

Abstract

Naringenin (Nar) and its structural isomer, naringenin chalcone (ChNar), are two natural phytophenols with beneficial health effects belonging to the flavonoids family. A direct discrimination and structural characterization of the protonated forms of Nar and ChNar, delivered into the gas phase by electrospray ionization (ESI), was performed by mass spectrometry-based methods. In this study, we exploit a combination of electrospray ionization coupled to (high-resolution) mass spectrometry (HR-MS), collision-induced dissociation (CID) measurements, IR multiple-photon dissociation (IRMPD) action spectroscopy, density functional theory (DFT) calculations, and ion mobility-mass spectrometry (IMS). While IMS and variable collision-energy CID experiments hardly differentiate the two isomers, IRMPD spectroscopy appears to be an efficient method to distinguish naringenin from its related chalcone. In particular, the spectral range between 1400 and 1700 cm–1 is highly specific in discriminating between the two protonated isomers. Selected vibrational signatures in the IRMPD spectra have allowed us to identify the nature of the metabolite present in methanolic extracts of commercial tomatoes and grapefruits. Furthermore, comparisons between experimental IRMPD and calculated IR spectra have clarified the geometries adopted by the two protonated isomers, allowing a conformational analysis of the probed species.
Keywords: flavanones, structural elucidation, conformational analysis, tandem mass spectrometry, isomeric discrimination, IRMPD action spectroscopy, naringenin
Introduction
A comprehensive identification of metabolites in complex biological matrices is of utmost importance to identify the molecular basis of their nutraceutical, biological, and pharmaceutical properties.1 To this end, numerous analytical techniques are exploited.2 Among them, mass spectrometry (MS) represents the method of choice for metabolite profiling, offering high accuracy, specificity, and sensitivity.3,4 However, MS analysis is not always able to distinguish between isomeric forms, so a multidimensional approach is required to obtain a clear assignment of the observed species. This notion appears particularly true concerning families of compounds such as flavonoids, flavones, and related molecules, which are highly populated in biological matrices and possess a vast isomeric variability.5 Flavonoids are an important class of phytochemicals, ubiquitous in plants, fruits, vegetables, flowers, cereals, and plant derivatives such as wines, teas, honey, and bee pollen.6 These natural polyphenols possess an extensive range of pharmacological activities, comprising antioxidative, anti-inflammatory, anticancer, antimutagenic, antiviral, and anti-ischemic effects. They are mainly present as glycosides, but free aglycones are also found in many plants. Due to their wide range of biological activities and the existence of numerous natural isomers, many efforts have aimed at differentiating and characterizing individual aglycones by tandem mass spectrometry.7−10 In particular, collision-induced dissociation (CID) mass spectrometry studies have successfully characterized several isomeric flavonoids belonging to distinct subgroups, through their different fragmentation patterns.11 However, it was not possible to apply this powerful technique to the direct discrimination of isomeric chalcones and flavanones due to their similar behavior under CID.7,12 Particularly, the discrimination of naringenin (Nar) and its structural isomer, naringenin chalcone (ChNar), in complex mixtures by MS appears to be a difficult task, without preliminary separation. In fact, the reference CID mass spectra for Nar and ChNar recorded either in positive or negative mode show identical fragmentation patterns.13,14
Nar (4′,5,7-trihydroxyflavanone) belongs to the flavanone group, and is a natural antioxidant abundant, especially in citrus fruits with numerous health-promoting effects.15,16 Nar is produced by stereo-specific isomerization catalyzed by chalcone isomerase from ChNar ((2E)-3-(4-hydroxyphenyl)-1-(2,4,6-trihydroxilphenyl)-2-propen-1on).17,18 Thus, the two species can be both present in many vegetables. Structurally, Nar shows the typical C6–C3–C6 flavan nucleus, formed by two aromatic rings (A and B) joined by a 2,3-dihydro-4-pyrone ring (C), as shown in Figure 1. There are numerous theoretical and experimental studies in the literature that have described the structure of neutral naringenin.19−21 The C-ring adopts a flattened chair-type conformation, in which the chiral C2 atom deviates from the plane defined by the other five atoms of the ring and binds mainly in the equatorial position of the B-ring. In nature, Nar is found predominantly as the (S)-enantiomer.17 On the other hand, ChNar is an open-chain flavonoid, in which the two aromatic rings A and B are joined by an α,β-unsaturated carbonyl system (Figure 1). The α,β-double bond of natural ChNar is always in trans configuration, while the cis isomer is reported to be extremely unstable. With respect to Nar, the absence of the central C-ring in ChNar allows internal rotations around the single bonds C1′–Cγ, Cγ–Cα, and Cβ–C1. From a conformational and energetic point of view, the most important rotation is around the Cγ–Cα single bond, leading to two distinct conformers referred to as s-cis and s-trans, depending on the relative position of the keto group, Cγ=O, and the double bond Cα=Cβ with respect to the single bond Cγ–Cα. Previous theoretical conformational studies on neutral chalcones have indicated that the s-cis conformation is the most stable geometry, with an energy gap to the s-trans species of approximately 24.5 kJ mol–1.22,23 Generally, chalcones in the s-cis conformation assume a relatively planar geometry. In contrast, the s-trans conformers are definitely nonplanar.23 In both Nar and ChNar, there is a strong intramolecular hydrogen bond between the carbonyl oxygen atom and the H atom of the adjacent hydroxyl group on the A ring.
Figure 1.
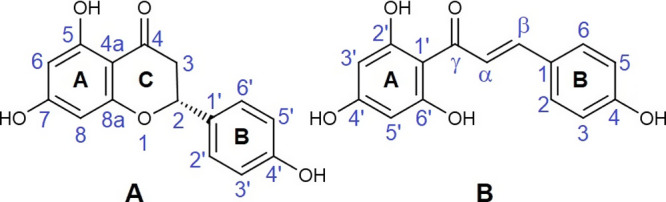
Schematic representation and numbering schemes of (A) naringenin and (B) naringenin chalcone. The numbering of the atoms is reported according to convention.
Because Raman and IR spectra of neutral Nar and ChNar reported in the literature are different,24 it was decided to sample the protonated forms of both isomers by IR multiple-photon dissociation (IRMPD) spectroscopy. IRMPD is an “action” spectroscopy technique that allows one to obtain IR spectra of ionic species mass-isolated in the storage cell of a mass spectrometer.25−28 Comparison of experimental spectra with those obtained by DFT calculations of plausible structures can be used to discriminate isomers and investigate molecular properties, conformational analysis, and binding interactions. This strategy has been applied to a large diversity of biomolecular ions and metal adducts, such as nucleotides,29−31 modified amino acids and peptides,32−34 metabolites,35−37 polyphenols,38−42 and chalcones.43
The main purpose of this contribution is to determine structural features and conformational distribution of protonated Nar and ChNar isolated in the gas phase by exploiting a combined approach based on electrospray ionization (ESI) coupled to (high-resolution) mass spectrometry, IRMPD action spectroscopy, theoretical DFT calculations, and ion mobility-mass spectrometry (IMS). This study was carried out on protonated species to avoid possible naringenin/naringenin chalcone isomerization during the ionization processes in the ESI source or solution. It has in fact been reported that an equilibrium in chalcone—flavanone—isomerization occurs promptly in neutral and alkaline media, thus behaving as proteiform species.12,24,44 In the proposed equilibrium mechanism, deprotonation on the C2′ hydroxyl group of naringenin chalcone is followed by the cyclization reaction yielding the enolate form of naringenin.24 Furthermore, IRMPD spectroscopy was also used to investigate the nature of the metabolite present in methanolic extracts of commercial tomatoes and grapefruits, chosen for their distinct flavonoid content.
Materials and Methods
Chemical Reagents and Materials
Naringenin and Naringenin chalcone (purity ≥ 95%) are research-grade commercial products (Merck Life Science S.r.l. Milan, Italy) and were used as received. Italian cherry tomatoes (Solanum lycopersicum L.) cultivar “datterino” and red grapefruits (Citrus Paradisi) cultivar “Star Ruby” were purchased from a local grocery store in Rome, Italy. Gaseous ions [Nar + H]+ and [ChNar + H]+, both at m/z 273, were obtained by electrospray ionization mass spectrometry (ESI-MS) in positive ion mode, through direct infusion by a syringe pump of a solution (10 μM) of either Nar or ChNar in acidified (1% HCOOH) methanol to favor protonation. Naringenin and Naringenin chalcone has been extracted by maceration from grapefruit and tomato peel.45−47 Details of the tomato and grapefruit sample preparation are described in the Supporting Information.
