

Abstract
Attention depends on cholinergic stimulation of nicotinic and muscarinic acetylcholine receptors in the medial prefrontal cortex. Pyramidal neurons in layer VI of this region express cholinergic receptors of both families and play an important role in attention through their feedback projections to the thalamus. Here, we investigate how nicotinic and muscarinic cholinergic receptors affect the excitability of these neurons using whole-cell recordings in acute brain slices of prefrontal cortex. Since attention deficits have been documented in both rodents and humans having genetic abnormalities in nicotinic receptors, we focus in particular on how the cholinergic excitation of layer VI neurons is altered by genetic deletion of either of two key nicotinic receptor subunits, the accessory α5 subunit or the ligand-binding β2 subunit. We find that the cholinergic excitation of layer VI neurons is dominated by nicotinic receptors in wild-type mice and that the reduction or loss of this nicotinic stimulation is accompanied by a surprising degree of plasticity in excitatory muscarinic receptors. These findings suggest that disrupting nicotinic receptors fundamentally alters the mechanisms and timing of excitation in prefrontal attentional circuitry.
Introduction
Prefrontal acetylcholine (ACh) release increases with attentional effort (Passetti et al., 2000; Dalley et al., 2004) and correlates with detection of cues on attention tasks (Parikh et al., 2007). The loss of prefrontal ACh afferents, by contrast, substantially lowers cue detection in attention tasks (McGaughy et al., 1996). Attentional processing depends on both ionotropic nicotinic receptors (Bailey et al., 2010; Guillem et al., 2011) and metabotropic muscarinic receptors (Robbins et al., 1998), types of cholinergic receptors that may have synergistic effects (Ellis et al., 2006). The cortico-thalamic neurons of layer VI are very sensitive to nicotinic stimulation (Kassam et al., 2008; Bailey et al., 2010). These neurons are thought to control the attentional “search light” of the brain through their various feedback projections to the thalamus (Crick, 1984; Zikopoulos and Barbas, 2006; Briggs and Usrey, 2011). Layer VI neurons express the relatively rare α5 nicotinic accessory subunit (Wada et al., 1990; Salas et al., 2003), in addition to the α4 and β2 subunits that form the high-affinity nicotinic receptors. In layer VI neurons, the α5 subunit is incorporated into the α4β2 subtype of nicotinic receptors, greatly enhancing their conductance (Ramirez-Latorre et al., 1996) and currents (Bailey et al., 2010). Of particular interest, loss of the α5 nicotinic receptors results in attention deficits in mice (Bailey et al., 2010). As layer VI neurons also express muscarinic ACh receptors (Buckley et al., 1988), we investigate the combined effects of nicotinic and muscarinic ACh stimulation on the excitability of these neurons. Since genetic abnormalities in nicotinic receptors are linked to attentional dysfunction (Rigbi et al., 2008; Bailey et al., 2010; Guillem et al., 2011), we examine how the cholinergic excitation of layer VI neurons is altered by genetic deletion of two key nicotinic receptor subunits: the α5 subunit, which increases the conductance of the high-affinity nicotinic receptor, or the β2 subunit, which is required for assembly and function of these receptors.
Materials and Methods
Homozygous mice derived from heterozygous parents were used to generate α5 (α5+/+, Salas et al., 2003) and β2 (β2−/−, Picciotto et al., 1995) knock-out mice for experiments. Wild-type (WT) mice were bred in this manner from both α5 and β2 lines. Neurons from both WT groups were combined for analysis since no statistically significant differences were observed in our experiments.
Adult male mice from postnatal day (P) 60 to P180 were used to prepare 400-μm-thick coronal slices of the prefrontal cortex (2.34 to 1.34 mm anterior to bregma, Paxinos and Franklin, 2001) using a protocol approved by the University of Toronto Animal Care and Use Committee. In brief, the excised brain was cooled with 4°C oxygenated sucrose-based ACSF before slicing with a Dosaka Linear Slicer. Slices were transferred to 30°C oxygenated ACSF (128 mM NaCl, 10 mM D-glucose, 24 mM NaHCO2, 2 mM CaCl2, 2 mM MgSO4, 3 mM KCl, 1.25 mM NaH2PO4; pH 7.4). For recordings, slices were placed in a chamber on the stage of an Olympus BX50WI microscope. Oxygenated ACSF at room temperature flowed over the slice at 3–4 ml/min.
