

Abstract
Multiple cancers regulate oxidative stress by activating the transcription factor NRF2 through mutation of its negative regulator KEAP1. NRF2 has been studied extensively in KEAP1-mutant cancers, however the role of this pathway in cancers with wildtype KEAP1 remains poorly understood. To answer this question, we induced NRF2 via pharmacological inactivation of KEAP1 in a panel of 50+ non-small cell lung cancer cell lines. Unexpectedly, marked decreases in viability were observed in >13% of the cell lines—an effect that was rescued by NRF2 ablation. Genome-wide and targeted CRISPR screens revealed that NRF2 induces NADH-reductive stress, through the upregulation of the NAD+-consuming enzyme ALDH3A1. Leveraging these findings, we show that cells treated with KEAP1 inhibitors or those with endogenous KEAP1 mutations are selectively vulnerable to Complex I inhibition, which impairs NADH oxidation capacity and potentiates reductive stress. Thus, we identify reductive stress as a metabolic vulnerability in NRF2-activated lung cancers.
Keywords: NRF2-KEAP1 pathway, Reductive Stress, non-small cell lung cancer, functional genomic, oxidative phosphorylation, NADH/NAD+
Graphical Abstract
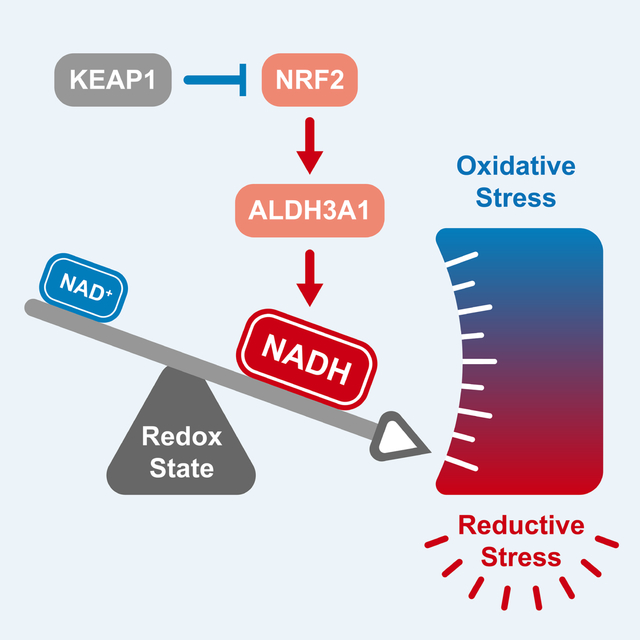
A subset of lung cancer cell lines is sensitive to activation of the transcription factor NRF2. The metabolic state of a cancer cell dictates sensitivity to NRF2, whose activation results in a NADH/NAD+ imbalance. As a result, blockade of mitochondrial complex I is a synthetic lethality in NRF2-activated lung cancers.
Introduction
To support their rapid proliferation, tumors must adapt their metabolism to an ever-growing list of cell intrinsic and extrinsic pressures1–5. One such pressure is the maintenance of redox homeostasis as a pre-requisite for tumor proliferation6,7. Tumors have biochemically rewired core metabolic pathways to maintain elevated levels of biosynthetic molecules and consequently generate increased reactive oxygen species (ROS) compared to normal cells8–12. High levels of ROS modify nucleic acids, proteins and lipids—and can provoke lethal cellular effects through multiple mechanisms, including creating DNA damage, impairing function of mitochondria and other organelles and disrupting the integrity of cell membranes6,13.
While redox imbalance in cancer cells has been investigated extensively in the context of oxidative stress, the converse of oxidative stress, reductive stress and in particular its impact on malignant cells is poorly understood14. Reductive stress is induced by excessive levels of antioxidants and high concentrations of reduced nucleotide cofactors, such as NADH, required for antioxidant and detoxification reactions. An overly reductive cell state15–19 can affect vital cellular processes such as oxidative protein folding in the ER20. Indeed, recent studies have demonstrated that reductive stress is just as harmful to cell proliferation as oxidative stress21–24 with high levels of NADH leading to disruption of de novo lipid, amino acid and nucleotide biosynthesis as a result of decreased electron acceptors25–30.
To counter oxidative stress, tumors rely on NRF2, the central transcriptional regulator of the antioxidant response31–35. Under conditions of low oxidative stress, NRF2 binds to KEAP1, a tumor suppressor and ROS sensing protein, that leads NRF2 to its rapid proteasomal degradation36,37. Under high ROS levels, key ROS-sensing cysteines in the backbone of KEAP1 are modified resulting in the dissociation and nuclear translocation of NRF238,39, and the consequent induction of several hundred genes involved in antioxidant response31,40. NRF2 is activated via genetic inactivation of KEAP1 in many cancers, including ~30% of non-small cell lung cancers (NSCLCs)41. In KEAP1-mutant cancers, NRF2 is absolutely required for tumor growth functioning as a potent oncogene32,42–45.
To date, a significant body of our knowledge about the role of the NRF2-KEAP1 pathway in cancer comes from the discovery that NRF2 functions as an oncogene in the context of KEAP1-mutations in lung cancer31,45–50. We hypothesized that NRF2 activity might benefit the proliferation of NSCLCs when KEAP1 is not mutated. By activating NRF2 in a panel of NSCLCs we found, surprisingly, that rather than promote cell proliferation, activation of NRF2 via acute pharmacologic or genetic inhibition of KEAP1 potently blocks the growth of >13% of NSCLC cell lines –characterizing them as KEAP1-dependent. This dependency stems from a cell’s intrinsic preference for glucose utilization– with KEAP1-dependent cells characterized by lower levels of glycolysis and sensitivity to Complex I inhibitors. Mechanistically, we find that NRF2 activation results in an increase in NADH levels in KEAP1-dependent but not KEAP1-independent cells. We demonstrate through manipulation of glycolysis and NADH oxidation rates, that NADH levels are both necessary and sufficient to mediate sensitivity to NRF2 activation. We identify that ALDH3A1, a dehydrogenase involved in antioxidant response, has a primary role in increasing NADH levels and sensitizing KEAP1-dependent cells to NRF2 activation. Finally, we demonstrate that increased NADH/NAD+ ratio due to treatment with KEAP1-inhibitors or the presence of KEAP1-mutations confers exquisite sensitivity to a clinical grade Complex I inhibitor, which overwhelms NADH homeostasis in these cells, leading to reductive stress. Thus, we reveal how over-activation of an antioxidant signaling pathway leads to a reduced cellular state that can create “oxidative addiction” and synthetic lethal opportunities within a subset of lung cancers.
Results
Identification of KEAP1-dependent NSCLC cell lines
Multiple studies have identified a proliferation benefit from NRF2 activation in the context of KEAP1-mutant NSCLC cell lines44–46,51. We hypothesized that additional NSCLC cell lines that are KEAP1-wildtype (WT) would also gain a proliferative advantage following NRF2 activation. To test this hypothesis, we treated a panel of 50+ genetically diverse NSCLC cell lines (with a majority of lines WT for KEAP1, NRF2, and CUL3) with KI696, a potent and specific inhibitor of KEAP1-NRF2 interactions52,53, which leads to NRF2 stabilization and activation (Figure 1A). Proliferation was not altered in most lines, including KEAP1-mutants (e.g., H2122), and was increased in a limited subset (Figure 1B). Surprisingly, we find that >13% of NSCLC cell lines have a substantial block in proliferation following KEAP1 inhibition (Figure 1B). In support of NRF2 activation mediating the proliferation block with KI696, treatment of a subset of NSCLC lines with bardoxolone, a well-established NRF2 activator, also potently blocked proliferation (Figures S1A–B). Expression of KEAP1-targeting sgRNAs or doxycycline (DOX)-inducible shRNAs strongly blocked the proliferation of KI696-sensitive cell lines in comparison to non-targeting sgRNA/shRNAs, characterizing these cells as KEAP1-dependent (Figures 1C, S1C–E). Proliferation was not impacted in KI696-insensitive cells following genetic depletion of KEAP1, marking them as KEAP1-independent (Figures 1C, S1C–E). KEAP1-dependence could be rescued by re-expressing a sgKEAP1-mutant KEAP1 cDNA under a DOX-repressible element, and we observed a proliferation block in KI696-sensitive lines following DOX treatment and subsequent depletion of KEAP1 (Figures S1F–G).
Figure 1: Identification of KEAP1-dependent NSCLC cell lines.
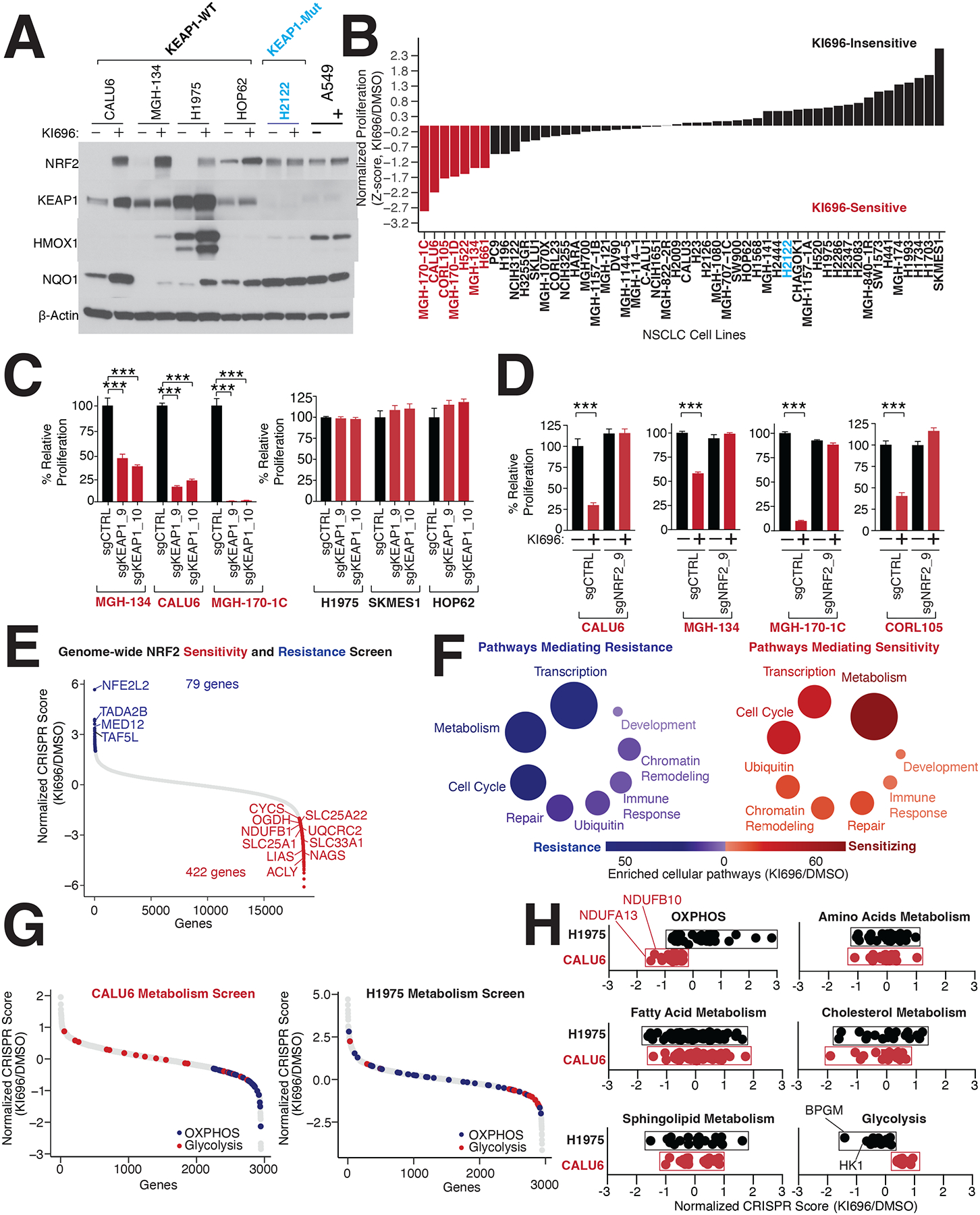
(A) Representative immunoblot analysis of NSCLC cell lines following treatment with the NRF2-activator KI696 (1μM) for 48 hrs. (B) NRF2 activation in a panel of 50+ NSCLC cell lines identifies KI696-sensitive cell lines. Cells were pre-treated with KI696 (1 μM) for 48 hrs and proliferation was determined by crystal violet staining following another 6 days of treatment. (C) Depletion of KEAP1 blocks proliferation of KI696-sensitive cell lines. KI696-sensitive (herein referred to as KEAP-dependent) and KI696-insensitive (KEAP1-independent) cell lines expressing sgRNAs targeting KEAP1 or a non-targeting control were analyzed for proliferation defects as described in (B) (Data are represented as a mean ± SEM, n=5 biological replicates). (D) Depletion of NRF2 rescues KI696-sensitivity. NSCLC cell lines expressing the indicated sgRNAs were treated with KI696 (1 μM) and proliferation was determined as described in (B) (Data are represented as a mean ± SEM, n=5 biological replicates). (E) Genome-wide CRISPR screen identifies genes mediating resistance and sensitivity to KEAP1-dependency. Highlighted genes are key mediators of sensitivity (red) or resistance (blue) (see also Table S1). (F) Genes localized to metabolic pathways function as key mediators of sensitivity to NRF2 activation in CALU6 cells (see also Table S1). (G) Metabolism-focused CRISPR screen identifies metabolic regulators of NRF2 sensitivity. KEAP1-dependent CALU6 (red) and KEAP1-Independent (black) H1975 cells were infected with a metabolism-focused sgRNA library and treated as described in (E) (see also Table S2). (H) Inactivation of oxidative phosphorylation (OXPHOS) sensitizes KEAP1-dependent cells to NRF2 activation whereas blockage of glycolysis sensitizes KEAP1-independent cells to NRF2 (see also Figure S2E). Statistical significance was determined by One-way ANOVA with Sidak’s corrections for multiple comparisons. *** indicates p-values < 0.0001.
The strong reliance on KEAP1 for proliferation was quite unexpected given the canonical characterization of KEAP1 as a recurrently mutated tumor suppressor in lung cancer31,41,45,47,51,54–56. In KEAP1-dependent cells, KI696 treatment leads to a stabilization of NRF2 and expression of NRF2 target genes (Figures 1A, S1A), suggesting that KEAP1-dependency is associated with NRF2 activation. Depleting NRF2 in four KEAP1-dependent cell lines did not alter proliferation at baseline (Figures 1D, S1H), but completely reversed the proliferation arrest following pharmacological or genetic inhibition of KEAP1 (Figures 1D, S1H–J).
By analyzing cancer essentiality data from genome-wide CRISPR screens across 800+ cancer cell lines57,58, we find that multiple cancer cell lines of different origins are sensitive to loss of KEAP1, including breast and skin cancers which share a similar rate of dependency as NSCLCs (Figure S1K). These results illustrate that KEAP1-dependency is broadly observed in multiple cancer subtypes highlighting the complex role of established oncogenes when their corresponding tumor suppressors are not mutated59,60.
Functional genomic interrogation of KEAP1 dependency
We did not find a correlation between KEAP1-dependency and mutational status of other oncogenic pathways (e.g., LKB1, PI3K, p53, KRAS). To search for genes which may predict sensitivity or resistance to NRF2 activation, we performed a genome-wide CRISPR screen in the KEAP1-dependent cell line CALU6. Following infection with the sgRNA library (10 sgRNAs/gene), cells were grown for 11 population doublings in the presence of 1 μM of KI696 or vehicle control. For each gene, we calculated a CRISPR score by comparing the relative fold change between corresponding sgRNAs enriched in KI696 vs. vehicle. This analysis identified 79 genes mediating resistance and 422 genes mediating sensitivity to KI696 (Figures 1E, S2A, Table S1). Validating our screening approach, we identified NRF2 as the top-scoring gene that mediated resistance when depleted in KEAP1-dependent cell lines (Figure 1E). We also identified that depleting members of the mediator complex (e.g., TAF5L and TADA2B), results in resistance to NRF2 (Figure S2B), likely by supporting the NRF2 transcriptional program. Consistent with this premise, we also found that depletion of MYC, a master regulator of transcription61, decreases NRF2-target gene expression and mediates resistance to KI696 (Figures S2C–D). Moreover, MYC transcript levels are lower in KEAP1-dependent cells compared to KEAP1-independent (Figure S2E).
There was a striking enrichment of metabolic genes identified as mediators of KI696 sensitivity in the CRISPR screen, including many genes belonging to mitochondrial metabolic pathways (Figure 1F, Table S1). To better define metabolic mechanisms of sensitivity to NRF2 activation, we next undertook a metabolism-focused CRISPR screen, encompassing ~2000 genes connected to different metabolic processes28 in KEAP1-dependent (CALU6) and KEAP1-independent (H1975) cells following KI696 treatment (Figure 1G, Table S2). We found an enrichment for a subset of glycolytic genes mediating sensitivity in KEAP1-independent cells, whereas those genes enriched in oxidative phosphorylation (OXPHOS) sensitized KEAP1-dependent cells (Figures 1H, S2E). The absence of additional glycolytic or OXPHOS genes scoring in this screen most likely stems from their pan-essential nature.