High-Resolution FT-ICR-MS
High-resolution mass analyses were performed using a Bruker BioApex 4.7T Fourier transform ion cyclotron resonance (FT-ICR) mass spectrometer equipped with an Apollo I ESI source, (FT-ICR lab, Sapienza Università di Roma), with internal calibration (ensuring Δm < 2 ppm).48
CID Experiments
CID experiments at variable energy were made using a 2000 Q-Trap instrument (Applied Biosystems), a commercial hybrid triple quadrupole linear ion trap mass spectrometer with a Q1q2QLIT configuration. The operating procedures are reported in the Supporting Information. Phenomenological threshold energies (TEs) of the various fragment ions can be obtained by CID at variable collision energy as described previously48−51 (see the Supporting Information for more details).
Ion Mobility Experiments
Traveling wave ion mobility-mass spectrometry (TWIMS-MS) experiments were performed with a Synapt G2 HDMS quadrupole/time-of-flight mass spectrometer (Waters, Manchester, UK) equipped with an ESI source. The operating details are described in the Supporting Information.
IRMPD Experiments
Sample solutions were directly infused in a 3D quadrupole ion trap (QIT) mass spectrometer (Bruker, Amazon Speed ETD, Bremen, Germany) coupled to the beamline of the free-electron laser for Infrared eXperiments (FELIX),52 as explained in the Supporting Information. During an IRMPD “action” spectroscopy experiment, the absorption of multiple resonant IR photons leads to an increase in the internal energy of the analyte, causing its unimolecular dissociation and, consequently, the appearance of fragment ions in the mass spectrum. To prevent excessive depletion of the parent ions and to minimize the formation of fragment ions below the low mass cut of the QIT, the IR spectra were recorded at several levels of laser pulse energy attenuation.53
Computational Details
To obtain the lowest energy conformations of candidate isomers of the sampled [Nar + H]+ and [ChNar + H]+ ions, two different strategies were adopted as illustrated in the Supporting Information. Optimized geometries, thermodynamic properties (electronic energy values, zero point energy (ZPE) and thermal corrections, entropies, and free energies at 298 K), and harmonic vibrational frequencies were calculated in the gas phase using density functional theory with the B3LYP functional and 6-311++G(d,p) basis set.54,55 All quantum chemical calculations were performed with the Spartan′16 software package. CCSs were calculated from the B3LYP-D3/6-311++G(d,p) optimized structures with the MobCal software as appropriately modified by Kim et al. for mobility separations employing N2 as drift gas.56,57
Results and Discussion
HR-MS, CID Experiments
MS is extensively used for the characterization of flavonoid compounds in foods. However, discriminating between naringenin (Nar) and its chalcone isomer (ChNar), directly infused into a mass spectrometer without prior (U)HPLC separation, is a challenging task.13,58−60 The high-resolution mass spectrum of protonated naringenin recorded in the FT-ICR mass spectrometer displays the monoisotopic peak at m/z 273.07647 in good agreement with the calculated elemental composition [C15H13O5]+ (error + 2.6 ppm), as reported in Figure S1.
Under CID conditions, the MS/MS spectrum of protonated naringenin [Nar + H]+ reported in Figure S2 shows four fragment ions assigned to characteristic cleavages of the C-ring in the flavanone, as presented in Scheme S1.61,62 The two primary fragment ions at m/z 153 and 147 correspond to the 1,3A+ and 1,4B+–2H cleavages, respectively. Two other ions, namely m/z 119 and 91, are due to subsequent losses of one and two CO molecules from the1,4B+–2H fragment. Threshold energy values are not directly measurable;49,63,64 however, phenomenological TEs of the two competitive and sequential fragmentation paths can be obtained from comparative analysis, as shown in the breakdown curves of Figure S3 and reported in Table S1. The relatively low TE values of 0.70 and 1.0 eV found for the formation of m/z 147 and 153, respectively, confirm the facile cleavage of the C-ring in flavanones.61 Subsequent losses of CO require more energy, as indicated by TE values of 2.30 and 3.30 eV found for ions at m/z 119 and 91, respectively. As expected from the literature, the behavior of [ChNar + H]+ under CID displays quite identical characteristics.13,14 The MS/MS spectrum of [ChNar + H]+ reveals the same four fragment ions observed in the case of [Nar + H]+ (Figure S2, orange trace), namely ions at m/z 153, 147, 119, and 91 with phenomenological TEs (Figure S4 and Table S1) of 1.15, 1.00, 2.50, and 3.60 eV, respectively. All TE values found for the fragmentation paths of ChNar are slightly higher than those obtained for Nar; however, these small differences do not produce substantial differences in the spectra of the two isomers.
IRMPD and Computational Study
Comparison of IRMPD Spectra for [Nar + H]+ and [ChNar + H]+
To discriminate between the protonated forms of the two isomers, ions at m/z 273, generated by ESI-MS from the methanolic solution of naringenin or naringenin chalcone, were isolated in the mass spectrometer and interrogated by IRMPD spectroscopy in the fingerprint region (700–1850 cm–1). Comparison of the IRMPD spectra of [Nar + H]+ and [ChNar + H]+ shown in Figure 2 indicates the spectral range between 1400 and 1700 cm–1 to be specific and appropriate to distinguish between the two isomers. In this range, the IRMPD spectrum of protonated ChNar displays only one strong absorption at 1550 cm–1, while the experimental spectrum of [Nar + H]+ presents four major bands at 1460, 1513, 1550, and 1623 cm–1. In particular, this latter absorption, predominant in the IR spectrum of [Nar + H]+, together with the band at 1460 cm–1 can be chosen as vibrational signatures of naringenin in foodstuff analysis. Conversely, a predominant feature at 1550 cm–1 could be indicative of the presence of ChNar. In contrast, the spectral range between 700 and 1400 cm–1 is not suitable for differentiating between the two species. In this interval, both isomers present similar characteristics, namely [Nar + H]+ exhibits three pronounced absorptions at 1170, 1240, and 1280 cm–1, which are superimposable onto the IR bands observed at 1160, 1233, and 1293 cm–1 in the spectrum of [ChNar + H]+. Weaker features at ca. 840, 957, 1040, and 1326 cm–1 are also present in the IRMPD spectra of both isomers. The assignment of the experimental vibrational modes of [Nar + H]+ and [ChNar + H]+ will be discussed in the next two sections.
Figure 2.

Comparison between the IRMPD spectra of protonated naringenin (light blue trace) and naringenin chalcone (red trace, magnified by a factor of 2, as indicated, in the yellow trace).
Another distinctive aspect of the IRMPD investigations of Nar and ChNar lies in the different photofragmentation yields, R, of the two isomers. Upon IRMPD of both species, the fragment ions observed are identical to the ones seen in the CID experiments. Nonetheless, the R value of protonated chalcone is five times higher than that of naringenin, despite only slightly higher TE values of [ChNar + H]+. Of course, this finding is not a conclusively discriminating facet, but it could be taken into consideration in food analysis as a valuable parameter to distinguish between the two isomers.
