

Abstract
Introduction:
High-grade gliomas (HGG) are the most common malignant primary brain tumors in adults, with a median survival of ~18 months. The standard of care (SOC) is maximal safe surgical resection, and radiation therapy with concurrent and adjuvant temozolomide. This protocol remains unchanged since 2005, even though HGG median survival has marginally improved.
Areas covered:
Gene therapy was developed as a promising approach to treat HGG. Here we review completed and ongoing clinical trials employing viral and non-viral vectors for adult and pediatric HGG, as well as the key supporting preclinical data.
Expert opinion:
These therapies have proven safe, and pre- and post-treatment tissue analyses demonstrated tumor cell lysis, increased immune cell infiltration, and increased systemic immune function. Although viral therapy in clinical trials has not yet significantly extended survival of HGG, promising strategies are being tested. Oncolytic HSV vectors have shown promising results both for adult and pediatric HGG. A recently published study demonstrated that HG47Δ improved survival in recurrent HGG. Likewise, PVSRIPO has shown survival improvement compared to historical controls. It is likely that further analysis of these trials will stimulate the development of new administration protocols, and new therapeutic combinations which will improve HGG prognosis.
Keywords: Gene therapy, Viral vectors, Glioblastoma, High-grade glioma
1. Introduction
In 2022, it is estimated that High-grade glioma (HGG) will lead to 18,000 deaths in the United States [1]. Decades of ongoing preclinical and clinical research have led to an improved understanding of the biological mechanism of HGG and some improvements in clinical outcomes [2–5]. Presently, patients with newly diagnosed HGG will live, on average, between one and two years after being diagnosed. The current clinical standard of care (SOC) for patients with newly diagnosed HGG involves maximal safe surgical resection and radiation therapy (RT) with concurrent temozolomide (TMZ) followed by adjuvant TMZ. This regimen resulted from a randomized-controlled Phase 3 study conducted by Stupp and colleagues, comparing RT combined with concomitant and adjuvant TMZ therapy to RT alone in patients with newly diagnosed HGG, published in 2005 [6]. The experimental arm in this study resulted in a median overall survival (OS) of 14.6 months in the TMZ plus RT arm vs. 12.1 months in the RT alone arm. In follow-up of this patient cohort, the 5-year OS of the experimental group was 9.8% vs. 1.9% in the control group [7].
While certain tumors – i.e., harboring methylguanine methyltransferase (MGMT) promoter methylation – may benefit from alkylating chemotherapy (TMZ) [8], RT has anti-tumor activity regardless of molecular subtype and has been shown to improve progression-free survival (PFS) as well as OS among newly diagnosed patients.
The critical role of RT in the treatment of HGG was stregthed over the past several decades, with numerous studies showing that radiation (if delivered in a “sufficiently high” dose) is responsible for improving multiple disease-related endpoints. Historically, to improve treatment HGG, early clinical trials demonstrated that (beyond surgery) RT was more beneficial than agents such as BCNU (carmustine), CCNU (lomustine), and bleomycin [9]. Compared to supportive care, RT was also noted to be superior in improving outcomes (BTSG 69-01)[9]. Several decades later, this was again demonstrated by a more modern French trial (which concluded in 2005) examining elderly patients; the study was terminated early when it was determined that patients not undergoing RT were significantly more likely to die from their disease sooner [10]. Importantly, clinical trials also showed that a cumulative dose of up to 60 Gy was associated with the best overall outcomes and large fields such as whole-brain radiotherapy (WBRT) were unnecessary [11]. Since then, dose escalation attempts have shown no benefit with doses higher than 60 Gy, such as in clinical trials like RTOG 7401, RTOG 9006, and RTOG 9305 (which utilized stereotactic radiosurgery as a “boost”) [12–14]. For most patients, modern fractionated radiotherapy is undertaken to utilize “standard fractionation” (i.e. ≤ 2Gy/daily fraction of treatment) to a cumulative dose of 60 Gy delivered in 6 weeks. For patients who are older or with poor performance status, there are also clinical trial data that support the use of “hypofractionated” approaches (i.e., abbreviated courses with up to 3.5Gy/day) finishing in ≤ 3 weeks [15–18].
Modern treatment planning utilizes intensity-modulated highly-conformal RT which delivers radiation to the tumor/cavity plus surrounding areas at risk of microscopic involvement. After three-dimensional definition, software-assisted expansion (typically of 2 cm) is used to define the target/treatment volume [19]. Depending on the approach utilized, this may or may not incorporate expansions beyond any non-enhancing abnormality seen on MRI T2/FLAIR sequences; if the latter is incorporated into the treatment approach, a lower dose of 46–50 Gy may be utilized to comprehensively cover areas of non-enhancing disease. After target volume definition, sophisticated computer-assisted optimization helps enhance treatment metrics to minimize irradiation of normal critical structures.
Subsequently, the addition of tumor-treating fields (TTFields), a low-intensity alternating electric field delivered via transducers on the scalp, to maintenance TMZ in the up-front treatment setting led to an increase in median OS increase from 16.0 months to 20.9 months and PFS from 4.0 months to 6.7 months when compared to maintenance TMZ alone [20]. Though some barriers remain in the use of TTFields in large numbers of patients with HGG globally, including access to the device, device cost, and inconvenience of use, it has been suggested that the use of TTFields treatment be discussed with all newly diagnosed patients with HGG [21]. Additionally, meta-analysis of several clinical trials revealed that the addition of bevacizumab to RT led to improved PFS, but similar improvement was not seen in the median OS or OS at 6 months (OS6) from diagnosis [6,22].
Numerous recent Phase III studies have been conducted with cytotoxic chemotherapy, targeted therapy, and immunotherapy. However, these studies have failed to meet their primary efficacy endpoints and recommended up-front treatment for patients with HGG remains unchanged [23–27]. Despite advances in surgical, imaging, and therapeutic techniques, the median survival for patients with HGG has improved marginally at best during the past several decades, highlighting the need to expand our clinical/translational research efforts to improve outcomes.
Pediatric diffuse HGG (pHGG) is a particularly devastating childhood cancer, accounting for 10–25% of all pediatric central nervous system (CNS) tumors, with few patients surviving more than two years after diagnosis [28,29]. Although clinical presentation and tumor histology of HGG and pHGG are often very similar, these two entities are distinct in the genetic and epigenetic alterations found in these two tumor types [30–32].
The most recent updates to the WHO Classification of Tumors of the CNS (5th edition, 2021) have led to significant changes in how pediatric CNS tumors are classified, shifting from the use of histologic features to a predominantly molecularly based classification system [33]. According to this updated classifier, pediatric-type HGGs are divided into four subtypes: diffuse midline glioma (DMG), H3K27 altered; diffuse hemispheric glioma, H3G34 mutant; diffuse pHGG, H3 wildtype and IDH wildtype; and infant-type hemispheric glioma [33]. Each subtype varies in its frequently concurrent genetic alterations and overall prognosis while DMG remains incurable, infant-type hemispheric gliomas display a less aggressive clinical behavior and has frequent mutations in receptor tyrosine kinases that might become targetable with tyrosine kinase inhibitors [34,35].
Treatment approaches that have become SOC in adult HGG patients have not led to equivalent improvements for pediatric patients, despite several parallel treatment regimens studied in children and adolescents [32,36–38]. There remains some debate about the SOC treatment of pHGG, in part because it is now understood that pHGG includes several distinct subtypes and, therefore, previous clinical trials included very heterogenous patient populations. Currently, non-DMG pHGG is treated with focal RT combined with concomitant and adjuvant cytotoxic chemotherapy (TMZ +/− lomustine) or adjuvant targeted therapy based on tumor-specific mutations. A distinct entity of pHGG, unique to the pediatric and adolescent population, is DMG, which occurs in midline structures, most commonly the pons and thalamus [39]. Traditional cytotoxic chemotherapy has been studied in several variations in patients with DMG with no improvement in outcome when combined with focal RT compared to focal RT alone, and therefore focal RT remains the standard clinical approach for patients with DMG [40–43].
Due to treatment resistance and infiltrative nature of HGG, gene therapy arises as a promising approach. Gene therapy employs genetic material to transduce in tumor cells to fight disease. A variety of viral vectors have been proposed for HGG and are currently ongoing Phase I and II clinical trials studying safety and efficacy in both adult and pediatric HGG. This review covers clinical trials using gene therapy for the treatment of HGG. We used ClinicalTrial.gov (accession date: October 2022) database to create a summary table containing the vector used, ID number, phase, status, and age of the patients (Table 1). We have also included key supporting pre-clinical data for these studies and the published clinical trial studies results.
Table I.
Clinical trials employing gene therapy for HGG.
| Type of Vector | ID Number | Therapy | Phases | Status | Age |
|---|---|---|---|---|---|
| AdV | NCT00870181 | AdV-TK+GCV | 2 | Completed | Adult, Older Adult |
| AdV | NCT00589875 | AdV-TK + Valacyclovir | 2 | Completed | Adult, Older Adult |
| AdV | NCT00634231 | AdV-TK + Valacyclovir | 1 | Completed | Child, Adult |
| AdV | NCT00751270 | AdV-TK + Valacyclovir | 1 | Completed | Adult, Older Adult |
| AdV | NCT03596086 | AdV-TK + Valacyclovir | 1|2 | Recruiting | Adult, Older Adult |
| AdV | NCT03603405 | AdV-TK + Valacyclovir | 1|2 | Recruiting | Adult, Older Adult |
| AdV | NCT01811992 | Ad-hCMV-TK + Ad-hCMV-Flt3L | 1 | Completed | Adult, Older Adult |
| AdV | NCT03576612 | AdV-TK + Valacyclovir + Nivolumab | 1 | Active, not recruiting | Adult, Older Adult |
| AdV | NCT00004041 | Ad5CMV-p53 | 1 | Completed | Adult, Older Adult |
| AdV | NCT00004080 | SCH-58500 | 1 | No Results Available | Adult, Older Adult |
| AdV | NCT02026271 | Ad-RTS-hIL-12 + Veledimex | 1 | Completed | Adult, Older Adult |
| AdV | NCT03679754 | Ad-RTS-hIL-12 + Veledimex | 1 | Completed | Adult, Older Adult |
| AdV | NCT03636477 | Ad-RTS-hIL-12 + Veledimex + Nivolumab | 1 | Completed | Adult, Older Adult |
| AdV | NCT04006119 | Ad-RTS-hIL-12 + Veledimex + Cemiplimab-Rwlc | 2 | Completed | Adult, Older Adult |
| AdV | NCT03330197 | Ad-RTS-hIL-12 + Veledimex | 1|2 | Terminated | Child, Adult |
| AdV | NCT00031083 | BG00001 | 1 | Completed | Adult, Older Adult |
| AdV | NCT01260506 | VB-111 + Bevacizumab | 1|2 | Completed | Adult, Older Adult |
| AdV | NCT02511405 | VB-111 + Bevacizumab | 3 | Completed | Adult, Older Adult |
| AdV | NCT04406272 | VB-111 + Bevacizumab | 2 | Recruiting | Adult, Older Adult |
| Oncolytic AdV | NCT00805376 | DNX-2401 | 1 | Completed | Adult, Older Adult |
| Oncolytic AdV | NCT01582516 | delta-24-RGD adenovirus | 1|2 | Completed | Adult, Older Adult |
| Oncolytic AdV | NCT02798406 | DNX-2401 + Pembrolizumab | 2 | Completed | Adult, Older Adult |
| Oncolytic AdV | NCT01956734 | DNX2401 | 1 | Completed | Adult, Older Adult |
| Oncolytic AdV | NCT03714334 | DNX-2440 | 1 | Recruiting | Adult, Older Adult |
| Oncolytic AdV | NCT03178032 | DNX-2401 | 1 | Unknown status | Child, Adult |
| Oncolytic AdV | NCT02197169 | DNX-2401 + Interferon-gamma | 1 | Completed | Adult, Older Adult |
| Oncolytic HSV-1 | NCT00157703 | G207 | 1 | Completed | Adult, Older Adult |
| Oncolytic HSV-1 | NCT00028158 | G207 | 1|2 | Completed | Adult, Older Adult |
| Oncolytic HSV-1 | NCT02457845 | G207 | 1 | Active, not recruiting | Child, Adult |
| Oncolytic HSV-1 | NCT04482933 | G207 | 2 | Not yet recruiting | Child, Adult |
| Oncolytic HSV-1 | NCT03911388 | G207 | 1 | Recruiting | Child, Adult |
| Oncolytic HSV-1 | NCT02062827 | M032 (NSC 733972) | 1 | Active, not recruiting | Adult, Older Adult |
| Oncolytic HSV-1 | NCT05084430 | M032 + Pembrolizumab | 1|2 | Active, not recruiting | Adult, Older Adult |
| Oncolytic HSV-1 | NCT02031965 | HSV-1716 + Dexamethasone | 1 | Terminated | Child, Adult |
| Oncolytic HSV-1 | NCT05095441 | C5252 | 1 | Recruiting | Adult, Older Adult |
| Oncolytic HSV-1 | NCT03657576 | C134 | 1 | Active, not recruiting | Adult, Older Adult |
| Oncolytic HSV-1 | UMIN000002661 | G47delta | 1|2 | Completed | Adult, Older Adult |
| Oncolytic HSV-1 | UMIN000015995 | G47delta | 2 | Completed | Adult, Older Adult |
| Oncolytic HSV-1 | NCT03152318 | rQNestin + Cyclophosphamide | 1 | Active, not recruiting | Adult, Older Adult |
| Retrovirus | NCT00001328 | G1TKSVNa.53 Producer Cell Line + Cytovene | 1 | Completed | Adult, Older Adult |
| Retrovirus | NCT04105374 | Toca 511 vector + Toca FC | 2|3 | Withdrawn | Adult, Older Adult |
| Retrovirus | NCT01156584 | Toca 511 vector + Toca FC | 1 | Completed | Adult, Older Adult |
| Retrovirus | NCT01985256 | Toca 511 vector + Toca FC | 1 | Completed | Adult, Older Adult |
| Retrovirus | NCT02598011 | Toca 511 vector + Toca FC | 1 | Withdrawn | Adult, Older Adult |
| Retrovirus | NCT02414165 | Toca 511 vector + Toca FC + Lomustine + Bevacizumab | 2|3 | Terminated | Adult, Older Adult |
| Retrovirus | NCT04327011 | Toca 511 vector + Toca FC | 1 | Terminated | Adult, Older Adult |
| Oncolytic NDV | NCT01174537 | New Castle Disease Virus | 1|2 | Withdrawn | Child, Adult, Older Adult |
| Oncolytic MV | NCT00390299 | Carcinoembryonic Antigen-Expressing Measles Virus | 1 | Completed | Adult, Older Adult |
| Oncolytic MV | NCT02962167 | Modified Measles Virus | 1 | Recruiting | Child, Adult |
| Oncolytic Reovirus | NCT00528684 | REOLYSIN | 1 | Completed | Adult, Older Adult |
| Oncolytic Reovirus | NCT02444546 | Wild-type Reovirus + Sargramostim | 1 | Active, not recruiting | Child, Adult |
| Oncolytic PVSRIPO | NCT01491893 | PVSRIPO | 1 | Completed | Adult, Older Adult |
| Oncolytic PVSRIPO | NCT02986178 | PVSRIPO | 2 | Active, not recruiting | Adult, Older Adult |
| Oncolytic PVSRIPO | NCT03973879 | PVSRIPO + Atezolizumab | 1|2 | Withdrawn (Re-submission Planned) | Adult, Older Adult |
| Oncolytic PVSRIPO | NCT03043391 | PVSRIPO | 1 | Active, not recruiting | Child, Adult |
| Oncolytic PVSRIPO | NCT04479241 | Lerapolturev + Pembrolizumab | 2 | Active, not recruiting | Adult, Older Adult |
| Oncolytic Parvovirus | NCT01301430 | H-1PV | 1|2 | Completed | Adult, Older Adult |
| Oncolytic Vaccinia | NCT03294486 | Combination of TG6002 and 5-FC | 1|2 | Unknown status | Adult, Older Adult |
| Liposome | NCT00470613 | SGT-53 | 1 | Completed | Adult, Older Adult |
| Liposome | NCT02340156 | SGT-53 | 2 | Terminated | Adult, Older Adult |
| Liposome | NCT03554707 | SGT-53 | Early 1 | Not yet recruiting | Child, Adult |
| Nanoparticle | NCT03020017 | NU-0129 | Early 1 | Completed | Adult, Older Adult |
| NSC | NCT01172964 | E. coli CD-expressing genetically modified NSC + Flucytosine | 1 | Completed | Child, Adult, Older Adult |
| NSC | NCT02015819 | E. coli CD-expressing genetically modified NSC + Flucytosine + Leucovorin | 1 | Completed | Adult, Older Adult |
| NSC | NCT02192359 | Carboxylesterase-expressing Allogeneic NSC + Irinotecan | 1 | Completed | Adult, Older Adult |
| NSC + AdV | NCT03072134 | NSC-CRAd-Survivin-pk7 | 1 | Completed | Adult, Older Adult |
| NSC + AdV | NCT05139056 | NSC-CRAd-Survivin-pk7 | 1 | Not yet recruiting | Adult, Older Adult |
| NSC + AdV | NCT04758533 | AloCELYVIR | 1|2 | Recruiting | Child, Adult |
| MSC + Oncolytic AdV | NCT03896568 | Allogenic MSC loaded with Ad5-DNX-2401 | 1 | Recruiting | Adult, Older Adult |
| HSC + Lentivirus | NCT01269424 | Autologous HSC modified with retroviral vector encoding for MGMTP140K + O6-benzylguanine | 1 | Active, not recruiting | Adult, Older Adult |
AdV: adenovirus vector, TK: thymidine kinase, GCV: ganciclovir, HSV: herpes simplex virus, NDV: Newcastle disease virus, MV: Measles virus, NSC: Neural Stem Cells, MSC: Mesenchymal Stem Cells, HSC: Hematopoietic Stem Cells, CD: cytosine deaminase, 5-FC: 5-flucytosine.