Anaerobic metabolism mediates resistance to NRF2 activation
To further investigate the metabolic basis for KEAP1-dependency, we focused on the opposing forms of glucose utilization that were differentially essential following KI696 treatment in KEAP1-dependent and –independent cells. The ratio of extracellular acidification rate (ECAR) to oxygen consumption rate (OCR) is used to characterize the preference for aerobic vs. anaerobic metabolism62. By comparing the OCR/ECAR ratio of 12 NSCLC models to their corresponding sensitivity for KI696, we found a strong correlation between a preference for aerobic metabolism and NRF2 sensitivity, whereas cells with high glycolytic rates were insensitive to NRF2 activity (Figure 2A). These results were further substantiated by comparing KEAP1-dependency to a glycolytic gene signature; NSCLCs marked by a high glycolytic gene signature possessed a lower sensitivity to NRF2 activation (Figure 2B, Table S3). Interestingly, we found that KEAP1-independent cells had on average higher lactate dehydrogenase (LDH) activity compared to their KEAP1-dependent counterparts (Figure S2F). Moreover, KEAP1-independent cell lines had higher levels of phosphofructokinase (PFK), one of the rate-limiting enzymes in glycolysis (Figures 2C, S2G). The higher glycolytic rates characterizing KEAP1-independent cells suggested this form of glucose utilization might be a mechanism to adapt to NRF2 activation. Therefore, we over-expressed PFK in KEAP1-dependent CALU6 cells, and observed not only an increase in glycolysis as measured by lactate secretion but a partial rescue of proliferation following NRF2 activation (Figures 2D, S2H–I).
Figure 2: Metabolic requirements for NRF2 sensitivity.
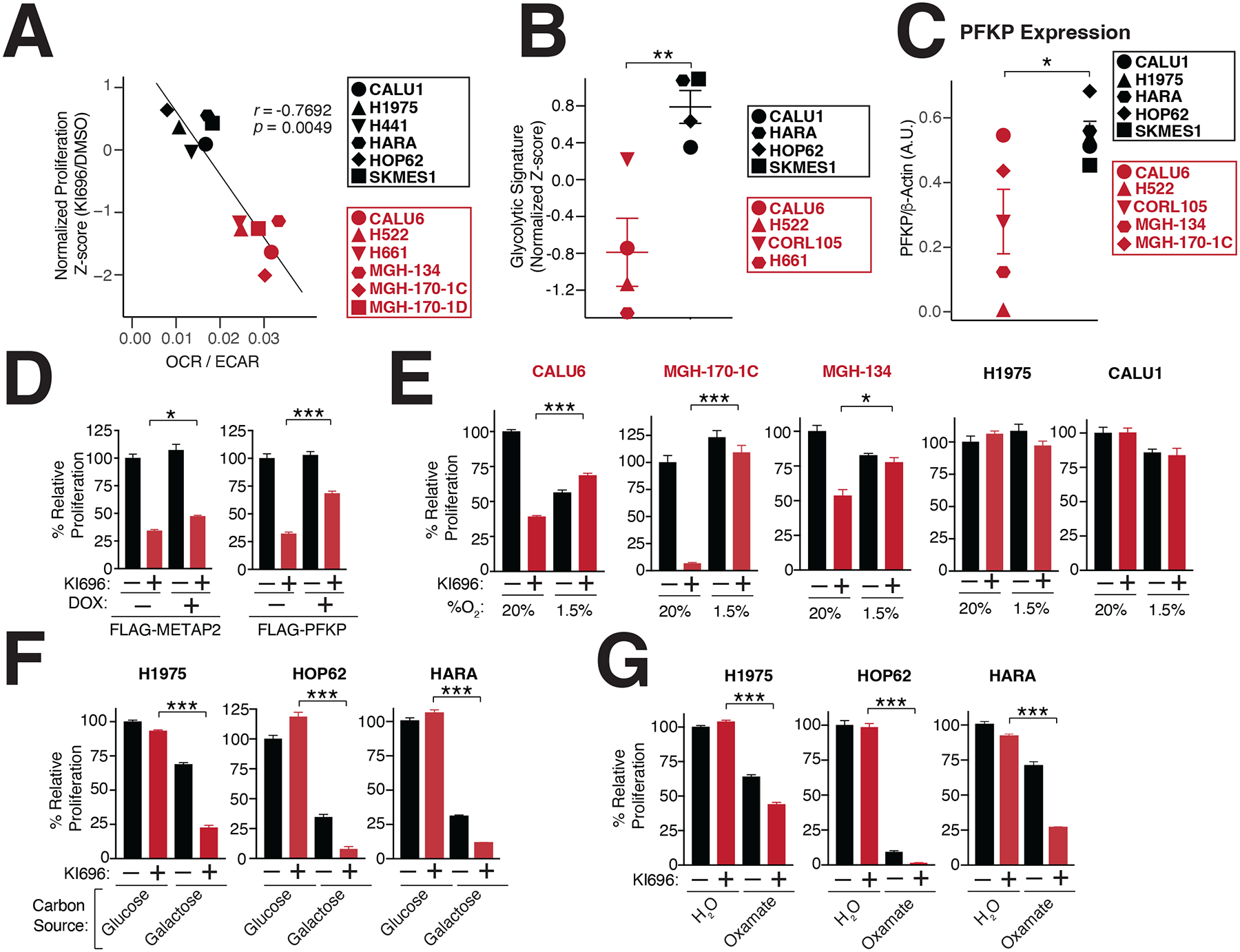
(A) NRF2 sensitivity correlates with higher levels of oxidative metabolism. Oxygen consumption rate (OCR) and extracellular acidification (ECAR) were measured in a panel of NSCLC cells and the OCR/ECAR for each cell line was plotted against its corresponding sensitivity to NRF2 activation (Data are represented as a mean ± SEM, n=6–8 biological replicates). (B) KEAP1-dependent cells have a lower glycolytic gene signature (see also Table S3). (C) The rate-limiting glycolytic enzyme phosphofructokinase, platelet isoform (PFKP), is highly expressed in KEAP1-dependent cells. Quantification of PFKP levels relative to β-actin (see also Figure S2G). (D) PFKP overexpression restores proliferation following NRF2 activation. Relative proliferation of CALU6 cells expressing FLAG-PFKP or FLAG-METAP2 (control) was determined by crystal violet staining following doxycycline (DOX) (100 nM) and KI696 (1 μM) treatment (Data are represented as a mean ± SEM, n=5 biological replicates). (E) Hypoxia rescues NRF2 sensitivity. Relative proliferation in a panel of NSCLC cell lines following treatment with KI696 (1 μM) and culture in normoxic (20% O2) or hypoxic conditions (1.5% O2) was determined as in (D) (Data are represented as a mean ± SEM, n= 5 biological replicates). (F-G) Glycolytic inhibition sensitizes cells to NRF2 activation. KEAP1-independent cells were treated with KI696 (1 μM) and cultured in media containing glucose (10 mM) or galactose (10 mM) (F) or co-treated with sodium oxamate (10 mM) (G) and relative proliferation was determined as in (D) (Data are represented as a mean ± SEM, n=5 biological replicates). (H) NRF2 activation decreases maximal respiration in KEAP1-dependent cells. Maximal respiration was determined in a panel of NSCLC lines following treatment with KI696 (1 μM) for 48 hrs (Data are represented as a mean ± SEM, n=6–8 biological replicates). * indicates p-values < 0.05, *** indicates p-values < 0.0001. One-way ANOVA with Sidak’s post-hoc correction and two-tailed student’s t-test were used to determine statistical significance.
Given the well-established connection between hypoxia and glycolytic reprogramming63, we induced hypoxia in KEAP1-dependent cells, and observed increased expression in the glycolytic enzyme HK2 (Figure S2J). Importantly, induction of hypoxia largely rescued NRF2 sensitivity in KEAP1-dependent cells, but had no effect on KEAP1-independent cells (Figure 2E). Our results suggest that high glycolytic rates are sufficient to overcome NRF2 sensitivity. Thus, we next asked whether decreasing glycolysis within KEAP1-independent cells results in NRF2-sensitivity. To this end, we blocked the initial and terminal steps of glycolysis by growing KEAP1-independent cells in galactose-containing media or treating cells with sodium oxamate, an LDH inhibitor64, respectively. As expected, both treatments resulted in a strong sensitization to NRF2 activation in KEAP1-independent cells (Figures 2F–G). These results indicate that upregulation of glycolysis is both necessary and sufficient to rescue NRF2 sensitivity.
NRF2 activation disrupts mitochondrial metabolism in KEAP1-dependent cells which are hypersensitive to ETC disruption
We next investigated the impact of NRF2 activation on mitochondrial respiration65, finding a substantial decrease in maximal respiratory capacity in KEAP1-dependent cells following KI696 treatment (Figure 3A). This defect in respiration was the result of NRF2, as loss of the transcription factor completely rescued OCR following KI696 treatment (Figure 3B). Inhibition of mitochondria function also extended to mitochondrial metabolism, where we found that TCA metabolites were largely downregulated following NRF2 activation in KEAP1-dependent cell lines and are consistent with previous studies53,65 (Figure 3C Table S4). Importantly, KI696-mediated defects in TCA metabolism are completely dependent on NRF2 in CALU6 cells (Figure S3A Table S4). Curiously, we did not observe substantial changes in mitochondrial gene or protein expression in KEAP1-dependent cells following NRF2 activation (Figures S3B–C) in comparison to canonical NRF2 targets, suggesting NRF2-regulation of mitochondrial function is posttranslational or dependent on a change in metabolite(s) levels. These results indicate that NRF2 activation can be particularly detrimental to cancer cells which are reliant on mitochondrial function for their proliferation.
Figure 3: NRF2 activation decreases mitochondrial activity in KEAP1-dependent cells.
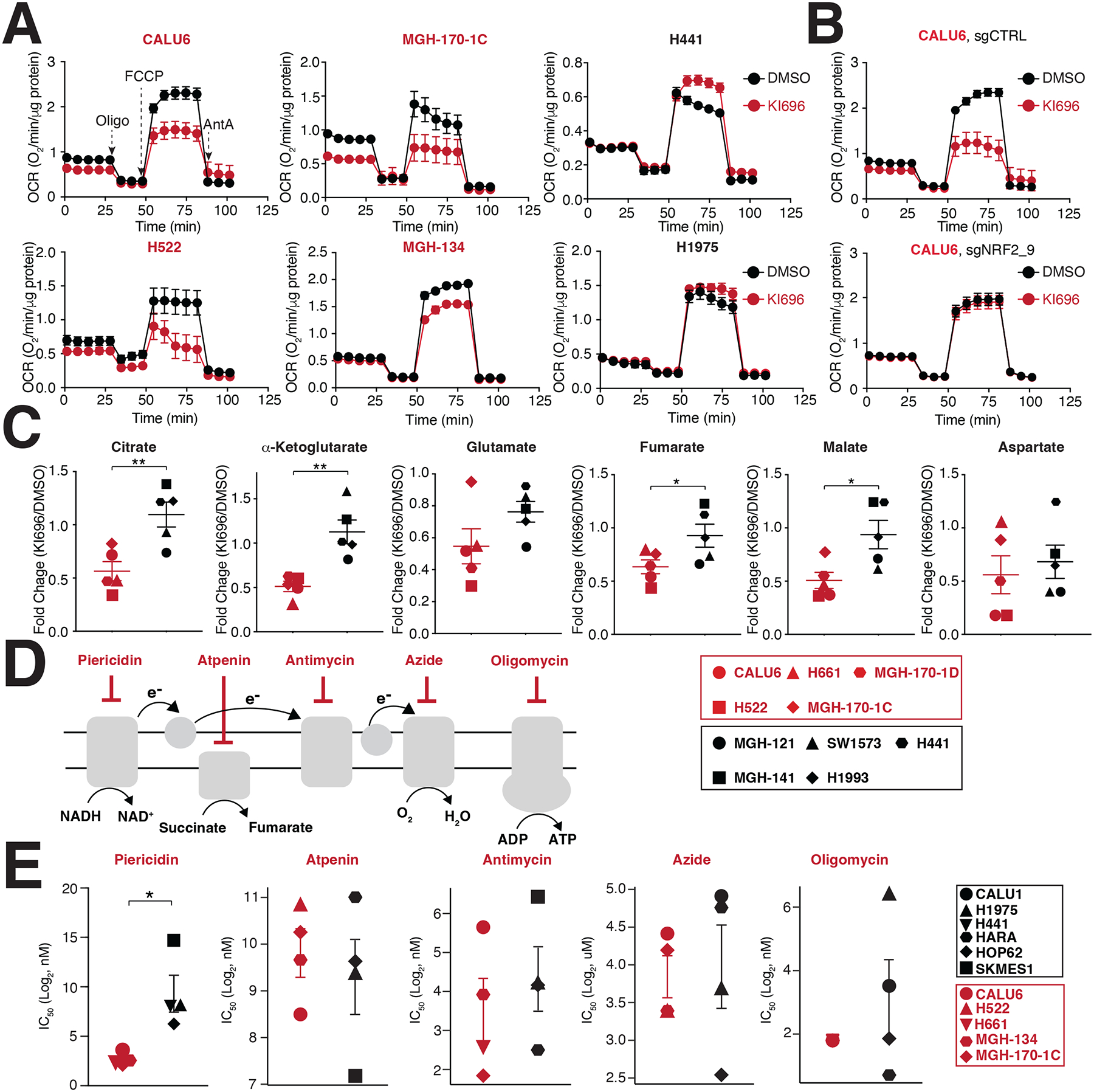
(A) NSCLC cell lines were treated with KI696 (1 μM) for 48 hrs and the oxygen consumption rate (OCR) was determined using a Seahorse Bioflux analyzer (Data are represented as a mean ± SEM, n= 6–8 biological replicates). (B) NRF2 regulates OCR in KEAP1-dependent cells. CALU6 cells expressing the indicated sgRNA targeting NRF2 or a control, were treated with KI696 (1 μM) for 48 hrs and OCR was determined as described in (A) (Data are represented as a mean ± SEM, n=6–8 biological replicates). (C) NRF2 activation decreases TCA metabolites in KEAP1-dependent cell lines. NSCLC cell lines were treated with KI696 (1 μM) for 48 hrs and the levels of the indicated metabolites were determined by GCMS (see methods). Fold change (KI696/DMSO) is depicted in the plots (Data are represented as a mean, n= 5 samples per group with 4 biological replicates per sample). (D-E) Complex I inhibition is selectively toxic to KEAP1-dependent cell lines. Schematic of different ETC inhibitors used in this study (D). IC50-values (E) were determined for a panel of NSCLC cell lines (Data are represented as a mean ± SEM, n= 4–5 samples/group measured in 4–6 biological replicates). * indicates p-values < 0.05, ** indicates p-values < 0.01, *** indicates p-values < 0.0001. Statistical significance was determined by Student’s t-test and corrected for multiple hypotheses by False Discovery Rate (FDR), see Methods.
Given the prominent role of the electron transport chain (ETC) in regulating oxygen consumption, we asked if there was altered sensitivity to inhibition of ETC complexes between KEAP1-dependent and KEAP1-independent cells. In general, we found comparable IC50 values for ETC inhibitors targeting complexes II-V between dependent and independent cells (Figures 3D–E). Interestingly, KEAP1-dependent cells demonstrated a striking sensitivity to the Complex I inhibitors piericidin, rotenone, and phenformin (Figures 3D–E, S3D–E). Because pyrimidine biosynthesis is compromised following complex III-V inhibition66,67, we hypothesized that a depletion in uridine levels might mask differential sensitivity between the two cell populations for Complex III-V inhibitors. Indeed, supplementation with uridine resulted in an increased sensitivity of KEAP1-dependent cells to Complex III-V inhibition compared to KEAP1-independent cells (Figure S3F). Collectively, these results establish that a cell’s intrinsic preference for glucose utilization (aerobic vs. anaerobic fermentation) dictates its sensitivity to NRF2 activation, with inhibition of ETC complexes, a particular liability for KEAP1-dependent cells.
