

Abstract
Purpose:
Neoadjuvant chemotherapy is increasingly administered to patients with resectable or borderline resectable pancreatic ductal adenocarcinoma (PDAC), yet its impact on the tumor immune microenvironment is incompletely understood.
Experimental design:
We employed quantitative, spatially-resolved multiplex immunofluorescence and digital image analysis to identify T-cell subpopulations, macrophage polarization states and myeloid cell subpopulations in a multi-institution cohort of up-front resected primary tumors (n=299) and in a comparative set of resected tumors after FOLFIRINOX-based neoadjuvant therapy (n=36) or up-front surgery (n=30). Multivariable-adjusted Cox proportional hazards models were used to evaluate associations between the immune microenvironment and patient outcomes.
Results:
In the multi-institutional resection cohort, immune cells exhibited substantial heterogeneity across patient tumors and were located predominantly in stromal regions. Unsupervised clustering using immune cell densities identified four main patterns of immune cell infiltration. One pattern, seen in 20% of tumors and characterized by abundant T cells (T cell-rich) and a paucity of immunosuppressive granulocytes and macrophages, was associated with improved patient survival. Neoadjuvant chemotherapy was associated with a higher CD8:CD4 ratio, greater M1:M2-polarized macrophage ratio, and reduced CD15+ARG1+ immunosuppressive granulocyte density. Within neoadjuvant-treated tumors, 64% showed a T-cell-rich pattern with low immunosuppressive granulocytes and macrophages. M1-polarized macrophages were located closer to tumor cells after neoadjuvant chemotherapy and colocalization of M1-polarized macrophages and tumor cells was associated with greater tumor pathologic response and improved patient survival.
Conclusion:
Neoadjuvant chemotherapy with FOLFIRINOX shifts the PDAC immune microenvironment towards an anti-tumorigenic state associated with improved patient survival.
Keywords: macrophage, myeloid immune cell, neoadjuvant treatment, pancreatic cancer, spatial analysis, tumor microenvironment
INTRODUCTION
Pancreatic ductal adenocarcinoma (PDAC) is the third leading cause of cancer-related death in the US1, with a 5-year survival rate of only 11%1. Patients who present with localized disease are increasingly treated with neoadjuvant chemotherapy to facilitate margin-negative surgical resection and potentially improve long-term survival2–5. Beyond the direct cytotoxic effect of chemotherapeutic agents on cancer cells, chemotherapy may alter the PDAC immune microenvironment6–8. The PDAC microenvironment is composed of abundant stroma and a diverse immune cell infiltrate, enriched with immunosuppressive cells, including regulatory CD4+ T cells, M2-polarized macrophages and myeloid-derived suppressor cells (MDSCs). The admixture of these suppressive cells with effector and cytotoxic T cells is thought to play an important role in facilitating immune tolerance and tumor evasion of immunologic clearance9–13. Furthermore, several studies have suggested that immune cell densities are associated with patient outcomes, including survival after surgical resection for localized disease14–18. However, despite the increasing use of neoadjuvant therapy, few studies have investigated its potential effects on the PDAC immune microenvironment, and these studies have either focused on limited immune cell subsets or included patients receiving diverse neoadjuvant treatment programs7,8,19–23.
To investigate the impact of neoadjuvant therapy on the PDAC immune microenvironment, we developed quantitative, spatially-resolved, multiplex immunofluorescence assays to characterize T-cell subpopulations, macrophage subtypes and MDSCs. We first defined the immune landscape in a large cohort of primary, previously-untreated resected PDAC and then conducted a paired assessment with tumors resected after treatment with neoadjuvant FOLFIRINOX (5-fluorouracil, folinic acid, irinotecan, oxaliplatin) chemotherapy. Immune cell densities and spatial organization were then evaluated with patient outcomes, tumor genomic features, histologic response to neoadjuvant therapy, and additional clinicopathologic data to identify and determine the significance of major immune cell infiltration patterns in neoadjuvant-treated PDAC.
MATERIAL AND METHODS
Study population and specimens
To characterize the baseline PDAC immune landscape, we studied tumors from our existing cohort of patients treated with up-front surgery. This primary resection cohort, which has undergone prior molecular characterization and tissue microarray-based myeloid cell profiling17,24, included patients from three academic cancer centers: Dana-Farber/Brigham and Women’s Cancer Center (DF/BWCC), University of Rochester Medical Center (URMC), and Stanford Cancer Institute (SRI). Clinicopathological data including sex, age at surgery, type of pancreatic resection, post-operative chemotherapy and radiotherapy, tumor location, AJCC (8th ed.) pT and pN stages, histologic grade, lymphovascular invasion, and resection margin status were collected from the medical records (Supplementary Table S1), as previously described24. After relevant exclusions, 299 patients were included in the primary resection cohort (Supplementary Figure S1).
To assess changes in immune cell composition after neoadjuvant treatment, we selected representative whole slide tumor sections from 36 patients treated with neoadjuvant FOLFIRINOX chemotherapy at DF/BWCC that had available tumor blocks and residual tumor histologically confirmed in the pancreas. These tumors were staged as borderline resectable (70%), locally advanced (19%) or up-front resectable (11%), as per convention at Dana-Farber/Brigham and Women’s Cancer Center25. For comparison, we randomly selected representative whole slide tumor sections from 30 patients that were also included in the multi-institutional resection cohort that underwent up-front surgery at DF/BWCC. In the neoadjuvant cohort, 24 patients were treated with neoadjuvant chemotherapy alone, 8 with neoadjuvant chemotherapy followed by stereotactic body radiotherapy, and 4 with neoadjuvant chemotherapy followed by long-course chemoradiation (Supplementary Table S1).
The institutional review board (IRB) from each institution approved this study. Patients treated at the DF/BWCC signed a written informed consent, and the informed consent was waived at URMC and SCI, as patients were identified retrospectively according to IRB-exempt protocols. The study was conducted in accordance with the U.S. Common Rule.
Assessment of histological response to neoadjuvant treatment
We assessed hematoxylin and eosin-stained slides from the neoadjuvant-treated cohort and estimated the treatment effect using both the Modified Ryan Scheme for Tumor Regression Score26, as recommended by the College of American Pathologists, and the MD Anderson grading system 27,28. Using the Modifier Ryan scheme, we further classified tumors that were scored 1 as “good responders” and tumors that scored 2 or 3 as “poor responders” (Supplementary Figure S2).
Multiplexed immunofluorescence assays
A customized multiplex immunofluorescence (mIF) panel was designed to characterize T-cell subsets and included: CD3 (pan-T cell marker), CD4 (helper T cells), CD8 (cytotoxic T cells), CD45RO (PTPRC isoform, memory T cells), and FoxP3 (regulatory T cells) (Supplementary Table S2). Two mIF panels previously developed for PDAC were used to characterize myeloid cell subsets (CD14, CD15, ARG1, CD33, and HLA-DR) and macrophage polarization (CD68, CD163, IRF5, CD86, and CD206)17. All panels included 2-(4-amidinophenyl)-1H-indole-6-carboxamidine (DAPI) as a nuclear marker and cytokeratin to identify epithelial cells (Figure 1).
Figure 1. Overview of study cohort and analysis approach.
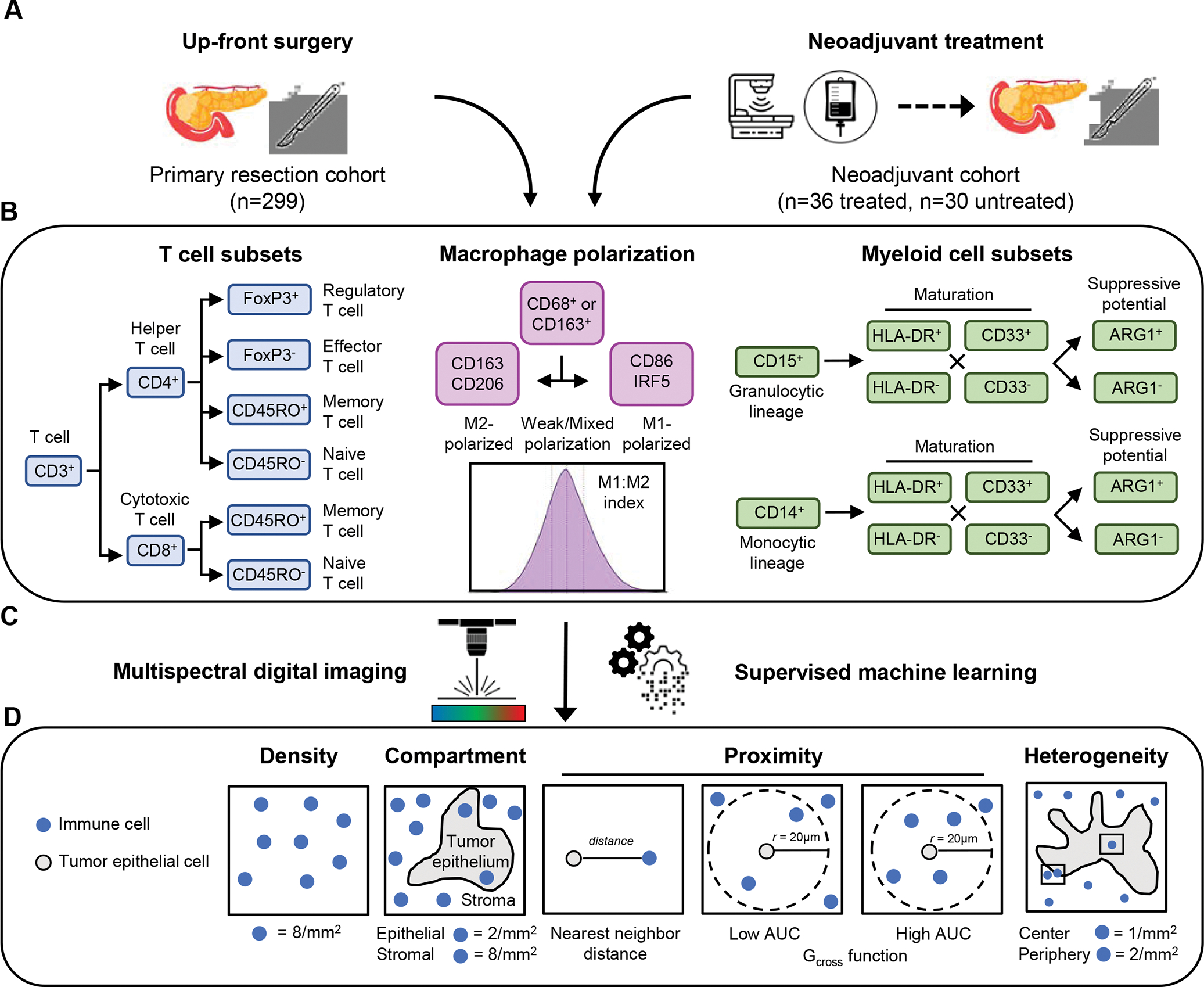
Two pancreatic ductal adenocarcinoma tissue cohorts were analyzed, including a primary resection cohort and a neoadjuvant-treatment cohort (A). T cell subsets (helper and cytotoxic T cells, including regulatory activity and naïve/memory status), differentially-polarized macrophages (M1- and M2-polarized macrophages, including strength of polarization) and myeloid cell subsets (granulocytes and monocytes, including maturity and ARG1 immunosuppressive activity) were evaluated using three multiplexed immunofluorescence panels (B). Combinatorial protein expression and cytomorphology data were analyzed by multispectral digital image analysis and supervised machine learning to identify specific cell types (C). Quantification of immune cell abundance was performed to measure immune cell densities overall and within separate tissue compartments (tumor epithelium or stroma); immune cell proximity to tumor cells was assessed by nearest neighbor distance (NND) and the Gcross function; and regions of interest were analyzed to assess immune cell heterogeneity (D).