2. Gene therapy vectors
Unprotected nucleic acids (RNA or DNA) are not stable for long periods within a biological environment. Furthermore, RNA needs to reach the cell cytoplasm, to perform its function, and DNA must enter the cell nucleus to be transcribed and edit the cell’s genome. Therefore, the major challenge in gene therapy is the delivery of effectors to cells. There is a wide variety of delivery methods, but they can broadly be divided into two groups: Viral and non-viral vectors.
2.1. Viral Vectors
Viral vectors employ a wide range modified viral species (Fig. 1 highlights the viral vectors employed in clinical trials for HGG). Generally, they employ strong promoters to induce high-level gene expression. The nucleic acid content of each virus greatly influences its gene expression signature. Single-stranded RNA (ssRNA) genomes exhibit rapid initiation of transgene expression, but they are transient. DNA-based vectors are also transient, but typically generate long-term expression based on their ability to integrate into the host genome. Likewise, retroviruses, carrying RNA genomes, can establish long-term expression through chromosomal integration. In addition, viral vectors can have oncolytic features, and therefore trigger immunogenic cell death (ICD) (Fig. 2). Oncolytic viruses are weakly pathogenic that have been genetically modified, or naturally occurring, to replicate in host cancer cells, without harming normal cells, resulting in enhanced anti-cancer effects.
Figure 1.
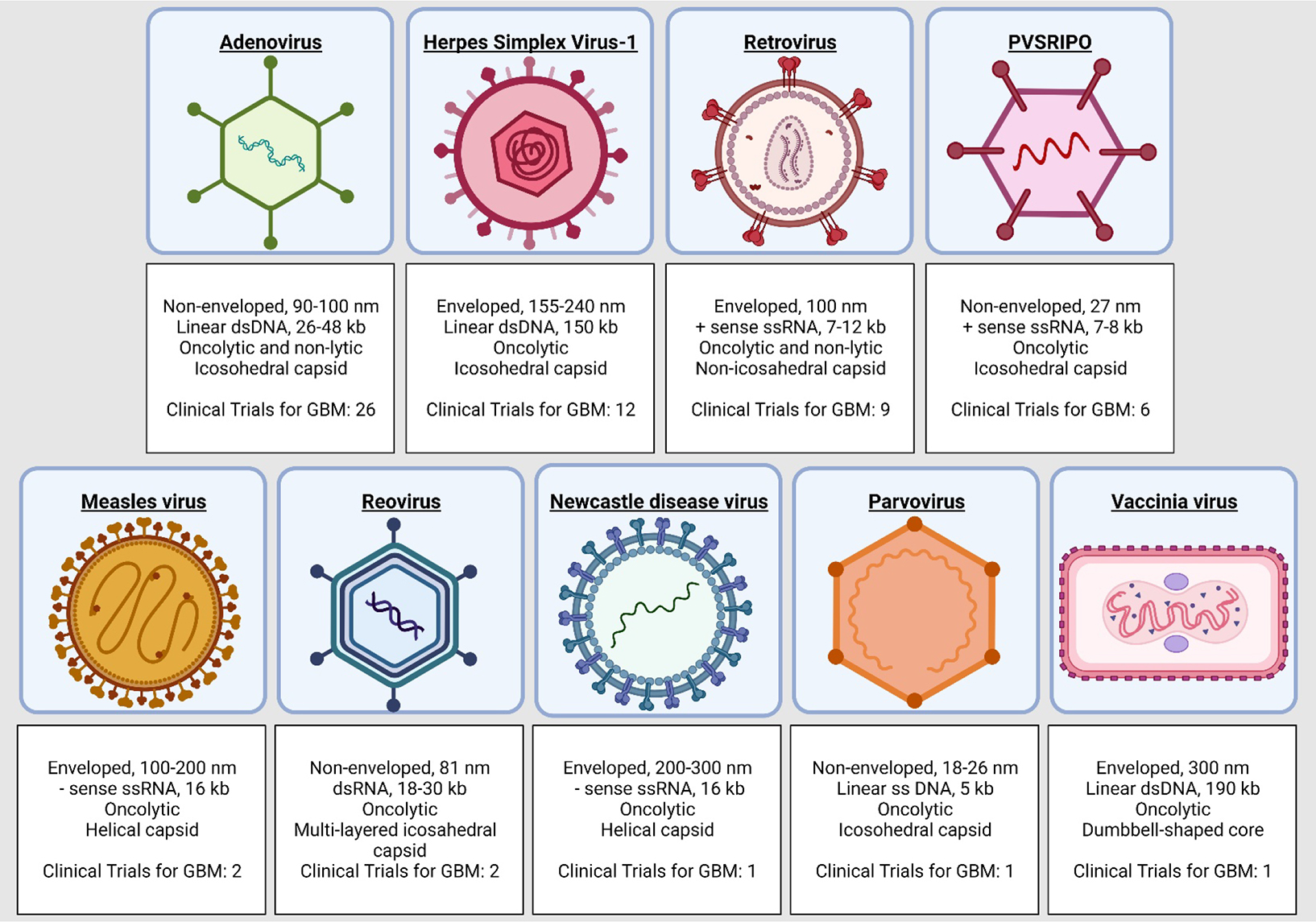
Viral vectors being tested in clinical trials for HGG.
Figure 2.
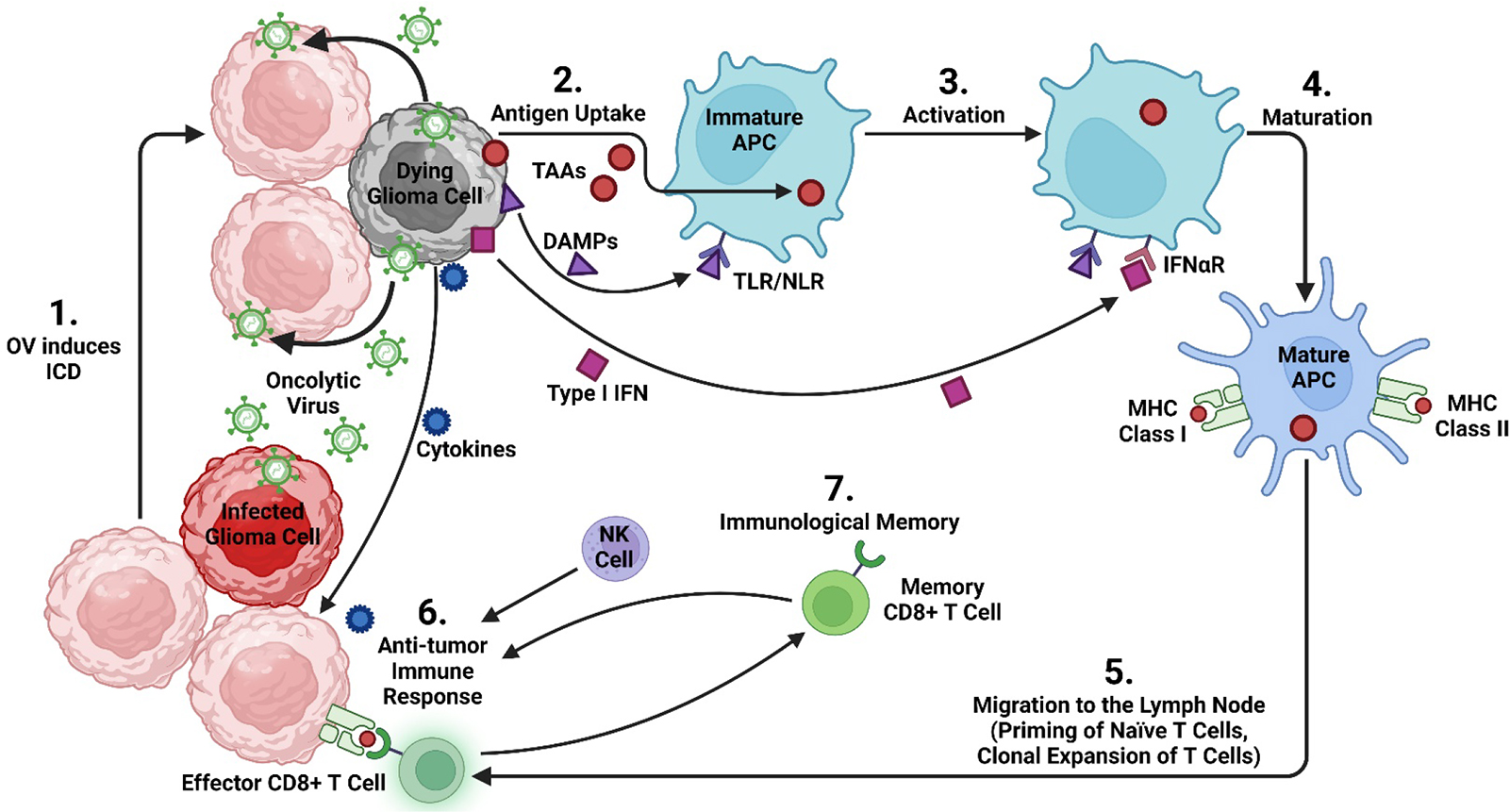
(1) Oncolytic viruses (OV) infect glioma cells, initiating the process of immunogenic cell death (ICD). Dying glioma cells release damage associated molecular patterns (DAMP) and type I interferons, molecules that stimulate the host’s immune response. Following the virus-mediated lysis of the cell, oncolytic virus is released into the surrounding tissue and can infect adjacent glioma cells. (2) Tumor-associated antigens (TAA) recruit immature antigen-presenting cells (APC) to the tumor. (3) APC become activated following antigen-uptake and begin interacting with DAMP via pattern recognition receptors (toll-like receptors (TLR) and nod-like receptors (NLR)). (4) Following this interaction, immature antigen-capturing APC become mature APC, which are able to form and transport peptide-loaded MHC complexes to the cell surface. (5) Mature APC migrate to a regional lymph node where they prime naïve T cells. This is followed by clonal expansion and then release of cytotoxic CD8+ T lymphocytes (CTL). (6) These tumor specific CTL are attracted to the tumor by cytokines released from dying glioma cells. Natural killer (NK) cells, part of the innate immune system, are attracted to the tumor by chemokine. Altogether, this generates an anti-tumor immune response. (7) Continuous exposure of TAA to CTL promotes immunological memory, generating memory CD8+ T cells.