Protonation Site and Conformational Analysis
Protonated Naringenin
Due to its structure, naringenin presents several possible protonation sites, namely the carbonyl oxygen in C4, the three hydroxyl oxygens in C5, C7, and C4′, and carbon atoms such as C6 and C8 of the A-ring. To elucidate the [Nar + H]+ structure, its experimental spectrum is compared with the calculated IR spectra of potential protonated naringenin structures. The structures, relative free energies, and IR calculated spectra of selected isomers of [Nar + H]+ are presented in Figure 3 together with the IRMPD spectrum of [Nar + H]+. A complete set of optimized geometries investigated in this study is reported in the Supporting Information Figures S5–S9. Focusing first on the isomers protonated on the carbonyl oxygen (4–O), three families of conformers are conceivable, differing by the orientation of the hydroxyl groups in C4 and C5. The Nar_1 family (Figures 3, S5, and S6) is characterized by an anticlockwise orientation of both hydroxyl groups (4 and 5). This orientation allows the formation of an intramolecular hydrogen bond between the hydrogen atom of the 4–OH and the oxygen in C5. Conversely, the clockwise alignment of both hydroxyl group in C4 and C5, typical of the Nar_2 conformers (Figures 3 and S7, upper panel) yields a hydrogen bond between the hydrogen atom in C5 and the oxygen in C4, as observed in neutral naringenin.20,21 All Nar_1 conformers are more stable than Nar_2 species (Nar_2a, the more stable conformer among the Nar_2 family, lies 25.0 kJ mol–1 higher in energy relative to the global minimum Nar_1). A possible explanation for this difference in stability is that the counterclockwise orientation allows for a stronger intramolecular hydrogen bond than the clockwise arrangement, as previously observed in the case of protonated o-hydroxybenzaldehyde, where the most stable geometry is stabilized by the intramolecular H-bond occurring between the proton on the formyl group and the phenolic oxygen atom.65 In fact, in the counterclockwise orientation, it is the best hydrogen-bonding acceptor atom, i.e., the phenolic oxygen sp3, that binds to the hydrogen of the best hydrogen-bonding donor atom, i.e., the carbonyl oxygen sp2.66,67 Moreover, the calculated intramolecular hydrogen bond lengths of the Nar_1 conformers are between 1.786 and 1.796 Å, which are significantly shorter than those for the Nar_2 species varying between 1.878 and 1.883 Å. Finally, the Nar_3 family, lying over 28.0 kJ mol–1 higher in energy relative to the ground-state minimum, presents the C5–OH and C4–OH group with anti-clockwise and clockwise orientation respectively (Figures 3 and S7, lower panel). This arrangement does not consent to any intramolecular H-bonding.
Figure 3.

IRMPD spectrum of [Nar + H]+ (bottom panel) compared with calculated IR spectra of Nar_1, Nar_C1a, Nar_C2, Nar_2a, and Nar_3, whose optimized structures are on the right. Relative free energies at 298 K are reported in kJ mol–1. Distances are given in Å. An open diamond indicates the calculated C4=O stretching mode at 1676 cm–1 for Nar_C1a and Nar_C2, and a black diamond indicates the C5–OH stretching mode expected at 1490 cm–1 for Nar_2a and Nar_3. The y-scale of calculated IR intensity ranges up to 1200 km mol–1.
Protonation on aromatic carbon C8 or C6 yields stable isomers, Nar_C1 and Nar_C2 (Figures 3 and S8), with comparable stabilities. They possess the shorter intramolecular hydrogen bond, (computed lengths 1.652–1.644 Å) and the most stable conformers of these types of isomers, namely Nar_C1_a and Nar_C2, are located at only 9.6 and 10.3 kJ mol–1, respectively, above the global minimum. As already reported for neutral naringenin,20 the C-ring of all considered conformers adopts a chair-type conformation where only the C2 atom is located out of the plane defined by the A and C rings. Bound to C2, the B-ring prefers the equatorial position with a dihedral angle τNar1, formed by C3–C2–C1′–C2′ atoms, varying between 50 and 60° in good agreement with the value of 60.18° measured by X-ray crystallography.20 The torsional angles τNar2, formed by C6–C7–O(7)–H(7) atoms, and τNar3 formed by C3′–C4′–O(4′)–H(4′), adopt values of either 0 or 180°, approximately. A complete scheme with all the species investigated (showing relative free energies, dihedral angles τNar1, τNar2, and τNar3, hydrogen bonding length and C–O lengths, total electronic energies, and thermal corrections) is provided in the Supporting Information (Tables S2–S4), together with the XYZ coordinates of the most stable conformer of each family of rotamers/isomers.
Looking at Figure 3, the IRMPD spectrum is well interpreted by the computed spectrum of Nar_1. Conversely, any contribution of ring-protonated species in the sampled population can be excluded. As shown in Figures 3 and S10, the computed IR spectra for the Nar_C1 and Nar_C2 optimized structures are very different from the experiment. Moreover, the typical feature at 1676 cm–1 due to the stretching mode of the carbonyl involved in H-bonding, is missing. We may discard protonation on 7-OH or 4′-OH since the calculated free energy of Nar_4 and Nar_5 (Figure S9), representative isomers protonated on 7–OH and 4′–OH, are higher in energy compared with the global minimum (over 169 kJ mol–1). Moreover, their computed spectra are not compatible with the experimental IRMPD spectrum. To complete the study, also the protonation at the O1 position was examined; however, all efforts to optimize this geometry yielded an open C-ring structure, that was not further investigated. Comparison of the calculated spectra with the experimental one of [Nar + H]+, reported in Figures 3 and S11, leads us to exclude also any contributions from Nar-2 and Nar-3 family conformers in the ion population of [Nar + H]+. The calculated spectra of all conformers of Nar_2 and Nar_3 types present an important feature at ca. 1490 cm–1, corresponding to the C5–OH stretching, which does not appear in the experimental spectrum and in the calculated spectra of the Nar_1 conformers. This vibrational mode occurs red-shifted at 1225 cm–1 for Nar_1 conformers, as a consequence of the H-bond between 5–O and 4–HO. The anticlockwise alignment of Nar-1 conformers elongates the C5–OH bond. The calculated bond lengths of C5–OH are 1.360, 1.332, and 1.337 Å for Nar_1, Nar_2, and Nar_3, respectively. On the other hand, to allow the H-bond between 4–O and 5–HO, the C4–O bond length of Nar_2 conformers is increased compared to the Nar_1 species. As a result, the C4–OH stretching mode for Nar_1, preserving its partial double bond character (1.305 Å), is expected at 1504 cm–1 and clearly visible in the experimental IRMPD spectrum of [Nar + H]+, while it is red-shifted at 1340 cm–1 in the calculated spectra of Nar_2 (1.335 Å). Furthermore, the distinct experimental absorption band at 1623 cm–1 is well interpreted by the CC stretching mode (A ring) of Nar_1 predicted at 1636 cm–1, while it appears blue-shifted at 1653 cm–1 in the calculated spectra of Nar_2 species.
Having therefore confirmed that protonated naringenin adopts the anticlockwise alignment typical of Nar_1 conformers, it is possible to estimate a conformational analysis on these geometries. Each family of structures includes multiple conformers differing in the arrangement of the hydroxyl group bound in C7–O or C4′–O in which the dihedral angles τNar2 and τNar3 can assume a value of 0 and 180°. Throughout this paper, the notation na indicates that the value of dihedral angle τNar2 is about 0° while the notation n′ means that the dihedral τNar3 has a the value of 0° (Scheme 1).
Scheme 1. Exemplary Notation of Conformers.

In general, for all conformers considered, the 180° rotation of C4′–OH has little influence on relative energies, yielding species with similar free energy as observed for neutral naringenin.20 Furthermore, in agreement with similar findings in an IRMPD study on deprotonated genistein68and with an IR study on neutral naringenin,69 the calculated IR spectra for each pair of such conformers are completely superimposable as can be noted in the calculated spectra IR collected in the Supporting Information (Figures S10–S12).