All antibodies were first optimized using chromogenic single-plex staining protocols and then using single-plex immunofluorescence. Subsequently, antibodies were combined into a multiplexed IF assay in which sequential rounds of antigen retrieval, antigen detection, and fluorescent labeling via tyramide signal amplification were performed on 4μm sections of formalin-fixed paraffin-embedded samples using a Leica BOND RX Research Stainer (Leica Biosystems, Buffalo, IL) (Supplementary Figure S3 and S4).
Evaluation of immune cell subsets in multiplexed immunofluorescence images
To analyze stained slides, digital images were acquired at 200x magnification using an automated multispectral imaging system (Vectra 3.0, PerkinElmer, Hopkinton, MA)29. After image acquisition and spectral unmixing, each digital image was visually inspected by a trained pathologist to ensure the presence of invasive PDAC and to exclude regions of non-neoplastic epithelium and tissue processing artifacts. For tissue microarray (TMA) analysis, only tumor cores with malignant cells comprising at least 5% of the total tissue area were included. For the whole slide section analysis, six regions of interest were selected, three from the tumor center and three from the periphery, to account for tumor heterogeneity18. Tertiary lymphoid structures (TLS), if present, were also selected for analysis (Supplementary Figure S5). Following quality control procedures, images were then processed using supervised machine learning (inForm 2.4.1, PerkinElmer) to segment each region of interest into tumor epithelial and stromal areas.
After detecting single cells and performance of subcellular segmentation, supervised machine learning was used to identify T cells based upon a combination of cytomorphology and subcellular T-cell marker expression patterns, allowing identification of the following phenotypes: CD3+CD4+, CD3+CD8+, and CD3+CD4−CD8−. In addition, other immune cells (CD3−CD45RO+), tumor cells (CK+) and stromal cells (CK−CD3−CD4−CD8−CD45RO−) were identified (Supplementary Figure S6). The resultant single-cell level data were further analyzed using R v.4.0 (R Foundation for Statistical Computing, Vienna, Austria).
Assessment of immune cell densities and spatial features
For each tumor, immune cell densities were separately calculated for the tumor epithelial area, stromal area, and combined tissue area. Proximity of immune cells to tumor cells was assessed using nearest neighbor distance (NND) and the Gcross function, using the spatstat package v.2.2.0 in R. Briefly, NND computes the distance (μm) between each point i (i.e., immune cell) to its nearest neighbor point j (i.e., tumor cell), whereas the Gcross function estimates the probability of finding at least one point j (i.e., immune cell) within a specified radius (μm) of any point i (i.e., tumor cell), thereby estimating the degree of tumor-immune cell co-localization (Figure 1, Supplementary Figure S7).
Statistical analysis
For survival analyses, Cox proportional hazards regression models were used to compute hazard ratios (HRs) and 95% confidence intervals (CIs). Overall survival (OS) was defined as time between surgery and death for any reason or last follow-up if alive. Disease-free survival (DFS) was defined as time between surgery and disease recurrence or last follow-up if no recurrence. For the primary resection cohort, immune cell densities and spatial measurements were categorized into quartiles by cohort. For the neoadjuvant cohort, median values were used to dichotomize for two-group comparisons. The proportional hazards assumption was satisfied by including a product of the exposure variable and time as a time-dependent variable in the model (all P>0.05). Trend tests were performed by including an ordinal score for each quartile in Cox models. In multivariable analyses, models were adjusted for potential prognostic factors, including age, sex, pathologic N stage, tumor grade, lymphovascular invasion, resection margin status, and receipt of perioperative treatment. The Kaplan–Meier method was used to generate survival curves, and the log-rank test was used for estimating statistical significance. We performed unsupervised clustering using the k-means function (stats package v.4.2.0 in R) based on normalized overall densities of non-overlapping immune cells subsets. Statistical analysis was performed with SAS 9.4 software (SAS Institute) and R software (version 4.0). Two-sided P values <0.05 were considered statistically significant.
Data Availability
The data generated in this study are available upon request from the corresponding author.
RESULTS
Characterization of immune microenvironment in resected pancreatic cancer
To define the landscape of myeloid and lymphoid cell types in PDAC, we began by assessing T-cell populations and spatial distribution in the primary resection cohort using a custom-designed mIF panel, digital image analysis and supervised machine learning for tissue segmentation and cell phenotyping. Across 299 tumors, we identified 2.4 million specifically phenotyped cells, including 819,000 cytokeratin-positive tumor cells and 1.6 million CD3+ T cells. More than 90% of CD3+ T cells were positive for CD4 or CD8. CD45RO+ memory T cells were significantly more common than naïve T cells (average of 2.1 memory T cells for every naïve T cell), and FoxP3 expression was identified in only 11% of CD3+CD4+ helper T cells (Supplementary Table S3). While 94.8% of T cells were located within stromal regions, 5.2% of T cells were identified within tumor epithelial areas, indicating very close proximity to tumor cells. Across tumors, T-cell densities varied greatly, with a median (IQR) of 320 (144–612) CD3+ cells/mm2 and a range of 10 to 5014 CD3+ cells/mm2 (Supplementary Table S3). To ensure that the TMA analysis was representative of larger tumor areas that can be evaluated using whole slide sections (WSS), we compared immune cell density measurements from a subset of tumors in the TMA set with those derived from WSS of the same tumors. This analysis revealed correlation coefficients of 0.55 for CD3+ T cells, 0.55 for CD3+CD4+ T cells and 0.54 for CD3+CD8+ T cells (Supplementary Figure S8).
We next combined T-cell results with myeloid cell data drawn from two additional mIF panels17 (Figure 1), resulting in 270 tumors with data available across all three panels. These additional myeloid cell-focused panels cumulatively provided information for 2.6 million macrophages, 219,379 CD14+ cells and 159,033 CD15+ cells. Integration of T-cell and myeloid cell densities revealed significant heterogeneity in aggregate cell densities across the 270 tumors, with no individual immune cell population serving as a dominant determinant of overall immune cell density (Figure 2A). Analysis of combined tissue area densities showed that T-cell densities were positively correlated with macrophage and CD14+ monocyte densities but not correlated with CD15+ granulocyte densities, including immunosuppressive CD15+ARG1+ granulocytes (Figure 2B). Across all immune cell populations, densities were higher in stromal areas than in tumor epithelial areas17 (Figure 2C). However, this difference was more pronounced for T-cell and macrophage subsets, suggesting that different immune cell subtypes are preferentially located within specific locations in the larger microenvironment.
Figure 2. Immune cell landscape in up-front resected pancreatic ductal adenocarcinoma (PDAC).