2.1.1. Adenovirus vectors
Adenoviruses (AdV) are non-enveloped double-stranded DNA viruses. Their genome is a linear molecule of 26–45 kb with an icosahedral protein capsid. They have the ability to remain stable to physical and chemical changes [44]. AdV are very effective vectors for gene delivery, due to their easy genetic manipulation, effortless transfection, and high transduction efficiency. They can deliver transgenes of ~8 kb, and up to 30 kb in the newer generation helper-dependent vectors. However, their main disadvantage is that they induce immune reactivity [45].
AdV have genes controlling two stages of the virus life cycle: Early and Late. Early genes are involved in entering the host cell, delivering the genome to the nucleus, and replicating the viral DNA (regions E1 to E4). Late genes orchestrate transcription/translation of the structural parts of the virus (regions L1 to L5). First generation AdV vectors have a deletion of the E1a and E1b genes and can have an insertion cassette of up to 3–4 kb of foreign DNA, or deletion of E1 and E3, with the ability to hold 5–6 kb of DNA. However, because of the induced immunogenicity against transduced cells, second generation AdV were created with the deletion of E1 and E2 or E1 and E4 genes. Second generation of AdV are more complex to produce but have a decreased hepatotoxicity and a greater stability in vivo. Nonetheless, they still induce immunological challenges [44]. The last generation of AdV are called gutless or helper virus-dependent, where the entire viral coding sequences are removed, therefore reducing immunogenicity and enhancing safety. These vectors have the end sequences of the genome, the ITRs sequence required for DNA replication and the packaging signal (Ψ) of the virus [44–46].
One of the main strategies involving AdV is suicide gene therapy. This method induces the expression of an enzyme that can convert a nontoxic prodrug to an active toxic compound, therefore causing the death of transduced cells. This approach is lethal for replicating cells, which allows targeting brain tumor cells, without harming non-dividing brain cells. AdV inducing the expression of herpes simplex virus-thymidine kinase (HSV-TK) is accompanied by a pro-drug, ganciclovir (GCV), that can be activated by this enzyme. HSV-TK phosphorylates GCV, and monophosphorylated GCV gets further phosphorylated into triphosphate-GCV (3P-GCV). When cells initiate replication, 3P-GCV is incorporated in de novo DNA strands, which causes DNA replication to be halted, followed by apoptosis.
The viral therapy using AdV-TK combined with GCV was studied in a randomized Phase II clinical trial (NCT00870181) for patients with recurrent grade III and IV malignant gliomas. The study was deemed to be safe and displayed improvement of PFS at 6 months, PFS and OS in patients treated with AdV-TK compared with SOC treatment [47]. To further study this therapy, a prospective Phase II clinical trial (NCT00589875) for newly diagnosed malignant gliomas studied gene-mediated cytotoxic immunotherapy (GMCI). The patients received AdV-TK along with valacyclovir (valine ester of GCV, that has improved bioavailability in the brain), combined with standard radiation and chemotherapy. Reports showed improvement in patients with greater tumor resection, with a median OS of 25 months of the combined therapy versus 16.9 months for SOC alone and 3-year survival of 32% vs 6%, respectively [48]. Moreover, a Phase I clinical trial for pediatric brain tumors (NCT00634231) combining AdV-TK intratumoral injection and GMCI along with SOC, proved to be safe and a survival of more than 24 months was obtained by 3 of the 8 patients, including 2 patients with no tumor progression at 37.3 and 47.7 months after therapy [49].
There are several completed (NCT00751270) or ongoing clinical trials (NCT03596086 and NCT03603405) for newly diagnosed or recurrent HGG using AdV-HSV1-TK/valacyclovir. Also, there are trials exploring the combination of AdV-HSV1-TK/valacyclovir with AdV-Flt3L (NCT01811992), or with immune check point inhibitors drug (Nivolumab) (NCT03576612). However, further studies are needed using AdV in combination with other therapies to assess the actual efficacy of these approaches.
Another approach of viral therapy is AdV-p53, which employs a replication deficient Ad to restore wild type p53 gene function. This is the case of the first gene therapy product approved by a government agency, Gendicine. It was approved in 2003 by the China Food and Drug Administration (CFDA), to treat head and neck cancer. There have been two completed clinical trials of AdV-p53 for glioma in the United States (NCT00004041 and NCT00004080). However, the only one that published its results shows that at the dose and schedule employed, transduced cells were found only close to the injection site [50].
Non-replicative AdV therapies have also been used as immune boosting therapies to enhace the antitumor immune response. A new strategy has been developed using an AdV expressing interleukin-12 (IL-12) under the control of the RheoSwitch System® (Ad-RTS-hiL-12) combined with the activator Veledimexin (VDX) [51]. This approach has been tested in a Phase I clinical trial (NCT02026271) for recurrent HGG and proved to be safe and showed promising preliminary results, with increased lymphocyte tumor infiltration, possibly a response to IL-12 [52]. Nonetheless, corticosteroids reduced the OS. This resulted in a Phase I sub-study (NCT03679754) to analyze IL-12 as a monotherapy in patients with GBM. This viral strategy was also employed in Phase I dose escalation clinical trial (NCT03636477), where Ad-RTS-hIL-12/VDX was administered in combination with Nivolumab for recurrent glioblastomas. The results showed a safe outcome of the combination treatment and is now headed to a Phase II clinical trial using Ad-RTS-hIL-12 plus Veledimex in combination with Cemiplimab-rwlc for patients with recurrent glioblastoma (NCT04006119) [53]. In the pediatric field, a Phase-I clinical trial (NCT03330197) explored the effect of Ad-RTS-hIL-12/VDX therapy for pediatric brain tumors but it was terminated in 2021 due to slow accrual.
Also, AdV have been used to induce the expression of interferon beta (IFN-β). A Phase I clinical trial for recurrent glioma administered a pretreatment of a stereotactic injection of Ad-IFN-β, followed 4–8 days later by surgical removal of tumor and additional injections of Ad.hIFN-β into the tumor bed (NCT00031083). Resected tumor exhibited necrosis, and more infiltration of neutrophils, macrophages, and monocytes compared with pretreated tumors. One subject developed confusion and presented swelling and edema and died 83 days post initial injection. Postmortem examination showed hemorrhagic necrosis and inflammation involving the leptomeninges, consistent with direct contact with cerebrospinal fluid (CSF) containing virus [54].
Another approach employing Ad vectors is VB-111. This non-replicating AdV carries a proapoptotic Fas-chimera transgene (Fas and human TNF receptor 1) under the control of a modified murine pre-proendothelin promoter (PPE-1-3x) [55]. VB-111 targets and damages blood vessels. A Phase I/II study for recurrent GBM evaluated the safety, tolerability, and efficacy of single and multiple doses of intravenous infusion of VB-111 with and without bevacizumab (anti- Vascular endothelial growth factor A) (NCT01260506). Patients treated with VB-111 monotherapy that continued to be treated with bevacizumab after progression showed significant survival and PFS advantage [56]. This was followed by a Phase III clinical trial, where patients were randomized 1:1 to receive VB-111 in combination with bevacizumab or bevacizumab monotherapy. In this study, upfront concomitant administration of VB-111 and bevacizumab failed to improve outcomes in recurrent GBM. (NCT02511405)[57]. Currently VB-111 is being analyzed under a different administration protocol in a Phase II clinical trial (NCT04406272) where a group of patients receive VB-111 before and after the surgery, while the other group only receives it after surgery.
Oncolytic adenovirus therapy is another approach investigated for the treatment of glioblastomas. This therapy is based on the engineered modification of AdV to conditionally replicate in tumor cells and induce cell lysis, exposing cancer cell antigens and finally stimulating the immune response [58,59]. The oncolytic adenovirus DNX-2401 (Tasadenoturev) was generated to target replicating tumor cells with retinoblastoma pathway mutations [60]. Several clinical trials have been recently completed or are ongoing with this agent. The first clinical trial administering DNX-2401 was a Phase I, dose-escalation study for patients with recurrent HGG (NCT00805376). It was reported that 20% of patients survived for >3 years, and 3/37 patients presented >95% tumor reduction. DNX-2401 treatment generated an antitumor response, possibly due to its oncolytic effects [61].
Similarly, another Phase I/II clinical trial using DNX-2401, intratumoral administration directly into the tumor mass, has been performed in the Netherlands (NCT01582516). The cerebrospinal fluid (CSF) analysis from these patients showed that DNX-2401 treatment increased cytokines levels such as interleukin 6 (IL-6), tumor necrosis factor alpha (TNF-α), and Interferon gamma (IFN-γ). Follow up experiments performed in vitro evaluated the effects of patients’ CSF on macrophages and showed an increment of CD64 (M1 pro-inflammatory polarization)[62]. Another strategy is the combination of DNX-2401 with immune therapies. Ongoing Phase II clinical trial (CAPTIVE, NCT02798406) is studying the effect of Pembrolizumab, an anti-PD-1 antibody, treatment in combination with the oncolytic DNX-2401. In addition, a Phase I trial combining oncolytic DNX-2401 therapy and TMZ is ongoing for recurrent glioblastoma (NCT01956734). Further, a first Phase I trial is testing a third generation of DNX-2440 expressing the co-stimulatory molecule OX40L, for recurrent glioblastoma (NCT03714334). For pediatric brain tumors (DIPG), a Phase I clinical trial evaluating DNX-2401 treatment (NCT03178032) is currently ongoing. Another combination studied was intratumoral administration DNX-2401 alone versus DNX-2401 with interferon gamma (IFN). This Phase Ib study results showed the addition of IFN did not improve survival [63].
2.1.2. HSV vectors
Herpes virus simplex 1 (HSV-1) are human infectious virus from the Herpesviridae family. They are enveloped virus with a 152 kb viral genome of double-stranded DNA. The genome of HSV contains 74 known ORFs and two unique regions called long region (UL) containing 56 viral genes and a short region (US) containing 12 genes. HSV are highly infectious and can infect several cell types; many genes are not necessary for growth in vitro and can be removed to create space to insert exogenous transgenes. HSV-1 can be grown with minimal requirements for replication and packaging (Patel, 2011). HSV-1 has neurotropic properties that help for targeting neural pathologies. Furthermore, the deletion of the γ34.5 gene reduces the neurovirulence and neuropathogenicity. HSV-1 induces tumor cell death, and also initiates and improves the antitumor immune response [64,65].
The first oncolytic virus engineered to treat brain tumors was an HSVvirus [66]. HSV-G207 is a vector carrying the deletion of two copies of γ34.5 and the insertion of E. Coli ß-galactosidase gene (LacZ) in the UL39 gene, thus inhibiting replication of HSV-1 in normal cells. Also, this vector expresses the thymidine-kinase (TK) gene, whose cytotoxicity can be activated with systemic GCV. This therapy is based on the ability to induce glioma cell death by oncolysis, while also enhancing the tumor infiltration of cytotoxic immune effectors (Fig. 2). There have been two clinical trials, Phase I and II, (NCT00157703 and NCT00028158), employing a single dose of HSV-G207 for glioma treatment. Results proved the treatment to be safe and indicated a potential treatment response [67–69]. For pediatric brain tumors, the use of the oncolytic HSV-G207 is currently ongoing. A Phase I clinical trial (NCT02457845) administrating HSV-G207 with radiation showed no toxic effects, clinical, radiographic, and pathological responses in 11 of 12 patients showed significant increased tumor infiltrating lymphocyte [70]. There are two ongoing studies aiming to describe the effect of this vector: a Phase II clinical trial administering HSV-G207 with a single radiation dose in pediatric recurrent HGG (NCT04482933); and a Phase I clinical trial in recurrent cerebellar brain tumors (NCT03911388).
Further evolution of HSV-207 includes a modification (M032-HSV-1) by inserting the proinflammatory cytokine IL-12 to increase the activation of the adaptive immune response[71]. A Phase I clinical trial (NCT02062827) using this approach for the treatment of recurrent HGGs is currently in progress, as well as a Phase I/II in combination with Pembrolizumab (NCT05084430). Several ongoing trials are testing HSV variants in patients with GBM, such as HSV-1716 (NCT02031965), MVR-C252 (NCT05095441), C134 (NCT03657576) have been conducted or are ongoing in patients with GBM.
Another oncolytic HSV-1 is G47Δ. This is a triple mutated third generation HSV-1, generated by deleting the α47 gene and overlapping US11 promoter from parental second generation G207, that carried deletions in both copies of the γ34.5 gene and an inactivation of the ICP6 gene [72]. G47Δ was proved to be safe when administered to patients with GBM in Japan (UMIN-CTR: UMIN000002661). Follow up, a Phase II clinical trial was (UMIN-CTR: UMIN000015995) for adult patients with residual or recurred HGG after initial therapy of surgery, radiation and temozolomide. The protocol of administration was intratumoral injection of G47Δ, and repeatedly for up to six doses, employing MRI-guided stereotactic surgery at intervals of 5–14 days for the initial and second doses, and up to six doses at intervals of 4 ± 2 weeks for the subsequent doses. Results indicate that HSV G47Δ was able to improve survival, keeping a safety profile. OS was 20.2 (16.8–23.6) months after treatment initiation and 28.8 (20.1–37.5) months from the initial surgery [73]. This trial was submitted in 2014, by then glioma patients were not tested on regular basis for isocitrate dehydrogenase 1 (IDH1) mutation, therefore the status of IDH1 mutation was not included as a covariate in this study. Pos hoc analysis determined that 6 of 19 patients had IDH1 mutation. Regardless, authors did not find a statical difference considering the IDH status in the OS [73].
A different strategy employing HSV is a vector that was generated by including one deletion of the γ34.5 gene and a second copy of this gene under the control of the nestin promoter (rQNestin γ34.5v2). This construct enables viral replication in tumor cells that have a high expression of nestin. Thus, viral replication occurs in glioma tumor cells sparing normal cells [74]. There is an active Phase I clinical trial employing rQNestin γ34.5v2 (NCT03152318) for recurrent HGG. Treatment is administered intratumorally during surgery, with and without cyclophosphamide. Preliminary results showed this treatment mediated NOTCH induced infiltration of myeloid derived suppressor cells (MDSCs) and increased CCL2 and IL-10, marking suppressed anti-tumor immunity. This led the authors to propose a combination with pharmacologic blockade of NOTCH signaling to overcome MDSC recruitment and immune suppression [75].
