

Abstract
In addition to their lipid-lowering functions, statins elicit additional pleiotropic effects on apoptosis, angiogenesis, inflammation, senescence, and oxidative stress. Many of these effects have been reported in cancerous and noncancerous cells like endothelial cells (ECs), endothelial progenitor cells (EPCs) and human umbilical vein cells (HUVCs). Not surprisingly, statins' effects appear to vary largely depending on the cell context, especially as pertains to modulation of cell cycle, senescence, and apoptotic processes. Perhaps the most critical reason for this discordance is the bias in selecting the applied doses in various cells. While lower (nanomolar) concentrations of statins impose anti-senescence, and antiapoptotic effects, higher concentrations (micromolar) appear to precipitate opposite effects. Indeed, most studies performed in cancer cells utilized high concentrations, where statin-induced cytotoxic and cytostatic effects were noted. Some studies report that even at low concentrations, statins induce senescence or cytostatic impacts but not cytotoxic effects. However, the literature appears to be relatively consistent that in cancer cells, statins, in both low or higher concentrations, induce apoptosis or cell cycle arrest, anti-proliferative effects, and cause senescence. However, statins’ effects on ECs depend on the concentrations; at micromolar concentrations statins cause cell senescence and apoptosis, while at nonomolar concentrations statins act reversely.
Keywords: Statins, Akt, Apoptosis, Cancer, Cytostatic, Endothelial cells, Senescence
Introduction
Statins are the most widely used medications in the management and treatment of hypercholesterolemia. Based on their polarity, statins are categorized in two groups, lipophilic and hydrophilic statins. Lipophilic statins readily cross the cell membrane, whereas hydrophilic ones employ carrier proteins to gain intracellular access. Lipophilic statins include atorvastatin, lovastatin, simvastatin, pitavastatin, and fluvastatin, while pravastatin and rosuvastatin are hydrophilic ones [1–4]. Considering their simple diffusion through membrane, the pleiotropic effects of lipophilic statins in extrahepatic tissues are extensively studied [5–18]. In particular, simvastatin has been widely used in cancer studies [19]. Owing to their inability to readily cross cell membranes, hydrophilic statins do not have pleiotropic effects in extrahepatic cells or their effects are not significant (see Tables 1 and 2) [20–22]. This could explain why they are less investigated in cancer studies as compared to lipophilic ones.
Table 1.
The effects of statins on apoptosis and senescence in different vascular cardiovascular cell lines
| Statin | Dose/concentration | Cell type | Mechanism of effect | Effect on apoptosis |
|---|---|---|---|---|
|
Fluvastatin Pravastatin |
3 µM 10–20 µM |
Cardiac myocytes of Sprague–Dawley rats | Inhibition of the RhoA localization in the membrane; pravastatin did not have effect [101] | Increased |
| Simvastatin Fluvastatin Pravastatin |
Animal Rat fluvastatin 50 mg/kg per day |
L6 fibroblast cells and Rats in vivo | Inhibition of the RhoA localization in the membrane; pravastatin did not have effect [102] |
Increased (cell line) Myotoxicity (Rat) |
| Simvastatin | 71.6 µM, 143.3 µM | L6 myoblasts |
Induction of tyrosine phosphorylation, 60 µM of pravastatin did not have effect on apoptosis [103] |
Increased (71.6 µM), 143 µM cause necrosis |
| Simvastatin | 71.6 µM | L6 myoblasts | Inhibition of the Ras isoprenylation and its downstream pathway Raf1/MEK and PI3-k [104] | Increased |
| Simvastatin | Oral administration (50 mg/kg/day) for 2 weeks, | Animal rabbit cardiac fibers | Raised serum CK and myopathy was induced by lesions of the muscle surface membrane [105] | Increased (myopathy) |
| Simvastatin, Simvastatin-acid form, and pravastatin |
Simvastatin 47.8 µM, 60 µM, 71.6 µM simvastatin-acid 401 μM pravastatin μg/ml |
L6 rat myoblasts | The mechanism of cell damage may relate to the [Ca2 +]i releasing and lipophilicity: Pravastatin caused little or no change in [Ca2 +]i and cell damage while simvastatin induced Apoptosis [106] | Increased by simvastatin Lipophilic statin but not pravastatin hydrophilic statin |
|
Fluvastatin Pitavastatin Pravastatin |
1–10 µM | Synoviocytes | Blocking geranylgeranylation of RhoA and subsequently activation of caspase 3; pravastatin had no effect [107] | Increased |
| Atorvastatin Simvastatin | 100 μM | Vascular smooth muscle cells from Rat thoracic aorta | Blocking the prenylation of RhoA and downregulating the expression of Bcl-2 [78] | Increased |
|
Simvastatin Fluvastatin Pravastatin |
0.5–5 μM |
Primary human adult cardiac myocytes |
Downregulating both mRNA/protein of Mcl-1 (an inhibitor of apoptosis); pravastatin had no effect [108] | Increased |
|
Atorvastatin Mevast |
0.01 to 0.1 µM | HUVECs | Activate the endothelial Ras and promote Akt and mediate activation of eNOS [69] | Increased |
|
Atorvastatin Mevastatin |
> 0.1 µM | EPCs and Mononuclear cells | Halt angiogenesis and induce endothelial cell apoptosis [69] | Not seen even at high doses |
| Atorvastatin Mevastatin | 0.1, 0.05, and 0.01 µM | EPCs and Mononuclear cells |
Inhibit senescence [81] Regulation of cell cycle regulatory genes [81] |
decreased |
| Atorvastatin, Pravastatin Pitavastatin | nanomolar concentrations | HUVEC | Inhibit senescence via activation of Akt and then upregulation of eNOS, SIRT1, and catalase [83] | Decreased |
| Simvastatin pravastatin | (0.1 µM) |
In vivo: rabbit In vitro: HUVEC, COS-7 cells |
Activation of Akt/eNOS and consequently induction of angiogenesis [109] | |
| Lovastatin | 2, 10, and 50 μM |
In vitro Mononuclear cells (MNCs) CD34 + isolated from human umbilical cord |
Lovastatin reverses the survival and function of EPCs by regulating the Akt/eNOS signaling pathway and the gene transcription of eNOS. Coincubation of 50 µM simvastatin with Triciribine induced apoptosis [110] | Inhibition of the apoptosis induced by oxLDL |
|
Atorvastatin Rosuvastatin |
0.01–1 μM | EPCs isolated from peripheral blood | Atorvastatin suppressed homocysteine-induced ROS accumulation and EPCs apoptosis. It also antagonized Hcy-induced activation of NADPH oxidase and overexpression of Nox4 mRNA and p-p38MAPK protein. Nox4 siRNA transfected EPCs showed a similar result [111, 112] | Decreased |
| Pravastatin | 0.002, 0.02, 0.2, 2 μM | Endothelial colony-forming cells (ECFCs) | Akt- and eNOS-phosphorylation were augmented. Further, expression levels of HO-1, VEGF-A, and PlGF were increased, whereas expression levels of sFlt-1 and Eng were decreased [113] | Proliferation, migration, and tube formation of ECFCs were enhanced by pravastatin |
|
Atorvastatin Mevastatin |
0.1 μM 0.1 μM |
EPCs | Upregulation of the telomere repeat-binding factor TRF2 [114] | Statins enhance the migratory capacity of EPCs |
|
Atorvastatin Mevastatin |
0.1 μM | EPCs | Atorvastatin or mevastatin dose-dependently inhibited the onset of EPC senescence in culture. Moreover, atorvastatin increased the proliferation of EPCs. Atorvastatin modulated the expression of cell cyclins while downregulating the cell cycle inhibitor, p27Kip1. The effects of statins on the senescence were independent of NO, ROS, telomerase, and Rho kinase but dependent on GGPP [115] | statins inhibited senescence of EPCs |
Table 2.