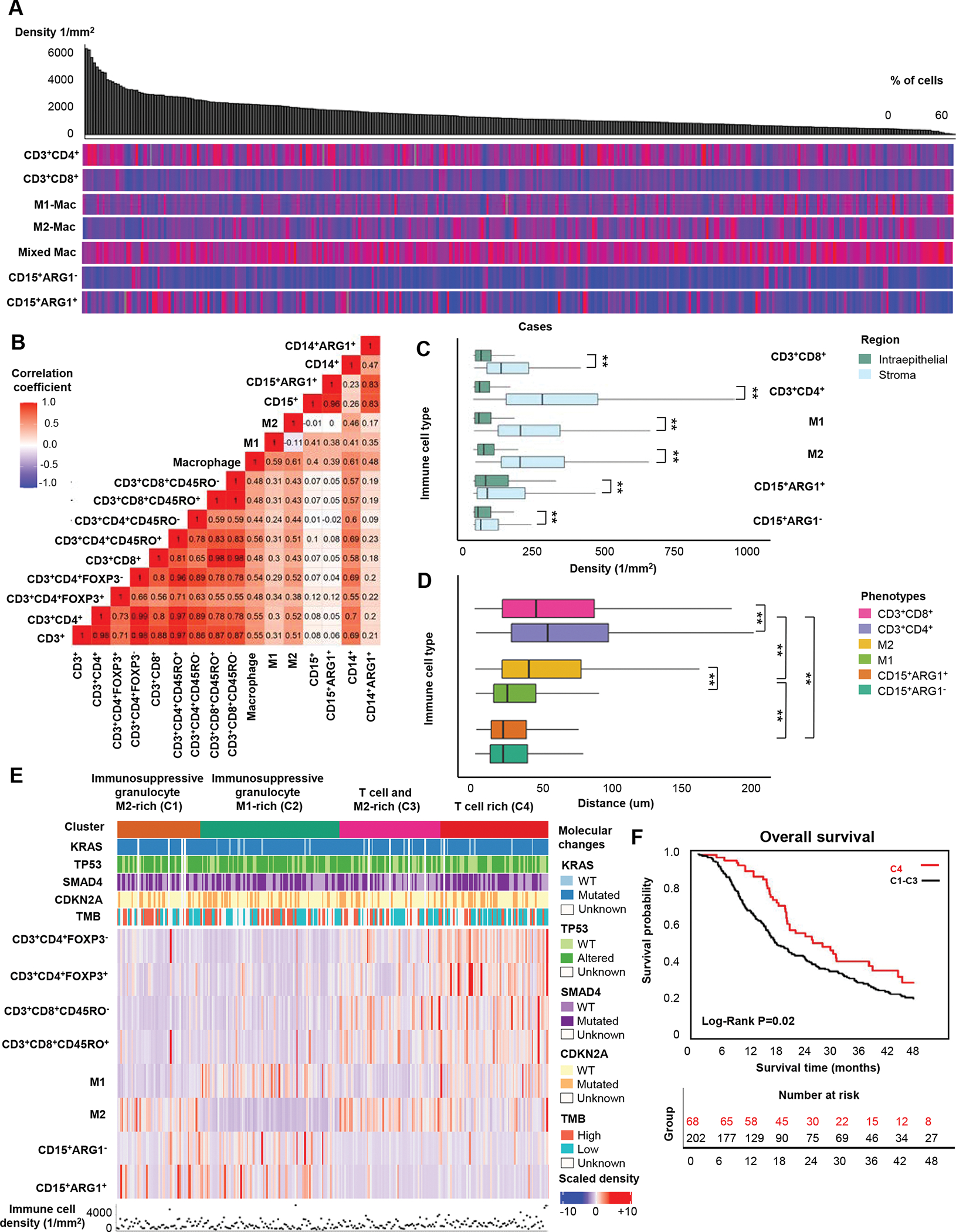
Immune cell distributions across 270 PDACs organized by decreasing total cell density (top grey bar plot). Heatmaps display the relative distributions (0–100%) of the non-overlapping immune cell populations within the total cell count. Subsets included in this analysis are displayed on the left. Macrophages with minimal polarization towards M1- or M2-phenotypes are classified as mixed (A). Spearman correlation coefficients for densities of major immune cell types and subtypes (B). Immune cell densities in tumor intraepithelial and stromal regions (C). Boxplots depicting distances between individual immune cells and the closest tumor cell based on a total of 886,315 immune cells (D). Unsupervised k-means clustering analysis of immune cell densities and their associations with the four main PDAC genetic alterations and tumor mutational burden (E). Kaplan-Meier survival curves comparing the T cell-rich cluster (C4) to a combined group of clusters C1-C3 (F). P values were calculated with the Wilcoxon rank-sum test and Kruskal-Wallis test. ** P <0.005
Although higher T-cell densities and, more specifically, cytotoxic CD8+ T-cell densities are associated with better patient outcomes in many tumor types, data in pancreatic cancer are conflicting10,11,30. In our primary resection cohort, higher overall CD3+ T-cell density was associated with improved OS and DFS in multivariable-adjusted models (Supplementary Table S4). When comparing patient tumors in the top versus bottom quartiles of CD3+ T-cell density, multivariable-adjusted Cox regression models demonstrated a hazard ratio (HR) of 0.58 (95% CI, 0.39–0.87, Ptrend = 0.01) for DFS and 0.69 (95%CI, 0.48–1.03; Ptrend = 0.03) for OS. Interestingly, these associations were driven largely by CD3+CD4+ helper T-cell densities, rather than CD3+CD8+ cytotoxic T-cell densities. Furthermore, CD3+ T-cell densities were not associated with alterations in the main PDAC driver genes, including KRAS, CDKN2A, SMAD4, and TP53, as assessed by next generation sequencing and immunohistochemistry24, nor with tumor mutational burden (TMB), classified as high or low according to the median cohort TMB of 6.7 mutations per megabase (data not shown).
Beyond density, immune cell spatial configuration in relation to tumor cells is likely important for the anti-tumor immune response and has been associated with patient outcomes for several immune cell types11,17. Across our integrated lymphoid and myeloid data set, most immune cells were localized within 50 μm of a tumor cell (Figure 2D). Although immune cell distance distributions partially overlapped, granulocytes were closest to tumor cells, followed by macrophages and then T cells. In our prior work, we found that M1-macrophages were closer to tumor cells than M2 macrophages, and that greater proximity of M2 macrophages to tumor cells was associated with worse patient outcomes17. We analyzed T cells in a similar fashion and found that CD3+CD8+ T cells were closer to tumor cells than CD3+CD4+ T cells. However, the degree of co-localization between tumor cells and either CD3+CD4+ or CD3+CD8+ T cells did not harbor prognostic significance (data not shown). Taken together, these results indicate that numerous immune cell subtypes exhibit spatial organization with respect to tumor cells and that this organization may reflect the effectiveness of the anti-tumor immune response (Figure 2D).
Immune cell composition identifies PDAC subtypes with prognostic significance
We previously reported that primary PDAC could be clustered into groups based on immune cell abundance (hypo- or hyper-inflamed) and lineage dominance (myeloid or lymphoid)18. However, no prognostic significance was identified for these different groups, potentially due to the limited number of tumors included in the study. Using our new integrated data set, which also includes some prognostically relevant immune cell subtypes not characterized in the prior study, we performed unsupervised clustering using densities of the major, non-overlapping immune cell types to identify patterns in immune cell infiltration (Figure 2E). This analysis was performed in 270 patients and revealed four clusters (C1-C4). The first two clusters were enriched in immunosuppressive granulocytes, depleted of T cells, and differed according to predominance of M1-polarized (C1) or M2-polarized macrophages (C2). The third cluster (C3) was characterized by high CD3+CD8+ T cell and M2-polarized macrophage densities, while the fourth cluster (C4) was distinguished by high densities of multiple T cell subtypes, moderate M1-polarized macrophage density, and low M2-polarized macrophage and granulocyte densities. Alterations in the four PDAC driver genes were not associated with these clusters, nor was TMB (Figure 2E).
We next considered whether these immune-based clusters were associated with patient outcomes, hypothesizing that patients within the “T cell-rich” cluster would have improved survival. Using this cluster as the reference group, each of the other three clusters was associated with shorter OS and demonstrated largely overlapping survival curves by Kaplan-Meier analysis (Supplementary Figure S9). Given these similarities, we combined cases from these three clusters for outcome analyses. Using the “T cell-rich” cluster (C4) as a reference, the combined clusters associated with shorter DFS (HR of 1.65 (95% CI, 1.15–2.35, Ptrend 0.01) and shorter OS (HR of 1.61 (95% CI, 1.14–2.28, Ptrend 0.01) (Figure 2F, Table 1).
Table 1.
Cox regression models for overall survival and disease-free survival according to immune cell clusters in the primary resection cohort.
| Overall survival (N=270) a | ||||||||
|
| ||||||||
| Immune cell cluster b | No. of patients | Median survival (mo) | 3-yr OS rate | 5-yr OS rate | Univariable HR (95% CI) | P | Multivariable HR (95% CI) | P |
|
| ||||||||
| C4 | 68 | 26.0 | 36% | 19% | 1.00 (reference) | 1.00 (reference) | ||
| C1-C3 | 202 | 17.7 | 27% | 16% | 1.49 (1.06–2.07) |
0.02 | 1.61 (1.14–2.28) |
0.01 |
|
| ||||||||
| Disease-free survival (N=270) a | ||||||||
|
| ||||||||
| Immune cell cluster b | No. of patients | Median survival (mo) | Univariate HR (95% CI) | P | Multivariable HR (95% CI) | P | ||
|
| ||||||||
| C4 | 68 | 17.5 | 1.00 (reference) | 1.00 (reference) | ||||
| C1-C3 | 202 | 11.6 | 1.41 (1.00–1.97) | 0.04 | 1.67 (1.16–2.39) | 0.01 | ||
Cox proportional hazards regression model adjusted for age, sex, pathologic N stage (N0, N1, N2), tumor grade (well/moderately differentiated, poorly differentiated, unknown), lymphovascular invasion (negative, positive, unknown), resection margin status (R0, R1, R2, unknown), and receipt of adjuvant treatment.
Immune cell clusters were defined by unsupervised k-means clustering analysis of 270 tumors from the primary resection cohort and based on normalized immune cell densities from non-overlapping immune cells. C4 represents “T cell-rich” cluster and “C1-C3” refers to combination of the three remaining clusters: C1 (“Immunosuppressive granulocyte and M2-polarized macrophage-rich”), C2 (“Immunosuppressive granulocyte and M1-polarized macrophage-rich”), and C3 (“T cell and M2-polarized macrophage-rich”).