Electrophysiology
Pipettes (3–4 MΩ) containing 120 mM K-gluconate, 5 mM KCl, 2 mM MgCl2, 4 mM K2-ATP, 0.4 mM Na2-GTP, 10 mM Na2-phosphocreatine, and 10 mM HEPES buffer (adjusted to pH 7.3 with KOH) were used to patch layer VI pyramidal neurons in prelimbic cortex (Paxinos and Franklin, 2001). These neurons were conservatively selected based on their proximity to white matter (<200 μm; Alves et al., 2010) together with known morphological distinguishing features of layer VI (on average, the pyramidal neurons are smaller and closer together than in layer V). Neurons were recorded in current clamp using an EPC10 (HEKA) and corrected for the liquid junction potential.
Across the genotypes, there were no significant differences in resting potential (F (2,177) = 0.43, p = 0.6), input resistance (F(2,177) =1.2, p = 0.3), or spike amplitude (F(2,177) = 1.6, p = 0.2). Recordings were made either at resting membrane potential or with current injected to elicit a steady ~1 Hz action potential firing at baseline. ACh-induced depolarizations were measured relative to the resting potential. Changes in action potential frequency were determined using Clampfit software (Molecular Devices) by comparing the rate of firing over a 30 s period during the peak of the ACh response to that at baseline. As a control experiment, a pharmacologically identified subgroup of layer VI pyramidal neurons was selected based on their response to hypocretin (Bayer et al., 2004). There were no significant differences in cell properties between neurons responsive to hypocretin and the general population of layer VI pyramidal neurons.
Pharmacology
We probed cholinergic currents by applying 1 mM ACh to the bath after a period of baseline recording. Either atropine (200 nM), or a combination of DHβE (3 μM) and MLA (10 nM) were applied in ACSF to examine nicotinic and muscarinic effects, respectively. No antagonists were used in cholinergic experiments examining combined nicotinic and muscarinic effects. The subset of experiments testing responses to hypocretin 2 (100–300 nM), in addition to ACh, were conducted at 32°C (Bayer et al., 2004). All compounds were obtained from Sigma, Phoenix Pharmaceuticals, or Tocris Bioscience.
Statistical analysis
Effects of ACh on membrane potential, rate of action potential firing, and the kinetics of these responses were assessed for all genotypes with one-way ANOVAs, as were comparisons of cell properties across genotypes. We used Fisher’s exact tests to evaluate genotype differences in the proportions of neurons depolarized to threshold by ACh. Unpaired t tests were used to compare components of the ACh response. The effect of atropine on the muscarinic response was analyzed by paired t test. The interaction between nicotinic and muscarinic effects across genotypes was analyzed by two-way ANOVA, and the difference within each genotype was assessed with Bonferroni post hoc tests.
Results
Nicotinic excitability in layer VI is reduced in α5−/− mice and eliminated in β2−/− mice
First, we examined the nicotinic depolarization of layer VI pyramidal neurons in WT, α5−/−, and β2−/− mice since these subunits are known to be the principal constituents of the nicotinic receptors expressed in this layer along with the α4 subunit. In the presence of atropine (200 nM) to block muscarinic receptors, stimulation with ACh (1 mM, 15 s) resulted in significantly different depolarization across the genotypes (F(2,49) = 47.89, p = 0.0001). As illustrated in Figure 1, ACh depolarized layer VI neurons in WT mice to the greatest extent (n = 24), depolarized α5−/− neurons to a lesser degree (n = 21), and did not alter the membrane potential in β2−/− neurons (n = 7). Nicotinic excitation was sufficient to elicit action potentials in the majority of WT neurons, fewer α5−/− neurons (p < 0.05), and none of the β2−/− neurons.
Figure 1.

