

Abstract

We describe the first catalytic generation of Fischer-type acyloxy Rh(II)-carbenes from carboxylic acids and Rh(II)-carbynoids. This novel class of transient donor/acceptor Rh(II)-carbenes evolved through a cyclopropanation process providing access to densely functionalized cyclopropyl-fused lactones with excellent diastereoselectivity. DFT calculations allowed the analysis of the properties of Rh(II)-carbynoids and acyloxy Rh(II)-carbenes as well as the characterization of the mechanism.
The discovery and development of transient donor/acceptor Rh(II)-carbenes generated from dirhodium paddlewheel catalysts and diazo compounds has represented a breakthrough in the field of carbene transfer catalysis (Figure 1A).1−3 Aryl, heteroaryl, alkenyl, or alkynyl substituents used as donor groups modulate the reactivity by decreasing the electrophilicity of the Rh(II)-carbene, which often offers exceptional levels of site- and stereoselectivity in intermolecular alkene cyclopropanations and C–H bond insertions (Figure 1A). However, the introduction of heteroatomic functionalities such as acyloxy, hydroxy, alkoxy, or amino as donor groups remains a long-standing problem, due to the lack of appropriate diazo compounds or carbene precursors.4
Figure 1.

Generation of novel donor/acceptor Rh(II)-carbenes.
Novel strategies to generate Rh(II)-carbenes catalytically with such heteroatomic functionalities promise to offer new synthetic applications and advance the field of carbene chemistry. An unexplored strategy to circumvent this problem could involve the attack of heteroatomic nucleophiles to the carbene carbon atom of Rh(II)-carbenes substituted with a leaving group (Figure 1B). If successful, this strategy would also provide a novel approach for the generation of Fischer metal-carbenes,5 a class of organometallic species that have been rather challenging to generate in a catalytic fashion.6 There is a small number of catalytic reactions where Fischer carbenes were proposed, and these include the seminal reports by McDonald,7 Wipf,8 and Barluenga9 with W(0) catalysts and recent examples by Kakiuchi,10 Kusama,11 Wang,12 and Tobisu13 with Rh(I), Cu(I), and Pd(0) catalysts, respectively.14
Herein, we present the discovery and development on the use of alkenyl carboxylic acids as nucleophiles for the first catalytic generation of transient acyloxy Rh(II)-carbenes (Figure 1C). The latter species evolved through intramolecular diastereoselective cyclopropanations, leading to complex cyclopropyl-fused lactones. Key to this work was the generation of Rh(II)-carbynoids, a class of Rh(II)-carbenes substituted with a hypervalent iodine(III) as a leaving group.
Our group has pioneered the generation of Rh(II)-carbynoids as carbyne transfer species by selective diazo activation of hypervalent iodine reagents15 with Rh(II) carboxylate catalysts (Figure 2, int-I).16 We have demonstrated that Rh(II)-carbynoids were able to transfer the carbynoid ligand to alkenes or alkynes, generating cyclopropyl- or cyclopropenyl-I(III) species that evolved, through the departure of the I(III) group, to synthetically useful allyl or cyclopropenium cations, respectively. Considering the outstanding nucleofuge ability of the hypervalent iodine(III)17 and high electrophilicity of the Rh-carbynoid (int-I), we recently hypothesized that alkenyl carboxylic acids 1 could be sufficiently nucleophilic to displace the iodine(III) group and provide a Fischer-type acyloxy Rh-carbene (Figure 2, int-II). The latter species could then evolve through (a) intramolecular alkene cyclopropanation that leads to complex cyclopropyl-fused δ-lactones 3 or (b) nucleophilic attack at the carbene carbon atom by acid 1 that would provide acetal 4.
Figure 2.

Mechanistic hypothesis.
Our envisaged carbyne transfer process with carboxylic acids was initially tested by adding 2 equiv of 3-phenyl-4-pentenoic acid (1a) and pseudocyclic hypervalent iodine reagent 2a as a limiting reagent to a solution of Rh2(OAc)4 (3 mol %) in CH2Cl2 at −50 °C (Table 1). Unfortunately, lactone 3a was not detected, and instead, acetal 4a was isolated in 65% yield (Table 1, entry 1). This result suggested the generation of a Fischer-type Rh(II)-carbene that underwent nucleophilic attack by 1a. Remarkably, no products derived from an alkene cyclopropanation were observed.16a
Table 1. Optimization Studiesa.


Performed at 0.1 mmol scale by addition of 1a and 2 over the Rh catalyst in CH2Cl2 at −50 °C during 30 min and then warmed to rt in 3 h.
1H NMR yields used CH2Br2 as an internal standard; a single diastereoisomer of 3a was observed (>20:1) in each entry, and 4a was obtained as an equimolecular mixture of four diastereoisomers.
NaHCO3 was added (2 equiv). oct = octanoate. adc = 1-adamantylcarboxylate. esp = α,α,α′,α′-tetramethyl-1,3-benzenedipropanoate.