The effects of statins on the apoptosis and senescence in different cancer cell lines
| Statin | IC50 values | Cell line | Mechanism of effect | Effect on apoptosis |
|---|---|---|---|---|
| Fluvastatin | 10 μM | MCF10A |
The anticancer effect of statins is independent of prenylation of RAS family proteins and is associated with a cancer cell EMT phenotype [126] Inhibition of the RAS prenylation was uncoupled from fluvastatin-induced-apoptosis [126] |
Induction of apoptosis |
| Simvastatin Atorvastatin | Atorvastatin 0.3–49.1 µM, Simvastatin 0.2- 40.8 µM | Triple-negative breast cancer (TNBC) | MVA rescued the effects of these statins [127] | Induction of apoptosis |
| Simvastatin | 12 and 8 μM | 12 and 8 μM in PC3 and LNCaP cell lines respectively | Induces subG1/G1 arrest [128] | Induction of apoptosis |
| 4.06 µM | breast cancer cell lines BoM-1833 (BoM) derived from MDA-231, MCF7/BoM, and T47D | Growth inhibition [129] | Induction of apoptosis | |
| Simvastatin | 0.481 µM | Adrenal carcinoma SW13 vimentin-positive (SW13-vim+) | Simvastatin targeting of vimentin may promote apoptotic cell death [130] | Induction of apoptosis |
| Simvastatin | 60 µM | T47D breast cancer cell | Decreased the cyclin D1 expression and cell growth [131] | Induction of apoptosis |
| Atorvastatin | 1.16 μM to 4.3 μM | MDA-MB-231 cells | Atorvastatin sensitivity correlates with decreased cholesterol levels in atorvastatin-treated cell [132] | |
| Simvastatin, fluvastatin |
PC9 and PC9 GR4 (simvastatin:4 µM, fluvastatin: 2 µM) H460, H358, and PC9 BrM3 (simvastatin:12 µM, fluvastatin: 4 µM) |
Decreases metastatic lung cancer cell survival in vitro synergistically with ABL tyrosine kinases inhibitor. Isoprenoid and mevalonate rescued the effects of statin [133] | Induction of apoptosis | |
| Fluvastatin | 5.3 µM | human A549 lung adenocarcinoma cells | PI3K inhibition [134] | Induction of apoptosis |
| Lovastatin | Anaplastic thyroid cancer cells | Blocking the membrane localization of RhoA and Rac. Mevalonate, GGPP rescued the effects of these statin [135] | Induction of apoptosis | |
| Lipophilic statins | Osteosarcoma cells | Blocking RhoA-p42/p44 MAPKs-Bcl-2 survival pathway. RhoA agonist rescued the effects of statins [122] | Induction of apoptosis | |
| Lovastatin | 0.3 µM | Human prostate cancer cells | Induce senescence and cause G1 cell cycle arrest. GGPP/mevalonate, but not FPP were able to rescue the effects of statin [124] | Induction of apoptosis |
| Pitavastatin | 10 μM | Breast cancer and melanoma tumors | Enhance effects of radiation on cellular senescence of radiation. 5 mM mevalonate rescued the effects of Pitavastatin [30] | Induction of apoptosis |
| Simvastatin | 0.1 µM |
Primary prostatic Normal epithelial cell lines RWPE-1 and PWR-1E |
Exert cytostatic and senescent effects and partially induced apoptosis [125] | Induction of apoptosis |
| Simvastatin | 10 μM |
Primary prostatic normal epithelial cell lines RWPE-1 and PWR-1E cancer cells |
In contrast, simvastatin had a cytotoxic effect both on normal and cancer cells. Combination of LDL-C and mevalonate rescued the effects of statin [125] | Induction of apoptosis |
| Lovastatin | 0.3 µM | Prostate cancer cells: PC-3, DU-145, LNCaP | Senescence and G1 cell cycle arrest. GGPP, mevalonate, constitutively active RhoA (caRhoA) rescued the effects of lovastatin [124] | Induction of apoptosis |
In addition to inhibiting cholesterol synthesis, statins can also reduce non-sterol products of the mevalonate pathway, such as isoprenoids [23–30]. Farnesyl pyrophosphate (FPP) and geranylgeranyl pyrophosphate (GGPP), are the major isoprenoids involved in prenylation of proteins [31]. Several proteins ranging from heterotrimeric G protein subunits to nuclear lamins have been found to undergo prenylation. However, the Ras superfamily of small GTPases is the most widely-known group of these proteins [32]. In this paper, we discuss some of the most important signaling pathways that are modulated by statins.
The Ras group of proteins plays a significant role in cell growth, proliferation, and survival [33–36]. Two major Ras-driven signaling cascades are the MAPK (Raf/MEK/ERK) and PI3K/Akt/mTOR pathways, both of which regulate cellular proliferation and differentiation [34, 37–40]. The MAPK pathway promotes proliferation by activating several transcription factors and kinases including AP-1, Myc, Jun, Fos, p90RSK1, Elk, Ets, and MNK [34, 41–44]. Moreover, it regulates survival and apoptosis by modulating the activity of several proteins like including JNK, SAPK, 14-3-3, and NF-Kβ [45, 46].
The second major downstream pathway of Ras is PI3K/Akt [34, 47]. PI3K is a lipid kinase that consists of a regulatory subunit, p85, and a catalytic subunit, p110. Ras interacts with p110 and recruits it to the cell membrane resulting in the activation of PI3-K, which in turn recruits phosphoinositide-dependent kinase-1 (PDK1). PDK1 or the mammalian target of rapamycin mTOR can then activate Akt [48]. Consequently, phosphorylated Akt drives several pathways that promote cellular growth and evasion of apoptosis [40] (Fig. 1).
Fig. 1.

Activation of PI3K: Binding of an external ligand leads to the dimerization of receptor monomers and the heterologous autophosphorylation. Depending on the receptor, different proteins may bind to a phosphorylated domain. The insulin receptor substrate-1(IRS-1) binds to the activated IGF-1 receptor. IRS-1 serves as a binding and activation site for PI3K. In addition, PI3K may bind directly to a phosphorylated receptor tyrosine kinase, a completely different mechanism of PI3K. Activation begins with the small membrane bound GTPase Ras. By binding to active GTP-bound Ras, PI3K is activated, and migrates to the inner side of the cell membrane where it binds to phosphatidylinositol bisphosphate or PIP2. PI3K can phosphorylate PIP2 to PIP3, which can activate protein kinase B, also known as Akt. Akt binds to BAX and hinders its ability to form pores in the outer mitochondrial membrane, thus suppressing apoptosis. Moreover, Akt phosphorylates BAD leading to the release and activation of death inhibitory protein, which, as the name implies, inhibits apoptosis. Akt can also promote protein synthesis by first activating Rheb, which activates mTOR. mTOR itself interacts with and activates the translation factor S6K, thereby promoting mRNA translation and protein synthesis. In addition, Akt-induced phosphorylation of FOXO promotes the transfer of ubiquitin peptides onto the protein causing FOXO to undergoe proteasomal degradation
Extensive studies have established the effects of statins on cell cycle progression, apoptosis, and senescence in different types of cells especially cancer cells and endothelial cells [49]. Interestingly, the cell-context appears to be a critical factor in determining the effect of statins on cell behavior. For instance, in endothelial cells, statins inhibit cytostatic and pro-apoptotic effects (see Table 1), whereas in cancer cells, the opposite effects are reported (Table 2). While the type of cells is an important parameter to consider in these discordant results, the concentrations/doses used could be the factor that tips the balance. Indeed, there is a large discrepancy among statin concentrations used in various studies. Pleiotropic effects of statins have been reported to appear at concentrations of 1–50 μM. However, at therapeutic doses, the mean concentration of statins in human serum only ranges from 1 to 15 nM. Additionally, 95–99% of statins in the blood are bound to proteins, therefore only 0.01–0.5 nM of them is the free fraction, and hence the pharmacologically active. Similarly, the therapeutic dosage in humans is approximately 0.1–1 mg/kg bodyweight, while doses of 1–100 or even 500 mg/kg body weight have been used in most studies in rodents. Indeed, statins concentrations used to induce pleiotropic effects in animal studies are much greater than those used in patients, reaching 1000 fold higher in some studies [50]. In this review, we discuss the effects of statins on endothelial cells (as models of non-cancerous cells) as well as on a battery of cancer cells.
Cytoprotective effects of statins
In cells of cardiovascular origins such as endothelial cells, statins appear to protect against oxidative damages and apoptosis through a series of mechanisms.
Cytoprotective effects of statins via reduction of free radicals’ production
The antioxidative activities of statins have been primarily ascribed to downregulation of ROS-generating enzymes such as NADPH oxidase and upregulating HO-1 rather than superoxide scavenging [51].
Halting NADPH oxidase activity
ROS play critical roles in modulating various cellular processes and phenotypes [52, 53]. NADPH oxidase is considered the primary source of ROS, particularly superoxide radicals, in a multitude of cells [54]. The functional structure of NADPH oxidase consists of two membrane-bound components: gp91phox (Nox2) and p22phox, in addition to other cytosolic regulatory subunits including p40phox, p47phox, p67phox, and Rac. Phosphorylation and subsequent membrane translocation of the cytosolic subunits followed by interaction with p22phox and Nox2 are crucial steps in the activation of NADPH oxidase [55, 56]. The small GTP-binding protein Rac-1, a member of the Rho protein subfamily, plays a pivotal role in the assemblage and activation of the NADPH oxidase [57, 58]. In various cells including macrophages, human and rat smooth muscle cells, human vascular endothelial cells, cardiovascular cells, neuronal cells, cancer cells, and THP-1 derived monocytes statins have been shown by blocking prenylation of Ras and Rho families prevent the formation of NADP-oxidase subunits into a functional unit [59–67]. This reduces the production of ROS, and hence, a cytoprotective effect is achieved.
Induction of HO-1 activity
The role of HO-1 as a cardio-vasculoprtective player has been established. It appears that by virtue of its ability to induce anti-inflammatory, anti-proliferative, anti-apoptotic, and antioxidative activities in the vasculature, HO-1 protects vessels from a multitude of pathologic conditions, prime of which is atherosclerosis [61]. HO-1 acts by catalyzing the oxidative degradation of heme to carbon monoxide, biliverdin, and free iron [68]. Importantly, statins have been shown to induce HO-1 activity, apparently via p38- and PI3K/Akt-dependent mechanism, as inhibition of these two pathways seem to abrogate statin-induced HO-1 expression. [59–61]. This is in line with other reports showing that statins act via PI3K/Akt to stabilize HO-1 mRNA [62, 65].