Focusing on the Nar_1 family, the global minimum Nar_1 shows an anticlockwise orientation of the three hydroxyl substituents of the A and C rings. The 180° rotation of the dihedral angle τNar3 leads to conformer Nar_1′ lying only 1.1 kJ mol–1 higher in energy. The calculated energy barrier for the interconversion from Nar_1 to Nar_1′ is 16.5 kJ mol–1 as shown in Figure S13, where the potential energy surface (PES) of the isomerization paths between selected geometries of Nar_1 is reported. This low rotational barrier allows isomerization between these minima in protonated naringenin as in the case of the neutral molecule. The single bond character of C4′–OH (calculated length of 1.355 Å), associated with the CO stretching mode computed and found in the IRMPD spectrum of [Nar + H]+ at 1275 and 1280 cm–1 respectively, further supports these findings. Nar_1a and Nar_1a′ display a clockwise orientation of the C7 hydroxyl group and are at 5.3 and 6.3 kJ mol–1 in relative energy. The relatively high energy (30.1 kJ mol–1) of the transition state for Nar_1/Nar_1a interconversion, via rotation of the C7–OH bond (Figure S13), reflects the presence of an extensive π conjugation through the A ring between the protonated carbonyl and the para hydroxyl substituent, as recently observed for protonated p-hydroxybenzaldehyde.65 It follows that, compared to the C4′–OH, the C7–OH bond features an increased double-bond character with a calculated length of 1.332 Å, leading the CO stretching mode to be blue-shifted at 1459 cm–1, in good agreement with the experimental feature at 1460 cm–1 in the IRMPD spectrum of [Nar + H]+. Nevertheless, a comparison between the calculated spectra of Nar_1 and Nar_1a with the IRMPD spectrum of protonated naringenin, presented in Figure 4, confirms the presence of both conformers in the ion population. In general, both calculated spectra of Nar_1 and Nar_1a are in good agreement with the experimental IRMPD spectrum of [Nar + H]+. The experimental features at 1240 cm–1 (highlighted in gray in Figure 4), well predicted in Nar_1 and Nar_1′ computed spectra and not present in the a counterpart, and at 1356 cm–1 (highlighted in pink), matching only with the calculated spectra of Nar_1a and Nar_1a′ rotamers, can be taken as vibrational signatures of both pairs of conformers. The vibrational modes of [Nar + H]+ can be assigned as described in Table S5, which lists the experimental IRMPD features together with the calculated IR bands of the global minimum Nar_1 with the exception of the absorption at 1356 cm–1 explained with the calculated mode of Nar_1a. To complete the conformational analysis of the Nar_1 family, the conformers bearing the B-ring in the axial position were also considered. With respect to their equatorial counterparts, they differ only in the position of the C2 atom relative to the plane defined by the C and A ring and in the value of the dihedral angle τNar1 that falls between −172.02 and −176.30°. As in the case of neutral naringenin, the axial arrangement is less stable (>8.8 kJ mol–1) as compared to the Nar_1 equatorial conformers. The optimized geometries are reported in Figure S6. The IR computed spectra of Nar_1_Ax (Figures 4 and S12) are quite similar to those of the Nar_1 equatorial conformer with the exception of a small band calculated at 1415 cm–1 (highlighted in light violet in Figure 4) corresponding to the CH2 scissoring vibrational mode. This feature is not discernible in the experimental spectrum. The axial configuration does not appear to be present in the ion population, in agreement with a predicted Boltzmann distribution which indicate the equatorial conformers to be the main contributor (over 98%) to the ground-state population of neutral S-Naringenin.20
Figure 4.

IRMPD spectrum of [Nar + H]+ (light blue profile) compared with calculated IR spectra of Nar_1, Nar_1a, and Nar_1_Ax (gray, red, and violet profiles, respectively), whose optimized structures are reported on the right. Distances are given in Å. Relative free energies at 298 K are reported in kJ mol–1. The y-scale of calculated IR intensity ranges up to 1200 km mol–1.
Protonated Naringenin Chalcone
Compared with naringenin, the presence of the α,β unsaturated bond and of an additional hydroxyl group, increases the possible sites of protonation of naringenin chalcone, whose structure is depicted in Figure 1. In addition to the carbonyl oxygen in Cγ, the three hydroxyl groups in C2′, C4′ (of the A-ring), and C4 (of the B-ring), and the aromatic 3′ and 5′ carbon atoms of the A ring, already considered in the case of naringenin, also the hydroxyl in C6′ and the double bond Cα=Cβ can be envisioned as protonation sites. The complete survey of all geometries considered is reported in the Supporting Information (Figures S14–18). Figure 5 shows the optimized geometries and their IR calculated spectra of selected isomers of [ChNar + H]+ protonated on the carbonyl oxygen (Cγ) plotted together with the IRMPD spectrum of [ChNar + H]+. Similarly to Naringenin, protonation on the carbonyl oxygen (Cγ) yields three different families of conformers distinguished by the motif of the hydroxyl groups at Cγ and C2′. The ChNar_1 family (Figures 5 and S14 and S15) displays the anticlockwise arrangement of both hydroxyl groups, forming an intramolecular hydrogen bond between the hydrogen atom of the Cγ–OH and the O atom in C2′. On the other hand, hydrogen bonding occurs between the C2′OH group and the Cγ–O atom in the ChNar_2 species characterized by the clockwise alignment of these two hydroxyl groups (Figures 5 and S16). In the ChNar_3 conformers, the orientation of these neighboring hydroxyl groups does not allow any hydrogen bonding (Figures 5 and S16). As observed for naringenin, each family of conformers comprises multiple rotamers with comparable energies, depending on the disposition of the hydroxyl group in C4′ or C4 around the C–O bond. To be consistent with naringenin, the notation na indicates that the value of dihedral angle τCh4, formed by C3′–C4′–O(4′)–H(4′) atoms of the A ring, is about 0° while the notation n′ means that the dihedral τCh5, formed by C3–C4–O(4)–H(4) atoms of the B-ring, has a value of 0°.
Figure 5.

IRMPD spectrum of [ChNar + H]+ (bottom panel, in red) compared with calculated IR spectra of ChNar_1, ChNar_1a_sT, ChNar_2a, and ChNar_3a, whose optimized structures are reported on the right. Distances are given in Å. Relative free energies at 298 K are reported in kJ mol–1. The y-scale of calculated IR intensity ranges up to 3500 km mol–1.
Focusing on the s-cis configuration, again the anticlockwise pattern of hydroxyl groups in Cγ and C2′ increases the stability of the ion. The energy difference is more than 20 kJ mol–1 in favor of ChNar_1 conformers versus the ChNar_2 family. The enhanced intramolecular hydrogen bond, favored also by the short distance between H(γ) and O(2′) of about 1.665 Å, and the substantial planarity of the entire geometry with dihedral angles of about −10° for τCh1 (formed by C2′–C1′–Cγ–O(γ), −1,7° for τCh2 (O(γ)–Cγ–Cα–Cβ) and 0.3° for τCh3 (Cα–Cβ–C1–C2), play an important role in the global stabilization of the ChNar_1 species. The complete survey with all species investigated (listing relative free energies, dihedral angles τCh1, τCh2, τCh3, τCh4, and τCh5 values and hydrogen bonding length, total electronic energies and thermal corrections, together with the cartesian coordinates of the most stable conformer of each family of rotamers/isomers) is provided in the Supporting Information (Tables S6 and S7). The clockwise motif increases the intramolecular hydrogen bond length, with the calculated distance between H(2′) and O(γ) for the ChNar_2 conformers being ca. 1.760 Å. Furthermore, the co-planarity between the A- and B-ring is lost in both protonated ChNar_2 and ChNar_3 geometries characterized by the OH (γ) in clockwise orientation, due to steric hindrance between the two hydrogen atoms in γ and β position, as shown by τCh1, τCh2, τCh3 values reported in Table S6. Again, the lack of an intramolecular hydrogen bond in ChNar_3 makes decreases their stability further, the whole conformers lying ca. 34 kJ mol–1 higher in energy. ChNar_1_sT conformers are the s-trans counterpart of ChNar_1. The nonplanarity of the A-ring relative to the rest of the molecule in the s-trans conformation is essentially due to steric repulsion between the O and H atoms in 6′ and β position and illustrated by a τCh1 and τCh2 values of about −33 and 160°, respectively. The elongation of the intramolecular H-bond (1.900 Å) in ChNar_1_sT is a consequence of the A-ring nonplanarity, leading to a loss in stability of ca. 24 kJ mol–1. As in the case of naringenin, the combination of high free energy values (lying over 200 kJ mol–1 over the global minimum) and the noncorrespondence between computed and experimental spectra of ChNar_4 and ChNar_5 (Figure S17) allow us to exclude protonation on the hydroxyl oxygen atom in 4′- or 4- position. All attempts to optimize 6′–O protonated geometries resulted in a proton shift to the double bond. The so-formed isomers, which lie over 59 kJ mol–1 in energy with respect to the global minimum, are protonated at Cα as can be seen in ChNar_C3 (Figure S18). Protonation on the aromatic 3′ or 5′ carbon atoms generates isomers with different stability depending on the orientation of the hydroxyl group ortho-positioned relatively to the protonated carbon (Figure S18). Among them, the most stable ChNar_C1 and ChNar_C2a lie at 16.3 and 27.0 kJ mol–1 above the global minimum, ChNar_1. However, the poor agreement between the experimental and computed spectrum of the carbon-protonated isomers (Figure S19) allows us to exclude their contribution to the ion population.