NRF2 activation results in NADH reductive stress in KEAP1-dependent cells
We hypothesized that NRF2 sensitivity arises from a shared metabolic activity in KEAP1-dependent and –independent cells and only reveals itself upon NRF2 activation because it is limiting in dependent cells but not in independent cells. Our metabolic characterization of KEAP1-dependence revealed a heightened sensitivity to ETC inhibition and that an increase in glycolytic rates could reverse NRF2 sensitivity. Because one of the shared biochemical activities of ETC and glycolysis is regulating NADH oxidation (Figure S4A), we investigated whether NRF2 activation results in heightened NADH levels, which can block multiple cellular reactions dependent on NAD+ 25–30. We constructed a panel of NSCLC cell lines that stably express SoNar, a genetically encoded NADH/NAD+ reporter68 that measures differences in the relative levels of NADH and NAD+. At baseline, we found that KEAP1-dependent and KEAP1-independent cells had a similar ratio of NADH/NAD+ (Figure S4B). However, following KI696 treatment we found a consistently higher NADH/NAD+ ratio generated in KEAP1-dependent cells compared to KEAP1-independent cells that were mirrored by traditional methods to measure NADH/NAD+ (Figure 4A, S4C–E). Furthermore, genetic depletion of KEAP1 using DOX-inducible shRNAs also mediated an increased NADH/NAD+ ratio in KEAP1-dependent but not KEAP1-independent cells (Figures S4F–I). The increase in NADH/NAD+ ratio was completely reliant on NRF2, as depletion of the transcription factor blocked the KI696-mediated increase in the NADH/NAD+ ratio (Figures 4B, S4J). These results demonstrate NRF2 activation is both necessary and sufficient to mediate high NADH/NAD+ ratio in KEAP1-dependent cells.
Figure 4: NRF2 induces NADH-reductive stress in KEAP1-dependent cells.
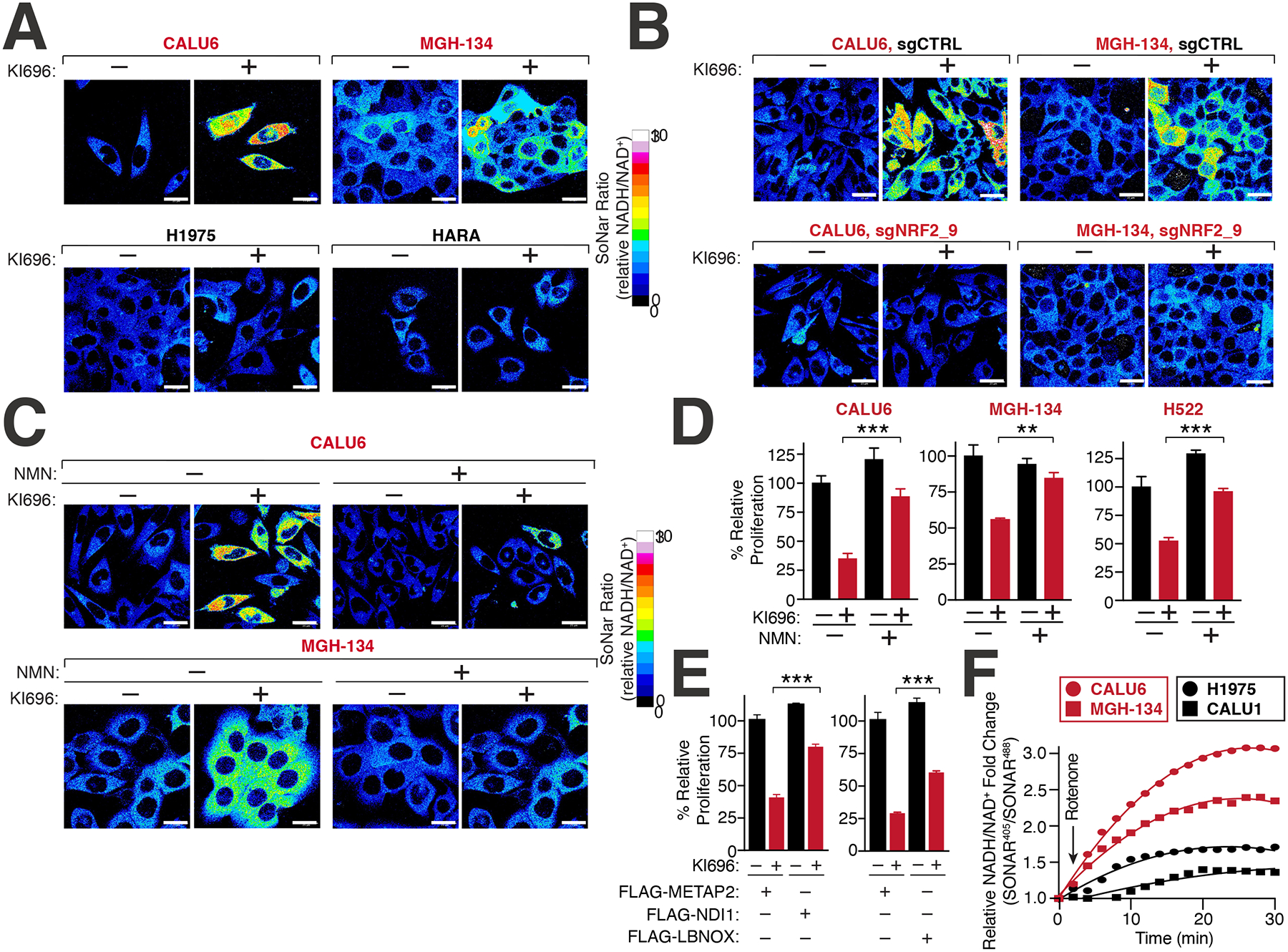
(A) NRF2 activation increases the NADH/NAD+ ratio in KEAP1-dependent but not KEAP1-independent cells. Immunofluorescence analysis of NSCLC cell lines stably expressing the NADH/NAD+ reporter SoNar following treatment with KI696 (1 μM) for 48 hrs. The representative NADH/NAD+ ratiometric image was constructed by taking the ratio of the emission intensity of 405 (NADH binding) vs 488 (NAD+ binding) for SONAR (see also Figures S4D–E). (B) NRF2 depletion rescues KI696-mediated NADH/NAD+ increase. NSCLC cells expressing SoNar as in (A) and corresponding sgRNAs targeting NRF2 or a control were treated with KI696 and cells were analyzed as in (A) (see also Figure S4J). (C-D) Supplementation with NMN restores the NADH/NAD+ ratio following NRF2 activation and rescues proliferation in KEAP1-dependent cells. KEAP1-dependent NSCLCs were treated with KI696 and NMN (1mM) where indicated and analyzed as in (A) or assayed for a change in proliferation by crystal violet staining 6 days post treatment (D) (Data are represented as a mean ± SEM, n= 4–5 biological replicates) (see also Figure S4K). (E) Over-expression of NADH oxidizing enzymes partially rescues NRF2 activation. CALU6 cells stably expressing NDI1, LbNOX or METAP2 (control) were treated with KI696 and assayed for proliferation as described in (D) (Data are represented as a mean ± SEM, n= 5 biological replicates). (F) KEAP1-dependent cells have a higher rate of Complex I NADH oxidation compared to KEAP1-independent NSCLCs. NSCLC cell lines stably expressing SoNar were treated with rotenone (0.5 μM) and analyzed by flow cytometry taking the ratio of the emission intensity at λem 530nm after excitation at λex 405 nm (NADH binding) or λex 488 nm (NAD+ binding).
To determine whether NADH/NAD+ imbalance was a primary mechanism underlying NRF2 sensitivity, we treated cells with β-nicotinamide mononucleotide (NMN), an NAD+ precursor69, which substantially rescued both high NADH/NAD+ ratio upon NRF2 activation and the concomitant proliferation block in KEAP1-dependent cells (Figures 4C–D, S4K). Supplementation with NMN reversed the KI696-mediated defects in respiration in KEAP1-dependent cells (Figure S4L), most likely by providing the required NAD+ equivalents for the TCA cycle. Furthermore, KEAP1-dependent cells expressing the NADH oxidizing enzymes LbNOX or NDI170–72 were partially protected from NRF2-mediated defects in proliferation and respiration in comparison to cells expressing a control protein (METAP2) (Figures 4E, S4M–N). Consistent with our findings that increasing glycolysis through induction of hypoxia or over-expression of PFK rescued NRF2 activation, we also found these perturbations decreased the overall magnitude change in NADH/NAD+ following NRF2 activation (Figures S5A–B). Conversely, KEAP1-independent cells treated with a pyruvate kinase inhibitor led to the activation of pyruvate dehydrogenase which increases the NADH/NAD+ ratio by shunting pyruvate away from lactate production73, resulting in increased sensitivity to NRF2 activation (Figure S5C). We also wondered whether NRF2 sensitivity in KEAP1-dependent cells might be due to high levels of GSH, however decreasing GSH levels with the GCLM inhibitor buthionine sulphoximine (BSO) did not rescue NRF2 activation in CALU6 cells (Figure S5D). These results suggested that upon activation of NRF2 in KEAP1-dependent cells, NADH-reductive stress is induced, leading to a block in proliferation.
Our findings suggested that KEAP1-dependency may arise, in part, from a metabolic preference to utilize ETC for NADH oxidation, resulting in a corresponding sensitivity to Complex I blockade previously observed (Figure 3E). To probe this hypothesis, we measured NADH/NAD+ ratio in KEAP1-dependent and KEAP1-independent cells following sequential treatments with rotenone and oxamate. KEAP1-dependent cells possessed a higher NADH/NAD+ ratio following Complex I inhibition compared to KEAP1-independent cells (Figure 4F). However, LDH-based NADH oxidation, accounted for the majority of the NADH oxidation in both cell types and was indeed higher in KEAP1-independent cells (Figure S5E). Collectively, our findings provide one explanation by which cells with lower glycolytic rates are unable to cope with NADH reductive stress brought upon NRF2 activation.
ALDH3A1 partially underlies NADH-reductive stress in KEAP1-dependent cells
To explore the mechanisms by which NRF2 activation increases NADH levels in KEAP1-dependent cells, we focused on 105 enzymes that utilize NAD+ and are up-regulated following NRF2 activation. We further filtered corresponding NAD+-utilizing enzymes based on their ability to mediate resistance to NRF2 activation when depleted, identifying ALDH3A1 as a compelling candidate (Figure 5A). ALDH3A1 is an NRF2-responsive gene (Figures S3B–C) that functions in antioxidant defense, specifically by converting reactive aldehydes to their corresponding carboxylic acids74,75. The dehydrogenase activity of ADLH3A1 results in the conversion of NAD+ to NADH, and depletion of this gene led to a substantial decrease in the NADH/NAD+ ratio following NRF2 activation in KEAP1-dependent cells (Figures 5B, S5F–G). ALDH3A1 over-expression was sufficient to increase the NADH/NAD+ ratio in CALU6 cells (Figure 5C). Consistent with the role of MYC in regulating the NRF2 transcription program, depleting MYC reduced both the levels of ALDH3A1 and NADH/NAD+ following NRF2 activation (Figures S2C, S5H). Importantly, depletion of ALDH3A1 in KEAP1-dependent cells overcame the proliferation block and restored respiration and TCA metabolite levels following NRF2 activation (Figures 5D, S5I–K).
Figure 5: Identification of ALDH3A1 as a mediator of NRF2 reductive stress.
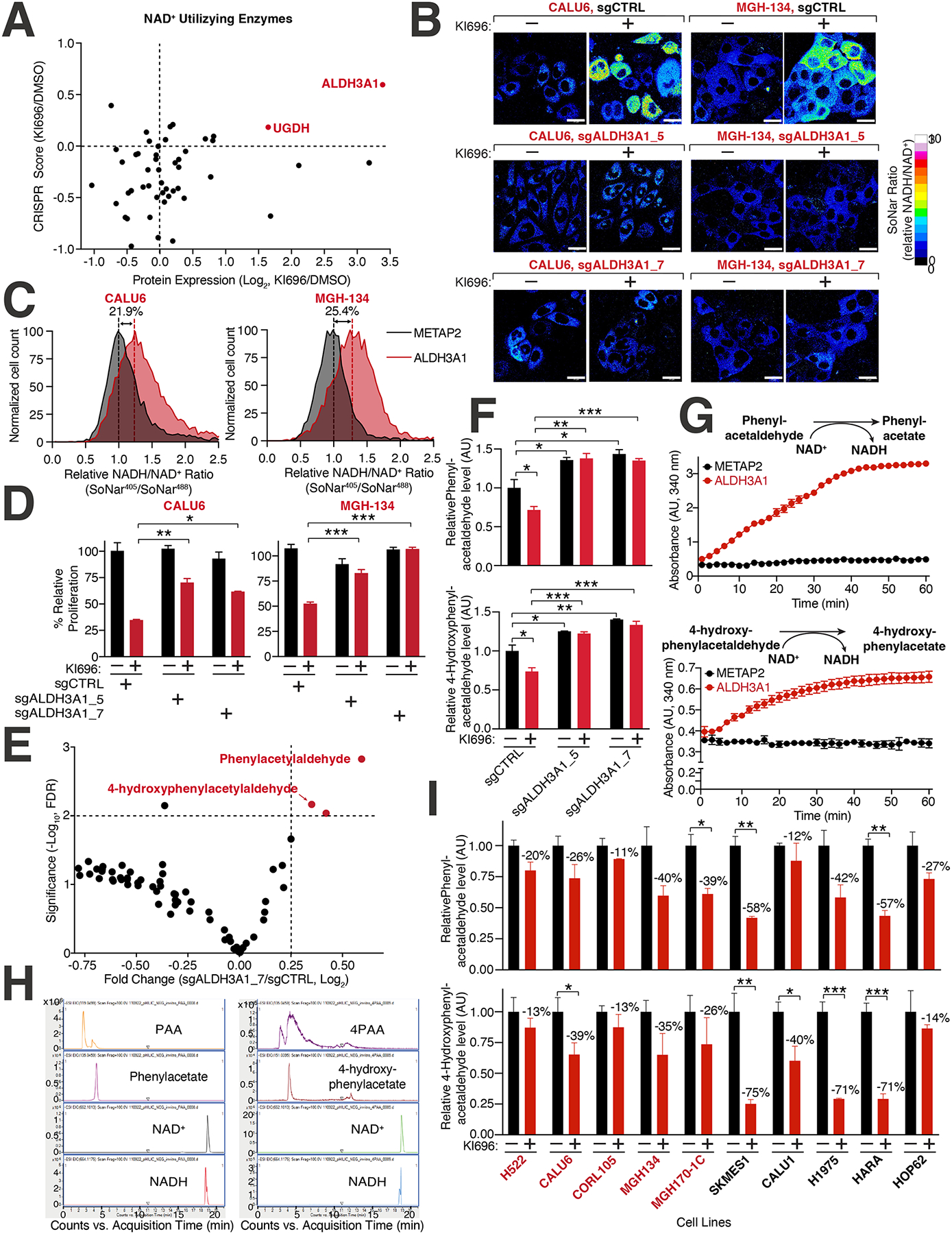
(A) Expression and dependency of NAD+ utilizing enzymes following NRF2 activation in CALU6 cells. Scatter plot of NAD+-utilizing enzymes identified in metabolism-focused CRISPR screen (see also Table S2) and proteomics (see also Figure S3C) following KI696 treatment. (B) ALDH3A1 depletion rescues high NADH levels following NRF2 activation. CALU6 and MGH-134 cells stable expressing SoNar and the indicated sgRNAs were treated with KI696 for 48 hrs. The representative NADH/NAD+ ratiometric image was constructed by taking the ratio of the emission intensity of 405 nm (NADH binding) vs 488 nm (NAD+ binding) for SoNar (see also Figure S5G). (C) ALDH3A1 is sufficient to increase NADH/NAD+ ratio in KEAP1-dependent cells. The NADH/NAD+ ratio CALU6 or MGH-134 cells expressing SoNar and over-expressing ALDH3A1 or Metap2 (control) was determined by flow cytometry. (D) Loss of ALDH3A1 rescues proliferation following NRF2 activation. KEAP1-dependent NSCLC cells expressing the indicated sgRNAs were treated with KI696 (1 μM) and proliferation was determined as described in (D) (Data are represented as a mean ± SEM, n= 5 biological replicates per condition). (E) ALDH3A1 regulates aldehyde levels in KEAP1-dependent cells. Plot compares fold change with corresponding significance for mass spectrometry analysis of CALU6 cells expressing the indicated sgRNAs. Each data point corresponds to an m/z value consistent with an aldehyde identified in human cells92 (see also Table S5, Methods). (F) PAA and 4PAA are regulated by ALDH3A1. CALU6 cells expressing the indicated sgRNAs were treated with KI696 for 48 hrs and the relative levels of metabolites consistent with PAA and 4PAA were analyzed as described in (E) (Data are represented as a mean ± SEM, n= 3 biological replicates). (G) PAA and 4PAA are ALDH3A1 substrates. PAA and 4PAA and NAD+ were added to highly purified ALDH3A1 or a control protein (METAP2) and NADH accumulation was determined in vitro by monitoring its absorbance at 340 nm (Data are represented as a mean ± SEM, n= 3 biological replicates). (H) Liquid chromatography mass spectrometry (LCMS)-based fragmentation corresponding to substrates (NAD+, PAA, 4PAA) and products (NADH, phenylacetate, 4-hydroxyphenylacetate) from ALDH3A1 reaction conducted in (G) (see also Methods). (I) PAA and 4PAA levels are regulated by NRF2. The indicated cell lines were treated with KI696 for 48 hrs and metabolites consistent with PAA or 4PAA were determined as described in (E) (Data are represented as a mean ± SEM, n= 3 biological replicates and normalized to DMSO treated samples) * indicates p-values < 0.05, ** indicates p-values < 0.01, *** indicates p-values < 0.0001. One-way ANOVA with Sidak’s post-hoc correction was used to determine statistical significance. Scale bar: 25 μm.
