

Abstract
Thousands of long non-coding RNAs (lncRNAs) are encoded in mammalian genomes, yet most remain uncharacterized. Here, we functionally characterized a mouse lncRNA named U90926. Analysis of U90926 RNA levels revealed minimal expression across multiple tissues at steady state. However, the expression of this gene was highly induced in macrophages and dendritic cells by toll-like receptor activation, in a p38 MAP kinase- and MyD88-dependent manner. To study the function of U90926, we generated U90926-deficient (U9-KO) mice. Surprisingly, we found minimal effects of U90926 deficiency in cultured macrophages. Given the lack of macrophage-intrinsic effect, we investigated the subcellular localization of U90926 transcript and its protein-coding potential. We found that U90926 RNA localizes to the cytosol, associates with ribosomes, and contains an open reading frame that encodes a novel glycosylated protein (termed U9-ORF), which is secreted from the cell. An in vivo model of endotoxic shock revealed that, in comparison with WT mice, U9-KO mice exhibited increased sickness responses and mortality. Mechanistically, serum levels of IL-6 were elevated in U9-KO mice, and IL-6 neutralization improved endotoxemia outcomes in U9-KO mice. Taken together, these results suggest that U90926 expression is protective during endotoxic shock, potentially mediated by the paracrine and/or endocrine actions of the novel U9-ORF protein secreted by activated myeloid cells.
Keywords: LncRNA, macrophages, toll-like receptors, endotoxic shock, IL-6
Introduction
Macrophages are the prototypical innate immune cell, strategically positioned to encounter infectious organisms, detect them via an array of microbial pattern recognition receptors (PRRs), and rapidly respond by producing pro-inflammatory mediators that alert the immune system. While this response is required for efficient clearance of infectious agents, an over exuberant response can drive immune-mediated pathology and fatal systemic shock(1). Therefore, regulatory mechanisms underlying these responses could be harnessed for treating conditions such a septic shock, which annually causes ~270,000 deaths in the U.S. alone(2).
Gene expression in mammals is a highly dynamic, complex, and tightly regulated process. This is particularly evident in innate immune cells, which can completely reprogram their entire gene expression repertoire within minutes of encountering an inflammatory stimulus. The central dogma of molecular biology dictates that information encoded in the genome is transcribed into RNA, followed by translation into protein, with the latter carrying out most cellular functions. However, recent advances in genomics have challenged this notion by demonstrating that protein-coding transcripts may in fact be outnumbered by non-canonical transcripts that do not encode proteins(3–5). Emerging evidence suggests that a large class of such transcripts, the so-called long non-coding RNAs (lncRNAs), defined as greater than 200 nucleotides in length and lacking a conserved open reading frame (ORF)(3), play unique cell type-specific roles in numerous cellular processes, including innate immune cell effector function(6–8).
Several early landmark studies have identified specific lncRNAs, such as lincRNA-Cox2, THRIL, and lincRNA-EPS, that function as positive or negative regulators of macrophage function and pro-inflammatory gene expression(9–11). These lncRNAs control gene expression and cellular functions in diverse ways, including interaction with proteins involved in gene expression regulation or signaling, interactions with chromatin, interactions with mRNAs or miRNAs, and others(6, 7). Each lncRNA appears to have its own unique mode of action, and novel mechanisms continue to emerge. Among these is the emerging evidence of pervasive translation of lncRNAs(12, 13), including two recent examples in which the protein products control macrophage inflammatory functions(14, 15).
Our previous studies(16–18) have focused on a central signaling pathway activated in macrophages downstream of PRRs, the p38 MAP kinase (MAPK) pathway, in which we identified an uncharacterized putative lncRNA named U90926, which was among the most highly downregulated genes in p38α MAPK-deficient murine macrophages(16, 18). Since the initial annotation of U90926 lncRNA by a genome exploration research group(19), there was little known about the function of this gene. However, several studies have begun to shed some light on the biological function of U90926, including a possible role as a repressor of adipogenesis in vitro, a viral replication co-factor in retinal cells in vitro, and a pro-inflammatory factor in microglia during experimental stroke(20–22). Here, we focused on the function of U90926 in macrophages. We demonstrate that the expression of this gene is highly inducible in myeloid cells by lipopolysaccharide (LPS) and other toll-like receptor (TLR) agonists in a MyD88- and p38α-dependent manner. To study the role of this gene in vivo, using CRISPR-Cas9, we generated the first (to our knowledge) mouse completely lacking the U90926 locus (U9-KO). We demonstrate that, surprisingly, U90926 deficiency has minimal intrinsic effects on macrophage pro-inflammatory gene expression in vitro. Unexpectedly, we found that U90926 RNA localizes to the cytoplasm, associates with ribosomes, and contains an ORF that encodes a novel 87-amino acid-long protein that is secreted from the cell. In vivo, U90926 deficiency exacerbates endotoxic shock through the upregulation IL-6 production. Taken together, our results suggest that TLR-mediated induction of U90926 in macrophages may regulate inflammatory responses through paracrine production of the novel secreted protein encoded in this locus. These findings challenge the “non-coding” paradigm of lncRNA function, warranting a careful examination of the coding potential of ORFs contained in each of these genes.
Materials and Methods
Animals
Wild type C57BL/6J (B6) mice and wild-derived PWD/PhJ (PWD) mice were purchased from Jackson Laboratories (Bar Harbor, Maine, USA) and were acclimated at the animal facility at UVM for at least 2 weeks prior to any experimentation, including breeding. p38αCKOLysM mice were generated in our laboratory previously(16), by crossing LysM-Cre mice (B6.129P2-Lyz2tm1(cre)Ifo/J)(23), and p38αf/f (floxed) mice (B6.129-Mapk14tm1.2OtsuOtsuRbrc)(24). U90926-deficient mice (C57BL6/J-U90926em1Uvm) were generated as described below. 8–12 weeks old male and/or female mice were used in all experiments. The experimental procedures were approved by the Institutional Animal Care and Use Committee of the University of Vermont.
Generation of U90926-deficient mice
U90926-deficient mice (U9-KO) were generated by CRISPR/Cas9 in B6 background. Using the CRISPOR tool(25), we designed two custom single guide RNAs (sgRNAs) targeting cut sites ~200 bp upstream of the first exon (site 1: 5’-ATACGGATGCAGCCTGTACA -3’) and ~200 bp downstream of the last exon (site 2: 5’- GCGGCCCACGTACCATGCGT-3’), to delete the entire ~5.5 Kb U90926 locus (Fig. 2a). The sgRNAs and recombinant Cas9 enzyme were prepared by Synthego, Inc (Redwood City, CA). Microinjection of single cell embryos was performed at the University of Rochester Mouse Genome Editing Resource, on a fee-for-service basis. Two primer sets were used for the initial PCR-based genotyping. One set of primers (forward primer: 5’-GCTGAAAGAGAACACATTGCCTT-3’ and reverse primer: 5’-GGGCTAAGATGTTGCTTCCTGT-3’) targets the 5’ WT region and produces a 500 bp amplicon, which should be absent in KO. A second set of primers (forward primer: 5’-GCTGAAAGAGAACACATTGCCTT-3’ and reverse primer: 5’- ACTGAGAGAAGCCAAAGCTCA -3’) was designed flanking the entire region, which only yields product if the deletion is present (in between 200–500 bp, depending on size of deletion). A single male U9-KO mouse was chosen as the F0 founder to generate the U9-KO colony. To eliminate any potential off-target deletions made by CRISPR/Cas9 elsewhere in the genome, U9-KO mice were backcrossed with WT B6 mice. A single male KO founder was bred to two WT B6 females. Resulting heterozygous (HET) U9-KO were backcrossed to B6 for 2 more generations and subsequently bred as HET ˣ HET to generate all subsequent KO animals and WT controls. An optimized PCR genotyping reaction was designed to identify U9-KO and WT genotypes. Three primers were used at in a single reaction: Forward primer (5’-GAGCCTTTGTGCAAGGCTTAT-3’) was designed to bind upstream of the sgRNA site 1, KO reverse primer (5’-CAGCCCAAGTACACCTGCT-3’) binds downstream of sgRNA site 2 (should yield a 152 bp product only if U90926 whole locus is deleted), and a WT reverse primer (5’-ATGGATTCCCCACCTCATTCC-3’) binds downstream of sgRNA site 1 and near the exon 1 (only yields a 346 bp product when U90926 is not deleted). HET mice produce both the products. In all the PCR reactions, Accuris 2x Taq Red Dye Master Mix (Accuris instruments, Dublin, Ireland) was used. Annealing temperature was 550C for the primers and total 35 cycles were run in the Biometra Trio thermal cycler (Analytik Jena, Upland, CA). Backcrossed U9-KO mice were also validated by Sanger sequencing at the Vermont Integrative Genomics Resource core facility at UVM. A representative U9-KO genomic sequence (assembled contigs 5’ to 3’) showing the deletion is given below (SgRNA binding sites are underlined):
Figure 2. Generation and validation of U9-KO mice.
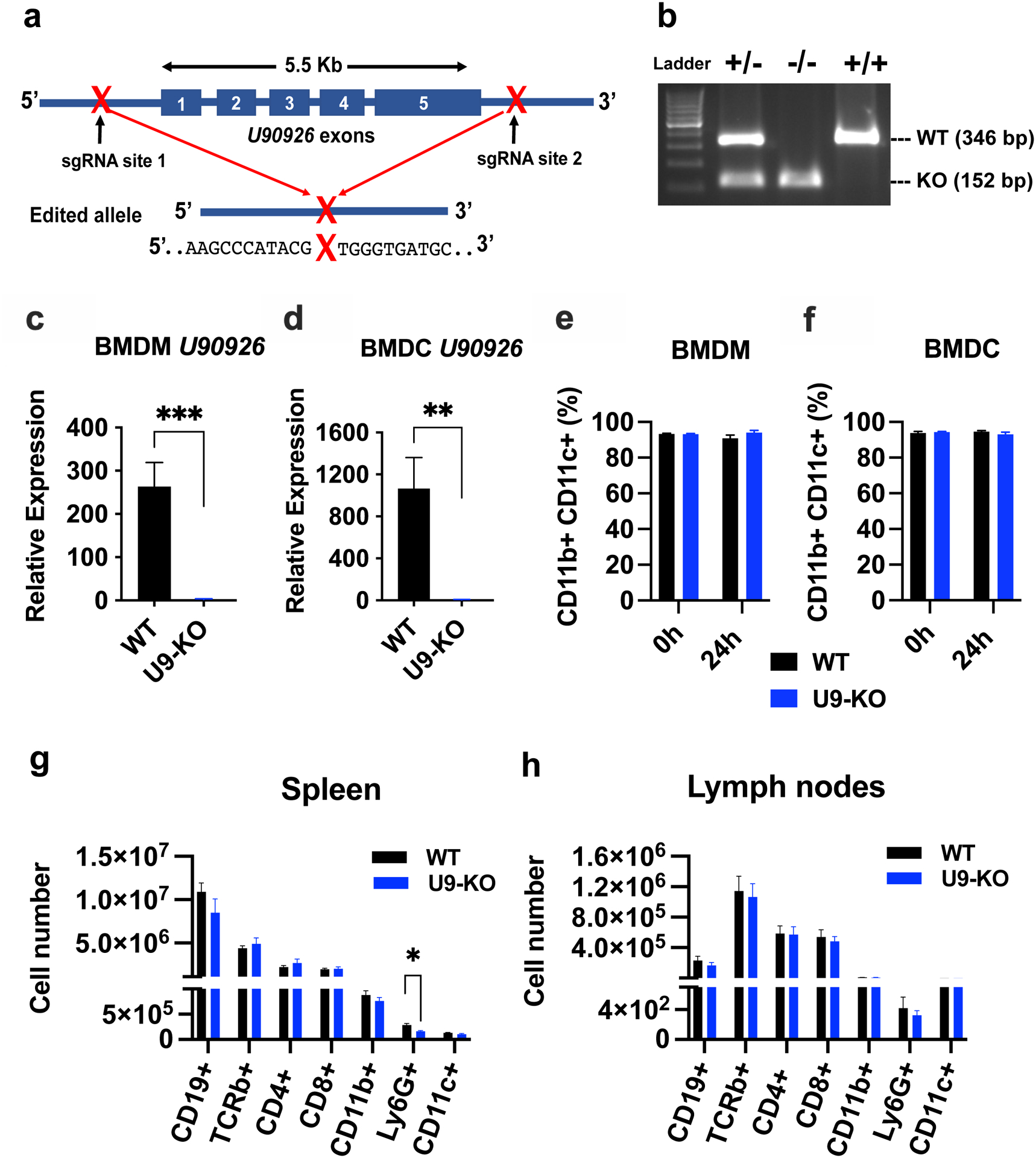
(a) Schematic of CRISPR targeting of the U90926 locus and the repaired double-strand break site (designated as “X”) flanked by the indicated sequences, as identified by Sanger sequencing. (b) PCR-based genotyping of U9-KO mice. Three primers were used in a single reaction: Forward primer was upstream of the sgRNA site 1, KO reverse primer downstream of sgRNA site 2 (yields a product only if U90926 whole locus is deleted), and a WT reverse primer downstream of sgRNA site 1 and near the exon 1 (only yields a product when U90926 is not deleted). (c, d) BMDM and BMDC were isolated from WT and U9-KO mice (n=6 for each group) and stimulated with LPS (100 ng/ml) for 4h, followed by RT-qPCR to measure U90926 expression. (e, f) BMDM and BMDC isolated from WT and U9-KO mice (n=8 for each group) were stimulated with LPS (100ng/ml) for 24h and analyzed by flow cytometry using CD11b and CD11c differentiation markers. (g, h) Cells were isolated from spleen and lymph nodes of naïve WT and U9-KO mice (n=8 for each group). Flow cytometry and cell counting was used to enumerate the following cell subsets: CD19+ (B cells; CD19+ CD11b− TCRβ−), TCRβ+ ((T cells; CD19− CD11b− TCRβ+), CD4+ (CD4 T cells; CD19− CD11b− TCRβ+ CD4+) and CD8+ (CD8 T cells; CD19− CD11b− TCRβ+ CD8+) and different myeloid cells markers, CD11b+ (CD19− CD11b+ TCRβ− CD11c−), CD11c+ (CD19− CD11b− TCRβ− CD11c+) and Ly6G+ (CD19− CD11b+ TCRβ− CD11c− Ly6G+). Data are represented as the mean ± SEM. Significance was determined by Student’s t test in panel (c) and (d). Two-way ANOVA was performed in panel (e) and (f) and Student’s t test was performed for panel (g) and (h). P values are represented as follows: ns= >0.05, *= ≤0.05, **= ≤0.01, ***= ≤0.001.
