

Abstract
Immune-mediated bile duct epithelial injury and toxicity of retained hydrophobic bile acids drive disease progression in fibrosing cholangiopathies such as biliary atresia or primary sclerosing cholangitis. Emerging therapies include pharmacological agonists to farnesoid X receptor (FXR), the master regulator of hepatic synthesis, excretion, and intestinal reuptake of bile acids. Unraveling the mechanisms of action of pharmacological FXR agonists in the treatment of sclerosing cholangitis (SC), we found that intestinally restricted FXR activation effectively reduced bile acid pool size but did not improve the SC phenotype in MDR2−/− mice. In contrast, systemic FXR activation not only lowered bile acid synthesis but also suppressed proinflammatory cytokine production by liver-infiltrating inflammatory cells and blocked progression of hepatobiliary injury. The hepatoprotective activity was linked to suppressed production of IL1β and TNFα by hepatic macrophages and inhibition of TH1/TH17 lymphocyte polarization. Deletion of FXR in myeloid cells caused aberrant TH1 and TH17 lymphocyte responses in diethoxycarbonyl-1,4-dihydrocollidine–induced SC and rendered these mice resistant to the anti-inflammatory and liver protective effects of systemic FXR agonist treatment. Pharmacological FXR activation reduced IL1β and IFNγ production by liver- and blood-derived mononuclear cells from patients with fibrosing cholangiopathies. In conclusion, we demonstrate FXR to control the macrophage-TH1/17 axis, which is critically important for the progression of SC. Hepatic macrophages are cellular targets of systemic FXR agonist therapy for cholestatic liver disease.
INTRODUCTION
Sclerosing cholangitis (SC) results from progressive destruction of large or small bile ducts, liver inflammation, and biliary fibrosis. Several pediatric conditions can present with this liver disease phenotype including extrahepatic biliary atresia (EHBA) in neonates, progressive familial intrahepatic cholestasis from multidrug resistance 3 (MDR3) deficiency in younger children, or primary SC (PSC) in adolescents with inflammatory bowel disease. Although the etiologic factors vary between these conditions, toxic bile and T lymphocyte–mediated cholangiocyte injury have emerged as key drivers of the SC phenotype (1). Hydrophobic bile acids (BAs), such as taurochenodeoxycholate (TCDCA) or glycochenodeoxycholate, which are retained in the liver and plasma of patients with EHBA or PSC, were shown to induce hepatocellular apoptosis and to reduce bile flow in perfused rat livers (2–4). Under both conditions, aberrant activation of cytotoxic CD8 and effector T helper (TH) 1 and TH17 lymphocytes is critical for initiation and progression of bile duct epithelial injury (5–8). These lymphocyte responses, including production of type 1 cytokines and chemokines, are at least in part controlled by the inflammasome and monocyte-derived proinflammatory cytokines (9, 10). Although we currently lack therapies controlling the specific etiologic factors underlying EHBA or PSC, complications from impaired bile flow through an injured biliary tree and progression of fibrosis have been the focus of preclinical investigations and clinical trials. Molecular targets of these emerging therapies include the nuclear BA receptor FXR (farnesoid X receptor), master regulator for key enzymes of BA homeostasis. FXR activation in hepatocytes leads to up-regulation of its target gene small heterodimer protein (SHP), which represses expression of CYP7A1, a rate-limiting enzyme of BA synthesis. Fibroblast growth factor 19 (FGF19; Fgf15 in mice) is a gastrointestinal hormone released by enterocytes into the portal circulation upon FXR-mediated sensing of intestinal BA and down-regulates hepatic BA synthesis via signaling though the receptor FGF4R on hepatocytes. Pharmacological FXR agonists were shown to reduce serum alkaline phosphatase (ALP) activity, a surrogate marker for biliary injury, in clinical trials in PSC (11–13). Recently, longer-term follow-up studies on obeticholic acid (OCA), a BA-derived FXR agonist and U.S. Food and Drug Administration–approved therapy for primary biliary cholangitis (PBC), raised concerns about drug accumulation causing pruritus and hepatic decompensation in patients with cirrhosis (14). These safety concerns in conjunction with an evolving field of FXR agonists with various chemical structures contributing to substantial differences in pharmacokinetics prompted us to investigate whether intestinally restricted FXR (iFXR) agonists would be as efficacious as, but safer than, systemic ones in the treatment of cholestatic liver disease.
The goal of our study was to explore the role of synthetic FXR agonist tropism in controlling the SC phenotype and to better understand the mechanism of action of pharmacological FXR activation in the treatment of fibrosing cholangiopathies. To this end, we used FXR agonists with systemic and intestinal tropism that were derived from the nonsteroidal FXR agonist fexaramine (15–17). Fexaramine, which has a short half-life and primarily activates FXR in the intestine, improves obesity and metabolic functions in preclinical models. MET409, a fexaramine derivative with sustained systemic FXR (sFXR) activation, was recently shown to improve steatosis in a short-term clinical trial in patients with nonalcoholic steatohepatitis (NASH) (16, 17). We studied systemic and intestinal FXR activation in two murine models of fibrosing cholangiopathies. The first model is the MDR2 knockout (MDR2−/−) mouse, which lacks biliary phospholipids leading to toxic BA accumulation and displays diminished bicarbonate umbrella protecting cholangiocytes, sterile inflammation, and rapid progression of biliary injury and fibrosis (18). In this model, bile duct epithelial injury is modulated by TH1-polarized and regulatory CD4 lymphocytes, and interferon-γ (IFNγ)–producing CD8+ and natural killer lymphocytes promote fibrosis (19, 20). In the second model, mice are fed with 3,5-diethoxycarbonyl-1,4-dihydrocollidine (DDC), which causes SC via the formation of intraductal porphyrin plugs (21). Both models recapitulate various aspects of intrahepatic disease progression in patients with EHBA after Kasai portoenterostomy or PSC, including liver histopathological changes such as ductal proliferation and biliary fibrosis and the involvement of various innate inflammatory and adaptive lymphocyte responses. Here, we tested whether hepatic macrophages and lymphocytes that drive sterile inflammation and immune-mediated bile duct destruction, respectively, are direct cellular targets of pharmacological FXR agonists.
RESULTS
Intestinal FXR activation represses BA synthesis but fails to protect from progression of SC
We reasoned that if control of BA pool size was the primary mechanism of action for pharmacological FXR agonists in the treatment of SC, then sFXR (M345 or M044) and iFXR agonists (M379) should be similarly effective in blocking hepatobiliary injury if both reduced hepatic retention of BA. To test this, MDR2−/− mice were treated with either of these compounds for 7 days. Compared with vehicle treatment, administration of an sFXR, but not an iFXR agonist, up-regulated hepatic expression of the FXR target gene Shp, confirming the minimal systemic effects of the iFXR agonist (Fig. 1A). Both agonists induced Shp and Fgf15 expression in the terminal ileum. Both agonists also significantly lowered plasma concentrations of C4 (P < 0.01), a biomarker of hepatic BA de novo synthesis, and similarly decreased total liver BA concentrations in MDR2−/− mice, demonstrating similar anticholestatic effects (Fig. 1B) (22). However, only administration of an sFXR, but not an iFXR agonist, significantly reduced serum biomarkers of hepatobiliary injury that were all highly elevated in MDR2−/− compared with age- and sex-matched noncholestatic MDR2+/− mice, including ALP and total bilirubin (TB; P < 0.05) (Fig. 1C). Liver sections from MDR2−/− mice of the three treatment groups underwent immunohistochemical (IHC) staining for cytokeratin-19 (CK19), a tissue biomarker for biliary mass and injury, and subsequent image analysis (Fig. 1D). The CK19+ area was decreased after administration of sFXR compared with vehicle, corroborating the results of the analysis of serum liver biochemistries. Although the focus of this investigation was to delineate the effects of short-term treatment with FXR agonists on biliary epithelial injury, we observed reduced liver fibrosis, as assessed by Sirius Red staining of liver sections, and decreased expression of Spp1 encoding osteopontin, a profibrogenic cytokine implicated in SC pathogenesis, after treatment with an sFXR agonist (fig. S1).
Fig. 1. Intestinally restricted FXR agonist reduces bile acid synthesis but fails to improve sclerosing cholangitis in MDR2−/− mice.
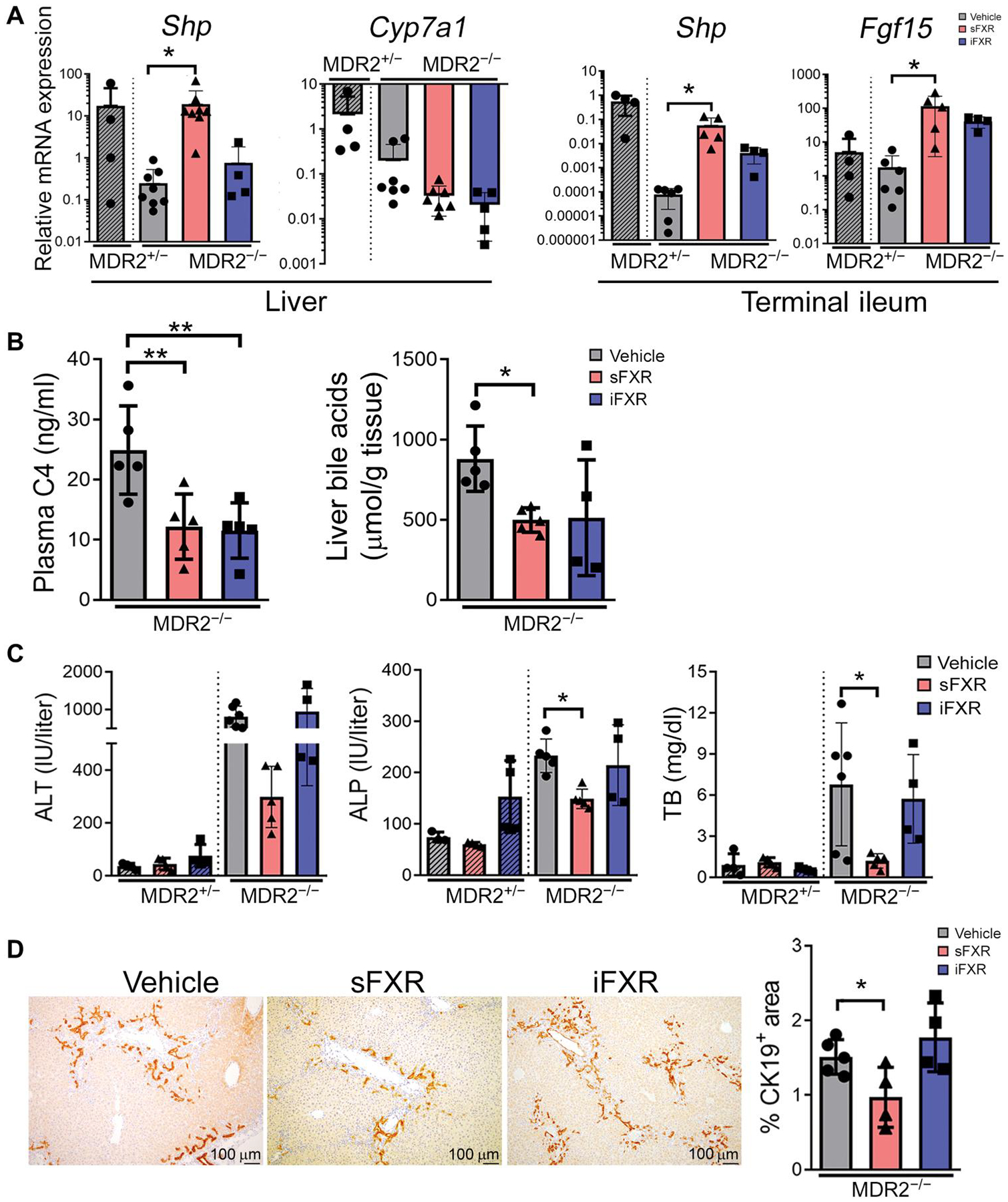
(A) MDR2−/− mice were subjected to 7 days of treatment with vehicle, systemic FXR agonist (sFXR; M345), or intestinally restricted FXR (iFXR) agonist (M379) before collection of tissues to quantitate relative mRNA expressions by qPCR for FXR target genes in the liver and terminal ileum. Vehicle-treated, noncholestatic MDR2+/− mice served as controls. (B) End of treatment plasma 7α-hydroxy-4-cholesten-3-one (C4) concentrations were measured by a stable isotope dilution tandem mass spectrometry method. Liver tissues were extracted with organic solvents, and bile acid concentrations were determined by colorimetric assay. (C) Serum samples were collected after 7 days of treatment and assayed for alanine aminotransferase (ALT), alkaline phosphatase (ALP), and total bilirubin (TB) by colorimetry. (D) Liver sections from mice of the three groups at the end of treatment were subjected to CK19 immunohistochemistry and subsequent image analysis. Results are means ± SD; each dot represents an individual mouse. Multiplicity adjusted P values were determined by one-way ANOVA with Dunnett’s post hoc test compared to mean of the vehicle. *P < 0.05; **P < 0.01.
