

Abstract
Neonatal ventral hippocampus (nVH) lesion in rats is a useful model to study developmental origins of adult cognitive deficits and certain features of schizophrenia. NVH lesion-induced reorganization of excitatory and inhibitory neurotransmissions within prefrontal cortical (PFC) circuits is widely believed to be responsible for many of the behavioral abnormalities in these animals. Here, we provide evidence that development of an aberrant medial PFC (mPFC) alpha-1 adrenergic receptor (α-1AR) function following neonatal lesion markedly affects glutamatergic synaptic plasticity within PFC microcircuits and contributes to PFC-related behavior abnormalities. Using whole-cell patch-clamp recording, we report that norepinephrine (NE)- induced α-1AR-dependent long-term depression (LTD) in a subset of corticocortical glutamatergic inputs is strikingly diminished in mPFC slices from nVH lesioned rats,. The LTD impairment occurs in conjunction with completely blunted α-1AR signaling through extracellular signal regulated kinase 1/2 (ERK1/2). These α-1AR abnormalities have functional significance in a mPFC-related function, i.e., extinction of conditioned fear memory. Post-pubertal animals with nVH lesion show significant resistance to extinction of fear by repeated presentations of the conditioned tone stimulus. Medial PFC infusion of an α-1AR antagonist (benoxathian) or LTD blocking peptide (Tat-GluR23Y) impaired fear extinction in sham controls, but had no significant effect in the lesioned animals. The data suggest that impaired α-1 adrenergic regulation of cortical glutamatergic synaptic plasticity may be an important mechanism in cognitive dysfunctions reported in neurodevelopmental psychiatric disorders.
INTRODUCTION
Neonatal ventral hippocampus (nVH) lesion induced by bilateral injection of the excitotoxin ibotenic acid in postnatal day 7 (PD7) rats has repeatedly been shown to result in post-pubertal abnormalities in cognitive, social, motivational and motor behaviors (Marcotte et al. 2001; Tseng et al. 2009). Striking resemblance of the nature and temporal course of these behaviors to schizophrenia led to the suggestion that nVH lesioned animals serve as a model to test the neurodevelopmental hypothesis of schizophrenia. Converging evidence suggest that the neonatal lesions significantly compromise the development and functions of medial PFC (mPFC)(Flores et al. 2005; O’Donnell et al. 2002; Ryan et al. 2013), a brain region that receives direct excitatory projections from the VH (Thierry et al. 2000; Carr and Sesack 1996). For example, we and other investigators have shown that nVH lesion leads to decrease in dendritic spines of pyramidal neurons (Flores et al. 2005; Ryan et al. 2013; Marquis et al. 2008), changes in the functional properties of glutamatergic and GABAergic synaptic inputs (Ryan et al. 2013) and altered regulation of pyramidal and putative interneurons in the PFC (O’Donnell et al. 2002; Gruber et al. 2010; Tseng et al. 2008). Aberrations in PFC functions, while considered to be key mechanisms in schizophrenia, are shared by many behavioral disorders (Volk and Lewis 2010; Gamo and Arnsten 2011); accordingly, it is being recognized that nVH lesion offers a unique paradigm to study mechanisms of PFC dysfunctions arising from a developmentally compromised brain, without regard to psychiatric diagnosis.
Work in our laboratory has shown that nVH lesion induces alterations in adult PFC α-1 adrenergic receptor (α-1AR) expression and function, with no significant change in α-2AR (Bhardwaj et al. 2004; Kamath et al. 2008; Al-Khairi et al. 2009). We have observed a reduced activation of protein kinase C (PKC) by α-1AR agonists in the mPFC of nVH lesioned animals ((Al-Khairi et al. 2009). Considerable evidence supports a role of noradrenergic system in the modulation of PFC-dependent attentional and cognitive functions (Berridge and Waterhouse 2003; Aston-Jones et al. 2000; Ramos and Arnsten 2007). Findings suggest that the plasticity of glutamate synapses in PFC could be an important mechanism by which adrenergic inputs regulate PFC activity and function. For example, norepinephrine (NE) acting on α-1 AR produces a long-term depression (LTD) of AMPA receptor mediated excitatory postsynaptic responses recorded from neurons in the mPFC (Marzo et al. 2010). LTD has been associated with PFC-related cognitive functions, such as behavioral flexibility (Nicholls et al. 2008).
Here we worked on the hypothesis that altered mPFC α-1AR plasticity may mediate some of the cognitive abnormalities in the nVH lesioned rats by disrupting PFC synaptic plasticity. We focused on fear memory extinction as a paradigm to assess potential cognitive correlate of abnormal α-1AR signaling as this behavior is associated with adrenergic transmission within the mPFC (Hugues et al. 2007; Mueller et al. 2008; Do-Monte et al. 2010). We report that α-1AR signaling through extracellular signal regulated kinase 1/2 (ERK1/2) and α-1AR-mediated LTD are markedly disrupted in the mPFC of nVH lesioned rats. We also find that the α-1AR abnormality and LTD deficits may be related to abnormal extinction of conditioned fear displayed by the lesioned animals.
METHODS
Neonatal VH lesion
Animal care and surgical procedures were according to guidelines of the Canadian Council of Animal Care and were approved by the McGill University Animal Care Committee. Timed pregnant Sprague–Dawley rats were obtained from Charles River Canada, and gave birth in our animal facility. NVH lesion was performed on PD7 male pups according to our previously described procedure (Flores et al. 1996; Ryan et al. 2013). Pups were anesthetized by hypothermia by covering in crushed ice for 18–20 min, and secured on a modified platform fixed to Kopf stereotaxic apparatus. The “lesion” group received bilateral VH infusions of 0.3μl ibotenic acid (Sigma, 10 μg/μl in 0.1 M PBS) over a period of 2 min, whereas the “sham” group received the same volume of PBS (coordinates: AP −3.0 mm from Bregma, ML +3.5 mm from midline, DV −5.0 mm from dura). After surgery, pups were placed on a heating pad until full recovery and returned to their respective mothers. After weaning (PD25), they were pair-housed (a sham and a lesioned per cage).