Looking at Figure 5, ChNar_1 displays a theoretical IR spectrum in very good agreement with the experimental features of [ChNar + H]+. Additionally, the computed spectrum of ChNar_1a_sT shows similar features to the experimental one. The two distinct absorptions, expected at 1510 and 1576 cm–1 for ChNar_1a_sT, can be accounted for in the intense experimental feature at 1550 cm–1 typical of [ChNar + H]+. Consequently, a contribution of s-trans conformers with anti-clockwise alignment cannot be excluded in the sampled [ChNar + H]+ population. Rotations around the C4–OH and C4′–OH bonds do not change the IR spectra of individual rotamers of a given family, as can be seen in the spectra depicted in Figures S20 and S21 in the Supporting Information. The calculated energy barriers for the interconversion from ChNar_1 to either ChNar_1′ or ChNar_1a are shown in Figure S22, where the potential energy surface (PES) of the isomerization paths between selected geometries of ChNar_1 is reported.
In the calculated spectra of the geometries with OH(γ) clockwise oriented, namely ChNar_2 and _3, a feature is predicted at 1485 cm–1, which does not find a counterpart in the experimental spectrum. This band corresponds to the bending mode of the OH group in the γ position coupled with CC stretches. The same vibrational mode is expected at 1434 and 1439 cm–1 in the calculated spectra of ChNar_1 and ChNar_1-sT and appears in the experimental spectra of [ChNar + H]+. Moreover, the vibrational Cα=Cβ stretching mode coupled to the bending of the OH group in γ position is predicted at 1283 cm–1 for all the ChNar optimized geometries protonated on the carbonyl oxygen (Cγ) and is particularly evident in the computed spectra of ChNar_2 and _3. The calculated intensity of this vibrational mode, however, seems better interpreted in the ChNar_1 spectrum. These considerations lead us to exclude a significant contribution of ChNar_2 and _3 in the analyzed population. Therefore, from the analysis of the IRMPD spectrum, similar to our findings for naringenin, one may conclude that the two adjacent hydroxyl groups in the 2′ and γ positions adopt an anticlockwise arrangement in protonated naringenin chalcone. The vibrational modes of [ChNar + H]+ can be assigned as described in Table S8, which lists the experimental IRMPD features together with the calculated IR bands of the global minimum ChNar_1. With this OH arrangement, while [Nar + H]+ adopts the equatorial conformation, [ChNar + H]+ may assume both the s-cis and the s-trans conformations. To confirm these conclusions, an ion mobility analysis was performed on [ChNar + H]+ and [Nar + H]+.
Ion Mobility Experiments
Considering the possible different conformations of Nar_1, Nar_1′Ax, ChNar_1, and ChNar_1a_sT, ion mobility-mass spectrometry (IMS) could be useful to further characterize our samples. However, the two investigated compounds could not be discriminated via our IMS experiments. ESI (+)-IMS performed on an equimolar mixture of [Nar + H]+ and [ChNar + H]+ shows a unique drift time value, DT (mixture) = 2.63 ms (Figure S20 upper panel). In addition, both species when analyzed separately revealed identical DT values in the mobility diagram, with an arrival time of 2.63 ms for both [Nar + H]+ and [ChNar + H]+. However, the peak width observed in the mobilogram appears slightly different for the two isomers, analyzed in the same μ-molar concentration. While the mobility diagram of [Nar + H]+ shows a narrow peak, the [ChNar + H]+ one appears slightly broader. A similar broadening is observed for the [Nar + H]+/[ChNar + H]+mixture. This observation suggests that two distinct conformations might be present for [ChNar + H]+.
To support this hypothesis, the drift times were correlated to the collisional cross-section (CCS) of both isomers, using polyalanine oligomers to calibrate the mobility data,70 and compared with the calculated CCS for Nar_1, Nar_1′Ax, ChNar_1, and ChNar_1a_sT. The results are reported in Table S9. The CCS values of 160.8 and 160.1 Å2 calculated for Nar_1 and ChNar_1, respectively, are in good agreement with the identical values of 160.9 Å2 obtained for [Nar + H]+ and [ChNar + H]+, supporting that IMS analysis does not allow for differentiation between the two isomers. The calculated values for the axial conformer of Nar_1 (162.4 Å2 for Nar_1Ax) and s-trans conformer of ChNar_1 (158.3 Å2 for ChNar_1_sT) appear slightly different. There is a difference of ±1.8 Å2 from their more stable conformer. Clearly, this small difference is not sufficient to separate the two conformers in arrival time distribution data but may cause the broadening of the peak observed for [ChNar + H]+ but not for [Nar + H]+.
Analysis of Metabolites in Complex Matrix
Considering the spectroscopic discrimination obtained for the standard solutions, we decided to assay by IRMPD spectroscopy the methanolic extracts of tomato peel (TP) and grapefruit albedo (GA), chosen for their high flavonoid content. Among the Citrus species, grapefruit is recognized as one of the fruits with the highest concentration of naringenin.16,71 In particular, its albedo is rich in naringin (naringenin-7-neohesperoside), one of the glycoside forms of naringenin. In contrast, fresh tomatoes are particularly interesting for their high content of naringenin chalcone, especially concentrated especially in the peel.45 However, also naringenin was found in tomato skin.72,73 For both phytochemical extracts, IRMPD spectroscopy was carried out on the ion at m/z 273 mass-selected in the ion trap from the mass spectrum of the complex matrix solutions directly electrosprayed into the mass spectrometer. Unfortunately, the full scan spectrum of both natural samples exhibited a low abundance of the compound at m/z 273. Despite the low signal-to-noise ratio of the mass-selected ion in the natural samples, we were able to record both IRMPD spectra, using higher pulse energies to increase the photofragmentation yield. For comparative purpose, knowing that IRMPD spectra may look different depending on the power level due to saturation effects, we also recorded the spectra of the reference compounds again at increased laser pulse energy as shown in Figure S24 where the high-power IRMPD spectra of the standard are plotted together with their IRMPD spectra recorded at lower laser energy. The resulting high-power IRMPD spectra of [Nar + H]+ and [ChNar + H]+ are shown in Figure 6 (in light blue and red, respectively), and compared with the IRMPD spectra of the ion at m/z 273 isolated from the TP extract (upper panel, black trace) and the methanolic solution of GA (lower panel, green trace). Focusing first on the IRMPD spectra of the two standards performed at low and high laser pulse energy, for [Nar + H]+, the improvements led to an overall increase in photofragmentation yield, R (by a factor of 5) without changing the spectrum profile (light blue trace versus blue trace, lower panel Figure S24). For [ChNar + H]+, on the other hand, due to the very high IRMPD yield (R) observed in the low-laser pulse energy spectrum, saturation is observed on the strong absorption at 1550 cm–1 in the high laser power spectrum. Consequently, all other bands appear to relatively increase as shown in the upper panel of Figure S24. Switching to the phytochemical extracts, the species at m/z 273 from TP extract yields the same photofragments at m/z 153, 147, 119, and 91, as observed for both [Nar + H]+ and [ChNar + H]+. To determine the presence of one or both isomers in the TP extract, the three IRMPD spectra can be compared, focusing mainly on the absorptions in the 1400–1800 cm–1 spectral range (Figure 6, upper panel). At first glance, the entire spectrum of the m/z 273 species from TP is very similar to the high laser energy spectrum of the ChNar standard (Figure 6, panel a). In particular, the pronounced absorption at 1550 cm–1 confirms the presence of ChNar in the sample of TP. However, the two absorptions typical of naringenin at 1460 and 1623 cm–1 emerge a little more in the spectrum of TP when compare to the ChNar standard spectrum. Therefore, in this particular sample of tomato skin, both isomers appear to contribute to the ion at m/z 273, with a higher prevalence of ChNar. Looking the IRMPD spectrum of the mass-selected ion at m/z 273 from GA extract, it appears similar to that of the [Nar + H]+ standard and shows clearly the two vibrational features at 1463 and 1617 cm–1, suggesting a predominant contribution of naringenin in GA (Figure 6, panel d). The unexpectedly intense feature at 1520 cm–1 found in the GA IRMPD spectrum can be attributed to some contribution of ChNar in the mass-selected ion population at m/z 273 (Figure 6, panel c). In conclusion, in the natural extracts analyzed by IRMPD, the presence of Nar and ChNar is likely. However, our experiments also suggest a clear prevalence of naringenin chalcone in the TP, while the GA contains mainly naringenin.