After this, by reversing the stoichiometry (ratio 1a:2a, 1:1.3), we were delighted to observe 30% yield of the desired lactone 3a accompanied by a 20% yield of 4a (entry 2). 3a was obtained as a single diastereoisomer (>20:1), where the ester group from the Rh(II)-carbynoid, the aryl ring, and the internal olefinic C–H bond from 1a are in the syn disposition. Then, we argued that a way to increase the efficiency of the reaction, by decreasing 4a formation, could rely on the use of sterically demanding rhodium catalysts. While the use of Rh2(Oct)4 provided similar results to Rh2(OAc)4 (entry 3), Rh2(Adc)4 and Du Bois’ catalyst Rh2(esp)218 increased the efficiency of the cyclization while disfavoring formation of 4a (entries 4, 5). With Rh2(esp)2, we evaluated the effect of the counteranion and structural nature of 2 and found that while triflate and tetrafluoroborate reagents 2b,c and linear reagent 2d were less efficient (entries 6–8), cyclic derivative 2e provided no conversion to 3a (entry 9). In addition, we observed that sodium bicarbonate (2 equiv) slightly improved the efficiency of the reaction (entry 10).19 Finally, alternative diazo compounds substituted with sulfonium,20,21 ammonium,20 or bromide22 (2f–h, entries 11–13) were ineffective in promoting formation of 3a/4a.
Control experiments showed that tetrabutylammonium salt or methyl ester derivative derived from 3-phenylpent-4-enoic acid were unreactive, which underlines a key role of the carboxylic acid functionality for the generation of the acyloxy Rh(II)-carbene. Further controls showed that no reaction takes place between 1a and 2a, which indicates that generation of int-II from a presumable α-acyloxy diazoacetate derivative is highly unlikely.23
With the optimized conditions, we investigated the scope of this carbyne transfer reaction with a range of alkenyl carboxylic acids (Table 2A–C). We found that 4-pentenoic acids with para-substituted aromatic rings at C3 provided δ-lactones 3b–f with excellent diastereoselectivity (>20:1). Moreover, meta- and ortho-substitution on the aromatic rings was tolerated as well as alkyl substitution at C3 (3i). It was believed that geminal substitution at C3 could be detrimental in the cyclopropanation event for steric reasons. In contrast to our beliefs, we were glad to observe that lactones 3j,k were obtained with high efficiency and that could be explained by a Thorpe–Ingold effect. On the other hand, C2 substitution with alkyl or protected amino groups provided 3l,m as single diastereoisomers, and the unsubstituted derivative 4-pentenoic acid led to 3n in moderate yield.
Table 2. Scope of the Catalytic Carbyne Transfer with Alkenyl Carboxylic Acidsa,b.
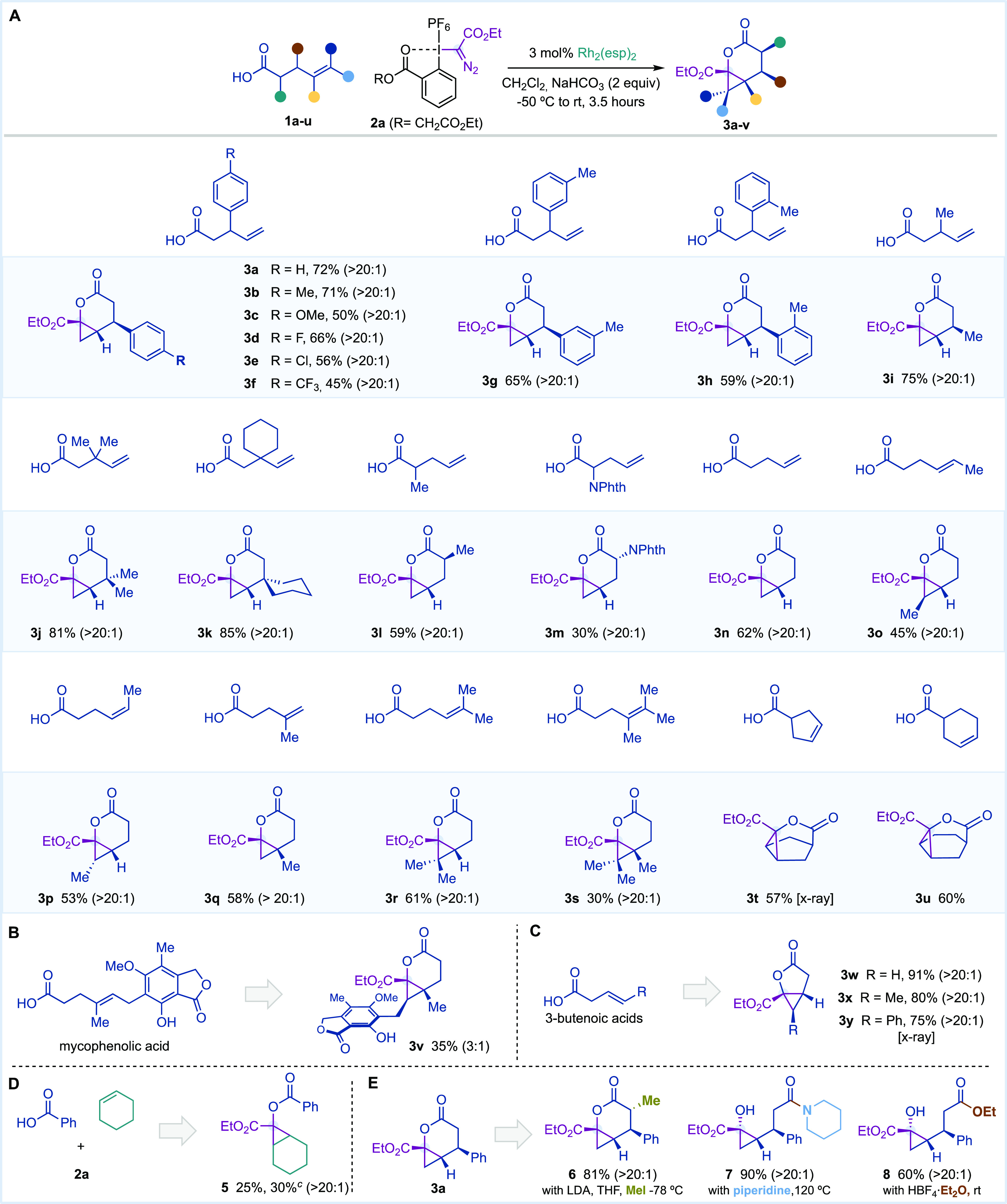
Performed with 1 (0.20 mmol, 1 equiv), 2a (0.26 mmol, 1.3 equiv), and Rh2(esp)2 (0.006 mmol, 3 mol %) in CH2Cl2 (0.08 M). Yields are reported on the basis of isolated pure product.