GGATGGGACCCCCCCAACACCTGAGGCAACATGACCATGCTGAGGTTGGACAGGACAGGGGAGCCTTTGTGCAAGGCTTATACATCTATGCCAAGCTTCTGTCTCCCGCCCAGTGAGACCCAGGGCAAAGCCCATACG……………………………………………………………TGGGTGATGCTATAGTGATATAGCAGGTGTACTTGGGCTG
Cell culture experiments
Bone marrow-derived macrophages (BMDMs) and bone marrow-derived dendritic cells (BMDCs) were generated from mouse bone marrow using a standard 7-day M-CSF (10ng/ml, Peprotech) and GM-CSF (20ng/ml, Peprotech) differentiation protocol, respectively as previously described(17, 26). BMDMs were cultured in complete DMEM media containing 10% FBS, 2 mM L-glutamine, 100 Units/ml penicillin, and 100 μg/ml streptomycin. BMDCs were cultured in complete RPMI 1640 media containing 10% FBS, 2 mM L-glutamine, 100 Units/ml penicillin, 100 μg/ml streptomycin, and 55 μM beta-mercaptoethanol. At day 7, differentiated BMDM and BMDC were harvested and plated (1 million cells/well in a 12-well dish or 200K/well in a 96-well dish) for different in vitro experiments with indicated treatments. Cells from peritoneal cavity were isolated by lavage as described previously(16). Immortalized MyD88-KO macrophages(15) were generously provided by the Fitzgerald lab (UMass Medical School, MA) and cultured in complete DMEM with 10% FBS and penicillin and streptomycin. RAW 264.7 macrophages, HEK 293T cells, HeLa cells, 3T3 fibroblasts, and 3T3-L1 preadipocytes, originally obtained from ATCC and donated by various investigators, all were cultured in DMEM supplemented with 10% FBS and sodium pyruvate. AML12 cells were obtained from ATCC and cultured in DMEM F12: HEPES with 10%FBS, ITS-G 100X (Gibco) and 100nM dexamethasone (Gibco). For co-culture, AML12 and primary BMDMs were used at 4:1 ratio and cultured in AML12 media described above. For all the in vitro lipopolysaccharide (LPS) stimulation studies, we used 100 ng/ml of E. coli LPS O26:B6 (Sigma Aldrich, St. Louis, MO). Select agonists from the mouse TLR1–9 Agonist kit (InvivoGen, San Diego, CA) were used to activate TLR1/2, TLR3, TLR5, and TLR9 and cytokine receptors were activated using recombinant mouse TNFα and IL-1β (Peprotech). For the p38 MAPK inhibition experiment, SB203580 (Tocris Bioscience, Bristol, UK) was used at 10 μM. For all in vitro experiments, biological replicates were used, with each replicate representing independent BMDM cultures from individual mice.
RNA extraction and quantitative reverse transcription PCR (RT-qPCR)
For most of the in vitro experiments, 12-well plates were seeded with 1×106 cells per well and stimulated at different time points. RNA was extracted according to the manufacturer’s instructions using the RNeasy plus Mini Kit by Qiagen (Hilden, Germany). For in vivo experiments, Direct-zol RNA Microprep Kits (Zymo Research, Irvine, CA) were used for RNA extraction as per manufacturer’s protocol. Whole liver tissue samples were collected into 2 ml screw cap tubes containing 1/3 full of 1.0mm diameter Silicon-Carbide particles (Biospec Products, Bartlesville, OK) and 1 ml of TRIzol reagent (Invitrogen, Waltham, MA) per tube. Then, the samples were homogenized using a Mini-Beadbeater instrument by Biospec Products (Bartlesville, OK) for total 2 min by 4 cycles, and placing tubes on ice-water bath in each interval to avoid over-heating. Then, tubes were centrifuged at 13,000 rpm for 2 min to pellet any debris, 200μL of homogenate were collected and 100μL of fresh TRIzol was added, followed by extraction using the Direct-zol RNA Microprep Kit as above.
RNA concentration was determined by Nanodrop (Thermo Scientific NanoDrop 2000 Spectrophotometer) and cDNA synthesis reaction by reverse transcription was performed according to the manufacturer’s instructions using the qScript cDNA Super MIX kit (QuantBio, Beverly, MA). The qPCR reaction was performed according to the manufacturer’s instructions using the DyNAmo ColorFlash SYBR Green kit by Thermo Fisher Scientific (Waltham, MA), using target-specific primers. The qPCR was run on a Quant Studio 3 Real-Time PCR System by Applied Biosystems (Thermo Fisher Scientific, Waltham, MA). Target gene expression was normalized by the expression of the housekeeping gene B2m (β-2-Microglobulin) and calculated by a comparative Ct method formula 2^-(deltaCt) and multiplied by a factor of 10,000 for ease of visualization, i.e. 2^-(Cttarget gene – CtB2m)*10,000. We did not normalize relative expression to a reference sample (i.e. delta-deltaCt).
Flow cytometry
For in vitro experiments, BMDMs and BMDCs differentiated for 7 days were seeded at 1×106 cells/well in a 12-well dish. Cells were treated with LPS, and 0 and 24 hours and after incubation cells were removed using non-enzymatic cell stripper liquid (Corning Inc., Corning, NY) and subjected to staining for flow cytometry. For in vivo experiments, spleen and lymph nodes were isolated from mice single cell suspensions were prepared followed by passing through a 70 μm strainer. Red blood cell (RBC) lysis was performed by incubating the cells in 0.8% ammonium chloride solution (StemCell Technologies, Cambridge, MA) and cells were subjected to staining for flow cytometry analysis. Cells were stained with the UV LIVE/DEAD fixable stain (Invitrogen) and then surface labeled for different combinations of following markers: CD11b, CD11c, CD80, CD86, CD40, CD19, TCRβ, Ly6G, MHCII, CD4, and CD8 (Biolegend, San Diego, CA) and fixed with 1% paraformaldehyde (Sigma Aldrich, St. Louis, MO). Samples were analyzed on an LSRII cytometer (BD Biosciences) or an Aurora (Cytek Biosciences). All flow cytometry data analysis was performed using FlowJo Software version 10.6.1 (BD Biosciences).
Cytokine quantification by ELISA
For the detection of cytokines in the cell culture supernatants or sera, ELISAs were performed as described previously(17), using the primary capture mAbs anti-TNFα, anti-IL-6, and anti-IL-1β and their corresponding biotinylated detection mAbs (Biolegend, San Diego, CA). Other ELISA reagents included: recombinant mouse TNFα, IL-6, and IL-1β as standards (Biolegend, San Diego, CA), HRP-conjugated avidin D (Vector Laboratories, Burlingame, CA), and TMB microwell peroxidase substrate and stop solution (Kirkegaard and Perry Laboratories, Gaithersburg, MD). IL-10 cytokine was measured using Mouse IL-10 DuoSet ELISA kit (R&D Systems, Minneapolis, MN) according to manufacturer’s instructions. Biological replicates were used, representing independent BMDM cultures from individual mice.
RNAseq and bioinformatic analysis
Four biological replicates per genotype, representing independent BMDM cultures from individual mice, were used. BMDMs were seeded at 1×106 cells/well and treated with LPS for 4 and 24 hours. RNA was extracted as described above for in vitro experiments, followed by quantification, quality assessment, library preparation, and sequencing at the Vermont Integrative Genomics Resource core facility at UVM. cDNA libraries were synthesized using SMARTer Stranded Total RNA-Seq Kit v2 - Pico Input Mammalian (Takara, Japan), according to manufacturer protocol. Ribosomal RNA depletion was done to ensure recovery of a range of RNA species. Single-end 75 bp sequencing was performed on an Illumina HiSeq1500, generating around 30 million reads per sample.
For whole liver tissue RNAseq, RNA was extracted as described above, followed by quantification and quality assessment. Library preparation and sequencing were performed commercially by Novogene (Novogene Co, China), as follows. mRNA library preparation (poly A enrichment) was done by using NEB Next Ultra II Directional RNA Library Prep Kit for Illumina (New England Biolabs, Ipswich, MA) followed by paired end 150 bp sequencing using NovaSeq 6000 platform, to generate 20 million reads per sample.
Demultiplexed sequencing data was received from the sequencing facilities and quality control of the sequencing data was done using FastQC platform. Genomic alignment and transcript quantification from RNA-seq data was performed using Salmon tool(27). Differential gene-expression analyses were conducted using DESeq2(28) in R studio. Cutoffs for differential gene expression are described in the Results section. Gene ontology (GO) enrichment analysis on differentially expressed genes was performed using PANTHER(29). For upstream regulator analysis, Ingenuity Pathways Analysis (IPA) was used where we input list of DEGs and their fold change from cutoff logFC= |0.6| and Padj=<0.2.
RNA subcellular localization
WT BMDMs were stimulated with LPS for 4 and 24 hours to induce U90926 expression, followed by lysis and nuclear and cytoplasmic fractionation. Nuclear and cytoplasmic fractions were separated by NE-PER Nuclear and Cytoplasmic Extraction Reagents Kit (Thermo Fisher Scientific, Waltham, MA) according to manufacturer’s protocol. After separation, TRIzol LS reagents (Thermo Fisher Scientific, Waltham, MA) were added to lysates according to manufacturer’s suggested ratio to liquid sample. Then, we proceeded with the Direct-zol RNA micro prep kit for RNA extraction, followed by cDNA synthesis and RT-qPCR, as described above. Abundance of each transcript target in nuclear and cytoplasmic fractions was normalized to its respective abundance in an unfractionated input sample.
Plasmid constructs
U9-ORF cDNA was amplified by PCR from cDNA from LPS-stimulated BMDM. At the C-terminal end of the U9-ORF, before the stop codon, a hemagglutinin (HA) epitope tag (5’-TACCCATACGACGTGCCTGACTACGCC-3’) was added by PCR. Amplified cDNAs were cloned into the pcDNA3.1 vector (ThermoFisher, USA) using EcoRI and NotI restriction sites. The correct sequence of the U9-ORF-HA construct was confirmed by Sanger sequencing. The pcDNA3.1 TYK2-HA construct was previously generated in our laboratory(30). The U9-full cDNA-HA construct consists of full U90926 cDNA (522 bp) with HA tag sequence inserted just before the stop codon in the ORF, cloned into pBABE-puro vector (Addgene #1764). The U9-full cDNA-HA construct in pBABE-puro background was constructed commercially by insert synthesis and ligation at Agentide (Westfield, NJ).
Immunocytochemistry (ICC)
HeLa, 3T3, 3T3-L1, and RAW264.7 cells were cultured in 35 mm glass bottom dishes (MatTek, Ashland, MA). 80% confluent cells were transfected with different plasmids using Lipofectamine2000 (Invitrogen) according to manufacturer’s protocol. 48 hours post transfection, cells were fixed with 1% PFA/PBS for 20 min at RT. Cells were washed 3x with PBS, blocked and permeabilized for 10 minutes with 1% BSA/PBS containing 0.1% Saponin, and incubated for 1 hour with 1:1000 diluted anti-HA.11 epitope tag mouse antibody (Biolegend, San Diego, CA) and 1:500 diluted anti-golgin-97 rabbit (Molecular Probes, Eugene, OR) primary antibodies in 1% BSA/PBS. Cells were washed 2x with PBS, blocked with 1% BSA/PBS for 1 hour, and incubated for an additional hour with Alexa Fluor 488 goat anti-mouse IgG and Alexa Fluor 568 goat anti-rabbit IgG secondary antibodies (Invitrogen). 2x PBS wash was preformed and DAPI (1:10,000) in PBS was added for 10 min. The same protocols were followed for experiments with protein transport inhibitors in which inhibitors were added 24 hours post transfection, meaning 24 hours before collection. Therein, GolgiPlug containing brefeldin A (BD Biosciences) at 1:2000 dilution or GolgiStop containing monensin (BD Biosciences) at 1:1000 dilution was used. Cells were stored in PBS and imaged on a DeltaVision epifluorescence microscope (Cytiva/GE/Applied Precision, Issaquah, WA) with a Xenon illuminator, using an Olympus 60× PlanApo 1.42 NA oil immersion objective. Z-stacks of acquired images were deconvolved using softWoRx software using default settings (Applied Precision) to reassign out of focus light to the appropriate Z-slice. Microscopy images from individual channels were exported as TIFF files and further processed using Fiji software (National Institutes of Health), as follows. Each fluorescence channel image was adjusted for optimal visualization by adjusting the minimum and maximum levels. Adjustments were always applied to the entire image.
Immunoblot analysis
At 24 hours post-transfection, media was changed to serum free media and samples were collected at 48 hours post-transfection. Cell supernatants were collected and whole-cell lysates were prepared by lysing cells in RIPA buffer. For concentrating supernatants, StrataClean Resin (Agilent, Santa Clara, CA) was used according to manufacturer’s protocol. Samples were diluted and boiled in Laemmli SDS sample buffer for 5 minutes and loaded on 4–15% Mini-PROTEAN TGX Stain-Free Protein Gels (Bio-Rad laboratories, Hercules, CA). After separation via SDS-PAGE, proteins were transferred to methanol activated polyvinylidene difluoride (PVDF) membrane. Mouse anti-HA.11 epitope tag antibody (BioLegend, San Diego, CA) was used as primary antibody and horseradish peroxidase (HRP)-conjugated rat anti-mouse Ig, κ light chain (BD Biosciences, New Jersey) was used as secondary antibody. Membranes were imaged using chemiluminescence detection upon addition of Clarity western ECL substrate (Bio-Rad Laboratories) on the Syngene PXi imager.
Immunoprecipitation and deglycosylation
Whole cell extracts were incubated with 1.5 ug of HA antibody overnight at 4°C with rotation. Washed protein G Dynabeads (Thermo Fisher Scientific, Waltham, MA) were added to the samples and incubated for 2 hours at 4°C with rotation. Immunoprecipitated protein was washed 5 times in NP40 lysis buffer followed by 3 washes in 1X TBS before resuspending in water. The deglycosylation reaction was performed under denaturing conditions overnight at 37°C using the Protein Deglycosylation Mix II from New England Biolabs and subsequently analyzed by immunoblot, as described above.
Peptide preparation and mass spectrometry:
Gel regions indicated in Figure S4a were excised, diced to 1 mm cubes and transferred to separate microcentrifuge tubes. Gel pieces were washed with HPLC-grade H2O and then de-stained in 50 mM ammonium bicarbonate (NH4HCO3) and 50% acetonitrile (MeCN) at 37 °C for 30 minutes. De-stain was then removed, and gel pieces were dehydrated in 100% MeCN for 10 minutes. For reduction and alkylation of samples, dried gel pieces were incubated with 25 mM dithiothreitol solution (DTT) for 30 minutes at 55 °C, followed by 10 minutes at room temperature. Gel pieces were partially dehydrated with 100% MeCN. After a 5–10-minute incubation at room temperature, solvent was removed and 10 mM iodoacetamide in 50 mM NH4HCO3 (IAA) was added. Samples were incubated in the dark at room temperature for 45 minutes. The IAA solution was removed, and gel pieces were incubated in 50 mM NH4HCO3 for 5 minutes. Gel pieces were partially dehydrated in 100% MeCN for 5 minutes, then rehydrated in HPLC-grade H2O. De-staining, partial dehydration, and rehydration were repeated. Gel pieces were then completely dehydrated using 100% MeCN. On ice, dried gel pieces were rehydrated in 12 ng/μL sequencing grade modified trypsin in 50 mM NH4HCO3. After a 20-minute incubation at 4 °C, an additional equal volume of 50 mM NH4HCO3 was added to submerge gel pieces, followed by overnight incubation at 37 °C. After centrifugation, supernatants were transferred to new microcentrifuge tubes. Remaining peptides were extracted from gel pieces with the addition of 50% MeCN, 2.5% formic acid (FA). Supernatants were combined with the initial tryptic digest, and gel pieces were dehydrated in 100% MeCN. The final extraction was combined with the previous two extractions and peptides were dried in a speed-vac.