Cytoprotective and cytotoxic effects of statins via activation PI3K/Akt in endothelial cells
PI-3 K/Akt and AMPK are two extensively studied pathways underpinning the anti-apoptotic effects of statins in noncancerous cells such as EPCs and HUVEC. These cytoprotective effects of statins have been studied particularly in endothelial cells, largely because of the ability of these drugs to modulate angiogenesis. In regard to concentrations used, it is important to cautions that statins act as a double-edged sword in endothelial cells and angiogenesis [69]. While nanomolar concentrations of statins induce angiogenesis in human umbilical vein endothelial cells, micromolar concentrations exert reverse effects [70], which has been attributed to the ability of these drugs to inhibit proliferation and migration as well as inducing apoptosis in these cells [71, 72]. Importantly, Akt appears to mediate these effects via a endothelial nitric oxide synthase (eNOS), a key regulator of vascular homeostasis [73–76]. Ineed, activated Akt stimulates post-transcriptional phosphorylation, hence activation, of eNOS which in turn activates the VEGF-mediated migration of mature endothelial cells and subsequently stimulates angiogenesis [69, 70, 77–79] (see Fig. 2).
Fig. 2.

Low doses of statins (left-top) potentiate HO-1 activity via PI3K/Akt. In addition, PI3K/Akt stimulates eNOS, thereby causing increased catalase activity which prevents free radical-induced senescence. Moreover, eNOS upregulates VEGF, which is a main angiogenic factor. Low doses of statins induce cell cycle progression and reduce senescence by regulating the expression of several proteins including p27Kip1. They also activate caMKKβ which drives the activation of AMPK and LKB1. In addition, by virtue of its ability to stimulate Rac 1, AMPK can also regulate NAPDH oxidase and eNOS-mediated angiogenesis. An alternative pathway by which low doses of statins induce Rac-1 is by inhibiting Rho. On the other hand, high doses (top-right) induce apoptosis by inhibiting PI3K/Akt-modulated activity of death inhibitory protein (DIP) and BAX. Moreover, statins at high concentrations, can also induce apoptosis by inhibiting Rho-induced Bcl-2 or TNF-α-induced NFkB
Clinically relevant doses (0.01 to 0.1 µM) of atorvastatin activate endothelial Ras and promote Akt and eNOS phosphorylation activation. In contrast, higher concentrations (> 0.1 µM) of atorvastatin block angiogenesis and migration of endothelial cells by inducing apoptosis [69, 80].
In vitro, atorvastatin or mevastatin (0.1, 0.05, and 0.01 µM) inhibits the onset of endothelial progenitor cell senescence in a dose-dependent manner. Moreover, atorvastatin increases EPC proliferation and colony-forming capacity. GGPP, mevalonate, and FPP reverse the senescence inhibitory effect of atorvastatin. Contextually, atorvastatin’s anti-senescence effect appears to be to an increase in the expression of cell cycle-promoting genes including cyclins as well as suppression of p27Kip1, a cell cycle-inhibitory protein [81] (Figure 2). Likewise, pitavastatin induces migration, proliferation, and viability of human microvascular endothelial cells (HMVECs) at low concentrations (0.01 mM) but suppresses these cellular processes at higher concentrations (1 mM) [59, 82]. At nanomolar concentrations, atorvastatin, pravastatin, and pitavastatin suppressed hydrogen peroxide-induced senescence in human umbilical vein endothelial cells (HUVECs). This effect occurred via statin’s ability to activate Akt and subsequently upregule eNOS, SIRT1, and catalase. [83] (Figure 2).
Despite the aforementioned evidence, high concentrations of statins can evoke endothelial release of VEGF and an increase in endothelial apoptosis, most probably by inhibition of geranylgeranylation of Rho, which is known to play a paradoxical role in angiogenesis. Indeed, Rho modulates the activity of VEGFR-2, which is employed by VEGF to activate Rho GTPases [71, 72, 84]. Rho GTPases can then act via regulatory and effector proteins, most notable of which is ROCK, to influence angiogenic processes [85, 86] (Fig. 3).
Fig. 3.
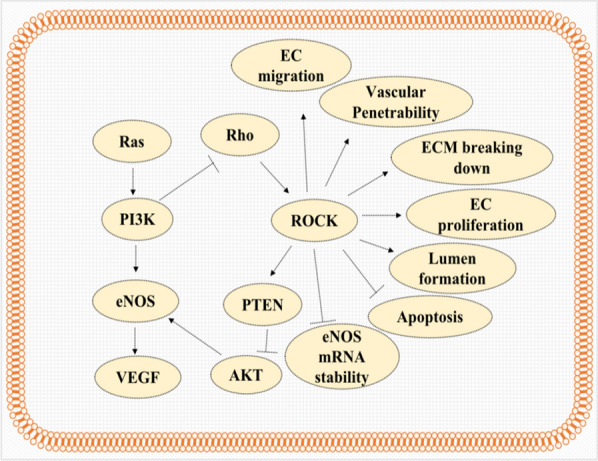
Role of Rho in angiogenesis in Endothelial cells. The activation of the Rho/ROCK pathway via induction of phosphatase activity of PTEN, which in turn inhibits the activation of Akt and via reducing the stability of eNOS mRNA abolishes the acute activation of angiogenesis by statins; however, this pathway induces all process involved in angiogenesis including endothelial (ES) migration, vascular permeability, extracellular matrix (ECM) degradation, endothelial cell (EC) proliferation, lumen formation, and inhibition of apoptosis; therefore in long term period, Rho/Rock pathway may increase angiogenesis by statins
Interestingly, statins upregulate the expression of eNOS via inhibition of Rho activity [77, 87–89]. Despite the angiogenic functions, Rho GTPase may downregulate eNOS expression via destabilizing its mRNA. As such, statins upregulate the expression of eNOS by prolonging its mRNA half-life through inhibiting the Rho/ROCK pathway [86]. Contextually, inhibition of the Rho/ROCK pathway activates PI3K/Akt and leads to the rapid phosphorylation (acute) activation of eNOS [86, 90]. This suppression of PI3K/Akt is assumed to arise from decreasing PTEN activity, since RhoA/ROCK is required for intracellular localization and phosphorylation of PTEN, which in turn is crucial for the phosphatase activity of PTEN that antagonizes the PI3K/Akt pathway [91]. Therefore, despite its pro-angiogenic role, the Rho/ROCK pathway may negatively regulate the acute activation of angiogenesis via two distinct mechanisms, namely, eNOS expression and eNOS activity [86].
Cytoprotective effects of statins through activating AMPK in endothelial cells
AMPK (AMP-activated protein kinase) is a cellular energy sensor that is activated in response to an increase in the intracellular AMP: ATP ratio. It stimulates ATP- producing catabolic pathways and inhibits ATP- consuming anabolic pathways. For instance, AMPK inhibits fatty acid and cholesterol synthesis through direct phosphorylation of anabolic enzymes including Acetyl-CoA carboxylase (ACC) and HMG-CoA reductase (HMGR) [92]. While the AMPK pathway is traditionally thought of as a regulator of metabolism, recent studies have demonstrated that AMPK may play a significant part in maintaining normal endothelial function through AMP-independent activation of AMPK [75, 93–96]. LKB1 and the calcium/calmodulin-dependent kinase (CaMKK) are protein kinases that phosphorylate AMPK. The pathways that regulate LKB1 remain elusive. CaMKK seems to play a key role in modulating effects of statins by phosphorylation of LKB1 and AMPK, as well as by direct activation of Rac-1 [97](see Fig. 2).
In cultured vascular endothelial cells (e.g. ovine aortic endothelial cells), simvastatin (10 μM) increased activity of Rac-1 via CaMKKβ, AMPK and LKB1. The importance of Rac-1 aictivation becomes evidence in light of the notion that Rac-1 plays a key role in eNOS activation. Indeed, siRNA-mediated AMPK knockdown was shown to suppress Rac-1 activation and subsequently prevents activation of eNOS. Interestingly, it has been shown that Rac-1 in turn regulates LKB1 phosphorylation [98].
Activation of small GTPases like Rho and Rac-1 requires geranylgeranylation and subsequent translocation to the cell membrane. Interestingly, some studies showed that statins, by inhibiting, prenylation, paradoxically activate Rac-1 [99]. For example, simvastatin (10 μM) caused a 34-fold increase in Rac-1 activation in endothelial cells despite its inhibition of prenylation and activity of Rho [98]. One plausible explanation for this paradoxical observation is that statins, by reducing geranylgeranylation, preferentially inhibit Rho, which is a tonic inhibitor of Rac-1. This inhibition effectively leads to Rac-1 activation [100]. Alternatively, it is possible that statins dissociate the inhibitory interaction of Rac-1 with guanine nucleotide dissociation inhibitors (RhoGDI) [98].
Cytotoxic (apoptotic) and cytostatic effects of statins in cancer cells
Statins can induce apoptosis via different mechanisms [39]. Indeed, they can activate the intrinsic pathway of apoptosis via disturbing the mitochondrial membrane potential and releasing the second mitochondria-derived activator of caspases (Smac/DIABLO) [116]. Moreover, upregulation of proapoptotic proteins Bax and Bim besides the downregulation of antiapoptotic protein Bcl-2 are considered the main mechanisms of induction of apoptosis by statins. Statins have also been revealed to activate procaspase 3, 7, 8, and 9 [78, 116–118].