The diastereoselectivity ratio indicated in parentheses was determined by 1H NMR analysis of the reaction crude mixture. NPhth = phthalimide.
Reaction performed at 0 °C.
After this, we evaluated the alkene substitution and observed that the stereochemistry of the double bond is preserved in the final products (3o,p). Tri- and tetrasubstituted alkenes typically undergo carbene allylic C–H bond insertion for alternative donor/acceptor Rh(II)-carbenes.24,25 However, our acyloxy Rh(II)-carbene preferred to evolve via cyclopropanation and delivered penta- and hexasubstituted cyclopropanes 3r,s. Furthemore, carboxylic acids substituted with cyclic alkenes, such as cyclopentene or cyclohexene, were converted to complex tricyclic lactones 3t,u. Moreover, our carbyne transfer process was applied in the late-stage functionalization of mycophenolic acid (Table 2B, 3v), an immunosuppressant agent used to prevent rejection following organ transplantation. Furthermore, 3-butenoic acids could be converted into γ-lactones 3w–y with high efficiency (Table 2C). Under the optimized reaction conditions, we were glad to observe intermolecular cyclopropanation using benzoic acid, cyclohexene, and reagent 2a albeit in low yield (5, 25–30%, >20:1; Table 2D). Attempts to improve the efficiency by changing the stoichiometry, temperature, base, or catalyst were unsuccessful (see the Supporting Information for details).26
Derivatizations of 3a were performed by methylation via lithium enolate to provided 6 as a single diastereoisomer27 and by amidation or esterification to provided cyclopropanols 7 and 8, respectively (Table 2E).28 The latter results formally represent the cyclopropanation of an alkene with a Rh(II)-carbene substituted with a hydroxy as a donor group.29
Later, we studied computationally the mechanism for the formation of lactone 3a. We applied a B3LYP-D3BJ method in solvent to the study of the reaction with acid 1a reagent 2a* and Rh2(esp)2 as the catalyst. The full system was considered in the calculations, with the only exception that the pendant R group in 2a, (R = CH2CO2Et, see Table 2A) was replaced by a methyl group for the sake of conformational simplicity. A simplified version of the resulting free energy profile is shown in Figure 3, and full computational details are supplied in the Supporting Information.30