Effect of adrenergic drugs on ERK1/2 expression
PFC coronal slices (200μm, 2.2–3.7 mm anterior to bregma) from PD60 animals were obtained on a Leica vibratome in chilled carbogenated (95% O2/5% CO2) artificial cerebrospinal fluid (ACSF; (in mM): NaCl 125, KCl 2.5, NaHCO3 26, NaH2PO4 1.25, MgCl2 1, CaCl2 2, and D-glucose 25 and 0.1% protease inhibitor cocktail (Sigma cat P-8340), pH 7.35 and osmolarity 310–320.). Separate slices were incubated with either ACSF, or α-1AR agonist phenylephrine (PE, 20μM, Sigma) or PE + α-1AR antagonist prazosin (20 μM; Sigma) for 10 min at room temp. The area corresponding to the prelimbic (PL) and infralimbic (IL) mPFC was rapidly dissected and homogenized in buffer containing protease and phosphorylation inhibitors (0.2 mM PMSF, 1 mM leupeptin, 1 mM pepstatin, 0.2mM sodium orthovanadate, pH 7.4). Western blotting was done as described previously (Al-Khairi et al. 2009). The blotted membranes were incubated with anti-p-ERK1/2 antibody (1:1000, rabbit polyclonal, Cell Signaling, MA), followed by anti-rabbit IgG: horseradish peroxidase (HRP) second antibody. The blots were developed using chemiluminescence system (Perkin-Elmer). After exposure to X-ray film, the blot was stripped and reprobed with a polyclonal antibody against total-ERK 1/2 (1:2500, Cell Signaling). The membranes were re-stripped and probed using anti-tubulin antibody (1:5000, Sigma). Relative optical densities (ROD) of bands were analyzed on image analysis system (MCID-4, Imaging Research). ROD of p-ERK was normalized with t-ERK, and ROD of t-ERK was normalized with tubulin. The data were analyzed separately using 2-way analysis of variance (ANOVA) with lesion and drug treatment as independent variables, followed by post-hoc tukey’s test with p set at 0.05.
Electrophysiological recording
Coronal 400 μm PFC slices (PD42-57) were obtained as above and recovered at 32°C for 1 h in carbogenated ACSF. Slices were transferred to a submerged-type recording chamber and perfused with ACSF at a flow rate of 1.5 ml/min at room temperature. The recordings were done essentially as described by us previously (Bagot et al. 2012). Inhibitory synaptic function was reduced by the GABA-A receptor antagonist bicuculline (1μM, Sigma) in all recordings. Higher concentration of bicuculline was not used to avoid epileptiform discharges.
Whole cell recording of excitatory postsynaptic potential (EPSP) was performed on layer V pyramidal neurons as previously described (Ryan et al. 2013). Briefly, patch pipettes were pulled from borosilicate glass capillaries and filled with intracellular solution (pH 7.25, 280–290 mOsm) composed of (in mM): 120 mM K-gluconate, 17.5 KCl, 10 HEPES, 2 MgCl2, 0.5 ethylene glycol tetraacetic acid (EGTA), 4 ATP and 5 QX-314 (pH 7.2). After breakthrough, cortical neurons were injected with several 200 ms long hyperpolarizing pulses to estimate the input resistance (179.3 ± 15.7 MΩ). EPSPsuperficial and EPSPdeep were evoked by stimulating the superficial (layer I/II) and deep cortical layers (layer VI) via constant current pulses (0.08 ms) delivered through tungsten bipolar electrodes. EPSPsuperficial corresponds to excitatory inputs from pyramidal neurons in layer II/III pyramidal neurons, which relay information from layer VI neurons that received thalamocortical synaptic inputs, while EPSPdeep are mediated by collaterals between deep layer pyramidal neurons (Thomson and Deuchars 1997). NE (20 μM, Sigma) was added into the perfusing solution to induce LTD of EPSP for 10min. In experiments when two independent pathways were sequentially stimulated from superficial and deep cortical layers, the inter-stimulation intervals were set to be 700 ms. Access resistance of the whole-cell recording was monitored continuously so that only recordings with low (< 15 MΩ) and stable access resistance (< 20% change) were kept for analysis.
Fear Conditioning and Extinction
PD60 sham and lesioned animals were used in the standard paradigm of fear conditioning essentially as described previously (Bhardwaj et al. 2009). Two minutes after placement in the boxes (Kinder Scientific, CA), the animals received 4 pairings of tone and footshock with a variable inter-trial interval (ITI) averaging 120s. The tone (85 dB) had duration of 30 s; during the last second of the tone, a foot shock (0.5 mA, 1s duration) was delivered. Contextual fear memory was tested the next day in the same chamber without any foot shock or tone. On day 3, auditory cued fear was determined in a novel box. After 3 min of habituation, animals were exposed to 3 min of continuous auditory tone of 85dB. The behavior of animals was videotaped and analyzed for the proportion of time spent freezing, in 5-s bin, by a person blind to experimental status. Freezing was defined as a lack of body movement, including head movement, and not in resting position.
Extinction of fear memory was assessed in a new cohort of rats essentially as described by Mueller et al (Mueller et al. 2008). Twenty four hours after tone-shock conditioning, extinction learning was evaluated in the conditioning box. After 2 min acclimatization, animals were exposed to a series of 14 tones (85dB, 30s duration) with a variable ITI (90–180s). Freezing behavior was analyzed as above. On day 3, recall (retrieval) of extinction memory was similarly evaluated by 6 presentations of the tone. The data is presented as block of two trials (averaged) during extinction learning and extinction recall.
MPFC microinfusion of α-1AR antagonist benoxathian and GluR23Y peptide
A cohort of animals (PD50-60) were anesthetized with isoflurane and implanted with a bilateral 26-gauge stainless-steel guide cannula (Plastics One, Inc.) aimed at infra-limbic PFC region [AP +2.9; ML ±0.75; and DL −4.1] (Paxinos and Watson 2007). After 7d, animals were fear conditioned as described above. A day later, animals were microinjected bilaterally with a water soluble α-1AR antagonist benoxathian (Sigma) (10 nmol in 0.3 μl/side) or ACSF to create 4 groups of animals: sham-ACSF, sham-benoxathian, lesion-ACSF and lesion-benoxathian. Thirty minutes later, fear extinction was assessed by giving 14 tone-alone trials as described in the previous section. Twenty four hours later, recall of fear was evaluated in the same animals.. Another group of conditioned animals (sham and lesioned) were similarly microinjected with either Tat-GluR23Y peptide or scrambled peptide (15 pmol each; Sheldon Biotechnology, McGill), Fear extinction and recall were assessed as described above. Drug doses were selected based on previous reports indicating effective blockade of α-1AR in mPFC (Nicniocaill and Gratton 2007) and AMPAR endocytosis (Brebner et al. 2005). Data were analyzed by two or three-way repeated measure ANOVA followed by post-hoc tukey’s test. Student’s t-test was used for the analysis of the contextual memory.
Lesion and cannula verification
After experiment, animals were sacrificed and 35-μm coronal sections at the level of the ventral hippocampus or mPFC were stained with 0.5% Cresyl Violet. The extent of hippocampal damage was ascertained using a light microscope and MCID system.