To probe the role of ALDH3A1 in cells, we analyzed metabolites with mass spectrometry for m/z values consistent with aldehydes found in human cells, the predominant substrates for aldehyde dehydrogenases76,77, including ALDH3A1. We identified a significant depletion in metabolites consistent with m/z values for phenylacetaldehyde (PAA) and 4-hydroxy-phenylacetaldehyde (4PAA) in ALDH3A1-depleted CALU6 cells (Figures 5E–F, Table S5). High concentrations of PAA are known to block cancer cell proliferation through a ROS-based mechanism78. Our cellular data suggests that ALDH3A1 has a role in aldehyde clearance and to provide additional evidence, we established an ALDH3A1 in vitro assay finding that both PAA and 4PAA function as substrates of ALDH3A1 resulting in an increase in NADH absorbance following addition of either aldehyde (Figure 5G). Using LCMS, we further confirmed the carboxylate products of PAA and 4PAA in the ALDH3A1 in vitro assay (Figure 5H). Following activation of NRF2 and a concomitant increase in ALDH3A1 levels, we observed a decrease in metabolites consistent with PAA and 4PAA in a panel of NSCLC cells (Figure 5I).
As we identified ALDH3A1 as a key driver of the NADH/NAD+ ratio following NRF2 activation, we wondered whether NADH-reductive stress is solely due to NAD+ consumption or arises from defects in NADH turnover due to ETC dysfunction. To address this question, we localized SoNar to both the cytosol and mitochondrial matrix and found heightened levels of NADH in both locations following NRF2 activation (Figure S5L). Our findings suggest that NRF2 mediates NADH-reductive stress through NAD+ consumption and ETC dysfunction, and further illustrates how in the absence of oxidative stress, over-expression of antioxidant defense genes reshapes the cellular redox state to an overly reduced environment leading to a block in proliferation.
Disruption of Complex I activity is a metabolic liability in KEAP1-mutant cells
Our results suggest that NRF2 can result in an imbalance of NADH/NAD+, constituting a liability that may be exploited in cellular states with hyperactive NRF2 signaling. We tested this hypothesis by treating a panel of KEAP1-dependent, KEAP1-independent, and KEAP1-mutant cells with IACS-010759 (IACS), a potent Complex I inhibitor79, finding that KEAP1-mutant cells had a substantial increase in NADH/NAD+ following treatment (Figures 6A, S6A). Consistent with our hypothesis, KEAP1-independent cells, that were co-treated with KI696 and IACS also had substantial increases in NADH levels (Figures 6A, S6A). The increase in NADH levels following IACS treatment, directly translated to lower IC50 values in KEAP1-mutant and KEAP1-dependent cell lines compared to KEAP-independent cells (Figure 6B). Moreover, we found IACS potently blocked the growth of KEAP1-dependent and NRF2-activated cells in soft agar but not KEAP1-independent cells (Figures 6C, S6B–C). We next asked whether IACS would be effective in targeting KEAP1-mutant tumors in vivo, finding that IACS strongly blocked the growth of a KEAP1-mutant patient-derived xenograft (PDX) but had no effect on a KEAP1-WT PDX (Figures 6D–G, S6D–E). By depleting ALDH3A1 in KEAP1-mutant cells we found a partial rescue of IACS treatment (Figures S6F–G). Over-expression of LbNOX and NDI1 in H460 cells or treatment with NMN, also led to a partial rescue of IACS treatment (Figures S6H–I). These results reveal that the NADH/NAD+ ratio is at its tipping point in KEAP1-mutant cells and that a slight disruption in NADH oxidation through Complex I inhibition coordinated with hyper-active NRF2 signaling overwhelms NADH homeostasis leading to reductive stress and a blockage in cell proliferation (Figure 6H). Collectively, our results illustrate how reductive stress can be leveraged as a synthetic lethal opportunity within a genetically– and metabolically-defined subset of cancers.
Figure 6: Inducing NADH-reductive stress selectively blocks proliferation of NRF2-activated NSCLCs.
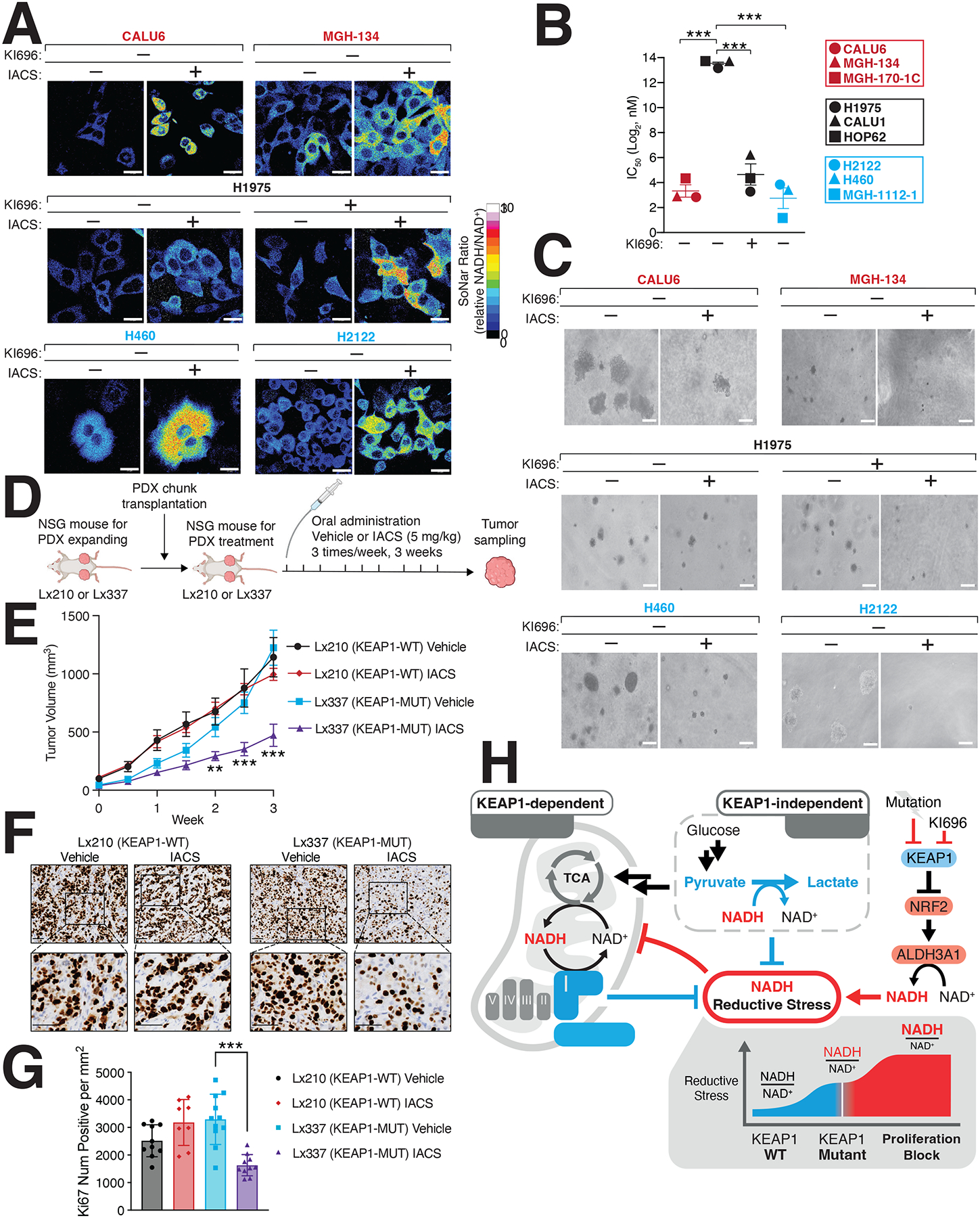
(A) IACS-010759 (IACS) a Complex-I inhibitor, selectively increases NADH/NAD+ ratio in KEAP1-dependent (red) and KEAP1-mutant cells (blue) but not KEAP1-independent cells. NSCLC cells stably expressing SoNar were treated with IACS and analyzed by immunofluorescence. The representative NADH/NAD+ ratiometric image was constructed by taking the ratio of the emission intensity of 405 nm (NADH binding) vs 488 nm (NAD+ binding) for SoNar (see also Figure S6A). (B) IACS selectively blocks NSCLC proliferation following NRF2 activation. IC50 values were calculated for each cell line and where indicated cells were also treated with KI696 (1 μM) (Data are represented as a mean ± SEM, n= 6 biological replicates). (C) IACS selectively inhibits the anchorage-independent growth of NSCLCs with hyperactivate NRF2 signaling. Representative images of NSCLC cell lines grown in soft agar following treatment with IACS-017509 (200 nM) or co-treated with KI696 (1 μM) as indicated (Data are represented as a mean ± SEM, n= 6–8 biological replicates) (see also Figure S6C). (D) Schematic for in vivo study. (E) IACS selectively blocks the growth of KEAP1-mutant tumors. Relative tumor growth of subcutaneous patient-derived xenografts (PDX) WT or mutant (MUT) for KEAP1 receiving vehicle or IACS (5 mg/kg) (Data are normalized to first treatment, n=10 KEAP1-WT, Vehicle; 12 KEAP1-WT IACS, 14 KEAP1-MUT Vehicle, 18 KEAP1-MUT IACS (see also Figure S6E). (F-G) Representative immunohistochemistry staining (F) and quantification of Ki67 serial sections taken from KEAP1-WT and KEAP1-MUT PDX tumors treated with IACS or vehicle. (H) Model. NRF2 activation following pharmacologic inhibition or mutation of KEAP1 increases ALHD3A1 resulting in NADH reductive stress. KEAP1-dependent and -independent cells utilize different NADH oxidation pathways to counter reductive stress. * indicates p-values < 0.05, ** indicates p-values < 0.01, *** indicates p-values < 0.0001. One-way ANOVA with Sidak’s post-hoc correction was used to determine statistical significance. Scale-bar: 25 μm for immunofluorescence and 50 μM for soft agar and immunohistochemistry.
Discussion
Activation of NRF2 has been widely regarded as a favorable event in cancer growth via the activation of oxidative stress-responsive genes. Our study identifies a previously unappreciated role for NRF2 in NADH-reductive stress. Herein, we provide one explanation for the metabolic cost that cancer must pay to harbor high NRF2 activity, providing the metabolic context required to support this pathway. By pharmacologically profiling a large panel of genetically diverse NSCLC cell lines in combination with functional genomic analysis, we uncovered, that NRF2 activity can only be tolerated under certain metabolic contexts required to offset high NADH levels. These findings indicate that NADH levels are both necessary and sufficient for NRF2 sensitivity and that the precise cellular pathways used for NADH oxidation are a key determinant for enduring NRF2 activation. Cells with high glycolytic rates tolerate NRF2 activation and do not incur high levels of NADH-reductive stress compared with their counterparts which rely more heavily on Complex I for NADH oxidation. As a result, cells with low glycolytic rates suffer an NADH/NAD+ imbalance imposed by NRF2 activity.
Our study highlights a growing appreciation for the role of NADH-reductive stress in cell biology21–24 and directly demonstrates how high levels of reductive stress mediated through an imbalance in NADH/NAD+ directly impact cancer cell growth. Overcoming oxidative stress has been at the forefront of understanding why cancers activate NRF2. Excitingly, our results illustrate that for a subset of NSCLCs, overcoming NADH-reductive stress is a critical determinant for cell proliferation. Cancer dependency (DEPMAP) analysis revealed the essential role of KEAP1 across multiple cancers, suggesting that KEAP1 regulation of NADH-reductive stress may be a general requirement for proliferation. Based on our dissection of this dependency in NSCLCs, we anticipate that many KEAP1-dependent cancers will have lower rates of glycolysis and a high dependence on Complex I for NADH oxidation. Our investigation of NRF2-mediated reductive stress strongly suggests that the aldehyde dehydrogenase, ALDH3A1, an NRF2-target gene has a substantial role in the regulation of NADH/NAD+ in KEAP1-dependent cells through its control of the aldehydes PAA and 4PAA. Metabolism of phenylalanine has been suggested to give rise to PAA78,80, and although its role in tumors is still emerging, recent studies have demonstrated that at high levels PAA increases ROS and is cytotoxic to breast cancer cells78.
Given the central role of the mitochondria in generating cellular ROS81, it is not surprising that NRF2 would modulate mitochondrial activity as one mechanism to downregulate ROS levels. Our study indicates a key role in the manipulation of NADH levels as a mechanism by which NRF2 controls mitochondrial function and supports a growing body of literature connecting NRF2 with mitochondrial regulation53,65. NRF2 launches two parallel campaigns to quelch a rise in oxidative stress by: 1) increasing antioxidant levels and 2) decreasing the cellular sources of oxidative stress, in this case mitochondrial respiration. Our work suggests that NRF2 does not directly modulate mitochondrial gene or protein levels, rather relying on a yet to be described posttranslational mechanism to regulate mitochondria. Given the reductive cellular environment brought upon by NRF2 activation that we describe, it would not be surprising if reductive protein stress brought about by incorrect disulfide bond formation is an additional mechanism by which NRF2 stymies mitochondrial function and cancer growth.
While an abundance of cancer genomics studies indicate that NRF2 activation is a common event in NSCLCs, our work begins to illuminate when this activation may be permissible. Loss of KEAP1 is not sufficient to initiate tumor growth and is thought to function in a supportive role following oncogene activation such as KRAS or loss of p5332,42,45. Our results suggest that a cancer cell may only be able to support NRF2 activation once its glycolytic rate is increased to overcome the burden of NRF2-induced reductive stress. While loss of NRF2 is not lethal, loss of KEAP1 leads to postnatal lethality due to esophageal hyperkeratinization, disruption of renal function and bone mineralization82–84. Whether these dysfunctions arise from NADH-reductive stress remains to be determined but underscores that NRF2 activation can only be tolerated in certain metabolic contexts. Undoubtedly, new therapies aimed at activating NRF285 in non-glycolytic tissues may have unexpected side effects caused by NRF2-induced reductive stress. In particular, there is growing interest in activating NRF2 to treat patients with mitochondrial diseases86,87 and our work would suggest caution should be taken, given its impact on respiration. Future studies treating NSUFS4-mouse models of Leigh syndrome88 with NRF2 activators, may go a long way in determining the therapeutic potential of activating NRF2 in this set of diseases.
The sensitivity of KEAP1-mutant cells to Complex I inhibition may arise from their limited capacity to oxidize additional NADH. The partial rescue of IACS-010759 following depletion of ALDH3A1 suggests that there are likely additional mechanisms by which NRF2 regulates NADH levels in KEAP1-mutant cells or there are off-targets associated with this inhibitor. While clinical reports suggest that IACS-010759 is tolerable in humans89,90, its anti-cancer efficacy is limited because of toxicity. While this clinical trial did not rely on biomarkers to guide patient selection our finding suggests IACS-010759 may be useful use in patients harboring KEAP1-mutations.
In summary, our findings provide a complex picture for the role of metabolic tumor suppressors in controlling the proliferation of cancer cells. They provide one explanation for how metabolism-centered oncogenic pathways are supported and the metabolic rewiring required for proliferation upon their activation. Importantly, this study identifies a cellular signaling pathway whose activation directly controls reductive stress within cancer cells. Our findings are in line with previous studies demonstrating the critical role that NAD+ plays in multiple cellular pathways and how maintenance of NADH homeostasis is required for tumor progression25–30. Moreover, the discovery that AMPK, long considered to harbor tumor suppressive functions, is paradoxically required for the growth of tumors by regulating lysosomal gene expression, supports the essentiality of some tumor suppressors91. Collectively, our study provides a metabolic context by which NRF2 activation can create synthetic lethal opportunities through the generation of NADH-reductive stress, forming the basis to exploit this form of stress in treating KEAP1 mutant cancers.
Limitations of Study
In this study we identified that a subset of NSCLC cell lines is hypersensitive to NRF2 activation. NRF2 activation results in NADH-reductive stress through the over-expression of ALDH3A1 leading to ETC dysfunction. However, our study does not address the root cause of this mitochondrial dysfunction. Although depletion of ALDH3A1 or increasing NAD+ levels following treatment with NMN restored oxygen consumption following NRF2 activation, they did not do so fully, suggesting additional mechanisms. We still know very little about how an overly reductive cell state regulates different organellar pathways, and whether this occurs at the level of nucleotide co-factors as described herein or at the level of reduced proteins or lipids. Thus, more studies are needed to understand the impact of reductive stress so that it may be exploited for the development of anti-cancer therapies.
STAR Methods
RESOURCE AVAILABILITY
Lead contact
Further information and requests for reagents should be directed to the Lead Contact, Liron Bar-Peled (LBAR-PELED@mgh.harvard.edu).