The effects of statins’ concentrations on Bcl-2 have been studied widely. Indeed, in high concentrations, statins may induce apoptosis by reducing Bcl-2 level, while in lower concentrations, they tend to suppress apoptosis and cell death by increasing Bcl-2 expression [119]. (Fig. 2). As is known, the expression of Bcl-2 gene has been shown to be upregulated by NF-κB. In this context, it is noteworthy that high concentrations of simvastatin (50 μM) were found to reduce Bcl-2 protein levels through inhibition of TNF-α, protein that is for NF-κB activation [120]. Similarly, simvastatin (20 μM) was shown to reduce Bcl-2 mRNA and induce apoptosis in a battery of human cancer cell lines including MCF7 breast cancer cells, NCI-N87 human gastric cancer cells, HepG2 human hepatocellular carcinoma and non-small cell lung carcinoma (NCH lung) cells. However, normal cells (SAEC human normal small airway epithelial cells) did not seem to exhibit the same response [121]. Other lipophilic statins (1 μM for cerivastatin and 10 μM for atorvastatin and simvastatin) promoted apoptotic programs by inhibiting RhoA activity, which caused decreased phospho-p42/p44-MAPK and Bcl-2 levels [122].
Cytostatic effects of statins on cancer cells have also been reported. These effects occur mainly via the upregulation of cell cycle inhibitors including p21WAF1/CIP1 or p27KIP1 [42]. Simvastatin (10 μM) downregulates the transcriptional activity of ATF-2 and c-jun, which then causes a dramatic decrease in the proliferative capacity of glioma cells [123].
There are many studies indicating the major mechanism underlying the cytotoxic and cytostatic effects of statins on cancer cells arise from reduction of geranylgeranyl pyrophosphate (GGPP) which is crucial for membrane localization and activation of small G proteins like Rho. These studies report that supplying cells with mevalonate or GGPP reverses the inhibitory effect of statins and prevent induction of apoptosis or cell cycle arrest by statins. Lovastatin (0.3 µM) was shown to induce senescence and G1 cell cycle arrest in human prostate cancer cells, and supplementation with GGPP or mevalonate, but not FPP, reversed cell cycle arrest and senescence. In addition, constitutively active RhoA (caRhoA) reversed lovastatin-induced senescence in caRhoA-transfected PC-3 cells. This indicates that statins could act through the inhibition of Rho activity to induce cytotoxic effects, at least in this cell line [124].
Pitavastatin (10 μM) enhanced the effects of radiation on cellular senescence in breast cancer and melanoma tumors. However, 5 mM mevalonic acid was sufficient to restore these effects of pravastatin [30]. Likewise, simvastatin (100 nM) was shown to exert cytostatic and senescent effects and partially induce apoptosis in prostate epithelial cells. In contrast, 10 μM simvastatin had a cytotoxic effect both on normal and cancer cells. A combination of LDL-cholesterol and mevalonate supplementation was able to rescue the cytostatic/cytotoxic of 10 μM simvastatin [125]. Others report that lovastatin (0.3 µM) causes senescence and G1 cell cycle arrest in human prostate cancer cells. GGPP or mevalonate, but not FPP, reversed the cell cycle arrest and cell senescence induced by lovastatin. Moreover, constitutively active RhoA (caRhoA) abolishes the senescence induced by statins in caRhoA-transfected PC-3 cells [124]. Table 2 provides highlights and an overview of further studies regarding the effect of statins on cancer cells.
Conclusion and perspective
The effects of statins on endothelial cells can be either protective via boosting cytoprotective effects such as antioxidant levels in the cells, or destructive via inhibition of growth signaling and induction of apoptosis. Although the type of effect depends on the applied concentration, an absolute threshold/dose could not be definitively determined. However, it seems that at lower doses -particularly in nanomolar concentrations—statins act more in line with cytoprotection rather than halting the cell cycle or inducting apoptosis in ECs. Moreover, it seems that the effects of different concentrations of statins on endothelial cells largely depends on their effects on the Rho activity. Negative feedback mechanisms may be yet another underlying mechanism that could explain the contradictory effects of statins in cancer cells versus EPCs. Statins block the production of isoprenoid units required for the prenylation of proteins, which ultimately results in the deceleration of cell cycle progression. The reduced rate of cell cycle progression is sensed by the cell, which then reduces the expression and activity of cell cycle inhibitory proteins as a feedback response. Afterward, the cell cycle is regulated by the positive inducers (proto-oncogenes) like the Ras superfamily rather than negative regulators (tumor-suppressors) such as p53 and p27.
Cancer cells normally have a high proliferation rate and, as a result, there may be permanently increased levels of cytostatic proteins; however, cancer cells are not responding to high levels of these proteins. Therefore, low concentrations of statins probably are not able to exert the same effects in EPCs and cancer cells. However, how low concentrations of statins can induce apoptosis in cancer cells is a question yet to be answered. As a possible mechanism, cancer cells may be more dependent on cell cycle stimulatory factors and even the low concentration of statins can impose cytostatic effects. To determine the precise effects of statins on cancer cells, the effects of the nanomolar concentrations of these medications on these cells should be further studied.
Author contributions
Conceptualization: YA, AS; Writing-original draft: YA, JKF, DG; Writing-review and editing: AHE, AS; Approval of the final content: all authors. All authors read and approved the final manuscript.
Funding
No funding was received for this study.
Availability of data and materials
No primary data exists for this review article.
Declarations
Ethics approval and consent to participate
Not applicable.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Yasin Ahmadi, Email: ahmadi.bchemistry@yahoo.com.
Amirhossein Sahebkar, Email: amir_saheb2000@yahoo.com.
References
- 1.Althanoon Z, Faisal IM, Ahmad AA, Merkhan MM, Merkhan MM. Pharmacological aspects of statins are relevant to their structural and physicochemical properties. Syst Rev Pharm. 2020;11(7):167–171. [Google Scholar]
- 2.Niedzielski M, Broncel M, Gorzelak-Pabiś P, Woźniak E. New possible pharmacological targets for statins and ezetimibe. Biomed Pharmacother. 2020;129:110388. doi: 10.1016/j.biopha.2020.110388. [DOI] [PubMed] [Google Scholar]
- 3.Miller BF, Thyfault JP. Exercise-pharmacology interactions: metformin, statins, and healthspan. Physiology. 2020;35(5):338–347. doi: 10.1152/physiol.00013.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ahmadi Y, Ghorbanihaghjo A, Argani H. The effect of statins on the organs: similar or contradictory? J Cardiovasc Thorac Res. 2017;9(2):64–70. doi: 10.15171/jcvtr.2017.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bahrami A, Bo S, Jamialahmadi T, Sahebkar A. Effects of 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors on ageing: Molecular mechanisms. Ageing Res Rev. 2020;58:101024. doi: 10.1016/j.arr.2020.101024. [DOI] [PubMed] [Google Scholar]
- 6.Bahrami A, Parsamanesh N, Atkin SL, Banach M, Sahebkar A. Effect of statins on toll-like receptors: a new insight to pleiotropic effects. Pharmacol Res. 2018;135:230–238. doi: 10.1016/j.phrs.2018.08.014. [DOI] [PubMed] [Google Scholar]
- 7.Kouhpeikar H, Delbari Z, Sathyapalan T, Simental-Mendia LE, Jamialahmadi T, Sahebkar A. The Effect of Statins through Mast Cells in the Pathophysiology of Atherosclerosis: a Review. Curr Atheroscler Rep. 2020;22(5):19. doi: 10.1007/s11883-020-00837-9. [DOI] [PubMed] [Google Scholar]