Figure 6.

Comparison between the IRMPD spectra of mass-selected ions at m/z 273 from (1) the methanolic extract of tomato peel (upper panel (a and b), black trace) and (2) the methanolic extract of grapefruit albedo (lower panel (c and d), green trace) with standard solution of naringenin chalcone (on the left (a and c), red trace) and of naringenin (on the right (b and d) light blue trace) recorded at higher energy.
In summary, while IMS and CID experiments hardly differentiate the two isomers, IRMPD spectroscopy turned out as an efficient method for distinguishing naringenin from its chalcone. The present results confirm IRMPD spectroscopy in combination with DFT calculations to be a powerful tool not only to determine the structure and conformational space of food metabolites but also to design new approaches for targeted metabolomics.
Acknowledgments
The research leading to these results has received funding from the Università degli Studi di Roma “La Sapienza”—Progetti medi Ateneo 2021, grant number RM12117A8514BC20, from LASERLAB-EUROPE (grant agreement no. 871124, European Union’s Horizon 2020 Research and Innovation Programme) and the EU Horizon 2020 Programme (EU_FT-ICR_MS, under grant number 731077, respectively). We gratefully acknowledge the Nederlandse Organisatie voor Wetenschappelijk Onderzoek (NWO) for the support of the FELIX Laboratory.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jafc.2c07453.
Breakdown curves, optimized geometries, comparison between experimental and calculated IR spectra, potential energy surface (PES), operating details, complete survey of all geometries and Table of IRMPD absorptions and calculated vibrational frequencies (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Harvey A. L.; Edrada-Ebel R.; Quinn R. J. The re-emergence of natural products for drug discovery in the genomics era. Nat. Rev. Drug Discov. 2015, 14, 111–129. 10.1038/nrd4510. [DOI] [PubMed] [Google Scholar]
- Dunn W. B.; Bailey N. J. C.; Johnson H. E. Measuring the metabolome: current analytical technologies. Analyst 2005, 130, 606–625. 10.1039/b418288j. [DOI] [PubMed] [Google Scholar]
- Lisec J.; Schauer N.; Kopka J.; Willmitzer L.; Fernie A. R. Gas chromatography mass spectrometry–based metabolite profiling in plants. Nat. Protoc. 2006, 1, 387–396. 10.1038/nprot.2006.59. [DOI] [PubMed] [Google Scholar]
- Dettmer K.; Aronov P. A.; Hammock B. D. Mass spectrometry-based metabolomics. Mass Spectrom. Rev. 2007, 26, 51–78. 10.1002/mas.20108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsao R. Chemistry and biochemistry of dietary polyphenols. Nutrients 2010, 2, 1231–1246. 10.3390/nu2121231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panche A. N.; Diwan A. D.; Chandra S. R. Flavonoids: an overview. J. Nutr. Sci. 2016, 5, e47 10.1017/jns.2016.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W.-Z.; Ye M.; Qiao X.; Wang Q.; Bo T.; Guo D.-A. Collision-induced dissociation of 40 flavonoid aglycones and differentiation of the common flavonoid subtypes using electrospray ionization ion-trap tandem mass spectrometry and quadrupole time-of-flight mass spectrometry. Eur. J. Mass Spectrom. 2012, 18, 493–503. 10.1255/ejms.1206. [DOI] [PubMed] [Google Scholar]
- Zhang J. M.; Brodbelt J. S. Structural characterization and isomer differentiation of chalcones by electrospray ionization tandem mass spectrometry. J. Mass Spectrom. 2003, 38, 555–572. 10.1002/jms.472. [DOI] [PubMed] [Google Scholar]
- Ma Y. L.; Li Q. M.; Van den Heuvel H.; Claeys M. Characterization of flavone and flavonol aglycones by collision-induced dissociation tandem mass spectrometry. Rapid Commun. Mass Spectrom. 1997, 11, 1357–1364. 10.1002/(SICI)1097-0231(199708)11:12<1357::AID-RCM983>3.0.CO;2-9. [DOI] [Google Scholar]
- Kuhn F.; Oehme M.; Romero F.; Abou-Mansour E.; Tabacchi R. Differentiation of isomeric flavone/isoflavone aglycones by MS2 ion trap mass spectrometry and a double neutral loss of CO. Rapid Commun. Mass Spectrom. 2003, 17, 1941–1949. 10.1002/rcm.1138. [DOI] [PubMed] [Google Scholar]
- Pinheiro P. F.; Justino G. C.. Structural analysis of flavonoids and related compounds-a review of spectroscopic applications; IntechOpen; 2012, 33 −56. [Google Scholar]
- Zhang Y.; Zhang P.; Cheng Y. Structural characterization of isoprenylated flavonoids from Kushen by electrospray ionization multistage tandem mass spectrometry. J. Mass Spectrom. 2008, 43, 1421–1431. 10.1002/jms.1423. [DOI] [PubMed] [Google Scholar]
- Wani T. A.; Pandith S. A.; Gupta A. P.; Chandra S.; Sharma N.; Lattoo S. K. Molecular and functional characterization of two isoforms of chalcone synthase and their expression analysis in relation to flavonoid constituents in Grewia asiatica L. PLoS One 2017, 12, e0179155 10.1371/journal.pone.0179155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerlich M.; Neumann S. MetFusion: integration of compound identification strategies. J. Mass Spectrom. 2013, 48, 291–298. 10.1002/jms.3123. [DOI] [PubMed] [Google Scholar]
- Venkateswara Rao P.; Kiran S. D. V. S.; Rohini P.; Bhagyasree P. Flavonoid: a review on naringenin. J. Pharmacogn. Phytochem. 2017, 6, 2778–2783. [Google Scholar]
- Khan M. K.; Zill-E-Huma; Dangles O. A comprehensive review on flavanones, the major citrus polyphenols. J. Food Compos. Anal. 2014, 33, 85–104. 10.1016/j.jfca.2013.11.004. [DOI] [Google Scholar]
- Halbwirth H. The creation and physiological relevance of divergent hydroxylation patterns in the flavonoid pathway. Int. J. Mol. Sci. 2010, 11, 595–621. 10.3390/ijms11020595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waki T.; Mameda R.; Nakano T.; Yamada S.; Terashita M.; Ito K.; Tenma N.; Li Y.; Fujino N.; Uno K.; Yamashita S.; Aoki Y.; Denessiouk K.; Kawai Y.; Sugawara S.; Saito K.; Yonekura-Sakakibara K.; Morita Y.; Hoshino A.; Takahashi S.; Nakayama T. A conserved strategy of chalcone isomerase-like protein to rectify promiscuous chalcone synthase specificity. Nat. Commun. 2020, 11, 870. 10.1038/s41467-020-14558-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin W.; Lah M. Structure of (R,S)-naringenin. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 1986, 42, 626–628. 10.1107/S010827018609515X. [DOI] [Google Scholar]
- Nesterov V. V.; Zakharov L. N.; Nesterov V. N.; Calderon J. G.; Longo A.; Zaman K.; Kaneez Choudhury F.; Farrell W.; Shulaev V.; Richmond M. G. 5,7-Dihydroxy-2-(4-hydroxyphenyl)chroman-4-one (naringenin): X-ray diffraction structures of the naringenin enantiomers and DFT evaluation of the preferred ground-state structures and thermodynamics for racemization. J. Mol. Struct. 2017, 1130, 994–1000. 10.1016/j.molstruc.2016.10.068. [DOI] [Google Scholar]
- Ávila E. P.; Mendes L. A. O.; De Almeida W. B.; Dos Santos H. F.; De Almeida M. V. Conformational analysis and reactivity of naringenin. J. Mol. Struct. 2021, 1245, 131027 10.1016/j.molstruc.2021.131027. [DOI] [Google Scholar]