Figure 3.
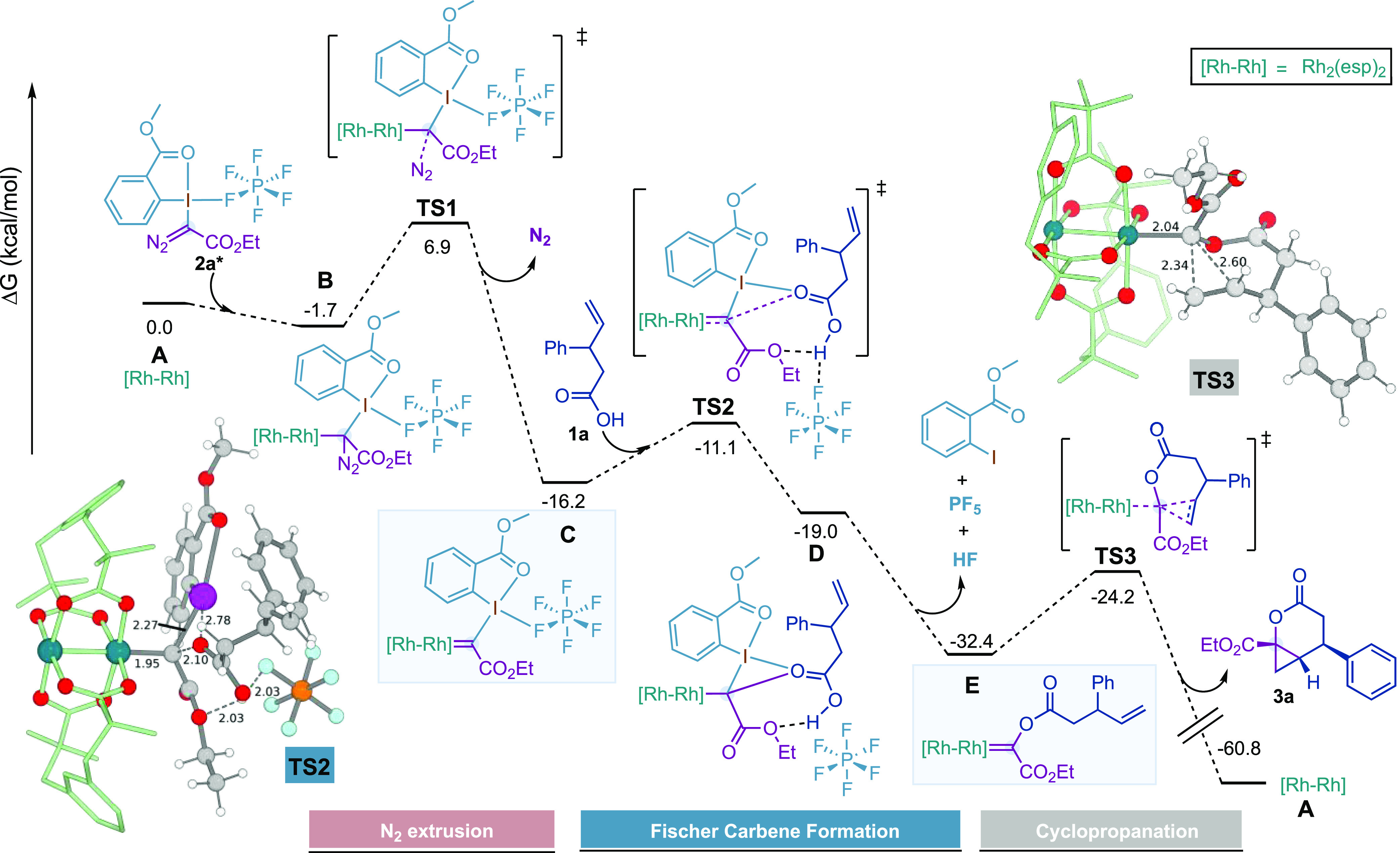
Free energy profile for the catalyzed reaction between 1a and 2a* with Rh2(esp)2.
The initial reaction between 2a* and Rh2(esp)2 follows the well-established path for the formation of metallocarbenic species either with dirhodium complexes31 or coinage metal complexes.32 A dinitrogen moiety is extruded from the precursor, and the metal-carbene bond is formed, resulting in the Rh-carbynoid C.
The 3D structure of C, presented in Figure 4 from two different views, has reasonable geometrical parameters, with Rh–C distances and C–I distances of 1.92 and 2.19 Å, respectively. Remarkably, the PF6– counterion and the carbonyl ester group in the ortho position of the Ar–I(III) moiety remain attached to the iodine center (F–I, 2.78 Å; O–I, 2.73 Å) in the nonpolar medium, with a distorted square planar arrangement around iodine and the carbonyl group. The two substituents on the carbene are nearly eclipsed with the nearest O–Rh bond of the bimetallic core, with 12.5 and −5.3° dihedral angles for CO2Et and the Ar–I(III) substituent, respectively (Figure 4b). Furthermore, the ethyl group of CO2Et and the aryl group of the Ar–I(III) substituent are placed away from the esp units. The angle between Rh–Rh–Ccarbene is 166.9°, and the carbene carbon is slightly pyramidal (∠Rh–Ccarbene–CCO2Et–IIAr = 165°), which may be due to steric interaction of the Ar–I(III) group with the esp ligand. It is also worth mentioning that the two phenyl groups in the esp ligands are by no means in a symmetric arrangement, with one of them much closer to the carbynoid ligand.
Figure 4.
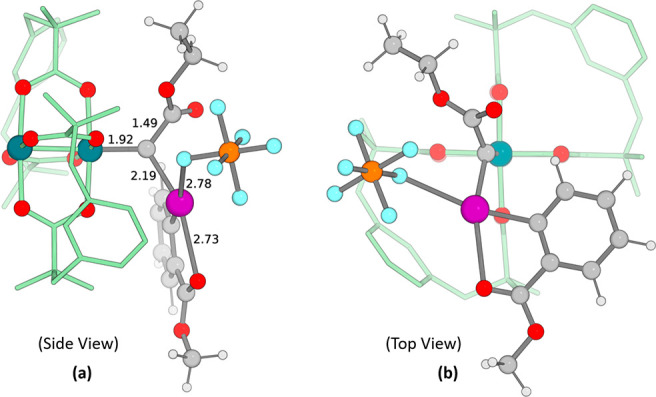
Computed geometry of Rh-carbynoid C with (a) side and (b) top views. For structural clarity, hydrogens are omitted, and carbon atoms are shown in tube format in the esp ligand.
Rh(II)-carbynoid C interacts with 1a through transition state TS2 and evolves to intermediate D. The latter can go through a series of low barrier fragmentations detailed in the Supporting Information and evolve finally to the Fischer carbene E. From here, the cyclopropane formation occurs in a single step through TS3 (8.2 kcal/mol barrier) where the phenyl ring from the carboxylate moiety and the dirhodium are displayed in favorable pseudo-equatorial positions to prevent steric clashes.