Abbreviations: CI: confidence interval; HR: hazard ratio; IQR: interquartile range; mo: months; OS, overall survival
Neoadjuvant therapy is associated with an altered immune TME
Having characterized the baseline immune landscape of localized pancreatic cancer, we next investigated whether treatment with FOLFIRINOX chemotherapy prior to surgery altered the immune microenvironment. Across whole slide tumor sections from 36 patients treated with neoadjuvant FOLFIRINOX and 30 patients treated with up-front surgical resection, we identified 315,345 CD3+ T cells, 358,922 macrophages, and 104,060 CD15+ granulocytes. As in the primary resection cohort, marked inter-tumor heterogeneity was observed for aggregate immune cell density and relative immune cell composition in both groups (Figure 3A, Supplementary Table S5).
Figure 3. Associations of neoadjuvant therapy with immune cell profiles in the pancreatic cancer microenvironment.
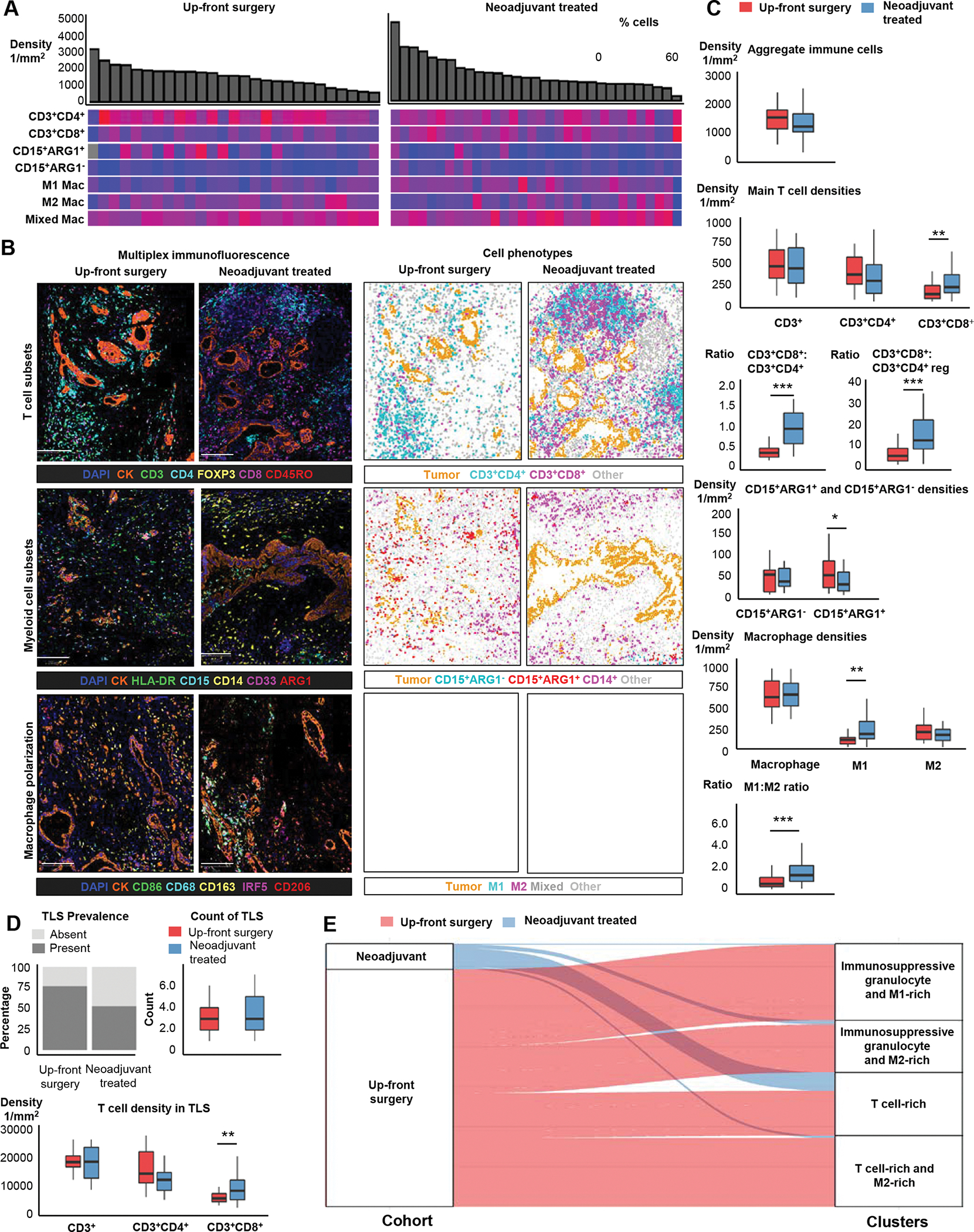
Immune cell composition of 30 up-front resected pancreatic ductal adenocarcinomas (PDAC) and 36 cases that underwent neoadjuvant treatment (A). Examples of multiplex immunofluorescence images and corresponding phenoplots for T-cell subsets, myeloid cell subsets and macrophage polarization panel in up-front resected and neoadjuvant-treated PDACs (B). Boxplots depicting the distribution of overall (combined intraepithelial and stromal areas) immune cell densities in 30 up-front resected PDACs and 36 neoadjuvant-treated cases (C). Immune cell composition of tertiary lymphoid follicles in 23 up-front resected PDACs and 25 neoadjuvant-treated cases (D). Sankey plot depicting the relationship between neoadjuvant treatment status and the four main patterns of immune cell infiltration detected using unsupervised k-means clustering analysis across both the up-front resected and neoadjuvant cohorts (E). Scale bars represent 200μm. P values were calculated with Wilcoxon rank-sum test. ***: P <0.001, **: P <0.005, *: P <0.05
Comparison of neoadjuvant-treated tumors to those treated with upfront surgery did not identify a significant difference in aggregate immune cell density (Figure 3B–C). However, large differences in cell density were observed for multiple immune cell subtypes (Figure 3B–C, Table 2, Supplementary Figure S10). Neoadjuvant therapy was associated with a significantly higher CD3+CD8+ T-cell density as compared to up-front surgery, with increased CD3+CD8+ T-cell density in both the intraepithelial and stromal compartments. This increase was driven by both naïve (CD3+CD8+CD45RO−) and memory (CD3+CD8+CD45RO+) CD8+ T cells. In contrast, CD3+CD4+ T-cell density was decreased in the neoadjuvant therapy group (Supplementary Table S5). These differences translated into a significantly increased CD3+CD8+:CD3+CD4+ ratio (median ratio 0.94 vs. 0.35; P<0.005) and an increased cytotoxic to regulatory T-cell (CD3+CD8+:CD3+CD4+FoxP3+) ratio (median ratio 13.8 vs. 5.1; P<0.005) in neoadjuvant-treated patients, indicating a large shift in T-cell functional status despite similar total T-cell numbers.
Table 2.
Immune cell densities within intraepithelial and stromal regions in patients who underwent up-front surgery or received neoadjuvant chemotherapy prior to surgery
| Intraepithelial Regions (cells/mm2) | |||
|
| |||
| Immune cell subset | Up-front surgery (n=30) a | Neoadjuvant treated (n=36) a | P b |
|
| |||
| CD3+ T cells | 115 (54–243) | 136 (85–311) | 0.71 |
| CD3+CD4+ | 73 (31–152) | 43 (25–116) | 0.08 |
| CD3+CD8+ | 27 (16–89) | 82 (36–127) | 0.02 |
|
| |||
| CD15+ granulocytes | 74 (32–237) | 124 (50–256) | 0.17 |
| CD15+ARG1+ | 33 (12–205) | 28 (12–78) | 0.30 |
| CD15+ARG1− | 23 (5–54) | 60 (31–179) | <0.01 |
| CD14+ monocytes | 130 (36–200) | 263 (130–490) | <0.01 |
|
| |||
| Macrophages | 105 (82–198) | 647 (350–1071) | <0.01 |
| M1-polarized macrophages | 30 (16–61) | 290 (141–501) | <0.01 |
| M2-polarized macrophages | 13 (8–34) | 42 (13–69) | 0.02 |
|
| |||
| Stromal Regions (cells/mm2) | |||
|
| |||
| Immune cell subset | Up-front surgery (n=30) a | Neoadjuvant treated (n=36) a | P b |
|
| |||
| CD3+ T cells | 619 (407–997) | 600 (337–912) | 0.91 |
| CD3+CD4+ | 462 (305–719) | 329 (146–548) | 0.09 |
| CD3+CD8+ | 132 (80–264) | 225 (147–416) | 0.01 |
|
| |||
| CD15+ granulocytes | 95 (59–539) | 73 (34–190) | 0.15 |
| CD15+ARG1+ | 73 (39–464) | 38 (12–66) | <0.01 |
| CD15+ARG1− | 51 (11–79) | 36 (19–105) | 0.42 |
| CD14+ monocytes | 604 (331–1001) | 524 (382–824) | 0.76 |
|
| |||
| Macrophages | 888 (662–1038) | 782 (586–1153) | 0.90 |
| M1-polarized macrophages | 120 (57–160) | 159 (101–381) | 0.02 |
| M2-polarized macrophages | 231 (146–353) | 189 (98–268) | 0.03 |
Median (interquartile range, IQR)
P values were calculated with the Wilcoxon rank-sum test
Within the myeloid immune cell populations, the overall density of CD15+ granulocytes expressing the immunosuppressive marker ARG1 was lower in neoadjuvant-treated tumors (Figure 3C, Table 2, Supplementary Table S5, Supplementary Figure S10). While overall CD14+ monocyte density was not significantly different, the intraepithelial density of CD14+ monocytes was two-fold higher in neoadjuvant-treated tumors (P<0.005; Table 2). Deeper analysis of CD14+ monocytes using markers for early (CD33+) and late stages of maturation/differentiation (HLA-DR+) revealed a three-fold increase in mature/differentiated CD14+HLA-DR+CD33− monocytes in neoadjuvant-treated tumors, an effect that was most pronounced within intraepithelial regions (Supplementary Table S5).