RESULTS
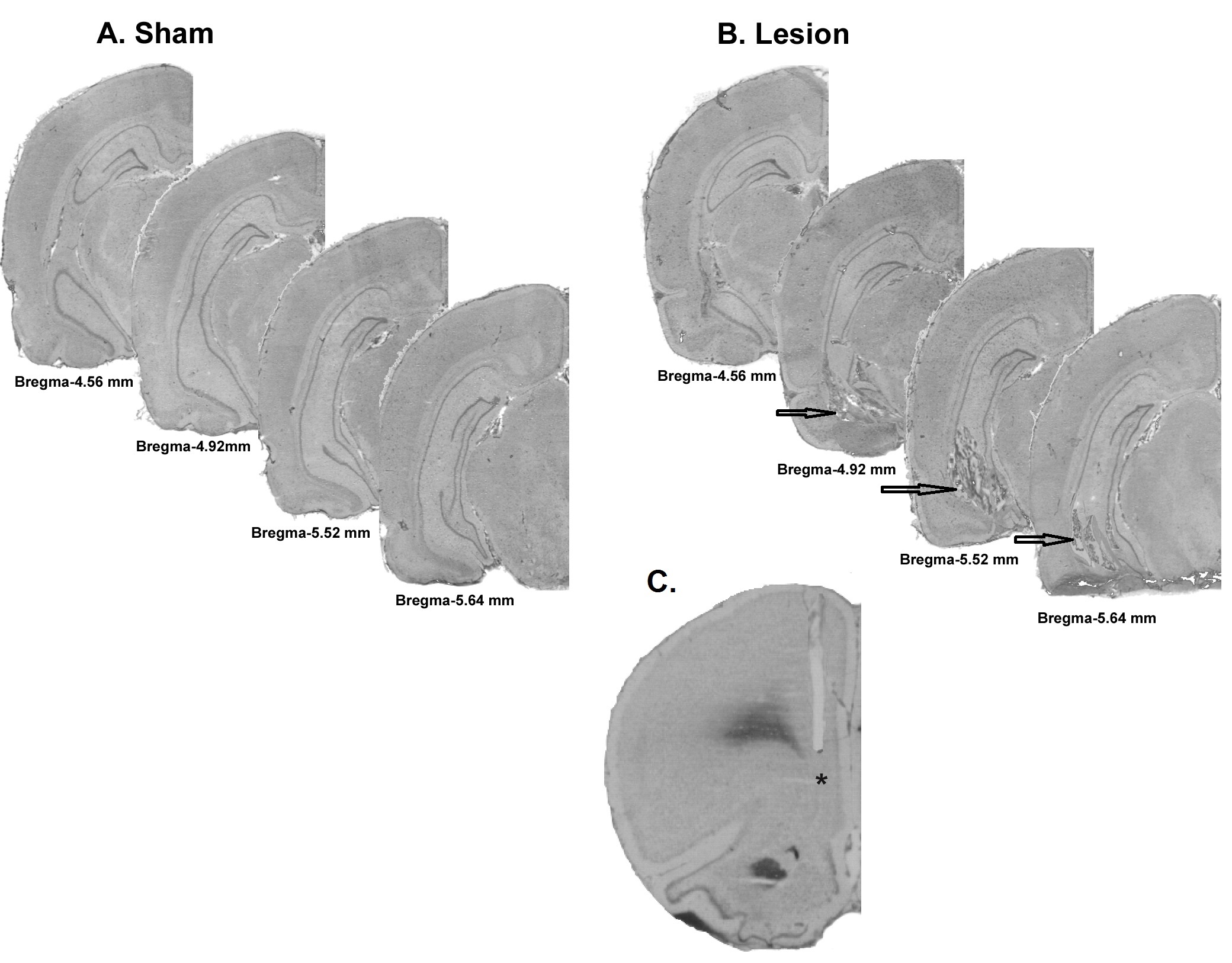
As reported earlier (Flores et al. 1996; Bhardwaj et al. 2004), nVH lesioned animals showed bilateral neuronal loss, retraction, and cavitations in the ventral half of the hippocampus including the CA1 (Fig S1). Only animals showing bilateral neuronal loss and atrophy of the VH with no significant damage to dorsal hippocampus or adjacent nuclei were included in data analyses.
Effect of nVH lesion on ERK1/2 activation in PFC slices
We previously reported on a blunted α-1 activation of PKC in nVH lesioned animals (Al-Khairi et al. 2009). Here, we evaluated mPFC ERK1/2 activation by a specific α-1AR agonist PE (20μM) in sham and lesioned animals (n=7each). Fig 1A show representative Western blots of phospho-ERK1/2 (p-ERK1/2) and total ERK1/2 (t-ERK1/2). Two-way ANOVA of p-ERK1/2 level (ratio of p-ERK1/2 to total ERK1/2, Fig 1B) showed a significant lesion x drug interaction (F2,36=4.73; p=0.015). Incubation with PE increased ERK1/2 phosphorylation by over 2.5 fold (p=0.001) in slices from sham animals, which was blocked by co-incubation with the antagonist prazosin. Notably, however, PE completely failed to increase p-ERK1/2 level in the lesioned animals, suggesting an uncoupling between α-1AR and ERK1/2 signaling. NVH-lesioned animals without agonist application (ACSF group) although appear to have increased basal p-ERK1/2 level compared to sham-ACSF; however, this is not statistically significant (p=0.14). Lastly, total ERK1/2 level (ratio of t-ERK1/2 vs α-tubulin) was not significantly different between sham and lesioned groups (F2,36=0.22; p=0.80) (Fig 1C).
Figure 1.

Effect of nVH lesion on ERK 1/2 activation in PFC slices. A, representative western blot signals using antibodies for (phospho) p-ERK 1/2, (total) t-ERK 1/2 and alpha-tubulin. B and C show mean ± SEM ratio of relative optical density (ROD) of p-ERK1/2 vs t-ERK 1/2, as well as t-ERK 1/2 vs tubulin. Incubation with α-1AR agonist phenylephrine (PE) stimulates p-ERK 1/2 the sham but not lesioned animals (**p=0.001). PE-induced p-ERK1/2 stimulation is α-1AR dependent as it is completely blocked by antagonist prazosin (Pr) (##p=0.001, sham PE vs sham PE+Pr).