Peptides were resuspended in 2.5% MeCN/2.5% FA. Peptides were separated on a reverse-phase HPLC column (length=15 cm × 100 μm) packed in house with C18 resin (1.8 μm 120 Å, UChrom C18) prior to analysis via a linear ion trap-orbitrap (Orbitrap Eclipse; resolution=120,000) mass spectrometer fitted with an Easy-nLC 1200 (both instruments from Thermo Scientific; Waltham, MA, USA). Following loading (flow rate=300 nL/min) onto the C18 column in Solvent A (2.5% MeCN/0.1% FA), peptides were eluted using a 0–25% gradient of Solvent B (80% MeCN, 0.1% FA) over 60 min followed by an elution to 0–40% gradient of Solvent B over 10 minutes electrosprayed (1.9 kV) into the mass spectrometer. This gradient was followed by 9 minutes at 95% Solvent B before a 5 minute equilibration in 5% Solvent B.The precursor scan (375–1600 m/z) was followed by ten low energy collision-induced dissociation (CID) tandem mass spectra. Dynamic exclusion settings: repeat count=3, exclusion duration=15 sec, exclusion width= ±5 ppm).
SEQUEST searches were performed using a forward and reverse protein database which included known contaminants (such as human keratins and porcine trypsin) and the sequence for U9-ORF with its HA-tag as shown in Figure S4b. Tryptic peptides were required, 2 missed cleavages permitted, a precursor mass tolerance of ± 5 PPM and a fragment ion tolerance of ± 0.01 Da. The following differential modifications were permitted: oxidation of methionine (+15.9949 Da), carboxyamidomethylation of cysteine (+57.0215 Da), and acrylamidation of cysteine (+71.0371 Da). Peptides were filtered by cross-correlation (XCorr) scores dependent on charge state (charge states of 2 to 4 were considered): (XCorrz=+2 > or = 1.8; XCorrz=+3 > or = 2.0; XCorrz=+4 > or = 2.2) and only proteins with three or more identified peptides were further considered. These parameters resulted in a false discovery rate (FDR) <0.01%.
In vivo LPS challenge experiments
Age‐ and sex‐matched male and female U9-KO or WT mice were injected intraperitoneally (i.p.) with E. coli LPS O26:B6 (Sigma Aldrich, St. Louis, MO) at 15 mg/kg body weight (low dose) or 60 mg/kg (high dose). LPS-challenged mice were monitored for survival and semi-quantitative evaluation of clinical signs induced by the sickness responses for 72 hours. A clinical scoring system was adapted from previous study(31) with minor modifications, as follows, 0 = no abnormal clinical signs, 1 = ruffled fur but lively, 2 = ruffled fur, moving slowly, hunched, and sick, 3 = ruffled fur, squeezed eyes, hardly moving, down, and very sick, 4 = same as 3 but with incontinence, 5 = moribund or dead. Blood and tissues were collected at different time points and analyzed. Blood was incubated for 30 min at room temperature to allow clotting, followed by storage on ice for 1–2 hours, centrifugation, and collection of serum. For the in vivo IL-6 neutralization experiments, immediately after LPS injection, 100 μg/mouse of mAb isotype control (anti-trinitrophenol) or anti-IL-6 antibodies (Bio × Cell, Lebanon, NH) were administered by i.p injection.
Statistical analyses
Statistical analyses not pertaining to RNA-seq data were carried out using GraphPad Prism software version 9.2.0 (GraphPad Software, San Diego, CA, USA). The specific tests used to assess the significance of the observed differences are detailed in the figure legends. All center values represent the mean, and error bars represent the standard error of the mean. A P-value of <0.05 was considered significant.
Results
LncRNA U90926 is upregulated in macrophages and dendritic cells in response to TLR-ligands in a MyD88/p38α MAPK dependent manner
U90926 is a lncRNA located on mouse chromosome 5, between two protein-coding genes, Uso1 and Ppef2. The U90926 gene is encoded in a ~5.5 Kbp locus, comprising 5 exons encoding a 522 bp polyadenylated transcript(32). Using microarray analysis, we originally discovered U90926 as a highly downregulated gene in p38α MAP kinase (MAPK)-deficient peritoneal macrophages stimulated in vitro with heat-killed Mycobacterium tuberculosis(16), and more recently confirmed these data by RNAseq in LPS-activated bone marrow derived macrophages (BMDM)(18). To determine the cell/tissue type specificity of this transcript, mining transcriptomic data across >100 tissue and cell types, we identified the highest expression of this gene in LPS-stimulated BMDMs and thioglycolate-elicited peritoneal macrophages, with minimal expression in other cells/tissues at steady state (Fig. 1a). Leveraging published kinetic transcriptomic data from bone marrow derived dendritic cells (BMDCs) stimulated with an array of toll-like receptor (TLR) ligands(33), we found that U90926 RNA expression was maximally induced ~4–6 hours after activation of TLR1/2, TLR4, TLR7, and TLR9, but not TLR3, and sustained for at least 24 hours (Fig. 1b). Using RT-qPCR, we confirmed that U90926 RNA is upregulated with comparable kinetics in BMDMs and BMDCs (Fig. 1c) and determined that the expression of this gene in BMDM is induced by activation of TLR1/2, TLR4, TLR5, and TLR9, but not TLR3 (Fig. 1d). These results demonstrate that the expression of U90926 is rapidly inducible in macrophages and dendritic cells by TLR signals. Given the documented heterogeneity of BMDC preparations(26), we focused the majority of our subsequent in vitro experiments on BMDM.
Figure 1. Expression of U90926 is induced in mouse macrophage and dendritic cells by TLR signals in a MyD88- and p38α MAPK-dependent manner.
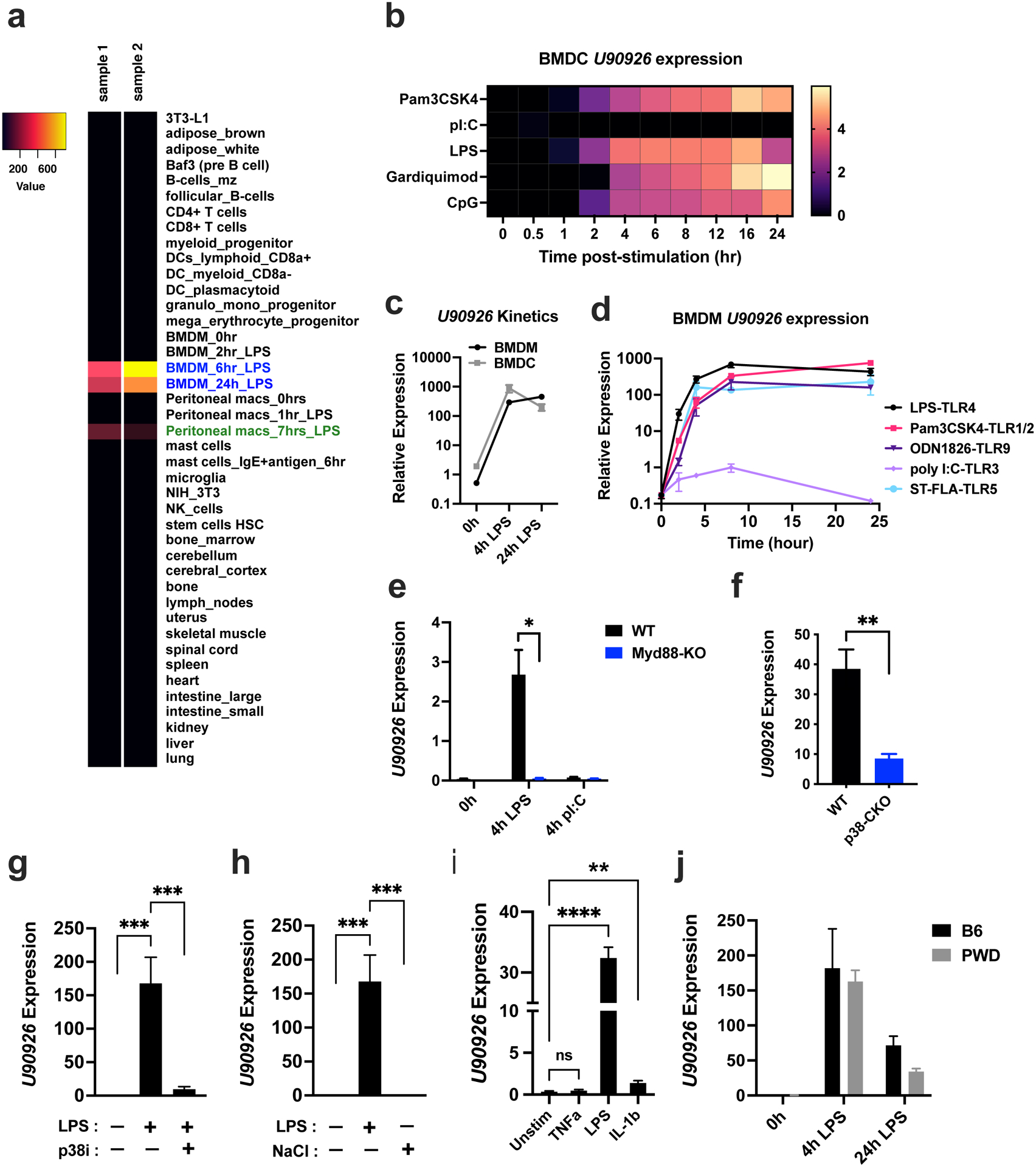
(a) Microarray gene expression data for U90926 were downloaded from BioGPS.org using the geneatlas_MOE430 dataset. A subset of representative tissues is shown in a heatmap. (b) U90926 expression in BMDC stimulated with the indicated TLR ligands was extracted from a published Nanostring gene expression dataset(33). (c) BMDMs and BMDCs were isolated from B6 mice and stimulated with LPS (100 ng/ml) for 4h and 24h, followed by RT-qPCR for U90926 expression measurement. (d) B6 WT BMDMs were stimulated with the TLR-ligands LPS (100 ng/ml), Pam3CSK4 (1 μg/ml), ODN1826 (1 μM), polyI:C (10 μg/ml), or ST-FLA (1 μg/ml) for the indicated times, followed by RT-qPCR to measure U90926 expression. (e) Immortalized BMDMs from WT and MyD88-KO B6 mice were stimulated with LPS (100 ng/ml) or polyI:C (10 μg/ml) for 4 hours, and U90926 expression was measured by RT-qPCR. (f) BMDMs were isolated from p38αCKOLysm (p38-CKO) and p38αflox/flox Cre-negative littermate control (WT) B6 mice and stimulated with LPS in vitro for 4hrs, followed by RT-qPCR to measure U90926 expression. (g) BMDMs were isolated from WT B6 mice and stimulated with LPS with or without pretreatment with the p38 MAPK inhibitor SB203580 (p38i, 10μM) for 4 hrs, followed by RT-qPCR. (h) BMDMs isolated from WT B6 mice were treated with LPS (100 ng/ml) or NaCl (40 mM) for 4 hours, and U90926 expression was measured by RT-qPCR. (i) BMDMs isolated from WT B6 mice were treated with TNFα (10 ng/ml), IL-10(10 ng/ml), and LPS (100 ng/ml) as positive control for 4 hours, followed by RT-qPCR. (j) BMDMs were isolated from B6 and PWD mice and stimulated with LPS (100 ng/ml) for 4 and 24 hours, followed by RT-qPCR. All RT-qPCR data are expressed relative to the housekeeping gene Beta-2-microglobulin (B2m) and calculated by a comparative Ct method formula 2^-(deltaCt), and multiplied by a factor of 10,000 for ease of visualization. Data are represented as the mean ± SEM. Significance was determined by two-way ANOVA for panel (e) and two-tailed Student’s t test for panel (f). One-way ANOVA was performed for more than two groups comparison in panel (g) and (h). P values are represented as follows: ns>0.05, *=≤0.05, **=≤0.01, ***=≤0.001, ****=≤0.0001.
Next, we assessed the signals downstream of TLR activation required for the induction of U90926 expression. Since myeloid differentiation factor 88 (MyD88) is a required adaptor protein for signaling downstream of all of the TLRs above with the exception of TLR3(34, 35), we investigated the necessity of MyD88 for U90926 expression, using WT and MyD88-KO immortalized BMDM cell lines(15). U90926 expression was induced by LPS in WT but not in MyD88-deficient cells, whereas TLR3 activation by polyI:C failed to induce the expression of U90926 in either WT or MyD88-KO cells, as expected (Fig. 1e). Our previous transcriptomic findings implicated p38α MAPK, a major signaling pathway downstream of MyD88, in the expression of U90926(16, 18). We confirmed these findings using RTq-PCR, demonstrating that induction of U90926 was significantly, though not completely, impaired in p38α-deficient BMDMs (Fig. 1f). Consistently, pretreatment of WT BMDM with SB203580, a pharmacological inhibitor of p38 MAPK, also highly reduced U90926 expression after LPS stimulation (Fig. 1g). To investigate if p38 MAPK activation is sufficient to induce U90926 expression independent of TLR-activation, we used NaCl-induced osmotic stress as a known activator of p38 MAPK in BMDMs and other cell types(36–38). NaCl-induced hyperosmotic stress through the addition of an extra 40 mM NaCl did not induce U90926 expression (Fig. 1h), suggesting that p38 MAPK activation alone is not sufficient. We also investigated U90926 expression in response to cytokine stimuli, including TNFα, which induces p38 MAPK activation(39), and IL-1β, which signals via MyD88 and p38 MAPK(40, 41). We found that TNFα did not stimulate U90926 expression, but IL-1β stimulation induced significant U90926 expression in WT BMDM, albeit very modest compared with LPS stimulation (Fig. 1i). Taken together, these findings demonstrate that the expression of U90926 is robustly induced by TLR signals in a MyD88- and p38α-dependent manner.
Our original studies and the experiments above were performed utilizing the most widely used laboratory strain of mice, C57BL/6J (B6), which represents a single unique genotype, artificially selected in a captive setting for over a hundred years. However, we recently demonstrated that certain aspects of the LPS response in B6 myeloid cells may be idiosyncratic to classic inbred strains, and not conserved in the genetically divergent wild-derived inbred strain of mice, PWD/PhJ (PWD)(42). To investigate the conservation of U90926 regulation across divergent genotypes, we used BMDM from PWD mice. BMDM from PWD and B6 mice demonstrated comparable induction of U90926 in response to LPS stimulation (Fig. 1j), suggesting that regulation of U90926 expression is conserved across divergent murine genotypes and not unique to the B6 strain. Given these findings, we continued our studies in the genetically tractable B6 model.
Generation of U90926-deficient mice (U9-KO)
To study the function of U90926 in vivo and ex vivo, we generated U90926-deficient mice (U9-KO) by CRISPR-Cas9 on the C57BL/6J background. Two single guide RNAs (sgRNAs), targeted upstream of the 5’ and downstream of 3’ ends of the gene, were used to delete the entire ~5.5 Kb U90926 locus (Fig. 2a) by microinjection together with Cas9 enzyme into single cell B6 embryos. We obtained multiple successful founders, all of which were confirmed by polymerase chain reaction (PCR) genotyping, showing deletion of all 5 U90926 exons, with some variability around the cut sites, as expected from non-homologous end joining repair (Figure S1a). We selected a single founder to generate the U9-KO mouse line (Pup ID# 11 in Figure S1a). To eliminate any potential off-target deletions made by CRISPR/Cas9 elsewhere in the genome(43, 44), we backcrossed the U9-KO to WT B6 mice, starting with a single male U9-KO founder and followed by 2 more backcrosses. The resulting heterozygous (HET) mice were bred HET × HET to establish homozygous KO and WT control progeny in parallel, which were used in all subsequent experiments. Backcrossed U9-KO mice were validated by Sanger sequencing (see materials and methods) and PCR genotyping (Fig. 2b) to confirm deletion of the entire U90926 genomic sequence.