Materials availability
All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and code availability
Source data for graphs can be found in Data S1. The RNA-seq data have been deposited in GEO: GSE221194 and the proteomics data have been deposited in PRIDE: PXD039027 and both are publicly available. The accession numbers are provided in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze data reported in this paper is available from the lead contact.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| NRF2 | Abcam | Cat#: ab62352 |
| KEAP1 | Cell Signaling Tech | Cat#: 7705 |
| HMOX1 | Proteintech | Cat#: 10701–1-AP |
| NQO-1 | Cell Signaling Tech | Cat#: 3187S |
| HK2 | Cell Signaling Tech | Cat#: 2867T |
| ALDH3A1 | Novus Biologicals | Cat#: NBP2–15340 |
| FLAG | Sigma | Cat#: F7425 |
| β-ACTIN | Cell Signaling Tech | Cat#: 4970 |
| HRP-labeled anti-mouse | Cell Signaling Tech | Cat#: 7076 |
| HRP-labeled anti-rabbit | Cell Signaling Tech | Cat#: 7074 |
| Bacterial and Virus Strains | ||
| DH5α | Fisher Scientific | Cat#:18265017 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| DMEM | Corning | Cat#:15–013-CV |
| RPMI-1640 | Corning | Cat#: ICN1646454 |
| FBS | Corning | Cat#: 35–010-CV |
| GlutaMax | Invitrogen | Cat#: 35050061 |
| Penicillin-Streptomycin | Millipore | Cat#: P0781 |
| Blasticidin | Fisher Scientific | Cat#: 50712728 |
| Puromycin | Sigma-Aldrich | Cat#: P8833 |
| Sequencing grade modified trypsin | Promega | Cat#: V5111 |
| DMSO | Fisher Scientific | Cat#: MT-25950CQC |
| Urea | VWR Intl | Cat#: 97063–798 |
| CHAPS hydrate | Sigma-Aldrich | Cat#: C3023 |
| DTT | Fisher Scientific | Cat#: BP1725 |
| Protease Inhibitor Cocktail | Sigma-Aldrich | Cat#: 5892791001 |
| Polybrene | Sigma-Aldrich | Cat#: TR-1003-G |
| X-tremegene HP transfection reagent | Sigma-Aldrich | Cat#: 6366236001 |
| Cell Titer Glo reagent | Promega | Cat#: 88836 |
| KI696 | MedChemExpress | Cat#: HY-101140 |
| Bardoxolone-methyl | MedChemExpress | Cat#: HY-13324 |
| Piericidin A | Cayman | Cat#: 15379 |
| Rotenone | Sigma-Aldrich | Cat#: R8875 |
| Phenformin | Cayman | Cat#: 14997 |
| Atpenin A5 | Cayman | Cat#: 11898 |
| Antimycin A | Cayman | Cat#: 19433 |
| Sodium azide | Sigma-Aldrich | Cat#: S8032 |
| Oligomycin A | Cayman | Cat#: 11342 |
| IACS-010759 | Cayman | Cat#: 25867 |
| 2-Deoxyglucose | Sigma-Aldrich | Cat#: D8375 |
| β-Nicotinamide mononucleotide | MedChemExpress | Cat#: HY-F0004 |
| Sodium oxamate | Cayman | Cat#: 19057 |
| AZD7545 | Cayman | Cat#: 19282 |
| Phenylacetaldehyde | Sigma | Cat#: 107395 |
| 4-Hydroxyphenylacetaldehyde | This Study | N/A |
| Critical Commercial Assays | Cat#: UN3334 | |
| Seahorse XFe96 FluxPac | Agilent | Cat# 102601–100 |
| NAD/NADH-Glo™ Assay kit | Promega | Cat#: 103344–100 |
| Deposited data | ||
| RNA-seq data | This study | GEO: GSE221194 |
| Proteomics data | This study | PRIDE: PXD039027 |
| Recombinant DNA | ||
| pLenti:SoNar-NES | This study | |
| pCW57.1: FLAG-METAP2 | This study | N/A |
| pCW57.1: FLAG-LbNOX | This study | N/A |
| pCW57.1: FLAG-NDI-1 | This study | N/A |
| pCW57.1: FLAG-PFKP | This study | N/A |
| pCW57.1:FLAG-KEAP1-PAM mutant | This study | N/A |
| pLENTI-CRISPRv2:sgCTRL | This study | N/A |
| pLENTI-CRISPRv2:sgNRF2_9 | This Study | N/A |
| pLENTI-CRISPRv2:sgNRF2_10 | This Study | N/A |
| pLENTI-CRISPRv2:sgKEAP1_9 | This Study | N/A |
| pLENTI-CRISPRv2:sgKEAP1_10 | This Study | N/A |
| pLENTI-CRISPRv2:sgALDH3A1_5 | This Study | N/A |
| pLENTI-CRISPRv2:sgALDH3A1_7 | This Study | N/A |
| pLKO-Tet-On: shGFP | 111 | |
| pLKO-Tet-On: shKEAP1_1 | This Study | N/A |
| Human CRISPR Metabolic Gene Knockout Library | 28 | 110066 |
| Human Activity-Optimized CRISPR Knockout Library | 97 | 1000000100 |
| Software and Algorithms | ||
| Prism (v7.0e) | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| FlowJo (v10.0.7) | Treestar Inc. | https://www.flowjo.com/ |
| ImageJ | NIH | https://imagej.net/software/fiji/ |
| R version (4.0.3) | The R Project for Statistical Computing | https://www.r-project.org |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
All cell lines were grown in RPMI-1640 (Corning) media containing 10% FBS (Corning), Penicillin-Streptomycin (100 mg/ml, Millipore) and 1% GlutaMax (Gibco). All cell lines were tested at least once for Mycoplasma and if not noted elsewhere were obtained from American Tissue Type Collection (ATCC). All MGH cell lines were kindly provided by Dr. Aaron N Hata. Whenever thawed, cells were passaged at least three times before being used in experiments. For normoxia and hypoxia experiments, cell lines were maintained at 5% CO2 with the indicated oxygen concentrations.
Animal Studies
Patient-derived xenografts (PDX) used in this study were previously described in45. PDX tumors were stored in cryo-tubes in 10% dimethyl sulphoxide (DMSO) containing Dulbecco’s Modified Eagle Medium (DMEM) media containing 10% FBS and 20 mg/ml Gentamicin. NOD-SCID IL2R gammanull (NSG; NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ) mice were purchased from Jackson laboratories (Strain #: 005557).
METHOD DETAILS
Lentivirus virus production
Mammalian lentiviral particles harboring sgRNA-encoding plasmids, shRNA-encoding plasmids, or cDNA-encoding plasmids were co-transfected with the psPAX2 envelope and VSV-G packaging plasmids into actively growing HEK-293T cells (ATCC) using Xtremegene-HP (Sigma) transfection reagent as previously described93. Virus-containing supernatants were collected 48 hours after transfection, filtered to eliminate cells and target cells were infected in the presence of 8 μg/ml polybrene (Millipore) at a concentration of 2.5 × 105 cells/well. 24 hours later, cells were selected with puromycin (Sigma-Aldrich) or blasticidin (Sigma-Aldrich) and analyzed 3–10 days after selection was initiated. The sequences of sgRNAs and shRNAs used in this study can be found in Table S6.
cDNA cloning and mutagenesis
cDNAs were amplified using Q5 High-Fidelity 2X master mix (NEB) and subcloned into pCW57.1-DOX on or pCW57.1-DOX off (Addgene) or pLentiCRISPRv2 (Addgene) by T4 ligation or Gibson cloning (NEB). Site directed mutants were generated using QuikChange XLII site-directed mutagenesis (Agilent), using primers containing the desired mutations. The sequences of primers used in this study can be found in Table S6. The SoNar cDNA is previously described68. All constructs were verified by DNA sequencing.
Cell lysis
The indicated cell lines were rinsed once with ice-cold PBS and lysed with Triton lysis buffer (1% Triton X-100, 40 mM HEPES pH 7.4, 2.5 mM MgCl2 and 1 tablet of EDTA-free protease inhibitor (Roche) (per 25 ml buffer)) and gentle sonication using a QSonica 700A water-chilled sonicator. The soluble fractions of cell lysates were isolated by centrifugation at 13,000 rpm in a microcentrifuge for 10 min, normalized and proteins were denatured by the addition of 5X sample buffer and boiling for 5 min as described94. Samples were resolved by 8%–16% SDS-PAGE and analyzed by immunoblotting.
Monolayer proliferation assay
Unless otherwise noted, cells were pre-treated with KI696 for 3 days in 6-well plates, prior to the onset of proliferation assays. For cell lines harboring Doxycycline (DOX)-inducible or repressible cDNAs or shRNAs, cells were treated with DOX 3 days in 6-well plates prior to the onset of proliferation assays. For all other compounds, pre-treatment occurred in a 6-well plate for 2 days with the indicated concentrations listed in the figure legends. At the onset of a proliferation assay, cells were cultured in 96-well plates at 2.5 × 103 cells per well in 100 μl of RPMI and the indicated compound or vehicle control was added for compounds where pre-treatment was required. For all other compounds, agents were administered 24 hours after cell seeding. Where indicated (Figure S3F), tissue culture media was supplemented with 400 μM uridine. To quantify cell proliferation crystal violet staining was used as described in95 with slight modification. Briefly, culture media was removed and 50 μL of crystal violet stain (0.5% in 25% methanol) was added to cells for 30 min at room temperature. After removal of crystal violet, cells were washed with water and dried overnight before quantification. Cell viability was quantified in ImageJ (NIH v2.0.3) as previously described in96 on threshold images. To calculate half maximal inhibitory concentrations (IC50) cells were cultured at 2.5 × 103 cells per well in 100 μL RPMI media and compounds were added the following day. Cell viability was assessed on day six of treatment using crystal violet staining. IC50 values were calculated using log(inhibitor) vs % normalized response formula in Prism v.7 (GraphPad). For Figure S1I, cells transduced with the indicated sgRNAs were seeded in a 96-well plate in 100 μL of media and 50 μl of Cell Titer Glo reagent (Promega) was added to each well and the luminescence read on the SpectraMax M5 plate reader (Molecular Devices).
Anchorage-independent growth assay
Multiple NSCLC cell lines were tested for their ability to form colonies in soft agar. Cells were seeded at concentrations between 3.0–6.0 × 104 cells/well in a 6-well plate, cell concentrations required to form viable colonies in a 3-week time frame. CALU6, H460 and H2122 were seeded at 3.0 × 104 per well. MGH-134 and H1975 were seeded at 6.0 × 104 cells/well. Where indicated, cells were pre-treated with 1 μM of KI696 for 3 days prior to the onset of anchorage independent growth. For all assays, an equal number of cells from each comparison group was embedded in a solution of 0.4% Noble agar solution (Difco Labs) and the indicated agents or vehicle control were added to the 0.4% Noble agar solution before solidification. Cells were then placed on top of hardened layer of 0.6% agar in a 6-well plate. Cells were grown for 14–20 days at 37°C with 5% CO2. Fresh media (200 μL) was added every 5 days. Bright field images were obtained using light microscopy (Nikon Eclipse Ti) with x10 objective lens and colony formation area was measured in ImageJ (NIH, v2.0.3) as described in96.
Animal Studies
All animal studies described were approved by the NYU Langone Medical Center Institutional Animal Care and Use Committee. Animals were housed according to IACUC guidelines in ventilated cages in a specific pathogen-free (SPF) animal facility. Patient-derived xenograft (PDX) tumors were stored in cryo-tubes in 10% dimethyl sulphoxide (DMSO) containing Dulbecco’s Modified Eagle Medium (DMEM) media containing 10% FBS and 20 mg/ml Gentamicin. Upon thawing, PDX tumors were trimmed to approximately 3 mm3 and surgically transplanted subcutaneously in the flanks of 6–8 week old male and female NSG mice. Vetbond was used to close wounds. Engraftment was checked every 3 days after transplantation. After the establishment of tumors, mice harboring tumors with volumes of 50–100 mm3 were randomized and assigned to a treatment group. Subcutaneous tumor volumes were calculated according to the following formula: volume (mm3) = (a2 × b) × (π/6), in which a is the smaller dimension and b is the larger dimension. Animals either received 5 mg/kg IACS-010759 or vehicle 0.5% Methyl cellulose (100 μl per dose) 3 times a week through oral gavage for a total of 3 weeks. Tumor growth was tracked for a minimum of 8 tumors per experimental group.
Genome-wide and metabolism-focused CRISPR screens
CRISPR screens were conducted as previously described in97. Briefly, CALU6 cells were infected with a genome-wide CRISPR library (Addgene,97) or CALU6 and H1975 cells were infected with a metabolism-focused CRISPR library (Addgene,28) ensuring a multiplicity of infection ~0.3 following 3 days puromycin selection. Cells were allowed to recover for 1 day and an initial input was taken with the number of infected cells corresponding to 1000X the size of the library. The screen was initiated by treating cells (~200 × 106 cells for genome-wide and 30 × 106 for metabolism-focused) with DMSO or 1 μM KI696, maintaining this cell number and compound for 11 population doublings. At the end of the screen, cells were harvested, and genomic DNA was extracted using Macherey Nagel Blood XL kit (Macherey). Libraries were generated from each sample by PCR based amplification of the sgRNA amplicon from 200 μg of genomic DNA using custom PCR primers harboring an index primer and illumina 5’ and 3’ adaptors. Libraries were pooled and analyzed on a NextSeq500 (Illumina) using single end 75bp reads. sgRNAs were mapped and quantified as described in97 with modifications as described. Raw sgRNA counts for each screen were filtered to remove guides with poor representation in the library input. Read counts were then normalized using a “median ratio method” adapted from98.To calculate a “CRISPR Score” for each gene, log2 normalized DMSO and KI696 sgRNA values were subtracted from corresponding input values and the median sgRNA value for each gene was determined. The difference between DMSO and KI696 CRISPR scores is presented.
Gene set enrichment analysis (GSEA) and metabolic pathway enrichment
GSEA99 was carried out using pre-ranked lists from genome-wide CRISPR-score values using the fast GSEA package in100 Gene sets were collected from MSigDB version 7.4 and top/bottom 250 gene categories were then stratified into subcategories as described in Figure 1F. To identify KI696 mediated sensitivity from different metabolic pathways, genes were curated into different metabolic pathways as described in101 and a CRISPR value for each pathway was determined by calculating the aggregate CRISPR-score for each gene comprising this pathway (see Figures 1H, S2A).
Glycolytic signature
A glycolytic gene signature was calculated for NSCLC cell lines according to102 with slight modifications. Briefly, the mean expression of glycolytic genes, as defined in KEGG glycolysis gene set V7.5.1, was calculated for NSCLC cell lines by using publicly available transcriptomic data (20Q4) from DEPMAP103. Expression values were then Z-score normalized to get glycolytic gene-score = (xi-μ)/σ, where xi is the mean expression value of glycolytic genes of the ith sample (NSCLC cell line), μ is the average expression of glycolytic gene across all samples and σ is the standard deviation of glycolytic gene expression across all samples.
Seahorse Flux Analyses
Oxygen consumption rates (OCR) and extracellular acidification rates (ECAR) were measured using XFe96 Extracellular Flux Analyzer (Agilent) as described previously with slight modifications1. Briefly, all cell lines were plated on a poly-L-lysine coated 96-well Seahorse plates (Agilent) at 10 × 103 cells/well, with the exception of H522 and H661 that were seeded at 15 × 103 and 5 × 103 cells/well, respectively. Cells were treated with KI696 (1 μM) or DMSO for 48 hours in RPMI media. To analyze OCR and ECAR, the media was changed to RPMI supplemented with 2 mM L-glutamine and 10 mM D-glucose, but lacking sodium bicarbonate. Measurements of OCR were carried out at baseline and after injections of oligomycin (1.5 μM), FCCP/Na Pyruvate (3 μM/1 mM), and Antimycin (1 μM). To measure ECAR, the assay media was modified to RPMI supplemented with 2 mM L-glutamine and ECAR was measured following the injections of D-glucose (10 mM), oligomycin (1.5 μM), and 2-Deoxyglucose (100 mM). OCR/ECAR measurements were carried out at baseline. All OCR and ECAR values were normalized to total protein content as measured by BCA (Pierce) according to manufacturer’s instructions.
Confocal imaging of cell lines expressing SoNar reporter
NSCLC cell lines expressing the indicated the SoNar reporter were seeded on poly-lysine coated 8-well chamber (iBidi) at 10 × 103 cells per well and treated with compounds as described in the text. For KI696 treatments, cells were pre-treated for 2 days with 1 μM KI696 or vehicle control prior to seeding on glass bottom dishes. Dishes were firmly mounted the stage adaptor of Zeiss 710 Laser Scanning Confocal microscope (Carl Zeiss Inc.). Constant temperature (37°C), humidity, and 5% CO2 atmosphere are maintained throughout the duration of cell imaging. Images were acquired using a 63X oil objective. Relative NAD+ levels were determined by exciting SoNar expressing cells with a 488 nm laser and measuring emission at 500–520 nm range. Relative NADH levels were determined by exciting SoNar expressing cells with a 405 nm laser and measuring emission at 500–545 nm range. Acquisition parameters were kept identical between samples. Images were acquired using a 63X oil objective. Ratiometric images of SONAR were processed using ImageJ (NIH, v2.0.3) to 32-bit images and presented in 16 colors mode. Threshold images were quantified for mean fluorescence intensity in ImageJ.