Impairment of α-1AR-induced LTD in the mPFC of nVH lesioned rats
NE acting on α-1AR produces a LTD of AMPAR-mediated excitatory postsynaptic responses in the cerebral cortex (Kirkwood et al. 1999), hippocampus (Scheiderer et al. 2004) and PFC (Marzo et al. 2010), and this LTD is mediated by ERK1/2. Here, we tested if changes in α-1AR mediated ERK1/2 activation in nVH lesioned rats could affect NE-related synaptic plasticity in the mPFC. Under current clamp mode, excitatory postsynaptic potentials (EPSPs) evoked by stimulating either the superficial (layer I/II; EPSPsuperficial) (Fig 2A) or the deep cortical layer (layer VI; EPSPdeep) (Fig 2B) were recorded from layer V pyramidal neurons in the mPFC. In sham operated rats (n=4), NE (20 μM, 10 min) induced LTD of EPSPs evoked by stimulating superficial cortical layers (changes in EPSPsuperficial at 30 min after NE application: −33.2 ± 9.4%; vs. baseline: t(5) = 3.53, p = 0.017, n = 6 slice) (Fig 2C). Moreover, NE failed to induce LTD of EPSPs evoked by stimulating deep cortical layers (changes in EPSPdeep : −6.2 ± 12.5%; vs. baseline: t(6) = 0.50, p = 0.637, n = 7 slice) (Fig 2D). Surprisingly, no depression of EPSPsuperficial was produced by NE in nVH lesioned rats (n=5). In fact, student’s t-test comparison revealed a trend of enhancement of EPSPsuperficial at 30 min after NE treatment (27.3 ± 11.7%; vs. baseline: t(6) = 2.34, p = 0.057, n = 7 slices) (Fig 2E). Similar to its effect on sham operated control rats, NE did not affect EPSPdeep in nVH lesioned rats (−9.6 ± 7.2%; vs. baseline: t(5) = 1.34, p = 0.239, n = 6 slice) (Fig 2F). Two-way ANOVA on the data of lesion and inputs (superficial vs. deep) on NE-LTD of EPSP and found significant interaction between inputs and lesion (F(1,19) = 5.67; p = 0.028). Post hoc Tukey’s test revealed significant difference of the slope of EPSPsuperficial between sham and nVH lesioned rats (p = 0.012).
Figure 2.

Norepinephrine (NE)-induced LTD in the PFC is abolished by nVH-lesion. Figure A left shows the infralimbic region of the prefrontal cortex (dotted line circle) that was examined in this study. A and B show the schematic diagrams depicting the placement of recording and stimulating electrodes. Whole-cell EPSPs evoked by stimulating either the layer I/II (EPSPsuperficial) or layer VI cortical inputs (EPSPdeep) were recorded from layer V pyramidal neurons. C, A brief NE treatment (20 μM, 10-min) induced LTD of EPSPsuperficial in slices obtained from sham-operated rats. D, Note that similar NE treatment did not produce long-lasting change of EPSPdeep. E, F, NE failed to induce LTD of EPSPsuperficial and EPSPdeep in slices obtained from nVH lesion rats. * p < 0.05 vs. baseline EPSP before NE application. G, Using slices obtained from control rats, NE-LTD of EPSPsuperficial can be abolished by α-1 adrenergic receptor antagonist prazosin (10 μM). H, A short Tat-GluR23Y peptide, which is known to inhibit the endocytosis of AMPA receptor, was delivered to layer V pyramidal neurons recorded from slices of control rats through whole-cell recording. NE failed to induce LTD of EPSPsuperficial in peptide-treated neurons.
NE-LTD was abolished by an α-1 AR antagonist prazosin (changes in EPSPsuperficial at 30 min after NE application: 4.7 ± 9.1%; vs. baseline: t(3) = 0.51, p = 0.866, n = 4, two rats) (Fig 2G), suggesting the involvement of α-1 AR. AMPA receptor endocytosis is an important expression mechanisms for activity-dependent LTD in both the hippocampus and mPFC (Collingridge et al. 2010). In support of the requirement of AMPAR endocytosis for LTD in mPFC, it has been shown that an interfering peptide GluA23Y, that disrupts tyrosine phosphorylation of GluA2 subunit (Ahmadian et al. 2004), impairs LTD formation (Van den Oever et al. 2008). To examine if NE-LTD expression is mediated by AMPAR endocytosis, we added the GluA23Y peptide into intracellular solution (100 ng/ml) and found that NE failed to induce LTD (changes in EPSPV at 30 min after NE application: 2.2 ± 10.2%; vs. baseline: t(3) = 0.21, p = 0.845, n = 4, from 4 rats) (Fig 2H).
NVH lesion leads to impaired fear extinction
Two-way repeated measure ANOVA of conditioning data (n=7 each group) showed no significant effect of lesion (F1,48= 0.55, p=0.47) or lesion x trial interaction (F(4,,48= 2.08, p=0.1)(Fig 3A). Student’s t- test of contextual fear memory showed no significant difference the sham and lesioned animals (p=0.70) (Fig 3B). Similarly, two-way ANOVA of auditory fear memory revealed no significant effect of lesion (F(1,11)=0.73, p=0.41) or lesion x tone interaction (F(1,11)=0.52,p=0.49) (Fig 3C).
Figure 3.

Fear memory and extinction in sham and nVH lesioned animals. A, Mean (± SEM) fear behavior (% time freezing) during fear conditioning is not significantly different between sham and nVH lesioned rats. B, The two groups of rats also do not differ in contextual fear memory assessed by exposure to the conditioning context 24 h following fear conditioning. C, Auditory fear memory by the presentation of conditioned stimulus (tone) 48 h following fear conditioning also did not differ in the sham and nVH lesioned rats. D, nVH lesion leads to impaired extinction of fear memory as lesioned animals show increased fear behavior compared to sham animals during presentation of repeated tone trials. E, Lesioned animals show impaired recall of fear memory as assessed by tone presentation, a day after extinction learning trials. The extinction training and recall data are presented as pool of 2 tone trials. *Two-way ANOVA of extinction training and recall showing significant effect of lesion (p=0.04 and p=0.008 respectively).
Analysis of extinction data (n=7 each) showed that nVH lesion causes a significant impairment in the extinction of conditioned fear memory. During extinction trials, sham animals showed reduction in freezing which decreased to approximately 20% after 14 trials, whereas lesioned animals showed only about 50% reduction at the end of the trials. Two-way ANOVA of extinction learning trials showed a significant effect of lesion (F(1,72)=5.34, p=0.04) and tone (F(6,72)=5.76, p=0.0001) but no lesion x tone interaction (F(6,72)=0.85,p=0.53) (Fig 3D). Analysis of recall of fear memory 24h after extinction also showed a significant main effect of lesion (F(1,24)=10.24, p=0.008) but not tone trials (F(2,24)=0.44, p=0.064) or lesion x tone interaction (F(2,24)=1.76,p=0.193) (Fig 3E).