Nicotinic excitability of layer VI pyramidal neurons is reduced in α5−/− and eliminated in β2−/−mice. A, Stimulation of only nicotinic receptors following blockade of muscarinic receptors by atropine (200 n ) resulted in significant differences in depolarization across all genotypes (p <0.0001), with markedly less depolarization of α5−/− and β2−/− compared to WT neurons (**p <0.001). B, Sample traces show nicotinic responses in neurons across all genotypes, including the percentages of neurons with suprathreshold (top) and subthreshold (bottom) responses. A smaller proportion of neurons were depolarized to threshold in α5−/− and β2−/− neurons than in WT (α5−/−: p <0.05, β2−/−: p <0.01). C, In neurons already firing action potentials by current injection, nicotinic stimulation affects spiking frequency differently across genotypes (p <0.0001), with a smaller change in action potential frequency in α5−/− and β2−/− compared to WT neurons (*p <0.05, **p <0.001). D, Sample traces show nicotinic responses in neurons depolarized to fire action potentials by current injection. Examples (2 s in duration) of baseline and peak responses are shown above each trace. Data in all figures are shown as mean ±SEM.
To examine nicotinic effects on the excitability of already-depolarized neurons, we injected neurons with positive current to elicit action potentials (~1 Hz) at baseline. The current required did not differ significantly across genotypes, consistent with the lack of significant difference in input resistance. As illustrated in Figure 1C, ACh increased the frequency of action potential firing in a genotype-dependent manner (F(2,31) = 11.78, p = 0.0002). This treatment resulted in a large increase in the firing frequency of WT (n =15) and α5 −/− neurons (n =14) but not β2 −/− neurons (n = 5).
Cholinergic excitation of layer VI cortical neurons primarily involves nicotinic receptors unless nicotinic drive is impaired
The striking differences across genotypes in the response to nicotinic stimulation raised the question of how layer VI neurons normally respond to ACh in the absence of atropine, when it can stimulate both nicotinic and muscarinic receptors together. This experiment is shown in Figure 2 (we use “cholinergic” to identify conditions where both nicotinic and muscarinic receptors are stimulated). In layer VI neurons from WT mice, we find that cholinergic depolarization does not differ greatly from nicotinic depolarization alone (cholinergic: 20.3 ± 1.3 mV, n = 23; nicotinic: 19.2 ± 1.0 mV, n = 24; t = 0.7, p = 0.5). By contrast, layer VI neurons from α5 −/− mice show greater depolarization when both nicotinic and muscarinic receptors are stimulated (cholinergic: 15.3 ± 1.1 mV, n = 26; nicotinic: 11.9 ± 1.1 mV, n = 21; t =2.2, p = 0.03). More strikingly in β2 −/− mice, the muscarinic component appears dominant since the response to nicotinic stimulation is negligible in comparison with the response to cholinergic stimulation (cholinergic: 4.3 ±0.6 mV, n =23; nicotinic: 0.1 ± 0.2 mV, n = 7; t = 3.8, p = 0.0005). Yet, there are significant differences in the magnitude of depolarization elicited by cholinergic stimulation across the genotypes (F(2,69) = 63.12, p = 0.0001). Compared to nicotinic stimulation alone, cholinergic stimulation had a significantly greater effect on the ability to elicit action potentials from rest in neurons from α5 −/− mice, and was able to elicit spiking in some neurons from β2 −/− mice.
Figure 2.

Cholinergic (nicotinic and muscarinic receptor) stimulation depolarizes WT neurons most, but it increases spike frequency similarly across genotypes. A, Cholinergic stimulation depolarizes neurons to a different degree across genotypes (p <0.0001), with less depolarization in α5 −/− and β2 −/− compared to WT neurons (*p <0.01, **p <0.001). B, Sample traces showing cholinergic responses in neurons of all genotypes. C, Cholinergic stimulation increases action potential firing frequency to a similar degree across all genotypes (p =0.5). D, Sample traces showing cholinergic responses in neurons of all genotypes depolarized to fire action potentials by current injection. B, D, Note the slower onset and prolonged peak response in β2 −/− neurons.
In contrast to the differences in nicotinic effects in layer VI neurons across genotypes, cholinergic stimulation increased the frequency of action potential firing similarly across all three groups. As illustrated in Figure 2C, we observed no significant differences in the percentage increase in action potential frequency at the peak of the cholinergic response between layer VI neurons from WT (n =22), α5 −/−(n =17), and β2 −/−(n =24) mice (F(2,60) = 0.67, p = 0.5). The similar effects of ACh across genotypes on spike frequency cannot be attributed to a ceiling effect, since injected depolarizing current was consistently able to elicit faster spiking across the genotypes (paired t test, n =59, t = 20.7, p < 0.0001).