Systemic FXR activation represses proinflammatory hepatic cytokine responses
Probing for mechanisms by which sFXR agonist treatment attenuated the SC phenotype, hepatic mRNA expression for inflammatory genes was determined by quantitative polymerase chain reaction (qPCR). As expected, mRNA expression of the genes interleukin-1β (Il1b), tumor necrosis factor-α (Tnfa), and Il6, all previously associated with SC pathogenesis (23), was higher in vehicle-treated MDR2−/− mice compared with MDR2+/− mice serving as age- and sex-matched, noncholestatic controls (Fig. 2A). Compared with vehicle treatment, hepatic expression of Il1b and Tnfa was significantly lower in MDR2−/− mice after treatment with sFXR (P < 0.05) but not with iFXR agonists. To determine whether the differential gene expression between treatment groups originated at least in part from direct effects of the sFXR agonist on liver-infiltrating inflammatory cells, liver mononuclear cells (LMNCs) were isolated from wild-type (WT) and MDR2−/− mice, cultured, and stimulated with lipopolysaccharide (LPS), a pathogen-associated molecular pattern (PAMP), in the presence of various concentrations of sFXR agonist. Dose-dependent induction of the FXR target gene Shp was observed in LMNCs from MDR2−/− mice upon culture with sFXR agonist (Fig. 2B). Conditioning of the culture medium with sFXR agonist resulted in dose-dependent repression of LPS-induced gene expression for IL1β and type 1 cytokines IFNγ and TNFα. Expression of NLR family pyrin domain containing 3 (Nlrp3), which encodes the inflammasome complex that cleaves pro-IL1β to become biologically active, was also repressed by FXR activation. Because liver-infiltrating T lymphocytes are an important source of type 1 cytokine production in cholestatic liver disease, we examined the effects of FXR activation on LPS-induced IFNγ production by CD4 and CD8 lymphocytes via intracellular flow cytometry (ICF) after restimulation with phorbol 12-myristate 13-acetate (PMA)/ionomycin. Incubation of LMNCs from MDR2−/− mice with sFXR agonist abrogated LPS-induced IFNγ production by both T cell populations in vitro (Fig. 2C and fig. S2 with gating strategy).
Fig. 2. FXR activation represses proinflammatory cytokine expression in liver mononuclear cells.
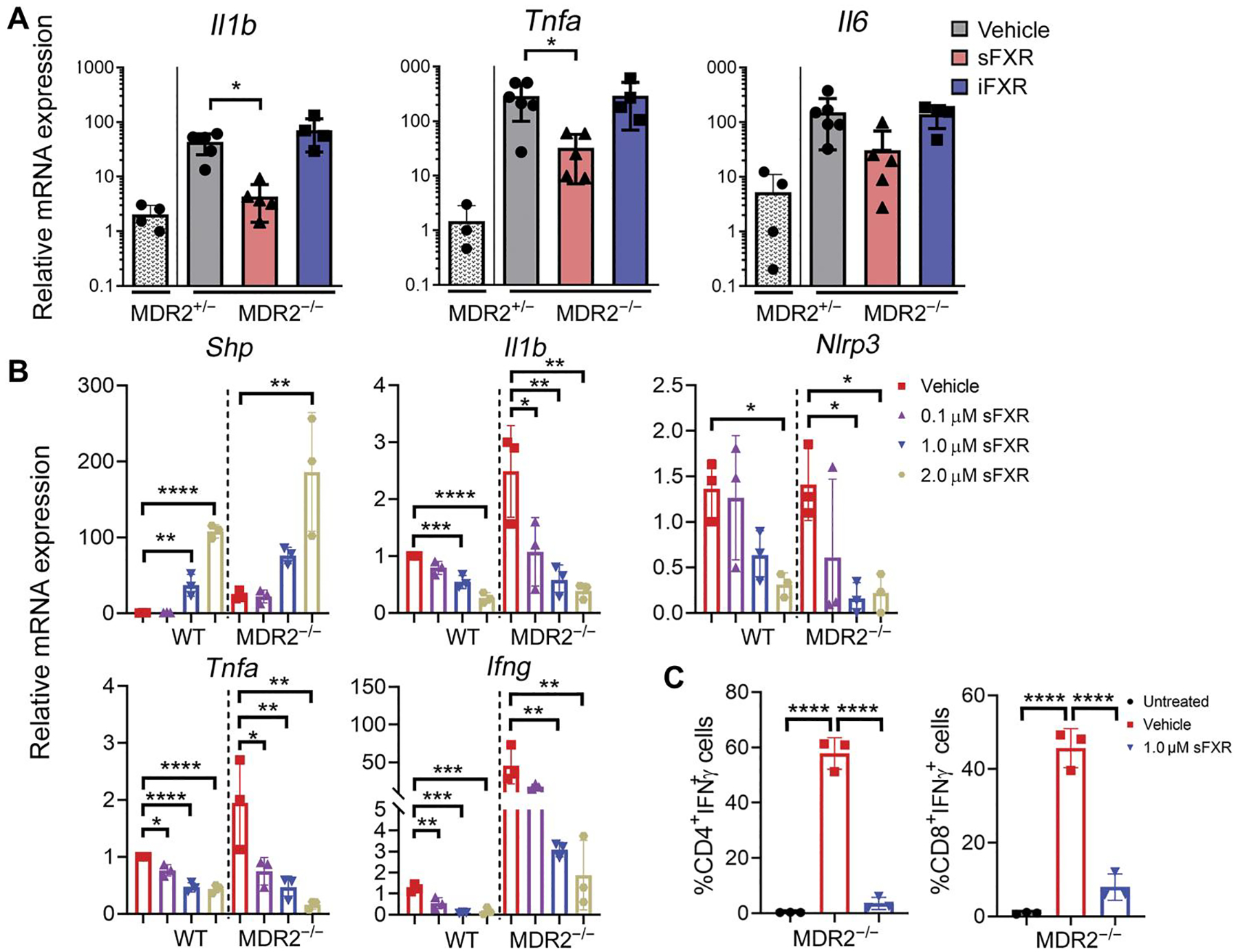
(A) Gene expression of proinflammatory cytokines in whole livers from MDR2−/− mice treated for 1 week with vehicle (corn oil), sFXR (M345), or iFXR (M379) was quantitated by TaqMan qPCR. Vehicle-treated MDR2+/− mice served as controls. (B) Liver mononuclear cells (LMNCs) isolated from WT or MDR2−/− mice were pretreated with various concentrations of FXR agonist (M044) and cultured in the presence of LPS (100 ng/ml) for 16 hours. mRNA abundance of FXR target Shp and proinflammatory genes was quantitated by qPCR in cultured LMNCs. (C) LMNCs isolated form MDR2−/− mice were stimulated with LPS in the presence of FXR agonist (M044) or vehicle before intracellular flow cytometry (ICF) to determine expression of IFNγ by CD4+ and CD8+ T cells. Cultured LMNCs without exposure to LPS or FXR agonist served as untreated controls. Results are means ± SD; n = 3 to 5 mice per group. Each dot represents an individual mouse. Multiplicity adjusted P values were determined using one-way ANOVA with Dunnett’s post hoc test compared to mean of the vehicle. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
FXR controls macrophage and T lymphocyte effector cytokine production
IFNγ and IL17A drive immune-mediated bile duct injury in PSC and EHBA (19, 24). To elucidate whether T lymphocytes are direct cellular targets of FXR agonists, we used an established model of DDC-induced SC (21). Seven days of exposure to 0.1% DDC admixed to the diet resulted in liver inflammation and bile duct proliferation in this model accompanied by a rise in serum alanine aminotransferase (ALT), ALP, and TB (fig. S3), all consistent with SC and similar to the phenotype observed in MDR2−/− mice. LMNCs were isolated from DDC-fed mice and cultured in the presence of LPS and FXR agonist. Conditioning of the culture medium with FXR agonist reduced IFNγ and IL17A production by CD4+ and CD8+ lymphocytes (Fig. 3A). To find out whether T lymphocytes were directly targeted by FXR agonist, LMNCs were separated into CD45+CD3+CD4+ and CD45+CD3+CD8+ T lymphocyte populations by fluorescence-activated cell sorting (FACS) and subsequently cultured in the presence of LPS and FXR agonist. sFXR treatment did not significantly (P ≥ 0.05) decrease IFNγ production in purified CD4+ or CD8+ lymphocytes, indicating that T lymphocytes were not the direct cellular targets of FXR agonist (Fig. 3B). Testing the hypothesis that macrophages were targeted by FXR agonist, we cultured bone marrow–derived macrophages (BMDMs) and stimulated them with LPS. FXR activation repressed LPS-induced transcription of Il1b, Nlrp3, and Tnfa in cultured BMDMs (Fig. 3C). Next, BMDMs from WT and FXR−/− mice were stimulated with either LPS or TCDCA, a hydrophobic BA implicated as danger-associated molecular pattern (DAMP) in cholestatic liver disease (25). The concentration of 100 μM for TCDCA is similar to what was found in the plasma of patients with EHBA (4). Both danger signals induced IL1β secretion by BMDMs from WT and FXR−/− mice, which was reduced upon FXR activation (Fig. 3D). FXR agonism failed to reduce IL1β production by BMDM from FXR−/− mice stimulated with LPS or TCDCA, demonstrating the specificity of the synthetic ligand M044 for FXR. FXR activation significantly reducedNLRP3 protein expression in WT-BMDM stimulated with LPS (P < 0.0001) or TCDCA (P < 0.01). Genetic deletion of Takeda G protein–coupled receptor 5 (TGR5), the surface BA receptor on macrophages, did not abrogate TCDCA-induced IL1β production by BMDMs (fig. S4).
Fig. 3. A synthetic FXR agonist targets macrophages and indirectly controls effector cytokine production by T lymphocytes.