Blockade of α-1AR in mPFC impairs fear extinction in sham animals, but does not exacerbate extinction deficit in lesioned animals
We investigated if abnormality in mPFC α-1AR function could be related to extinction deficits of nVH animals, by microinfusing α-1AR antagonist benoxathian (n=6–7 each group) (Fig 4A). Three-way ANOVA showed a significant effect of lesion (F(1,126)=12.37, p=0.002) as well benoxathian x lesion interaction (F(1,126)=10.92, p=0.003). A 2-way ANOVA comparing average % freezing at the last block of 2 trials indicates that sham animals with benoxathian infusion, compared to ACSF infusion, show higher freezing, i.e, resistance to extinction (lesion x drug interaction, F(1,21)=4.40,p=0.043; post-hoc p=0.002) (Fig 4B). The lesioned animals with ACSF infusion showed impaired extinction compared to sham-ACSF group (p=0.004) as observed earlier (Fig 3D). However, α-1AR antagonist infusion had no further effect in lesioned animals. Taken together with our observations of abnormal α-1AR signaling and LTD, these data showing similar impairments of extinction in lesioned animals without drug and controls with mPFC α-1 blockade, indicate a relationship between dysfunctional α-1AR and impaired extinction after nVH lesion.
Figure 4.

Effect of mPFC α-1 adrenergic blockade on extinction memory in sham and nVH lesioned animals. A, shows extinction learning curve of rats infused with either ACSF or benoxathian (Ben) bilaterally into infralimbic PFC, 30 minutes before the session (arrow sign). B, Bar diagram of mean (± SEM) freezing response at the last block of 2 trials in the four groups of animals. Sham animals with mPFC benoxathian infusion, compared to ACSF, show higher freezing indicating resistance to extinction learning. However, the α-1AR antagonist has no significant effect in nVH lesioned animals, i.e., do not exacerbate extinction deficit of lesioned animals (*p=0.002 and #p=0.004 compared to sham-ACSF group. C, Freezing responses of the animals during the extinction recall. D, Mean ± SEM of freezing response at the last block of 2 tone trials during recall (#p=0.0003 compared to sham-ACSF).
The effect of benoxathian infusion in sham and lesioned animals during recall of extinction memory parallels its effect seen during extinction learning (Fig 4C,D). Three-way ANOVA showed a significant effects of lesion (F(1,42)=23.71, p=0.001) and lesion x drug (F(1,42)=7.36, p=0.013)(Fig 4C). A 2-way ANOVA of % freezing during the last block of trial showed a significant lesion x drug interaction (F(1,20)=4.74, p=0.042). Sham-benoxathian animals freeze at higher levels at the end of recall session compared to sham-ACSF animals (23.4% vs. 11.1%) (Fig 4D); however, this change did not reach statistical significance. Lesioned -ACSF animals also show impaired recall memory compared to sham-ACSF group. However, as observed for extinction learning, benoxathian infusion had no significant effect in the lesioned animals.
MPFC blockade of AMPA receptor endocytosis disrupts fear extinction in sham animals, but has no significant effect in lesioned animals
We examined the effect of infralimbic infusion of Tat-GluR23Y peptide given 30 minutes before extinction (n=9–13). Three-way ANOVA of extinction trials showed a significant effect of lesion (F(1,222)=7.25, p=0.01) and lesion x peptide interaction (F(1,222)=6.54, p=0.014), but not of peptide alone (F(1,222)=0.57, p=0.45) (Fig 5A). Comparing % freezing at the last block of trial indicates that sham animals with Tat-GluR23Y infusion, compared to scrambled peptide infusion, show significant disruption in extinction learning (lesion x peptide interaction: F(1,37)=11.00, p=0.002; pair-wise post hoc p=0.005). Lesion-scambled peptide animals showed significantly higher freezing than sham-scrambled peptide group (p<0.001); however, similar to the observations with benoxathian, Tat-GluR23Y peptide had no significant effect in the lesioned animals, (Fig 5B).
Figure 5.

Effect of mPFC infusion of AMPA receptor endocytosis blocker Tat-GluR23Y on extinction memory in sham and nVH lesioned animals. A, shows extinction learning curve of rats infused with either scrambled peptide (Scr Pep, 15 pmoles) or Tat-GluR23Y peptide (Tat-Pep, 15 pmoles) bilaterally into infralimbic PFC, 30 minutes before the session (arrow sign). B, Bar diagram of mean (± SEM) freezing response at the last block of 2 trials in the four groups of animals. Lesion-Scr Pep animals show increased freezing compared to sham-Scr Pep (#p<0.001) indicating resistance to extinction. Tat-GluR23Y peptide infusion caused increased freezing in sham animals (*p=0.005 vs sham-Scrambled peptide). However, the Tat-GluR23Y peptide has no significant effect in lesioned animals. C, Freezing responses of the animals during the extinction recall session. D, Mean ± SEM of freezing responses at the last block of 2 tone trials during recall. Lesioned animals with scrambled peptide infusion show impaired recall memory compared to sham animals with scrambled peptide (*, main effect of lesion, p=0.0002). Tat-GluR23Y peptide had no significant effect on extinction recall in sham or nVH lesioned animals.
The effect of peptide in recall of extinction memory is also similar to its effect seen during extinction learning (Fig 5C,D) in that a three-way ANOVA showed a significant main effect of lesion (F(1,74)=11.99, p=0.001). However, no significant effect of Tat-peptide (F(1,74)=0.78, p=0.38) or lesion x peptide interaction was observed on extinction recall (F(1,74)=2.02, p=0.164) (Fig 5C). Analysis of last block of trials shows that scrambled peptide-infused nVH lesioned animals showed impaired fear recall compared to scrambled peptide-infused sham animals (F(1,137)=16.96, p=0.0002). However, Tat-peptide infusion in the sham or lesioned animals produced no significant effects on extinction recall (Fig 5D).
DISCUSSION
The principal finding of our study is that the lesion of ventral hippocampus at a developmentally critical age in rats leads to postpubertal alterations in prefrontal α-1AR-mediated signaling, synaptic plasticity and behavior. A key α-1AR mediated intracellular signaling mechanism, i.e.,ERK1/2 activation, is anomalous in the mPFC of lesioned animals. Incubation with a specific α-1AR agonist increased ERK1/2 phosphorylation in the control mPFC as expected; however, this activation was not observed in the lesioned animals. There was no significant change in the level of total ERK1/2 in the lesioned group. Agonist activation of α1-AR leads to intracellular signaling cascade that includes, among others, PKC and ERK1/2 (Koshimizu et al. 2003). We have previously reported that PKC activation by α-1AR is also blunted in the prefrontal slices of nVH lesioned animals (Al-Khairi et al. 2009). Thus, taken together, our data point to a severe dysregulation of cortical α-1AR function in the lesioned animals.