2.1.3. Retroviral vectors
Retroviruses are single-stranded RNA oncolytic enveloped viruses. Their genome has two copies of highly condensed RNA molecules of 7–12 kb [76]. One of the most common retroviral used is the murine leukemia virus-derived replication-competent gamma-retroviruses, that allows the incorporation of up to 8 kb of foreign DNA and has de ability to transfect only replicating cells [77]. The first genetic delivery systems tested in the clinic to treat brain tumors were retroviral vectors, encoding for HSV-TK plus systemic administration of cytovene (GCV) (NCT00001328) [78–80]. The preliminary results revealed an anti-tumor activity in patients with smaller tumors and recurrent glioma [79]. A Phase III, multicenter, randomized, open-label, controlled trial for newly diagnosed HGG. However, the treatment efficiency was lower than expected, and OS did not support further studies. The failure of the protocol was attributed to a presumably poor rate of delivery of the HSV-TK gene to tumor cells. The authors proposed that in the future delivery should be improved[78].
Another approach using retrovirus was Vocimagene amiretrorepvec (Toca 511), a tumor-targeted, replication-competent gamma-retrovirus that induces the expression of cytosine deaminase (CD). The prodrug 5-flucytosine (5-FC) is metabolized by CD into 5-fluorouracil (5-FU). 5-FU interferes with thymidylate synthase and incorporation into RNA and DNA, which leads to cell death [81]. Toca 511 plus 5-FC was tested in combination with TMZ and was able to improve treatment in TMZ-sensitive glioma [82–84]. This combined treatment (Toca 511 + 5-FC) was reported to sensitize glioma to radiation therapy [85]. Several clinical trials tested this combination for the treatment of patients with HGG (NCT04105374; NCT01156584; NCT01985256; NCT02598011; NCT02414165; NCT04327011). In a Phase I trial, patients treated with Toca 511 plus Toca FC exhibited a promising response, therefore this treatment was further evaluated in a randomized Phase III clinical trial [86]. Unfortunate, treatment failed to meet expectations, since there were no significant differences against the active control group (mOS: 11.10 vs. 12.22 months) [57].
2.1.4. Newcastle disease virus
Newcastle disease virus (NDV) is a highly pathogenic avian single-strand RNA virus. NDV-based vectors have oncolytic and immunostimulatory capabilities, with a natural tropism for tumor cells [87]. Recombinant NDV vectors were generated by reverse genetics to be used as a potential cancer therapy agent [88]. The Hebrew University Jerusalem (HUJ) strain of NDV (NDV-HUJ) was tested in a Phase I/II trial for patients with recurrent HGG, in a protocol of intravenous administration (NCT01174537) [89], however this trial was withdrawn, and there are no published results.
2.1.5. Measles virus
Measles virus (MV) is an enveloped single-strand RNA oncolytic virus. MV hemagglutinin glycoprotein has a high affinity for receptors such as nectin-4 and CD46 on host cells, and its fusion protein mediates cell-cell fusion, inducing apoptosis of these cells [90,91]. CD46 is a receptor overexpressed by neoplastic cells [90]. The highly attenuated Edmonston strain (MV-Edm) genetically engineered to produce carcinoembryonic antigen (CEA) has potential therapeutic efficacy in gliomas [92,93]. A Phase I dose-escalating clinical trial of MV-CEA exhibit no dose-limiting toxicities in patients with recurrent HGG (NCT00390299) [93]. Another approach is a modification of MV-Edm genetically engineered to produce human thyroidal sodium iodide symporter (NIS). Expression of NIS allows cells to actively transport iodide ions into the cell. MV-NIS-infected cells express NIS, and can uptake radioiodine, thus providing the basis for in vivo radioiodine imaging studies to reveal the location of MV-NIS-infected cells. MV-NIS has been evaluated in a Phase I clinical trial (NCT02962167) for children and young adults with recurrent medulloblastoma or recurrent atypical teratoid rhabdoid tumors (ATRT) [58,94].
2.1.6. Reovirus vectors
Reovirus is a double-stranded RNA oncolytic virus. It is generally nonpathogenic in humans, with oncolytic properties having been observed in a broad range of tumor types, including carcinoma, myeloma, and melanoma, and the wild-type strain pelareorep (Reolysin) has received orphan drug designation by the FDA. They are able to infect and lyse tumor cells but not normal cells because Reovirus employs activated Ras signaling pathway present in glioma cells to elicit proteolytical viral disassembly [95]. A Phase I trial using single intratumoral injections of reovirus to treat recurrent malignant glioma patients showed it was well tolerated. Ten patients exhibited tumor progression, one had stabilization, and one was not evaluable for response. Median survival was 21 weeks (range, 6–234) (NCT00528684)[96]. Another active trial is employing this virus in combination with Sargramostim (recombinant granulocyte macrophage colony-stimulating factor (GM-CSF)) (NCT02444546).
2.1.7. Poliovirus vectors
Polioviruses (PV) are single-stranded RNA viruses in the Enterovirus genus and Picornaviridae family responsible for poliomyelitis. PVSRIPO is an oncolytic recombinant nonpathogenic polio–rhinovirus chimera. It nonlethally infects antigen-presenting cells, eliciting proinflammatory cytokine responses and enabling T-cell stimulation while simultaneously causing lethal cytotoxicity in neoplastic cells, illustrating that this gene therapy could be a potential counter to tumor immunosuppression as well as a method to initiate antitumor immunity [97]. PVSRIPO was initially tested in a Phase I clinical trial for the treatment of recurrent HGG (NCT01491893). This study reported no neurovirulent potential and an increased OS of PVSRIPO-treated patients in comparison with historical controls. Patients who received PVSRIPO had a 24- and 36-month survival rate was 21% (11 out of 33 patients)[97]. An active Phase II clinical trial is now trying to establish its efficacy as a monotherapy for HGG treatment (NCT02986178). Combination of PVSRIPO with Atezolizumab, an anti-PD-L1 monoclonal antibody, has been proposed in a Phase Ib/II for adult HGG (NCT03973879). Although this trial was withdrawn, it is registered that a re-submission is planned. There are two currently active PVSRIPO clinical trials, a Phase I for pediatric grade III/IV malignant gliomas (NCT03043391), and a Phase II multi-institutional clinical trial for adult HGG (NCT04479241).
2.1.8. Parvovirus
Parvoviruses are single-stranded DNA viruses in the Parvoviridae family. Parvovirus H-1(H1PV) infects rats and is nonpathogenic to humans. Preclinical data of oncolytic H1PV infection of glioma cells demonstrated strong cytotoxicity [98]. H1PV has been employed in a Phase I/II clinical trial for adults with recurrent HGG (NCT01301430). It had 2 groups, group 1 received an intratumoral injection and a follow up administration into the walls of the tumor cavity during tumor resection; In group 2 H1PV was administered intravenously and afterwards, into the surrounding brain tissue during tumor resection. In the 6-month follow-up period, 12/18 of the subjects had recurrent disease or died. H-1PV was confirmed to have crossed the blood brain barrier (BBB) and, subjects with the higher dose had an increase in antibodies [99].
2.1.9. Vaccinia virus
Vaccinia viruses (VACV) are enveloped double-stranded DNA oncolytic viruses in the Poxviridae family. They have a large genome that can carry transgenes to enhance its oncolytic ability [100].
Several solid tumors have been clinically treated with different VACV, with promising results [101]. A highly attenuated variant VACV, called TG6002, was developed by deleting two genes from Copenhagen strain VACV. TG6002 expresses fusion suicide gene (FCU1) that encodes for a bifunctional fusion protein combining cytosine deaminase (CD) and uracil phosphoribosyl transferase activity (UPRTase)[102]. FCU1 can convert 5-FC into toxic metabolites 5-FU and 5-fluoro-uridilyl monophosphate (5-FUMP), which induce cytotoxicity due to the inhibition of DNA and protein synthesis. There is currently a Phase I/II clinical trial for HGG using TG6002 plus 5-FC in combination with SOC (NCT03294486), but it has no published results yet.
2.1.10. Lentiviral vectors
Lentiviruses are positive-sense single-stranded RNA viruses [103]. Lentiviral vectors (LV) integrate into the host genome, and can replicate in non-dividing cells. [104]. In comparison to gamma-retroviruses, LV area able to hold a substantially bigger transgenes cassette (up to 18 kb) [105], and third-generation HIV-based vectors present an increased transduction efficacy. These vectors can be engineered to have tissue tropism through pseudotyping. They have also been shown to have a low immunogenicity because they lack viral protein synthesis expression [103].
Glioma cells present a higher transduction efficiency than other brain cells, when infected with LV pseudotyped with Lymphocytic Choriomeningitis Virus glycoproteins [106,107]. LV are widely used for silencing RNA [108] or to induce T cells specific chimeric antigen receptors expression against glioma antigens [109]. A LV with a p2A peptide enabled a multi expression system of tumor suppressor proteins, growth arrest-specific (GAS)-1, and Phosphatase and tensin homolog (PTEN), under the control of a CMV promoter [110]. This vector inhibited human glioma cell growth in vitro, and diminished progression a murine xenograft model of human HGG [110]. LV encoding a shRNA for the orphan nuclear receptor TLX (NR2E1), inhibited tumorigenicity of human glioma stem cell in mice. This receptor plays key roles in neurogenesis during early embryogenesis and is crucial to maintain stemness and control the differentiation of adult neural stem cells in the central nervous system.
The Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) and CRISPR-associated (Cas) 9 systems have also been encoded using LV. This approach has been used for the ablation of the Transcriptional Enhancer Factor 1 (TEAD1) in human HGG cells, which showed a decreased movement of the cells and altered the migratory and epithelial-mesenchymal transition (EMT) transcriptome signatures[111].
In pre-clinical gene therapy models, LV have been used for the expression of several RNA molecules, e.g., miRNAs, AntagomiRs, siRNAs, and shRNAs, with potencially encouraging results for their application in humans [112–117]. LV-mediated silencing of the vascular endothelial growth factor (VEGF)/VEGF receptor (VEGFR) signaling pathway correlated with smaller tumor size, decreased matrix metalloproteinase 9 (MMP9) and increased tumor necrosis, in xenograft models of glioma[118].
2.1.11. Adeno-associated virus vectors
Adeno-associated viruses (AAV) are small ssDNA non-enveloped viruses with replication defects that belong to the non-pathogenic parvovirus family [119–121]. AAV are non-autonomous, and require a helper virus to replicate inside the host cell [122]. AAV are able to infect a wide variety of dividing and nondividing cells with high efficiency [123], and their small size allow them to better infiltrate solid tumors like gliomas [124]. In the absence of helper virus, AAV genomes enter into latency and remain largely as episomes. Only a small percentage of AAV genomes integrate into the host cell’s genome, therefore copy number is reduced with each cell division, making them preferable for short-term transgenic expression [119,125,126]. In pre-clinical models of HGG, the AAV-mediated genetic alteration of tumor cells by inducing the expression of anti-tumor proteins has demonstrated encouraging outcomes. The main advantages of AAV are the reduced immunogenicity compared to AdV, the high titer production, and the option of pseudotyping. The encouraging supporting pre-clinical gene therapy research employing AAV promotes the clinical testing of these vectors for the treatment of HGG[127]. In human and mouse HGG models, intracranial injection of AAV encoding human IFN-β induced glioma cell death and produced long-term survival [128,129]. In a xenograft mouse model of invasive glioma, intracranial administration of AAV engineered to produce soluble tumor necrosis factor-related apoptosis-inducing ligand (sTRAIL), coupled with lanatoside C prolonged median survival [130]. Newly engineered CNS-tropic AAV was developed by genetically inserting 14 cell-penetrating peptides into the AAV9 capsid, overcoming the inability of AAV9 to penetrate the BBB. This molecule was able to reach a fivefold-to-tenfold greater efficiency in penetrating the BBB than AAV9 in primates[131]. This modifications on AAV9 will facilitate the clinical translation of gene therapies for the CNS.
2.1.12. Baculovirus vectors
Baculovirus (BV) are enveloped dsDNA viruses, that generally infect insects and do not replicate in human cells, making them very safe [132]. The BV genome is approximately 134 kb, can harbor large transgenes, and is comparatively easy to engineer [132,133]. There is no pre-existing anti-BV immunity in humans, proposing them as a potentially effective therapy option, but there are no clinical trials employing them yet [134]. The BV can avoid some of the problems carried by mammalian viral vectors, while also providing another option for cancer gene therapy. BV encoding diphtheria toxin A gene has acceptable enhanced efficacy against malignant glioma [135].
2.2. Non-viral Vectors
Non-viral vectors have been frequently studied in recent years. Compared to viral vectors, they are less cytotoxic, immunogenic, and mutagenic. However, they face significant challenges such as gene transfer efficiency, specificity, duration of gene expression, and safety. In the last decade, non-viral vectors have become a fast-paced research topic in gene delivery. One of the major challenges of treating HGG is that therapeutic agent must cross the BBB and be in sufficient concentrations to be effective. To achieve this, synthetic non-viral vectors are developed with these features. On the other hand, cell therapies are able to migrate to tumor sites and cross the BBB, making them excellent tools for gene delivery. These properties make them also a good delivery system for viral therapies.
2.2.1. Nanoparticles and Liposomes
Liposomes, nanoparticles, and polymeric vectors have been proposed for HGG therapies since they present a low risk of immunogenicity and cytotoxicity[136]. Liposomes are non-toxic, biocompatible, and biodegradable 0.4–2.5 μm vesicles that containing a liquid phase [137]. Nanoparticles have a diameter of 1–1000 nm and are often synthesized from biocompatible polymers. They are also uniquely able to target multiple ligands (e.g., antibodies, peptides, small molecules, cell surface proteins); later they degrade on site over the course of weeks. These vectors are fabricated to able to cross the BBB and therefore reach HGG tumors [138]. Although there are no non-viral vectors currently FDA approved for the treatment of HGG, several have entered clinical trials as described below.