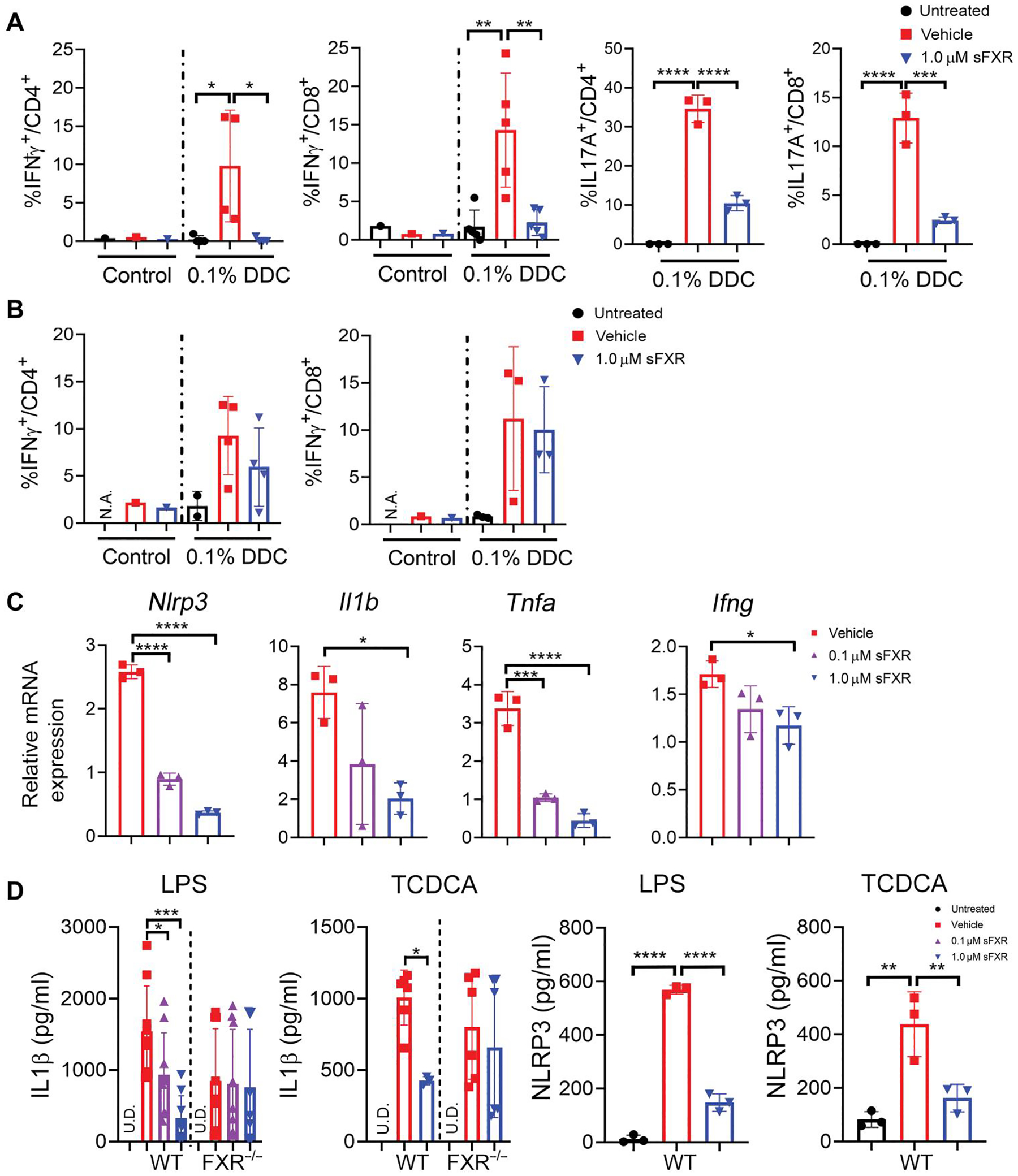
(A) LMNCs were isolated from mice fed either a diet containing 0.1% DDC or control diet for 7 days. Cells were either left untreated or stimulated with LPS (100 ng/ml) in the presence or absence of 1.0 μM FXR agonist (M044) for 16 hours before ICF to determine expression of IFNγ and IL17A by CD4+ and CD8+ T cells. (B) CD4+ and CD8+ T cells were separated from LMNCs by FACS. Purified lymphocytes were cultured under the same conditions as described in (A) before ICF analysis. Results are means ± SD; n = 3 to 5 mice per group. Each dot represents the result from an individual mouse. Multiplicity adjusted P values were determined using one-way ANOVA with Dunnett’s post hoc test compared to mean of the vehicle. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. (C) Bone marrow–derived macrophages (BMDMs) from WT mice were cultured, pretreated with vehicle or FXR agonist (M044) at 0.1 or 1 μM concentrations, stimulated with LPS (100 ng/ml), and subjected to RNA extraction for subsequent TaqMan qPCR. (D) BMDMs from WT or FXR−/− mice were cultured, pretreated with FXR agonist (M044) or vehicle, and stimulated with LPS (100 ng/ml) or 100 μM TCDCA. IL1β concentration was measured in the supernatant by TR-FRET, and NLRP3 protein concentration was measured in lysed cells by enzyme-linked immunosorbent assay (ELISA). Results are means ± SD. Each dot represents an individual culture well, and results are pooled from two independent experiments. Multiplicity adjusted P values were determined using one-way ANOVA with Dunnett’s post hoc test compared to mean of the vehicle. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. U.D., undetected.
FXR controls innate cytokine production by hepatic macrophages
To investigate whether responses of BMDMs to danger signals and FXR activation recapitulate functions of hepatic macrophages as key drivers of liver inflammation, we performed experiments with macrophages isolated from livers of WT, FXR−/−, and FXRΔMC mice, in which FXR was deleted in LysM-cre–expressing myeloid cells. Expression of Cre in the endogenous M lysozyme locus of myeloid cells was previously reported to delete loxP-flanked target genes in 83 to 98% of mature macrophages and in 100% of granulocytes (26). Using immunofluorescence (IF) and confocal microscopy, we found that only CD11b+ macrophages from WT, but not from global or conditional FXR-deficient mice, expressed FXR protein (Fig. 4A). When hepatic macrophages from WT mice were cultured and stimulated with LPS, pharmacological FXR activation up-regulated Shp and repressed transcription of proinflammatory genes Nlrp3, Il1b, and Tnfa (Fig. 4B). Expression of these genes was not significantly altered in hepatic macrophages from FXR−/− or FXRΔMC mice when cultured in the presence of FXR agonist. FXR activation reduced both LPS- and TCDCA-induced secretion of IL1β and TNFα by macrophages (Fig. 4, C and D). FXR agonist did not reduce viability of Kupffer cells (percentage live cell count before and after treatment with 1.0 μM FXR; WT, 93 ± 1.5% and 87 ± 2.5%; FXR−/−, 90 ± 3.5% and 84 ± 5.5%; n = 3 independent experiments). Regulation was specific to FXR activation; both LPS- and TCDCA-induced secretion of IL1β and TNFα by hepatic macrophages from FXR−/− mice was not suppressed by sFXR treatment. To test whether other myeloid cells that play a role in the pathogenesis of SC express FXR, neutrophils were isolated from the bone marrow of WT and FXRΔMC mice and expanded in culture. Confocal imaging showed no expression of FXR in Gr-1–positive mature neutrophils (fig. S5A). Neutrophils were also not responsive to FXR agonist treatment when stimulated with LPS (fig. S5B).
Fig. 4. FXR activation induces Shp in hepatic macrophages and blocks IL1β and TNFα responses to danger signals.
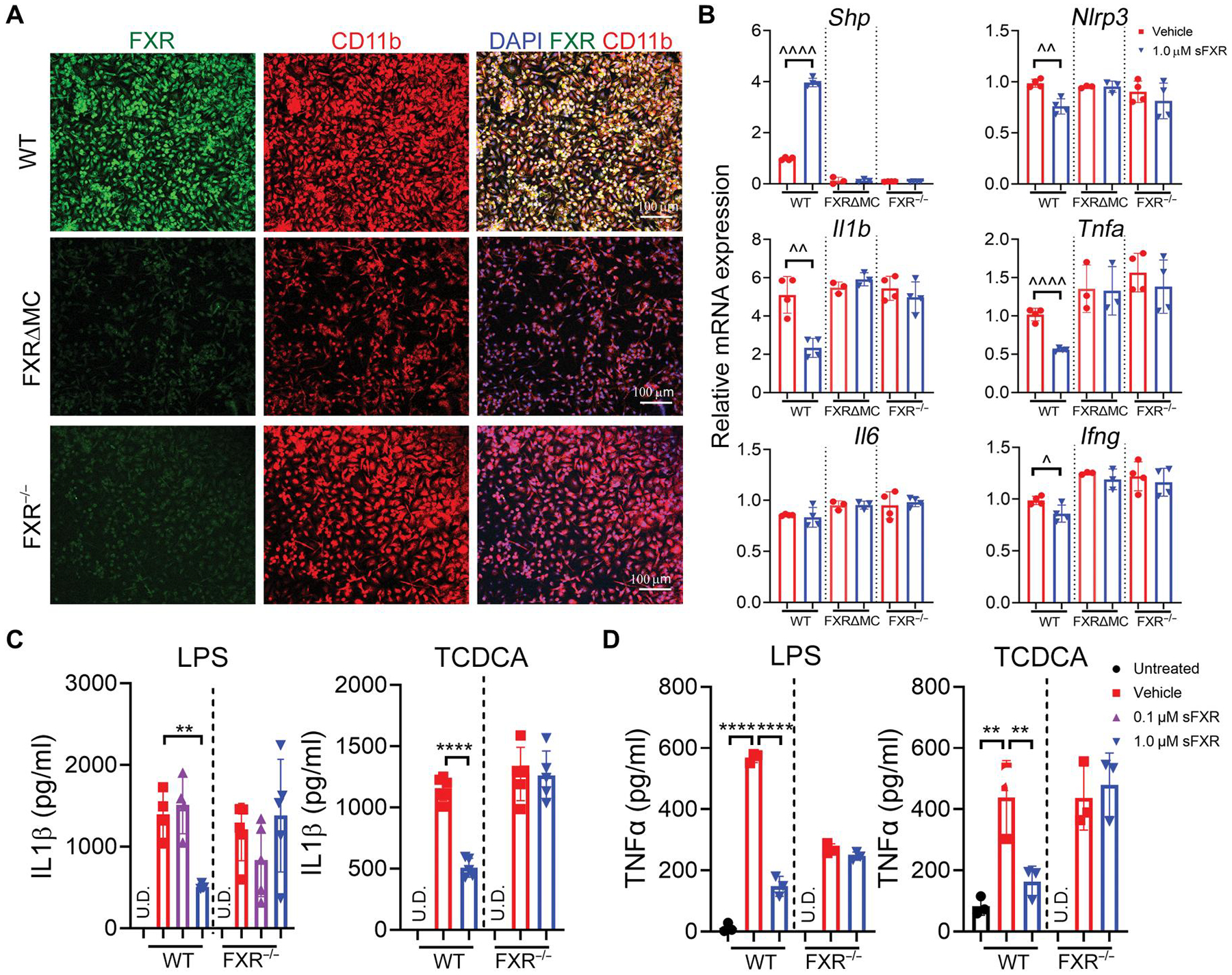
(A) Hepatic macrophages were isolated from WT, FXR
IL1β controls TH17 polarization and liver phenotype in xenobiotic-induced SC
Macrophages are established drivers of liver injury in the DDC model of SC (27). IL1β secreted by macrophages have previously been shown to act as licensing signal for effector IFNγ cytokine production by T lymphocytes expressing IL1 receptor α (IL1Rα) (28). In addition, monocyte-derived IL1β and IL6 were found to polarize TH17 lymphocytes in PSC (29). We investigated the macrophage–IL1β–T lymphocyte axis and its relevance for the SC phenotype in the model of DDC-induced SC (21). LMNCs were isolated from DDC-treated and control mice and cultured in the presence of LPS and various concentrations of an IL1Rα antagonist. IFNγ expression by T lymphocyte subsets was measured by ICF (Fig. 5A). LPS induced higher IFNγ production in T lymphocytes among LMNCs from DDC-treated versus control mice, indicating that a DDC-induced priming of effector lymphocytes functions in vivo. An IL1Rα antagonist reduced LPS-induced IFNγ expression by CD4+ and CD8+ lymphocytes from DDC-treated mice in a dose-dependent fashion. To examine the role of IL1β in control of hepatic immune activation and phenotype of SC in vivo, WT and Il1r1−/− mice, in which IL1β signaling was disrupted, were challenged with 0.1% DDC diet for 7 days. Compared with WT, LMNCs from DDC-fed Il1r1−/− mice displayed slightly decreased IFNγ and nearly abrogated IL17A production by CD4+ or CD8+ lymphocytes upon LPS stimulation (Fig. 5B). Liver injury was less severe in Il1r1−/− mice compared with WT mice upon DDC challenge based on lower serum ALT, ALP, and TB concentrations and less ductal proliferation (Fig. 5, C and D). Collectively, the findings demonstrate that the IL1β/IL1R axis shapes licensing of T lymphocyte responses and determines the SC phenotype in mice challenged with DDC.
Fig. 5. IL1β signaling mediates injury responses in xenobiotic sclerosing cholangitis.