Based on the reported roles of α-1AR and ERK1/2 in PFC LTD (Marzo et al. 2010) and our recent observation of decreased number of dendritic spines in mPFC pyramidal neurons in nVH lesioned animals (Ryan et al. 2013), we asked whether mPFC neurons of lesioned animals show changes in α-1AR-mediated plasticity of glutamate synapses. Our results suggest that the influence of α-1AR on the plasticity of glutamatergic transmission is dramatically diminished in nVH lesioned rats. Incubation of PFC slices of sham-operated animals with NE led to an LTD of excitatory postsynaptic response of layer V pyramidal cells evoked by stimulating superficial layers of mPFC, which can be blocked by an α-1AR antagonist prazosin. This form of α-1 dependent NE-LTD (α-1AR LTD) has been previously demonstrated in other brain regions as well (Kirkwood et al. 1999; Scheiderer et al. 2004; McElligott and Winder 2008). Most interestingly, we found that the α-1AR-LTD was completely absent in the mPFC of nVH lesioned rats. This failure in LTD induction in the lesioned animals may be related to our finding of the failure of α-1AR to trigger downstream signaling such as ERK1/2. It is important to note that the short term depression of EPSPsuperficial during and shortly after NE application was intact in slices from control and lesioned rats, suggesting that only LTD of EPSPsuperficial was affected. While NE-LTD is likely mediated by AMPAR endocytosis that is sensitive to Tat-GluR23Y peptide, mechanism underlying NE-induced short term depression seems to be related to α2 adrenergic receptor activation (Marzo et al., 2010). These findings are consistent with our previous report showing selective changes in α-1AR, but not α-2AR in nVH lesioned rats (Bhardwaj et al. 2004).
Our focus on fear memory extinction as a paradigm to assess cognitive correlate of abnormal α-1AR signaling and plasticity was predicated on the reasoning that this behavior is associated with adrenergic transmission and long-term plasticity within the mPFC neurons (LeDoux 2000; Quirk and Mueller 2008; Mueller and Cahill 2010). Further, while impaired fear extinction is common in conditions of altered adrenergic responsivity, such as posttraumatic stress disorder and stress (Holmes and Wellman 2008), a deficit in fear extinction memory has also been shown in individuals suffering from schizophrenia (Holt et al. 2009). Fear extinction is widely believed to be a new form of learning, and is modulated by inputs from medial PFC to the amygdala. Increased IL PFC neuronal activity is associated with retrieval of extinction memory (Milad and Quirk 2002; Santini et al. 2008), and mPFC NE (Hugues et al. 2007) and activation of α-1 as well as β-AR associated PKA signaling in the IL PFC appear necessary for fear extinction (Mueller et al. 2008; Do-Monte et al. 2010).
Our data show that nVH lesioned animals have significant impairments in learning to extinguish fear memory and consolidating extinction memory. These impairments are apparently not due to altered anxiety-like behaviors of lesioned animals as previous studies have shown no significant change in anxiety-like behaviors (Wood et al. 2003). It may be argued that these deficits are due to altered functioning of amygdala or the hippocampus; however, due to normal expression of fear during conditioning or in the tests of contextual and auditory fear memory, it seems less likely. Further, adult hippocampus inactivation does not significantly affect acquisition of extinction (Corcoran et al. 2005).
We also find that, unlike in controls, mPFC infusion of α-1AR antagonist in nVH lesioned animals is without any significant effect, i.e., the extinction learning and memory impairments are not exacerbated. We believe that impaired fear extinction in nVH lesioned animals is due to mPFC α-1AR abnormality as these receptors in nVH animals are non-responsive to agonist stimulation, and because α-1AR-dependent LTD in mPFC circuit is unable to form.
LTD-related mechanisms have been reported to facilitate extinction and consolidation of extinction memory in rats. Systemic administration of Tat-GluR23Y peptide that blocks AMPAR endocytosis, a key mechanism in LTD formation, interferes with learning of extinction as well as recall of fear memory (Dalton et al. 2008). Our data showing deficits in extinction learning and recall by mPFC microinfusion of Tat-GluR23Y peptide in sham animals suggest that LTD in the mPFC is necessary for normal extinction learning, but not for recall. Further, similar to the results with α-1AR antagonist benoxathian, mPFC microinfusion of Tat-GluR23Y peptide in the lesioned animals did not exacerbate extinction learning or recall. This data, suggestive of mPFC LTD deficit in lesioned animals is consistent with our in vitro data showing a loss of α-1AR mediated LTD in layer1/2→5 of the mPFC.
Tat-GluR23Y peptide would also affect LTD that is induced by non adrenergic stimuli, e.g., glutamate NMDA receptor (Ge et al. 2010). However, similar effects of this peptide and α-1AR antagonist on fear extinction may suggest a common cellular mechanism targeted by these two distinct pharmacological reagents, i.e.,α-1AR-LTD in the mPFC. Previous studies suggest that extinction is associated with increased excitability of IL PFC excitatory neurons that project to amygdala to inhibit fear (Quirk and Beer 2006). While it seems counterintuitive that LTD, which could reduce neuronal excitability, promotes fear extinction, it is important to note that only a subset of cortico-cortical inputs from the superficial layer I/II to layer V pyramidal neurons can be depressed by NE. Selective α-1 adrenergic suppression of these inputs could reduce synaptic noises from intrinsic cortical network, which could facilitate detection of stimuli from other cortical and more important extrinsic inputs that could influence layer V pyramidal neuronal excitability. This increase in signal/noise ratio mediated by α-1AR could have significant impact on enhancing extinction that requires detection of dissociation between salient cues and noxious stimuli. Diminution of this α-1AR function, we believe, may be responsible for extinction learning/memory deficit in nVH lesioned animals. At the same time, it is possible that other PFC abnormalities described in the nVH lesioned animals may also be involved in extinction deficits in these animals. In particular, mPFC GABAergic interneuron deficit described in the lesioned animals (Tseng et al. 2008)could contribute to extinction deficits by altering excitability of the mPFC pyramidal cells. It has been shown that GABA-A receptor agonist muscimol infusion in the IL PFC before extinction training results in long-term facilitation of extinction (Akirav et al. 2006). Further, extinction of reward seeking behavior, which shares PFC neural circuits with fear extinction (Peters et al. 2009), was recently shown to be facilitated by optogenetic stimulation of fast spiking parvalbumin interneurons in the PL mPFC of mice (Sparta et al. 2014).
NE is increasingly being recognized as an important modulator of prefrontal cognitive functions, impairments in which is a hallmark of schizophrenia and other psychiatric disorders (Arnsten 2011). Whereas excessive α-1 activation impairs working memory functions (Arnsten 2000; Arnsten et al. 1999), noradrenergic activity at mPFC α1-receptors facilitates attentional and cognitive performance of rats, e.g., behavioral flexibility (Lapiz and Morilak 2006; Sirvio and MacDonald 1999). Our present set of data linking impaired α-1 adrenergic regulation of cortical glutamatergic synaptic plasticity and cognition in nVH lesion model may represent novel mechanisms of prefrontal dysfunctions observed in neurodevelopmental mental disorders.