- 8.Koushki K, Shahbaz SK, Mashayekhi K, Sadeghi M, Zayeri ZD, Taba MY, et al. Anti-inflammatory action of statins in cardiovascular disease: the role of inflammasome and Toll-Like receptor pathways. Clin Rev Allergy Immunol. 2021;60(2):175–199. doi: 10.1007/s12016-020-08791-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Parizadeh SM, Azarpazhooh MR, Moohebati M, Nematy M, Ghayour-Mobarhan M, Tavallaie S, et al. Simvastatin therapy reduces prooxidant-antioxidant balance: results of a placebo-controlled cross-over trial. Lipids. 2011;46(4):333–340. doi: 10.1007/s11745-010-3517-x. [DOI] [PubMed] [Google Scholar]
- 10.Sahebkar A, Kotani K, Serban C, Ursoniu S, Mikhailidis DP, Jones SR, et al. Statin therapy reduces plasma endothelin-1 concentrations: a meta-analysis of 15 randomized controlled trials. Atherosclerosis. 2015;241(2):433–442. doi: 10.1016/j.atherosclerosis.2015.05.022. [DOI] [PubMed] [Google Scholar]
- 11.Sahebkar A, Serban C, Mikhailidis DP, Undas A, Lip GY, Muntner P, et al. Association between statin use and plasma D-dimer levels. A systematic review and meta-analysis of randomised controlled trials. Thromb Haemost. 2015;114(3):546–57. doi: 10.1160/TH14-11-0937. [DOI] [PubMed] [Google Scholar]
- 12.Sahebkar A, Serban C, Ursoniu S, Mikhailidis DP, Undas A, Lip GY, et al. The impact of statin therapy on plasma levels of von Willebrand factor antigen. Systematic review and meta-analysis of randomised placebo-controlled trials. Thromb Haemost. 2016;115(3):520–32. doi: 10.1160/th15-08-0620. [DOI] [PubMed] [Google Scholar]
- 13.Serban C, Sahebkar A, Ursoniu S, Mikhailidis DP, Rizzo M, Lip GY, et al. A systematic review and meta-analysis of the effect of statins on plasma asymmetric dimethylarginine concentrations. Sci Rep. 2015;5:9902. doi: 10.1038/srep09902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bland AR, Payne FM, Ashton JC, Jamialahmadi T, Sahebkar A. The cardioprotective actions of statins in targeting mitochondrial dysfunction associated with myocardial ischaemia-reperfusion injury. Pharmacol Res. 2022;175:105986. doi: 10.1016/j.phrs.2021.105986. [DOI] [PubMed] [Google Scholar]
- 15.Mahjoubin-Tehran M, De Vincentis A, Mikhailidis DP, Atkin SL, Mantzoros CS, Jamialahmadi T, et al. Non-alcoholic fatty liver disease and steatohepatitis: State of the art on effective therapeutics based on the gold standard method for diagnosis. Mol Metab. 2021;50:101049. doi: 10.1016/j.molmet.2020.101049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sahebkar A, Chew GT, Watts GF. Recent advances in pharmacotherapy for hypertriglyceridemia. Prog Lipid Res. 2014;56:47–66. doi: 10.1016/j.plipres.2014.07.002. [DOI] [PubMed] [Google Scholar]
- 17.Shakour N, Ruscica M, Hadizadeh F, Cirtori C, Banach M, Jamialahmadi T, et al. Statins and C-reactive protein: in silico evidence on direct interaction. Arch Med Sci. 2020;16(6):1432–1439. doi: 10.5114/aoms.2020.100304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sohrevardi SM, Nasab FS, Mirjalili MR, Bagherniya M, Tafti AD, Jarrahzadeh MH, et al. Effect of atorvastatin on delirium status of patients in the intensive care unit: a randomized controlled trial. Arch Med Sci. 2021;17(5):1423–1428. doi: 10.5114/aoms.2019.89330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Di Bello E, Zwergel C, Mai A, Valente S. The innovative potential of statins in cancer: new targets for new therapies. Front Chem. 2020;8:516. doi: 10.3389/fchem.2020.00516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Climent E, Benaiges D, Pedro-Botet J. Hydrophilic or lipophilic statins? Fron Cardiovasc Med. 2021;8:491. doi: 10.3389/fcvm.2021.687585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Beckwitt CH, Shiraha K, Wells A. Lipophilic statins limit cancer cell growth and survival, via involvement of Akt signaling. PLoS ONE. 2018;13(5):e0197422. doi: 10.1371/journal.pone.0197422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ahmadi Y, Mahmoudi N, Yousefi B, Karimian A. The effects of statins with a high hepatoselectivity rank on the extra-hepatic tissues. New Funct Stat Pharmacol Res. 2020;152:104621. doi: 10.1016/j.phrs.2019.104621. [DOI] [PubMed] [Google Scholar]
- 23.Liao JK, Laufs U. Pleiotropic effects of statins. Annu Rev Pharmacol Toxicol. 2005;45:89–118. doi: 10.1146/annurev.pharmtox.45.120403.095748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhou Q, Liao JK. Pleiotropic effects of statins. Circ J. 2010;74(5):818–826. doi: 10.1253/circj.CJ-10-0110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Davignon J. Beneficial cardiovascular pleiotropic effects of statins. Circulation. 2004 doi: 10.1161/01.CIR.0000131517.20177.5a. [DOI] [PubMed] [Google Scholar]
- 26.Oesterle A, Laufs U, Liao JK. Pleiotropic effects of statins on the cardiovascular system. Circ Res. 2017;120(1):229–243. doi: 10.1161/CIRCRESAHA.116.308537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kavalipati N, Shah J, Ramakrishan A, Vasnawala H. Pleiotropic effects of statins. Indian J Endocrinol Metab. 2015;19(5):554. doi: 10.4103/2230-8210.163106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sadowitz B, Maier KG, Gahtan V. Basic science review: Statin therapy-Part I: the pleiotropic effects of statins in cardiovascular disease. Vasc Endovasc Surg. 2010;44(4):241–251. doi: 10.1177/1538574410362922. [DOI] [PubMed] [Google Scholar]
- 29.Bedi O, Dhawan V, Sharma P, Kumar P. Pleiotropic effects of statins: new therapeutic targets in drug design. Naunyn Schmiedebergs Arch Pharmacol. 2016;389(7):695–712. doi: 10.1007/s00210-016-1252-4. [DOI] [PubMed] [Google Scholar]
- 30.Palaniswamy C, Selvaraj DR, Selvaraj T, Sukhija R. Mechanisms underlying pleiotropic effects of statins. Am J Ther. 2010;17(1):75–78. doi: 10.1097/MJT.0b013e31819cdc86. [DOI] [PubMed] [Google Scholar]
- 31.McTaggart S. Isoprenylated proteins. Cell Mol Life Sci CMLS. 2006;63(3):255–267. doi: 10.1007/s00018-005-5298-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jeong A, Suazo KF, Wood WG, Distefano MD, Li L. Isoprenoids and protein prenylation: implications in the pathogenesis and therapeutic intervention of Alzheimer's disease. Crit Rev Biochem Mol Biol. 2018;53(3):279–310. doi: 10.1080/10409238.2018.1458070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Downward J. Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer. 2003;3(1):11–22. doi: 10.1038/nrc969. [DOI] [PubMed] [Google Scholar]
- 34.Macaluso M, Russo G, Cinti C, Bazan V, Gebbia N, Russo A. Ras family genes: an interesting link between cell cycle and cancer. J Cell Physiol. 2002;192(2):125–130. doi: 10.1002/jcp.10109. [DOI] [PubMed] [Google Scholar]
- 35.Raghavaraju G, Liu H-S. Ha-ras Oncogene and Anticancer Drug Resistance. Genomic Med, Biomark, Health Sci. 2011;3(1):39–48. doi: 10.1016/S2211-4254(11)60006-X. [DOI] [Google Scholar]
- 36.Wennerberg K, Rossman KL, Der CJ. The Ras superfamily at a glance. J Cell Sci. 2005;118(5):843–846. doi: 10.1242/jcs.01660. [DOI] [PubMed] [Google Scholar]
- 37.Nussinov R, Tsai CJ, Jang H. Oncogenic Ras Isoforms Signaling Specificity at the Membrane. Cancer Res. 2018;78(3):593–602. doi: 10.1158/0008-5472.CAN-17-2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stephen AG, Esposito D, Bagni RK, McCormick F. Dragging ras back in the ring. Cancer Cell. 2014;25(3):272–281. doi: 10.1016/j.ccr.2014.02.017. [DOI] [PubMed] [Google Scholar]
- 39.Wong WW, Dimitroulakos J, Minden M, Penn L. HMG-CoA reductase inhibitors and the malignant cell: the statin family of drugs as triggers of tumor-specific apoptosis. Leukemia. 2002;16(4):508. doi: 10.1038/sj.leu.2402476. [DOI] [PubMed] [Google Scholar]
- 40.Zenonos K, Kyprianou K. RAS signaling pathways, mutations and their role in colorectal cancer. World J Gastrointest Oncol. 2013;5(5):97. doi: 10.4251/wjgo.v5.i5.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cox AD, Der CJ. Ras family signaling: therapeutic targeting. Cancer Biol Ther. 2002;1(6):599–606. doi: 10.4161/cbt.306. [DOI] [PubMed] [Google Scholar]
- 42.Gazzerro P, Proto MC, Gangemi G, Malfitano AM, Ciaglia E, Pisanti S, et al. Pharmacological actions of statins: a critical appraisal in the management of cancer. Pharmacol Rev. 2012;64(1):102–146. doi: 10.1124/pr.111.004994. [DOI] [PubMed] [Google Scholar]