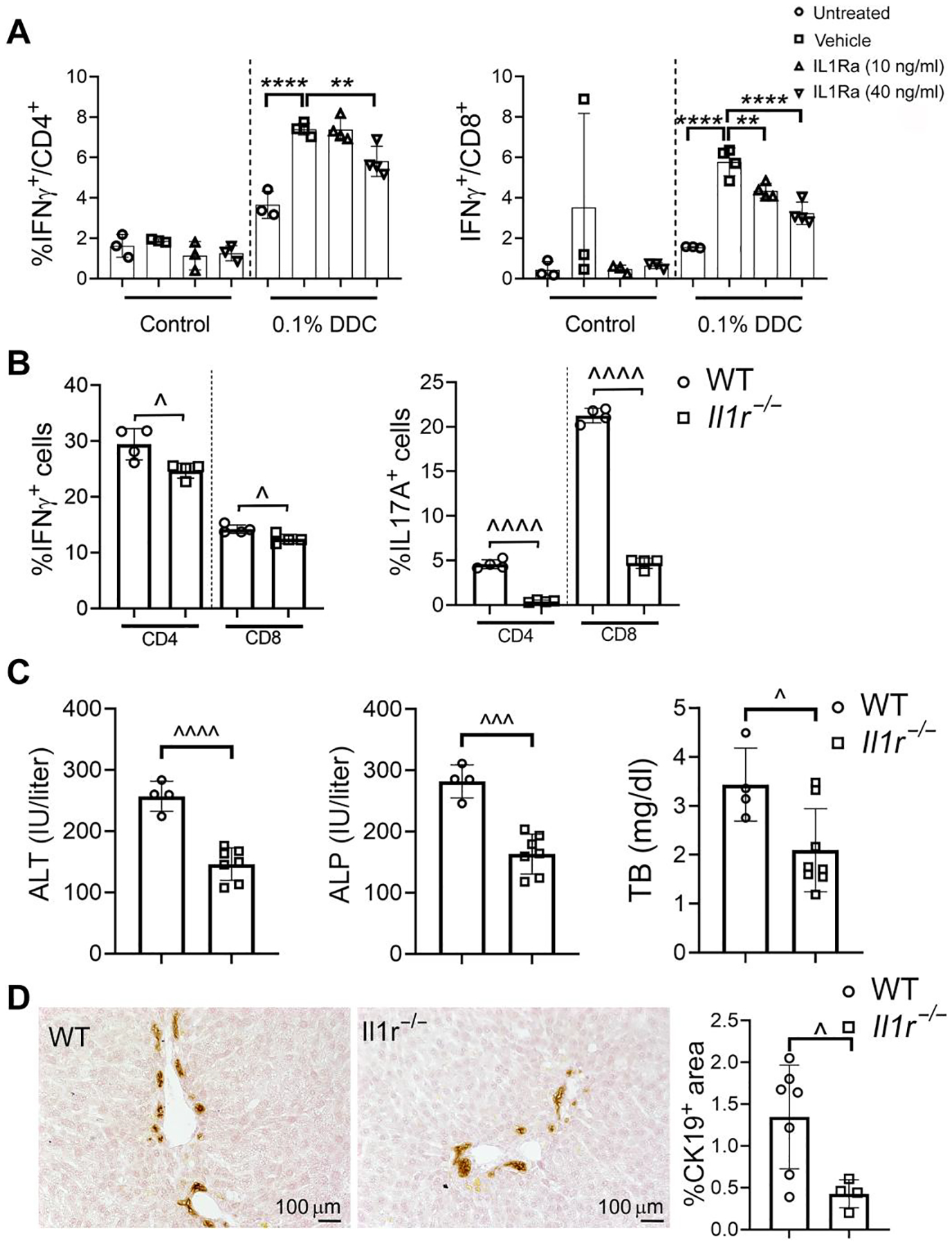
(A) LMNCs were isolated from WT mice fed with either 0.1% DDC or control chow. Cells were cultured, treated with various concentrations of IL1Rα antagonist or vehicle, stimulated with LPS (100 ng/ml), and subjected to ICF analysis to determine IFNγ expression by CD4+ and CD8+ T cells. Dots represent results from individual wells of two independent experiments. Multiplicity adjusted P values were determined using one-way ANOVA with Dunnett’s post hoc test compared to mean of the vehicle. **P < 0.01; ****P < 0.0001. (B) LMNCs were isolated from WT and Il1r1−/− mice after treatment with DDC for 7 days, stimulated with LPS, and subjected to ICF to determine expression of IFNγ and IL17A by CD4+ and CD8+ T lymphocyte subsets. (C) Serum samples were collected from WT and Il1r1−/− mice after 7 days of DDC challenge and assayed for ALT, ALP, and TB. (D) Ductal proliferation was assessed by CK19 IHC and image analysis on liver sections from mice exposed to DDC for 7 days. In (B) to (D), results are represented as means ± SD with each dot denoting results from individual mice. P values were calculated using unpaired, two-tailed t tests with ^P < 0.05; ^^^P < 0.001; ^^^^P < 0.0001.
FXR signaling in myeloid cells controls severity of murine SC
To directly test the hypothesis that FXR suppression of hepatic macrophage function mediates the hepatoprotective effects of FXR agonist treatment in preclinical SC, we examined the relationship between BA homeostasis, T lymphocyte cytokine production, and SC phenotype in WT and FXRΔMC mice exposed to DDC and pharmacological FXR agonist. First, we confirmed that FXR deletion was selective for myeloid cells in FXRΔMC mice leaving FXR signaling in hepatocytes intact. Hepatocytes were isolated from WT, FXRΔMC, and FXR−/− mice, maintained in short-term cultures, and subjected to IF and qPCR studies. Primary hepatocytes from mice of the three genotypes expressed hepatocyte nuclear factor-4 alpha (HNF4α), the master regulator of hepatocyte differentiation, which localized to the 4′,6-diamidino-2-phenylindole (DAPI)–stained nuclei. FXR was expressed and colocalized with HNF4α in hepatocytes from WT and FXRΔMC mice, but not from FXR−/− mice (Fig. 6A). FXR activation by the synthetic agonist M044 up-regulated Shp and down-regulated Cyp7a1 in primary WT and FXRΔMC hepatocytes (Fig. 6B), confirming intact FXR signaling in hepatocytes from conditional FXR knockout mice. FXR activation minimally reduced LPS-induced Il1b expression by hepatocytes from WT mice and had no effect on expression of chemokine ligand Cxcl1 in hepatocytes of either of the three genotypes. Compared with vehicle-treated mice exposed to DDC, hepatic mRNA expression of Shp increased and that of Cyp7a1 decreased after treatment of WT and FXRΔMC mice with sFXR agonist for 7 days (Fig. 6C). Consequently, total liver BA concentrations were significantly decreased upon sFXR treatment in both groups (WT, P < 0.001; FXRΔMC, P < 0.05) (Fig. 6D), confirming intact FXR regulation of BA homeostasis in FXRΔMC mice.
Fig. 6. FXR-mediated control of bile acid homeostasis is preserved in FXRΔMC mice with targeted deletion of FXR in myeloid cells.
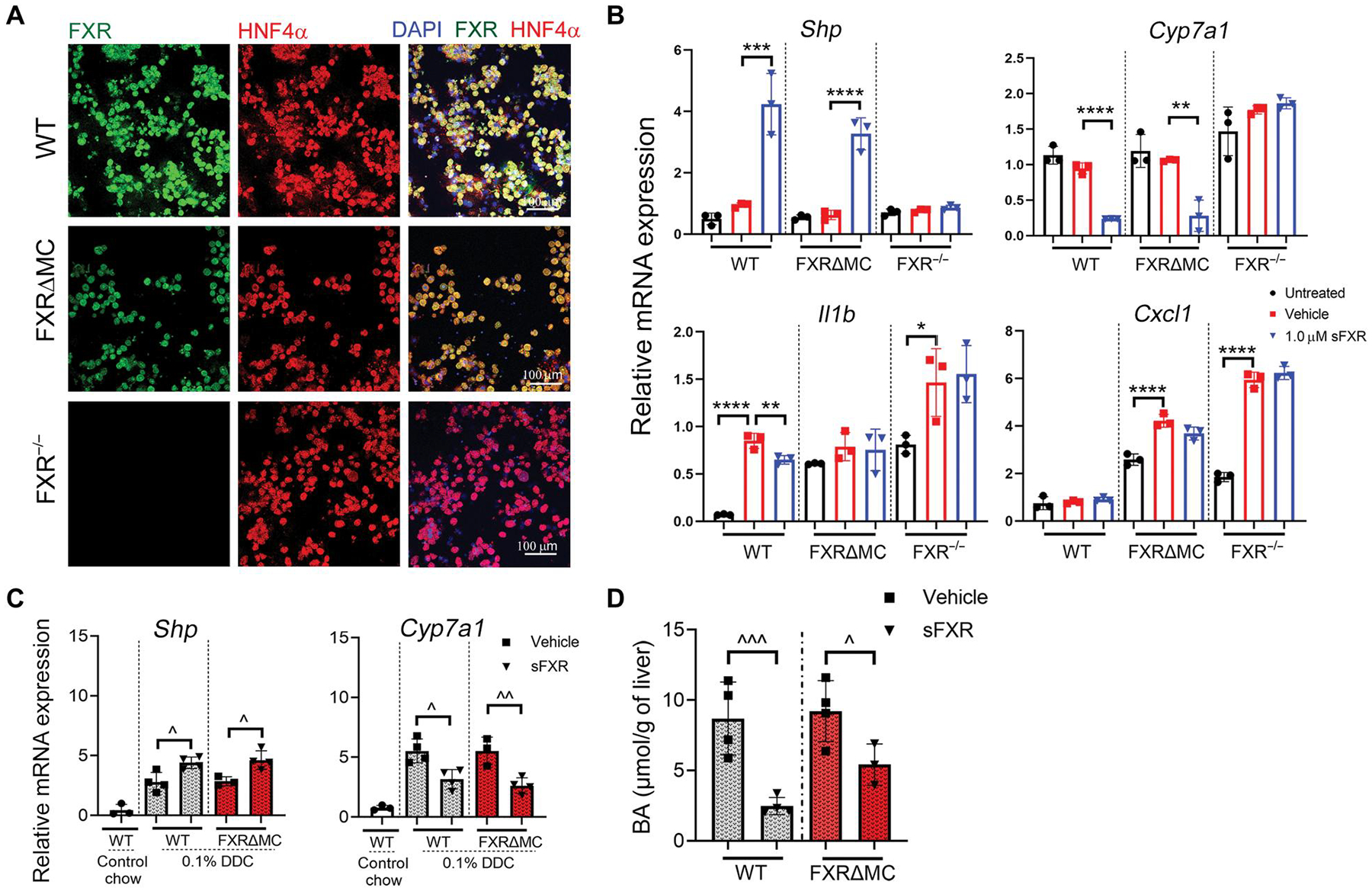
(A) Primary hepatocytes were isolated from WT, FXRΔMC, and FXR−/− mice, cultured, and subjected to dual IF for FXR (green) and HNF4α+ (red) with DAPI staining nuclei. (B) Hepatocytes from WT, FXRΔMC, or FXR−/− mice were cultured, pretreated with 1.0 μM FXR agonist M044 or vehicle, stimulated with LPS (100 ng/ml), followed by isolation of total RNA and TaqMan qPCR of FXR target, bile acid synthesis, and proinflammatory genes. Dots represent individual wells. Multiplicity adjusted P values were determined using one-way ANOVA with Dunnett’s post hoc test compared to mean of the vehicle. * P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. (C) WT and FXRΔMC were challenged with 0.1% DDC for 7 days and treated with either vehicle or sFXR agonist M044 for 6 days before collection of livers for total RNA isolation and quantitation of FXR target gene expression by qPCR. (D) Liver tissue extracts from these mice were subjected to colorimetric assays to measure total bile acid concentrations. Results are means ± SD with each dot denoting individual mice. P values were calculated using unpaired, two-tailed t tests with ^P < 0.05; ^^P < 0.01; ^^^P < 0.001.
Next, we examined whether selective deletion of FXR in myeloid cells rendered FXRΔMC mice more susceptible to xenobiotic-induced liver injury, despite intact regulation of BA homeostasis in hepatocytes. Compared with WT mice, FXRΔMC mice challenged with DDC displayed up-regulated hepatic gene expression for proinflammatory cytokines, increased IFNγ and IL17A production by LPS-stimulated CD4+ and CD8+ T lymphocytes, elevated serum ALT, ALP, and TB concentrations, and aggravated periportal inflammation and ductal proliferation on hematoxylin and eosin–and CK19-stained liver sections, respectively (fig. S6, A to D). We concluded that not only pharmacological but also endogenous FXR ligands tamed proinflammatory cytokine responses by hepatic macrophages and influenced the liver phenotype in xenobiotic SC.