We next examined macrophage density and polarization status across an M1-M2 spectrum, as previously described17. Neoadjuvant treatment was associated with a six-fold higher number of intraepithelial macrophages compared to up-front resected tumors (Table 2, Supplementary Table S5, Supplementary Figure S10). In both stromal and intraepithelial areas, neoadjuvant therapy shifted the balance of M1 and M2 polarized macrophages towards an M1 phenotype, as reflected by a significantly higher M1:M2 density ratio in neoadjuvant-treated tumors. Although neoadjuvant treatment was associated with alterations in both M1 and M2-polarized macrophage densities, the shift towards an M1 phenotype was largely driven by higher M1 density, rather than lower M2 polarized macrophage density (Table 2).
Finally, we compared immune cell density measurements from the tumor center to the periphery while carefully excluding the tertiary lymphoid structures (TLS) that are often present at the tumor periphery. Although we observed a trend towards higher densities at the tumor periphery for most immune cell subsets, these differences were not statistically significant (Supplementary Figure S11). We next investigated TLS, which are ectopic lymphoid aggregates that can form in non-lymphoid tissues at sites of malignancy, and whose presence has been associated with prognosis and immunotherapy response for some cancer types, including PDAC17–19. Although a smaller proportion of neoadjuvant tumors harbored TLS, no differences were identified in median TLS count between up-front surgery and neoadjuvant-treated tumors (Figure 3D, Supplementary Figure S12). No differences were identified in median TLS count between the up-front surgery and neoadjuvant-treated groups (Figure 3D, Supplementary Figure S12). However, when we further stratified TLS by intratumoral or peritumoral location, we found that intratumoral TLS were slightly more abundant in neoadjuvant-treated tumors (43% of TLS) than up-front resected tumors (34% of TLS), although this difference was not statistically significant. Similar to immune infiltrates present within tumor epithelial regions, neoadjuvant therapy was associated with a higher CD3+CD8+ T-cell density and higher CD3+CD8+ to CD3+CD4+ ratio within TLS as compared to tumors that underwent up-front surgery (Figure 3D, Supplementary Table S6). Although CD3+CD4+ T-cell density was slightly decreased in TLS from neoadjuvant-treated tumors, this change was not statistically significant, also mirroring results seen in tumor epithelial regions. Overall, most immune cell densities did not differ by TLS location. The remaining immune cell subsets (e.g., macrophages and myeloid cell subsets) were not analyzed as they were not major TLS cellular components.
Based on the above results, neoadjuvant treatment was associated with a tumor immune microenvironment that included more cytotoxic T cells, fewer immunosuppressive granulocytes, and a shift towards M1-polarization within macrophages. To understand how these features related to the immune landscape of up-front resected pancreatic cancer, we expanded our unsupervised clustering analysis of the primary resection cohort by including the neoadjuvant-treated tumors. This analysis yielded four clusters that closely resembled those identified initially (Figure 3E, Supplementary Figure S13). Neoadjuvant-treated tumors were enriched in the T cell-rich cluster, with 64% of neoadjuvant-treated tumors included in this cluster. In contrast, only 20% of tumors that underwent up-front resection were included in this cluster, suggesting that the immune microenvironment seen after neoadjuvant FOLFIRINOX therapy was similar to that which is associated with longer overall survival in patients treated with up-front surgery.
Radiotherapy after neoadjuvant FOLFIRINOX is associated with few differences in the immune microenvironment
Radiotherapy is commonly administered following chemotherapy and prior to PDAC resection; however, the clinical effectiveness and immune modulating effects of this therapy remain unclear31–34. We therefore compared the immune microenvironment among patients who received FOLFIRINOX alone (N=24) and those who received FOLFIRINOX followed by radiotherapy (N=12) prior to surgery (Supplementary Table S7, Supplementary Figure S14). Overall, very few differences in immune cell population densities were identified, with the most notable difference being a reduction in CD3+CD8+ cytotoxic T-cell density in patients treated with radiotherapy. Although limited by modest case numbers, these results suggest that radiotherapy may not have a major impact on the immune microenvironment when administered following FOLFIRINOX chemotherapy.
Neoadjuvant therapy is associated with altered immune cell spatial distribution
Neoadjuvant therapy may influence the spatial configuration of immune cells with respect to tumor cells35–38. To investigate this possibility, we first assessed immune cell spatial localization within WSS from patients that underwent up-front resection. Similar to results from TMA analysis of the multi-institution resection cohort, most immune cells were co-localized within 50 μm of the closest tumor cell (Supplementary Figure S15A). We next evaluated whether co-localization between tumor cells and the main immune cell populations was altered in neoadjuvant-treated tumors. Given the large differences in intraepithelial macrophage densities between up-front resected and neoadjuvant-treated tumors (Figure 4A), we focused first on macrophage co-localization. Using the Gcross function, which measures the probability of co-localization between a tumor cell and immune cells within a specified radius, we found a greater likelihood of co-localization between tumor cells and M1-polarized macrophages in neoadjuvant-treated tumors as compared to those treated with up-front surgery (Figure 4B). Analysis using a nearest neighbor (NND) approach, which provides a complementary measure of spatial proximity, confirmed these results and showed that M1-polarized macrophages were, on average, 48% closer to tumor cells in neoadjuvant-treated tumors (Figure 4C). In contrast, no differences were observed for M2-polarized macrophages using either the Gcross function or NND. Beyond macrophages, T-cell spatial organization was also associated with neoadjuvant therapy. In both Gcross function and NND analyses, CD3+CD8+ cytotoxic T cells were located closer to tumor cells in neoadjuvant-treated tumors as compared to up-front resected tumors. This association was specific for CD3+CD8+ cells and was not observed for CD3+CD4+ cells (Supplementary Figure S15B–C). Overall, these results indicate that the spatial configuration of multiple prominent immune cell populations in the PDAC immune microenvironment is altered in tumors from patients treated with neoadjuvant therapy.
Figure 4. The association of neoadjuvant treatment with macrophage spatial composition in the pancreatic ductal adenocarcinoma (PDAC) microenvironment.
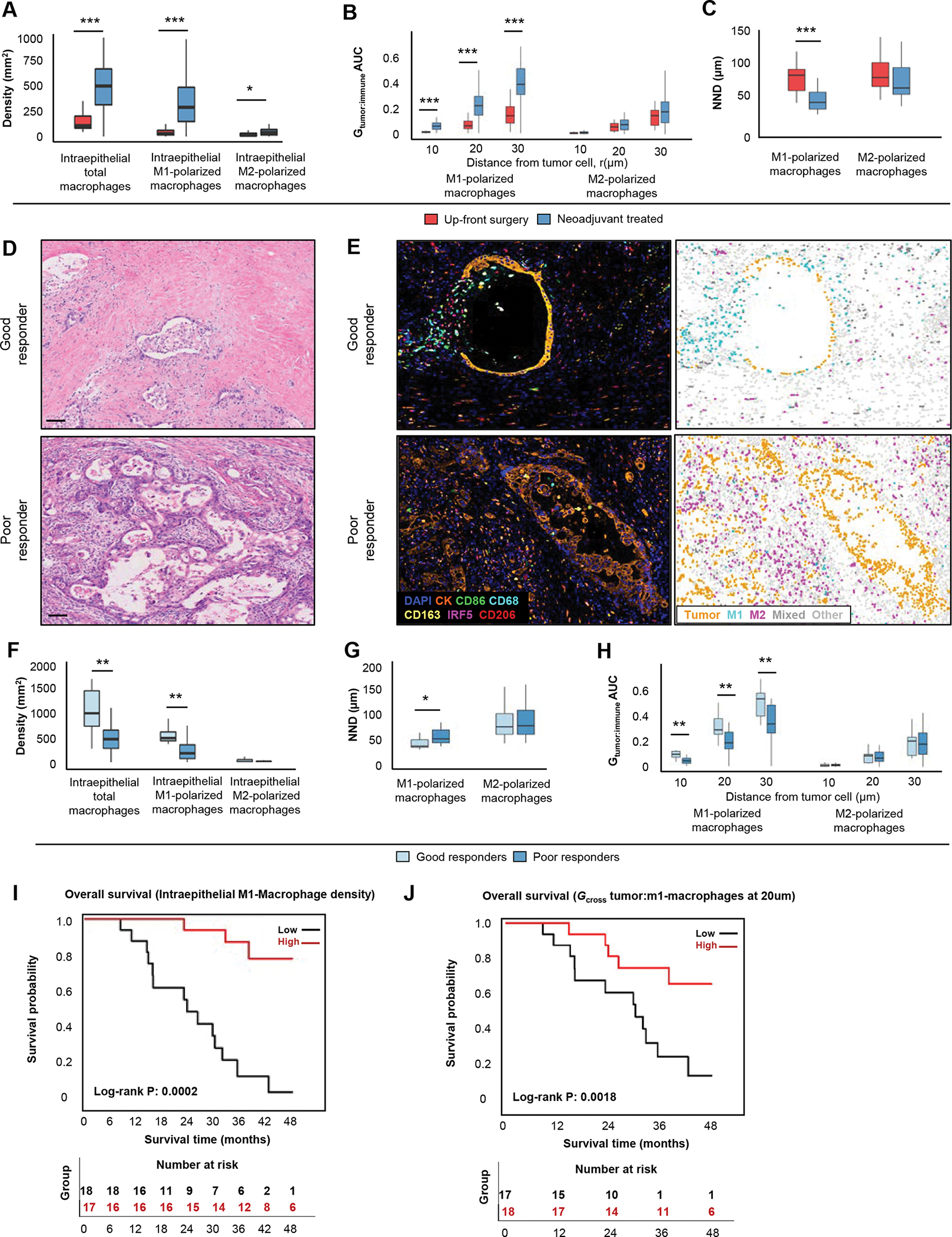
Macrophage densities in 29 up-front resected and 35 neoadjuvant-treated PDACs (A). Distribution of the immune cell-tumor cell Gcross proximity analysis and nearest neighbor distance (NND) in 29 up-front resected and 35 neoadjuvant-treated cases (B and C). Hematoxylin and eosin (D) and mIF stained tumors (E) show examples of good and poor histological response to treatment. Immune cell densities (F) and spatial analysis metrics in 10 cases with a good histologic response and 25 cases with a poor histologic response (G-H). Kaplan-Meier survival curves according to density of intraepithelial M1-polarized macrophages (I) and co-localization between tumor cells and M1-macrophages at 20 um using the Gcross function (J). Scale bars represent 100μm. P values were calculated with the Wilcoxon rank-sum test. ***: P <0.001, **: P <0.005, *: P <0.05
Histological response to neoadjuvant treatment is associated with macrophage localization and polarization status
Pancreatic cancer exhibits variable histologic response to neoadjuvant therapy39,40 and can be measured according to several grading systems28. In our cohort, overall degree of histological response in neoadjuvant-treated tumors was similar when assessed by both the Modified Ryan scheme and MD Anderson grading system (Supplementary Table S8). Since the Modified Ryan scheme is widely used for clinical reporting, we selected this scheme for further analyses. Given that the immune TME may both govern and be influenced by histologic response, we sought to determine whether immune cell configuration was associated with tumor histological response (Figure 4D–E, Supplementary Table S9). We found that intraepithelial macrophage density was strongly associated with better tumor histological response, an association that was driven by higher densities of intraepithelial M1-polarized macrophages (Figure 4F). Further evaluation of M1-polarized macrophage spatial configuration revealed greater co-localization between tumor cells and M1-polarized macrophages in tumors with a good histologic response as compared to those with a poor histologic response (Figure 4G–H).