- 43.Steinmüller L, Cibelli G, Vinson C, Thiel G. Regulation and composition of activator protein 1 (AP-1) transcription factors controlling collagenase and c-Jun promoter activities. Biochem J. 2001;360(3):599–607. doi: 10.1042/bj3600599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Karin M, Liu Z-g, Zandi E. AP-1 function and regulation. Curr Opin cell Biol. 1997;9(2):240–6. doi: 10.1016/S0955-0674(97)80068-3. [DOI] [PubMed] [Google Scholar]
- 45.McCubrey JA, Steelman LS, Abrams SL, Chappell WH, Russo S, Ove R, et al. Emerging MEK inhibitors. Expert Opin Emerg Drugs. 2010;15(2):203–223. doi: 10.1517/14728210903282760. [DOI] [PubMed] [Google Scholar]
- 46.Molina JR, Adjei AA. The Ras/Raf/MAPK pathway. J Thorac Oncol. 2006;1(1):7–9. doi: 10.1016/S1556-0864(15)31506-9. [DOI] [PubMed] [Google Scholar]
- 47.Brown AJ. Cholesterol, statins and cancer. Clin Exp Pharmacol Physiol. 2007;34(3):135–141. doi: 10.1111/j.1440-1681.2007.04565.x. [DOI] [PubMed] [Google Scholar]
- 48.Ahmadi Y, Ghorbanihaghjo A, Argani H. The balance between induction and inhibition of mevalonate pathway regulates cancer suppression by statins: a review of molecular mechanisms. Chem Biol Interact. 2017;273:273–285. doi: 10.1016/j.cbi.2017.06.026. [DOI] [PubMed] [Google Scholar]
- 49.Ayad MT, Taylor BD, Menon R. Regulation of p38 mitogen-activated kinase-mediated fetal membrane senescence by statins. Am J Reprod Immunol. 2018;80(4):e12999. doi: 10.1111/aji.12999. [DOI] [PubMed] [Google Scholar]
- 50.Björkhem-Bergman L, Lindh JD, Bergman P. What is a relevant statin concentration in cell experiments claiming pleiotropic effects? Br J Clin Pharmacol. 2011;72(1):164. doi: 10.1111/j.1365-2125.2011.03907.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Inoue I, Goto S-i, Mizotani K, Awata T, Mastunaga T, Kawai S-i, et al. Lipophilic HMG-CoA reductase inhibitor has an anti-inflammatory effect: reduction of mRNA levels for interleukin-1β, interleukin-6, cyclooxygenase-2, and p22phox by regulation of peroxisome proliferator-activated receptor α (PPARα) in primary endothelial cells. Life Sci. 2000;67(8):863–76. doi: 10.1016/S0024-3205(00)00680-9. [DOI] [PubMed] [Google Scholar]
- 52.Badran A, Nasser SA, Mesmar J, El-Yazbi AF, Bitto A, Fardoun MM, et al. Reactive oxygen species: modulators of phenotypic switch of vascular smooth muscle cells. Int J Mol Sci. 2020 doi: 10.3390/ijms21228764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shaito A, Aramouni K, Assaf R, Parenti A, Orekhov A, Yazbi AE, et al. Oxidative stress-induced endothelial dysfunction in cardiovascular diseases. Front Biosci (Landmark Ed) 2022;27(3):105. doi: 10.31083/j.fbl2703105. [DOI] [PubMed] [Google Scholar]
- 54.Paravicini TM, Touyz RM. NADPH oxidases, reactive oxygen species, and hypertension. Diabetes Care. 2008;31(Supplement 2):S170–S180. doi: 10.2337/dc08-s247. [DOI] [PubMed] [Google Scholar]
- 55.Minakami R, Sumimoto H. Phagocytosis-coupled activation of the superoxide-producing phagocyte oxidase, a member of the NADPH oxidase (nox) family. Int J Hematol. 2006;84(3):193–198. doi: 10.1532/IJH97.06133. [DOI] [PubMed] [Google Scholar]
- 56.Vignais P. The superoxide-generating NADPH oxidase: structural aspects and activation mechanism. Cell Mol Life Sci. 2002;59(9):1428–1459. doi: 10.1007/s00018-002-8520-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wassmann S, Laufs U, Bäumer AT, Müller K, Konkol C, Sauer H, et al. Inhibition of geranylgeranylation reduces angiotensin II-mediated free radical production in vascular smooth muscle cells: involvement of angiotensin AT1 receptor expression and Rac1 GTPase. Mol Pharmacol. 2001;59(3):646–654. doi: 10.1124/mol.59.3.646. [DOI] [PubMed] [Google Scholar]
- 58.Sawma T, Shaito A, Najm N, Sidani M, Orekhov A, El-Yazbi AF, et al. Role of RhoA and Rho-associated kinase in phenotypic switching of vascular smooth muscle cells: implications for vascular function. Atherosclerosis. 2022;358:12–28. doi: 10.1016/j.atherosclerosis.2022.08.012. [DOI] [PubMed] [Google Scholar]
- 59.Gueler F, Park J-K, Rong S, Kirsch T, Lindschau C, Zheng W, et al. Statins attenuate ischemia-reperfusion injury by inducing heme oxygenase-1 in infiltrating macrophages. Am J Pathol. 2007;170(4):1192–1199. doi: 10.2353/ajpath.2007.060782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Moneta G. Simvastatin Induces Heme Oxygenase-1: a novel mechanism of vessel protection Lee TS, Chang CC, Zhu Y, et al (Univ of Californa, Riverside) Circulation 110. Year Book Vasc Surg. 2006;2006:19–20. doi: 10.1016/S0749-4041(08)70021-6. [DOI] [PubMed] [Google Scholar]
- 61.Ali F, Hamdulay S, Kinderlerer A, Boyle J, Lidington E, Yamaguchi T, et al. Statin-mediated cytoprotection of human vascular endothelial cells: a role for Kruppel-like factor 2-dependent induction of heme oxygenase-1. J Thromb Haemost. 2007;5(12):2537–2546. doi: 10.1111/j.1538-7836.2007.02787.x. [DOI] [PubMed] [Google Scholar]
- 62.Hinkelmann U, Grosser N, Erdmann K, Schröder H, Immenschuh S. Simvastatin-dependent up-regulation of heme oxygenase-1 via mRNA stabilization in human endothelial cells. Eur J Pharm Sci. 2010;41(1):118–124. doi: 10.1016/j.ejps.2010.05.021. [DOI] [PubMed] [Google Scholar]
- 63.Wu M-L, Ho Y-C, Yet S-F. A central role of heme oxygenase-1 in cardiovascular protection. Antioxid Redox Signal. 2011;15(7):1835–1846. doi: 10.1089/ars.2010.3726. [DOI] [PubMed] [Google Scholar]
- 64.Hsieh C-H, Jeng S-F, Hsieh M-W, Chen Y-C, Rau C-S, Lu T-H, et al. Statin-induced heme oxygenase-1 increases NF-κB activation and oxygen radical production in cultured neuronal cells exposed to lipopolysaccharide. Toxicol Sci. 2008;102(1):150–159. doi: 10.1093/toxsci/kfm298. [DOI] [PubMed] [Google Scholar]
- 65.Chen J-C, Huang K-C, Lin W-W. HMG–CoA reductase inhibitors upregulate heme oxygenase-1 expression in murine RAW264. 7 macrophages via ERK, p38 MAPK and protein kinase G pathways. Cell Signal. 2006;18(1):32–9. doi: 10.1016/j.cellsig.2005.03.016. [DOI] [PubMed] [Google Scholar]
- 66.Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer. 2009;9(11):798–809. doi: 10.1038/nrc2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Delbosc S, Morena M, Djouad F, Ledoucen C, Descomps B, Cristol J-P. Statins, 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors, are able to reduce superoxide anion production by NADPH oxidase in THP-1-derived monocytes. J Cardiovasc Pharmacol. 2002;40(4):611–617. doi: 10.1097/00005344-200210000-00015. [DOI] [PubMed] [Google Scholar]
- 68.Dulak J, Józkowicz A. Carbon monoxide-a" new" gaseous modulator of gene expression. Acta Biochimica Pol. 2003;50(1):31–48. doi: 10.18388/abp.2003_3712. [DOI] [PubMed] [Google Scholar]
- 69.Urbich C, Dernbach E, Zeiher AM, Dimmeler S. Double-edged role of statins in angiogenesis signaling. Circ Res. 2002;90(6):737–744. doi: 10.1161/01.RES.0000014081.30867.F8. [DOI] [PubMed] [Google Scholar]
- 70.Dulak J, Loboda A, Jazwa A, Zagorska A, Dörler J, Alber H, et al. Atorvastatin affects several angiogenic mediators in human endothelial cells. Endothelium. 2005;12(5–6):233–241. doi: 10.1080/10623320500476559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dulak J, Józkowicz A. Anti-angiogenic and anti-inflammatory effects of statins: relevance to anti-cancer therapy. Curr Cancer Drug Targets. 2005;5(8):579–594. doi: 10.2174/156800905774932824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Undas A, Celinska-Lowenhof M, Stepien E, Nizankowski R, Tracz W, Szczeklik A. Effects of simvastatin on angiogenic growth factors released at the site of microvascular injury. Thromb Haemost. 2006;95(6):1045. doi: 10.1160/TH06-01-0022. [DOI] [PubMed] [Google Scholar]
- 73.Gomez-Cerezo J, Pagan-Munoz B, Lopez-Rodriguez M, Estebanez-Munoz M, Barbado-Hernandez F. The role of endothelial progenitor cells and statins in endothelial function: a review. Cardiovasc Hematol Agent Med Chem. 2007;5(4):265–72. doi: 10.2174/187152507782109836. [DOI] [PubMed] [Google Scholar]
- 74.Liu Y, Wei J, Hu L, Hu S. Beneficial effects of statins on endothelial progenitor cells. Am J Med Sci. 2012;344(3):220–226. doi: 10.1097/MAJ.0b013e31824998f9. [DOI] [PubMed] [Google Scholar]