The role of the usually inert PF6– counterion as a proton abstraction agent deserves some comment. We are in no way claiming that HF will be a side product of the reaction, as it will immediately react with the HCO3– excess present in the medium to regenerate PF6–, in a step that has not been included in the profile for simplicity. We consider however that it emphasizes a defining feature of Rh(II)-carbynoid C in abstracting the proton from carboxylic acids.33
We finally analyzed computationally the Rh(II)-carbynoids and Fischer Rh(II)-carbene complexes and compared their properties with a donor/acceptor derivative substituted with a phenyl ring (Table 3). The metal moiety was slightly simplified from that used in the mechanistic study above, as the Rh2(OAc)4 was used instead of Rh2(esp)2. We first analyzed the metal complexes through charge decomposition analysis (CDA), which separates the metal-carbene bonding interaction into ligand to metal σ donation and metal to ligand π back-donation.34 Remarkably, for the carbynoid, the σ donation (0.245) is the lowest, and the back-donation (0.123) is the highest. This means that the ligand transfers less electronic density to the dirhodium core of the system in comparison with the Rh-carbenes substituted with phenyl or acetoxy. However, the Rh(II)-carbynoid complex is a stronger electrophile, as shown by the much more negative LUMO energy, and the significantly higher electrophilicity index ω.35 This seeming contradiction is explained by the presence of the formally cationic hypervalent iodine attached to carbon. In contrast, the acetoxy Rh(II)-carbene is the least electrophilic of the three complexes and has the highest LUMO energy.
Table 3. Calculated Values of Charge Donation (d) and Back Donation (b)a from the Carbene Ligand to Rhodium, LUMO Energies (eV), and Electrophilicity Indices (ω)a.

d and b values are obtained by charge decomposition analysis. [Rh] = Rh2(OAc)4.
In summary, we have disclosed a carbyne transfer process that catalytically transforms alkenyl carboxylic acids into complex cyclopropyl-fused lactones by the formation of two single C–C bonds and one C–O bond at the carbyne carbon atom. Relevant on this work was the catalytic generation, from a Rh(II)-carbynoid, of a new class of donor/acceptor Rh(II)-carbene substituted with an acyloxy group that can be considered a Fischer-type carbene. This new way of accessing Fischer-type carbenes in a catalytic fashion is remarkably different to recent approaches,10−13 and DFT calculations unveiled key features of both Rh(II)-carbynoid and acyloxy Rh(II)-carbene.
Acknowledgments
European Research Council (ERC-CoG 2019, 865554) Agencia Estatal de Investigación (AEI) of the Ministerio de Ciencia e Innovación (PID2019-104101GB-I00, PID2020-112825RB-I00, Severo Ochoa Excellence Accreditation 2020–2023-CEX2019-000925-S), ICIQ Foundation, and the CERCA Programme (Generalitat de Catalunya) are gratefully acknowledged for financial support. We thank the European Union for a Marie Skłodowska-Curie Individual Fellowship (794815) (to L.J.), the AEI for a FPI predoctoral fellowship (PRE2020-092989) (to E.P.), and AGAUR for a Beatriu de Pinós fellowship (2019-BP-00190) (to A.K.S.).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.3c00012.
Experimental procedures and spectral data (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- For a perspective:; a Davies H. M. L. Finding Opportunities from Surprises and Failures. Development of Rhodium-Stabilized Donor/Acceptor Carbenes and Their Application to Catalyst-Controlled C-H Functionalization. J. Org. Chem. 2019, 84, 12722–12745. 10.1021/acs.joc.9b02428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For selected reviews:; a Doyle M. P.; Forbes D. C. Recent Advances in Asymmetric Catalytic Metal Carbene Transformations. Chem. Rev. 1998, 98, 911–936. 10.1021/cr940066a. [DOI] [PubMed] [Google Scholar]; b Davies H. M. L.; Beckwith R. E. J. Catalytic Enantioselective C-H Activation by Means of Metal-Carbenoid-Induced C-H Insertion. Chem. Rev. 2003, 103, 2861–2903. 10.1021/cr0200217. [DOI] [PubMed] [Google Scholar]; c Davies H. M. L.; Manning J. R. Catalytic C-H Functionalization by Metal Carbenoid and Nitrenoid Insertion. Nature 2008, 451, 417–424. 10.1038/nature06485. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Davies H. M. L.; Morton D. Guiding Principles for Site Selective and Stereoselective Intermolecular C–H Functionalization by Donor/Acceptor Rhodium Carbenes. Chem. Soc. Rev. 2011, 40, 1857–1869. 10.1039/c0cs00217h. [DOI] [PubMed] [Google Scholar]; e Davies H. M. L.; Liao K. Dirhodium Tetracarboxylates as Catalysts for Selective Intermolecular C–H Functionalization. Nat. Rev. Chem. 2019, 3, 347–360. 10.1038/s41570-019-0099-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For characterization and isolation of donor/acceptor Rh(II)-carbenes:; a Kornecki K. P.; Briones J. F.; Boyarskikh V.; Fullilove F.; Autschbach J.; Schrote K. E.; Lancaster K. M.; Davies H. M. L.; Berry J. F. Direct Spectroscopic Characterization of a Transitory Dirhodium Donor-Acceptor Carbene Complex. Science 2013, 342, 351–354. 