Myeloid cells mediate hepatoprotective effects of pharmacological FXR agonists
Having demonstrated that FXR signaling in hepatocytes and FXR control of BA homeostasis were preserved in FXRΔMC mice, we examined to what extent loss of FXR in myeloid cells in these mice determined their response to pharmacological FXR agonist treatment of DDC-induced SC. Daily administration of sFXR agonist repressed hepatic expression of Il1b and Tnfa and significantly reduced IFNγ and IL17A production by CD4+ and CD8+ lymphocytes in WT mice challenged with DDC (P < 0.001); none of these proinflammatory cytokine responses were altered by sFXR agonist treatment in FXRΔMC mice (Fig. 7, A and B). sFXR agonist lowered serum ALT, ALP, and TB concentrations and decreased bile duct proliferation only in WT and not in FXRΔMC mice (Fig. 7, C and D). Further supporting a critical role of liver FXR activation in the treatment of SC, iFXR failed to improve the SC phenotype in WT mice challenged with DDC, which was consistent with findings in the MDR2−/− model (fig. S7). In summary, FXR in myeloid cells controlled innate cytokine production upon stimulation with extrinsic and intrinsic danger signals, restrained macrophage-dependent licensing of T lymphocytes, and limited bile duct epithelial injury. Myeloid cells mediated the therapeutic effects of sFXR agonists in xenobiotic-induced SC.
Fig. 7. Genetic deletion of FXR in myeloid cells confers resistance to FXR-directed treatment of models of sclerosing cholangitis.
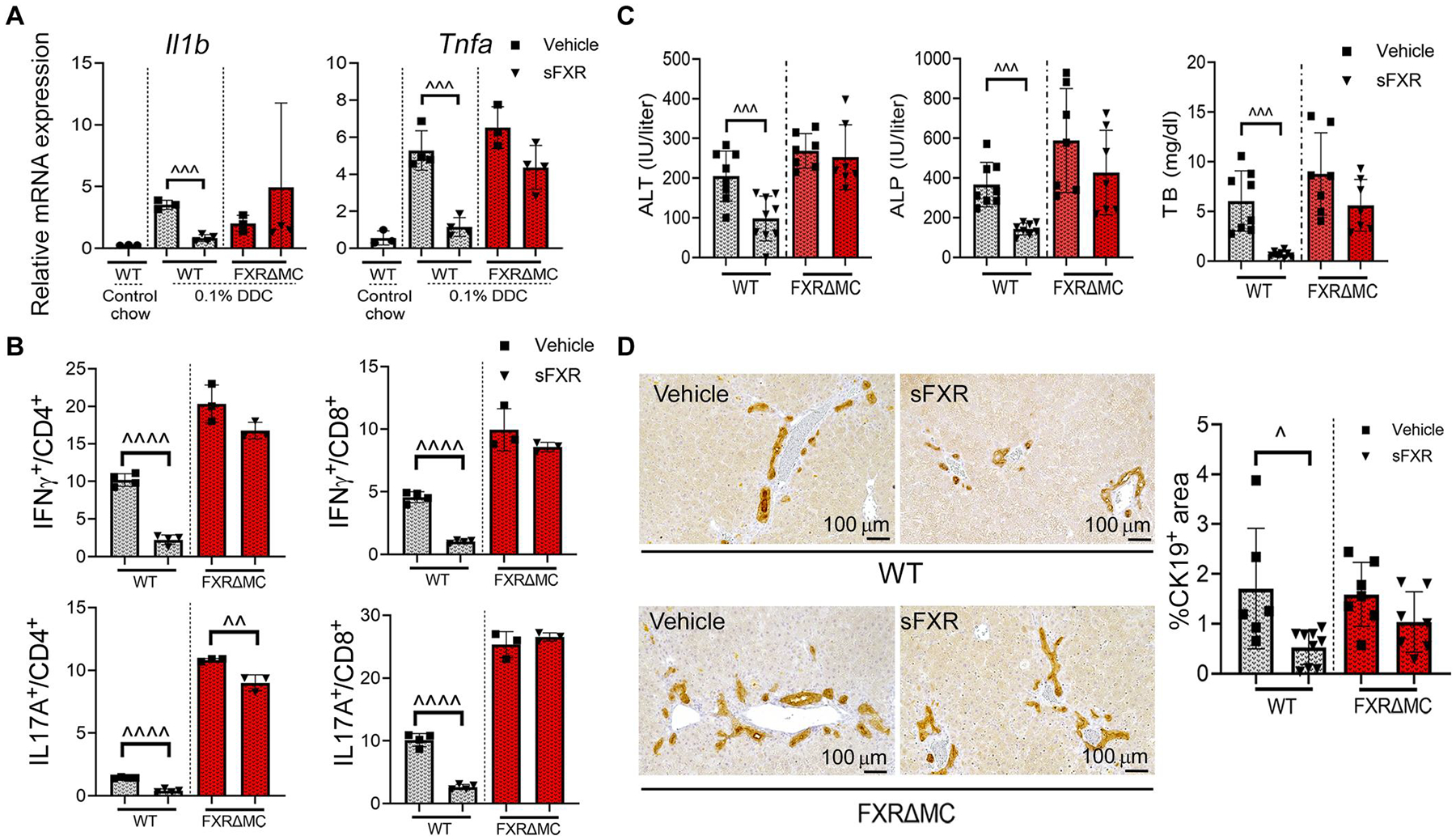
WT and FXRΔMC mice were challenged with 0.1% DDC for 7 days and treated with sFXR agonist M044 or vehicle for 6 days before collection of liver tissue and serum samples. (A) Whole liver mRNA expression of proinflammatory genes was quantitated by qPCR. (B) LMNCs were separated, cultured, stimulated with LPS, and subjected to ICF to determine expression of IFNγ and IL17A by CD4+ and CD8+ lymphocytes. (C) Serum liver biochemistries were measured by colorimetric assays. (D) Liver sections were subjected to IHC for CK19 followed by image analysis. Representative photomicrographs depict the degree of bile duct proliferation within the treatment groups. Results are means ± SD with each dot representing results from individual mice. P values were calculated using unpaired, two-tailed t tests with ^P < 0.05; ^^^P < 0.001; ^^^^P < 0.0001.
FXR activation blocks innate and adaptive cytokine responses in hepatic and circulating mononuclear cells from patients with fibrosing cholangiopathies
To validate our findings on the effects of FXR activation on T lymphocyte–derived cytokine production in preclinical models of SC, we separated mononuclear cells from liver tissue and peripheral blood of patients with fibrosing cholangiopathies and assessed the effects of FXR agonist on in vitro cytokine production. Details of the clinical characteristics of the study participants are included in table S1. LMNCs were isolated from explanted livers of two patients with advanced biliary fibrosis from EHBA, cultured, stimulated with LPS in the presence or absence of FXR agonist, and subjected to ICF analysis. In LMNCs from both patients, LPS-induced IFNγ production by CD4+ and CD8+ lymphocytes was partly blocked when the medium was conditioned with FXR agonist (Fig. 8A). To assess the effects of FXR agonist on innate cytokine production by circulating mononuclear cells from pediatric patients with PSC or from age-matched healthy controls (HCs), peripheral blood mononuclear cells (PBMCs) were cultured in the presence of various concentrations of FXR agonist, stimulated with LPS or TCDCA, and IL1β production was assayed by time-resolved fluorescence resonance energy transfer (TR-FRET). IL1β in culture supernatants was below detection limit without stimulation, LPS induced higher IL1β production in PBMCs from patients with PSC compared with HCs, and TCDCA induced variable amounts of IL1β in patients and controls (fig. S8). FXR agonist inhibited LPS-induced IL1β production in a dose-dependent fashion in PBMCs from patients with PSC and partly blocked TCDCA-induced innate cytokine production. To assess the impact of the FXR agonist on effector cytokine production by circulating T lymphocytes, PBMCs were stimulated with LPS or TCDCA in the presence of FXR agonist, and cells were subjected to ICF to determine cell-specific IFNγ expression (gating strategy in fig. S9A). High percentages of T lymphocytes from patients with PSC produced IFNγ upon stimulation with LPS (means ± SD: 23 ± 12% of CD8+ and 16 ± 12% of CD4+ cells) or TCDCA (19 ± 4% of CD8+ and 13 ± 3% of CD4+ cells), as compared to less than 2% of these lymphocyte subsets from HCs when treated under identical conditions (Fig. 8B). Among liver biochemistries that were determined at the time of collection of PBMCs from patients with PSC, serum gamma-glutamyl transferase (GGT) activity, a marker of biliary injury, correlated with frequencies of IFNγ+CD8+ cells after TCDCA stimulation (table S2 and fig.S9B). The highest expression of IFNγ in CD8+ lymphocytes after stimulation with LPS or TCDCA was observed in a patient with PSC-related end-stage liver disease and inflammatory bowel disease, who was listed for liver transplantation (participant #3). Incubation with 1.0 μM FXR agonist blocked an LPS- or TCDCA-induced rise in IFNγ-expressing CD8+ cells by a mean of 79%. FXR activation also reduced IFNγ expression by CD4+ lymphocytes but with higher variability. Percent reduction of IFNγ expression by T lymphocytes was not correlated with liver biochemistries. Collectively, these studies using LMNCs and PBMCs from patients with fibrosing cholangiopathies corroborated findings in murine SC, showing that pharmacological FXR activation blocks LPS- and hydrophobic BA-induced innate cytokine production and licensing of effector T lymphocytes.
Fig. 8. FXR activation represses IFNγ production by T lymphocytes in liver and peripheral blood mononuclear cells from patients with fibrosing cholangiopathies.
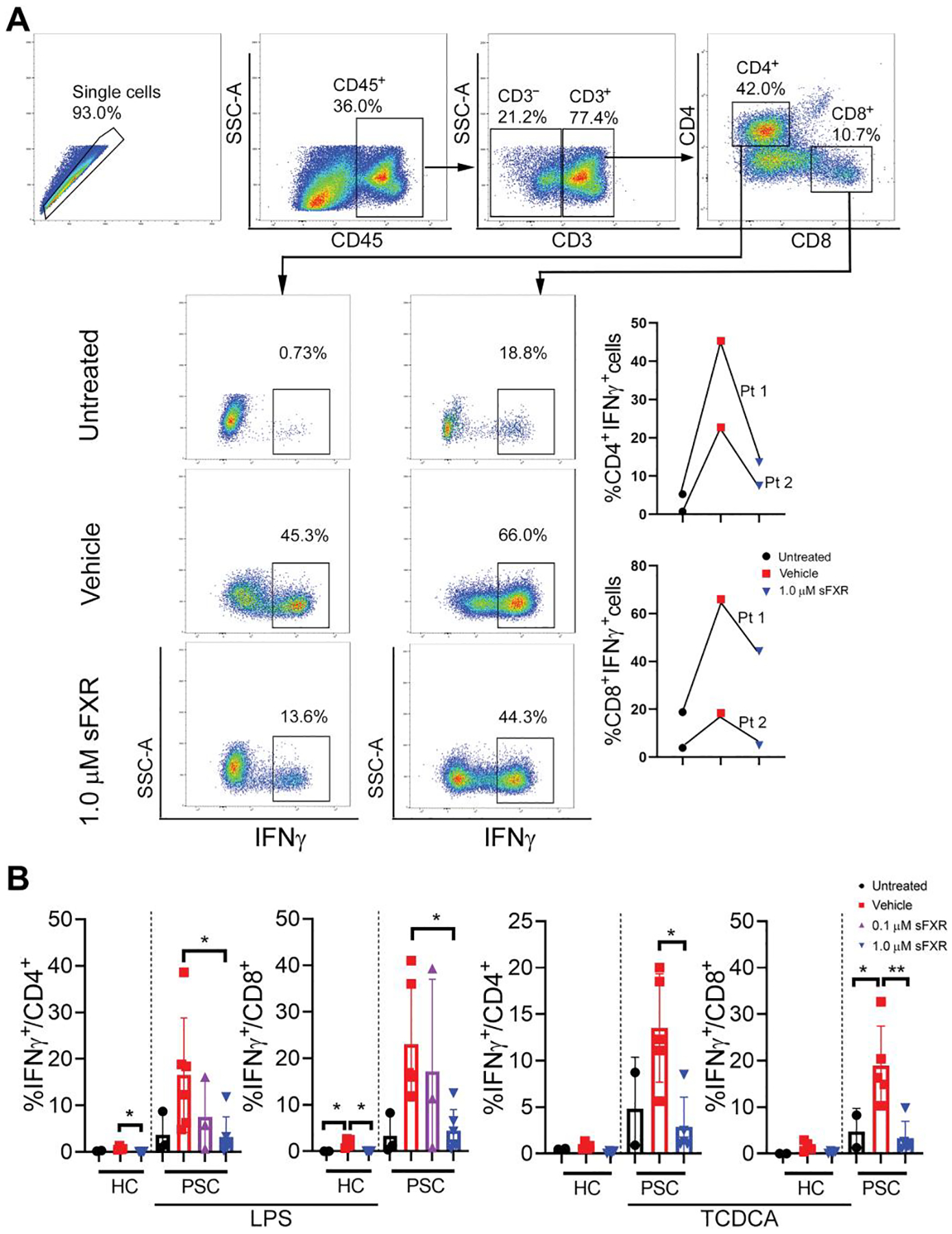
(A) LMNCs were freshly isolated from explanted livers of two patients with extrahepatic biliary atresia, cultured, stimulated with LPS (100 ng/ml) in the presence of 1.0 μM FXR agonist M044 or vehicle, and subjected to ICF to determine IFNγ expression by T lymphocytes. Representative gating strategy used to gate liver mononuclear cell population. First row: Doublets were excluded on a side scatter area (SSC-A) versus side scatter height (SSC-H) plot. Live cells were gated as Live/Dead ultraviolet negative. Leukocytes were then gated as to exclude debris, red blood cells, and platelets. Subpopulations were gated as CD4+T cells (CD3+CD4+) or CD8+ T cells (CD3+CD8+). Second, third, and fourth rows: Representative panels and images showing percentage of immune cells positive for IFNγ after ex vivo stimulation of LMNCs with LPS (100 ng/ml). (B) Cryopreserved PBMCs from patients with PSC and from healthy controls (HCs) were thawed, cultured, stimulated with LPS (100 ng/ml) in the presence of 1.0 μM FXR agonist M044 or vehicle, and subjected to ICF analysis to determine IFNγ expression on T lymphocytes. Results are means ± SD. Dots represent results from individual study participants. Multiplicity adjusted P values were determined using one-way ANOVA with Dunnett’s post hoc test compared to mean of the vehicle. *P < 0.05; **P < 0.01.