SGT-53 is a tumor-targeted nanomedicine, that employs a nanocomplex containing a plasmid encoding human wild-type p53 (wtp53)[139]. SGT-53 can cross the BBB to deliver p53 cDNA to HGG cells, to introduce the expression of wtp53. A Phase I clinical trial (NCT00470613) in 2013 exhibited minimal side effects of SGT-53 therapy in patients with solid tumors of several types of cancers [139]. It showed a strong antitumor activity in 7 of 11 patients and expression of p53 transgene only in the targeted metastatic tumors but not normal skin tissue. Recent work showcased SGT-53 in combination with TMZ to improve chemosensitivity, resulting in a sensibilization of tumor cells to TMZ, and apoptosis in a HGG mouse model [140]. A Phase II clinical trial of SGT-53 combined with SOC for the treatment of recurrent HGG began in 2015 but was terminated in 2021 due low enrollment (NCT02340156). Currently, there is an active, but not yet recruiting, Phase I clinical trial exploring SGT-53 in combination with SOC, for pediatric malignancies of central nervous system (NCT03554707).
NU-0129 is a spherical gold nanoparticle containing silencing RNA (siRNA) targeting the oncoprotein BCL-2-like protein 12 (Bcl2L12) [139,141]. A 2019 Phase 0 clinical trial of HGG patients demonstrated that NU-0129 cross the BBB, accumulate in the tumor, and dimmish Bcl2L12 protein expression, without short or long-term toxicity (NCT03020017).
A synthetic protein nanoparticle (SPNP) made of polymerized human serum albumin (HSA) and oligo ethylene glycol (OEG), containing siRNA targeting Signal Transducer and Activation of Transcription 3 factor (STAT3i), and equipped with the cell-penetrating peptide iRGD, engineered to target brain tumors. In HGG mouse models, this nanoparticle penetrated the BBB, and delivered siRNA against STAT3 throughout the tumor. SPNP in combination with irradiation resulted in tumor regression and long-term survival in 87.5% of mice [142].
2.2.2. Stem cells
Stem cells (SC) are undifferentiated multipotent progenitor cells, which have the capacity to self-renew, migrate, and differentiate. There are different types of SC; mainly they can be clustered into 2 groups: Embryonic Stem Cells (ESC) and Adult Stem Cells (ASC).
ASC are undifferentiated precursors that are present in all differentiated tissue and can repopulate damaged tissue [143,144]. This group of cells includes Hematopoietic Stem Cells (HSC), Mesenchymal Stem Cells (MSC), and Neural Stem Cells (NSC). HSC can be obtained from peripheral blood, umbilical cord blood, or bone marrow, being the most accessible adult SC. They can give rise to both myeloid and lymphoid lineages [145]. MSC are stromal cell that can be isolated from many different tissues (e.g., umbilical cord, bone marrow, adipose tissue). NSC are the only endogenous brain SC. This population is mainly located within the subventricular zone (SVZ) of the forebrain, and hippocampus. In rodents they give rise to neurons, astrocytes, and oligodendrocytes [146].
SC are part of cell repair in local tissue, but they have also showcased the ability to migrate to tumor site, even crossing the BBB, making them a great tool for gene delivery [147,148]. Both MSC and NSC exhibit a tropism for malignant gliomas [147,149].
Glioma extra cellular matrix (ECM), as well as secreted factors, induce cell migration of several cell types [150]. Tumors display similarities with chronic injury, like hypoxia, and inflammation [151]. Both, angiogenic and proinflammatory factors are produced by microglia and astrocytes in brain tumors [152].The chemokines produced by them, as well as tumor cells and infiltrating tumor cells, such as VEGF, HIF1α, CCL-25, IL-6, CXCL16, and CXCL12, attract SC [153–155]. Studies using mouse models have demonstrated that both MSC and NSC migrate to brain tumors [156]. NCS express CCR2, which induces migration to Monocyte chemoattractant protein-1(MCP-1/ CCL2), which is highly expressed in brain neoplastic lesions [157]. The lack of major histocompatibility complex type II (MHC II) in NSC allows the implantation of these cells which are able to escape host immune responses upon transplantation [158]. Although there are several chemokines mediating SC tropism to brain tumors, there is still much to learn, and the molecular mechanisms involved. Nevertheless, the migratory potential of SC strongly supports their therapeutic use, especially to possibly target tumor cells located far from the main tumor mass.
One of the most explored pathways for SC-based induced cytotoxicity is the use of tumor necrosis factor-related apoptosis inducing ligand (TRAIL) interaction with death receptor (DR), which is expressed by tumor cells, and when ligated activates caspase-mediated apoptosis. This is supported by the fact that HGG exhibits TRAIL-mediated apoptosis [159–162]. Delivery of soluble TRAIL (sTRAIL) deployed by NSCs in xenograft models of glioma showed an improved survival with a significant reduction of tumor burden [163]. Although TRAIL has been studied in animal models it has not reached yet human trials.
SC have been proposed for suicide gene therapy, transducing SC to express the enzyme capable of pro-drug activation. NSC can be genetically modified to express HSV1-TK, allowing them to catalyze conversion of GCV into 3P-GCV, or to express CD allowing the conversion of 5-FC pro-drug into 5-FU. Both mechanisms were described above. Another combination of enzyme pro-drug activation is the rabbit carboxylesterase, that converts irinotecan into toxic topoisomerase-1 inhibitor SN-38 [164].
The only SC procedure approved by the FDA is HSC administration, which is employed for the treatment of multiple myelomas, leukemia, and blood disorders [165]. The first human study in neuro-oncology employing SC for was performed by Portnow et al.[166]. Fifteen patients with recurrent HGG, received a single dose injected intracranially of genetically modified NSC, expressing CD, plus prodrug 5-FC, as a single dose (NCT01172964). It displayed no difference in OS. But this study establishes the safety of NSC, and effectiveness of these cells to locally produce chemotherapy, 5-FU [166]. Brain autopsy determined that NSC were not tumorigenic and also that they had migrated to distant tumor sites. Next, a dose-escalation, with multiple-treatment rounds of NSC-CD plus 5-FC, in combination with 5-FC and folinic acid (Leucovorin) (NCT02015819), was performed HGG patients to determine a safe dose to take forward to Phase II.
Administration of NSC modified to express carboxylesterase plus the prodrug irinotecan is being studied in a currently active, not yet recruiting, Phase I clinical trial for recurrent HGG. There will be two administrations of carboxylesterase-expressing allogeneic NSC on days 1 and 15 (NCT02192359).
SC-based therapy can also be employed to load these cells with oncolytic virus, and therefore take advantage of SC tropism to the tumor. In a xenograft murine model of glioma, human MSC transfected with replication-competent oncolytic adenovirus (CRAd) were able to reach the brain and deliver CRAd to glioma cells, when injected away from tumor site [167]. SC-based delivery could improve oncolytic virotherapy for HGG.
A Phase I clinical trial (NCT03072134) was conducted for newly diagnosed HGG employing NSC engineered to deliver an oncolytic adenovirus, CRAd-S-pk7. NSC-CRAd-S-pk7 were administered intracranially after initial surgery. The results from this study prove the safety of the treatment [168]. Moving forward, the not yet recruiting Phase I clinical trial (NCT05139056) will employ NSC-CRAd-S-pk7 in recurrent HGG, administered intracerebrally once weekly for up to 4 doses. In the pediatric field, a currently recruiting Phase I/II clinical trial for Diffuse Intrinsic Pontine Glioma (DIPG) uses AloCELYVIR as a treatment. AloCELYVIR is a bone marrow-derived allogenic MSC infected with an oncolytic adenovirus ICOVIR-5 (NCT04758533). The administration will be a weekly infusion of AloCELYVIR for 8 weeks. Results should be forthcoming.
Another viral therapy combined with SC is bone marrow derived MSC loaded with oncolytic adenovirus DNX-2401. This approach was studied in Phase I clinical trial (NCT03896568) for patients with recurrent HGG. To date, this trial has not published any results.
Currently, active Phase I clinical trial MGMT unmethylated HGG employs autologous CD34+HSC modified with a lentiviral vector.to express an MGMT mutant (MGMT-P140K) characterized by normal methyltransferase activity coupled with low affinity for the MGMT inhibitor O6-benzylguanine (BG). This would enable patients to receive and tolerate a higher dose of TMZ and BG with minimal toxicity (NCT01269424). Reported results registered moderate toxicity, and viral transduction range was from 3–75%[169].
3. Conclusion
There is an enormous variety of viral vectors that is currently being tested in clinical trials for HGG (Fig. 1, Table I), There is also a large set of pre-clinical data supporting its promising for application to this disease. Different therapeutic approaches using viral vectors have been proposed: suicide gene therapy, introduces a foreign gene coding for an enzyme to convert a prodrug into a toxic metabolite; oncolytic viruses, targeting glioma cells and potentially replicating throughout tumors; and transduction of immune stimulatory molecules to reduce gliomas immune suppression.
Oncolytic viruses can induce ICD eliciting a potent and long-lasting anti-cancer immunity (Fig. 2). ICD in combination with SOC has a solid mechanistic basis for a synergistic effect, since many viruses inhibit cellular DNA repair pathways, as a defense mechanism. Oncolytic viruses can induce radiosensitivity selectively and enhanced on glioma cells, sparing normal tissue. There is evidence indicating suicide gene therapy induces also ICD, and therefore also can be synergistic with radiotherapy.
The Phase I/II clinical trials reviewed here were able to demonstrate that viral therapies are safe, while exhibiting much promise for the treatment of HGG. Nonetheless, the Phase III trials failed to generate an efficient and approved therapy.
4. Expert Opinion
HGG are highly aggressive tumors that possess inter- and intratumor heterogeneity. The current SOC for HGG, which has remained unchanged since 2005, is maximal safe surgical resection, RT plus TMZ followed by adjuvant TMZ. Since 2005 there has been no significant improvement in survival for HGG patients. The ability of glioma cells to infiltrate healthy tissue makes it impossible to eliminate tumor growth by surgical resection alone, leading to tumor recurrence and poor long-term prognosis. Despite the field constantly thriving to improve surgical and imaging techniques, survival has not improved.
Gene therapy is a versatile and promising technique to be employed in neuro-oncology. Viral vectors used for gene therapy can be administered locally during initial surgery and reach infiltrating cells that cannot be resected, therefore overcoming therapeutic resistance and reducing recurrence. The high infection efficiency of viral vectors has made them widely popular for gene therapy in HGG clinical trials. Furthermore, several different vectors employing a variety of genome editing strategies have been proven to be safe. Histopathological analyses of pre- and post-treatment tissue has demonstrated not only virus activity in vivo, but also lysis of tumor cells and an increase of immune cell infiltration and activation. Despite these promising data, several Phase III clinical trials have been conducted with suicide gene therapy, targeted therapy, and immunotherapy without achieving an increase in survival for patients suffering from HGG.
Going forward, efforts to improve viral therapy efficacy will be directed to achieving a tumor microenvironment more favorable to the immune system, potentiating immunogenicity and synergizing with immune system anti-tumor response. One of the main downsides of viral therapy is the biodistribution and diffusion of virus within the tumor. Studies concerning the physical barriers of infections, such as the extracellular matrix, necrosis areas, calcification areas, and hypoxia, among others, are now being considered as factors contributing to reduced diffusion throughout the tumor. A combination of viral therapies and extracellular matrix modifiers (such as metalloproteinases) might allow a better viral distribution, allowing the vectors to reach deep infiltrating cells.
Concomitant stimulation of the immune system has been a goal of the latest clinical trials. Checkpoint blockade such as PD-1/PD-L1 have been proven to work successfully in several different types of tumors. A synergistic approach of viral therapy and checkpoint blockades, aiming to overcome immune suppression and potentiate anti-tumor response of effectors cells, has been used in HGG. These Phase I and II clinical trials have shown that this combination has an impact on the immune system response, yet they have not achieved an increase in survival. Future Phase III trials will determine if there is an effective synergistic treatment for HGG.
Even though viral therapy has exhibited a remarkable potential for treatment of HGG in both pre-clinical and Phase I/II clinical data, current protocols are not able to transition into approved treatments. Undeniably, viral gene therapy provided a new therapeutic approach and perspective in HGG treatment. HGG
Ongoing oncolytic HSV-1 derived viral therapies are showing much promise. G207 administration showed encouraging results both for adults and pediatric HGG. rQNestin modification of HSV-1 is proposing a much safer treatment option, with strong supporting pre-clinical data. HSV-1 G47Δ was able to improve survival, and therefore, has received governmental approval as a new drug in Japan.
Also, new virus have aroused as possible therapies for HGG. PVSRIPO has showed survival improvement in HGG treated adults compared to historical controls. Now it is being evaluated in pediatric HGG, as well as a Phase II for adults with HGG. Strong preclinical data support MV as a therapy for HGG, and now being tested both for pediatric and adult HGG.
Deeper analysis of these trials and following ones will bring light to viral therapy, new protocols of administration, and/or new combinations which will allow these treatments to improve HGG prognosis.
Article highlights.
High-grade gliomas (HGG) standard of care (SOC) remains unchanged since 2005, even though HGG median survival has marginally improved.
There are currently over 20 clinical trials using gene therapy to treat HGG.
Administration of HSV-1 G207 showed promising results in HGG and pediatric HGG.
HSV-1 rQNestin offers a safer treatment option with strong supporting preclinical data.
HSV-1 G47Δ was able to improve survival and received government approval in Japan.
PVSRIPO showed an improvement in HGG survival compared to the historical controls.
Acknowledgments
This work was supported by National Institutes of Health, National Institute of Neurological Disorders and Stroke (NIH/NINDS) grants: R37-NS094804, R01-NS105556, R21-NS107894, R21-NS091555; R01-NS074387 to M.G.C.; National Institute of Neurological Disorders and Stroke (NIH/NINDS) grants: R01-NS076991, R01-NS096756, R01-NS082311 to P.R.L.; National Institute of Biomedical Imaging and Bioengineering (NIH/NIBI): R01-EB022563; National Cancer Institute (NIH/NCI) U01CA224160; Rogel Cancer Center at The University of Michigan G023089 to M.G.C. Ian’s Friends Foundation grant G024230, Leah’s Happy Hearts Foundation grant G013908, Pediatric Brain Tumor Foundation grant G023387 and ChadTough Foundation grant G023419 to P.R.L. RNA Biomedicine grant: F046166 to M.G.C.