- Kozlowski D.; Trouillas P.; Calliste C.; Marsal P.; Lazzaroni R.; Duroux J.-L. Density functional theory study of the conformational, electronic, and antioxidant properties of natural chalcones. J. Phys. Chem. A. 2007, 111, 1138–1145. 10.1021/jp066496+. [DOI] [PubMed] [Google Scholar]
- Bakarić D.; Baranović G. The conformational equilibrium and vibrational properties of chalcone. J. Mol. Struct. 2019, 1196, 429–438. 10.1016/j.molstruc.2019.06.098. [DOI] [Google Scholar]
- González Moreno A.; Prieto P.; Ruiz Delgado M. C.; Domínguez E.; Heredia A.; de Cózar A. Structure, isomerization and dimerization processes of naringenin flavonoids. Phys. Chem. Chem. Phys. 2021, 23, 18068–18077. 10.1039/D1CP01161H. [DOI] [PubMed] [Google Scholar]
- Rijs A.M.; Oomens J.. IR spectroscopic techniques to study isolated biomolecules. In: Rijs A.; Oomens J. (eds) Gas-Phase IR Spectroscopy and Structure of Biological Molecules. Topics in Current Chemistry; Springer: Cham, 2014, 364. [DOI] [PubMed] [Google Scholar]
- Roithová J. Characterization of reaction intermediates by ion spectroscopy. J. Chem. Soc. Rev. 2012, 41, 547–559. 10.1039/C1CS15133A. [DOI] [PubMed] [Google Scholar]
- Carlo M. J.; Patrick A. L. Infrared multiple photon dissociation (IRMPD) spectroscopy and its potential for the clinical laboratory. J. Mass Spectrom. Adv. Clin. Lab. 2022, 23, 14–25. 10.1016/j.jmsacl.2021.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fridgen T. D. Infrared consequence spectroscopy of gaseous protonated and metal ion cationized complexes. Mass Spectrom. Rev. 2009, 28, 586–607. 10.1002/mas.20224. [DOI] [PubMed] [Google Scholar]
- Corinti D.; Crestoni M. E.; Chiavarino B.; Fornarini S.; Scuderi D.; Salpin J.-Y. Insights into cisplatin binding to uracil and thiouracils from IRMPD spectroscopy and tandem mass spectrometry. J. Am. Soc. Mass Spectrom. 2020, 31, 946–960. 10.1021/jasms.0c00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu R. R.; Yang B.; Frieler C. E.; Berden G.; Oomens J.; Rodgers M. T. Diverse mixtures of 2,4-dihydroxy tautomers and O4 protonated conformers of uridine and 2′-deoxyuridine coexist in the gas phase. Phys. Chem. Chem. Phys. 2015, 17, 25978–25988. 10.1039/C5CP02227D. [DOI] [PubMed] [Google Scholar]
- Chiavarino B.; Crestoni M. E.; Fornarini S.; Scuderi D.; Salpin J.-Y. Undervalued N3 coordination revealed in the cisplatin complex with 2′-deoxyadenosine-5′-monophosphate by a combined IRMPD and theoretical study. Inorg. Chem. 2017, 56, 8793–8801. 10.1021/acs.inorgchem.7b00570. [DOI] [PubMed] [Google Scholar]
- Maitre P.; Scuderi D.; Corinti D.; Chiavarino B.; Crestoni M. E.; Fornarini S. Applications of infrared multiple photon dissociation (IRMPD) to the detection of posttranslational modifications. Chem. Rev. 2020, 120, 3261–3295. 10.1021/acs.chemrev.9b00395. [DOI] [PubMed] [Google Scholar]
- Corinti D.; De Petris A.; Coletti C.; Re N.; Chiavarino B.; Crestoni M. E.; Fornarini S. Cisplatin primary complex with l-histidine target revealed by IR multiple photon dissociation (IRMPD) spectroscopy. ChemPhysChem 2017, 18, 318–325. 10.1002/cphc.201601172. [DOI] [PubMed] [Google Scholar]
- Paciotti R.; Corinti D.; De Petris A.; Ciavardini A.; Piccirillo S.; Coletti C.; Re N.; Bellina B.; Barran P.; Chiavarino B.; Crestoni M. E.; Fornarini S. Cisplatin and transplatin interaction with methionine: bonding motifs assayed by vibrational spectroscopy in the isolated ionic complexes. Phys. Chem. Chem. Phys. 2017, 19, 26697–26707. 10.1039/C7CP05203K. [DOI] [PubMed] [Google Scholar]
- Martens J.; Koppen V.; Berden G.; Cuyckens F.; Oomens J. Combined liquid chromatography-infrared ion spectroscopy for identification of regioisomeric drug metabolites. Anal. Chem. 2017, 89, 4359–4362. 10.1021/acs.analchem.7b00577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens J.; Berden G.; Bentlage H.; Coene K. L. M.; Engelke U. F.; Wishart D.; van Scherpenzeel M.; Kluijtmans L. A. J.; Wevers R. A.; Oomens J. Unraveling the unknown areas of the human metabolome: the role of infrared ion spectroscopy. J. Inherit. Metab. Dis. 2018, 41, 367–377. 10.1007/s10545-018-0161-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cismesia A. P.; Bell M. R.; Tesler L. F.; Alves M.; Polfer N. C. Infrared ion spectroscopy: an analytical tool for the study of metabolites. Analyst 2018, 143, 1615–1623. 10.1039/C8AN00087E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lincke K.; Langeland J.; Madsen A. Ø.; Kiefer H. V.; Skov L.; Gruber E.; Mikkelsen K. V.; Andersen L. H.; Nielsen M. B. Elucidation of the intrinsic optical properties of hydrogen-bonded and protonated flavin chromophores by photodissociation action spectroscopy. Phys. Chem. Chem. Phys. 2018, 20, 28678–28684. 10.1039/C8CP05368E. [DOI] [PubMed] [Google Scholar]
- Corinti D.; Maccelli A.; Crestoni M. E.; Cesa S.; Quaglio D.; Botta B.; Ingallina C.; Mannina L.; Tintaru A.; Chiavarino B.; Fornarini S. IR ion spectroscopy in a combined approach with MS/MS and IM-MS to discriminate epimeric anthocyanin glycosides (cyanidin 3-O-glucoside and -galactoside). Int. J. Mass Spectrom. 2019, 444, 116179 10.1016/j.ijms.2019.116179. [DOI] [Google Scholar]
- Corinti D.; Crestoni M. E.; Fornarini S.; Dabbish E.; Sicilia E.; Gabano E.; Perin E.; Osella D. A multi-methodological inquiry of the behavior of cisplatin-based Pt(IV) derivatives in the presence of bioreductants with a focus on the isolated encounter complexes. J. Biol. Inorg. Chem. 2020, 25, 655–670. 10.1007/s00775-020-01789-w. [DOI] [PubMed] [Google Scholar]
- Kurka O.; Roithová J.; Bednář P. Examination of small molecule losses in 5-methylpyranopelargonidin MS/MS CID spectra by DFT calculations. J. Mass Spectrom. 2014, 49, 1314–1321. 10.1002/jms.3466. [DOI] [PubMed] [Google Scholar]
- Corinti D.; Maccelli A.; Chiavarino B.; Maitre P.; Scuderi D.; Bodo E.; Fornarini S.; Crestoni M. E. Vibrational signatures of curcumin’s chelation in copper(II) complexes: an appraisal by IRMPD spectroscopy. J. Chem. Phys. 2019, 150, 165101 10.1063/1.5086666. [DOI] [PubMed] [Google Scholar]
- Chiavarino B.; Sinha R. K.; Crestoni M. E.; Corinti D.; Filippi A.; Fraschetti C.; Scuderi D.; Maitre P.; Fornarini S. Binding motifs in the naked complexes of target amino acids with an excerpt of antitumor active biomolecule: an ion vibrational spectroscopy assay. Chem. – Eur. J. 2021, 27, 2348. 10.1002/chem.202003555. [DOI] [PubMed] [Google Scholar]
- Shimokoriyama M. Interconversion of chalcones and flavanones of a phloroglucinol-type structure. J. Am. Chem. Soc. 1957, 79, 4199–4202. 10.1021/ja01572a057. [DOI] [Google Scholar]
- Slimestad R.; Verheul M. Properties of chalconaringenin and rutin isolated from cherry tomatoes. J. Agric. Food Chem. 2011, 59, 3180–3185. 10.1021/jf104045k. [DOI] [PubMed] [Google Scholar]