To validate U90926 deficiency at the RNA level, we generated BMDM and BMDC from WT B6 control mice and U9-KO mice, and stimulated them with LPS to induce expression of U90926. In contrast to WT cells, U9-KO BMDM and BMDC showed no detectable expression of U90926 upon LPS stimulation (Fig. 2cand d). We next investigated whether U90926 deletion alters BMDM and BMDC differentiation. WT and U9-KO BMDM and BMDC were assessed by flow cytometry to measure expression of the major myeloid differentiation markers CD11b and CD11c, which did not differ between WT and U9-KO mice, both at baseline and after 24h LPS stimulation (Fig. 2e and f). Next, we determined the effect of U90926 deficiency on major leukocyte populations in spleen and lymph nodes at the basal level, using flow cytometry. No major immunophenotypic differences were found in U9-KO mice compared with WT mice with the exception of a decrease in Ly6G+ cell number in the spleen (Fig. 2g and h), which was not replicated in a repeat experiment (data not shown). Taken together, these data validate our U9-KO model for functional characterization of U90926 gene function in vitro and in vivo, without any apparent confounding influences on leukocyte development.
Deletion of U90926 locus has minimal intrinsic effects on myeloid cell function in vitro
Given that U90926 is highly expressed in macrophages and dendritic cells upon TLR-stimulation, we hypothesized that this gene may be involved in cell-intrinsic regulation of inflammatory gene expression, similar to other myeloid lncRNAs(9, 11, 45). Macrophages and myeloid DCs are professional antigen presenting cells, whose capacity for activation of naïve T cells is enhanced by TLR stimulation. Therefore, WT and U9-KO BMDMs were stimulated with LPS for 24 hours, followed by assessment of expression of cell surface co-stimulatory molecules involved in antigen presentation. Compared with WT, U90926-deficient BMDMs exhibited no significant differences in the surface expression of CD80, CD86 and CD40 (Fig. 3 a–c). Similar results were obtained in BMDCs (Figure S1b–d). These results suggest that U90926 is not involved in the regulation of co-stimulatory molecule expression in macrophages and dendritic cells.
Figure 3. U90926-deficiency has minimal intrinsic effects on macrophage function in vitro.
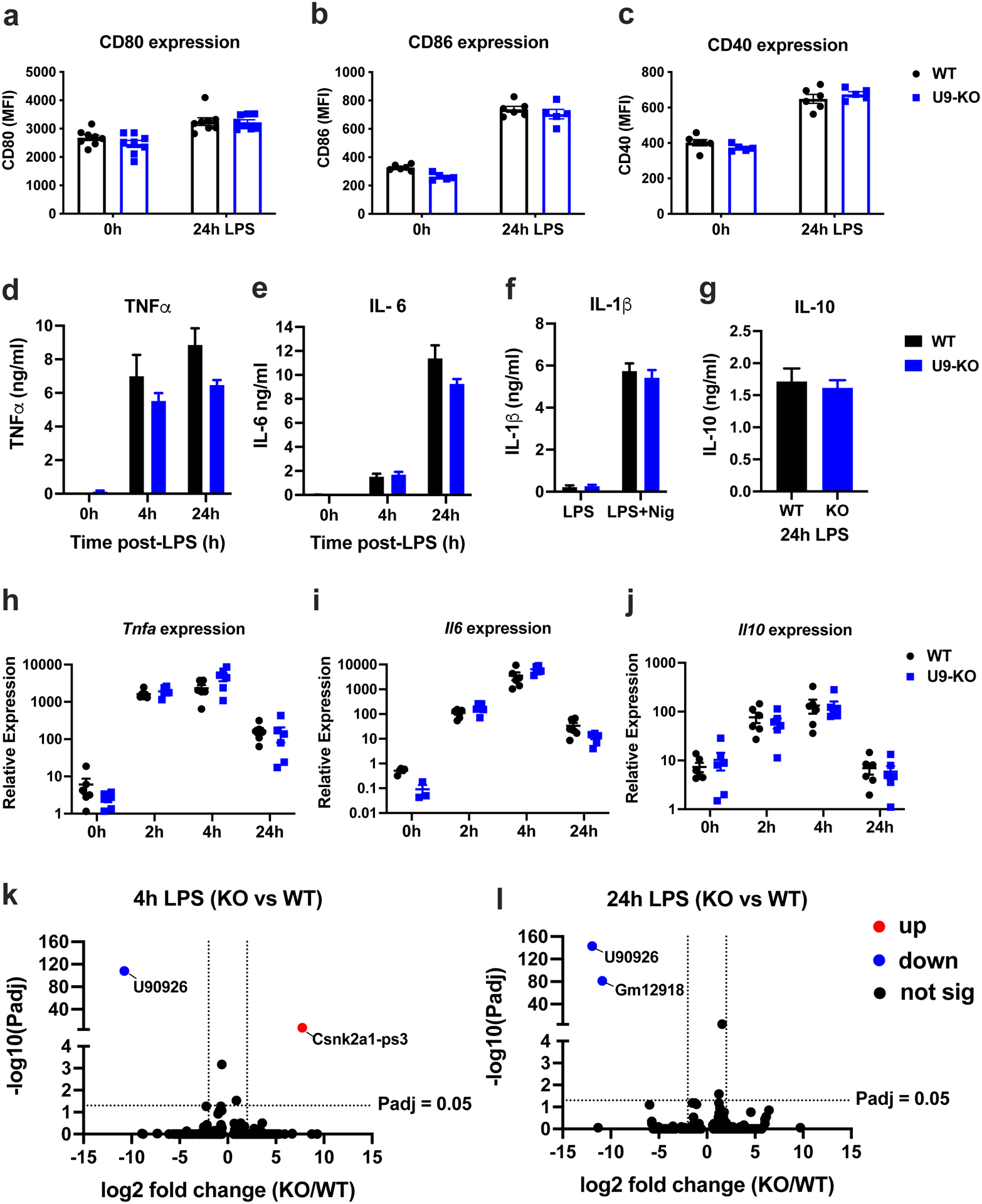
(a-c) WT and U9-KO BMDMs were stimulated with LPS (100 ng/ml) for 24h. Cell surface expression of the co-stimulatory markers CD80, CD86, and CD40 was measured by flow cytometry, and expressed as mean fluorescence intensity (MFI). (d-g) WT and U9-KO BMDMs were stimulated with LPS (100 ng/ml) for the indicated times, and cytokine production in the supernatants was analyzed by ELISA. In (f), BMDMs were stimulated with LPS (100 ng/ml) for 4h and followed by addition of nigericin (5 μM) for 1h. In (g), IL-10 secretion was measured at 24h post-LPS only, since it was not detectable at 4h by ELISA. (h-j) RNA was isolated from WT and U9-KO BMDMs stimulated with LPS (100 ng/ml) at 0h, 2h, 4h and 24h, followed by RT-qPCR to measure mRNA expression of the indicated cytokine genes. RT-qPCR data are expressed relative to the housekeeping gene, Beta-2-microglobulin (B2m), and multiplied by a factor of 10,000 for ease of visualization. Data are represented as the mean ± SEM. Significance was determined by two-tailed Student’s t test. (k, l) WT and U9-KO BMDMs (n=4 for each group) were stimulated with LPS (100 ng/ml) for 4h and 24h, and RNAseq was performed as indicated in Methods. DEseq2 analysis was used to identify differentially expressed genes. Volcano plots show differentially expressed genes passing a cutoff of |Log2 Fold Change|=2, Padj <0.05 (dotted lines on the plots), with up (red) = upregulated in U9-KO, down (blue) = downregulated in U9-KO.
Next, we assessed cytokine production and secretion, as another essential myeloid cell effector function upon TLR-stimulation. We measured the secretion and mRNA expression of key pro-inflammatory cytokines, TNFα and IL-6, and the anti-inflammatory cytokine, IL-10. Additionally, we assessed IL-1β secretion in response to NLRP3 inflammasome activation by LPS and nigericin stimulation. No significant differences in cytokine secretion as measured by ELISA were detected in U9-KO BMDM when compared with WT (Fig. 3 d–g). Similarly, we found no significant differences between WT and U9-KO BMDMs in Tnf, Il6, or Il10 cytokine mRNA expression (Fig. 3 h–j). These data suggest that in spite of being TLR-inducible, U90926 expression has no intrinsic effect on cytokine production and secretion by macrophages upon TLR-stimulation in vitro.
Next, we investigated the global effect of U90926 deficiency on the myeloid cell transcriptome in an unbiased manner by RNA sequencing (RNAseq). Since U90926 is expressed early after TLR-activation, we assessed gene expression at 4 hours, and 24 hours after LPS stimulation, to measure early and late effects in BMDMs generated from WT or U9-KO mice. Differential expression analyses using a standard cutoff for RNAseq, |Log2 Fold Change|>1 and Padj <0.05 revealed only two genes at each time point that passed filter, including U90926 itself (both time points), Csnk2a1-ps3 (at 4 hours), and Gm12918 (at 24 hours) (Fig. 3 k and l). Reanalysis using a relaxed cutoff of |Log2 Fold Change|>0.6, Padj <0.2 found only a total of 9 genes (at 4 hours) and 16 genes (at 24 hours) that were differentially expressed (Figure S1e). Most of these differentially expressed genes were either functionally uncharacterized pseudogenes or predicted genes, and/or not TLR-inducible inflammatory genes, suggesting that U90926 deficiency does not have a major impact on transcriptional regulation of macrophage inflammatory responses. Together with the results above, our findings suggest that U90926 has minimal cell-intrinsic effects on macrophage function.
U90926 RNA localizes to the cytoplasm, associates with ribosomes, and contains an ORF that encodes a secreted peptide
Given the lack of cell-intrinsic effect of U90926 on macrophage gene expression, we assessed the subcellular localization of U90926 transcript using subcellular fractionation and RT-qPCR. We found that U90926 RNA localized predominantly to the cytoplasm rather than the nucleus in BMDMs, similar to protein-coding mRNAs such as B2m, and unlike the small nucleolar RNA U6, which localized to the nucleus (Fig. 4a). Our in-silico analysis indicated that the U90926 transcript contains a 264 bp open reading frame (ORF) that has the potential to generate a ~10 kDa protein of 87 amino acids (Fig. 4b), henceforth referred to as U9-ORF. BLAST analysis of U9-ORF amino acid sequence revealed no significant homology to any known proteins (data not shown). Surprisingly, we identified a predicted endoplasmic reticulum (ER) signal peptide sequence of 16 amino acids at the N-terminus (Fig. 4b), which could make U9-ORF a secreted or organelle lumen-resident protein. Seeking additional evidence for translation, we mined publicly available ribosome-profiling data from a study of LPS-activated BMDCs(46). We observed global ribosome footprints that mapped to all five of the exons of U90926, as well as initiating ribosome footprints that mapped to exon 2 (Fig. 4c), where the start codon for U9-ORF lies, suggesting that the U90926 transcript associates with ribosomes and the U9-ORF may be translated.
Figure 4. U90926 RNA localizes to the cytoplasm, associates with ribosomes, and contains an ORF that encodes a secreted protein.
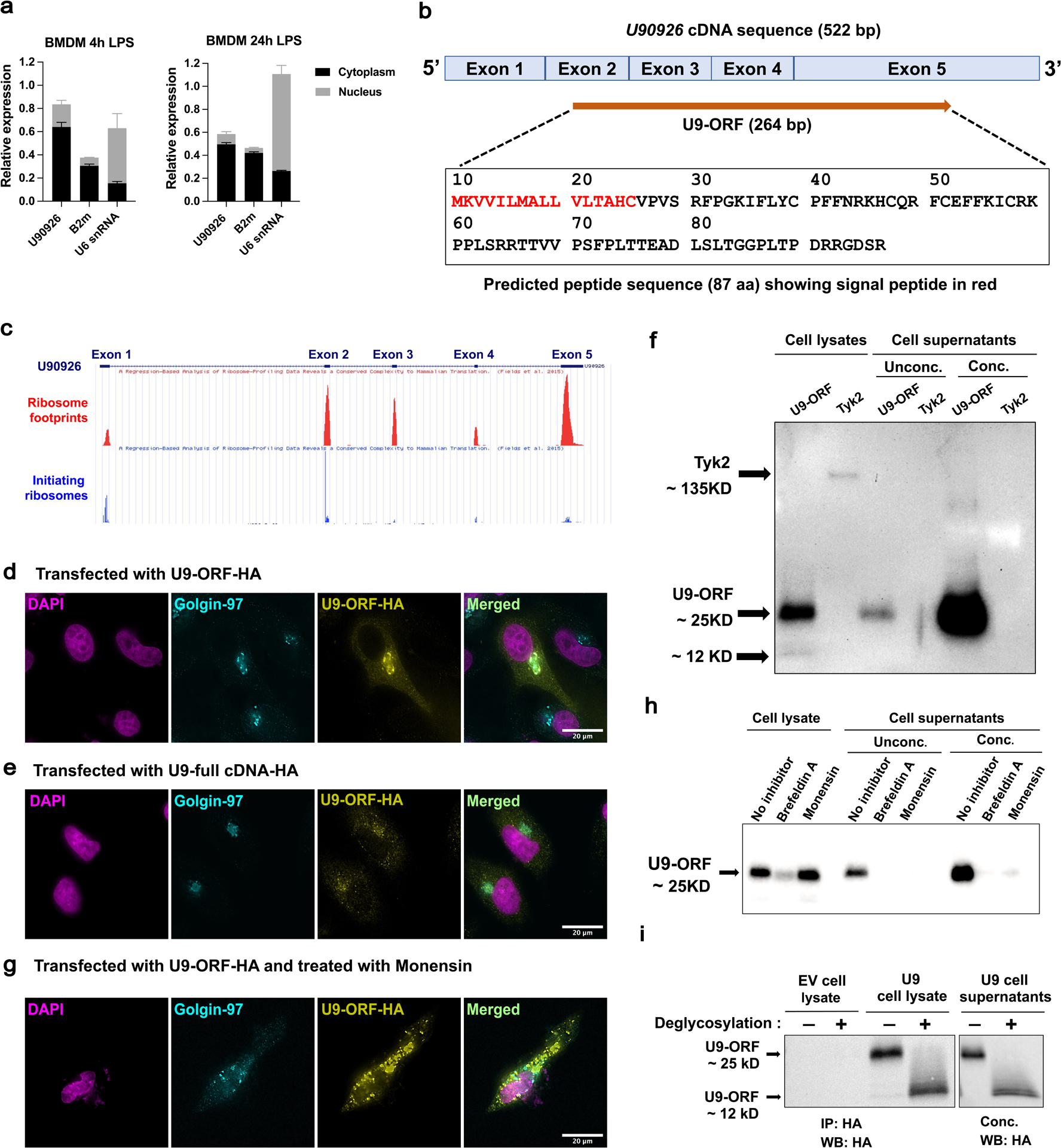
(a) WT BMDM were stimulated for 4h and 24h with LPS (100ng/ml), followed by isolation of nuclear and cytoplasmic fractions and RTq-PCR analysis of U90926, B2m (cytoplasmic control), and U6 SnRNA (nuclear control) expression (each normalized to unfractionated input as equal to 1). (b) A schematic diagram of U90926 cDNA sequence (522 bp) containing the 264 bp ORF spanning the exons is shown. Predicted amino acid sequences of the U9-ORF peptide (87 aa) and the signal peptide identified by SignalP software shown in red. (c) GWiPS was used to analyze of Ribo-seq data from LPS stimulated BMDC(46) at the U90926 locus. Global ribosome footprints or initiating ribosomes footprint data (in the presence of harringtonine) are shown, as indicated. (d, e) HeLa cells were transfected with U9-ORF-HA (d) and U9-full-lengthcDNA-HA (e) plasmids, followed by immunostaining for HA (yellow) and Golgin-97 (cyan; Golgi marker), and DAPI nuclear staining (magenta). (f) HeLa cells were transfected with U9-ORF-HA and TYK2-HA plasmids for 48 hours, followed by immunoblot analysis. Supernatants were loaded directly or concentrated using StrataClean resin, as indicated (Unconc. or Conc., respectively). (g) HeLa cells were transfected with U9-ORF-HA for total 48 hours, where protein transport inhibitor, monensin was added at 24 hours, and then cells were subjected to immunostaining at 48 hours of transfection, as above. (h) HeLa cells were transfected with U9-ORF-HA for a total of 48 hours, in which protein transport inhibitors, brefeldin A and monensin were added at 24 hours, and sample collection was done at 48 hours of transfection, followed by immunoblot analysis. Supernatants were analyzed as described above. (i) HeLa cells were transfected with empty vector control and U9-ORF-HA plasmid for 48 hours. Cell lysates were immunoprecipitated using an anti-HA antibody and supernatants were concentrated using StrataClean resin, followed by deglycosylation under denaturing conditions overnight at 37°C and analyzed by immunoblot.