Flow cytometry analysis
NSCLC cells expressing the indicated SoNar reporter were seeded at 0.25 × 106 cells/well in a 6-well plate for 48 hours. Cells were dislodged by trypsin digestion, resuspended in FACS buffer (PBS + 1% FBS, 2 mM EDTA) and treated sequentially at room temperature with 0.5 μM Rotenone (Sigma-Aldrich) followed by 5 mM sodium oxamate (Sigma-Aldrich). The fluorescent signal at 530 nm following excitation at 405 nm (NADH binding) or 488 nm (NAD binding) was measured every 2 min for a total of 30 min by an Aurora (Cytek) flow cytometer. The ratio of λex = 488 nm/λem = 530 nm to λex = 405 nm/λem = 530 nm signal was determined using Flowjo v10.6.
Lactate dehydrogenase (LDH) activity
LDH activity was determined as described in104. Briefly, cell pellets were lysed in water and the supernatant was cleared by centrifugation at 13,000 × rpm for 10 min at 4°C. Protein concentration was determined by Bradford (Bio-Rad). To initiate the NADH oxidation assay equal concentrations of cell lysate (20 μL) were added into 80 μL of a reaction buffer (1 mM sodium pyruvate, 0.5 mM NADH and 200 mM Tris pH 7.5). NADH oxidation rate was determined by kinetic absorbance at 340 nm which was collected every 2 min for 30 min using SpectraMax M5 plate reader (Molecular Devices). To determine LDH activity, NADH half-life was determined by one-phase exponential decay model using Prism v7 (GraphPad) and LDH activity is represented as 1/NADH(half-life).
Lactate measurement
Cell lines were cultured at 1 × 106 cells per well in a 6-well plate in 2 mL of RPMI culture media. The following day, culture media was replaced with 1 mL of serum-free RPMI after three successive washes with PBS. After 8 hours, the entirety of the supernatant (1 mL) was collected and mixed with a 50 μL of reaction buffer (30 mM Tris (Sigma-Aldrich) pH 7.4, 40 μM resazurin (Sigma-Aldrich), 0.01 U rLDH (Abcam), 0.05 U Diaphorase (Sigma-Aldrich), 1 mM NAD+ (Cayman). Reactions were carried out at room temp for 30 min in an opaque 96-well plate. The fluorescent signal corresponding to NADH oxidation was measured (550–585 nm) using SpectraMax M5 plate reader (Molecular Devices) and normalized to cell number.
In vitro ALDH3A1 assay
ALDH3A1 in vitro assay was conducted in 100 μl 0.1 M Tris buffer (pH 7.5) in a 96-well plate supplemented with 2.5 mM NAD+ (Cayman), 1 mM of phenylacetaldehyde (PAA, Sigma) or 4-hydroxyphenylacetaldehyde (4PAA, this study) and FLAG-purified METAP2 or ALDH3A1 from CALU6 cells. To initiate the assay, 4PAA or PAA was added immediately after the addition of enzyme, and the reaction was monitored by tracking NADH absorbance at 340 nm at 2-minute intervals on the SpectraMax M5 plate reader for 1 hour. For LCMS quantification of substrates and products from the in vitro ALDH3A1 assay samples were on a iHILIC column (5 μm, 150 mm × 2.1 mm I.D., Nest Group) coupled to an Agilent 6546 LC/Q-TOF with an ESI source operated in negative positive mode. The identity of each metabolite was confirmed by matching retention time and/or MS/MS fragmentation data to standard compounds and/or a database.
GCMS analysis
1 × 106 cells were cultured in a 6 cm dish in a total of 5 mL RPMI media and treated with DMSO or KI696 (1 μM) for 48 hours. Cells were washed with 0.9% NaCl solution and immediately flash frozen in liquid nitrogen. Metabolite extraction was undertaken by adding 800 μL ES1 (H2O:L-Norvalin (1 mg/mL) : Glutarate (1 mg/mL), 15 mL:37.5 μL:37.5 μL) (all from Sigma-Aldrich) followed by addition of chloroform (500 μL). Samples were vortexed, centrifuged at 13,000 × rpm for 10 min, 4 °C and the upper phase was dried and derivatized with TBDMS (Sigma-Aldrich) for downstream GC-MS analyses. Samples were analyzed on an Agilent 7890B GC system was coupled to 5977B single quadrupole mass spectrometer equipped with an electron ionization source. Automated injections were performed with an Agilent 7693 autosampler. The injector temperature was held constant at 270 °C. Injections of 1 μL were made in spitless mode. Chromatography was performed on a HP-5ms Ultra Inert Column (30 m × 0.25 mm, 0.25 μm film thickness, Agilent). Helium carrier gas was used at a constant flow of 1ml/min. The GC oven temperature program was 100 °C initial temperature with 3 min hold time and ramping at 10 °C/min to a final temperature of 300 °C with 12 min hold time. The transfer line temperature was 250 °C, and the source temperature was 230 °C. After a solvent delay of 5.5 min, mass spectra were acquired at 2.9 scans/s with a mass range of 50 to 550 m/z. Data processing was performed with MassHunter Workstation Software Quantitative Analysis (Version B.09.00 / Build 9.0.647.0, for GCMS and LCMS).
LCMS analysis
2.5 × 105 cells were cultured in a 6 well plate in 4 mL of RPMI and treated with DMSO or KI696 (1 μM) for 48 hours. On the day of sample collection, cells were washed three times with a 75 mM ammonium carbonate solution followed by extraction with 70% ethanol at 70°C for 3 min. The supernatant was cleared by centrifugation at 13,000 × rpm for 10 min at 4°C and immediately stored in −80°C until further processing by LCMS.
LCMS analysis was performed on a platform consisting of an Agilent 1260 Infinity II LC pump coupled to a Gerstel MPS autosampler (CTC Analytics, Zwingen, Switzerland) and an Agilent 6550 Series Quadrupole TOF mass spectrometer (Agilent, Santa Clara, CA, USA) with Dual AJS ESI source operating in negative mode as described previously (Fuhrer et al, 2011). The flow rate was 150 μl/min of mobile phase consisting of isopropanol:water (60:40, v/v) with 1 mM ammonium fluoride. For online mass axis correction, two ions in Agilent’s ESI-L Low Concentration Tuning Mix (G1969–85000) were used. Mass spectra were recorded in profile mode from m/z 50 to 1,050 with a frequency of 1.4 s for 2 × 0.48 min (double injection) using the highest resolving power (4 GHz HiRes).
All steps of mass spectrometry data processing and analysis were performed with MATLAB (The Mathworks, Natick, MA, USA) using functions embedded in the Bioinformatics, Statistics, Database, and Parallel Computing toolboxes as described previously in105. The resulting data included the intensity of each mass peak in each analyzed sample. Peak picking was done for each sample once on the total profile spectrum obtained by summing all single scans recorded over time, and using wavelet decomposition as provided by the Bioinformatics toolbox. In this procedure, a cutoff was applied to filter peaks of less than 5,000 ion counts (in the summed spectrum) to avoid detection of features that are too low to deliver meaningful insights. Centroid lists from samples were then merged to a single matrix by binning the accurate centroid masses within the tolerance given by the instrument resolution. Starting from the HMDB v4.0 database106, we generated a list of expected ions including deprotonated, fluorinated, and all major adducts found under these conditions. All formulas matching the measured mass within a mass tolerance of 0.001 Da were enumerated. As this method does not employ chromatographic separation or in-depth MS2 characterization, it is not possible to distinguish between compounds with identical molecular formula. The confidence of annotation reflects Level 4 but – in practice - in the case of intermediates of primary metabolism it is higher because they are the most abundant metabolites in cells biological extracts. The resulting matrix lists the intensity of each mass peak in each analyzed sample. An accurate common m/z was recalculated with a weighted average of the values obtained from independent centroiding.
RNAseq analysis
CALU6 cells were cultured in 6-well plates (2.5 × 105 cells/well) and treated with KI696 (1 μM) or vehicle control for 48 hrs. RNA was isolated by RNeasy Kit (Qiagen) following the manufacturer’s instructions and digested with DNase (Qiagen) from n=2 samples per condition. mRNA-seq libraries were prepared using NEBNext® Poly(A) mRNA Magnetic Isolation Module (NEB) kit according to manufacturer’s instructions. Libraries were then quantified by Kappa Library Quantification (Roche), pooled, and sequenced by single-end 75 base pairs using the Illumina NextSeq 500 platform. FASTQ files were then processed using RNA Express module (Illumina) and raw counts were further processed in R (v.4.0.3) and edgeR107 to obtain relative gene expression.
Proteome wide analysis of NRF2 activation
CALU6 cells were cultured in 6-well plates (0.25 × 106 cells) and treated with 1 μM KI696 for 48 hrs. Cells were washed once with ice-cold PBS, snap-frozen in liquid nitrogen and stored at −80°C until use. Frozen cell pellets were lysed in PBS supplemented with Benzonase (Santacruz) and protease inhibitors (Roche) using a chilled bath sonicator (QSONICA) and centrifuged for 3 min at 300 g. Proteins were quantified by BCA assay (Thermo Scientific) and a total of 50 μg of total protein extracts were used for each TMT channel. Protein extracts were reduced with 5 mM 5-Tris (2-carboxyethyl) phosphine hydrochloride (TCEP) (Sigma-Aldrich) for 2 min at room temperature, followed by alkylation using 20 mM Iodoacetamide (Sigma-Aldrich) for 30 min in the dark at room temperature. SP3 magnetic beads (Cytiva) were prewashed with LC-MS grade water and 250 μg combined SP3 beads (1:1 hydrophobic:hydrophilic) and LC-MS grade ethanol were added to each sample to reach a final concentration of 50% ethanol. SP3 protein binding occurred for 30 min at room temperature and beads were subsequently washed 3 times with 80% ethanol for resuspended with 175 ul of Trypsin/Lys-C (1 μg, Thermo Scientific A40009) in 200 mM EPPS (pH 8.4)/5 mM CaCl2. Proteins were digested overnight (16 h) at 37°C and peptides were dried using a Speedvac (Thermo Scientific). Peptides were desalted with stage tips using the following procedure: Peptides were reconstituted with 5% acetonitrile/0.1 % formic acid and loaded onto Empore C18 disks (3M) packed into a 200 μl pipette tips pre-equilibrated with LC-MS grade methanol and water containing 0.1% formic acid. C18 disk were washed 10 times with LC-MS grade water containing 0.1 % formic acid and subsequently eluted with 80% acetonitrile/0.1% formic acid and dried using a Speedvac (Thermo Scientific). Peptides were quantified with Quantitative Colorimetric Peptide Assay (Thermo Scientific) and 5 μg of total peptides were labeled using TMTpro16-plex reagents (Thermo Scientific). Briefly, peptides were reconstituted with 30% acetonitrile/70% 200 mM EPPS (pH 8.4) and labeled with 50 μg of TMT reagent per channel for 75 min at room temperature with rotation. Labeling was terminated by the addition of 5% hydroxylamine (Acros Organics) for 15 min, followed by supplementation with 10% formic acid. Samples were pooled and dried using a Speedvac (Thermo) and fractionated using high pH reversed phase fractionation (Thermo Scientific) following the manufactures directions.
Fractionated samples were reconstituted with 2% acetonitrile/0.1 % formic acid and subjected to liquid chromatography using an Easy nLC 1200. The columns used are an Acclaim PepMap trap Column (75 μm × 20 mm packed with 3-μm particles of C18 stationary phase) and an EASY-Spray analytical column (75 μm × 500 mm packed with 2-μm particles of C18 stationary phase). Peptides were loaded onto the trap column using 0.1 % formic acid in water. Peptides were separated over a 190 min gradient, consisting of 5–28% solvent B over 155 min, 28–43% solvent B over 25 min, 43–95% solvent B over 10 min, and 90% solvent B held for 10 min. Solvent A was 0.1% formic acid in water and solvent B was 80% acetonitrile and 0.1% formic acid in water. Peptides were ionized at 2300 V and analyzed on an Orbitrap Eclipse mass spectrometer coupled with a FAIMSpro system. Ionized peptides were separated using FAIMSpro (1.5 sec per cycle). MS1 was analyzed in the orbitrap at 120,000 resolution using 50 ms ion injection time (IIT) and 250% automatic gain control (AGC). Collision energy was set to 36% (CID) and fragmented ions were detected in the ion trap using default IIT and AGC. Real time search function was enabled to detect human peptides. Synchronous precursor selection was enabled and was set to isolate twenty notches. MS3 was analyzed in the orbitrap with the mass range at 100 – 500 Da and the resolution at 50,000.
Acquired RAW data were analyzed using ProteomeDiscoverer v2.5 (Thermo Scientific) using the SequestHT module. Data was search against the UniProtKB human universal database (UniProt UP000005640, downloaded May 2020) combined with the common Repository of Adventitious Proteins (cRAP, classes 1,2,3, and 5). Parameters were set as follows: MS1 tolerance of 10 ppm, MS/MS mass tolerance of 0.6 Da, trypsin (full) digestion with a maximum of zero missed cleavages, minimum peptide length of 6 and maximum of 144 amino acids. Cysteine carbamidomethylation (57.021 Da) and methionine oxidation (15.995 Da) were set as dynamic modifications. Lysine- and N-ternimous- TMTpro modification (304.207 Da) was set as static modification. A false discovery rate (FDR) of 1% was set for peptide-to-spectrum matches using the Percolator algorithm and for protein assignment. Reporter ion quantification was based on signal over noise (S/N) with the co-Isolation Threshold at 50, S/N threshold at 10, and SPS mass matches threshold at 50%. Abundances was normalized to total peptides. Protein ratio was calculated using PD2.5 pairwise ratio based algorithm and t-test was used for significance.
NADH/NAD+ measurement
0.25 × 106 CALU6 cells were treated with 1 μM KI696 or vehicle control in 6-well plates for 48 days. The ratio NADH/NAD+ was determined NAD/NADH-Glo™ Assay Kit (Promega) following the manufacturer’s protocol. Absorbance was measured using a SpectraMax M5 plate reader (Molecular Devices).
4-Hydroxyphenylacetalaldehyde Synthesis
![]()
To a flame dried round bottom flask, 4-Hydroxyphenethyl alcohol (0.7 g, 5.1 mM) was added under nitrogen atmosphere, then anhydrous DMSO (7.0 mL) was added. While stirring, TEA (1.4 mL, 10 mM) was added slowly. A solution of pyridine-sulfur trioxide complex (2.5 g, 16 mM) in anhydrous DMSO (5 mL) was added dropwise to the alcohol. After stirring for 1 h at room temperature, the reaction was diluted with CH2Cl2, then washed with ice-cold water. The organic layer was dried over Na2SO4, filtered, and concentrated to dryness. Purification using silica gel chromatography with hexane:ethyl acetate as eluent (5:1 to 2:1) yielded 147 mg (23%) of 4-hydroxyphenylacetaldehyde. 1H NMR (400 MHz, CDCl3): δ = 9.70 (t, J = 2.4Hz, 1H), 7.08–7.04 (m, 2H), 6.84–6.80 (m, 2H), 5.37 (br s, 1H), 3.61 (d, J = 2.4Hz, 1H); 13C NMR (100MHz, CDCl3): δ = 201.1, 155.2, 131.0, 123.9, 116.0, 49.8; LCMS (ESI): calcd for [C8H8O2]+H = 137.1; found 137.1. The spectroscopic data are in good agreement with those reported in the literature108,109 and can be found in Data S1.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis was performed using GraphPad Prism (v10.0.7) for Mac (GraphPad Software) or R statistical programing language (v4.0.3, R-project.org). Statistical values including the exact n, statistical test, and significance are reported in the Figure Legends. Statistical significance was defined as * p< 0.05 and unless indicated otherwise determined by 2-tailed Student’s t-test or one-way Anova. All post-hoc analyses are indicated in the figure legends. A False Discovery Rate (FDR) was calculated for Proteomics (using Proteome Discover, v2.5, Thermo), RNA-sequencing (EdgeR107) and metabolomics (Limma110) analysis to correct for multiple comparisons. CRISPR-scores were calculated as described in CRISPR screen section. For the KI696 and Bardoxolone small molecule screens, a Z-score was calculated using the following method: Z-Score= (xi-μ)/σ, where xi is the fold-change (KI696/DMSO) of the ith sample, μ is the mean fold-change (KI696/DMSO) across all samples and σ is the standard deviation of fold-change (KI696/DMSO) across all samples.