10.1126/science.1243200. [DOI] [PubMed] [Google Scholar]; b Werlé C.; Goddard R.; Fürstner A. The First Crystal Structure of a Reactive Dirhodium Carbene Complex and a Versatile Method for the Preparation of Gold Carbenes by Rhodium-to-Gold Transmetalation. Angew. Chem., Int. Ed. 2015, 54, 15452–15456. 10.1002/anie.201506902. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Werlé C.; Goddard R.; Philipps P.; Farès C.; Fürstner A. Structures of Reactive Donor/Acceptor and Donor/Donor Rhodium Carbenes in the Solid State and Their Implications for Catalysis. J. Am. Chem. Soc. 2016, 138, 3797–3805. 10.1021/jacs.5b13321. [DOI] [PubMed] [Google Scholar]
- During the preparation of this manuscript, the group of Davies reported an elegant approach for the generation of aryloxy-subsituted Rh(II)-carbenes:; Kubiak R. W.; Tracy W. F.; Alford J. S.; Davies H. M. L. Asymmetric Cyclopropanation with 4-Aryloxy-1-Sulfonyl-1,2,3-Triazoles: Expanding the Range of Rhodium-Stabilized Donor/Acceptor Carbenes to Systems with an Oxygen Donor Group. J. Org. Chem. 2022, 87, 13517–13528. 10.1021/acs.joc.2c00978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer E. O.; Maasböl A. On the Existence of a Tungsten Carbonyl Carbene Complex. Angew. Chem., Int. Ed. Engl. 1964, 3, 580–581. 10.1002/anie.196405801. [DOI] [Google Scholar]
- a Dötz K. H.; Stendel J. Fischer Carbene Complexes in Organic Synthesis: Metal-Assisted and Metal-Templated Reactions. Chem. Rev. 2009, 109, 3227–3274. 10.1021/cr900034e. [DOI] [PubMed] [Google Scholar]; b Barluenga J.; Aguilar E. Group 6 Metal Fischer Carbene Complexes: Versatile Synthetic Building Blocks. Adv. Organomet. Chem. 2017, 67, 1–150. 10.1016/bs.adomc.2017.04.001. [DOI] [Google Scholar]
- a McDonald F. E.; Reddy K. S.; Díaz Y. Stereoselective Glycosylations of a Family of 6-Deoxy-1,2-Glycals Generated by Catalytic Alkynol Cycloisomerization. J. Am. Chem. Soc. 2000, 122, 4304–4309. 10.1021/ja994229u. [DOI] [Google Scholar]; b McDonald F. E.; Subba Reddy K. Convergent Synthesis of Digitoxin: Stereoselective Synthesis and Glycosylation of the Digoxin Trisaccharide Glycal. Angew. Chem., Int. Ed. 2001, 40, 3653–3655. 10.1002/1521-3773(20011001)40:19<3653::AID-ANIE3653>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- Wipf P.; Graham T. H. Photoactivated Tungsten Hexacarbonyl-Catalyzed Conversion of Alkynols to Glycals. J. Org. Chem. 2003, 68, 8798–8807. 10.1021/jo034813s. [DOI] [PubMed] [Google Scholar]
- a Barluenga J.; Diéguez A.; Rodríguez F.; Fañanás F. J. Tandem [W(CO)5]-Catalyzed Cycloisomerization-Cyclopropanation Reactions Directed toward the Synthesis of Eight-Membered Carbocycles. Angew. Chem., Int. Ed. 2005, 44, 126–128. 10.1002/anie.200461414. [DOI] [PubMed] [Google Scholar]
- Takano S.; Shiomi R.; Morimoto Y.; Kochi T.; Kakiuchi F. Carbon–Carbon Bond Formation via Catalytically Generated Aminocarbene Complexes: Rhodium-Catalyzed Hydroaminative Cyclization of Enynes with Secondary Amines. Angew. Chem., Int. Ed. 2020, 59, 11754–11757. 10.1002/anie.202002710. [DOI] [PubMed] [Google Scholar]
- Takeuchi T.; Aoyama T.; Orihara K.; Ishida K.; Kusama H. Visible-Light-Induced In Situ Generation of Fischer-Type Copper Carbene Complexes from Acylsilanes and Its Application to Catalytic [4 + 1] Cycloaddition with Siloxydienes. Org. Lett. 2021, 23, 9490–9494. 10.1021/acs.orglett.1c03683. [DOI] [PubMed] [Google Scholar]
- Zheng L.; Guo X.; Li Y.-C.; Wu Y.; Xue X.-S.; Wang P. Cu/SaBox-Catalyzed Photoinduced Coupling of Acylsilanes with Alkynes. Angew. Chem., Int. Ed. 2023, 62, e202216373. 10.1002/anie.202216373. [DOI] [PubMed] [Google Scholar]
- Sakurai S.; Inagaki T.; Kodama T.; Yamanaka M.; Tobisu M. Palladium-Catalyzed Siloxycyclopropanation of Alkenes Using Acylsilanes. J. Am. Chem. Soc. 2022, 144, 1099–1105. 10.1021/jacs.1c11497. [DOI] [PubMed] [Google Scholar]
- For a recent stoichiometric synthesis of a Fischer-type Cu(I)-carbene from a non-Fischer carbene:; Álvarez M.; Besora M.; Molina F.; Maseras F.; Belderrain T. R.; Pérez P. J. Two Copper-Carbenes from One Diazo Compound. J. Am. Chem. Soc. 2021, 143, 4837–4843. 10.1021/jacs.1c01483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For selected reviews in hypervalent iodine chemistry:; a Zhdankin V. V.; Stang P. J. Chemistry of Polyvalent Iodine. Chem. Rev. 2008, 108, 5299–5358. 10.1021/cr800332c. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Yoshimura A.; Zhdankin V. V. Advances in Synthetic Applications of Hypervalent Iodine Compounds. Chem. Rev. 2016, 116, 3328–3435. 10.1021/acs.chemrev.5b00547. [DOI] [PubMed] [Google Scholar]; c Li Y.; Hari D. P.; Vita M. V.; Waser J. Cyclic Hypervalent Iodine Reagents for Atom-Transfer Reactions: Beyond Trifluoromethylation. Angew. Chem., Int. Ed. 2016, 55, 4436–4454. 10.1002/anie.201509073. [DOI] [PubMed] [Google Scholar]