6. References
*Highlighted bibliography of interest
** Highlighted bibliography of considerable interest.
- 1.Surveillance, Epidemiology, and End Results: Cancer Stat Facts: Brain and Other Nervous System Cancer. [Internet]. 2022.
- 2.Björkblom B, Wibom C, Eriksson M, et al. Distinct metabolic hallmarks of WHO classified adult glioma subtypes. Neuro-oncology. 2022. Sep 1;24(9):1454–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Galbraith K, Snuderl M. Molecular Pathology of Gliomas. Surg Pathol Clin. 2021. Sep;14(3):379–386. [DOI] [PubMed] [Google Scholar]
- 4.Molinaro AM, Taylor JW, Wiencke JK, et al. Genetic and molecular epidemiology of adult diffuse glioma. Nat Rev Neurol. 2019. Jul;15(7):405–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pekmezci M, Perry A. Genetic markers in adult high-grade gliomas. Semin Radiat Oncol. 2014. Oct;24(4):235–9. [DOI] [PubMed] [Google Scholar]
- 6.Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005. Mar 10;352(10):987–96. [DOI] [PubMed] [Google Scholar]
- 7.Stupp R, Hegi ME, Mason WP, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. The Lancet Oncology. 2009. May;10(5):459–66. [DOI] [PubMed] [Google Scholar]
- 8.Hegi ME, Liu L, Herman JG, et al. Correlation of O6-methylguanine methyltransferase (MGMT) promoter methylation with clinical outcomes in glioblastoma and clinical strategies to modulate MGMT activity. J Clin Oncol. 2008. Sep 1;26(25):4189–99. [DOI] [PubMed] [Google Scholar]
- 9.Walker MD, Alexander E Jr., Hunt WE, et al. Evaluation of BCNU and/or radiotherapy in the treatment of anaplastic gliomas. A cooperative clinical trial. J Neurosurg. 1978. Sep;49(3):333–43. [DOI] [PubMed] [Google Scholar]
- 10.Keime-Guibert F, Chinot O, Taillandier L, et al. Radiotherapy for glioblastoma in the elderly. N Engl J Med. 2007. Apr 12;356(15):1527–35. [DOI] [PubMed] [Google Scholar]
- 11.Shapiro WR, Green SB, Burger PC, et al. Randomized trial of three chemotherapy regimens and two radiotherapy regimens and two radiotherapy regimens in postoperative treatment of malignant glioma. Brain Tumor Cooperative Group Trial 8001. J Neurosurg. 1989. Jul;71(1):1–9. [DOI] [PubMed] [Google Scholar]
- 12.Nelson DF, Curran WJ Jr., Scott C, et al. Hyperfractionated radiation therapy and bis-chlorethyl nitrosourea in the treatment of malignant glioma--possible advantage observed at 72.0 Gy in 1.2 Gy B.I.D. fractions: report of the Radiation Therapy Oncology Group Protocol 8302. Int J Radiat Oncol Biol Phys. 1993. Jan 15;25(2):193–207. [DOI] [PubMed] [Google Scholar]
- 13.Ali AN, Zhang P, Yung WKA, et al. NRG oncology RTOG 9006: a phase III randomized trial of hyperfractionated radiotherapy (RT) and BCNU versus standard RT and BCNU for malignant glioma patients. J Neurooncol. 2018. Mar;137(1):39–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Souhami L, Seiferheld W, Brachman D, et al. Randomized comparison of stereotactic radiosurgery followed by conventional radiotherapy with carmustine to conventional radiotherapy with carmustine for patients with glioblastoma multiforme: report of Radiation Therapy Oncology Group 93-05 protocol. Int J Radiat Oncol Biol Phys. 2004. Nov 1;60(3):853–60. [DOI] [PubMed] [Google Scholar]
- 15.Roa W, Brasher PM, Bauman G, et al. Abbreviated course of radiation therapy in older patients with glioblastoma multiforme: a prospective randomized clinical trial. J Clin Oncol. 2004. May 1;22(9):1583–8. [DOI] [PubMed] [Google Scholar]
- 16.Roa W, Kepka L, Kumar N, et al. International Atomic Energy Agency Randomized Phase III Study of Radiation Therapy in Elderly and/or Frail Patients With Newly Diagnosed Glioblastoma Multiforme. J Clin Oncol. 2015. Dec 10;33(35):4145–50. [DOI] [PubMed] [Google Scholar]
- 17.Malmstrom A, Gronberg BH, Marosi C, et al. Temozolomide versus standard 6-week radiotherapy versus hypofractionated radiotherapy in patients older than 60 years with glioblastoma: the Nordic randomised, phase 3 trial. Lancet Oncol. 2012. Sep;13(9):916–26. [DOI] [PubMed] [Google Scholar]
- 18.Perry JR, Laperriere N, O’Callaghan CJ, et al. Short-Course Radiation plus Temozolomide in Elderly Patients with Glioblastoma. N Engl J Med. 2017. Mar 16;376(11):1027–1037. [DOI] [PubMed] [Google Scholar]
- 19.Niyazi M, Brada M, Chalmers AJ, et al. ESTRO-ACROP guideline “target delineation of glioblastomas”. Radiother Oncol. 2016. Jan;118(1):35–42. [DOI] [PubMed] [Google Scholar]
- 20.Stupp R, Taillibert S, Kanner A, et al. Effect of Tumor-Treating Fields Plus Maintenance Temozolomide vs Maintenance Temozolomide Alone on Survival in Patients With Glioblastoma: A Randomized Clinical Trial. Jama. 2017. Dec 19;318(23):2306–2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Burri SH, Gondi V, Brown PD, et al. The Evolving Role of Tumor Treating Fields in Managing Glioblastoma: Guide for Oncologists. American Journal of Clinical Oncology. 2018;41(2):191–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fu P, He Y-S, Huang Q, et al. Bevacizumab treatment for newly diagnosed glioblastoma: Systematic review and meta-analysis of clinical trials. Mol Clin Oncol. 2016;4(5):833–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stupp R, Hegi ME, Gorlia T, et al. Cilengitide combined with standard treatment for patients with newly diagnosed glioblastoma with methylated MGMT promoter (CENTRIC EORTC 26071-22072 study): a multicentre, randomised, open-label, phase 3 trial. The Lancet Oncology. 2014. Sep;15(10):1100–8. [DOI] [PubMed] [Google Scholar]
- 24.Weller M, Butowski N, Tran DD, et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): a randomised, double-blind, international phase 3 trial. The Lancet Oncology. 2017. Oct;18(10):1373–1385. [DOI] [PubMed] [Google Scholar]
- 25.Reardon DA, Brandes AA, Omuro A, et al. Effect of Nivolumab vs Bevacizumab in Patients With Recurrent Glioblastoma: The CheckMate 143 Phase 3 Randomized Clinical Trial. JAMA Oncol. 2020. Jul 1;6(7):1003–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Narita Y, Arakawa Y, Yamasaki F, et al. A randomized, double-blind, phase III trial of personalized peptide vaccination for recurrent glioblastoma. Neuro-oncology. 2019. Feb 19;21(3):348–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cloughesy TF, Brenner A, de Groot JF, et al. A randomized controlled phase III study of VB-111 combined with bevacizumab vs bevacizumab monotherapy in patients with recurrent glioblastoma (GLOBE). Neuro-oncology. 2020. May 15;22(5):705–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ostrom QT, Cioffi G, Gittleman H, et al. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2012–2016. Neuro-oncology. 2019. Nov 1;21(Suppl 5):v1–v100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Udaka YT, Packer RJ. Pediatric Brain Tumors. Neurol Clin. 2018 2018/August//;36(3):533–556. [DOI] [PubMed] [Google Scholar]
- 30.Mackay A, Burford A, Carvalho D, et al. Integrated Molecular Meta-Analysis of 1,000 Pediatric High-Grade and Diffuse Intrinsic Pontine Glioma. Cancer cell. 2017;32(4):520–537.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schwartzentruber J, Korshunov A, Liu XY, et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature. 2012. Jan 29;482(7384):226–31. [DOI] [PubMed] [Google Scholar]
- 32.Sturm D, Witt H, Hovestadt V, et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer cell. 2012. Oct 16;22(4):425–37. [DOI] [PubMed] [Google Scholar]
- 33.Louis DN, Perry A, Wesseling P, et al. The 2021 WHO Classification of Tumors of the Central Nervous System: a summary. Neuro-oncology. 2021. Aug 2;23(8):1231–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schwark K, Messinger D, Cummings JR, et al. Receptor tyrosine kinase (RTK) targeting in pediatric high-grade glioma and diffuse midline glioma: Pre-clinical models and precision medicine. Frontiers in oncology. 2022;12:922928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guerreiro Stucklin AS, Ryall S, Fukuoka K, et al. Alterations in ALK/ROS1/NTRK/MET drive a group of infantile hemispheric gliomas. Nat Commun. 2019. Sep 25;10(1):4343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Batra V, Sands SA, Holmes E, et al. Long-term survival of children less than six years of age enrolled on the CCG-945 phase III trial for newly-diagnosed high-grade glioma: a report from the Children’s Oncology Group. Pediatric blood & cancer. 2014. Jan;61(1):151–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cohen KJ, Pollack IF, Zhou T, et al. Temozolomide in the treatment of high-grade gliomas in children: a report from the Children’s Oncology Group. Neuro-oncology. 2011. Mar;13(3):317–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jakacki RI, Cohen KJ, Buxton A, et al. Phase 2 study of concurrent radiotherapy and temozolomide followed by temozolomide and lomustine in the treatment of children with high-grade glioma: a report of the Children’s Oncology Group ACNS0423 study. Neuro-oncology. 2016;18(10):1442–1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hayden E, Holliday H, Lehmann R, et al. Therapeutic Targets in Diffuse Midline Gliomas-An Emerging Landscape. Cancers (Basel). 2021. Dec 13;13(24). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bredlau AL, Korones DN. Diffuse intrinsic pontine gliomas: treatments and controversies. Adv Cancer Res. 2014;121:235–259. [DOI] [PubMed] [Google Scholar]
- 41.Frazier JL, Lee J, Thomale UW, et al. Treatment of diffuse intrinsic brainstem gliomas: failed approaches and future strategies. Journal of neurosurgery Pediatrics. 2009. Apr;3(4):259–69. [DOI] [PubMed] [Google Scholar]
- 42.Jalali R, Raut N, Arora B, et al. Prospective evaluation of radiotherapy with concurrent and adjuvant temozolomide in children with newly diagnosed diffuse intrinsic pontine glioma. International journal of radiation oncology, biology, physics. 2010. May 1;77(1):113–8. [DOI] [PubMed] [Google Scholar]
- 43.Aziz-Bose R, Monje M. Diffuse intrinsic pontine glioma: molecular landscape and emerging therapeutic targets. Current opinion in oncology. 2019. Nov;31(6):522–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Patel DH, Misra A. Gene delivery using viral vectors. Challenges in delivery of therapeutic genomics and proteomics: Elsevier Inc.; 2011. p. 207–270. [Google Scholar]
- 45.Bin Umair M, Akusa FN, Kashif H, et al. Viruses as tools in gene therapy, vaccine development, and cancer treatment. Arch Virol. 2022. Jun;167(6):1387–1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee CS, Bishop ES, Zhang R, et al. Adenovirus-Mediated Gene Delivery: Potential Applications for Gene and Cell-Based Therapies in the New Era of Personalized Medicine. Genes Dis. 2017. Jun;4(2):43–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ji N, Weng D, Liu C, et al. Adenovirus-mediated delivery of herpes simplex virus thymidine kinase administration improves outcome of recurrent high-grade glioma. Oncotarget. 2016. Jan 26;7(4):4369–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wheeler LA, Manzanera AG, Bell SD, et al. Phase II multicenter study of gene-mediated cytotoxic immunotherapy as adjuvant to surgical resection for newly diagnosed malignant glioma. Neuro Oncol. 2016. Aug;18(8):1137–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kieran MW, Goumnerova L, Manley P, et al. Phase I study of gene-mediated cytotoxic immunotherapy with AdV-tk as adjuvant to surgery and radiation for pediatric malignant glioma and recurrent ependymoma. Neuro Oncol. 2019. Mar 18;21(4):537–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lang FF, Bruner JM, Fuller GN, et al. Phase I Trial of Adenovirus-Mediated p53 Gene Therapy for Recurrent Glioma: Biological and Clinical Results. Journal of Clinical Oncology. 2003;21(13):2508–2518. [DOI] [PubMed] [Google Scholar]
- 51.Barrett JA, Cai H, Miao J, et al. Regulated intratumoral expression of IL-12 using a RheoSwitch Therapeutic System(®) (RTS(®)) gene switch as gene therapy for the treatment of glioma. Cancer Gene Ther. 2018. Jun;25(5–6):106–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chiocca EA, Yu JS, Lukas RV, et al. Regulatable interleukin-12 gene therapy in patients with recurrent high-grade glioma: Results of a phase 1 trial. Sci Transl Med. 2019. Aug 14;11(505). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Chiocca EA, Gelb AB, Chen CC, et al. Combined immunotherapy with controlled interleukin-12 gene therapy and immune checkpoint blockade in recurrent glioblastoma: An open-label, multi-institutional phase I trial. Neuro Oncol. 2022. Jun 1;24(6):951–963. *(Phase I clinical trial of Ad-RTS-hIL-12, that lead to ongoing Phase II)
- 54.Chiocca EA, Smith KM, McKinney B, et al. A Phase I Trial of Ad.hIFN-β Gene Therapy for Glioma. Molecular Therapy. 2008 2008/March/01/;16(3):618–626. [DOI] [PubMed] [Google Scholar]
- 55.Gruslova A, Cavazos DA, Miller JR, et al. VB-111: a novel anti-vascular therapeutic for glioblastoma multiforme. J Neurooncol. 2015. Sep;124(3):365–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Brenner AJ, Cohen YC, Vredenburgh JJ, et al. Ofranogene obadenovec (VB-111), an anti-cancer gene therapy in combination with bevacizumab to improve overall survival compared to bevacizumab monotherapy in patients with rGBM: A phase 2 historically controlled trial. Journal of Clinical Oncology. 2016;34(15_suppl):2074–2074. [Google Scholar]