DISCUSSION
Here, we show that fexaramine-related sFXR agonists block progression of murine SC via repression of innate cytokine production and macrophage-mediated licensing of effector T lymphocytes. This conclusion is derived from experiments in two murine models of SC comparing pharmacological FXR agonists with primarily intestinal versus liver and intestinal tropism and using transgenic mice with deletion of FXR in macrophages. FXR-mediated suppression of hepatic BA synthesis via transcriptional control of Cyp7a1 and reduction in liver and serum BA concentrations were not linked to efficacy of FXR-directed therapy in murine SC. These insights have the potential to change the design of future clinical trials of FXR-directed therapies for PSC, EHBA, or PBC.
Our findings challenge the premise that synthetic FXR agonists improve liver disease in fibrosing cholangiopathies by lowering the pool size of toxic BAs through repressed BA synthesis, increased bile flow, and reduced intestinal BA reclamation (11, 12). Instead, we report that systemic and iFXR agonists are similarly efficacious in suppressing BA de novo synthesis (measured by plasma C4 concentrations) and lowering hepatic BA concentrations, but only sFXR activation prevents liver injury in MDR2−/− mice and xenobiotic SC. Reduction of BA pool size is likely necessary, but it is not sufficient to achieve hepatoprotective effects of FXR-directed therapy in SC. The findings should caution against considering iFXR agonists as therapies for fibrosing cholangiopathies, although they are protective in models of noncholestatic liver disease (30). Our results may also explain why NGM282, an analog of FGF19, potently reduces serum C4 and total BA concentrations in patients with PSC but fails to lower serum ALP activity, a surrogate end point for biliary injury (31). We submit that FGF19 analogs, like iFXR agonists, lack hepatic FXR activation required for modulation of hepatic macrophage responses to prevent progression of SC.
Clinical trials in PSC have used serum C4 and BA to evaluate target engagement of FXR agonists and efficacy (11, 12). None of these studies investigated the effects of study drugs on liver inflammation or immune regulation beyond serum complement-reactive protein concentrations. Biomarkers of hepatic immune responses ought to be included in future clinical trials of anticholestatic therapies for cholangiopathies. Such biomarkers are yet to be defined but might include circulating IL1β or TNFα. Our results further nominate IFNγ expression by circulating T lymphocytes as a potential readout for anti-inflammatory activity of FXR agonists.
A state of chronic inflammation is characteristic for PSC; various intrinsic and extrinsic danger signals contribute to the production of IL1β, IL6, and TNFα by hepatic macrophages, monocytes, and other inflammatory cells. Recent single-cell genomic studies revealed that among liver myeloid cells, Kupffer cells displayed the highest mRNA expression of Nr1h4 encoding FXR (32). We show that FXR protein is present in cultured hepatic macrophages, and its activation inhibits production of IL1β and TNFα by macrophages in response to DAMPs (TCDCA) and PAMPs (LPS). Our observations that synthetic FXR agonist represses TCDCA-induced inflammasome activation and production of proinflammatory IL1β and TNFα by hepatic macrophages is particularly relevant for treatment of cholestatic liver diseases in which hydrophobic BAs drive sterile inflammation (25). Why TCDCA induces IL1β production by macrophages, whereas taurocholic acid failed to do so in peritoneal macrophages need further investigation (33). Most likely it is BA specificity of surface receptors on macrophages. A candidate is sphingosin-1 phosphate receptor 2 that was shown to mediate secondary BA-induced activation of extracellular signal–regulated kinase, release of cathepsin B, and production of IL1β in a murine macrophage cell line (34).
In a model of sepsis-induced cholestasis, it was shown that endogenous FXR ligand CDCA physically interacted with the NLRP3 inflammasome to constrain inflammation. The FXR agonists GW4064 and OCA did not alter transcription of proinflammatory cytokines in macrophages and failed to reduce mortality in this model. In contrast, we showed that fexaramine-derived FXR agonist potently induced Shp and repressed transcription of Nlrp3 and of proinflammatory cytokines in hepatic macrophages. Our results are consistent with reports showing FXR activation to reduce recruitment of nuclear factor κB and activator protein (AP)-1 to the promotors of proinflammatory genes (35). Conflicting results between our study and a previous study (36) may be explained by the emerging recognition that distinct chemotypes of FXR agonists exert different effects on target cells. Crystallography and nuclear magnetic resonance–based studies reveal that compared with the FXR ligands CDCA and GW4064, fexaramine-based compounds induce distinct conformational changes of FXR resulting in recruitment of a receptor/coactivator complex, causing specific transcriptional regulation (37). Different physical properties of various FXR agonists may also determine their uptake by hepatic macrophages. Collectively, our findings suggest that pharmacological FXR activation targets several putative drivers of progression of cholestatic liver disease, reduces hepatobiliary concentration of toxic BAs, and blocks the transcriptional proinflammatory program of hepatic macrophages responding to DAMPs and PAMPs (fig. S10).
Although there is emerging evidence for an influence of FXR on T lymphocyte homeostasis in the gut, little is known about FXR-dependent control of T lymphocyte polarization in the liver. Here, we show in murine models of SC that FXR antagonizes production of IFNγ and IL17A by hepatic T lymphocytes. These lymphocyte responses perpetuate inflammation, bile duct epithelial injury, and fibrosis in PSC and EHBA (5–7, 10, 19, 24). Our studies in Il1r−/− mice demonstrate that IL1β is nonredundant for differentiation of TH17 lymphocytes in murine SC. In contrast, TH1 polarization is only partly blocked in these animals, suggesting that other cytokines, for instance, TNFα, also control licensing of IFNγ-producing lymphocytes in SC. IL1β and TNFα have both been reported to promote TH1 responses in context-dependent manners (28, 38). Because FXR controls secretion of IL1β and TNFα by macrophages in SC in our studies, pharmacological FXR activation has the potential to disrupt macrophage-derived licensing of effector lymphocytes more potently than therapies targeting IL1β.
Unequivocal evidence for the critical role of FXR-expressing hepatic macrophages in determining the SC phenotype and response to FXR agonist therapy stems from our investigations in FXRΔMC mice. We show that among myeloid cells expressing the LysM-cre driver in FXRΔMC mice (39), it is only hepatic macrophages and not neutrophils that express FXR protein and respond to FXR agonist in vitro. Furthermore, FXR signaling in hepatocytes and control of BA homeostasis are preserved in these mice, as demonstrated in cultured hepatocytes and in vivo. Loss of FXR in macrophages renders FXRΔMC more susceptible to DDC-induced up-regulation of hepatic innate cytokine responses and TH1/TH17 polarization, suggesting roles for endogenous FXR ligands such as CDCA in constraining liver inflammation in murine SC. We note that although synthetic FXR agonist reduces BA synthesis and liver BA concentrations in DDC-challenged FXRΔMC mice, they are resistant to protection from SC progression by pharmacological FXR activation.
In validation studies using human samples, we find IFNγ production by T lymphocytes in LMNCs from explanted livers of patients with EHBA to be suppressed when stimulated with LPS in the presence of FXR agonist. Furthermore, IL1β secretion by PBMCs from patients with PSC is blocked by FXR agonist upon stimulation with the exogeneous or endogenous danger signals, LPS or TCDCA, respectively. More than 15% of circulating CD4+ and CD8+ lymphocytes from patients with PSC, but less than 2% of lymphocytes from HCs, produce IFNγ upon stimulation with LPS or TCDCA. IFNγ expression is diminished when PBMCs are cultured in the presence of an FXR agonist.
Several potential limitations and open questions remain. First, our studies in murine models of SC focused on the impact of FXR agonists on liver inflammation and bile duct epithelial injury. Longer treatment courses are needed to assess FXR-induced shifts in the gut microbiome, which is a known driver of SC, and to evaluate the effects on progression of liver fibrosis. Second, studies with fexaramine-derived agents revealed repression of macrophage-mediated proinflammatory cytokine production by this class of FXR agonist. Side-by-side investigations with other commonly used FXR agonists such as OCA or GW4064 are required to better understand the mechanism of action for each of these chemotypes in treatment of SC. Third, the reduction of IFNγ production by circulating T cells in experiments with PBMCs from patients with PSC was variable. Studies in larger cohorts of patients are needed to determine whether FXR responsiveness of PBMCs is linked to the stage of disease and predicts improvement of liver end points in clinical trials with FXR agonists. Together, investigation in murine models and on samples from patients with PSC or EHBA demonstrates that FXR activation in inflammatory cells reduces innate and adaptive immune responses, which mediate the therapeutic effects of FXR agonist therapy in SC.
MATERIAL AND METHODS
Study design
The overall goal of the study was to elucidate the role of FXR in hepatic nonparenchymal cells in controlling liver inflammation and SC phenotype. We first examined the effects of systemic and intestinal FXR agonist on liver injury and immune activation in MDR2−/− mice. Next, we investigated the impact of genetic deletion of FXR in myeloid cells on macrophage function and responsiveness of DDC-induced SC to FXR agonist treatment. Last, we performed validation studies on liver-infiltrating and circulating immune cells from patients with PSC or EHBA. Sample sizes for experimental groups were determined on the basis of power calculations using results from our prior investigations in mouse models of SC. End points were determined on the basis of the established disease model end points for SC. In vivo studies included up to three randomly assigned treatment groups vehicle, FXR agonist treated, and HCs. Animal in vivo studies were performed with n = 3 to 8 age- and sex-matched mice per group and in vitro studies on n = 3 to 5 cell cultures from two independent experiments. Human studies were performed on n = 2 to 6 cell cultures per group.
Human studies
Pediatric patients with PSC or EHBA and age-matched HCs were enrolled into prospective, cross-sectional, observational cohort studies at the Cincinnati Children’s Hospital Medical Center (CCHMC) following informed consent to Institutional Review Board (IRB)–approved protocols (IRB #2017–2284 and #2012–3320). Characteristics of the patients are summarized in table S1. Median ages of the individuals with EHBA (n = 2), PSC (n = 7), and HC (n = 6) were 8, 17, and 15 years, respectively. All nine patients had active liver disease at the time of blood collection, based on elevation of at least one of the serum liver biochemistries. The majority of patients with PSC had associated inflammatory bowel disease and displayed features overlapping with autoimmune hepatitis. PBMCs were purified by Ficoll gradient centrifugation (40).
In vitro effects of FXR agonists on human PBMCs
Cryopreserved PBMCs were thawed, resuspended in Dulbecco’s modified Eagle’s medium with 10% fetal bovine serum, and cultured in the presence of either 0.1 or 1 μM FXR agonist M044 (Metacrine Inc.) or vehicle (dimethyl sulfoxide) for 3 hours. The PBMCs were then stimulated with LPS (100 ng/ml; Sigma-Aldrich), 100 μM TCDCA (Cambridge Isotope Laboratories), or vehicle (phosphate-buffered saline) and cultured for 16 hours. Production of IL1β was measured by IL1β TR-FRET assay (Cisbio). IFNγ expression was measured by ICF after short-term activation with PMA/ionomycin (Sigma-Aldrich), as detailed in the Supplementary Materials.