- 75.Anwar MA, Samaha AA, Ballan S, Saleh AI, Iratni R, Eid AH. Salvia fruticosa Induces Vasorelaxation In Rat Isolated Thoracic Aorta: role of the PI3K/Akt/eNOS/NO/cGMP Signaling Pathway. Sci Rep. 2017;7(1):686. doi: 10.1038/s41598-017-00790-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Anwar MA, Samaha AA, Baydoun S, Iratni R, Eid AH, Rhus coriaria L. (Sumac) Evokes Endothelium-dependent Vasorelaxation of Rat Aorta: involvement of the cAMP and cGMP pathways. Front Pharmacol. 2018 doi: 10.3389/fphar.2018.00688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Elewa HF, El-Remessy AB, Somanath PR, Fagan SC. Diverse effects of statins on angiogenesis: new therapeutic avenues. Pharmacotherapy: J Human Pharmacol Drug Ther. 2010;30(2):169–76. doi: 10.1592/phco.30.2.169. [DOI] [PubMed] [Google Scholar]
- 78.Blanco-Colio LM, Villa A, Ortego M, Hernández-Presa MA, Pascual A, Plaza JJ, et al. 3-Hydroxy-3-methyl-glutaryl coenzyme A reductase inhibitors, atorvastatin and simvastatin, induce apoptosis of vascular smooth muscle cells by downregulation of Bcl-2 expression and Rho A prenylation. Atherosclerosis. 2002;161(1):17–26. doi: 10.1016/S0021-9150(01)00613-X. [DOI] [PubMed] [Google Scholar]
- 79.Walter DH, Zeiher AM, Dimmeler S. Effects of statins on endothelium and their contribution to neovascularization by mobilization of endothelial progenitor cells. Coron Artery Dis. 2004;15(5):235–242. doi: 10.1097/01.mca.0000131572.14521.8a. [DOI] [PubMed] [Google Scholar]
- 80.Sata M, Nishimatsu H, Suzuki E, Sugiura S, Yoshizumi M, Ouchi Y, et al. Endothelial nitric oxide synthase is essential for the HMG-CoA reductase inhibitor cerivastatin to promote collateral growth in response to ischemia. FASEB J. 2001;15(13):2530–2532. doi: 10.1096/fj.01-0415fje. [DOI] [PubMed] [Google Scholar]
- 81.Assmus B, Urbich C, Aicher A, Hofmann WK, Haendeler J, Rossig L, et al. HMG-CoA reductase inhibitors reduce senescence and increase proliferation of endothelial progenitor cells via regulation of cell cycle regulatory genes. Circ Res. 2003;92(9):1049–1055. doi: 10.1161/01.RES.0000070067.64040.7C. [DOI] [PubMed] [Google Scholar]
- 82.Katsumoto M, Shingu T, Kuwashima R, Nakata A, Nomura S, Chayama K. Biphasic effect of HMG-CoA reductase inhibitor, pitavastatin, on vascular endothelial cells and angiogenesis. Circ J. 2005;69(12):1547–1555. doi: 10.1253/circj.69.1547. [DOI] [PubMed] [Google Scholar]
- 83.Ota H, Eto M, Kano MR, Kahyo T, Setou M, Ogawa S, et al. Induction of endothelial nitric oxide synthase, SIRT1, and catalase by statins inhibits endothelial senescence through the Akt pathway. Arterioscler Thromb Vasc Biol. 2010;30(11):2205–2211. doi: 10.1161/ATVBAHA.110.210500. [DOI] [PubMed] [Google Scholar]
- 84.Xia Ma F, Han ZC. Statins, nitric oxide and neovascularization. Cardiovasc Drug Rev. 2005;23(4):281–292. doi: 10.1111/j.1527-3466.2005.tb00173.x. [DOI] [PubMed] [Google Scholar]
- 85.van der Meel R, Symons MH, Kudernatsch R, Kok RJ, Schiffelers RM, Storm G, et al. The VEGF/Rho GTPase signalling pathway: a promising target for anti-angiogenic/anti-invasion therapy. Drug Discov Today. 2011;16(5–6):219–228. doi: 10.1016/j.drudis.2011.01.005. [DOI] [PubMed] [Google Scholar]
- 86.Rikitake Y, Liao JK. Rho GTPases, statins, and nitric oxide. Circ Res. 2005;97(12):1232–1235. doi: 10.1161/01.RES.0000196564.18314.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Endres M. Statins and stroke. J Cereb Blood Flow Metab. 2005;25(9):1093–1110. doi: 10.1038/sj.jcbfm.9600116. [DOI] [PubMed] [Google Scholar]
- 88.Liao JK. Beyond lipid lowering: the role of statins in vascular protection. Int J Cardiol. 2002;86(1):5–18. doi: 10.1016/S0167-5273(02)00195-X. [DOI] [PubMed] [Google Scholar]
- 89.Park HJ, Kong D, Iruela-Arispe L, Begley U, Tang D, Galper JB. 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors interfere with angiogenesis by inhibiting the geranylgeranylation of RhoA. Circ Res. 2002;91(2):143–150. doi: 10.1161/01.RES.0000028149.15986.4C. [DOI] [PubMed] [Google Scholar]
- 90.Wolfrum S, Dendorfer A, Rikitake Y, Stalker TJ, Gong Y, Scalia R, et al. Inhibition of Rho-kinase leads to rapid activation of phosphatidylinositol 3-kinase/protein kinase Akt and cardiovascular protection. Arterioscler Thromb Vasc Biol. 2004;24(10):1842–1847. doi: 10.1161/01.ATV.0000142813.33538.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Li Z, Dong X, Wang Z, Liu W, Deng N, Ding Y, et al. Regulation of PTEN by Rho small GTPases. Nat Cell Biol. 2005;7(4):399. doi: 10.1038/ncb1236. [DOI] [PubMed] [Google Scholar]
- 92.Shackelford DB, Shaw RJ. The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nat Rev Cancer. 2009;9(8):563–575. doi: 10.1038/nrc2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Shackelford DB, Shaw RJ. The LKB1–AMPK pathway: metabolism and growth control in tumour suppression. Nat Rev Cancer. 2009;9(8):563–575. doi: 10.1038/nrc2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Choi HC, Song P, Xie Z, Wu Y, Xu J, Zhang M, et al. Reactive nitrogen species is required for the activation of the AMP-activated protein kinase by statin in vivo. J Biol Chem. 2008;283(29):20186–20197. doi: 10.1074/jbc.M803020200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 95.Choi HC, Son P, Xie Z, Wu Y, Xu J, Zhang M, et al. Withdrawal: reactive nitrogen species is required for the activation of the AMP-activated protein kinase by statin in vivo. J Biol Chem. 2019;294(27):10742. doi: 10.1074/jbc.W119.009748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Rossoni LV, Wareing M, Wenceslau CF, Al-Abri M, Cobb C, Austin C. Acute simvastatin increases endothelial nitric oxide synthase phosphorylation via AMP-activated protein kinase and reduces contractility of isolated rat mesenteric resistance arteries. Clin Sci. 2011;121(10):449–458. doi: 10.1042/CS20110259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Alessi DR, Sakamoto K, Bayascas JR. LKB1-dependent signaling pathways. Annu Rev Biochem. 2006;75(1):137–163. doi: 10.1146/annurev.biochem.75.103004.142702. [DOI] [PubMed] [Google Scholar]
- 98.Kou R, Sartoretto J, Michel T. Regulation of Rac1 by Simvastatin in Endothelial Cells Differential Roles of AMP-Activated Protein Kinase And Calmodulin-Dependent Kinase Kinase-β. J Biol Chem. 2009;284(22):14734–14743. doi: 10.1074/jbc.M808664200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kowluru A, Gleason NF. Underappreciated roles for Rho GDP Dissociation Inhibitors (RhoGDIs) in cell function: Lessons learned from the pancreatic islet β-cell. Biochem Pharmacol. 2021 doi: 10.1016/j.bcp.2021.114886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Jacobson JR, Dudek SM, Birukov KG, Ye SQ, Grigoryev DN, Girgis RE, et al. Cytoskeletal activation and altered gene expression in endothelial barrier regulation by simvastatin. Am J Respir Cell Mol Biol. 2004;30(5):662–670. doi: 10.1165/rcmb.2003-0267OC. [DOI] [PubMed] [Google Scholar]
- 101.Ogata Y, Takahashi M, Takeuchi K, Ueno S, Mano H, Ookawara S, et al. Fluvastatin induces apoptosis in rat neonatal cardiac myocytes: a possible mechanism of statin-attenuated cardiac hypertrophy. J Cardiovasc Pharmacol. 2002;40(6):907–915. doi: 10.1097/00005344-200212000-00012. [DOI] [PubMed] [Google Scholar]
- 102.Itagaki M, Takaguri A, Kano S, Kaneta S, Ichihara K, Satoh K. Possible mechanisms underlying statin-induced skeletal muscle toxicity in L6 fibroblasts and in rats. J Pharmacol Sci. 2009;109(1):94–101. doi: 10.1254/jphs.08238FP. [DOI] [PubMed] [Google Scholar]
- 103.Mutoh T, Kumano T, Nakagawa H, Kuriyama M. Involvement of tyrosine phosphorylation in HMG-CoA reductase inhibitor-induced cell death in L6 myoblasts. FEBS Lett. 1999;444(1):85–89. doi: 10.1016/S0014-5793(99)00031-9. [DOI] [PubMed] [Google Scholar]
- 104.Nakagawa H, Mutoh T, Kumano T, Kuriyama M. HMG-CoA reductase inhibitor-induced L6 myoblast cell death: involvement of the phosphatidylinositol 3-kinase pathway. FEBS Lett. 1998;438(3):289–292. doi: 10.1016/S0014-5793(98)01320-9. [DOI] [PubMed] [Google Scholar]
- 105.Nakahara K, Kuriyama M, Yoshidome H, Nagata K, Nagado T, Nakagawa M, et al. Experimental simvastatin-induced myopathy in rabbits. J Neurol Sci. 1992;113(1):114–117. doi: 10.1016/0022-510X(92)90273-N. [DOI] [PubMed] [Google Scholar]