Co-localization of M1-polarized macrophages and tumor cells is associated with better outcomes in neoadjuvant-treated patients
Given that M1-polarized macrophage density was higher in neoadjuvant-treated tumors and correlated with better histologic response, we hypothesized that higher M1-polarized macrophage density and greater co-localization with tumor cells would predict better outcomes among neoadjuvant-treated patients. In Kaplan-Meier and Cox regression analyses, higher intraepithelial M1-polarized macrophage density after neoadjuvant treatment was associated with longer OS (Figure 4I). Furthermore, greater co-localization of M1-polarized macrophages with tumor cells was also associated with longer OS (Figure 4J). Taken together, these results indicate that M1-polarized macrophages are enriched within intraepithelial areas after neoadjuvant therapy and that their greater density and proximity to tumor cells are associated with improved tumor histologic response and patient survival.
DISCUSSION
Patients with non-metastatic PDAC commonly receive neoadjuvant treatment with FOLFIRINOX followed in some cases by radiotherapy prior to resection. Although randomized data have just begun to emerge41,42, use of neoadjuvant therapy may be associated with longer overall survival compared to up-front resection5,43. Prior studies have suggested that the improved survival seen with neoadjuvant chemotherapy may be due, in part, to changes in the immune microenvironment20,22,44,45. However, existing studies have surveyed only a limited number of immune cell types and did not assess whether neoadjuvant therapy alters immune microenvironment spatial organization. Additionally, prior studies have largely relied upon singleplex immunohistochemistry and were unable to identify immune cell populations requiring multimarker analysis.
In the current study, we first characterized the immune microenvironment of a large multi-institutional cohort of previously-untreated resected PDAC. Consistent with our prior studies17,18, immune cell populations were highly variable in abundance between patients undergoing up-front resection, even when no selective pressure had been applied by neoadjuvant treatment. Several studies have found associations between specific immune cell densities and outcomes in PDAC; however, published data are inconsistent. In our study, survival analyses revealed improved survival for patients with higher T-cell densities, although these associations were largely due to higher CD4+ helper T-cell densities, rather than CD8+ T-cell densities. Interestingly, several pre-clinical studies have reported a potential direct anti-tumor effect driven by unique antigen-specific CD4+ helper T cells, in addition to a supportive role in orchestrating the CD8+ cytotoxic T-cell response46–48. Taken together, these results suggest that factors beyond T-cell density, such as functional capacity or the presence of immunosuppressive cells, may regulate T-cell response effectiveness in PDAC.
By conducting unsupervised clustering using immune cell densities, we identified four patterns of immune cell infiltration in resected PDAC. Furthermore, we found that the “T cell-rich” cluster, characterized by high abundance of T cells and low density of immunosuppressive granulocytes and M2-polarized macrophages, was associated with better survival. Overall, these results suggest that the relative balance between anti-tumorigenic immune cells, such as T cells and M1-polarized macrophages, and pro-tumorigenic myeloid cells, such as M2-polarized macrophages and ARG1+ granulocytes, is most important in defining patient prognosis in localized PDAC, rather than individual cell densities. This finding is consistent with our prior study18, which also noted that pancreatic tumors form clusters based on the relative balance between major immune cell subtypes.
The immune microenvironment of neoadjuvant-treated, resected pancreatic cancer has not been well characterized7,8,19–22,44. In the current study, we used mIF assays targeting a total of 15 immune cell markers across three 7-plex panels and a machine-learning-based analysis pipeline to analyze the distribution of immune cells in neoadjuvant-treated tumors. We confirmed prior findings that FOLFIRINOX-based neoadjuvant treatment was associated with higher cytotoxic CD3+CD8+ T-cell density49. We further found that neoadjuvant therapy was associated with higher M1-polarized macrophage density and decreased immunosuppressive granulocytes. Altogether, these results suggest that FOLFIRINOX-based neoadjuvant therapy may alter the PDAC immune microenvironment by changing the relative densities of specific immune cell subtypes, rather than globally increasing the immune cell infiltrate. Notably, these results from patient tumors are consistent with pre-clinical experimental studies that identify immune cell microenvironment alterations in chemotherapy-treated PDAC mouse models50,51.
Immune cell-tumor cell spatial interactions have been proposed as key factors in mediating an effective anti-tumor immune response11,52. While prior studies from our group and others have reported that proximity of CD8+ T cells and M2-polarized macrophages to tumor cells are associated with patient survival in up-front resected PDAC11,17, no studies have comprehensively analyzed tumor-immune cell spatial organization in neoadjuvant-treated tumors. We found that neoadjuvant-treated tumors exhibited an increased density of intraepithelial M1-polarized macrophages and greater co-localization of M1-polarized macrophages with tumor cells. Furthermore, higher density of intraepithelial M1-macrophages and greater co-localization of M1-polarized macrophages with tumor cells were both associated with better histological response to neoadjuvant therapy and longer survival. Importantly, cytotoxic T cells were also located closer to tumor cells in patients who received neoadjuvant chemotherapy compared to those who underwent up-front resection. Together, these results suggest that neoadjuvant treatment may alter spatial relationships between immune cells and tumor cells. Further investigation will be required to determine whether these changes are casually related to chemotherapy or whether they are a consequence of immunogenic cell death and may therefore represent a marker of therapeutic response20.
Our study has several limitations. While our multiplex immunofluorescence assays offer high sensitivity and highly quantitative data coupled with subcellular resolution for characterizing immune cells in fixed tissue, this approach requires marker and cell type pre-specification. We did not profile all immune cell types of potential interest, and our markers are not identical to those utilized in other studies, potentially limiting comparability of results. We were also unable to analyze matched pre- and post-neoadjuvant therapy specimens for individual patients, requiring us to infer changes due to neoadjuvant therapy at the cohort level, rather than at the level of individual patients. Finally, due to the typical 2–6 week time interval between completion of neoadjuvant therapy and subsequent surgical resection, we were not able to formally distinguish between direct effects of neoadjuvant therapy versus secondary responses to tumor cell death that may reshape the tumor microenvironment.
Our study has important strengths. Our cohort of up-front resected PDAC was derived from multiple institutions across the U.S. and had extensive clinicopathologic and molecular annotation. Our mIF panels were designed to analyze markers specifically selected for pancreatic cancer relevance and enabled phenotyping of immune cell subsets defined by combinatorial marker expression, thereby overcoming limitations of singleplex IHC studies. Supervised machine learning algorithms provided data at the single cell level, allowing for more accurate phenotype determination than with simple pixel or threshold classification approaches and enabled reporting of immune cell densities per square millimeter of tissue. In comparison to qualitative or non-standardized assessments, our measurements provide a benchmark for future pancreatic cancer tissue profiling studies independent of analysis platform. For evaluation of neoadjuvant-treated cases, every individual block from the tumor bed was reviewed to select the most representative tumor-containing block, enhancing reproducibility for future analyses. Finally, our spatially-resolved approach enabled us to identify a coordinated macrophage response in neoadjuvant-treated tumors characterized not just by increased M1-polarized macrophage stromal density, but also by greater proximity of M1-polarized macrophages to tumor cells. These prognostically-relevant shifts in macrophage localization would not have been possible to infer by dissociative approaches such as flow cytometry or single cell RNA sequencing.