- Victor M. M.; David J. M.; Sakukuma M. C. K.; França E. L.; Nunes A. V. J. A simple and efficient process for the extraction of naringin from grapefruit peel waste. Green Process Synth. 2018, 7, 524–529. 10.1515/gps-2017-0112. [DOI] [Google Scholar]
- Sudto K.; Pornpakakul S.; Wanichwecharungruang S. An efficient method for the large scale isolation of naringin from pomelo (Citrus grandis) peel. Int. J. Food Sci. Technol. 2009, 44, 1737–1742. 10.1111/j.1365-2621.2009.01989.x. [DOI] [Google Scholar]
- Corinti D.; Crestoni M. E.; Fornarini S.; Pieper M.; Niehaus K.; Giampà M. An integrated approach to study novel properties of a MALDI matrix (4-maleicanhydridoproton sponge) for MS imaging analyses. Anal. Bioanal. Chem. 2019, 411, 953–964. 10.1007/s00216-018-1531-7. [DOI] [PubMed] [Google Scholar]
- Schröder D.; Schwarz H.; Aliaga-Alcalde N.; Neese F. Fragmentation of the (cyclam-acetato)iron azide cation in the gas phase. Eur. J. Inorg. Chem. 2007, 6, 816–821. 10.1002/ejic.200600805. [DOI] [Google Scholar]
- Milko P.; Roithova J.; Schroder D.; Lemaire J.; Schwarz H.; Holthausen M. C. The phenoxy/phenol/copper cation: a minimalistic model of bonding relations in active centers of mononuclear copper enzymes. Chem. – Eur. J. 2008, 14, 4318–4327. 10.1002/chem.200800052. [DOI] [PubMed] [Google Scholar]
- Bouchoux G.; Salpin J.-Y.; Leblanc D. A relationship between the kinetics and thermochemistry of proton transfer reactions in the gas phase. Int. J. Mass Spectrom. Ion Process. 1996, 153, 37–48. 10.1016/0168-1176(95)04353-5. [DOI] [Google Scholar]
- Martens J.; Berden G.; Gebhardt C. R.; Oomens J. Infrared ion spectroscopy in a modified quadrupole ion trap mass spectrometer at the FELIX free electron laser laboratory. Rev. Sci. Instrum. 2016, 87, 103108 10.1063/1.4964703. [DOI] [PubMed] [Google Scholar]
- Berden G.; Derksen M.; Houthuijs K. J.; Martens J.; Oomens J. An automatic variable laser attenuator for IRMPD spectroscopy and analysis of power-dependence in fragmentation spectra. Int. J. Mass Spectrom. 2019, 443, 1–8. 10.1016/j.ijms.2019.05.013. [DOI] [Google Scholar]
- Lee C.; Yang W.; Parr R. G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. 10.1103/PhysRevB.37.785. [DOI] [PubMed] [Google Scholar]
- Becke A. D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. 10.1063/1.464913. [DOI] [Google Scholar]
- Campuzano I.; Bush M. F.; Robinson C. V.; Beaumont C.; Richardson K.; Kim H.; Kim H. I. Structural characterization of drug-like compounds by ion mobility mass spectrometry: comparison of theoretical and experimentally derived nitrogen collision cross sections. Anal. Chem. 2012, 84, 1026–1033. 10.1021/ac202625t. [DOI] [PubMed] [Google Scholar]
- Kim H.; Kim H. I.; Johnson P. V.; Beegle L. W.; Beauchamp J. L.; Goddard W. A.; Kanik I. Structural experimental and theoretical investigation into the correlation between mass and ion mobility for choline and other ammonium cations in N2. Anal. Chem. 2008, 80, 1928–1936. 10.1021/ac701888e. [DOI] [PubMed] [Google Scholar]
- Mintz-Oron S.; Mandel T.; Rogachev I.; Feldberg L.; Lotan O.; Yativ M.; Wang Z.; Jetter R.; Venger I.; Adato A.; Aharoni A. Gene expression and metabolism in tomato fruit surface tissues. Plant Physiol. 2008, 147, 823–851. 10.1104/pp.108.116004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribas-Agustí A.; Cáceres R.; Gratacós-Cubarsí M.; Sárraga C.; Castellari M. A. A validated HPLC-DAD method for routine determination of ten phenolic compounds in tomato fruits. Food Anal. Methods 2012, 5, 1137–1144. 10.1007/s12161-011-9355-5. [DOI] [Google Scholar]
- Krause M.; Galensa R. Determination of naringenin and naringenin-chalcone in tomato skins by reversed phase HPLC after solid phase extraction. Z. Lebensm. Unters. Forsch. 1992, 194, 29–32. 10.1007/BF01191036. [DOI] [Google Scholar]
- Wolfender J.-L.; Waridel P.; Ndjoko K.; Hobby K. R.; Major H. J.; Hostettmann K. Evaluation of Q-TOF-MS/MS and multiple stage IT-MSn for the dereplication of flavonoids and related compounds in crude plant extracts. Analusis 2000, 28, 895–906. 10.1051/analusis:2000280895. [DOI] [Google Scholar]
- Zhou D.-Y.; Xu Q.; Xue X.-Y.; Zhang F.-F.; Jing Y.; Liang X.-M. Rapid qualitative and quantitative analyses of flavanone aglycones in Fructus aurantii by HPLC ion-trap MS. J. Sep. Sci. 2007, 30, 858–867. 10.1002/jssc.200600466. [DOI] [PubMed] [Google Scholar]
- Armentrout P. B. Guided ion beam studies of transition metal–ligand thermochemistry. Int. J. Mass Spectrom. 2003, 227, 289–302. 10.1016/S1387-3806(03)00087-3. [DOI] [Google Scholar]
- Zocher E.; Sigrist R.; Chen P. Threshold CID Investigation of Isomeric Cu(I) Azabox Complexes. Inorg. Chem. 2007, 46, 11366–11370. 10.1021/ic701688t. [DOI] [PubMed] [Google Scholar]
- Chiavarino B.; Dopfer O.; Crestoni M. E.; Corinti D.; Maître P.; Fornarini S. IRMPD spectra of protonated hydroxybenzaldehydes: evidence of torsional barriers in carboxonium ions. ChemPhysChem 2020, 21, 749–761. 10.1002/cphc.202000041. [DOI] [PubMed] [Google Scholar]
- Ahmed M.; Jelsch C.; Guillot B.; Lecomte C.; Domagała S. Relationship between stereochemistry and charge density in hydrogen bonds with oxygen acceptors. Cryst. Growth Des. 2013, 13, 315–325. 10.1021/cg3014656. [DOI] [Google Scholar]
- Nagy P. I. Competing intramolecular vs. intermolecular hydrogen bonds in solution. Int. J. Mol. Sci. 2014, 15, 19562–19633. 10.3390/ijms151119562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paciotti R.; Chiavarino B.; Coletti C.; Scuderi D.; Re N.; Corinti D.; Rotari L.; Fornarini S.; Crestoni M. E. IRMPD spectroscopy of bare mono-deprotonated genistein, an antioxidant flavonoid. ACS Omega 2022, 7, 19535–19544. 10.1021/acsomega.2c01236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbate S.; Burgi L. F.; Castiglioni E.; Lebon F.; Longhi G.; Toscano E.; Caccamese S. Assessment of configurational and conformational properties of naringenin by vibrational circular dichroism. Chirality 2009, 21, 436–441. 10.1002/chir.20616. [DOI] [PubMed] [Google Scholar]
- Smith D. P.; Knapman T. W.; Campuzano I.; Malham R. W.; Berryman J. T.; Radford S. E.; Ashcroft A. E. Deciphering drift time measurements from traveling wave ion mobility spectrometry-mass spectrometry studies. Eur. J. Mass Spectrom. 2009, 15, 113–130. 10.1255/ejms.947. [DOI] [PubMed] [Google Scholar]
- Erlund I. Review of the flavonoids quercetin, hesperetin, and naringenin. Dietary sources, bioactivities, bioavailability, and epidemiology. Nutr. Res. 2004, 24, 851–874. 10.1016/j.nutres.2004.07.005. [DOI] [Google Scholar]
- Hunt G. M.; Baker E. A. Phenolic constituents of tomato fruit cuticles. Phytochemistry 1980, 19, 1415–1419. 10.1016/S0031-9422(00)82090-7. [DOI] [Google Scholar]
- Slimestad R.; Verheul M. Review of flavonoids and other phenolics from fruits of different tomato (Lycopersicon esculentum Mill.) cultivars. J. Sci. Food Agric. 2009, 89, 1255–1270. 10.1002/jsfa.3605. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.