is under dominant control of one TNF receptor type, TNF-R55. J Exp Med 177: 1277–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Madge LA, and May MJ. 2010. Classical NF-kappaB activation negatively regulates noncanonical NF-kappaB-dependent CXCL12 expression. J Biol Chem 285: 38069–38077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Madge LA, Kluger MS, Orange JS, and May MJ. 2008. Lymphotoxin-alpha 1 beta 2 and LIGHT induce classical and noncanonical NF-kappa B-dependent proinflammatory gene expression in vascular endothelial cells. J Immunol 180: 3467–3477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ueda Y, Yang K, Foster SJ, Kondo M, and Kelsoe G. 2004. Inflammation controls B lymphopoiesis by regulating chemokine CXCL12 expression. J Exp Med 199: 47–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ren Z, Spaargaren M, and Pals ST. 2021. Syndecan-1 and stromal heparan sulfate proteoglycans: key moderators of plasma cell biology and myeloma pathogenesis. Blood 137: 1713–1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Akhmetzyanova I, McCarron MJ, Parekh S, Chesi M, Bergsagel PL, and Fooksman DR. 2020. Dynamic CD138 surface expression regulates switch between myeloma growth and dissemination. Leukemia 34: 245–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lam WY, Jash A, Yao CH, D’Souza L, Wong R, Nunley RM, Meares GP, Patti GJ, and Bhattacharya D. 2018. Metabolic and Transcriptional Modules Independently Diversify Plasma Cell Lifespan and Function. Cell reports 24: 2479–2492 e2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.De Gregorio E, Caproni E, and Ulmer JB. 2013. Vaccine adjuvants: mode of action. Front Immunol 4: 214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Korsholm KS, Petersen RV, Agger EM, and Andersen P. 2010. T-helper 1 and T-helper 2 adjuvants induce distinct differences in the magnitude, quality and kinetics of the early inflammatory response at the site of injection. Immunology 129: 75–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Medina F, Segundo C, Campos-Caro A, Gonzalez-Garcia I, and Brieva JA. 2002. The heterogeneity shown by human plasma cells from tonsil, blood, and bone marrow reveals graded stages of increasing maturity, but local profiles of adhesion molecule expression. Blood 99: 2154–2161. [DOI] [PubMed] [Google Scholar]
- 52.Chen G, and Goeddel DV. 2002. TNF-R1 signaling: a beautiful pathway. Science 296: 1634–1635. [DOI] [PubMed] [Google Scholar]
- 53.Pober JS, Lapierre LA, Stolpen AH, Brock TA, Springer TA, Fiers W, Bevilacqua MP, Mendrick DL, and Gimbrone MA Jr. 1987. Activation of cultured human endothelial cells by recombinant lymphotoxin: comparison with tumor necrosis factor and interleukin 1 species. J Immunol 138: 3319–3324. [PubMed] [Google Scholar]
- 54.Zhou Z, Connell MC, and MacEwan DJ. 2007. TNFR1-induced NF-kappaB, but not ERK, p38MAPK or JNK activation, mediates TNF-induced ICAM-1 and VCAM-1 expression on endothelial cells. Cell Signal 19: 1238–1248. [DOI] [PubMed] [Google Scholar]
- 55.Lu ZY, Chen WC, Li YH, Li L, Zhang H, Pang Y, Xiao ZF, Xiao HW, and Xiao Y. 2016. TNF-alpha enhances vascular cell adhesion molecule-1 expression in human bone marrow mesenchymal stem cells via the NF-kappaB, ERK and JNK signaling pathways. Mol Med Rep 14: 643–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sneed TB, Stanley DJ, Young LA, and Sanderson RD. 1994. Interleukin-6 regulates expression of the syndecan-1 proteoglycan on B lymphoid cells. Cell Immunol 153: 456–467. [DOI] [PubMed] [Google Scholar]
- 57.Fitzgerald ML, Wang Z, Park PW, Murphy G, and Bernfield M. 2000. Shedding of syndecan-1 and −4 ectodomains is regulated by multiple signaling pathways and mediated by a TIMP-3-sensitive metalloproteinase. J Cell Biol 148: 811–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Manz RA, Hauser AE, Hiepe F, and Radbruch A. 2005. Maintenance of serum antibody levels. Annu Rev Immunol 23: 367–386. [DOI] [PubMed] [Google Scholar]
- 59.Radbruch A, Muehlinghaus G, Luger EO, Inamine A, Smith KG, Dorner T, and Hiepe F. 2006. Competence and competition: the challenge of becoming a long-lived plasma cell. Nature reviews. Immunology 6: 741–750. [DOI] [PubMed] [Google Scholar]
- 60.Wilson RP, McGettigan SE, Dang VD, Kumar A, Cancro MP, Nikbakht N, Stohl W, and Debes GF. 2019. IgM Plasma Cells Reside in Healthy Skin and Accumulate with Chronic Inflammation. J Invest Dermatol 139: 2477–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mallison SM, Smith JP, Schenkein HA, and Tew JG. 1991. Accumulation of plasma cells in inflamed sites: effects of antigen, nonspecific microbial activators, and chronic inflammation. Infect Immun 59: 4019–4025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li H, Willingham SB, Ting JP, and Re F. 2008. Cutting edge: inflammasome activation by alum and alum’s adjuvant effect are mediated by NLRP3. J Immunol 181: 17–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.McKee AS, Munks MW, MacLeod MK, Fleenor CJ, Van Rooijen N, Kappler JW, and Marrack P. 2009. Alum induces innate immune responses through macrophage and mast cell sensors, but these sensors are not required for alum to act as an adjuvant for specific immunity. J Immunol 183: 4403–4414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Meads MB, Hazlehurst LA, and Dalton WS. 2008. The bone marrow microenvironment as a tumor sanctuary and contributor to drug resistance. Clin Cancer Res 14: 2519–2526. [DOI] [PubMed] [Google Scholar]
- 65.Robinson MJ, Webster RH, and Tarlinton DM. 2020. How intrinsic and extrinsic regulators of plasma cell survival might intersect for durable humoral immunity. Immunological reviews 296: 87–103. [DOI] [PubMed] [Google Scholar]
- 66.Agarwal D, Allman D, and Naji A. 2020. Novel therapeutic opportunities afforded by plasma cell biology in transplantation. Am J Transplant 20: 1984–1991. [DOI] [PubMed] [Google Scholar]
- 67.Hiepe F, Dorner T, Hauser AE, Hoyer BF, Mei H, and Radbruch A. 2011. Long-lived autoreactive plasma cells drive persistent autoimmune inflammation. Nat Rev Rheumatol 7: 170–178. [DOI] [PubMed] [Google Scholar]
- 68.Wang YL, Chou FC, Chen SJ, Lin SH, Chang DM, and Sytwu HK. 2011. Targeting pre-ligand assembly domain of TNFR1 ameliorates autoimmune diseases - an unrevealed role in downregulation of Th17 cells. J Autoimmun 37: 160–170. [DOI] [PubMed] [Google Scholar]
- 69.Li P, Zheng Y, and Chen X. 2017. Drugs for Autoimmune Inflammatory Diseases: From Small Molecule Compounds to Anti-TNF Biologics. Front Pharmacol 8: 460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Williams SK, Fairless R, Maier O, Liermann PC, Pichi K, Fischer R, Eisel ULM, Kontermann R, Herrmann A, Weksler B, Romero N, Couraud PO, Pfizenmaier K, and Diem R. 2018. Anti-TNFR1 targeting in humanized mice ameliorates disease in a model of multiple sclerosis. Sci Rep 8: 13628. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.