In conclusion, we characterized the baseline immune microenvironment of resected pancreatic cancer in a large multi-institutional cohort and found significant heterogeneity in immune cell infiltration but also discrete patterns with prognostic significance. We also determined that FOLFIRINOX-based neoadjuvant treatment was associated with shifts in the balance of multiple lymphoid and myeloid cell subpopulations towards an anti-tumorigenic profile, resembling the features of up-front resected tumors with the most prognostically favorable immune cell infiltration pattern. Furthermore, we demonstrated that coordinated polarization shifts and spatial reorganization of macrophages are predictive of histologic response and prognosis in neoadjuvant-treated patients. Altogether, these results indicate that FOLFIRINOX-based neoadjuvant treatment modulates multiple aspects of the primary pancreatic cancer immune microenvironment. These findings may be leveraged in the design of new neoadjuvant treatment strategies.
Supplementary Material
Translational relevance:
Using multiplex immunofluorescence and digital image analysis of formalin-fixed, paraffin-embedded pancreatic cancer resections, we quantified and spatially characterized T-cell, macrophage, and myeloid cell populations in patients who underwent up-front surgery or received preoperative chemotherapy followed by surgery. In up-front resected tumors, unsupervised clustering identified four classes of immune cell infiltration, one of which was associated with improved patient survival and characterized by abundant T cells and a paucity of immunosuppressive granulocytes and macrophages. Tumors treated with neoadjuvant chemotherapy demonstrated increased cytotoxic T-cell and reduced immunosuppressive myeloid cell densities, along with greater proximity of cytotoxic T cells and M1-polarized macrophages to tumor cells. Understanding the changes to the tumor immune microenvironment induced by chemotherapy will be of critical importance as preoperative therapy is increasingly used for patients with resectable pancreatic cancers.
Funding:
S.A.V. is supported by the Finnish Cultural Foundation and Orion Research. S.K.D. is supported by Hale Center, NIH U01 CA224146-01, Novartis, BMS, and Genocea. A.J.A. is funded by the Lustgarten Foundation, Hale Family Center for Pancreatic Cancer Research, the Doris Duke Charitable Foundation, Pancreatic Cancer Action Network, and NIH grants K08 CA218420-02, P50CA127003, U01 CA224146, and U01 CA250549. B.M.W. is supported by the Hale Family Center for Pancreatic Cancer Research, Lustgarten Foundation Dedicated Laboratory program, NIH grant U01 CA210171, NIH grant P50 CA127003, Stand Up to Cancer - Lustgarten Foundation Pancreatic Cancer Dream Team Research Grant (Grant Number: SU2C-AACR-DT-20-16a), Pancreatic Cancer Action Network, Noble Effort Fund, Wexler Family Fund, Promises for Purple and Bob Parsons Fund. J.A.N. is supported by the Hale Family Center for Pancreatic Cancer Research, Lustgarten Foundation Dedicated Laboratory program, and NIH grants P50 CA127003, R01 CA248857, R01 CA205406, R01 CA169141, R35 CA197735, and U01 CA250549. The indicated Stand Up To Cancer (SU2C) research grant is administered by the American Association for Cancer Research, the scientific partner of SU2C.
Footnotes
Conflict of interests: S.K.D. is a co-founder and SAB member for Kojin Therapeutics. R.F.D. served on Advisory Boards for Exelixis Inc. and Helsinn Healthcare S.A. A.J.A. has consulted for Oncorus, Inc., Arrakis Therapeutics, Syros Pharmaceuticals, Mirati Therapeutics, Boehringer Ingelheim, T-knife Therapeutics, AstraZeneca, Servier, and Merck & Co., Inc, and has research funding from Mirati Therapeutics, Syros Pharmaceuticals, Bristol Myers Squibb, Revolution Medicines, Novartis, Deerfield, Inc., and Novo Ventures that is unrelated to this work. B.M.W. has research funding from Celgene and Eli Lilly and consulting for BioLineRx, Celgene, G1 Therapeutics, and GRAIL. J.A.N. has research funding from NanoString, Illumina, and Akoya Biosciences. The other authors declare no potential conflicts of interest.
REFERENCES
- 1.Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer Statistics, 2021. CA Cancer J Clin. 2021;71(1):7–33. doi: 10.3322/caac.21654 [DOI] [PubMed] [Google Scholar]
- 2.Nevala-Plagemann C, Hidalgo M, Garrido-Laguna I. From state-of-the-art treatments to novel therapies for advanced-stage pancreatic cancer. Nat Rev Clin Oncol. 2020;17(2):108–123. doi: 10.1038/s41571-019-0281-6 [DOI] [PubMed] [Google Scholar]
- 3.Strobel O, Neoptolemos J, Jäger D, Büchler MW. Optimizing the outcomes of pancreatic cancer surgery. Nat Rev Clin Oncol. 2019;16(1):11–26. doi: 10.1038/s41571-018-0112-1 [DOI] [PubMed] [Google Scholar]
- 4.Hu Q, Wang D, Chen Y, Li X, Cao P, Cao D. Network meta-analysis comparing neoadjuvant chemoradiation, neoadjuvant chemotherapy and upfront surgery in patients with resectable, borderline resectable, and locally advanced pancreatic ductal adenocarcinoma. Radiat Oncol. 2019;14(1):1–8. doi: 10.1186/s13014-019-1330-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chawla A, Ferrone CR. Neoadjuvant therapy for resectable pancreatic cancer: An evolving paradigm shift. Front Oncol. 2019;9(OCT):10–13. doi: 10.3389/fonc.2019.01085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ryan DP, Wo IIJY, Warshaw AL . Tumor microenvironment immune response in pancreatic ductal adenocarcinoma patients treated with neoadjuvant therapy. J Natl Cancer Inst. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reyes CM, Teller S, Muckenhuber A, et al. Neoadjuvant therapy remodels the pancreatic cancer microenvironment via depletion of protumorigenic immune cells. Clin Cancer Res. 2020;26(1):220–231. doi: 10.1158/1078-0432.CCR-19-1864 [DOI] [PubMed] [Google Scholar]
- 8.Farren MR, Sayegh L, Ware MB, et al. Immunologic alterations in the pancreatic cancer microenvironment of patients treated with neoadjuvant chemotherapy and radiotherapy. JCI Insight. 2020;5(1):1–13. doi: 10.1172/jci.insight.130362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thyagarajan A, Alshehri MSA, Miller KLR, Sherwin CM, Travers JB, Sahu RP. Myeloid-derived suppressor cells and pancreatic cancer: Implications in novel therapeutic approaches. Cancers (Basel). 2019;11(11). doi: 10.3390/cancers11111627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fukunaga A, Miyamoto M, Cho Y, et al. CD8+tumor-infiltrating lymphocytes together with CD4+tumor-infiltrating lymphocytes and dendritic cells improve the prognosis of patients with pancreatic adenocarcinoma. Pancreas. 2004;28(1):26–31. doi: 10.1097/00006676-200401000-00023 [DOI] [PubMed] [Google Scholar]
- 11.Carstens JL, De Sampaio PC, Yang D, et al. Spatial computation of intratumoral T cells correlates with survival of patients with pancreatic cancer. Nat Commun. 2017;8:1–13. doi: 10.1038/ncomms15095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mahajan UM, Langhoff E, Goni E, et al. Immune Cell and Stromal Signature Associated With Progression-Free Survival of Patients With Resected Pancreatic Ductal Adenocarcinoma. Gastroenterology. 2018;155(5):1625–1639.e2. doi: 10.1053/j.gastro.2018.08.009 [DOI] [PubMed] [Google Scholar]
- 13.Protti MP, De Monte L. Immune infiltrates as predictive markers of survival in pancreatic cancer patients. Front Physiol. 2013;4 AUG(August):1–6. doi: 10.3389/fphys.2013.00210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ino Y, Yamazaki-Itoh R, Shimada K, et al. Immune cell infiltration as an indicator of the immune microenvironment of pancreatic cancer. Br J Cancer. 2013;108(4):914–923. doi: 10.1038/bjc.2013.32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ligorio M, Sil S, Malagon-Lopez J, et al. Stromal Microenvironment Shapes the Intratumoral Architecture of Pancreatic Cancer. Cell. 2019;178(1):160–175.e27. doi: 10.1016/j.cell.2019.05.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Argyraki A, Markvart M, Stavnsbjerg C, et al. (生物用)HHS Public Access. 2015 IEEE Summer Top Meet Ser SUM 2015. 2018;10(1):1–13. doi: 10.1038/s41598-019-39414-9 [DOI] [Google Scholar]
- 17.Väyrynen SA, Zhang J, Yuan C, et al. Composition, Spatial Characteristics, and Prognostic Significance of Myeloid Cell Infiltration in Pancreatic Cancer. Clin Cancer Res. 2020:1069–1082. doi: 10.1158/1078-0432.ccr-20-3141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liudahl SM, Betts CB, Sivagnanam S, et al. Leukocyte Heterogeneity in Pancreatic Ductal Adenocarcinoma: Phenotypic and Spatial Features Associated with Clinical Outcome; 2021. doi: 10.1158/2159-8290.cd-20-0841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Michelakos T, Cai L, Villani V, et al. Tumor Microenvironment Immune Response in Pancreatic Ductal Adenocarcinoma Patients Treated with Neoadjuvant Therapy. J Natl Cancer Inst. 2021;113(2):182–191. doi: 10.1093/jnci/djaa073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Di Caro G, Cortese N, Castino GF, et al. Dual prognostic significance of tumour-Associated macrophages in human pancreatic adenocarcinoma treated or untreated with chemotherapy. Gut. 2015;65(10):1710–1720. doi: 10.1136/gutjnl-2015-309193 [DOI] [PubMed] [Google Scholar]