- 106.Nakahara K, Yada T, Kuriyama M, Osame M. Cytosolic Ca2+ increase and cell damage in L6 rat myoblasts by HMG-CoA reductase inhibitors. Biochem Biophys Res Commun. 1994;202(3):1579–1585. doi: 10.1006/bbrc.1994.2112. [DOI] [PubMed] [Google Scholar]
- 107.Nagashima T, Okazaki H, Yudoh K, Matsuno H, Minota S. Apoptosis of rheumatoid synovial cells by statins through the blocking of protein geranylgeranylation: a potential therapeutic approach to rheumatoid arthritis. Arthritis Rheum: J Am College Rheumatol. 2006;54(2):579–586. doi: 10.1002/art.21564. [DOI] [PubMed] [Google Scholar]
- 108.Demyanets S, Kaun C, Pfaffenberger S, Hohensinner PJ, Rega G, Pammer J, et al. Hydroxymethylglutaryl-coenzyme A reductase inhibitors induce apoptosis in human cardiac myocytes in vitro. Biochem Pharmacol. 2006;71(9):1324–1330. doi: 10.1016/j.bcp.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 109.Kureishi Y, Luo Z, Shiojima I, Bialik A, Fulton D, Lefer DJ, et al. The HMG-CoA reductase inhibitor simvastatin activates the protein kinase Akt and promotes angiogenesis in normocholesterolemic animals. Nat Med. 2000;6(9):1004–1010. doi: 10.1038/79510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ma F-X, Chen F, Ren Q, Han Z-C. Lovastatin restores the function of endothelial progenitor cells damaged by oxLDL. Acta Pharmacologica Sinica. 2009;30(5):545–52. doi: 10.1038/aps.2009.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Bao XM, Wu CF, Lu GP. Atorvastatin inhibits homocysteine-induced oxidative stress and apoptosis in endothelial progenitor cells involving Nox4 and p38MAPK. Atherosclerosis. 2010;210(1):114–121. doi: 10.1016/j.atherosclerosis.2009.11.032. [DOI] [PubMed] [Google Scholar]
- 112.Bao XM, Zheng HC, Wu CF, Lu GP. Rosuvastatin inhibits homocysteine-induced oxidative stress and apoptosis in endothelial progenitor cells involving Nox4. J Shanghai Jiaotong University (Medical Science) 2013;33(11):1436. [Google Scholar]
- 113.Meyer N, Brodowski L, Richter K, von Kaisenberg CS, Schröder-Heurich B, von Versen-Höynck F. Pravastatin Promotes Endothelial Colony-Forming Cell Function, Angiogenic Signaling and Protein Expression In Vitro. J Clin Med. 2021 doi: 10.3390/jcm10020183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Spyridopoulos I, Haendeler J, Urbich C, Brummendorf TH, Oh H, Schneider MD, et al. Statins enhance migratory capacity by upregulation of the telomere repeat-binding factor TRF2 in endothelial progenitor cells. Circulation. 2004;110(19):3136–3142. doi: 10.1161/01.CIR.0000142866.50300.EB. [DOI] [PubMed] [Google Scholar]
- 115.Assmus B, Urbich C, Aicher A, Hofmann WK, Haendeler J, Rossig L, Spyridopoulos I, Zeiher AM, Dimmeler S. HMG-CoA reductase inhibitors reduce senescence and increase proliferation of endothelial progenitor cells via regulation of cell cycle regulatory genes. Circ Res. 2003;92:1049–1055. doi: 10.1161/01.RES.0000070067.64040.7C. [DOI] [PubMed] [Google Scholar]
- 116.Cafforio P, Dammacco F, Gernone A, Silvestris F. Statins activate the mitochondrial pathway of apoptosis in human lymphoblasts and myeloma cells. Carcinogenesis. 2005;26(5):883–891. doi: 10.1093/carcin/bgi036. [DOI] [PubMed] [Google Scholar]
- 117.Dirks AJ, Jones KM. Statin-induced apoptosis and skeletal myopathy. Am J Physiol Cell Physiol. 2006;291(6):C1208–C1212. doi: 10.1152/ajpcell.00226.2006. [DOI] [PubMed] [Google Scholar]
- 118.Agarwal B, Bhendwal S, Halmos B, Moss SF, Ramey WG, Holt PR. Lovastatin augments apoptosis induced by chemotherapeutic agents in colon cancer cells. Clin Cancer Res. 1999;5(8):2223–2229. [PubMed] [Google Scholar]
- 119.Wood WG, Igbavboa U, Muller WE, Eckert GP. Statins, Bcl-2, and apoptosis: cell death or cell protection? Mol Neurobiol. 2013;48(2):308–314. doi: 10.1007/s12035-013-8496-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Viatour P, Bentires-Alj M, Chariot A, Deregowski V, de Leval L, Merville M-P, et al. NF-κB2/p100 induces Bcl-2 expression. Leukemia. 2003;17(7):1349–1356. doi: 10.1038/sj.leu.2402982. [DOI] [PubMed] [Google Scholar]
- 121.Spampanato C, De Maria S, Sarnataro M, Giordano E, Zanfardino M, Baiano S, et al. Simvastatin inhibits cancer cell growth by inducing apoptosis correlated to activation of Bax and down-regulation of BCL-2 gene expression. Int J Oncol. 2012;40(4):935–941. doi: 10.3892/ijo.2011.1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Fromigue O, Hay E, Modrowski D, Bouvet S, Jacquel A, Auberger P, et al. RhoA GTPase inactivation by statins induces osteosarcoma cell apoptosis by inhibiting p42/p44-MAPKs-Bcl-2 signaling independently of BMP-2 and cell differentiation. Cell Death Differ. 2006;13(11):1845–1856. doi: 10.1038/sj.cdd.4401873. [DOI] [PubMed] [Google Scholar]
- 123.Koyuturk M, Ersoz M, Altiok N. Simvastatin induces proliferation inhibition and apoptosis in C6 glioma cells via c-jun N-terminal kinase. Neurosci Lett. 2004;370(2):212–217. doi: 10.1016/j.neulet.2004.08.020. [DOI] [PubMed] [Google Scholar]
- 124.Lee J, Lee I, Park C, Kang WK. Lovastatin-induced RhoA modulation and its effect on senescence in prostate cancer cells. Biochem Biophys Res Commun. 2006;339(3):748–754. doi: 10.1016/j.bbrc.2005.11.075. [DOI] [PubMed] [Google Scholar]
- 125.Murtola TJ, Syvälä H, Pennanen P, Bläuer M, Solakivi T, Ylikomi T, et al. Comparative effects of high and low-dose simvastatin on prostate epithelial cells: the role of LDL. Eur J Pharmacol. 2011;673(1–3):96–100. doi: 10.1016/j.ejphar.2011.10.022. [DOI] [PubMed] [Google Scholar]
- 126.Yu R, Longo J, van Leeuwen JE, Mullen PJ, Ba-Alawi W, Haibe-Kains B, et al. Statin-induced cancer cell death can be mechanistically uncoupled from prenylation of RAS family proteins. Can Res. 2018;78(5):1347–1357. doi: 10.1158/0008-5472.CAN-17-1231. [DOI] [PubMed] [Google Scholar]
- 127.O'Grady S, Crown J, Duffy MJ. Anti-tumor effects of statins in triple-negative breast cancer: Apoptosis, chemosensitization and degradation of mutant-p53. Cancer Res. 2020;80(16_supplement):1775. doi: 10.1158/1538-7445.AM2020-1775. [DOI] [Google Scholar]
- 128.Mirzaei A, Rashedi S, Akbari MR, Khatami F, Aghamir SMK. Combined anticancer effects of simvastatin and arsenic trioxide on prostate cancer cell lines via downregulation of the VEGF and OPN isoforms genes. J Cell Mol Med. 2022;26(9):2728–40. doi: 10.1111/jcmm.17286. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 129.Thrasher J. Targeting Metastatic Breast Cancer with Statin Drugs and CoQ10. 2018.
- 130.Trogden KP, Battaglia RA, Kabiraj P, Madden VJ, Herrmann H, Snider NT. An image-based small-molecule screen identifies vimentin as a pharmacologically relevant target of simvastatin in cancer cells. FASEB J. 2018;32(5):2841–2854. doi: 10.1096/fj.201700663R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Putra B, Wahyuningsih MSH, Sholikhah EN. Cytotoxic activity of simvastatin in T47D breast cancer cell lines and its effect on cyclin D1 expression and apoptosis. J Med Sci. 2017;49(2):47–55. [Google Scholar]
- 132.Warita K, Warita T, Beckwitt CH, Schurdak ME, Vazquez A, Wells A, et al. Statin-induced mevalonate pathway inhibition attenuates the growth of mesenchymal-like cancer cells that lack functional E-cadherin mediated cell cohesion. Sci Rep. 2014;4(1):1–8. doi: 10.1038/srep07593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Luttman JH, Hoj JP, Lin KH, Lin J, Gu JJ, Rouse C, et al. ABL allosteric inhibitors synergize with statins to enhance apoptosis of metastatic lung cancer cells. Cell Rep. 2021;37(4):109880. doi: 10.1016/j.celrep.2021.109880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Levine Benjamin D, Cagan RL. Drosophila Lung Cancer Models Identify Trametinib plus Statin as Candidate Therapeutic. Cell Rep. 2016;14(6):1477–1487. doi: 10.1016/j.celrep.2015.12.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Zhong W-B, Wang C-Y, Chang T-C, Lee W-S. Lovastatin induces apoptosis of anaplastic thyroid cancer cells via inhibition of protein geranylgeranylation and de novo protein synthesis. Endocrinology. 2003;144(9):3852–3859. doi: 10.1210/en.2003-0098. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No primary data exists for this review article.