- a Wang Z.; Jiang L.; Sarró P.; Suero M. G. Catalytic Cleavage of C(sp2)–C(sp2) Bonds with Rh-Carbynoids. J. Am. Chem. Soc. 2019, 141, 15509–15514. 10.1021/jacs.9b08632. [DOI] [PubMed] [Google Scholar]; See also:; b Jiang L.; Sarró P.; Teo W. J.; Llop J.; Suero M. G. Catalytic Alkene Skeletal Modification for the Construction of Fluorinated Tertiary Stereocenters. Chem. Sci. 2022, 13, 4327–4333. 10.1039/D2SC00968D. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Tu H.-F.; Jeandin A.; Suero M. G. Catalytic Synthesis of Cyclopropenium Cations with Rh-Carbynoids. J. Am. Chem. Soc. 2022, 144, 16737–16743. 10.1021/jacs.2c07769. [DOI] [PMC free article] [PubMed] [Google Scholar]; See also:; Wang Z.; Herraiz A. G.; del Hoyo A. M.; Suero M. G. Generating Carbyne Equivalents with Photoredox Catalysis. Nature 2018, 554, 86–91. 10.1038/nature25185. [DOI] [PubMed] [Google Scholar]
- Okuyama T.; Takino T.; Sueda T.; Ochiai M. Solvolysis of Cyclohexenyliodonium Salt, a New Precursor for the Vinyl Cation: Remarkable Nucleofugality of the Phenyliodonio Group and Evidence for Internal Return from an Intimate Ion–Molecule Pair. J. Am. Chem. Soc. 1995, 117, 3360–3367. 10.1021/ja00117a006. [DOI] [Google Scholar]
- Espino C. G.; Fiori K. W.; Kim M.; Du Bois J. Expanding the Scope of C-H Amination through Catalyst Design. J. Am. Chem. Soc. 2004, 126, 15378–15379. 10.1021/ja0446294. [DOI] [PubMed] [Google Scholar]
- We believe that sodium bicarbonate may be acting as a base to quench the HPF6 (or HF) generated in the reaction. Sodium bicarbonate is highly insoluble in CH2Cl2 at −50 °C, and considering that 1a and 2a are added together and dropwise to a solution of the Rh catalyst, it is unlikely that could be deprotonating 1a at the first instance.
- a Weiss R.; Seubert D.-C. J.; Hampel F. α-Aryliodonio Diazo Compounds: SN Reactions at the α-C Atom as a Novel Reaction Type for Diazo Compounds. Angew. Chem., Int. Ed. Engl. 1994, 33, 1952–1953. 10.1002/anie.199419521. [DOI] [Google Scholar]; See also:; b Taylor M. T.; Nelson J. E.; Suero M. G.; Gaunt M. J. A Protein Functionalization Platform Based on Selective Reactions at Methionine Residues. Nature 2018, 562, 563–568. 10.1038/s41586-018-0608-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Li X.; Golz C.; Alcarazo M. α-Diazo Sulfonium Triflates: Synthesis, Structure, and Application to the Synthesis of 1-(Dialkylamino)-1,2,3-Triazoles. Angew. Chemie, Int. Ed. 2021, 60, 6943–6948. 10.1002/anie.202014775. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Wang X.; Tong W. Y.; Huang B.; Cao S.; Li Y.; Jiao J.; Huang H.; Yi Q.; Qu S.; Wang X. Convergent Synthesis of 1,4-Dicarbonyl Z-Alkenes through Three-Component Coupling of Alkynes, α-Diazo Sulfonium Triflate, and Water. J. Am. Chem. Soc. 2022, 144, 4952–4965. 10.1021/jacs.1c12874. [DOI] [PubMed] [Google Scholar]
- a Bonge H. T.; Pintea B.; Hansen T. Highly Efficient Formation of Halodiazoacetates and Their Use in Stereoselective Synthesis of Halocyclopropanes. Org. Biomol. Chem. 2008, 6, 3670–3672. 10.1039/b814374a. [DOI] [PubMed] [Google Scholar]; b Bonge H. T.; Hansen T. Intermolecular C-H and Si-H Insertion Reactions with Halodiazoacetates. Synthesis 2009, 2009, 91–96. 10.1055/s-0028-1083272. [DOI] [Google Scholar]; c Bonge H. T.; Hansen T. Halodiazoacetates as Useful Tools for Selective Halo-Functionalization. Pure Appl. Chem. 2011, 83, 565–575. 10.1351/PAC-CON-10-10-18. [DOI] [Google Scholar]; d Schnaars C.; Hennum M.; Bonge-Hansen T. Nucleophilic Halogenations of Diazo Compounds, a Complementary Principle for the Synthesis of Halodiazo Compounds: Experimental and Theoretical Studies. J. Org. Chem. 2013, 78, 7488–7497. 10.1021/jo401050c. [DOI] [PubMed] [Google Scholar]; e Mortén M.; Hennum M.; Bonge-Hansen T. Synthesis of Quinoline-3-Carboxylates by a Rh(II)-Catalyzed Cyclopropanation-Ring Expansion Reaction of Indoles with Halodiazoacetates. Beilstein J. Org. Chem. 2015, 11, 1944–1949. 10.3762/bjoc.11.210. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Mortén M.; Hennum M.; Bonge-Hansen T. On the Cause of Low Thermal Stability of Ethyl Halodiazoacetates. Beilstein J. Org. Chem. 2016, 12, 1590–1597. 10.3762/bjoc.12.155. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Peeters S.; Berntsen L. N.; Rongved P.; Bonge-Hansen T. Cyclopropanation–Ring Expansion of 3-Chloroindoles with α-Halodiazoacetates: Novel Synthesis of 4-Quinolone-3-Carboxylic Acid and Norfloxacin. Beilstein J. Org. Chem. 2019, 15, 2156–2160. 10.3762/bjoc.15.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An additional control experiment carried out with 3-phenyl-4-pentenoic acid (1a) and ethyl diazoacetate under the optimized reaction conditions did not afford the corresponding cyclopropyl carboxylic acid but instead the ester derivative 2-ethoxy-2-oxoethyl 3-phenylpent-4-enoate in 40% yield. See the Supporting Information for details.