- 21.Tsuchikawa T, Hirano S, Tanaka E, et al. Novel aspects of preoperative chemoradiation therapy improving anti-tumor immunity in pancreatic cancer. Cancer Sci. 2013;104(5):531–535. doi: 10.1111/cas.12119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Homma Y, Taniguchi K, Murakami T, et al. Immunological impact of neoadjuvant chemoradiotherapy in patients with borderline resectable pancreatic ductal adenocarcinoma. Ann Surg Oncol. 2014;21(2):670–676. doi: 10.1245/s10434-013-3390-y [DOI] [PubMed] [Google Scholar]
- 23.Shibuya KC, Goel VK, Xiong W, et al. Pancreatic ductal adenocarcinoma contains an effector and regulatory immune cell infiltrate that is altered by multimodal neoadjuvant treatment. PLoS One. 2014;9(5). doi: 10.1371/journal.pone.0096565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Qian ZR, Rubinson DA, Nowak JA, et al. Association of alterations in main driver genes with outcomes of patients with resected pancreatic ductal adenocarcinoma. JAMA Oncol. 2018;4(3):6–11. doi: 10.1001/jamaoncol.2017.3420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Perez K, Clancy TE, Mancias JD, Rosenthal MH, Wolpin BM. When, what, and why of perioperative treatment of potentially curable pancreatic adenocarcinoma. J Clin Oncol. 2017;35(5):485–489. doi: 10.1200/JCO.2016.70.2134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ryan R, Gibbons D, Hyland JMP, et al. Pathological response following long-course neoadjuvant chemoradiotherapy for locally advanced rectal cancer. Histopathology. 2005;47(2):141–146. doi: 10.1111/j.1365-2559.2005.02176.x [DOI] [PubMed] [Google Scholar]
- 27.Pylayeva-Gupta Y, Kelsey C. Martin Mhatre V. Ho J-AL. 基因的改变NIH Public Access. Bone. 2012;23(1):1–7. doi: 10.1002/cncr.26651.Histologic [DOI] [Google Scholar]
- 28.Wang H, Chetty R, Hosseini M, et al. Pathologic Examination of Pancreatic Specimens Resected for Treated Pancreatic Ductal Adenocarcinoma. Am J Surg Pathol. 2021;46(6):754–764. doi: 10.1097/PAS.0000000000001853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stack EC, Wang C, Roman KA, Hoyt CC. Multiplexed immunohistochemistry, imaging, and quantitation: A review, with an assessment of Tyramide signal amplification, multispectral imaging and multiplex analysis. Methods. 2014;70(1):46–58. doi: 10.1016/j.ymeth.2014.08.016 [DOI] [PubMed] [Google Scholar]
- 30.Lohneis P, Sinn M, Bischoff S, et al. Cytotoxic tumour-infiltrating T lymphocytes influence outcome in resected pancreatic ductal adenocarcinoma. Eur J Cancer. 2017;83(2017):290–301. doi: 10.1016/j.ejca.2017.06.016 [DOI] [PubMed] [Google Scholar]
- 31.Kim R FOLFIRINOX: A new standard treatment for advanced pancreatic cancer? Lancet Oncol. 2011;12(1):8–9. doi: 10.1016/S1470-2045(10)70237-0 [DOI] [PubMed] [Google Scholar]
- 32.Oba A, Ho F, Bao QR, Al-Musawi MH, Schulick RD, Chiaro M Del. Neoadjuvant Treatment in Pancreatic Cancer. Front Oncol. 2020;10(February):1–10. doi: 10.3389/fonc.2020.00245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Faris JE, Blaszkowsky LS, McDermott S, et al. FOLFIRINOX in Locally Advanced Pancreatic Cancer: The Massachusetts General Hospital Cancer Center Experience. Oncologist. 2013;18(5):543–548. doi: 10.1634/theoncologist.2012-0435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nanda RH, El-Rayes B, Maithel SK, Landry J. Neoadjuvant modified FOLFIRINOX and chemoradiation therapy for locally advanced pancreatic cancer improves resectability. J Surg Oncol. 2015;111(8):1028–1034. doi: 10.1002/jso.23921 [DOI] [PubMed] [Google Scholar]
- 35.Parra ER, Villalobos P, Behrens C, et al. Effect of neoadjuvant chemotherapy on the immune microenvironment in non-small cell lung carcinomas as determined by multiplex immunofluorescence and image analysis approaches. J Immunother Cancer. 2018;6(1):1–11. doi: 10.1186/s40425-018-0368-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nawaz S, Heindl A, Koelble K, Yuan Y. Beyond immune density: Critical role of spatial heterogeneity in estrogen receptor-negative breast cancer. Mod Pathol. 2015;28(6):766–777. doi: 10.1038/modpathol.2015.37 [DOI] [PubMed] [Google Scholar]
- 37.2. Multiplexed Ion Beam Imaging (MIBI). Physiol Behav. 2019;176(3):139–148. doi: 10.1016/j.cell.2018.08.039.A [DOI] [Google Scholar]
- 38.Kather JN, Suarez-Carmona M, Charoentong P, et al. Topography of cancer-associated immune cells in human solid tumors. Elife. 2018;7:1–19. doi: 10.7554/eLife.36967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Matsuda Y, Ohkubo S, Nakano-Narusawa Y, et al. Objective assessment of tumor regression in post-neoadjuvant therapy resections for pancreatic ductal adenocarcinoma: comparison of multiple tumor regression grading systems. Sci Rep. 2020;10(1):1–12. doi: 10.1038/s41598-020-74067-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wittmann D, Hall WA, Christians KK, et al. Impact of Neoadjuvant Chemoradiation on Pathologic Response in Patients With Localized Pancreatic Cancer. Front Oncol. 2020;10(April):1–11. doi: 10.3389/fonc.2020.00460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Versteijne E, Suker M, Groothuis K, et al. Preoperative Chemoradiotherapy Versus Immediate Surgery for Resectable and Borderline Resectable Pancreatic Cancer: Results of the Dutch Randomized Phase III PREOPANC Trial. J Clin Oncol. 2020;38(16):1763–1773. doi: 10.1200/JCO.19.02274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Motoi F, Kosuge T, Ueno H, et al. Randomized phase II/III trial of neoadjuvant chemotherapy with gemcitabine and S-1 versus upfront surgery for resectable pancreatic cancer (Prep-02/JSAP05). Jpn J Clin Oncol. 2019;49(2):190–194. doi: 10.1093/jjco/hyy190 [DOI] [PubMed] [Google Scholar]
- 43.Michelakos T, Sekigami Y, Kontos F, et al. Conditional Survival in Resected Pancreatic Ductal Adenocarcinoma Patients Treated with Total Neoadjuvant Therapy. J Gastrointest Surg. 2021. doi: 10.1007/s11605-020-04897-9 [DOI] [PubMed] [Google Scholar]
- 44.Okubo S, Suzuki T, Hioki M, et al. The immunological impact of preoperative chemoradiotherapy on the tumor microenvironment of pancreatic cancer. Cancer Sci. 2021;112(7):2895–2904. doi: 10.1111/cas.14914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Matsuki H, Hiroshima Y, Miyake K, et al. Reduction of gender-associated M2-like tumor-associated macrophages in the tumor microenvironment of patients with pancreatic cancer after neoadjuvant chemoradiotherapy. J Hepatobiliary Pancreat Sci. 2021;28(2):174–182. doi: 10.1002/jhbp.883 [DOI] [PubMed] [Google Scholar]
- 46.Takeuchi A, Badr MESG, Miyauchi K, et al. CRT AM determines the CD4+ cytotoxic T lymphocyte lineage. J Exp Med. 2016;213(1):123–138. doi: 10.1084/jem.20150519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xie Y, Akpinarli A, Maris C, et al. Naive tumor-specific CD4+ T cells differentiated in vivo eradicate established melanoma. J Exp Med. 2010;207(3):651–667. doi: 10.1084/jem.20091921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Matsuzaki J, Tsuji T, Luescher IF, et al. Direct tumor recognition by a human CD4+ T-cell subset potently mediates tumor growth inhibition and orchestrates anti-tumor immune responses. Sci Rep. 2015;5(October):1–14. doi: 10.1038/srep14896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Michelakos T, Cai L, Villani V, et al. Tumor microenvironment immune response in pancreatic ductal adenocarcinoma patients treated with neoadjuvant therapy. JNCI J Natl Cancer Inst. June 2020. doi: 10.1093/jnci/djaa073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ye J, Mills BN, Zhao T, et al. Assessing the magnitude of immunogenic cell death following chemotherapy and irradiation reveals a new strategy to treat pancreatic cancer. Cancer Immunol Res. 2020;8(1):94–107. doi: 10.1158/2326-6066.CIR-19-0373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nywening TM, Belt BA, Cullinan DR, et al. Targeting both tumour-associated CXCR2+ neutrophils and CCR2+ macrophages disrupts myeloid recruitment and improves chemotherapeutic responses in pancreatic ductal adenocarcinoma. Gut. 2018;67(6):1112–1123. doi: 10.1136/gutjnl-2017-313738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McNamara KL, Caswell-Jin JL, Joshi R, et al. Spatial proteomic characterization of HER2-positive breast tumors through neoadjuvant therapy predicts response. Nat Cancer. 2021;2(4):400–413. doi: 10.1038/s43018-021-00190-z [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data generated in this study are available upon request from the corresponding author.