Notably, the time course of cholinergic effects was genotype dependent, with slower onset and substantially longer currents in layer VI neurons from β2 −/− mice. The 10–90% rise times of cholinergic responses in β2 −/− mice were slower for depolarization (WT: 13.3 ±1.1 s, α5 −/− : 14.7 ±0.6 s, β2 −/−: 25.8 ±2.9 s; F(2,45) =10.1, p =0.0002; p <0.01 for WT vs β2 −/−) and action potential frequency (WT: 14.6 ± 0.8 s, α5 −/−: 13.4 ± 1.3 s, β2 −/−: 20.7 ± 2.6 s; F(2,42) = 4.1, p = 0.02). Yet, the peak cholinergic effects lasted much longer in β2 −/− neurons, as shown by the τ (63% decay time) of depolarization (WT: 53.0 ± 8.3 s, α5 −/−: 53.4 ± 9.2 s, β2 −/−: 257.3 ± 22.8 s; F(2,45) = 48.3, p <0.0001; p <0.001 for WT vs β2 −/−) and action potential frequency (WT: 42.7 ± 4.4 s, α5 −/−: 56.1 ± 10.8 s, β2 −/−: 111.6 ± 25.5 s; F(2,38) = 5.5, p <0.01; p <0.01 for WT vs β2 −/−).
Muscarinic responses are enhanced in layer VI of α5−/− and β 2−/− compared to WT mice
The difference between responses to nicotinic-only and total cholinergic stimulation in layer VI neurons of α5 −/− and β2 −/− mice suggests potential plasticity in muscarinic-only ACh effects. To address this question, we tested the effects of muscarinic-only stimulation using ACh in the presence of nicotinic blockers (DHβE 3 μM, MLA 10 nM), as well as antagonists for the AMPA and NMDA glutamate receptors (CNQX 10 μM, APV 50 μM) to assess whether functional upregulation of muscarinic currents occurred in layer VI neurons lacking specific nicotinic receptor subtypes. Changes in membrane potential following muscarinic stimulation were significantly different across the genotypes (F(2,47) = 4.20, p = 0.02), as illustrated in Figure 3. The elicited depolarization in layer VI neurons from WT mice were small (n = 17) compared to the larger depolarization in those from α5 −/−(n =16; t =2.6, p =0.01) and β2 −/−(n =17) mice. The muscarinic antagonist atropine (200 nM, 10 min) suppressed these responses in all genotypes (n = 11, t = 3.4, p = 0.006). Furthermore, selective antagonists for the excitatory M1 and M3 muscarinic receptors (pirenzepine, 500 nM; J-104129 fumarate, 50 nM) almost completely suppressed the depolarization (22 ± 17% of that seen previously; n = 5).
Figure 3.

Muscarinic responses are enhanced in α5 −/− and β2 −/− compared to WT neurons. A, Stimulation of only muscarinic receptors on layer VI neurons following blockade of nicotinic receptors (3 μM DHBE, 10 nM MLA) and glutamate receptors (10 μM CNQX, 50 μM APV) resulted in significant differences in depolarization across genotypes (p <0.05), with greater muscarinic depolarization seen in α5 −/− than WT neurons (*p <0.05). B, Sample traces show the muscarinic response in neurons from all genotypes. No WT neurons, but some α5 −/− and β2 −/− neurons are depolarized to threshold. C, Muscarinic stimulation increases action potential firing differently across the genotypes (p <0.001), with action potential frequency increasing to a greater degree in β2 −/− compared to WT neurons (*p <0.05). D, Sample traces showing muscarinic responses in neurons depolarized to fire action potentials by current injection.
There were also significant genotype differences in the increase in firing frequency elicited by muscarinic stimulation (F(2,46) = 3.86, p =0.03). Muscarinic stimulation increased peak action potential firing in layer VI neurons from WT mice (n = 14), but this increase was significantly larger in layer VI neurons from both α5 −/−(n =16; t =2.1, p =0.04) and β2 −/−(n =19; t =3.3, p =0.002) mice. Again, these muscarinic responses were suppressed by atropine across the genotypes (n = 7, t = 6.5, p = 0.0006). Similarly, M1/M3 antagonists completely eliminated the increase in action potential firing (−5 ± 6% of that seen previously, n = 7).