- 57.Cloughesy TF, Petrecca K, Walbert T, et al. Effect of Vocimagene Amiretrorepvec in Combination With Flucytosine vs Standard of Care on Survival Following Tumor Resection in Patients With Recurrent High-Grade Glioma: A Randomized Clinical Trial. JAMA Oncol. 2020. Dec 1;6(12):1939–1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ghajar-Rahimi G, Kang KD, Totsch SK, et al. Clinical advances in oncolytic virotherapy for pediatric brain tumors. Pharmacol Ther. 2022. Apr 26;239:108193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kiyokawa J, Wakimoto H. Preclinical And Clinical Development Of Oncolytic Adenovirus For The Treatment Of Malignant Glioma. Oncolytic Virother. 2019;8:27–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fueyo J, Gomez-Manzano C, Alemany R, et al. A mutant oncolytic adenovirus targeting the Rb pathway produces anti-glioma effect in vivo. Oncogene. 2000. Jan 6;19(1):2–12. [DOI] [PubMed] [Google Scholar]
- 61. Lang FF, Conrad C, Gomez-Manzano C, et al. Phase I Study of DNX-2401 (Delta-24-RGD) Oncolytic Adenovirus: Replication and Immunotherapeutic Effects in Recurrent Malignant Glioma. J Clin Oncol. 2018. May 10;36(14):1419–1427. *(Phase I clinical trial of DNX-2401, showing histopathologic examination of immune markers)
- 62.van den Bossche WBL, Kleijn A, Teunissen CE, et al. Oncolytic virotherapy in glioblastoma patients induces a tumor macrophage phenotypic shift leading to an altered glioblastoma microenvironment. Neuro Oncol. 2018. Oct 9;20(11):1494–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lang FF, Tran ND, Puduvalli VK, et al. Phase 1b open-label randomized study of the oncolytic adenovirus DNX-2401 administered with or without interferon gamma for recurrent glioblastoma. Journal of Clinical Oncology. 2017;35(15_suppl):2002–2002. [Google Scholar]
- 64.Wilcox DR, Longnecker R. The Herpes Simplex Virus Neurovirulence Factor γ34.5: Revealing Virus-Host Interactions. PLoS Pathog. 2016. Mar;12(3):e1005449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Alayo QA, Ito H, Passaro C, et al. Glioblastoma infiltration of both tumor- and virus-antigen specific cytotoxic T cells correlates with experimental virotherapy responses. Sci Rep. 2020. Mar 20;10(1):5095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Martuza RL, Malick A, Markert JM, et al. Experimental therapy of human glioma by means of a genetically engineered virus mutant. Science. 1991. May 10;252(5007):854–6. [DOI] [PubMed] [Google Scholar]
- 67. Markert JM, Razdan SN, Kuo HC, et al. A phase 1 trial of oncolytic HSV-1, G207, given in combination with radiation for recurrent GBM demonstrates safety and radiographic responses. Mol Ther. 2014. May;22(5):1048–55. **(Phase I of HSV-1 G207, demostrating safety and potential for clinical response of single-dose oncolytic HSV in combination with radiation)
- 68. Markert JM, Liechty PG, Wang W, et al. Phase Ib trial of mutant herpes simplex virus G207 inoculated pre-and post-tumor resection for recurrent GBM. Mol Ther. 2009. Jan;17(1):199–207. **(Phase Ib of HSV G207reporting evidence of viral replication and safety)
- 69.Markert JM, Medlock MD, Rabkin SD, et al. Conditionally replicating herpes simplex virus mutant, G207 for the treatment of malignant glioma: results of a phase I trial. Gene Ther. 2000. May;7(10):867–74. ** [DOI] [PubMed] [Google Scholar]
- 70.Friedman GK, Johnston JM, Bag AK, et al. Oncolytic HSV-1 G207 Immunovirotherapy for Pediatric High-Grade Gliomas. N Engl J Med. 2021. Apr 29;384(17):1613–1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Patel DM, Foreman PM, Nabors LB, et al. Design of a Phase I Clinical Trial to Evaluate M032, a Genetically Engineered HSV-1 Expressing IL-12, in Patients with Recurrent/Progressive Glioblastoma Multiforme, Anaplastic Astrocytoma, or Gliosarcoma. Human gene therapy Clinical development. 2016;27(2):69–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Todo T, Martuza RL, Rabkin SD, et al. Oncolytic herpes simplex virus vector with enhanced MHC class I presentation and tumor cell killing. Proc Natl Acad Sci U S A. 2001. May 22;98(11):6396–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Todo T, Ito H, Ino Y, et al. Intratumoral oncolytic herpes virus G47Δ for residual or recurrent glioblastoma: a phase 2 trial. Nature Medicine. 2022 2022/August/01;28(8):1630–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Chiocca EA, Nakashima H, Kasai K, et al. Preclinical Toxicology of rQNestin34.5v.2: An Oncolytic Herpes Virus with Transcriptional Regulation of the ICP34.5 Neurovirulence Gene. Mol Ther Methods Clin Dev. 2020. Jun 12;17:871–893. **(Preclinical toxicity determiantion of rQNestin34.5v.2, supporting ongoing Phase I clinical trial)
- 75.Otani Y, Yoo JY, Lewis CT, et al. NOTCH-Induced MDSC Recruitment after oHSV Virotherapy in CNS Cancer Models Modulates Antitumor Immunotherapy. Clin Cancer Res. 2022. Apr 1;28(7):1460–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Goff SP. Genetics of retroviral integration. Annu Rev Genet. 1992;26:527–44. [DOI] [PubMed] [Google Scholar]
- 77.Tai CK, Kasahara N. Replication-competent retrovirus vectors for cancer gene therapy. Front Biosci. 2008. Jan 1;13:3083–95. [DOI] [PubMed] [Google Scholar]
- 78.Rainov NG. A phase III clinical evaluation of herpes simplex virus type 1 thymidine kinase and ganciclovir gene therapy as an adjuvant to surgical resection and radiation in adults with previously untreated glioblastoma multiforme. Hum Gene Ther. 2000. Nov 20;11(17):2389–401. [DOI] [PubMed] [Google Scholar]
- 79.Ram Z, Culver KW, Oshiro EM, et al. Therapy of malignant brain tumors by intratumoral implantation of retroviral vector-producing cells. Nat Med. 1997. Dec;3(12):1354–61. [DOI] [PubMed] [Google Scholar]
- 80.Klatzmann D, Valéry CA, Bensimon G, et al. A Phase I/II Study of Herpes Simplex Virus Type 1 Thymidine Kinase “Suicide” Gene Therapy for Recurrent Glioblastoma. Human Gene Therapy. 1998 1998/November/20;9(17):2595–2604. [DOI] [PubMed] [Google Scholar]
- 81.Longley DB, Harkin DP, Johnston PG. 5-Fluorouracil: mechanisms of action and clinical strategies. Nature Reviews Cancer. 2003 2003/May/01;3(5):330–338. [DOI] [PubMed] [Google Scholar]
- 82.Huang TT, Hlavaty J, Ostertag D, et al. Toca 511 gene transfer and 5-fluorocytosine in combination with temozolomide demonstrates synergistic therapeutic efficacy in a temozolomide-sensitive glioblastoma model. Cancer Gene Ther. 2013. Oct;20(10):544–51. [DOI] [PubMed] [Google Scholar]
- 83.Ostertag D, Amundson KK, Lopez Espinoza F, et al. Brain tumor eradication and prolonged survival from intratumoral conversion of 5-fluorocytosine to 5-fluorouracil using a nonlytic retroviral replicating vector. Neuro Oncol. 2012. Feb;14(2):145–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tai CK, Wang WJ, Chen TC, et al. Single-shot, multicycle suicide gene therapy by replication-competent retrovirus vectors achieves long-term survival benefit in experimental glioma. Mol Ther. 2005. Nov;12(5):842–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Takahashi M, Valdes G, Hiraoka K, et al. Radiosensitization of gliomas by intracellular generation of 5-fluorouracil potentiates prodrug activator gene therapy with a retroviral replicating vector. Cancer Gene Ther. 2014. Oct;21(10):405–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Cloughesy TF, Landolfi J, Vogelbaum MA, et al. Durable complete responses in some recurrent high-grade glioma patients treated with Toca 511 + Toca FC. Neuro Oncol. 2018. Sep 3;20(10):1383–1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Schirrmacher V, van Gool S, Stuecker W. Breaking Therapy Resistance: An Update on Oncolytic Newcastle Disease Virus for Improvements of Cancer Therapy. Biomedicines. 2019. Aug 30;7(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Vigil A, Park MS, Martinez O, et al. Use of reverse genetics to enhance the oncolytic properties of Newcastle disease virus. Cancer Res. 2007. Sep 1;67(17):8285–92. [DOI] [PubMed] [Google Scholar]
- 89.Freeman AI, Zakay-Rones Z, Gomori JM, et al. Phase I/II trial of intravenous NDV-HUJ oncolytic virus in recurrent glioblastoma multiforme. Mol Ther. 2006. Jan;13(1):221–8. [DOI] [PubMed] [Google Scholar]
- 90.Dorig RE, Marcil A, Chopra A, et al. The human CD46 molecule is a receptor for measles virus (Edmonston strain). Cell. 1993. Oct 22;75(2):295–305. [DOI] [PubMed] [Google Scholar]
- 91.Muhlebach MD, Mateo M, Sinn PL, et al. Adherens junction protein nectin-4 is the epithelial receptor for measles virus. Nature. 2011. Nov 2;480(7378):530–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Galanis E, Bateman A, Johnson K, et al. Use of viral fusogenic membrane glycoproteins as novel therapeutic transgenes in gliomas. Hum Gene Ther. 2001. May 1;12(7):811–21. [DOI] [PubMed] [Google Scholar]
- 93.Msaouel P, Opyrchal M, Domingo Musibay E, et al. Oncolytic measles virus strains as novel anticancer agents. Expert Opin Biol Ther. 2013. Apr;13(4):483–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Estevez-Ordonez D, Chagoya G, Salehani A, et al. Immunovirotherapy for the Treatment of Glioblastoma and Other Malignant Gliomas. Neurosurg Clin N Am. 2021. Apr;32(2):265–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Strong JE, Coffey MC, Tang D, et al. The molecular basis of viral oncolysis: usurpation of the Ras signaling pathway by reovirus. The EMBO journal. 1998;17(12):3351–3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Forsyth P, Roldán G, George D, et al. A phase I trial of intratumoral administration of reovirus in patients with histologically confirmed recurrent malignant gliomas. Mol Ther. 2008. Mar;16(3):627–32. [DOI] [PubMed] [Google Scholar]
- 97. Desjardins A, Gromeier M, Herndon JE, 2nd, et al. Recurrent Glioblastoma Treated with Recombinant Poliovirus. N Engl J Med. 2018. Jul 12;379(2):150–161. **(Phase I clinical trial of PVSRIPO, showing no neurovirulencdeand higher survival rate comapred with historical controls)
- 98.Herrero YCM, Cornelis JJ, Herold-Mende C, et al. Parvovirus H-1 infection of human glioma cells leads to complete viral replication and efficient cell killing. Int J Cancer. 2004. Mar;109(1):76–84. [DOI] [PubMed] [Google Scholar]
- 99.Geletneky K, Huesing J, Rommelaere J, et al. Phase I/IIa study of intratumoral/intracerebral or intravenous/intracerebral administration of Parvovirus H-1 (ParvOryx) in patients with progressive primary or recurrent glioblastoma multiforme: ParvOryx01 protocol. BMC Cancer. 2012. Mar 21;12:99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Haddad D. Genetically Engineered Vaccinia Viruses As Agents for Cancer Treatment, Imaging, and Transgene Delivery [Review]. Frontiers in Oncology. 2017 2017-May-23;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Béguin J, Foloppe J, Maurey C, et al. Preclinical Evaluation of the Oncolytic Vaccinia Virus TG6002 by Translational Research on Canine Breast Cancer. Molecular Therapy - Oncolytics. 2020 2020/December/16/;19:57–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Foloppe J, Kempf J, Futin N, et al. The Enhanced Tumor Specificity of TG6002, an Armed Oncolytic Vaccinia Virus Deleted in Two Genes Involved in Nucleotide Metabolism. Molecular Therapy - Oncolytics. 2019 2019/September/27/;14:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Del Vecchio C, Calistri A, Parolin C, et al. Lentiviral Vectors as Tools for the Study and Treatment of Glioblastoma. Cancers (Basel). 2019. Mar 24;11(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kay MA. State-of-the-art gene-based therapies: the road ahead. Nat Rev Genet. 2011. May;12(5):316–28. [DOI] [PubMed] [Google Scholar]
- 105.Kumar M, Keller B, Makalou N, et al. Systematic determination of the packaging limit of lentiviral vectors. Hum Gene Ther. 2001. Oct 10;12(15):1893–905. [DOI] [PubMed] [Google Scholar]
- 106.Miletic H, Fischer YH, Neumann H, et al. Selective transduction of malignant glioma by lentiviral vectors pseudotyped with lymphocytic choriomeningitis virus glycoproteins. Hum Gene Ther. 2004. Nov;15(11):1091–100. [DOI] [PubMed] [Google Scholar]
- 107.Huszthy PC, Giroglou T, Tsinkalovsky O, et al. Remission of invasive, cancer stem-like glioblastoma xenografts using lentiviral vector-mediated suicide gene therapy. PLoS One. 2009. Jul 20;4(7):e6314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Luan Y, Zhang S, Zuo L, et al. Overexpression of miR-100 inhibits cell proliferation, migration, and chemosensitivity in human glioblastoma through FGFR3. Onco Targets Ther. 2015;8:3391–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Yu S, Li A, Liu Q, et al. Chimeric antigen receptor T cells: a novel therapy for solid tumors. J Hematol Oncol. 2017. Mar 29;10(1):78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Sanchez-Hernandez L, Hernandez-Soto J, Vergara P, et al. Additive effects of the combined expression of soluble forms of GAS1 and PTEN inhibiting glioblastoma growth. Gene Ther. 2018. Sep;25(6):439–449. [DOI] [PubMed] [Google Scholar]