Murine experiments
MDR2-deficient mice in BALB/cJ background were a gift from F. Lammert (Homburg University, Homburg, Germany) (41). Mice were bred in-house and maintained in a pathogen-free animal facility with temperature regulation and a 12-hour dark/light cycle. FXR agonists (M345, M044, and M379) are derivatives of fexaramine, a nonsteroidal, neutral FXR agonist that lacks the carboxylic acid group found in endogenous FXR ligands or GW4064 (15). Compounds were provided by Metacrine Inc. and dissolved in corn oil using brief heating and sonication to ensure solubility for in vivo studies. sFXR agonists (M345 or M044) are precursors of MET409, which is a high-affinity synthetic ligand to FXR with a median effective concentration of 16 nM versus human FXR, sustained activation of liver FXR, and no cross-reactivity against TGR5, vitamin D receptor (VDR), or peroxisome proliferator–activated receptor isoforms that has recently been tested in clinical trials in HCs and patients with NASH (42). M379 is an FXR agonist with metabolic instability that transiently activates intestinal FXR upon compound absorption with limited or no activation of hepatic FXR. It is referred to as iFXR agonist. M345 was used as sFXR for experiments in Figs. 1 and 2A, and all other experiments used the chemically similar M044 (replacing the retired compound M345) as sFXR agonist for in vivo and in vitro studies. M345 and M044 exhibit similar effects on SC phenotype, as shown in fig. S1. M345 at 30 mg/kg per day, M379 at 100 mg/kg per day, or vehicle (corn oil) was administered to 45-day-old female MDR2−/− and age- and sex-matched, noncholestatic MDR2+/− mice by once daily oral gavage for 7 days. FXR-deficient (FXR−/−) and transgenic FXRfl/fl mice in C57BL6 background were gifts from F. Gonzalez [National Cancer Institute, National Institutes of Health (NIH)] (43). Conditional FXR knockout (FXRΔMC) mice lacking FXR expression in the myeloid cell lineage were generated by breeding FXRfl/fl with LysM-cre mice (Jackson Laboratories, stock #007214). IL1r1−/− and TGR5−/− mice in C57BL6 background were gifts from C. Pasare and G. Vassileva, respectively (28, 44). Fibrosing cholangiopathy was induced in nulliparous, 2- to 3-month-old female mice of the various genotypes by feeding chow containing 0.1% of DDC (Sigma-Aldrich) for 7 days (21). Mice received daily normal saline boluses of 500 μl by intraperitoneal injections. FXR agonists were administered by oral gavage for 6 days starting 1 day after beginning 0.1% DDC diet. The protocols were approved by the Animal Care and Use Committee of the Cincinnati Children’s Research Foundation (Cincinnati, OH; IACUC2013–0284). Further method details are provided in the Supplementary Materials.
Statistical analysis
Graphs were generated using GraphPad Prism version 8 (GraphPad). Values are expressed as means ± SD unless specified. P values were determined by unpaired, two-tailed t test or by one-way analysis of variance (ANOVA) with Dunnett’s post hoc test for grouped analysis, with multiplicity adjusted P values (GraphPad); P < 0.05 was considered significant. Details on statistical tests are provided in the figure legends.
Supplementary Material
Acknowledgments:
We would like to thank C. Woods from the Pathology Research Core and M. Kofron, the director of the Confocal Imaging Core at the Cincinnati Children’s Hospital, for help with the graphical abstract design and confocal imaging, respectively. We would also like to extend our gratitude to P. Shivakumar for help with experimental design, W. Balistreri for insightful comments on the manuscript, patients and families for participating in the observational studies, and G. Tiao and care coordinators for facilitating liver tissue sample collection.
Funding:
This work was supported by NIH R01DK095001-06 (to A.G.M.), Center for Autoimmune Liver Disease, and Center for Translational Fibrosis Research. This project was supported in part by NIH P30 DK078392 (Research Flow Cytometry Core) of the Digestive Diseases Research Core Center in Cincinnati.
Competing interest:
A.G.M. received research reagents (FXR agonist) from Metacrine Inc. and provided paid consulting services to Metacrine Inc. in 2018. B.W. is an employee of Metacrine Inc. A.G.M. receives research grants from Mirum Pharmaceuticals that are not related to the investigations presented in this report. The other authors declare that they have no competing interests.
Footnotes
Supplementary Materials
This PDF file includes:
Materials and Methods
Other Supplementary Material for this manuscript includes the following:
Data and materials availability:
All data associated with this study are present in the paper or the Supplementary Materials. FXR agonists including M345, M044, and M379 are available from Metacrine Inc. under a material transfer agreement with CCHMC. Data file S1 contains the raw data from figures.
REFERENCES AND NOTES
- 1.Jansen PLM, Ghallab A, Vartak N, Reif R, Schaap FG, Hampe J, Hengstler JG, The ascending pathophysiology of cholestatic liver disease. Hepatology 65, 722–738 (2017). [DOI] [PubMed] [Google Scholar]
- 2.Mohajeri S, Bezabeh T, Ijare OB, King SB, Thomas MA, Minuk G, Lipschitz J, Kirkpatrick I, Micflikier AB, Summers R, Smith ICP, In vivo 1H MRS of human gallbladder bile in understanding the pathophysiology of primary sclerosing cholangitis (PSC): Immune-mediated disease versus bile acid-induced injury. NMR Biomed. 32, e4065 (2019). [DOI] [PubMed] [Google Scholar]
- 3.Rust C, Bauchmuller K, Fickert P, Fuchsbichler A, Beuers U, Phosphatidylinositol 3-kinase-dependent signaling modulates taurochenodeoxycholic acid-induced liver injury and cholestasis in perfused rat livers. Am. J. Physiol. Gastrointest. Liver Physiol 289, G88–G94 (2005). [DOI] [PubMed] [Google Scholar]
- 4.Zhou K, Wang J, Xie G, Zhou Y, Yan W, Pan W, Che Y, Zhang T, Wong L, Kwee S, Xiao Y, Wen J, Cai W, Jia W, Distinct plasma bile acid profiles of biliary atresia and neonatal hepatitis syndrome. J. Proteome Res 14, 4844–4850 (2015). [DOI] [PubMed] [Google Scholar]
- 5.Katt J, Schwinge D, Schoknecht T, Quaas A, Sobottka I, Burandt E, Becker C, Neurath MF, Lohse AW, Herkel J, Schramm C, Increased T helper type 17 response to pathogen stimulation in patients with primary sclerosing cholangitis. Hepatology 58, 1084–1093 (2013). [DOI] [PubMed] [Google Scholar]
- 6.Lages CS, Simmons J, Maddox A, Jones K, Karns R, Sheridan R, Shanmukhappa SK, Mohanty S, Kofron M, Russo P, Wang YH, Chougnet C, Miethke AG, The dendritic cell-T helper 17-macrophage axis controls cholangiocyte injury and disease progression in murine and human biliary atresia. Hepatology 65, 174–188 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liaskou E, Jeffery LE, Trivedi PJ, Reynolds GM, Suresh S, Bruns T, Adams DH, Sansom DM, Hirschfield GM, Loss of CD28 expression by liver-infiltrating T cells contributes to pathogenesis of primary sclerosing cholangitis. Gastroenterology 147, 221–232.e7 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shivakumar P, Sabla G, Mohanty S, McNeal M, Ward R, Stringer K, Caldwell C, Chougnet C, Bezerra JA, Effector role of neonatal hepatic CD8+ lymphocytes in epithelial injury and autoimmunity in experimental biliary atresia. Gastroenterology 133, 268–277 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang L, Mizuochi T, Shivakumar P, Mourya R, Luo Z, Gutta S, Bezerra JA, Regulation of epithelial injury and bile duct obstruction by NLRP3, IL-1R1 in experimental biliary atresia. J. Hepatol 69, 1136–1144 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kunzmann LK, Schoknecht T, Poch T, Henze L, Stein S, Kriz M, Grewe I, Preti M, Hartl J, Pannicke N, Peiseler M, Sebode M, Zenouzi R, Horvatits T, Bottcher M, Petersen BS, Weiler-Normann C, Hess LU, Ahrenstorf AE, Lunemann S, Martrus G, Fischer L, Li J, Carambia A, Kluwe J, Huber S, Lohse AW, Franke A, Herkel J, Schramm C, Schwinge D, Monocytes as potential mediators of pathogen-induced T-Helper 17 differentiation in patients with primary sclerosing cholangitis (PSC). Hepatology 72, 1310–1326 (2020). [DOI] [PubMed] [Google Scholar]
- 11.Kowdley KV, Vuppalanchi R, Levy C, Floreani A, Andreone P, LaRusso NF, Shrestha R, Trotter J, Goldberg D, Rushbrook S, Hirschfield GM, Schiano T, Jin Y, Pencek R, MacConell L, Shapiro D, Bowlus CL; AESOP Study Investigators A randomized, placebo-controlled, phase II study of obeticholic acid for primary sclerosing cholangitis. J. Hepatol 73, 94–101 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Trauner M, Gulamhusein A, Hameed B, Caldwell S, Shiffman ML, Landis C, Eksteen B, Agarwal K, Muir A, Rushbrook S, Lu X, Xu J, Chuang JC, Billin AN, Li G, Chung C, Subramanian GM, Myers RP, Bowlus CL, Kowdley KV, The Nonsteroidal farnesoid X receptor agonist cilofexor (GS-9674) improves markers of cholestasis and liver injury in patients with primary sclerosing cholangitis. Hepatology 70, 788–801 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.de Vries EM, Wang J, Leeflang MM, Boonstra K, Weersma RK, Beuers UH, Geskus RB, Ponsioen CY, Alkaline phosphatase at diagnosis of primary sclerosing cholangitis and 1 year later: Evaluation of prognostic value. Liver Int. 36, 1867–1875 (2016). [DOI] [PubMed] [Google Scholar]
- 14.Chapman RW, Lynch KD, Obeticholic acid-A new therapy in PBC and NASH. Br. Med. Bull 133, 95–104 (2020). [DOI] [PubMed] [Google Scholar]
- 15.Downes M, Verdecia MA, Roecker AJ, Hughes R, Hogenesch JB, Kast-Woelbern HR, Bowman ME, Ferrer JL, Anisfeld AM, Edwards PA, Rosenfeld JM, Alvarez JG, Noel JP, Nicolaou KC, Evans RM, A chemical, genetic, and structural analysis of the nuclear bile acid receptor FXR. Mol. Cell 11, 1079–1092 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fang S, Suh JM, Reilly SM, Yu E, Osborn O, Lackey D, Yoshihara E, Perino A, Jacinto S, Lukasheva Y, Atkins AR, Khvat A, Schnabl B, Yu RT, Brenner DA, Coulter S, Liddle C, Schoonjans K, Olefsky JM, Saltiel AR, Downes M, Evans RM, Intestinal FXR agonism promotes adipose tissue browning and reduces obesity and insulin resistance. Nat. Med 21, 159–165 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harrison SA, Bashir MR, Lee KJ, Shim-Lopez J, Lee J, Wagner B, Smith ND, Chen HC, Lawitz EJ, A structurally optimized FXR agonist, MET409, reduced liver fat content over 12 weeks in patients with non-alcoholic steatohepatitis. J. Hepatol 75, 25–33 (2021). [DOI] [PubMed] [Google Scholar]
- 18.Fickert P, Pollheimer MJ, Beuers U, Lackner C, Hirschfield G, Housset C, Keitel V, Schramm C, Marschall HU, Karlsen TH, Melum E, Kaser A, Eksteen B, Strazzabosco M, Manns M, Trauner M; International PSC Study Group (IPSCSG), Characterization of animal models for primary sclerosing cholangitis (PSC). J. Hepatol 60, 1290–1303 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ravichandran G, Neumann K, Berkhout LK, Weidemann S, Langeneckert AE, Schwinge D, Poch T, Huber S, Schiller B, Hess LU, Ziegler AE, Oldhafer KJ, Barikbin R, Schramm C, Altfeld M, Tiegs G, Interferon-γ-dependent immune responses contribute to the pathogenesis of sclerosing cholangitis in mice. J. Hepatol 71, 773–782 (2019). [DOI] [PubMed] [Google Scholar]