To directly assess the protein coding potential of U90926, we cloned the 264 bp U9-ORF cDNA into a mammalian expression vector and added a hemagglutinin (HA) epitope tag at the C-terminus. The resulting U9-ORF-HA plasmid was transfected into HeLa cells, and the presence and localization of HA-tagged U9-ORF product was assessed by immunocytochemistry (ICC). We found that the U9-ORF-HA protein was translated and localized predominantly to the perinuclear Golgi apparatus in HeLa cells (Fig. 4d). Similar results were obtained by transfection of the U9-ORF-HA plasmid into RAW264.7 murine macrophages, 3T3 mouse embryonic fibroblasts, and 3T3-L1 mouse pre-adipocytes (Figure S2a). To confirm that U9-ORF protein could be translated in the context of its native full-length mRNA, we also cloned the full length U90926 cDNA (522bp) into an expression vector, inserted an HA tag at the C-terminus of the ORF, and transfected it into HeLa cells, followed by ICC. As before, we found that U9-ORF was translated and localized to Golgi apparatus (Fig. 4e), suggesting that this protein may be destined for exocytosis and secretion.
To determine whether U9-ORF can be secreted, we transfected HeLa cells with U9-ORF-HA or, as a negative control, an HA-tagged cytoplasmic kinase, TYK2(30), and analyzed cell lysates and supernatants by immunoblotting. While both proteins were present in the cell lysates, we found abundant U9-ORF-HA, but not TYK2-HA, protein in the supernatant, with or without concentration (Fig. 4f). We confirmed the correct identity of the U9-ORF protein in the U9-ORF-HA transfected cell supernatants by mass spectrometry (Figure S3 and Table 1). To confirm that U9-ORF protein is secreted via the canonical exocytic pathway, we treated cells with vesicular transport inhibitors, monensin and brefeldin A, after transfection and analyzed by the cells by ICC, and cell lysates and supernatants by immunoblotting. Monensin and brefeldin A treatment resulted in a notable sub-cellular relocalization of U9-ORF protein (Fig. 4g and Figure S2b). Similarly, by immunoblotting, we found that monensin and brefeldin A treatment almost completely inhibited U9-ORF secretion into the supernatant (Fig. 4h). Interestingly, immunoblot analysis indicated that both the predominant cell-associated and secreted forms of the U9-ORF-HA protein migrated at a molecular weight of 25 kDa (Fig. 4f and h), larger than its predicted mass of 12 kDa, which was visible as a minor band only in cell lysates (Fig.4f). This suggested the presence of possible post-translational modifications (PTMs), such as glycosylation, which is a common on PTM on secreted proteins(47). In order to determine if the U9-ORF protein is glycosylated, we performed deglycosylation reactions on the U9-ORF-HA transfected cell lysates and supernatants, followed by immunoblotting. We found that deglycosylation caused a clear shift in the U9-ORF protein band to the predicted ~12kDa position (Fig. 4i), indicating that U9-ORF protein is indeed glycosylated. Taken together, our results suggest that U90926 is a cytoplasmic RNA that encodes a novel U9-ORF protein, which is glycosylated, and secreted out from the cell. Together with the RNAseq data demonstrating minimal intrinsic effects of U90926 deficiency on macrophage gene expression, these results suggest a model whereby in vivo U9-ORF secreted by macrophages may act on other cell types in a paracrine and/or endocrine manner.
Table 1. Tryptic peptides identified from U9-ORF via LC-MS/MS.
Table includes charge states for each peptide (z), SrchName (indicating gel region from Figure S4a), measured precursor mass accuracy (Δ Error, in ppm), SEQUEST XCorr values, and Unique ΔCorr values for each peptide identified. “.” after arginine and lysine residues indicate the sites of tryptic cleavage. “C#” indicates carboxyamidomethylation on cysteine.
| Peptide | z | SrchName | PPM | XCorr | Uniq. ΔCorr |
|---|---|---|---|---|---|
| R.FC#EFFK.I | 2 | ORF_U1 | −0.24 | 1.99 | 0 |
| R.FC#EFFK.I | 2 | ORF_U1 | −0.23 | 1.85 | 0 |
| R.FC#EFFK.I | 2 | ORF_U1 | −0.13 | 1.95 | 0 |
| R.FC#EFFK.I | 2 | ORF_U1 | −0.01 | 2.05 | 0 |
| R.FC#EFFK.I | 2 | ORF_U1 | −0.01 | 2.30 | 0 |
| R.FC#EFFK.I | 2 | ORF_U1 | −0.01 | 1.99 | 0 |
| R.FC#EFFK.I | 2 | ORF_U1 | 0.25 | 1.84 | 0 |
| R.FC#EFFK.I | 2 | ORF_U1 | 0.33 | 1.93 | 0 |
| R.FC#EFFK.I | 2 | ORF_U1 | 0.39 | 1.92 | 0 |
| K.IFLYC#PFFNRK.H | 3 | ORF_U1 | 0.53 | 2.86 | 0.81 |
| K.IFLYC#PFFNRK.H | 3 | ORF_U1 | 0.53 | 3.21 | 0.85 |
| K.IFLYC#PFFNRK.H | 3 | ORF_U1 | 0.54 | 3.43 | 0.84 |
| K.IFLYC#PFFNR.K | 2 | ORF_U1 | 1.28 | 2.52 | 0.86 |
| K.IFLYC#PFFNR.K | 2 | ORF_U1 | 1.78 | 2.50 | 0.88 |
| K.IFLYC#PFFNR.K | 2 | ORF_U1 | 1.78 | 2.36 | 0.85 |
| K.IFLYC#PFFNR.K | 2 | ORF_U2 | −0.55 | 2.55 | 0.96 |
U90926 expression protects mice from LPS-induced endotoxic shock by modulating IL-6 levels
Our findings so far had suggested that TLR-inducible expression of U90926 in macrophages may exert non-macrophage intrinsic effects. To test this hypothesis in vivo, we employed the widely used LPS endotoxemia model, in which TLR4 signaling in macrophages promotes morbidity and mortality due to hypercytokinemia and systemic shock, by interacting with multiple other cell types(48),(49). We first determined whether U90926 expression is inducible in vivo by LPS endotoxemia. WT B6 mice were challenged with a high dose (15 mg/kg) of E. coli O26:B6 LPS by intraperitoneal (i.p.) injection, followed by tissue collection at several time points. We found that U90926 expression was induced within hours after systemic LPS administration, in peritoneal cells collected by lavage, liver tissue, and visceral adipose tissue, and its expression was sustained for up to 48 hours (Fig. 5a), similar to its kinetics in vitro in BMDM (Fig. 1d). To determine the contribution of U90926 in severe endotoxemia, U9-KO and WT mice were challenged i.p. with high dose LPS (15 mg/kg), followed by monitoring of survival and quantitative evaluation of clinical signs induced by the sickness responses, as previously described(31, 50), at individual time points, and as a cumulative variable using area under the curve (AUC) analysis. In WT mice, this dose of LPS resulted in acute clinical signs that peaked at 12 hours, followed by recovery and survival in 86% of the mice (Fig. 5b–d). In contrast, U9-KO mice exhibited significantly sustained sickness responses and and reduced survival of 33% (Fig. 5b–d). Together, these results suggest that expression of U90926 downstream of TLR4 activation in endotoxemia is protective in septic shock.
Figure 5. U90926 deficiency exacerbates LPS endotoxemia by elevation of IL-6 levels.
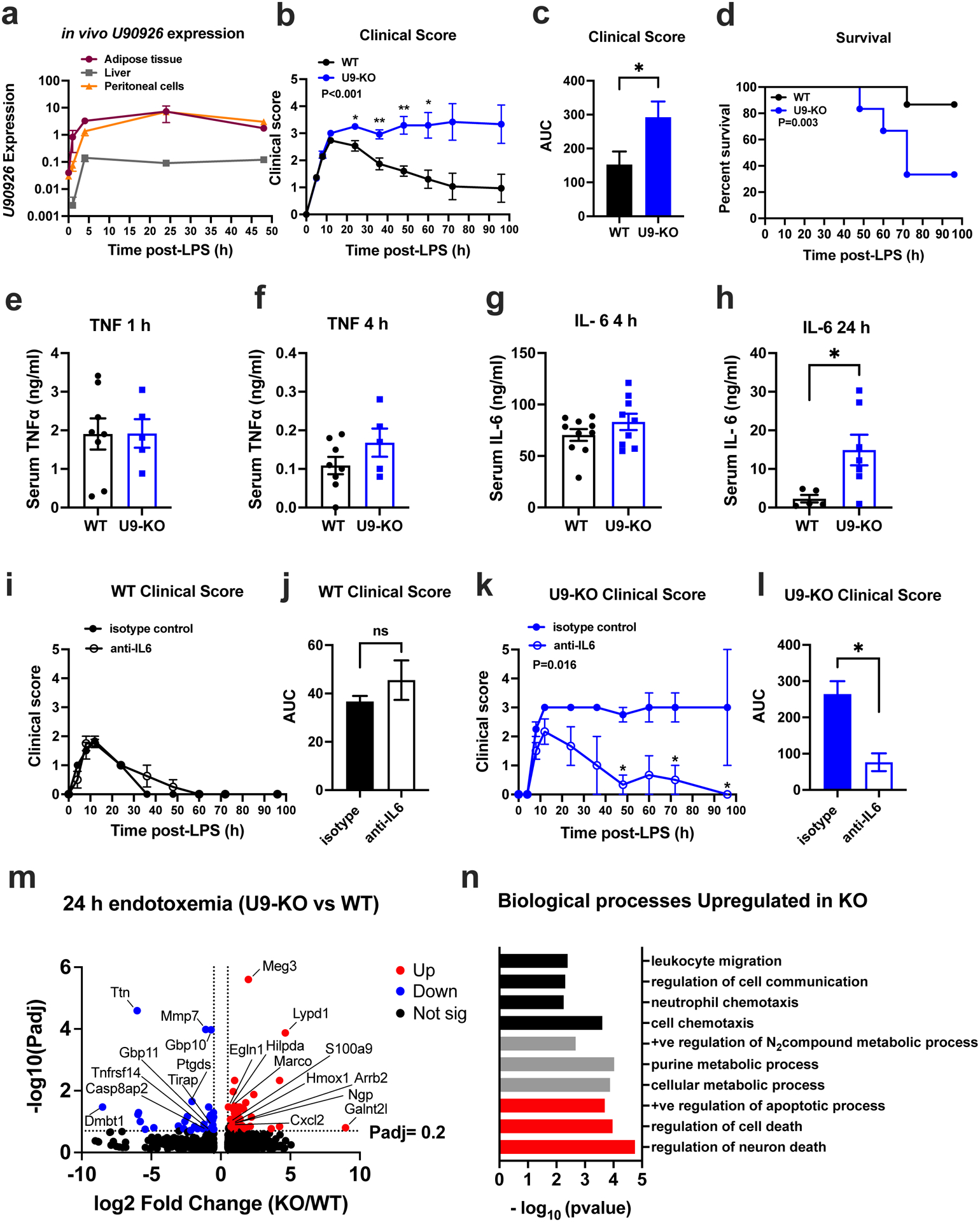
(a) WT B6 mice were challenged with 15mg/kg LPS i.p. and tissues were collected at the indicated time points, followed by RNA extraction and measurement of U90926 expression by RT-qPCR. B2m was used a normalizer for relative expression, as in Fig. 1. (b-d) WT (n=15) and U9-KO (n=12) mice were challenged with 15 mg/kg LPS i.p., followed by evaluation of sickness behavior (b, c) and survival (d). (c) Area under the curve (AUC) was calculated from kinetic data in (b), to calculate a cumulative measurement of the disease severity. Clinical score evaluation measurement is described in the Methods section. (e-h) Serum was collected from the LPS-challenged mice at indicated time points, and TNFα and IL-6 levels were analyzed by ELISA. (i-l) WT (n=7) and U9-KO (n=5) mice were challenged with LPS (15mg/kg) and simultaneously administerted isotype control (anti-trinitrophenol) or anti-IL-6 antibodies (100 μg/mouse) by i.p. injection, followed by evaluation of sickness behavior in (i) and (k). Area under the curve (AUC) calculated from data in (i) and (k) is shown in (j) and (l). Significance of differences was assessed using 2-way ANOVA (panels b, i, and k), Mantel-Cox test (panel d), and Welch’s T test (panel c, e-h, j, l). P values are represented as follows: ns= >0.05, *= ≤0.05, **=≤0.01, ***=≤0.001. (m) WT (n=4) and U9-KO (n=4) mice were challenged with 15mg/kg LPS i.p. for 24 h and RNAseq was performed on whole liver tissue RNA as indicated in Methods. DEseq2 analysis was used to identify differentially expressed genes. Volcano plots showing differentially expressed genes at a cutoff of |Log2 Fold Change|>0.6, Padj <0.2 (dotted lines on the plots) with up (red) = upregulated in U9-KO, down (blue) = downregulated in U9-KO. Selected genes of interest are labelled. (n) Differentially expressed genes upregulated in U9-KO were analyzed by GO enrichment analysis by PANTHER. A subset of select pathways are shown, falling into three major categories, indicated in black (cell migration/chemotaxis), grey (metabolic processes) and red (cell death/apoptosis).