Supplementary Material
Table S1. Genome-wide CRISPR screen in CALU6 cells following KI696 or DMSO treatment, related to Figure 1.
Table S2. Metabolism-focused CRISPR screen in CALU6 and H1975 cells following KI696 or DMSO treatment, related to Figure 1.
Table S3. Glycolytic signature gene list, related to Figure 2.
Table S4. MS analysis of metabolites after treatment with KI696, related to Figure 3.
Table S5. MS analysis of metabolites consistent with aldehydes in CALU6 cells expressing sgCTRL and sgALDH3A1, related to Figure 5.
Table S6. Primers and sgRNA/shRNAs used in this study, related to STAR Methods.
Data S1. Data underlying the display items in the manuscript, related to Figures 1,2,3,4,5,6 and S1–S6.
Highlights.
Small molecule and CRISPR screens identifies KEAP1 as a dependency in lung cancer
The glycolytic vs. oxidative nature of cancer cells underlies KEAP1 dependency.
NRF2 activation induces NADH-reductive stress through the upregulation of ALDH3A1
NADH-reductive stress is a metabolic liability in NRF2-activated cancers
Acknowledgements
We thank Yi Yang for the SoNar plasmid. We thank all members of the Bar-Peled Lab and David Sabatini for helpful suggestions. This work was supported by grants from the NIH (R00CA215249, R37CA260062, R21CA256082 to L.B-P, R01AR072304, R01AR043369; P01CA163222; R01CA222871 to D.E.F.; U01CA267827 and R01CA273461 to M.C.H.; R37CA222504 and R01CA227649 to T.P.; R01CA235412 and R01GM128448 to R.M.; R01CA215498 and R01CA219670 to N.B.). Damon Runyon Cancer Research Foundation Rachleff Innovator Award, Damon Runyon Cancer Research Fellowship (DG-2454-22 to C.H.A) AACR (19-20-45-BARP), Paula and Rodger Riney Foundation, the V Foundation, Melanoma Research Alliance, the Mary Kay Foundation to L.B-P, the Ludwig Cancer Center to L.B-P, A.N.H and M.C.H.), the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation to M.C.H, AACR to C.Y., American Cancer Society Research Scholar Grant (RSG-17-200-01 TBE) to T.P., and Uehara Memorial Fund to M.H.
DECLARATION OF INTERESTS
L.B-P is a founder, consultant and holds privately held equity in Scorpion Therapeutics. D.E.F. has a financial interest in Soltego, a company developing salt inducible kinase inhibitors for topical skin-darkening treatments that might be used for a broad set of human applications. The interests of L.B-P and D.E.F. were reviewed and are managed by Massachusetts General Hospital and Partners HealthCare in accordance with their conflict-of-interest policies. M.C.H. is on the SAB and holds equity in Alixia Therapeutics and is on the SAB of Minovia. T. P. is on the SAB of Faeth Therapeutics and consults for Dracen Pharmaceuticals. A.N.H receives research funding from Amgen, Blueprint Medicines, BridgeBio, Bristol-Myers Sqiub, C4 Therapeutics, Eli Lilly, Novartis, Nuvalent, Pfizer, Roche/Genentech, Scorpion Therapeutics and consults for Engine Biosciences, Nuvalent, TigaTx, Tolremo Therapeutics.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
INCLUSION AND DIVERSITY
We support inclusive, diverse and equitable conduct in research.
References
- 1.Kerk SA, Papagiannakopoulos T, Shah YM & Lyssiotis CA (2021). Metabolic networks in mutant KRAS-driven tumours: tissue specificities and the microenvironment. Nat Rev Cancer 21, 510–525, doi: 10.1038/s41568-021-00375-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Martinez-Reyes I & Chandel NS (2021). Cancer metabolism: looking forward. Nat Rev Cancer 21, 669–680, doi: 10.1038/s41568-021-00378-6. [DOI] [PubMed] [Google Scholar]
- 3.Cantor JR & Sabatini DM (2012). Cancer cell metabolism: one hallmark, many faces. Cancer Discov 2, 881–898, doi: 10.1158/2159-8290.CD-12-0345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vander Heiden MG & DeBerardinis RJ (2017). Understanding the Intersections between Metabolism and Cancer Biology. Cell 168, 657–669, doi: 10.1016/j.cell.2016.12.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Elia I & Haigis MC (2021). Metabolites and the tumour microenvironment: from cellular mechanisms to systemic metabolism. Nat Metab 3, 21–32, doi: 10.1038/s42255-020-00317-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chio IIC & Tuveson DA (2017). ROS in Cancer: The Burning Question. Trends Mol Med 23, 411–429, doi: 10.1016/j.molmed.2017.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chandel NS & Tuveson DA (2014). The promise and perils of antioxidants for cancer patients. N Engl J Med 371, 177–178, doi: 10.1056/NEJMcibr1405701. [DOI] [PubMed] [Google Scholar]
- 8.Vander Heiden MG, Cantley LC & Thompson CB (2009). Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324, 1029–1033, doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee AC et al. (1999). Ras proteins induce senescence by altering the intracellular levels of reactive oxygen species. J Biol Chem 274, 7936–7940, doi: 10.1074/jbc.274.12.7936. [DOI] [PubMed] [Google Scholar]
- 10.Irani K et al. (1997). Mitogenic signaling mediated by oxidants in Ras-transformed fibroblasts. Science 275, 1649–1652, doi: 10.1126/science.275.5306.1649. [DOI] [PubMed] [Google Scholar]
- 11.Gorrini C, Harris IS & Mak TW (2013). Modulation of oxidative stress as an anticancer strategy. Nature reviews. Drug discovery 12, 931–947, doi: 10.1038/nrd4002. [DOI] [PubMed] [Google Scholar]
- 12.Harris IS & DeNicola GM (2020). The Complex Interplay between Antioxidants and ROS in Cancer. Trends Cell Biol 30, 440–451, doi: 10.1016/j.tcb.2020.03.002. [DOI] [PubMed] [Google Scholar]
- 13.Sies H et al. (2022). Defining roles of specific reactive oxygen species (ROS) in cell biology and physiology. Nat Rev Mol Cell Biol 23, 499–515, doi: 10.1038/s41580-022-00456-z. [DOI] [PubMed] [Google Scholar]
- 14.Xiao W & Loscalzo J (2020). Metabolic Responses to Reductive Stress. Antioxid Redox Signal 32, 1330–1347, doi: 10.1089/ars.2019.7803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gores GJ et al. (1989). Swelling, reductive stress, and cell death during chemical hypoxia in hepatocytes. Am J Physiol 257, C347–354, doi: 10.1152/ajpcell.1989.257.2.C347. [DOI] [PubMed] [Google Scholar]
- 16.Narasimhan M & Rajasekaran NS (2015). Reductive potential - a savior turns stressor in protein aggregation cardiomyopathy. Biochim Biophys Acta 1852, 53–60, doi: 10.1016/j.bbadis.2014.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Korge P, Calmettes G & Weiss JN (2015). Increased reactive oxygen species production during reductive stress: The roles of mitochondrial glutathione and thioredoxin reductases. Biochim Biophys Acta 1847, 514–525, doi: 10.1016/j.bbabio.2015.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Manford AG et al. (2021). Structural basis and regulation of the reductive stress response. Cell 184, 5375–5390 e5316, doi: 10.1016/j.cell.2021.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Manford AG et al. (2020). A Cellular Mechanism to Detect and Alleviate Reductive Stress. Cell 183, 46–61 e21, doi: 10.1016/j.cell.2020.08.034. [DOI] [PubMed] [Google Scholar]
- 20.Yang Y, Song Y & Loscalzo J (2007). Regulation of the protein disulfide proteome by mitochondria in mammalian cells. Proc Natl Acad Sci U S A 104, 10813–10817, doi: 10.1073/pnas.0702027104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Perez-Torres I, Guarner-Lans V & Rubio-Ruiz ME (2017). Reductive Stress in Inflammation-Associated Diseases and the Pro-Oxidant Effect of Antioxidant Agents. Int J Mol Sci 18, doi: 10.3390/ijms18102098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ho HY, Lin YT, Lin G, Wu PR & Cheng ML (2017). Nicotinamide nucleotide transhydrogenase (NNT) deficiency dysregulates mitochondrial retrograde signaling and impedes proliferation. Redox Biol 12, 916–928, doi: 10.1016/j.redox.2017.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang H et al. (2012). Glutathione-dependent reductive stress triggers mitochondrial oxidation and cytotoxicity. FASEB J 26, 1442–1451, doi: 10.1096/fj.11-199869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goodman RP et al. (2020). Hepatic NADH reductive stress underlies common variation in metabolic traits. Nature 583, 122–126, doi: 10.1038/s41586-020-2337-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li Z et al. (2022). Cancer cells depend on environmental lipids for proliferation when electron acceptors are limited. Nat Metab, doi: 10.1038/s42255-022-00588-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Diehl FF, Lewis CA, Fiske BP & Vander Heiden MG (2019). Cellular redox state constrains serine synthesis and nucleotide production to impact cell proliferation. Nat Metab 1, 861–867, doi: 10.1038/s42255-019-0108-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Garcia-Bermudez J et al. (2018). Aspartate is a limiting metabolite for cancer cell proliferation under hypoxia and in tumours. Nat Cell Biol 20, 775–781, doi: 10.1038/s41556-018-0118-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Birsoy K et al. (2015). An Essential Role of the Mitochondrial Electron Transport Chain in Cell Proliferation Is to Enable Aspartate Synthesis. Cell 162, 540–551, doi: 10.1016/j.cell.2015.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sullivan LB et al. (2015). Supporting Aspartate Biosynthesis Is an Essential Function of Respiration in Proliferating Cells. Cell 162, 552–563, doi: 10.1016/j.cell.2015.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gui DY et al. (2016). Environment Dictates Dependence on Mitochondrial Complex I for NAD+ and Aspartate Production and Determines Cancer Cell Sensitivity to Metformin. Cell Metab 24, 716–727, doi: 10.1016/j.cmet.2016.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sporn MB & Liby KT (2012). NRF2 and cancer: the good, the bad and the importance of context. Nat Rev Cancer 12, 564–571, doi: 10.1038/nrc3278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.DeNicola GM et al. (2011). Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 475, 106–109, doi: 10.1038/nature10189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Itoh K et al. (1997). An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun 236, 313–322, doi: 10.1006/bbrc.1997.6943. [DOI] [PubMed] [Google Scholar]
- 34.Igarashi K et al. (1994). Regulation of transcription by dimerization of erythroid factor NF-E2 p45 with small Maf proteins. Nature 367, 568–572, doi: 10.1038/367568a0. [DOI] [PubMed] [Google Scholar]
- 35.Pillai R, Hayashi M, Zavitsanou AM & Papagiannakopoulos T (2022). NRF2: KEAPing Tumors Protected. Cancer Discov 12, 625–643, doi: 10.1158/2159-8290.CD-21-0922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Suzuki T & Yamamoto M (2015). Molecular basis of the Keap1-Nrf2 system. Free Radic Biol Med 88, 93–100, doi: 10.1016/j.freeradbiomed.2015.06.006. [DOI] [PubMed] [Google Scholar]
- 37.Baird L, Swift S, Lleres D & Dinkova-Kostova AT (2014). Monitoring Keap1-Nrf2 interactions in single live cells. Biotechnol Adv 32, 1133–1144, doi: 10.1016/j.biotechadv.2014.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang DD, Lo SC, Cross JV, Templeton DJ & Hannink M (2004). Keap1 is a redox-regulated substrate adaptor protein for a Cul3-dependent ubiquitin ligase complex. Mol Cell Biol 24, 10941–10953, doi: 10.1128/MCB.24.24.10941-10953.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kobayashi A et al. (2004). Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol Cell Biol 24, 7130–7139, doi: 10.1128/MCB.24.16.7130-7139.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Malhotra D et al. (2010). Global mapping of binding sites for Nrf2 identifies novel targets in cell survival response through ChIP-Seq profiling and network analysis. Nucleic Acids Res 38, 5718–5734, doi: 10.1093/nar/gkq212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cancer Genome Atlas Research, N. (2014). Comprehensive molecular profiling of lung adenocarcinoma. Nature 511, 543–550, doi: 10.1038/nature13385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Satoh H, Moriguchi T, Takai J, Ebina M & Yamamoto M (2013). Nrf2 prevents initiation but accelerates progression through the Kras signaling pathway during lung carcinogenesis. Cancer Res 73, 4158–4168, doi: 10.1158/0008-5472.CAN-12-4499. [DOI] [PubMed] [Google Scholar]
- 43.Rojo de la Vega M, Chapman E & Zhang DD (2018). NRF2 and the Hallmarks of Cancer. Cancer Cell 34, 21–43, doi: 10.1016/j.ccell.2018.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bar-Peled L et al. (2017). Chemical Proteomics Identifies Druggable Vulnerabilities in a Genetically Defined Cancer. Cell 171, 696–709 e623, doi: 10.1016/j.cell.2017.08.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Romero R et al. (2017). Keap1 loss promotes Kras-driven lung cancer and results in dependence on glutaminolysis. Nat Med 23, 1362–1368, doi: 10.1038/nm.4407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.DeNicola GM et al. (2015). NRF2 regulates serine biosynthesis in non-small cell lung cancer. Nat Genet 47, 1475–1481, doi: 10.1038/ng.3421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Leinonen HM, Kansanen E, Polonen P, Heinaniemi M & Levonen AL (2014). Role of the Keap1-Nrf2 pathway in cancer. Adv Cancer Res 122, 281–320, doi: 10.1016/B978-0-12-420117-0.00008-6. [DOI] [PubMed] [Google Scholar]
- 48.Chio II et al. (2016). NRF2 Promotes Tumor Maintenance by Modulating mRNA Translation in Pancreatic Cancer. Cell 166, 963–976, doi: 10.1016/j.cell.2016.06.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Goldstein LD et al. (2016). Recurrent Loss of NFE2L2 Exon 2 Is a Mechanism for Nrf2 Pathway Activation in Human Cancers. Cell Rep 16, 2605–2617, doi: 10.1016/j.celrep.2016.08.010. [DOI] [PubMed] [Google Scholar]
- 50.Fan Z et al. (2017). Nrf2-Keap1 pathway promotes cell proliferation and diminishes ferroptosis. Oncogenesis 6, e371, doi: 10.1038/oncsis.2017.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Singh A et al. (2006). Dysfunctional KEAP1-NRF2 interaction in non-small-cell lung cancer. PLoS Med 3, e420, doi: 10.1371/journal.pmed.0030420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Davies TG et al. (2016). Monoacidic Inhibitors of the Kelch-like ECH-Associated Protein 1: Nuclear Factor Erythroid 2-Related Factor 2 (KEAP1:NRF2) Protein-Protein Interaction with High Cell Potency Identified by Fragment-Based Discovery. J Med Chem 59, 3991–4006, doi: 10.1021/acs.jmedchem.6b00228. [DOI] [PubMed] [Google Scholar]
- 53.Ding H et al. (2021). Activation of the NRF2 antioxidant program sensitizes tumors to G6PD inhibition. Sci Adv 7, eabk1023, doi: 10.1126/sciadv.abk1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wu WL & Papagiannakopoulos T (2020). The Pleiotropic Role of the KEAP1/NRF2 Pathway in Cancer. Annual Review of Cancer Biology 4, 413–435, doi: 10.1146/annurev-cancerbio-030518-055627. [DOI] [Google Scholar]
- 55.Sanchez-Vega F et al. (2018). Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 173, 321–337 e310, doi: 10.1016/j.cell.2018.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kerins MJ & Ooi A (2018). A catalogue of somatic NRF2 gain-of-function mutations in cancer. Sci Rep 8, 12846, doi: 10.1038/s41598-018-31281-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tsherniak A et al. (2017). Defining a Cancer Dependency Map. Cell 170, 564–576 e516, doi: 10.1016/j.cell.2017.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lenoir WF, Lim TL & Hart T (2018). PICKLES: the database of pooled in-vitro CRISPR knockout library essentiality screens. Nucleic Acids Res 46, D776–D780, doi: 10.1093/nar/gkx993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Serrano M, Lin AW, McCurrach ME, Beach D & Lowe SW (1997). Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 88, 593–602, doi: 10.1016/s0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- 60.Davoli T et al. (2013). Cumulative haploinsufficiency and triplosensitivity drive aneuploidy patterns and shape the cancer genome. Cell 155, 948–962, doi: 10.1016/j.cell.2013.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lin CY et al. (2012). Transcriptional amplification in tumor cells with elevated c-Myc. Cell 151, 56–67, doi: 10.1016/j.cell.2012.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dar S et al. (2017). Bioenergetic Adaptations in Chemoresistant Ovarian Cancer Cells. Sci Rep 7, 8760, doi: 10.1038/s41598-017-09206-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tennant DA, Duran RV, Boulahbel H & Gottlieb E (2009). Metabolic transformation in cancer. Carcinogenesis 30, 1269–1280, doi: 10.1093/carcin/bgp070. [DOI] [PubMed] [Google Scholar]