With muscarinic-only stimulation, all three genotypes showed the longer 10–90% rise times and T characteristic of responses mediated only by G-protein-coupled receptors. These results contrast with the kinetics of the cholinergic responses where the fast, nicotinic responses dominated in the WT and α5 −/− mice, and the slow, muscarinic response dominated in the β2 −/− mice.
Hypocretin-responsive layer VI neurons show same pattern of muscarinic responses
The differences in the magnitude of the muscarinic responses across the genotypes raises the question of whether the layer VI neurons in the mice deleted for nicotinic subunits are indeed the same type of neurons as in WT mice. To address this question, we examined a pharmacologically identified subgroup of neurons within layer VI of cortex (Bayer et al., 2004). In these neurons excited by hypocretin (100–300 nM, 1 min), which were recorded at 32°C (based on Bayer et al., 2004), muscarinic responses showed the same pattern across genotypes as in the previous room temperature recordings of the general population of layer VI neurons (change in membrane potential: WT, 2.5 ± 0.6 mV, n =9; α5−/−, 8.7 ±2.3 mV, n =9; β2−/−, 7.9 ±1.4 mV, n =13; F(2,28) = 4.2, p = 0.02; change in spiking frequency: WT, 341 ± 94%, n =9; α5−/−, 763 ±160%, n =9; β2−/−, 651 ±113%, n = 10; F(2,25) = 2.9, p =0.07). These findings are consistent with the interpretation that layer VI neurons, which normally display large nicotinic responses to ACh, upregulate their muscarinic responses after loss or reduction of nicotinic excitation. To ascertain the specificity of this M1/M3 muscarinic upregulation, we compared the response to hypocretin across the genotypes since it is also mediated by a Gαq-coupled receptor. However, these responses did not differ by genotype (F(2,82) = 0.85, p = 0.4).
The balance of muscarinic to nicotinic excitation differs across genotypes
Differences in the responses to nicotinic and muscarinic stimulation across genotypes are illustrated in Figure 4. Analysis of changes in membrane potential shows a strong interaction between the responses to nicotinic and muscarinic stimulation (F(2,96) = 41.75, p = 0.0001) (Fig. 4A), with the genotypes varying greatly in the ratio of the muscarinic to the nicotinic response (WT: 0.2, α5−/−: 0.6, β2−/−: 61). As illustrated in Figure 4B, we observed a similar interaction in the increase in action potential frequency due to nicotinic and muscarinic stimulation (F(2,77) = 15.4, p =0.0001), with the genotypes varying substantially in the ratio of the muscarinic to the nicotinic response (WT: 0.5, α5−/−: 1.3, β2−/−: 4.1). There appears to be a negligible contribution of muscarinic receptors in WT mice, a balance between nicotinic and muscarinic contributions in α5−/−, and a predominant contribution of muscarinic receptors in β2−/− mice.
Figure 4.
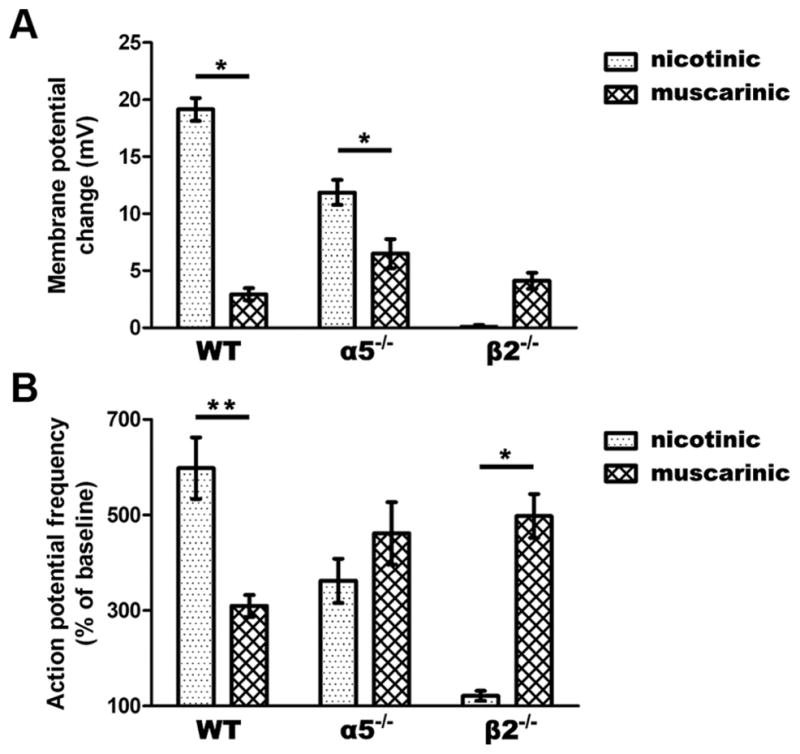
The balance of nicotinic to muscarinic excitation is shifted in α5 −/− and β2 −/− compared to WT neurons. A, There is a significant interaction in the degree of nicotinic and muscarinic depolarization across genotypes (p < 0.0001). Nicotinic stimulation contributes more to membrane depolarization in WT and α5 −/− neurons (*p <0.01). B, A significant interaction is found in the increase in action potential firing by nicotinic versus muscarinic stimulation across genotypes (p <0.0001). Nicotinic stimulation increases spiking frequency to a greater degree in WT neurons, while muscarinic stimulation makes a larger contribution in β2 −/− neurons (*p <0.01, **P <0.001).