To determine the inflammatory mechanisms underlying exacerbated endotoxemia in U9-KO mice, we assessed the serum levels of the key pro-inflammatory cytokines TNFα and IL-6 by ELISA, after LPS challenge. TNFα levels peaked at 1 h post-challenge and did not differ significantly between WT and U9-KO mice at both 1 h and 4 h (Fig. 5e–f). IL-6 levels peaked at 4 hours post-challenge at comparable levels between WT and U9-KO mice (Fig. 5g). However, at 24 hours, IL-6 levels remained elevated at a significantly higher level in U9-KO mice compared with WT (Fig. 5h).
To determine whether elevated IL-6 in U9-KO mice was directly responsible for their increased susceptibility to endotoxic shock, we blocked IL-6 using a neutralizing antibody in the LPS endotoxemia model. U9-KO and WT mice were challenged with LPS (15 mg/kg) and simultaneously treated with isotype control or anti-IL-6 antibody, followed by monitoring of clinical signs and survival as described above. In WT mice, neutralization of IL-6 had no significant effect on clinical symptoms (Fig. 5i–j), and 100% of the mice in both treatment groups survived the challenge. In contrast, IL-6 neutralization in U9-KO mice diminished the sustained clinical signs (Fig. 5k–l), with 100% percent of the anti-IL-6 treated U9-KO mice surviving, compared with 50% of the isotype control treated U9-KO mice (P= 0.172 by Mantel-Cox analysis). Given the published evidence that IL-6 blockade has a protective effect in WT mice during sepsis(51, 52), we tested whether IL-6 neutralization will also protect WT mice from higher LPS dose challenge, since KO but not WT mice displayed high morbidity and mortality when challenged with the lower dose of LPS in our previous experiments (Fig. 5b–l). We found that neutralization of IL-6 was indeed protective in WT mice when challenged with a higher dose of LPS (Figure S4a–c).
Taken together, the data above suggested that U90926 could play a protective role in septic shock at least in part by negatively regulating systemic IL-6 levels during LPS endotoxemia, possibly via secreted U9-ORF from macrophages acting in a paracrine and/or endocrine manner. This would be consistent with our findings of unchanged IL-6 production by U9-KO BMDM in vitro (Fig. 3e,i) and the critical role of hepatocytes in IL-6 production in response to systemic LPS(53). To determine whether BMDM-derived U9-ORF acts directly on hepatocytes to regulate IL-6 levels, we performed in vitro co-culture experiments between WT or U9-KO BMDMs and a mouse hepatocyte cell line, AML12 cells. We first tested whether hepatocytes express U90926 RNA in response to LPS, and found that unlike BMDM, hepatocytes do not express U90926 in response to LPS stimulation (Figure S4d). While hepatocytes cultured alone also produced minimal IL-6 in response to LPS, their presence in co-culture with WT or KO BMDM resulted a significant boost in IL-6 production compared with BMDM alone. However, there was no significant difference in IL-6 production between WT and KO BMDM co-cultures, indicating that macrophage-derived U9-ORF does not directly regulate IL-6 production by hepatocytes, at least in vitro (Figure S4e). Together with the findings that IL-6 neutralization is similarly protective in WT mice challenged with a higher dose of LPS (Figure S4a–c) as in KO mice challenged with a lower dose of LPS (Fig. 5i–j), these data suggest that U90926 deficiency may shift the threshold for endotoxic shock susceptibility, and subsequently causing elevated IL-6 levels downstream, rather than specifically and/or directly regulating IL-6 expression.
Given the critical involvement of both liver resident macrophages and hepatocytes in sepsis(54–58), to further examine the basis for the protective effect of U90926 in LPS endotoxemia, we performed RNAseq analysis on whole liver tissue isolated from WT and U9-KO mice after 24 h of systemic LPS challenge. Our analysis revealed 71 genes that were differentially expressed in the liver of U9-KO vs WT mice, including key immune response genes such as Cxcl2, Ngp, Arrb2, S100a9 and Ptgds (Fig. 5m). Gene Ontology (GO) enrichment analysis of differentially expressed genes identified that distinct pathways were upregulated and downregulated in U9-KO mice compared to WT. The pathways associated with genes upregulated in U9-KO fell into three major categories: leukocyte chemotaxis, metabolic processes, and cell death (Fig. 5n), suggesting that U90926 deficiency may exacerbate endotoxemia through augmented leukocyte recruitment, hepatocyte cell death, and/or dysregulated metabolism in the liver. In contrast, enrichment analysis of genes downregulated in U9-KO revealed much fewer significantly enriched pathways, but notably including defense response to Gram-positive and Gram-negative bacteria. To determine whether elevated IL-6 levels may have contributed to dysregulated gene expression in the liver of U9-KO mice, we performed upstream regulator analysis on genes differentially expressed in U9-KO liver tissue compared with WT (see Methods). We found significant enrichment of IL-6 as an upstream regulator (P=0.04) with a positive Z-score (0.161), with Cxcl3, Mmp7, S100a9, and Vldlr identified as target genes. Taken together, these data suggest that U90926 deficiency exacerbates endotoxic shock in association with elevated systemic IL-6 levels, resulting in (or caused by) dysregulated liver function, and ultimately, death.
Discussion
With recent advancement of high-throughput sequencing technologies and computational biology, the detection of novel lncRNA transcripts has become easier. However, functions of the most lncRNA remain largely unknown because in-depth experimental investigation of individual genes is needed to unravel their functions. Here, in this study we characterized a mouse putative lncRNA U90926. This gene lies between two conserved protein coding genes, Uso1 and Ppef2, on mouse chromosome 5. We became interested in U90926 when we identified it as one of the most highly downregulated genes in TLR-stimulated p38α MAPK-deficient macrophages in our transcriptomic analyses(16, 18). Given this discovery, we focused on identifying possible roles for U90926 in macrophages. We generated the first (to our knowledge) mouse model of U90926 deficiency and discovered that the induction of this gene by inflammatory signals plays a protective role in endotoxic shock. We also found evidence that U90926 encodes a novel secreted protein that could play paracrine roles in endotoxemia.
Prior to 2017, there had been no studies on the biological role of U90926. Since then, three independent groups have described possible functions for this gene. Using an in vitro model of adipocyte differentiation in 3T3-L1 pre-adipocyte cells, Chen et al discovered that 1) U90926 expression was downregulated during adipocyte differentiation, and 2) overexpression of U90926 inhibited adipocyte differentiation, while silencing had the opposite effect, thus implicating U90926 as a possible repressor of adipogenesis(20). Another in vitro study found that U90926 expression was highly upregulated in a murine retinal photoreceptor cell line 661W following herpes simplex virus 1 (HSV-1) infection, and silencing of this gene impaired HSV-1 replication, thus suggesting that this gene could serve as co-factor for DNA virus replication(21). Interestingly, the same group identified two human lncRNA transcripts syntenic with, and possibly homologous to, U90926, with the expression of one of these transcripts being elevated in the eye of individuals with acute retinal necrosis caused by HSV-1(59). Finally, a recent study demonstrated that U90926 expression was elevated in microglia during ischemic stroke in vitro and in vivo, and that silencing microglial U90926 reduced infarct volume and neutrophil recruitment in an experimental stroke model(22). These results are broadly consistent with our own findings, as discussed in more detail below, and suggest that expression of the U90926 transcript is induced by a variety of inflammatory signals, and it plays diverse and context-dependent roles in regulating downstream events. Importantly, given our discovery that this gene may encode a protein, it would be of interest to determine the role of the U9-ORF in these other models.
We demonstrated that expression of U90926, while minimal across multiple tissues at steady state, is highly inducible in bone marrow-derived macrophages and DCs upon stimulation with agonists of TLR1/2, TLR4, TLR5, TLR7, and TLR9. The latter is consistent with the findings of Shirahama et al that HSV-1 infection, which also triggers TLR9(60, 61), induced expression of this gene(21), although we note that in our study, we used a mimic of bacterial CpG DNA as an agonist. This also suggests that expression of U90926 may not be restricted to myeloid cells and may be present in other cell types that can respond to inflammatory signals such as TLRs, although we found no U90926 expression in LPS-stimulated mouse hepatocyte cell line, AML12 cells (Figure S4d). The findings of Xu and colleagues that ischemic stroke induced expression of U90926 in microglia(22) suggest that damage associated molecular patterns released in damaged CNS tissue, which can induce signaling through several TLRs(62), could be driving this activation. Alternatively, inflammatory cytokines, such as TNFα or IL-1β, which are abundantly produced during stroke(63), could potentially serve to induce expression of U90926, in particularly since the receptors for these cytokines induce signaling through MyD88 and/or p38α MAPK, two signals that we showed are required for TLR4-dependent induction of U90926. Consistent with this, there is microarray evidence that TNFα can induce U90926 expression in 3T3-L1 preadipocytes(64), although we did not detect any U90926 expression upon TNFα stimulation in BMDMs (Fig. 1i). However, we did find moderate U90926 expression in BMDMs upon IL-1b stimulation, which is consistent with the MyD88-dependence of IL-1R family(40). The findings of Chen et al that U90926 may be expressed by pre-adipocytes in the absence of inflammatory cues(20) suggest that there may be additional signals that control its expression. We note that we did find detectable, albeit low, expression of U90926 in whole adipose tissue, which was markedly increased upon systemic LPS challenge (Fig. 5a). Given the key role of tissue macrophages in adipose tissue homeostasis and inflammation(65–67), future studies could explore potential U90926-dependent cross-talk between adipocytes and adipose tissue macrophages.
Several previous studies have shown that expression of distinct lncRNAs either activated or repressed upon TLR-stimulation in macrophages plays an important regulatory role in controlling inflammation(9, 11, 45, 68). Surprisingly, we found that U90926 deficiency did not have any major intrinsic effect on macrophage inflammatory gene regulation in vitro (Fig. 3). This suggests that U90926 plays a role other than transcriptional regulation, unlike other macrophage lncRNA that are found in nucleus such as lincRNA-Cox2, lincRNA-EPS, Malat1 and MeXis(9, 11, 69, 70). Recent studies have shown that subcellular localization is critical in determining lncRNA function in the cell(71–73). Consistent with this, we found in our study that U90926 is localized predominantly in the cytoplasm rather than the nucleus of BMDM (Fig. 4a), which validates the lack of major intrinsic effect of U90926 deficiency on macrophage transcriptional regulation. Our findings on U90926 localization in BMDM is consistent with other groups, who showed that it is localized to the cytoplasm of 3T3-L1 preadipocytes and primary microglia(20, 22). In contrast, another study showed the U90926 RNA is localized primarily in nucleus in the HSV-1-infected 661W murine retinal photoreceptor cell line(21). It is possible that U90926 localization differs across cell types or cell states, since it has been known that lncRNA localization can differ in different cell types depending on splicing and functional predisposition(71, 74). Another potential explanation for the apparent nuclear localization in 661W cells is that it was measured early before HSV-1 infection, at a time point when the expression of this gene is only at ~10% of its maximal expression(21), and thus it may represent an intermediate accumulation of unspliced U90926 RNA prior to export out of the nucleus. Given our findings that the U90926 transcript may be translated, cell type-dependent localization of this RNA could represent differential non-coding RNA vs. protein effects, and thus deserves further investigation.
Several recent studies showed that many genes currently annotated as lncRNAs, are pervasively translated to small proteins(75, 76). In general, ORFs size restrictions and the absolute requirement for AUG as the sole initiator of translation have constrained the identification of potentially important transcripts with non-canonical protein-coding potential(76, 77). The concept of functional translation of lncRNAs in macrophages is strongly supported by two recent studies. A lncRNA called AW112010 was recently identified to encode a peptide, which is important in controlling mucosal immunity during both bacterial infection and colitis(14). Another recent study showed that a classically annotated lncRNA 1810058I24Rik encodes a mitochondrial micropeptide named Mm47, which regulated Nlrp3 inflammasome function in macrophages(15). Given the emerging evidence of lncRNA being translated and cytoplasmic location of U90926, we were prompted to identify the protein-coding potential of U90926. Despite having an ORF with a canonical AUG start site that can potentially encode a predicted 87- amino acid long protein, no prior study has addressed the question if the ORF in U90926 (U9-ORF) is functional. In this study, we found that not only did U90926 RNA localize to the cytoplasm, but it associated with ribosomes. Using cDNA transfection, we further showed that epitope-tagged U9-ORF is translated into a stable glycosylated protein that traffics to the Golgi and is secreted from the cell (Fig. 4 d–h), as predicted from the presence of a consensus ER signal peptide sequence at its N-terminus (Fig. 4b). Future studies will need to validate the expression and localization of endogenous U9-ORF in vivo.
The finding that U9-ORF is secreted potentially explains the lack of effect on macrophage-intrinsic function in vitro (Fig. 3), implicating paracrine cellular targets for this protein in vivo. In the LPS endotoxemia model, we found rapid induction of U90926 expression in immune cells, and in liver and adipose tissue. Moreover, we found that U9-KO mice exhibit significantly heightened susceptibility and mortality compared to WT controls. We also demonstrated that U9-KO mice have sustained levels of IL-6 at 24 hours and neutralization of lL-6 can rescue the phenotype of U9-KO mice in the LPS endotoxemia model (Fig. 5), albeit WT mice were similarly protected if challenged with a higher dose of LPS (Figure S4a–c). These data suggest that deletion of U90926 lowers the threshold for endotoxic shock, which is results in elevated systemic IL-6 levels, which drive morbidity and mortality. These effects could be possibly mediated via secreted U9-ORF from macrophages acting in a paracrine and/or endocrine manner although our in vitro experiments do not support a direct regulation of hepatocyte IL-6 production by macrophage derived U9-ORF (Figure S4e). An alternative possibility is that U9-ORF regulates IL-6 stability in vivo, which is an important way in which this cytokine is regulated(78, 79).
To further shed light on the basis of the protective role of U90926, we performed transcriptomic analysis on whole liver tissue, considering critical involvement of both the liver resident macrophages and hepatocytes during endotoxic shock(54, 55, 57, 58). We found that many immune genes and biological pathways are differentially regulated in U9-KO mice compared to WT, including pathways that are important in sepsis like cell chemotaxis, metabolic processes, and cell death(80–82), which were upregulated in U9-KO mice. This included an upregulation of neutrophil-specific genes Ngp and S100a9, and the chemokine Cxcl2, a neutrophil chemoattractant, suggesting enhanced neutrophil influx into the liver as a contributor to (or a result of) enhanced liver pathology in LPS-treated U9-KO mice, which is further consistent with the role of IL-6 in promoting neutrophilia in various inflammatory settings(83–85). Interestingly, silencing of U90926 in microglia resulted in the downregulation of Cxcl2 expression and decreased neutrophil recruitment to the CNS(22), which is the inverse of our results, but nonetheless implicates myeloid cell mediated neutrophil recruitment as a possible common mechanism. Of note, the putative mechanism of action of U90926 described in microglia involved its RNA form(22), and not the novel U9-ORF protein discovered in our study. Future studies will need to determine whether U90926 function in regulating inflammatory processes in vivo require its RNA form, its protein form, or both, as has been reported for other lncRNAs(86). Notably, we found a significant enrichment for IL-6-regulated genes in the genes differentially expressed in liver tissue of U9-KO mice, suggesting that elevated systemic IL-6 may be upstream of the dysregulated gene expression in the liver.