- 20.Taylor AE, Carey AN, Kudira R, Lages CS, Shi T, Lam S, Karns R, Simmons J, Shanmukhappa K, Almanan M, Chougnet CA, Miethke AG, Interleukin 2 promotes hepatic regulatory T cell responses and protects from biliary fibrosis in murine sclerosing cholangitis. Hepatology 68, 1905–1921 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fickert P, Stoger U, Fuchsbichler A, Moustafa T, Marschall HU, Weiglein AH, Tsybrovskyy O, Jaeschke H, Zatloukal K, Denk H, Trauner M, A new xenobiotic-induced mouse model of sclerosing cholangitis and biliary fibrosis. Am. J. Pathol 171, 525–536 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ogawa S, Zhou B, Kimoto Y, Omura K, Kobayashi A, Higashi T, Mitamura K, Ikegawa S, Hagey LR, Hofmann AF, Iida T, An efficient synthesis of 7α,12α-dihydroxy-4-cholesten-3-one and its biological precursor 7α-hydroxy-4-cholesten-3-one: Key intermediates in bile acid biosynthesis. Steroids 78, 927–937 (2013). [DOI] [PubMed] [Google Scholar]
- 23.Fickert P, Fuchsbichler A, Wagner M, Zollner G, Kaser A, Tilg H, Krause R, Lammert F, Langner C, Zatloukal K, Marschall HU, Denk H, Trauner M, Regurgitation of bile acids from leaky bile ducts causes sclerosing cholangitis in Mdr2 (Abcb4) knockout mice. Gastroenterology 127, 261–274 (2004). [DOI] [PubMed] [Google Scholar]
- 24.Oo YH, Banz V, Kavanagh D, Liaskou E, Withers DR, Humphreys E, Reynolds GM, Lee-Turner L, Kalia N, Hubscher SG, Klenerman P, Eksteen B, Adams DH, CXCR3-dependent recruitment and CCR6-mediated positioning of Th-17 cells in the inflamed liver. J. Hepatol 57, 1044–1051 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gong Z, Zhou J, Zhao S, Tian C, Wang P, Xu C, Chen Y, Cai W, Wu J, Chenodeoxycholic acid activates NLRP3 inflammasome and contributes to cholestatic liver fibrosis. Oncotarget 7, 83951–83963 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Clausen BE, Burkhardt C, Reith W, Renkawitz R, Förster I, Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 8, 265–277 (1999). [DOI] [PubMed] [Google Scholar]
- 27.Best J, Verhulst S, Syn WK, Lagaisse K, van Hul N, Heindryckx F, Sowa JP, Peeters L, Van Vlierberghe H, Leclercq IA, Canbay A, Dollé L, van Grunsven LA, Macrophage depletion attenuates extracellular matrix deposition and ductular reaction in a mouse model of chronic cholangiopathies. PLOS ONE 11, e0162286 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jain A, Song R, Wakeland EK, Pasare C, T cell-intrinsic IL-1R signaling licenses effector cytokine production by memory CD4 T cells. Nat. Commun 9, 3185 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kunzmann LK, Schoknecht T, Poch T, Henze L, Stein S, Kriz M, Grewe I, Preti M, Hartl J, Pannicke N, Peiseler M, Sebode M, Zenouzi R, Horvatits T, Böttcher M, Petersen BS, Weiler-Normann C, Hess LU, Ahrenstorf AE, Lunemann S, Martrus G, Fischer L, Li J, Carambia A, Kluwe J, Huber S, Lohse AW, Franke A, Herkel J, Schramm C, Schwinge D, Monocytes as potential mediators of pathogen-induced Th17 differentiation in patients with primary sclerosing cholangitis (PSC). Hepatology 72, 1310–1326 (2020). [DOI] [PubMed] [Google Scholar]
- 30.Hartmann P, Hochrath K, Horvath A, Chen P, Seebauer CT, Llorente C, Wang L, Alnouti Y, Fouts DE, Starkel P, Loomba R, Coulter S, Liddle C, Yu RT, Ling L, Rossi SJ, DePaoli AM, Downes M, Evans RM, Brenner DA, Schnabl B, Modulation of the intestinal bile acid/farnesoid X receptor/fibroblast growth factor 15 axis improves alcoholic liver disease in mice. Hepatology 67, 2150–2166 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hirschfield GM, Chazouilleres O, Drenth JP, Thorburn D, Harrison SA, Landis CS, Mayo MJ, Muir AJ, Trotter JF, Leeming DJ, Karsdal MA, Jaros MJ, Ling L, Kim KH, Rossi SJ, Somaratne RM, DePaoli AM, Beuers U, Effect of NGM282, an FGF19 analogue, in primary sclerosing cholangitis: A multicenter, randomized, double-blind, placebo-controlled phase II trial. J. Hepatol 70, 483–493 (2019). [DOI] [PubMed] [Google Scholar]
- 32.Guilliams M, Bonnardel J, Haest B, Vanderborght B, Wagner C, Remmerie A, Bujko A, Martens L, Thoné T, Browaeys R, De Ponti FF, Vanneste B, Zwicker C, Svedberg FR, Vanhalewyn T, Gonçalves A, Lippens S, Devriendt B, Cox E, Ferrero G, Wittamer V, Willaert A, Kaptein SJF, Neyts J, Dallmeier K, Geldhof P, Casaert S, Deplancke B, Ten Dijke P, Hoorens A, Vanlander A, Berrevoet F, Van Nieuwenhove Y, Saeys Y, Saelens W, Van Vlierberghe H, Devisscher L, Scott CL, Spatial proteogenomics reveals distinct and evolutionarily conserved hepatic macrophage niches. Cell 185, 379–396.e38 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cai SY, Ge M, Mennone A, Hoque R, Ouyang X, Boyer JL, Inflammasome is activated in the liver of cholestatic patients and aggravates hepatic injury in bile duct-ligated mouse. Cell. Mol. Gastroenterol. Hepatol 9, 679–688 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhao S, Gong Z, Du X, Tian C, Wang L, Zhou J, Xu C, Chen Y, Cai W, Wu J, Deoxycholic acid-mediated sphingosine-1-phosphate receptor 2 signaling exacerbates DSS-induced colitis through promoting cathepsin B release. J. Immunol. Res 2018, 2481418 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang CS, Kim JJ, Kim TS, Lee PY, Kim SY, Lee HM, Shin DM, Nguyen LT, Lee MS, Jin HS, Kim KK, Lee CH, Kim MH, Park SG, Kim JM, Choi HS, Jo EK, Small heterodimer partner interacts with NLRP3 and negatively regulates activation of the NLRP3 inflammasome. Nat. Commun 6, 6115 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kremoser C, FXR agonists for NASH: How are they different and what difference do they make? J. Hepatol 75, 12–15 (2021). [DOI] [PubMed] [Google Scholar]
- 37.Merk D, Sreeramulu S, Kudlinzki D, Saxena K, Linhard V, Gande SL, Hiller F, Lamers C, Nilsson E, Aagaard A, Wissler L, Dekker N, Bamberg K, Schubert-Zsilavecz M, Schwalbe H, Molecular tuning of farnesoid X receptor partial agonism. Nat. Commun 10, 2915 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sun W, Wu Z, Lin Z, Hollinger M, Chen J, Feng X, Young NS, Macrophage TNF-α licenses donor T cells in murine bone marrow failure and can be implicated in human aplastic anemia. Blood 132, 2730–2743 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Abram CL, Roberge GL, Hu Y, Lowell CA, Comparative analysis of the efficiency and specificity of myeloid-Cre deleting strains using ROSA-EYFP reporter mice. J. Immunol. Methods 408, 89–100 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Riedhammer C, Halbritter D, Weissert R, Peripheral blood mononuclear cells: Isolation, freezing, thawing, and culture. Methods Mol. Biol 1304, 53–61 (2016). [DOI] [PubMed] [Google Scholar]
- 41.Lammert F, Wang DQ, Hillebrandt S, Geier A, Fickert P, Trauner M, Matern S, Paigen B, Carey MC, Spontaneous cholecysto- and hepatolithiasis in Mdr2−/− mice: A model for low phospholipid-associated cholelithiasis. Hepatology 39, 117–128 (2004). [DOI] [PubMed] [Google Scholar]
- 42.Chen HC, Bischoff E, Lee K-J, Littler R, van de Wetering J, SAT-341-MET409, an optimized sustained FXR agonist, was safe and well-tolerated in a 14-day phase 1 study in healthy subjects. J. Hepatol 70, e789 (2019). [Google Scholar]
- 43.Sinal CJ, Tohkin M, Miyata M, Ward JM, Lambert G, Gonzalez FJ, Targeted disruption of the nuclear receptor FXR/BAR impairs bile acid and lipid homeostasis. Cell 102, 731–744 (2000). [DOI] [PubMed] [Google Scholar]
- 44.Vassileva G, Golovko A, Markowitz L, Abbondanzo SJ, Zeng M, Yang S, Hoos L, Tetzloff G, Levitan D, Murgolo NJ, Keane K, Davis HR Jr., J. Hedrick, E. L. Gustafson, Targeted deletion of Gpbar1 protects mice from cholesterol gallstone formation. Biochem. J 398, 423–430 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Miethke AG, Zhang W, Simmons J, Taylor AE, Shi T, Shanmukhappa SK, Karns R, White S, Jegga AG, Lages CS, Nkinin S, Keller BT, Setchell KDR, Pharmacological inhibition of apical sodium-dependent bile acid transporter changes bile composition and blocks progression of sclerosing cholangitis in multidrug resistance 2 knockout mice. Hepatology 63, 512–523 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chapman R, Fevery J, Kalloo A, Nagorney DM, Boberg KM, Shneider B, Gores GJ; American Association for the Study of Liver Diseases, Diagnosis and management of primary sclerosing cholangitis. Hepatology 51, 660–678 (2010). [DOI] [PubMed] [Google Scholar]
- 47.Yeoman AD, Westbrook RH, Al-Chalabi T, Carey I, Heaton ND, Portmann BC, Heneghan MA, Diagnostic value and utility of the simplified International Autoimmune Hepatitis Group (IAIHG) criteria in acute and chronic liver disease. Hepatology 50, 538–545 (2009). [DOI] [PubMed] [Google Scholar]
- 48.Ai W, Li H, Song N, Li L, Chen H, Optimal method to stimulate cytokine production and its use in immunotoxicity assessment. Int. J. Environ. Res. Public Health 10, 3834–3842 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Morsy MA, Norman PJ, Mitry R, Rela M, Heaton ND, Vaughan RW, Isolation, purification and flow cytometric analysis of human intrahepatic lymphocytes using an improved technique. Lab. Invest 85, 285–296 (2005). [DOI] [PubMed] [Google Scholar]
- 50.Lages CS, Simmons J, Chougnet CA, Miethke AG, Regulatory T cells control the CD8 adaptive immune response at the time of ductal obstruction in experimental biliary atresia. Hepatology 56, 219–227 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Finlon JM, Burchill MA, Tamburini BAJ, Digestion of the murine liver for a flow cytometric analysis of lymphatic endothelial cells. J. Vis. Exp, e58621 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li WC, Ralphs KL, Tosh D, Isolation and culture of adult mouse hepatocytes. Methods Mol. Biol 633, 185–196 (2010). [DOI] [PubMed] [Google Scholar]
- 53.Boxio R, Bossenmeyer-Pourié C, Steinckwich N, Dournon C, Nüsse O, Mouse bone marrow contains large numbers of functionally competent neutrophils. J. Leukoc. Biol 75, 604–611 (2004). [DOI] [PubMed] [Google Scholar]
- 54.Li PZ, Li JZ, Li M, Gong JP, He K, An efficient method to isolate and culture mouse Kupffer cells. Immunol. Lett 158, 52–56 (2014). [DOI] [PubMed] [Google Scholar]
- 55.Manzanero S, Generation of mouse bone marrow-derived macrophages. Methods Mol. Biol 844, 177–181 (2012). [DOI] [PubMed] [Google Scholar]
- 56.Malik A, Kondratov RV, Jamasbi RJ, Geusz ME, Circadian clock genes are essential for normal adult neurogenesis, differentiation, and fate determination. PLOS ONE 10, e0139655 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data associated with this study are present in the paper or the Supplementary Materials. FXR agonists including M345, M044, and M379 are available from Metacrine Inc. under a material transfer agreement with CCHMC. Data file S1 contains the raw data from figures.