Discussion
We have found that ACh predominantly excites layer VI pyramidal neurons in WT mice through nicotinic receptors. Impairment in nicotinic stimulation through genetic deletion of either the conductance-enhancing α5 subunit or the ligand-binding β2 subunit is accompanied by an increase in the cholinergic excitation of these neurons through the metabotropic muscarinic family of ACh receptors. This muscarinic excitation is suppressed by antagonists of M1 and M3 receptors and significantly alters the timing of the peak cholinergic response in mice deleted for the ligand-binding β2 subunit. Our results suggest that disrupting nicotinic receptor function can fundamentally alter the mechanisms and timing of excitation in prefrontal attentional circuitry.
Humans and rodents with aberrant expression or function of nicotinic receptors are at higher risk for attention deficits (Rigbi et al., 2008; Bailey et al., 2010; Guillem et al., 2011). Such changes in nicotinic receptors can result from genetic polymorphisms that reduce function (Bierut et al., 2008; Kuryatov et al., 2011) or through developmental exposure to the drug nicotine (Poorthuis et al., 2009). The observed upregulation of the typically smaller muscarinic component of cholinergic activation in neurons of α5−/− and β2−/− mice allows a substantial response to ACh despite the reduction or loss of nicotinic receptor function, and highlights the functional significance of nicotinic signaling within attention pathways. Our results suggest that humans with reduced prefrontal nicotinic receptor function may have a larger muscarinic contribution toward the overall cholinergic response in layer VI neurons. This plasticity of cholinergic signaling may, in fact, reduce the apparent severity of the attention deficits resulting from genetic or developmental alterations in nicotinic receptors. Indeed, in mice performing an attention task, α5−/− mice show only significantly lower accuracy compared to WT controls under challenging conditions (Bailey et al., 2010), while β2−/− mice demonstrate only a higher level of omissions but no decreases in accuracy (Guillem et al., 2011). Yet, having an atypical muscarinic component, which is slower and longer than the normal nicotinic excitation, may result in more complex changes to prefrontal attention circuitry and function than previously anticipated.
Our results indicate that synergistic effects of prefrontal nicotinic and muscarinic receptors on attentional performance (Ellis et al., 2006) do not normally reflect shared activation of layer VI neurons. Instead, these receptors may act predominantly on different cortical output layers. In mice with genetically induced nicotinic dysfunction, we observed increased excitatory muscarinic responses in layer VI neurons that act in a homeostatic manner to preserve their cholinergic response. However, our results suggest that this atypical muscarinic excitability of layer VI neurons is not uniformly upregulated. The ACh depolarization from rest is only partially rescued, yet the increase in action potential frequency of already-depolarized neurons appears completely normal. Since cortical muscarinic receptor binding is not altered in β2−/−mice (Zoli et al., 1999), the differential rescue of two aspects of cortical excitability suggests a locus of plasticity downstream of the muscarinic receptors.