- 111.Tome-Garcia J, Erfani P, Nudelman G, et al. Analysis of chromatin accessibility uncovers TEAD1 as a regulator of migration in human glioblastoma. Nat Commun. 2018. Oct 1;9(1):4020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Scherr M, Venturini L, Battmer K, et al. Lentivirus-mediated antagomir expression for specific inhibition of miRNA function. Nucleic Acids Res. 2007;35(22):e149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Sun BS, Dong QZ, Ye QH, et al. Lentiviral-mediated miRNA against osteopontin suppresses tumor growth and metastasis of human hepatocellular carcinoma. Hepatology. 2008. Dec;48(6):1834–42. [DOI] [PubMed] [Google Scholar]
- 114.Zeng LL, He XS, Liu JR, et al. Lentivirus-Mediated Overexpression of MicroRNA-210 Improves Long-Term Outcomes after Focal Cerebral Ischemia in Mice. CNS Neurosci Ther. 2016. Dec;22(12):961–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Scherr M, Venturini L, Eder M. Lentiviral vector-mediated expression of pre-miRNAs and antagomiRs. Methods Mol Biol. 2010;614:175–85. [DOI] [PubMed] [Google Scholar]
- 116.Song JL, Zheng W, Chen W, et al. Lentivirus-mediated microRNA-124 gene-modified bone marrow mesenchymal stem cell transplantation promotes the repair of spinal cord injury in rats. Exp Mol Med. 2017. May 19;49(5):e332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Alt EU, Barabadi Z, Pfnur A, et al. TRAF3IP2, a novel therapeutic target in glioblastoma multiforme. Oncotarget. 2018. Jul 3;9(51):29772–29788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Szabo E, Schneider H, Seystahl K, et al. Autocrine VEGFR1 and VEGFR2 signaling promotes survival in human glioblastoma models in vitro and in vivo. Neuro Oncol. 2016. Sep;18(9):1242–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Li C, Bowles DE, van Dyke T, et al. Adeno-associated virus vectors: potential applications for cancer gene therapy. Cancer Gene Ther. 2005. Dec;12(12):913–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Stilwell JL, Samulski RJ. Adeno-associated virus vectors for therapeutic gene transfer. Biotechniques. 2003. Jan;34(1):148–50, 152, 154 passim. [DOI] [PubMed] [Google Scholar]
- 121.McCown TJ. Adeno-associated virus (AAV) vectors in the CNS. Curr Gene Ther. 2005. Jun;5(3):333–8. [DOI] [PubMed] [Google Scholar]
- 122.van Putten EH, Dirven CM, van den Bent MJ, et al. Sitimagene ceradenovec: a gene-based drug for the treatment of operable high-grade glioma. Future Oncol. 2010. Nov;6(11):1691–710. [DOI] [PubMed] [Google Scholar]
- 123.Santiago-Ortiz JL, Schaffer DV. Adeno-associated virus (AAV) vectors in cancer gene therapy. J Control Release. 2016. Oct 28;240:287–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Enger PO, Thorsen F, Lonning PE, et al. Adeno-associated viral vectors penetrate human solid tumor tissue in vivo more effectively than adenoviral vectors. Hum Gene Ther. 2002. Jun 10;13(9):1115–25. [DOI] [PubMed] [Google Scholar]
- 125.Maier P, von Kalle C, Laufs S. Retroviral vectors for gene therapy. Future Microbiol. 2010. Oct;5(10):1507–23. [DOI] [PubMed] [Google Scholar]
- 126.Asad AS, Moreno Ayala MA, Gottardo MF, et al. Viral gene therapy for breast cancer: progress and challenges. Expert Opin Biol Ther. 2017. Aug;17(8):945–959. [DOI] [PubMed] [Google Scholar]
- 127.Riyad JM, Weber T. Intracellular trafficking of adeno-associated virus (AAV) vectors: challenges and future directions. Gene Ther. 2021. Dec;28(12):683–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.GuhaSarkar D, Neiswender J, Su Q, et al. Intracranial AAV-IFN-beta gene therapy eliminates invasive xenograft glioblastoma and improves survival in orthotopic syngeneic murine model. Mol Oncol. 2017. Feb;11(2):180–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Yoshida J, Mizuno M, Nakahara N, et al. Antitumor effect of an adeno-associated virus vector containing the human interferon-beta gene on experimental intracranial human glioma. Jpn J Cancer Res. 2002. Feb;93(2):223–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Crommentuijn MH, Maguire CA, Niers JM, et al. Intracranial AAV-sTRAIL combined with lanatoside C prolongs survival in an orthotopic xenograft mouse model of invasive glioblastoma. Mol Oncol. 2016. Apr;10(4):625–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Yao Y, Wang J, Liu Y, et al. Variants of the adeno-associated virus serotype 9 with enhanced penetration of the blood–brain barrier in rodents and primates. Nature Biomedical Engineering. 2022 2022/October/10. [DOI] [PubMed] [Google Scholar]
- 132.van Oers MM, Pijlman GP, Vlak JM. Thirty years of baculovirus-insect cell protein expression: from dark horse to mainstream technology. J Gen Virol. 2015. Jan;96(Pt 1):6–23. [DOI] [PubMed] [Google Scholar]
- 133.Dautzenberg IJC, van den Hengel SK, de Vrij J, et al. Baculovirus-assisted Reovirus Infection in Monolayer and Spheroid Cultures of Glioma cells. Sci Rep. 2017. Dec 15;7(1):17654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Ang WX, Zhao Y, Kwang T, et al. Local Immune Stimulation by Intravesical Instillation of Baculovirus to Enable Bladder Cancer Therapy. Sci Rep. 2016. Jun 8;6:27455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Wang CY, Li F, Yang Y, et al. Recombinant baculovirus containing the diphtheria toxin A gene for malignant glioma therapy. Cancer Res. 2006. Jun 1;66(11):5798–806. [DOI] [PubMed] [Google Scholar]
- 136.Caffery B, Lee JS, Alexander-Bryant AA. Vectors for Glioblastoma Gene Therapy: Viral & Non-Viral Delivery Strategies. Nanomaterials (Basel). 2019. Jan 16;9(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Malam Y, Loizidou M, Seifalian AM. Liposomes and nanoparticles: nanosized vehicles for drug delivery in cancer. Trends Pharmacol Sci. 2009. Nov;30(11):592–9. [DOI] [PubMed] [Google Scholar]
- 138.Wiwatchaitawee K, Quarterman JC, Geary SM, et al. Enhancement of Therapies for Glioblastoma (GBM) Using Nanoparticle-based Delivery Systems. AAPS PharmSciTech. 2021. Feb 11;22(2):71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Senzer N, Nemunaitis J, Nemunaitis D, et al. Phase I study of a systemically delivered p53 nanoparticle in advanced solid tumors. Mol Ther. 2013. May;21(5):1096–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Kim SS, Rait A, Kim E, et al. A tumor-targeting p53 nanodelivery system limits chemoresistance to temozolomide prolonging survival in a mouse model of glioblastoma multiforme. Nanomedicine. 2015. Feb;11(2):301–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Kumthekar P, Ko CH, Paunesku T, et al. A first-in-human phase 0 clinical study of RNA interference-based spherical nucleic acids in patients with recurrent glioblastoma. Sci Transl Med. 2021. Mar 10;13(584). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Gregory JV, Kadiyala P, Doherty R, et al. Systemic brain tumor delivery of synthetic protein nanoparticles for glioblastoma therapy. Nat Commun. 2020. Nov 10;11(1):5687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Prentice DA. Adult Stem Cells. Circulation Research. 2019;124(6):837–839. [DOI] [PubMed] [Google Scholar]
- 144.Clevers H What is an adult stem cell? Science. 2015;350(6266):1319–1320. [DOI] [PubMed] [Google Scholar]
- 145.Seita J, Weissman IL. Hematopoietic stem cell: self-renewal versus differentiation. Wiley Interdisciplinary Reviews: Systems Biology and Medicine. 2010;2(6):640–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Gage FH. Mammalian neural stem cells. Science. 2000. Feb 25;287(5457):1433–8. [DOI] [PubMed] [Google Scholar]
- 147.Aboody KS, Brown A, Rainov NG, et al. Neural stem cells display extensive tropism for pathology in adult brain: evidence from intracranial gliomas. Proc Natl Acad Sci U S A. 2000. Nov 7;97(23):12846–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Bechmann I, Galea I, Perry VH. What is the blood–brain barrier (not)? Trends in Immunology. 2007 2007/January/01/;28(1):5–11. [DOI] [PubMed] [Google Scholar]
- 149.Spaeth E, Klopp A, Dembinski J, et al. Inflammation and tumor microenvironments: defining the migratory itinerary of mesenchymal stem cells. Gene Therapy. 2008 2008/May/01;15(10):730–738. [DOI] [PubMed] [Google Scholar]
- 150.Carey-Ewend AG, Hagler SB, Bomba HN, et al. Developing Bioinspired Three-Dimensional Models of Brain Cancer to Evaluate Tumor-Homing Neural Stem Cell Therapy. Tissue Eng Part A. 2021. Jul;27(13–14):857–866. [DOI] [PubMed] [Google Scholar]
- 151.Dvorak HF. Tumors: Wounds That Do Not Heal. New England Journal of Medicine. 1986;315(26):1650–1659. [DOI] [PubMed] [Google Scholar]
- 152.Müller F-J, Snyder EY, Loring JF. Gene therapy: can neural stem cells deliver? Nature Reviews Neuroscience. 2006 2006/January/01;7(1):75–84. [DOI] [PubMed] [Google Scholar]
- 153.Jung Y, Kim JK, Shiozawa Y, et al. Recruitment of mesenchymal stem cells into prostate tumours promotes metastasis. Nature Communications. 2013 2013/April/30;4(1):1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Rattigan Y, Hsu J-M, Mishra PJ, et al. Interleukin 6 mediated recruitment of mesenchymal stem cells to the hypoxic tumor milieu. Experimental Cell Research. 2010 2010/December/10/;316(20):3417–3424. [DOI] [PubMed] [Google Scholar]
- 155.Xu J, Wang D, Liu D, et al. Allogeneic mesenchymal stem cell treatment alleviates experimental and clinical Sjögren syndrome. Blood. 2012;120(15):3142–3151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Stuckey DW, Shah K. Stem cell-based therapies for cancer treatment: separating hope from hype. Nature Reviews Cancer. 2014 2014/October/01;14(10):683–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Magge SN, Malik SZ, Royo NC, et al. Role of monocyte chemoattractant protein-1 (MCP-1/CCL2) in migration of neural progenitor cells toward glial tumors. Journal of Neuroscience Research. 2009;87(7):1547–1555. [DOI] [PubMed] [Google Scholar]
- 158.Molina-Holgado E, Molina-Holgado F. Mending the broken brain: neuroimmune interactions in neurogenesis. Journal of Neurochemistry. 2010;114(5):1277–1290. [DOI] [PubMed] [Google Scholar]
- 159.Sasportas LS, Kasmieh R, Wakimoto H, et al. Assessment of therapeutic efficacy and fate of engineered human mesenchymal stem cells for cancer therapy. Proceedings of the National Academy of Sciences. 2009;106(12):4822–4827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Hingtgen SD, Kasmieh R, van de Water J, et al. A Novel Molecule Integrating Therapeutic and Diagnostic Activities Reveals Multiple Aspects of Stem Cell-based Therapy. Stem Cells. 2010;28(4):832–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Menon LG, Kelly K, Yang HW, et al. Human Bone Marrow-Derived Mesenchymal Stromal Cells Expressing S-TRAIL as a Cellular Delivery Vehicle for Human Glioma Therapy. Stem Cells. 2009;27(9):2320–2330. [DOI] [PubMed] [Google Scholar]
- 162.Kauer TM, Figueiredo J-L, Hingtgen S, et al. Encapsulated therapeutic stem cells implanted in the tumor resection cavity induce cell death in gliomas. Nature Neuroscience. 2012 2012/February/01;15(2):197–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Balyasnikova IV, Ferguson SD, Han Y, et al. Therapeutic effect of neural stem cells expressing TRAIL and bortezomib in mice with glioma xenografts. Cancer Letters. 2011 2011/November/28/;310(2):148–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Gutova M, Goldstein L, Metz M, et al. Optimization of a Neural Stem-Cell-Mediated Carboxylesterase/Irinotecan Gene Therapy for Metastatic Neuroblastoma. Mol Ther Oncolytics. 2017. Mar 17;4:67–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Copelan EA. Hematopoietic Stem-Cell Transplantation. New England Journal of Medicine. 2006;354(17):1813–1826. [DOI] [PubMed] [Google Scholar]
- 166.Portnow J, Synold TW, Badie B, et al. Neural Stem Cell–Based Anticancer Gene Therapy: A First-in-Human Study in Recurrent High-Grade Glioma Patients. Clinical Cancer Research. 2017;23(12):2951–2960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Sonabend AM, Ulasov IV, Tyler MA, et al. Mesenchymal Stem Cells Effectively Deliver an Oncolytic Adenovirus to Intracranial Glioma. Stem Cells. 2008;26(3):831–841. [DOI] [PubMed] [Google Scholar]
- 168. Fares J, Ahmed AU, Ulasov IV, et al. Neural stem cell delivery of an oncolytic adenovirus in newly diagnosed malignant glioma: a first-in-human, phase 1, dose-escalation trial. The Lancet Oncology. 2021 2021/August/01/;22(8):1103–1114. *(Phase I clinical trial of NSC-CRAd-S-pk7 showing safety, and data supporting a phase 2/3 clinical trial).
- 169.Sloan AE, Roger L, Murphy C, et al. A phase I study of MGMT-P140K transfected hematopoetic progenitor cells (HPC) combined with TMZ/O6BG dose escalation for newly diagnosed, MGMT unmethylated glioblastoma: Tolerance and evidence of survival benefit. Journal of Clinical Oncology. 2019;37(15_suppl):2062–2062.31216227 [Google Scholar]