Supplementary Material
{kind=link}
Acknowledgments
FUNDING
The study was supported by a grant from the Canadian Institutes of Health Research (MOP-68922).
Footnotes
DISCLOSURE
The authors declare no competing financial interests.
Supplementary information is available at the Neuropsychopharmacology website.
Reference List
- Ahmadian G, Ju W, Liu L, Wyszynski M, Lee SH, Dunah AW, et al. Tyrosine phosphorylation of GluR2 is required for insulin-stimulated AMPA receptor endocytosis and LTD. EMBO J. 2004;23:1040–1050. doi: 10.1038/sj.emboj.7600126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akirav I, Raizel H, Maroun M. Enhancement of conditioned fear extinction by infusion of the GABAA agonist muscimol into the rat prefrontal cortex and amygdala. European Journal of Neuroscience. 2006;23:758–764. doi: 10.1111/j.1460-9568.2006.04603.x. [DOI] [PubMed] [Google Scholar]
- Al-Khairi I, Baharnoori M, Kamath A, Bhardwaj SK, Srivastava LK. Altered expression and alpha-1 adrenergic receptor mediated activity of protein kinase C in the prefrontal cortex of rats with neonatal ventral hippocampus lesions. Synapse. 2009;63:1051–1059. doi: 10.1002/syn.20691. [DOI] [PubMed] [Google Scholar]
- Arnsten AF. Stress impairs prefrontal cortical function in rats and monkeys: role of dopamine D1 and norepinephrine alpha-1 receptor mechanisms. Prog Brain Res. 2000;126:183–192. doi: 10.1016/S0079-6123(00)26014-7. [DOI] [PubMed] [Google Scholar]
- Arnsten AF. Catecholamine influences on dorsolateral prefrontal cortical networks. Biol Psychiatry. 2011;69:e89–e99. doi: 10.1016/j.biopsych.2011.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnsten AF, Mathew R, Ubriani R, Taylor JR, Li BM. Alpha-1 noradrenergic receptor stimulation impairs prefrontal cortical cognitive function. Biol Psychiatry. 1999;45:26–31. doi: 10.1016/s0006-3223(98)00296-0. [DOI] [PubMed] [Google Scholar]
- Aston-Jones G, Rajkowski J, Cohen J. Locus coeruleus and regulation of behavioral flexibility and attention. Prog Brain Res. 2000;126:165–182. doi: 10.1016/S0079-6123(00)26013-5. [DOI] [PubMed] [Google Scholar]
- Bagot RC, Tse YC, Nguyen HB, Wong AS, Meaney MJ, Wong TP. Maternal care influences hippocampal N-methyl-D-aspartate receptor function and dynamic regulation by corticosterone in adulthood. Biol Psychiatry. 2012;72:491–498. doi: 10.1016/j.biopsych.2012.03.016. [DOI] [PubMed] [Google Scholar]
- Berridge CW, Waterhouse BD. The locus coeruleus-noradrenergic system: modulation of behavioral state and state-dependent cognitive processes. Brain Res Brain Res Rev. 2003;42:33–84. doi: 10.1016/s0165-0173(03)00143-7. [DOI] [PubMed] [Google Scholar]
- Bhardwaj SK, Baharnoori M, Sharif-Askari B, Kamath A, Williams S, Srivastava LK. Behavioral characterization of dysbindin-1 deficient sandy mice. Behav Brain Res. 2009;197:435–441. doi: 10.1016/j.bbr.2008.10.011. [DOI] [PubMed] [Google Scholar]
- Bhardwaj SK, Quirion R, Srivastava LK. Post-pubertal adrenergic changes in rats with neonatal lesions of the ventral hippocampus. Neuropharmacology. 2004;46:85–94. doi: 10.1016/j.neuropharm.2003.08.001. [DOI] [PubMed] [Google Scholar]
- Brebner K, Wong TP, Liu L, Liu Y, Campsall P, Gray S, et al. Nucleus accumbens long-term depression and the expression of behavioral sensitization. Science. 2005;310:1340–1343. doi: 10.1126/science.1116894. [DOI] [PubMed] [Google Scholar]
- Carr DB, Sesack SR. Hippocampal afferents to the rat prefrontal cortex: synaptic targets and relation to dopamine terminals. J Comp Neurol. 1996;369:1–15. doi: 10.1002/(SICI)1096-9861(19960520)369:1<1::AID-CNE1>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Collingridge GL, Peineau S, Howland JG, Wang YT. Long-term depression in the CNS. Nat Rev Neurosci. 2010;11:459–473. doi: 10.1038/nrn2867. [DOI] [PubMed] [Google Scholar]
- Corcoran KA, Desmond TJ, Frey KA, Maren S. Hippocampal inactivation disrupts the acquisition and contextual encoding of fear extinction. J Neurosci. 2005;25:8978–8987. doi: 10.1523/JNEUROSCI.2246-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalton GL, Wang YT, Floresco SB, Phillips AG. Disruption of AMPA receptor endocytosis impairs the extinction, but not acquisition of learned fear. Neuropsychopharmacology. 2008;33:2416–2426. doi: 10.1038/sj.npp.1301642. [DOI] [PubMed] [Google Scholar]
- Do-Monte FH, Allensworth M, Carobrez AP. Impairment of contextual conditioned fear extinction after microinjection of alpha-1-adrenergic blocker prazosin into the medial prefrontal cortex. Behav Brain Res. 2010;211:89–95. doi: 10.1016/j.bbr.2010.03.014. [DOI] [PubMed] [Google Scholar]
- Flores G, Alquicer G, Silva-Gomez AB, Zaldivar G, Stewart J, Quirion R, et al. Alterations in dendritic morphology of prefrontal cortical and nucleus accumbens neurons in post-pubertal rats after neonatal excitotoxic lesions of the ventral hippocampus. Neuroscience. 2005;133:463–470. doi: 10.1016/j.neuroscience.2005.02.021. [DOI] [PubMed] [Google Scholar]