- a Davies H. M. L.; Ren P. Catalytic Asymmetric C-H Activation of Silyl Enol Ethers as an Equivalent of an Asymmetric Michael Reaction. J. Am. Chem. Soc. 2001, 123, 2070–2071. 10.1021/ja0035607. [DOI] [PubMed] [Google Scholar]; b Davies H. M. L.; Ren P.; Jin Q. Catalytic Asymmetric Allylic C-H Activation as a Surrogate of the Asymmetric Claisen Rearrangement. Org. Lett. 2001, 3, 3587–3590. 10.1021/ol0167255. [DOI] [PubMed] [Google Scholar]
- For a rare example of cyclopropanation of tetrasubsituted alkenes to access hexasubstituted cyclopropanes:; Nani R. R.; Reisman S. E. α-Diazo-β-Ketonitriles: Uniquely Reactive Substrates for Arene and Alkene Cyclopropanation. J. Am. Chem. Soc. 2013, 135, 7304–7311. 10.1021/ja401610p. [DOI] [PubMed] [Google Scholar]
- Analysis by GC-MS and 1H NMR of the reaction crude mixtures showed traces of side products that could not be identified in addition to the corresponding acetals 4 (<5% yield), unreactive carboxylic acid 1, and the Ar-I derived from 2a.
- Tomioka K.; Kawasaki H.; Yasuda K.; Koga K. A Model for the Diastereofacial Differentiation in the Alkylation of Endocyclic Enolate. J. Am. Chem. Soc. 1988, 110, 3597–3601. 10.1021/ja00219a039. [DOI] [Google Scholar]
- Sekiguchi Y.; Yoshikai N. Zinc-Catalyzed β-Functionalization of Cyclopropanols via Enolized Homoenolate. J. Am. Chem. Soc. 2021, 143, 18400–18405. 10.1021/jacs.1c10109. [DOI] [PubMed] [Google Scholar]
- For a recent selected synthesis of cyclopropanols:; Ni J.; Xia X.; Zheng W. F.; Wang Z. Ti-Catalyzed Diastereoselective Cyclopropanation of Carboxylic Derivatives with Terminal Olefins. J. Am. Chem. Soc. 2022, 144, 7889–7900. 10.1021/jacs.2c02360. [DOI] [PubMed] [Google Scholar]
- Data set collection of all computational results was uploaded to the ioChem-BD repository, and can be accessed via https://doi.org/10.19061/iochem-bd-1-266.; Álvarez-Moreno M.; de Graaf C.; Lopez N.; Maseras F.; Poblet J. M.; Bo C. Managing the Computational Chemistry Big Data Problem: the ioChem-BD Platform. J. Chem. Inf. Model. 2015, 55, 95–103. 10.1021/ci500593j. [DOI] [PubMed] [Google Scholar]
- a Nakamura E.; Yoshikai N.; Yamanaka M. Mechanism of C-H Bond Activation/C-C Bond Formation Reaction between Diazo Compound and Alkane Catalyzed by Dirhodium Tetracarboxylate. J. Am. Chem. Soc. 2002, 124, 7181–7192. 10.1021/ja017823o. [DOI] [PubMed] [Google Scholar]; b Qin C. M.; Boyarskikh V. H.; Hansen J. H.; Hardcastle K. I.; Musaev D. G.; Davies H. M. L. D2-Symmetric Dirhodium Catalyst Derived from a 1,2,2-Triarylcyclopropanecarboxylate Ligand: Design, Synthesis and Application. J. Am. Chem. Soc. 2011, 133, 19198–19204. 10.1021/ja2074104. [DOI] [PubMed] [Google Scholar]
- Rivilla I.; Sameera W. M. C.; Alvarez E.; Díaz-Requejo M. M.; Maseras M.; Pérez P. J. Catalytic Cross-Coupling of Diazo Compounds with Coinage Metal-based Catalysts: an Experimental and Theoretical Study. Dalton Trans. 2013, 42, 4132–4138. 10.1039/c2dt32439c. [DOI] [PubMed] [Google Scholar]
- A related feature of C is that it reacts with the carboxylic group rather than with the olefin group present in the substrate. We computed the corresponding alternative mechanism, and it has indeed a higher barrier. See the Supporting Information for details.
- Dapprich S.; Frenking G. Investigation of Donor-Acceptor Interactions: A Charge Decomposition Analysis Using Fragment Molecular Orbitals. J. Phys. Chem. 1995, 99, 9352–9362. 10.1021/j100023a009. [DOI] [Google Scholar]
- Parr R. G.; Szentpály L. v.; Liu S. Electrophilicity Index. J. Am. Chem. Soc. 1999, 121, 1922–1924. 10.1021/ja983494x. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.