- 64.Zhai X, Yang Y, Wan J, Zhu R & Wu Y (2013). Inhibition of LDH-A by oxamate induces G2/M arrest, apoptosis and increases radiosensitivity in nasopharyngeal carcinoma cells. Oncol Rep 30, 2983–2991, doi: 10.3892/or.2013.2735. [DOI] [PubMed] [Google Scholar]
- 65.Sayin VI et al. (2017). Activation of the NRF2 antioxidant program generates an imbalance in central carbon metabolism in cancer. Elife 6, doi: 10.7554/eLife.28083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Loffler M, Jockel J, Schuster G & Becker C (1997). Dihydroorotat-ubiquinone oxidoreductase links mitochondria in the biosynthesis of pyrimidine nucleotides. Mol Cell Biochem 174, 125–129. [PubMed] [Google Scholar]
- 67.Martinez-Reyes I et al. (2020). Mitochondrial ubiquinol oxidation is necessary for tumour growth. Nature 585, 288–292, doi: 10.1038/s41586-020-2475-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhao Y et al. (2015). SoNar, a Highly Responsive NAD+/NADH Sensor, Allows High-Throughput Metabolic Screening of Anti-tumor Agents. Cell Metab 21, 777–789, doi: 10.1016/j.cmet.2015.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Micheli V, Simmonds HA, Sestini S & Ricci C (1990). Importance of nicotinamide as an NAD precursor in the human erythrocyte. Arch Biochem Biophys 283, 40–45, doi: 10.1016/0003-9861(90)90609-3. [DOI] [PubMed] [Google Scholar]
- 70.McElroy GS et al. (2020). NAD+ Regeneration Rescues Lifespan, but Not Ataxia, in a Mouse Model of Brain Mitochondrial Complex I Dysfunction. Cell Metab 32, 301–308 e306, doi: 10.1016/j.cmet.2020.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Seo BB et al. (1998). Molecular remedy of complex I defects: rotenone-insensitive internal NADH-quinone oxidoreductase of Saccharomyces cerevisiae mitochondria restores the NADH oxidase activity of complex I-deficient mammalian cells. Proc Natl Acad Sci U S A 95, 9167–9171, doi: 10.1073/pnas.95.16.9167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Titov DV et al. (2016). Complementation of mitochondrial electron transport chain by manipulation of the NAD+/NADH ratio. Science 352, 231–235, doi: 10.1126/science.aad4017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Luengo A et al. (2021). Increased demand for NAD(+) relative to ATP drives aerobic glycolysis. Mol Cell 81, 691–707 e696, doi: 10.1016/j.molcel.2020.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wang B et al. (2017). Aldehyde dehydrogenase 1A1 increases NADH levels and promotes tumor growth via glutathione/dihydrolipoic acid-dependent NAD(+) reduction. Oncotarget 8, 67043–67055, doi: 10.18632/oncotarget.17688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pappa A, Chen C, Koutalos Y, Townsend AJ & Vasiliou V (2003). Aldh3a1 protects human corneal epithelial cells from ultraviolet- and 4-hydroxy-2-nonenal-induced oxidative damage. Free Radic Biol Med 34, 1178–1189, doi: 10.1016/s0891-5849(03)00070-4. [DOI] [PubMed] [Google Scholar]
- 76.Lindahl R (1992). Aldehyde dehydrogenases and their role in carcinogenesis. Crit Rev Biochem Mol Biol 27, 283–335, doi: 10.3109/10409239209082565. [DOI] [PubMed] [Google Scholar]
- 77.Shortall K, Djeghader A, Magner E & Soulimane T (2021). Insights into Aldehyde Dehydrogenase Enzymes: A Structural Perspective. Front Mol Biosci 8, 659550, doi: 10.3389/fmolb.2021.659550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Choi HS, Kim SL, Kim JH, Ko YC & Lee DS (2020). Plant Volatile, Phenylacetaldehyde Targets Breast Cancer Stem Cell by Induction of ROS and Regulation of Stat3 Signal. Antioxidants (Basel) 9, doi: 10.3390/antiox9111119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Molina JR et al. (2018). An inhibitor of oxidative phosphorylation exploits cancer vulnerability. Nat Med 24, 1036–1046, doi: 10.1038/s41591-018-0052-4. [DOI] [PubMed] [Google Scholar]
- 80.Tieman D et al. (2006). Tomato aromatic amino acid decarboxylases participate in synthesis of the flavor volatiles 2-phenylethanol and 2-phenylacetaldehyde. Proc Natl Acad Sci U S A 103, 8287–8292, doi: 10.1073/pnas.0602469103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Murphy MP (2009). How mitochondria produce reactive oxygen species. Biochem J 417, 1–13, doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yoshida E et al. (2018). Hyperactivation of Nrf2 leads to hypoplasia of bone in vivo. Genes Cells 23, 386–392, doi: 10.1111/gtc.12579. [DOI] [PubMed] [Google Scholar]
- 83.Suzuki T et al. (2017). Hyperactivation of Nrf2 in early tubular development induces nephrogenic diabetes insipidus. Nat Commun 8, 14577, doi: 10.1038/ncomms14577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wakabayashi N et al. (2003). Keap1-null mutation leads to postnatal lethality due to constitutive Nrf2 activation. Nat Genet 35, 238–245, doi: 10.1038/ng1248. [DOI] [PubMed] [Google Scholar]
- 85.Robledinos-Anton N, Fernandez-Gines R, Manda G & Cuadrado A (2019). Activators and Inhibitors of NRF2: A Review of Their Potential for Clinical Development. Oxid Med Cell Longev 2019, 9372182, doi: 10.1155/2019/9372182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Holmstrom KM, Kostov RV & Dinkova-Kostova AT (2016). The multifaceted role of Nrf2 in mitochondrial function. Curr Opin Toxicol 1, 80–91, doi: 10.1016/j.cotox.2016.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Esteras N, Dinkova-Kostova AT & Abramov AY (2016). Nrf2 activation in the treatment of neurodegenerative diseases: a focus on its role in mitochondrial bioenergetics and function. Biol Chem 397, 383–400, doi: 10.1515/hsz-2015-0295. [DOI] [PubMed] [Google Scholar]
- 88.Quintana A, Kruse SE, Kapur RP, Sanz E & Palmiter RD (2010). Complex I deficiency due to loss of Ndufs4 in the brain results in progressive encephalopathy resembling Leigh syndrome. Proc Natl Acad Sci U S A 107, 10996–11001, doi: 10.1073/pnas.1006214107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lemberg KM, Gori SS, Tsukamoto T, Rais R & Slusher BS (2022). Clinical development of metabolic inhibitors for oncology. J Clin Invest 132, doi: 10.1172/JCI148550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yap TA et al. (2019). Phase I trial of IACS-010759 (IACS), a potent, selective inhibitor of complex I of the mitochondrial electron transport chain, in patients (pts) with advanced solid tumors. Journal of Clinical Oncology 37, 3014–3014, doi: 10.1200/JCO.2019.37.15_suppl.3014. [DOI] [Google Scholar]
- 91.Eichner LJ et al. (2019). Genetic Analysis Reveals AMPK Is Required to Support Tumor Growth in Murine Kras-Dependent Lung Cancer Models. Cell Metab 29, 285–302 e287, doi: 10.1016/j.cmet.2018.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wishart DS et al. (2022). HMDB 5.0: the Human Metabolome Database for 2022. Nucleic Acids Res 50, D622–D631, doi: 10.1093/nar/gkab1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sarbassov DD, Guertin DA, Ali SM & Sabatini DM (2005). Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307, 1098–1101, doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 94.Kim D-H et al. (2002). mTOR Interacts with Raptor to Form a Nutrient-Sensitive Complex that Signals to the Cell Growth Machinery. Cell 110, 163–175. [DOI] [PubMed] [Google Scholar]
- 95.Feoktistova M, Geserick P & Leverkus M (2016). Crystal Violet Assay for Determining Viability of Cultured Cells. Cold Spring Harb Protoc 2016, pdb prot087379, doi: 10.1101/pdb.prot087379. [DOI] [PubMed] [Google Scholar]
- 96.Mehlem A, Hagberg CE, Muhl L, Eriksson U & Falkevall A (2013). Imaging of neutral lipids by oil red O for analyzing the metabolic status in health and disease. Nat Protoc 8, 1149–1154, doi: 10.1038/nprot.2013.055. [DOI] [PubMed] [Google Scholar]
- 97.Wang T et al. (2015). Identification and characterization of essential genes in the human genome. Science 350, 1096–1101, doi: 10.1126/science.aac7041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Li W et al. (2014). MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol 15, 554, doi: 10.1186/s13059-014-0554-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Subramanian A et al. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–15550, doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Korotkevich G et al. (2021). Fast gene set enrichment analysis. bioRxiv, 060012, doi: 10.1101/060012. [DOI] [Google Scholar]
- 101.Levy G et al. (2016). Nuclear receptors control pro-viral and antiviral metabolic responses to hepatitis C virus infection. Nat Chem Biol 12, 1037–1045, doi: 10.1038/nchembio.2193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Haynes WA et al. (2017). Empowering Multi-Cohort Gene Expression Analysis to Increase Reproducibility. Pac Symp Biocomput 22, 144–153, doi: 10.1142/9789813207813_0015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ghandi M et al. (2019). Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 569, 503–508, doi: 10.1038/s41586-019-1186-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zdralevic M et al. (2018). Double genetic disruption of lactate dehydrogenases A and B is required to ablate the “Warburg effect” restricting tumor growth to oxidative metabolism. J Biol Chem 293, 15947–15961, doi: 10.1074/jbc.RA118.004180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Fuhrer T, Heer D, Begemann B & Zamboni N (2011). High-throughput, accurate mass metabolome profiling of cellular extracts by flow injection-time-of-flight mass spectrometry. Anal Chem 83, 7074–7080, doi: 10.1021/ac201267k. [DOI] [PubMed] [Google Scholar]
- 106.Wishart DS et al. (2007). HMDB: the Human Metabolome Database. Nucleic Acids Res 35, D521–526, doi: 10.1093/nar/gkl923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Robinson MD, McCarthy DJ & Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140, doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Zhao T, Kurpiewska K, Kalinowska-Tluscik J, Herdtweck E & Domling A (2016). alpha-Amino Acid-Isosteric alpha-Amino Tetrazoles. Chemistry 22, 3009–3018, doi: 10.1002/chem.201504520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Sunazuka T et al. (1997). Syntheses and absolute structures of novel protein farnesyltransferase inhibitors, kurasoins A and B. J Antibiot (Tokyo) 50, 453–455, doi: 10.7164/antibiotics.50.453. [DOI] [PubMed] [Google Scholar]
- 110.Ritchie ME et al. (2015). limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43, e47, doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Patra KC et al. (2018). Mutant GNAS drives pancreatic tumourigenesis by inducing PKA-mediated SIK suppression and reprogramming lipid metabolism. Nat Cell Biol 20, 811–822, doi: 10.1038/s41556-018-0122-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Genome-wide CRISPR screen in CALU6 cells following KI696 or DMSO treatment, related to Figure 1.
Table S2. Metabolism-focused CRISPR screen in CALU6 and H1975 cells following KI696 or DMSO treatment, related to Figure 1.
Table S3. Glycolytic signature gene list, related to Figure 2.
Table S4. MS analysis of metabolites after treatment with KI696, related to Figure 3.
Table S5. MS analysis of metabolites consistent with aldehydes in CALU6 cells expressing sgCTRL and sgALDH3A1, related to Figure 5.
Table S6. Primers and sgRNA/shRNAs used in this study, related to STAR Methods.
Data S1. Data underlying the display items in the manuscript, related to Figures 1,2,3,4,5,6 and S1–S6.
Data Availability Statement
Source data for graphs can be found in Data S1. The RNA-seq data have been deposited in GEO: GSE221194 and the proteomics data have been deposited in PRIDE: PXD039027 and both are publicly available. The accession numbers are provided in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze data reported in this paper is available from the lead contact.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| NRF2 | Abcam | Cat#: ab62352 |
| KEAP1 | Cell Signaling Tech | Cat#: 7705 |
| HMOX1 | Proteintech | Cat#: 10701–1-AP |
| NQO-1 | Cell Signaling Tech | Cat#: 3187S |
| HK2 | Cell Signaling Tech | Cat#: 2867T |
| ALDH3A1 | Novus Biologicals | Cat#: NBP2–15340 |
| FLAG | Sigma | Cat#: F7425 |
| β-ACTIN | Cell Signaling Tech | Cat#: 4970 |
| HRP-labeled anti-mouse | Cell Signaling Tech | Cat#: 7076 |
| HRP-labeled anti-rabbit | Cell Signaling Tech | Cat#: 7074 |
| Bacterial and Virus Strains | ||
| DH5α | Fisher Scientific | Cat#:18265017 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| DMEM | Corning | Cat#:15–013-CV |
| RPMI-1640 | Corning | Cat#: ICN1646454 |
| FBS | Corning | Cat#: 35–010-CV |
| GlutaMax | Invitrogen | Cat#: 35050061 |
| Penicillin-Streptomycin | Millipore | Cat#: P0781 |
| Blasticidin | Fisher Scientific | Cat#: 50712728 |
| Puromycin | Sigma-Aldrich | Cat#: P8833 |
| Sequencing grade modified trypsin | Promega | Cat#: V5111 |
| DMSO | Fisher Scientific | Cat#: MT-25950CQC |
| Urea | VWR Intl | Cat#: 97063–798 |
| CHAPS hydrate | Sigma-Aldrich | Cat#: C3023 |
| DTT | Fisher Scientific | Cat#: BP1725 |
| Protease Inhibitor Cocktail | Sigma-Aldrich | Cat#: 5892791001 |
| Polybrene | Sigma-Aldrich | Cat#: TR-1003-G |
| X-tremegene HP transfection reagent | Sigma-Aldrich | Cat#: 6366236001 |
| Cell Titer Glo reagent | Promega | Cat#: 88836 |
| KI696 | MedChemExpress | Cat#: HY-101140 |
| Bardoxolone-methyl | MedChemExpress | Cat#: HY-13324 |
| Piericidin A | Cayman | Cat#: 15379 |
| Rotenone | Sigma-Aldrich | Cat#: R8875 |
| Phenformin | Cayman | Cat#: 14997 |
| Atpenin A5 | Cayman | Cat#: 11898 |
| Antimycin A | Cayman | Cat#: 19433 |
| Sodium azide | Sigma-Aldrich | Cat#: S8032 |
| Oligomycin A | Cayman | Cat#: 11342 |
| IACS-010759 | Cayman | Cat#: 25867 |
| 2-Deoxyglucose | Sigma-Aldrich | Cat#: D8375 |
| β-Nicotinamide mononucleotide | MedChemExpress | Cat#: HY-F0004 |
| Sodium oxamate | Cayman | Cat#: 19057 |
| AZD7545 | Cayman | Cat#: 19282 |
| Phenylacetaldehyde | Sigma | Cat#: 107395 |
| 4-Hydroxyphenylacetaldehyde | This Study | N/A |
| Critical Commercial Assays | Cat#: UN3334 | |
| Seahorse XFe96 FluxPac | Agilent | Cat# 102601–100 |
| NAD/NADH-Glo™ Assay kit | Promega | Cat#: 103344–100 |
| Deposited data | ||
| RNA-seq data | This study | GEO: GSE221194 |
| Proteomics data | This study | PRIDE: PXD039027 |
| Recombinant DNA | ||
| pLenti:SoNar-NES | This study | |
| pCW57.1: FLAG-METAP2 | This study | N/A |
| pCW57.1: FLAG-LbNOX | This study | N/A |
| pCW57.1: FLAG-NDI-1 | This study | N/A |
| pCW57.1: FLAG-PFKP | This study | N/A |
| pCW57.1:FLAG-KEAP1-PAM mutant | This study | N/A |
| pLENTI-CRISPRv2:sgCTRL | This study | N/A |
| pLENTI-CRISPRv2:sgNRF2_9 | This Study | N/A |
| pLENTI-CRISPRv2:sgNRF2_10 | This Study | N/A |
| pLENTI-CRISPRv2:sgKEAP1_9 | This Study | N/A |
| pLENTI-CRISPRv2:sgKEAP1_10 | This Study | N/A |
| pLENTI-CRISPRv2:sgALDH3A1_5 | This Study | N/A |
| pLENTI-CRISPRv2:sgALDH3A1_7 | This Study | N/A |
| pLKO-Tet-On: shGFP | 111 | |
| pLKO-Tet-On: shKEAP1_1 | This Study | N/A |
| Human CRISPR Metabolic Gene Knockout Library | 28 | 110066 |
| Human Activity-Optimized CRISPR Knockout Library | 97 | 1000000100 |
| Software and Algorithms | ||
| Prism (v7.0e) | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| FlowJo (v10.0.7) | Treestar Inc. | https://www.flowjo.com/ |
| ImageJ | NIH | https://imagej.net/software/fiji/ |
| R version (4.0.3) | The R Project for Statistical Computing | https://www.r-project.org |