The medial prefrontal cortex and its cholinergic afferents are essential for efficient attentional processing. Here, we show that the cholinergic excitation of layer VI neurons is of sufficient functional importance that compensatory processes provide this excitation even in the absence of the nicotinic receptors normally responsible in WT mice. While the upregulation of muscarinic excitability may ameliorate the severity of attention deficits resulting from alterations in nicotinic receptors, it would likely alter the speed of attentional processing and render layer VI attention circuitry vulnerable to the effects of anti-muscarinic medications.
Acknowledgments
This work was supported by grants to E.K.L. from the Canadian Institute of Health Research (MOP 89825), the Canada Research Chairs Program, and the Canadian Foundation for Innovation. M.R.P. was supported by Grants DA14241 and DA10455 from the National Institutes of Health. We thank Dr. Milton Charlton and Dr. Beverley Orser for constructive suggestions on the manuscript.
Footnotes
Author contributions: M.K.T., C.D.C.B., and E.K.L. designed research; M.K.T. and C.D.C.B. performed research; M.D. and M.R.P. contributed unpublished reagents/analytic tools; M.K.T. and E.K.L. analyzed data; M.K.T., C.D.C.B., M.D., M.R.P., and E.K.L. wrote the paper.
References
- Alves NC, Bailey CD, Nashmi R, Lambe EK. Developmental sex differences in nicotinic currents of prefrontal layer VI neurons in mice and rats. PLoS One. 2010;5:e9261. doi: 10.1371/journal.pone.0009261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey CD, De Biasi M, Fletcher PJ, Lambe EK. The nicotinic acetylcholine receptor α5 subunit plays a key role in attention circuitry and accuracy. J Neurosci. 2010;30:9241–9252. doi: 10.1523/JNEUROSCI.2258-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayer L, Serafin M, Eggermann E, Saint-Mleux B, Machard D, Jones BE, Mühlethaler M. Exclusive postsynaptic action of hypocretin-orexin on sublayer 6b cortical neurons. J Neurosci. 2004;24:6760–6764. doi: 10.1523/JNEUROSCI.1783-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bierut LJ, Stitzel JA, Wang JC, Hinrichs AL, Grucza RA, Xuei X, Saccone NL, Saccone SF, Bertelsen S, Fox L, Horton WJ, Breslau N, Budde J, Cloninger CR, Dick DM, Foroud T, Hatsukami D, Hesselbrock V, Johnson EO, Kramer J, et al. Variants in nicotinic receptors and risk for nicotine dependence. Am J Psychiatry. 2008;165:1163–1171. doi: 10.1176/appi.ajp.2008.07111711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briggs F, Usrey WM. Corticogeniculate feedback and visual processing in the primate. J Physiol. 2011;589:33–40. doi: 10.1113/jphysiol.2010.193599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckley NJ, Bonner TI, Brann MR. Localization of a family of muscarinic receptor mRNAs in rat brain. J Neurosci. 1988;8:4646–4652. doi: 10.1523/JNEUROSCI.08-12-04646.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crick F. Function of the thalamic reticular complex: the searchlight hypothesis. Proc Natl Acad Sci U S A. 1984;81:4586–4590. doi: 10.1073/pnas.81.14.4586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalley JW, Cardinal RN, Robbins TW. Prefrontal executive and cognitive functions in rodents: neural and neurochemical substrates. Neurosci Biobehav Rev. 2004;28:771–784. doi: 10.1016/j.neubiorev.2004.09.006. [DOI] [PubMed] [Google Scholar]
- Ellis JR, Ellis KA, Bartholomeusz CF, Harrison BJ, Wesnes KA, Erskine FF, Vitetta L, Nathan PJ. Muscarinic and nicotinic receptors synergistically modulate working memory and attention in humans. Int J Neuropsychopharmacol. 2006;9:175–189. doi: 10.1017/S1461145705005407. [DOI] [PubMed] [Google Scholar]
- Guillem K, Bloem B, Poorthuis RB, Loos M, Smit AB, Maskos U, Spijker S, Mansvelder HD. Nicotinic acetylcholine receptor β2 subunits in the medial prefrontal cortex control attention. Science. 2011;333:888–891. doi: 10.1126/science.1207079. [DOI] [PubMed] [Google Scholar]