Another important aspect of lncRNA biology is conservation across different species. Most lncRNA genes show low sequence conservation across species(7, 87), including many, if not most of the immune cell-specific lncRNAs identified to date that lack documented cross-species homologs(7). An emerging view of lncRNA conservation includes not only sequence, but structure, function and synteny(88). While we have so far been unable to find a human U90926 homolog using sequence homology, using synteny, Shirahama et al identified a putative human homolog of U90926, AC110615.1, as a potential biomarker of acute retinal necrosis(59). We will characterize this potential human homolog of U90926 in human macrophages in our future studies. Importantly, the lack of apparent conservation of the U9-ORF amino acid sequence in other species suggests that U90926 could represent an example of a dual-function lncRNA that evolved to lack its protein coding potential in other species.
In conclusion, we have identified U90926 as a key gene that is highly upregulated in activated macrophages and provided evidence for a protective role for this gene during endotoxic shock. Furthermore, we have discovered unexpected evidence for the translation of this putative lncRNA gene. These findings suggest manipulation of this gene and its products as potential experimental therapeutic approaches in sepsis, which remains a major unmet clinical need. More importantly, our findings yield provide important implications for the evolution of lncRNAs, their protein coding potential, and the physiological roles of this unique class of genes, which outnumbers canonically protein-coding genes.
Supplementary Material
Keypoints:
Acknowledgements
The authors are grateful to Dr. Markus Thali, Ph.D., (Department of Microbiology and Molecular Genetics, UVM) for access to the DeltaVision Deconvolution Microscope, Dr. Kate Fitzgerald (UMass Medical) for providing the MyD88-KO cells, Dr. Chris Sassetti and Dr. Michael Kiritsy (UMass Medical), for the provision of immortalized BMDC-like cell lines that were used in many optimization experiments, Dr. Susan Carpenter (UC Santa Cruz) for the provision of various reagents that were used in troubleshooting protocol, the UVM VIGR core facility and Scott Tighe, Korin Eckstrom, Dr. Seth Frietze, and Dr. Somen K. Mistri for their support of the RNA sequencing and analysis, Dr. Roxana del Rio-Guerra for the flow cytometry and cell sorting facility, and Dr. Cory Teuscher (Department of Medicine, UVM) for many helpful discussions and feedback.
This work was supported by grants from National MS Society (research grant RR-1602-07780) and the NIH NIAID (R21AI151116) to DNK.
Bibliography
- 1.Deutschman CS, Hellman J, Roca RF, De Backer D, and Coopersmith CM. 2020. The surviving sepsis campaign: basic/translational science research priorities. Intensive Care Med. Exp 8: 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rhee C, Dantes R, Epstein L, Murphy DJ, Seymour CW, Iwashyna TJ, Kadri SS, Angus DC, Danner RL, Fiore AE, Jernigan JA, Martin GS, Septimus E, Warren DK, Karcz A, Chan C, Menchaca JT, Wang R, Gruber S, and Klompas M. 2017. Incidence and Trends of Sepsis in US Hospitals Using Clinical vs Claims Data, 2009–2014. JAMA 318: 1241–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ponting CP, Oliver PL, and Reik W. 2009. Evolution and functions of long noncoding RNAs. Cell 136: 629–641. [DOI] [PubMed] [Google Scholar]
- 4.Ulitsky I, and Bartel DP. 2013. lincRNAs: genomics, evolution, and mechanisms. Cell 154: 26–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Taft RJ, Pheasant M, and Mattick JS. 2007. The relationship between non-protein-coding DNA and eukaryotic complexity. BioEssays News Rev. Mol. Cell. Dev. Biol 29: 288–299. [DOI] [PubMed] [Google Scholar]
- 6.Chen YG, Satpathy AT, and Chang HY. 2017. Gene regulation in the immune system by long noncoding RNAs. Nat. Immunol 18: 962–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Atianand MK, Caffrey DR, and Fitzgerald KA. 2017. Immunobiology of Long Noncoding RNAs. Annu. Rev. Immunol 35: 177–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sigdel KR, Cheng A, Wang Y, Duan L, and Zhang Y. 2015. The Emerging Functions of Long Noncoding RNA in Immune Cells: Autoimmune Diseases. J. Immunol. Res 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carpenter S, Aiello D, Atianand MK, Ricci EP, Gandhi P, Hall LL, Byron M, Monks B, Henry-Bezy M, Lawrence JB, O’Neill LAJ, Moore MJ, Caffrey DR, and Fitzgerald KA. 2013. A Long Noncoding RNA Mediates Both Activation and Repression of Immune Response Genes. Science 341: 789–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li Z, Chao T-C, Chang K-Y, Lin N, Patil VS, Shimizu C, Head SR, Burns JC, and Rana TM. 2014. The long noncoding RNA THRIL regulates TNFα expression through its interaction with hnRNPL. Proc. Natl. Acad. Sci. U. S. A 111: 1002–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Atianand MK, Hu W, Satpathy AT, Shen Y, Ricci EP, Alvarez-Dominguez JR, Bhatta A, Schattgen SA, McGowan JD, Blin J, Braun JE, Gandhi P, Moore MJ, Chang HY, Lodish HF, Caffrey DR, and Fitzgerald KA. 2016. A Long Noncoding RNA lincRNA-EPS Acts as a Transcriptional Brake to Restrain Inflammation. Cell 165: 1672–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ingolia NT, Brar GA, Stern-Ginossar N, Harris MS, Talhouarne GJS, Jackson SE, Wills MR, and Weissman JS. 2014. Ribosome Profiling Reveals Pervasive Translation Outside of Annotated Protein-Coding Genes. Cell Rep. 8: 1365–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li L-J, Leng R-X, Fan Y-G, Pan H-F, and Ye D-Q. 2017. Translation of noncoding RNAs: Focus on lncRNAs, pri-miRNAs, and circRNAs. Exp. Cell Res 361: 1–8. [DOI] [PubMed] [Google Scholar]
- 14.Jackson R, Kroehling L, Khitun A, Bailis W, Jarret A, York AG, Khan OM, Brewer JR, Skadow MH, Duizer C, Harman CCD, Chang L, Bielecki P, Solis AG, Steach HR, Slavoff S, and Flavell RA. 2018. The translation of non-canonical open reading frames controls mucosal immunity. Nature 564: 434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bhatta A, Atianand M, Jiang Z, Crabtree J, Blin J, and Fitzgerald KA. 2020. A Mitochondrial Micropeptide Is Required for Activation of the Nlrp3 Inflammasome. J. Immunol. Baltim. Md 1950 204: 428–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Krementsov DN, Noubade R, Dragon JA, Otsu K, Rincon M, and Teuscher C. 2014. Sex-specific control of central nervous system autoimmunity by p38 mitogen-activated protein kinase signaling in myeloid cells. Ann. Neurol 75: 50–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Raza A, Crothers JW, McGill MM, Mawe GM, Teuscher C, and Krementsov DN. 2017. Anti-inflammatory roles of p38α MAPK in macrophages are context dependent and require IL-10. J. Leukoc. Biol 102: 1219–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McGill MM, Sabikunnahar B, Fang Q, Teuscher C, and Krementsov DN. 2020. The sex-specific role of p38 MAP kinase in CNS autoimmunity is regulated by estrogen receptor alpha. J. Neuroimmunol 342: 577209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Okazaki Y, Furuno M, Kasukawa T, Adachi J, Bono H, Kondo S, Nikaido I, Osato N, Saito R, Suzuki H, Yamanaka I, Kiyosawa H, Yagi K, Tomaru Y, Hasegawa Y, Nogami A, Schönbach C, Gojobori T, Baldarelli R, Hill DP, Bult C, Hume DA, Quackenbush J, Schriml LM, Kanapin A, Matsuda H, Batalov S, Beisel KW, Blake JA, Bradt D, Brusic V, Chothia C, Corbani LE, Cousins S, Dalla E, Dragani TA, Fletcher CF, Forrest A, Frazer KS, Gaasterland T, Gariboldi M, Gissi C, Godzik A, Gough J, Grimmond S, Gustincich S, Hirokawa N, Jackson IJ, Jarvis ED, Kanai A, Kawaji H, Kawasawa Y, Kedzierski RM, King BL, Konagaya A, Kurochkin IV, Lee Y, Lenhard B, Lyons PA, Maglott DR, Maltais L, Marchionni L, McKenzie L, Miki H, Nagashima T, Numata K, Okido T, Pavan WJ, Pertea G, Pesole G, Petrovsky N, Pillai R, Pontius JU, Qi D, Ramachandran S, Ravasi T, Reed JC, Reed DJ, Reid J, Ring BZ, Ringwald M, Sandelin A, Schneider C, Semple C. a. M., Setou M, Shimada K, Sultana R, Takenaka Y, Taylor MS, Teasdale RD, Tomita M, Verardo R, Wagner L, Wahlestedt C, Wang Y, Watanabe Y, Wells C, Wilming LG, Wynshaw-Boris A, Yanagisawa M, Yang I, Yang L, Yuan Z, Zavolan M, Zhu Y, Zimmer A, Carninci P, Hayatsu N, Hirozane-Kishikawa T, Konno H, Nakamura M, Sakazume N, Sato K, Shiraki T, Waki K, Kawai J, Aizawa K, Arakawa T, Fukuda S, Hara A, Hashizume W, Imotani K, Ishii Y, Itoh M, Kagawa I, Miyazaki A, Sakai K, Sasaki D, Shibata K, Shinagawa A, Yasunishi A, Yoshino M, Waterston R, Lander ES, Rogers J, Birney E, Hayashizaki Y, FANTOM Consortium, and RIKEN Genome Exploration Research Group Phase I & II Team. 2002. Analysis of the mouse transcriptome based on functional annotation of 60,770 full-length cDNAs. Nature 420: 563–573. [DOI] [PubMed] [Google Scholar]
- 20.Chen J, Liu Y, Lu S, Yin L, Zong C, Cui S, Qin D, Yang Y, Guan Q, Li X, and Wang X. 2017. The role and possible mechanism of lncRNA U90926 in modulating 3T3-L1 preadipocyte differentiation. Int. J. Obes. 2005 41: 299–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shirahama S, Onoguchi-Mizutani R, Kawata K, Taniue K, Miki A, Kato A, Kawaguchi Y, Tanaka R, Kaburaki T, Kawashima H, Urade Y, Aihara M, and Akimitsu N. 2020. Long noncoding RNA U90926 is crucial for herpes simplex virus type 1 proliferation in murine retinal photoreceptor cells. Sci. Rep 10: 19406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen J, Jin J, Zhang X, Yu H, Zhu X, Yu L, Chen Y, Liu P, Dong X, Cao X, Gu Y, Bao X, Xia S, and Xu Y. 2021. Microglial lnc-U90926 facilitates neutrophil infiltration in ischemic stroke via MDH2/CXCL2 axis. Mol. Ther. J. Am. Soc. Gene Ther 29: 2873–2885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Clausen BE, Burkhardt C, Reith W, Renkawitz R, and Förster I. 1999. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 8: 265–277. [DOI] [PubMed] [Google Scholar]
- 24.Nishida K, Yamaguchi O, Hirotani S, Hikoso S, Higuchi Y, Watanabe T, Takeda T, Osuka S, Morita T, Kondoh G, Uno Y, Kashiwase K, Taniike M, Nakai A, Matsumura Y, Miyazaki J, Sudo T, Hongo K, Kusakari Y, Kurihara S, Chien KR, Takeda J, Hori M, and Otsu K. 2004. p38α Mitogen-Activated Protein Kinase Plays a Critical Role in Cardiomyocyte Survival but Not in Cardiac Hypertrophic Growth in Response to Pressure Overload. Mol. Cell. Biol 24: 10611–10620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR | Genome Biology | Full Text. . [DOI] [PMC free article] [PubMed]
- 26.Helft J, Böttcher J, Chakravarty P, Zelenay S, Huotari J, Schraml BU, Goubau D, and Reis e Sousa C. 2015. GM-CSF Mouse Bone Marrow Cultures Comprise a Heterogeneous Population of CD11c(+)MHCII(+) Macrophages and Dendritic Cells. Immunity 42: 1197–1211. [DOI] [PubMed] [Google Scholar]
- 27.Patro R, Duggal G, Love MI, Irizarry RA, and Kingsford C. 2017. Salmon: fast and bias-aware quantification of transcript expression using dual-phase inference. Nat. Methods 14: 417–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Love MI, Huber W, and Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15: 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mi H, Muruganujan A, Casagrande JT, and Thomas PD. 2013. Large-scale gene function analysis with PANTHER Classification System. Nat. Protoc 8: 1551–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boisson-Dupuis S, Ramirez-Alejo N, Li Z, Patin E, Rao G, Kerner G, Lim CK, Krementsov DN, Hernandez N, Ma CS, Zhang Q, Markle J, Martinez-Barricarte R, Payne K, Fisch R, Deswarte C, Halpern J, Bouaziz M, Mulwa J, Sivanesan D, Lazarov T, Naves R, Garcia P, Itan Y, Boisson B, Checchi A, Jabot-Hanin F, Cobat A, Guennoun A, Jackson CC, Pekcan S, Caliskaner Z, Inostroza J, Costa-Carvalho BT, de Albuquerque JAT, Garcia-Ortiz H, Orozco L, Ozcelik T, Abid A, Rhorfi IA, Souhi H, Amrani HN, Zegmout A, Geissmann F, Michnick SW, Muller-Fleckenstein I, Fleckenstein B, Puel A, Ciancanelli MJ, Marr N, Abolhassani H, Balcells ME, Condino-Neto A, Strickler A, Abarca K, Teuscher C, Ochs HD, Reisli I, Sayar EH, El-Baghdadi J, Bustamante J, Hammarström L, Tangye SG, Pellegrini S, Quintana-Murci L, Abel L, and Casanova J-L. 2018. Tuberculosis and impaired IL-23–dependent IFN-γ immunity in humans homozygous for a common TYK2 missense variant. Sci. Immunol 3: eaau8714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Okeke EB, Okwor I, and Uzonna JE. 2014. Regulatory T cells restrain CD4+ T cells from causing unregulated immune activation and hypersensitivity to lipopolysaccharide challenge. J. Immunol. Baltim. Md 1950 193: 655–662. [DOI] [PubMed] [Google Scholar]
- 32.Olarescu NC, Berryman DE, Householder LA, Lubbers ER, List EO, Benencia F, Kopchick JJ, and Bollerslev J. 2015. Growth Hormone Action Influences Adipogenesis of Mouse Adipose Tissue-Derived Mesenchymal Stem Cells. J. Endocrinol 226: 13–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Amit I, Garber M, Chevrier N, Leite AP, Donner Y, Eisenhaure T, Guttman M, Grenier JK, Li W, Zuk O, Schubert LA, Birditt B, Shay T, Goren A, Zhang X, Smith Z, Deering R, McDonald RC, Cabili M, Bernstein BE, Rinn JL, Meissner A, Root DE, Hacohen N, and Regev A. 2009. Unbiased Reconstruction of a Mammalian Transcriptional Network Mediating Pathogen Responses. Science 326: 257–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Akira S, and Hoshino K. 2003. Myeloid Differentiation Factor 88—Dependent and —Independent Pathways in Toll-Like Receptor Signaling. J. Infect. Dis 187: S356–63. [DOI] [PubMed] [Google Scholar]