- Kassam SM, Herman PM, Goodfellow NM, Alves NC, Lambe EK. Developmental excitation of corticothalamic neurons by nicotinic acetylcholine receptors. J Neurosci. 2008;28:8756–8764. doi: 10.1523/JNEUROSCI.2645-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuryatov A, Berrettini W, Lindstrom J. AChR alpha5 subunit variant associated with risk for nicotine dependence and lung cancer reduces (alpha4beta2)2alpha5 AChRs function. Mol Pharmacol. 2011;79:119–125. doi: 10.1124/mol.110.066357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGaughy J, Kaiser T, Sarter M. Behavioral vigilance following infusions of 192 IgG-saporin into the basal forebrain: selectivity of the behavioral impairment and relation to cortical AChE-positive fiber density. Behav Neurosci. 1996;110:247–265. doi: 10.1037//0735-7044.110.2.247. [DOI] [PubMed] [Google Scholar]
- Parikh V, Kozak R, Martinez V, Sarter M. Prefrontal acetylcholine release controls cue detection on multiple timescales. Neuron. 2007;56:141–154. doi: 10.1016/j.neuron.2007.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passetti F, Dalley JW, O’Connell MT, Everitt BJ, Robbins TW. Increased acetylcholine release in the rat medial prefrontal cortex during performance of a visual attentional task. Eur J Neurosci. 2000;12:3051–3058. doi: 10.1046/j.1460-9568.2000.00183.x. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ. The mouse brain in stereotaxic coordinates. San Diego: Academic; 2001. [Google Scholar]
- Picciotto MR, Zoli M, Léna C, Bessis A, Lallemand Y, Le Novère N, Vincent P, Pich EM, Brûlet P, Changeux JP. Abnormal avoidance learning in mice lacking functional high-affinity nicotine receptor in the brain. Nature. 1995;374:65–67. doi: 10.1038/374065a0. [DOI] [PubMed] [Google Scholar]
- Poorthuis RB, Goriounova NA, Couey JJ, Mansvelder HD. Nicotinic actions on neuronal networks for cognition: general principles and long-term consequences. Biochem Pharmacol. 2009;78:668–676. doi: 10.1016/j.bcp.2009.04.031. [DOI] [PubMed] [Google Scholar]
- Ramirez-Latorre J, Yu CR, Qu X, Perin F, Karlin A, Role L. Functional contributions of alpha5 subunit to neuronal acetylcholine receptor channels. Nature. 1996;380:347–351. doi: 10.1038/380347a0. [DOI] [PubMed] [Google Scholar]
- Rigbi A, Kanyas K, Yakir A, Greenbaum L, Pollak Y, Ben-Asher E, Lancet D, Kertzman S, Lerer B. Why do young women smoke? V. Role of direct and interactive effects of nicotinic cholinergic receptor gene variation on neurocognitive function. Genes Brain Behav. 2008;7:164–172. doi: 10.1111/j.1601-183X.2007.00329.x. [DOI] [PubMed] [Google Scholar]
- Robbins TW, Granon S, Muir JL, Durantou F, Harrison A, Everitt BJ. Neural systems underlying arousal and attention. Implications for drug abuse. Ann N Y Acad Sci. 1998;846:222–237. [PubMed] [Google Scholar]
- Salas R, Orr-Urtreger A, Broide RS, Beaudet A, Paylor R, De Biasi M. The nicotinic acetylcholine receptor subunit alpha 5 mediates short-term effects of nicotine in vivo. Mol Pharmacol. 2003;63:1059–1066. doi: 10.1124/mol.63.5.1059. [DOI] [PubMed] [Google Scholar]
- Wada E, McKinnon D, Heinemann S, Patrick J, Swanson LW. The distribution of mRNA encoded by a new member of the neuronal nicotinic acetylcholine receptor gene family (alpha 5) in the rat central nervous system. Brain Res. 1990;526:45–53. doi: 10.1016/0006-8993(90)90248-a. [DOI] [PubMed] [Google Scholar]
- Zikopoulos B, Barbas H. Prefrontal projections to the thalamic reticular nucleus form a unique circuit for attentional mechanisms. J Neurosci. 2006;26:7348–7361. doi: 10.1523/JNEUROSCI.5511-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoli M, Picciotto MR, Ferrari R, Cocchi D, Changeux JP. Increased neurodegeneration during aging in mice lacking high-affinity nicotine receptors. EMBO J. 1999;18:1235–1244. doi: 10.1093/emboj/18.5.1235. [DOI] [PMC free article] [PubMed] [Google Scholar]