- 35.Siednienko J, Gajanayake T, Fitzgerald KA, Moynagh P, and Miggin SM. 2011. Absence of MyD88 results in enhanced TLR3-dependent phosphorylation of IRF3 and increased IFN-β and RANTES production. J. Immunol. Baltim. Md 1950 186: 2514–2522. [DOI] [PubMed] [Google Scholar]
- 36.Zhang Z, and Cohen DM. 1996. NaCl but not urea activates p38 and jun kinase in mIMCD3 murine inner medullary cells. Am. J. Physiol 271: F1234–1238. [DOI] [PubMed] [Google Scholar]
- 37.Xu X, Wang Q, Long Y, Zhang R, Wei X, Xing M, Gu H, and Xie X. 2013. Stress-mediated p38 activation promotes somatic cell reprogramming. Cell Res. 23: 131–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Matthias J, Heink S, Picard F, Zeiträg J, Kolz A, Chao Y-Y, Soll D, de Almeida GP, Glasmacher E, Jacobsen ID, Riedel T, Peters A, Floess S, Huehn J, Baumjohann D, Huber M, Korn T, and Zielinski CE. 2020. Salt generates antiinflammatory Th17 cells but amplifies pathogenicity in proinflammatory cytokine microenvironments. J. Clin. Invest 130: 4587–4600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sabio G, and Davis RJ. 2014. TNF and MAP kinase signaling pathways. Semin. Immunol 26: 237–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Adachi O, Kawai T, Takeda K, Matsumoto M, Tsutsui H, Sakagami M, Nakanishi K, and Akira S. 1998. Targeted Disruption of the MyD88 Gene Results in Loss of IL-1- and IL-18-Mediated Function. Immunity 9: 143–150. [DOI] [PubMed] [Google Scholar]
- 41.Deguine J, and Barton GM. 2014. MyD88: a central player in innate immune signaling. F1000Prime Rep. 6: 97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Snyder JP, Gullickson SK, del Rio-Guerra R, Sweezy A, Vagher B, Hogan TC, Lahue KG, Reisz JA, D’Alessandro A, Krementsov DN, and Amiel E. 2022. Divergent Genetic Regulation of Nitric Oxide Production between C57BL/6J and Wild-Derived PWD/PhJ Mice Controls Postactivation Mitochondrial Metabolism, Cell Survival, and Bacterial Resistance in Dendritic Cells. J. Immunol 208: 97–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cho SW, Kim S, Kim Y, Kweon J, Kim HS, Bae S, and Kim J-S. 2014. Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res. 24: 132–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang X-H, Tee LY, Wang X-G, Huang Q-S, and Yang S-H. 2015. Off-target Effects in CRISPR/Cas9-mediated Genome Engineering. Mol. Ther. - Nucleic Acids 4: e264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Du M, Yuan L, Tan X, Huang D, Wang X, Zheng Z, Mao X, Li X, Yang L, Huang K, Zhang F, Wang Y, Luo X, Huang D, and Huang K. 2017. The LPS-inducible lncRNA Mirt2 is a negative regulator of inflammation. Nat. Commun 8: 2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fields AP, Rodriguez EH, Jovanovic M, Stern-Ginossar N, Haas BJ, Mertins P, Raychowdhury R, Hacohen N, Carr SA, Ingolia NT, Regev A, and Weissman JS. 2015. A Regression-Based Analysis of Ribosome-Profiling Data Reveals a Conserved Complexity to Mammalian Translation. Mol. Cell 60: 816–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Spiro RG 2002. Protein glycosylation: nature, distribution, enzymatic formation, and disease implications of glycopeptide bonds. Glycobiology 12: 43R–56R. [DOI] [PubMed] [Google Scholar]
- 48.Hoshino K, Takeuchi O, Kawai T, Sanjo H, Ogawa T, Takeda Y, Takeda K, and Akira S. 1999. Cutting edge: Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the Lps gene product. J. Immunol. Baltim. Md 1950 162: 3749–3752. [PubMed] [Google Scholar]
- 49.Greten FR, Arkan MC, Bollrath J, Hsu L-C, Goode J, Miething C, Göktuna SI, Neuenhahn M, Fierer J, Paxian S, Van Rooijen N, Xu Y, O’Cain T, Jaffee BB, Busch DH, Duyster J, Schmid RM, Eckmann L, and Karin M. 2007. NF-kappaB is a negative regulator of IL-1beta secretion as revealed by genetic and pharmacological inhibition of IKKbeta. Cell 130: 918–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gandhi R, Hayley S, Gibb J, Merali Z, and Anisman H. 2007. Influence of poly I:C on sickness behaviors, plasma cytokines, corticosterone and central monoamine activity: moderation by social stressors. Brain. Behav. Immun 21: 477–489. [DOI] [PubMed] [Google Scholar]
- 51.Riedemann NC, Neff TA, Guo R-F, Bernacki KD, Laudes IJ, Sarma JV, Lambris JD, and Ward PA. 2003. Protective Effects of IL-6 Blockade in Sepsis Are Linked to Reduced C5a Receptor Expression1. J. Immunol 170: 503–507. [DOI] [PubMed] [Google Scholar]
- 52.Honda S-I, Sato K, Totsuka N, Fujiyama S, Fujimoto M, Miyake K, Nakahashi-Oda C, Tahara-Hanaoka S, Shibuya K, and Shibuya A. 2016. Marginal zone B cells exacerbate endotoxic shock via interleukin-6 secretion induced by Fcα/μR-coupled TLR4 signalling. Nat. Commun 7: 11498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Norris CA, He M, Kang L-I, Ding MQ, Radder JE, Haynes MM, Yang Y, Paranjpe S, Bowen WC, Orr A, Michalopoulos GK, Stolz DB, and Mars WM. 2014. Synthesis of IL-6 by Hepatocytes Is a Normal Response to Common Hepatic Stimuli. PLoS ONE 9: e96053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nesseler N, Launey Y, Aninat C, Morel F, Mallédant Y, and Seguin P. 2012. Clinical review: The liver in sepsis. Crit. Care 16: 235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Triantafyllou E, Woollard KJ, McPhail MJW, Antoniades CG, and Possamai LA. 2018. The Role of Monocytes and Macrophages in Acute and Acute-on-Chronic Liver Failure. Front. Immunol 9: 2948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gregory SH, and Wing EJ. 2002. Neutrophil-Kupffer cell interaction: a critical component of host defenses to systemic bacterial infections. J. Leukoc. Biol 72: 239–248. [PubMed] [Google Scholar]
- 57.Koo DJ, Chaudry IH, and Wang P. 2000. Mechanism of hepatocellular dysfunction during sepsis: the role of gut-derived norepinephrine (review). Int. J. Mol. Med 5: 457–522. [DOI] [PubMed] [Google Scholar]
- 58.Paumelle R, Haas JT, Hennuyer N, Baugé E, Deleye Y, Mesotten D, Langouche L, Vanhoutte J, Cudejko C, Wouters K, Hannou SA, Legry V, Lancel S, Lalloyer F, Polizzi A, Smati S, Gourdy P, Vallez E, Bouchaert E, Derudas B, Dehondt H, Gheeraert C, Fleury S, Tailleux A, Montagner A, Wahli W, Van Den Berghe G, Guillou H, Dombrowicz D, and Staels B. 2019. Hepatic PPARα is critical in the metabolic adaptation to sepsis. J. Hepatol 70: 963–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shirahama S, Taniue K, Mitsutomi S, Tanaka R, Kaburaki T, Sato T, Takeuchi M, Kawashima H, Urade Y, Aihara M, and Akimitsu N. 2021. Human U90926 orthologous long non-coding RNA as a novel biomarker for visual prognosis in herpes simplex virus type-1 induced acute retinal necrosis. Sci. Rep 11: 12164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Krug A, Luker GD, Barchet W, Leib DA, Akira S, and Colonna M. 2004. Herpes simplex virus type 1 activates murine natural interferon-producing cells through toll-like receptor 9. Blood 103: 1433–1437. [DOI] [PubMed] [Google Scholar]
- 61.Zyzak J, Mitkiewicz M, Leszczyńska E, Reniewicz P, Moynagh PN, and Siednienko J. 2020. HSV-1/TLR9-Mediated IFNβ and TNFα Induction Is Mal-Dependent in Macrophages. J. Innate Immun 12: 387–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gong T, Liu L, Jiang W, and Zhou R. 2020. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat. Rev. Immunol 20: 95–112. [DOI] [PubMed] [Google Scholar]
- 63.Jayaraj RL, Azimullah S, Beiram R, Jalal FY, and Rosenberg GA. 2019. Neuroinflammation: friend and foe for ischemic stroke. J. Neuroinflammation 16: 142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ruan H, Hacohen N, Golub TR, Van Parijs L, and Lodish HF. 2002. Tumor necrosis factor-alpha suppresses adipocyte-specific genes and activates expression of preadipocyte genes in 3T3-L1 adipocytes: nuclear factor-kappaB activation by TNF-alpha is obligatory. Diabetes 51: 1319–1336. [DOI] [PubMed] [Google Scholar]
- 65.Lacasa D, Taleb S, Keophiphath M, Miranville A, and Clement K. 2007. Macrophage-secreted factors impair human adipogenesis: involvement of proinflammatory state in preadipocytes. Endocrinology 148: 868–877. [DOI] [PubMed] [Google Scholar]
- 66.Boutens L, and Stienstra R. 2016. Adipose tissue macrophages: going off track during obesity. Diabetologia 59: 879–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cheng H, Luan J, Mu D, Wang Q, Qi J, Li Z, and Fu S. 2019. M1/M2 Macrophages Play Different Roles in Adipogenic Differentiation of PDGFRα+ Preadipocytes In Vitro. Aesthetic Plast. Surg 43: 514–520. [DOI] [PubMed] [Google Scholar]
- 68.Vollmers AC, Covarrubias S, Kuang D, Shulkin A, Iwuagwu J, Katzman S, Song R, Viswanathan K, Vollmers C, Wakeland E, and Carpenter S. 2021. A conserved long noncoding RNA, GAPLINC, modulates the immune response during endotoxic shock. Proc. Natl. Acad. Sci. U. S. A 118: e2016648118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cui H, Banerjee S, Guo S, Xie N, Ge J, Jiang D, Zörnig M, Thannickal VJ, and Liu G. 2019. Long noncoding RNA Malat1 regulates differential activation of macrophages and response to lung injury. JCI Insight 4: 124522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sallam T, Jones M, Thomas BJ, Wu X, Gilliland T, Qian K, Eskin A, Casero D, Zhang Z, Sandhu J, Salisbury D, Rajbhandari P, Civelek M, Hong C, Ito A, Liu X, Daniel B, Lusis AJ, Whitelegge J, Nagy L, Castrillo A, Smale S, and Tontonoz P. 2018. Transcriptional regulation of macrophage cholesterol efflux and atherogenesis by a long noncoding RNA. Nat. Med 24: 304–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bridges MC, Daulagala AC, and Kourtidis A. 2021. LNCcation: lncRNA localization and function. J. Cell Biol 220: e202009045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chen L-L 2016. Linking Long Noncoding RNA Localization and Function. Trends Biochem. Sci 41: 761–772. [DOI] [PubMed] [Google Scholar]
- 73.Kopp F, and Mendell JT. 2018. Functional Classification and Experimental Dissection of Long Noncoding RNAs. Cell 172: 393–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Guo C-J, Ma X-K, Xing Y-H, Zheng C-C, Xu Y-F, Shan L, Zhang J, Wang S, Wang Y, Carmichael GG, Yang L, and Chen L-L. 2020. Distinct Processing of lncRNAs Contributes to Non-conserved Functions in Stem Cells. Cell 181: 621–636.e22. [DOI] [PubMed] [Google Scholar]
- 75.Andrews SJ, and Rothnagel JA. 2014. Emerging evidence for functional peptides encoded by short open reading frames. Nat. Rev. Genet 15: 193–204. [DOI] [PubMed] [Google Scholar]
- 76.Makarewich CA, and Olson EN. 2017. Mining for Micropeptides. Trends Cell Biol. 27: 685–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kozak M 1992. Regulation of Translation in Eukaryotic Systems. Annu. Rev. Cell Biol 8: 197–225. [DOI] [PubMed] [Google Scholar]
- 78.Tanaka T, Narazaki M, and Kishimoto T. 2014. IL-6 in Inflammation, Immunity, and Disease. Cold Spring Harb. Perspect. Biol 6: a016295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Adams R, Burnley RJ, Valenzano CR, Qureshi O, Doyle C, Lumb S, del Carmen Lopez M, Griffin R, McMillan D, Taylor RD, Meier C, Mori P, Griffin LM, Wernery U, Kinne J, Rapecki S, Baker TS, Lawson ADG, Wright M, and Ettorre A. 2017. Discovery of a junctional epitope antibody that stabilizes IL-6 and gp80 protein:protein interaction and modulates its downstream signaling. Sci. Rep 7: 37716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bantel H, and Schulze-Osthoff K. 2009. Cell death in sepsis: a matter of how, when, and where. Crit. Care 13: 173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Reddy RC, Narala VR, Keshamouni VG, Milam JE, Newstead MW, and Standiford TJ. 2008. Sepsis-induced inhibition of neutrophil chemotaxis is mediated by activation of peroxisome proliferator-activated receptor-{gamma}. Blood 112: 4250–4258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Carré JE, and Singer M. 2008. Cellular energetic metabolism in sepsis: The need for a systems approach. Biochim. Biophys. Acta BBA - Bioenerg 1777: 763–771. [DOI] [PubMed] [Google Scholar]
- 83.Florentin J, Zhao J, Tai Y-Y, Vasamsetti SB, O’Neil SP, Kumar R, Arunkumar A, Watson A, Sembrat J, Bullock GC, Sanders L, Kassa B, Rojas M, Graham BB, Chan SY, and Dutta P. 2021. Interleukin-6 mediates neutrophil mobilization from bone marrow in pulmonary hypertension. Cell. Mol. Immunol 18: 374–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rouault C, Pellegrinelli V, Schilch R, Cotillard A, Poitou C, Tordjman J, Sell H, Clément K, and Lacasa D. 2013. Roles of chemokine ligand-2 (CXCL2) and neutrophils in influencing endothelial cell function and inflammation of human adipose tissue. Endocrinology 154: 1069–1079. [DOI] [PubMed] [Google Scholar]
- 85.Fielding CA, McLoughlin RM, McLeod L, Colmont CS, Najdovska M, Grail D, Ernst M, Jones SA, Topley N, and Jenkins BJ. 2008. IL-6 regulates neutrophil trafficking during acute inflammation via STAT3. J. Immunol. Baltim. Md 1950 181: 2189–2195. [DOI] [PubMed] [Google Scholar]
- 86.Leygue E 2007. Steroid Receptor RNA Activator (SRA1): Unusual Bifaceted Gene Products with Suspected Relevance to Breast Cancer. Nucl. Recept. Signal 5: nrs.05006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Managadze D, Lobkovsky AE, Wolf YI, Shabalina SA, Rogozin IB, and Koonin EV. 2013. The Vast, Conserved Mammalian lincRNome. PLoS Comput. Biol 9: e1002917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Diederichs S 2014. The four dimensions of noncoding RNA conservation. Trends Genet. TIG 30: 121–123. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.