- Flores G, Barbeau D, Quirion R, Srivastava LK. Decreased binding of dopamine D3 receptors in limbic subregions after neonatal bilateral lesion of rat hippocampus. J Neurosci. 1996;16:2020–2026. doi: 10.1523/JNEUROSCI.16-06-02020.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamo NJ, Arnsten AF. Molecular modulation of prefrontal cortex: rational development of treatments for psychiatric disorders. Behav Neurosci. 2011;125:282–296. doi: 10.1037/a0023165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge Y, Dong Z, Bagot RC, Howland JG, Phillips AG, Wong TP, et al. Hippocampal long-term depression is required for the consolidation of spatial memory. Proc Natl Acad Sci U S A. 2010;107:16697–16702. doi: 10.1073/pnas.1008200107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruber AJ, Calhoon GG, Shusterman I, Schoenbaum G, Roesch MR, O’Donnell P. More Is Less: A Disinhibited Prefrontal Cortex Impairs Cognitive Flexibility. J Neurosci. 2010;30:17102–17110. doi: 10.1523/JNEUROSCI.4623-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes A, Wellman CL. Stress-induced prefrontal reorganization and executive dysfunction in rodents. Neurosci Biobehav Rev. 2008;33:773–783. doi: 10.1016/j.neubiorev.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt DJ, Lebron-Milad K, Milad MR, Rauch SL, Pitman RK, Orr SP, et al. Extinction memory is impaired in schizophrenia. Biol Psychiatry. 2009;65:455–463. doi: 10.1016/j.biopsych.2008.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hugues S, Garcia R, Lena I. Time course of extracellular catecholamine and glutamate levels in the rat medial prefrontal cortex during and after extinction of conditioned fear. Synapse. 2007;61:933–937. doi: 10.1002/syn.20448. [DOI] [PubMed] [Google Scholar]
- Kamath A, Al-Khairi I, Bhardwaj S, Srivastava LK. Enhanced alpha1 adrenergic sensitivity in sensorimotor gating deficits in neonatal ventral hippocampus-lesioned rats. Int J Neuropsychopharmacol. 2008;11:1085–1096. doi: 10.1017/S1461145708008845. [DOI] [PubMed] [Google Scholar]
- Kirkwood A, Rozas C, Kirkwood J, Perez F, Bear MF. Modulation of long-term synaptic depression in visual cortex by acetylcholine and norepinephrine. J Neurosci. 1999;19:1599–1609. doi: 10.1523/JNEUROSCI.19-05-01599.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koshimizu TA, Tanoue A, Hirasawa A, Yamauchi J, Tsujimoto G. Recent advances in alpha1-adrenoceptor pharmacology. Pharmacol Ther. 2003;98:235–244. doi: 10.1016/s0163-7258(03)00033-0. [DOI] [PubMed] [Google Scholar]
- Lapiz MDS, Morilak DA. Noradrenergic modulation of cognitive function in rat medial prefrontal cortex as measured by attentional set shifting capability. Neuroscience. 2006;137:1039–1049. doi: 10.1016/j.neuroscience.2005.09.031. [DOI] [PubMed] [Google Scholar]
- LeDoux JE. Emotion circuits in the brain. Annu Rev Neurosci. 2000;23:155–184. doi: 10.1146/annurev.neuro.23.1.155. [DOI] [PubMed] [Google Scholar]
- Marcotte ER, Pearson DM, Srivastava LK. Animal models of schizophrenia: a critical review. J Psychiatry Neurosci. 2001;26:395–410. [PMC free article] [PubMed] [Google Scholar]
- Marquis JP, Goulet S, Dore FY. Neonatal ventral hippocampus lesions disrupt extra-dimensional shift and alter dendritic spine density in the medial prefrontal cortex of juvenile rats. Neurobiol Learn Mem. 2008;90:339–346. doi: 10.1016/j.nlm.2008.04.005. [DOI] [PubMed] [Google Scholar]
- Marzo A, Bai J, Caboche J, Vanhoutte P, Otani S. Cellular mechanisms of long-term depression induced by noradrenaline in rat prefrontal neurons. Neuroscience. 2010;169:74–86. doi: 10.1016/j.neuroscience.2010.04.046. [DOI] [PubMed] [Google Scholar]
- McElligott ZA, Winder DG. Alpha1-adrenergic receptor-induced heterosynaptic long-term depression in the bed nucleus of the stria terminalis is disrupted in mouse models of affective disorders. Neuropsychopharmacology. 2008;33:2313–2323. doi: 10.1038/sj.npp.1301635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milad MR, Quirk GJ. Neurons in medial prefrontal cortex signal memory for fear extinction. Nature. 2002;420:70–74. doi: 10.1038/nature01138. [DOI] [PubMed] [Google Scholar]
- Mueller D, Cahill SP. Noradrenergic modulation of extinction learning and exposure therapy. Behav Brain Res. 2010;208:1–11. doi: 10.1016/j.bbr.2009.11.025. [DOI] [PubMed] [Google Scholar]
- Mueller D, Porter JT, Quirk GJ. Noradrenergic Signaling in Infralimbic Cortex Increases Cell Excitability and Strengthens Memory for Fear Extinction. J Neurosci. 2008;28:369–375. doi: 10.1523/JNEUROSCI.3248-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls RE, Alarcon JM, Malleret G, Carroll RC, Grody M, Vronskaya S, et al. Transgenic mice lacking NMDAR-dependent LTD exhibit deficits in behavioral flexibility. Neuron. 2008;58:104–117. doi: 10.1016/j.neuron.2008.01.039. [DOI] [PubMed] [Google Scholar]
- Nicniocaill B, Gratton A. Medial prefrontal cortical alpha1 adrenoreceptor modulation of the nucleus accumbens dopamine response to stress in Long-Evans rats. Psychopharmacology (Berl) 2007;191:835–842. doi: 10.1007/s00213-007-0723-1. [DOI] [PubMed] [Google Scholar]
- O’Donnell P, Lewis BL, Weinberger DR, Lipska BK. Neonatal hippocampal damage alters electrophysiological properties of prefrontal cortical neurons in adult rats. Cerebral Cortex. 2002;12:975–982. doi: 10.1093/cercor/12.9.975. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. Academic Press; New York: 2007. [Google Scholar]
- Peters J, Kalivas PW, Quirk GJ. Extinction circuits for fear and addiction overlap in prefrontal cortex. Learn Mem. 2009;16:279–288. doi: 10.1101/lm.1041309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quirk GJ, Beer JS. Prefrontal involvement in the regulation of emotion: convergence of rat and human studies. Curr Opin Neurobiol. 2006;16:723–727. doi: 10.1016/j.conb.2006.07.004. [DOI] [PubMed] [Google Scholar]
- Quirk GJ, Mueller D. Neural mechanisms of extinction learning and retrieval. Neuropsychopharmacology. 2008;33:56–72. doi: 10.1038/sj.npp.1301555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos BP, Arnsten AF. Adrenergic pharmacology and cognition: focus on the prefrontal cortex. Pharmacol Ther. 2007;113:523–536. doi: 10.1016/j.pharmthera.2006.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan RT, Bhardwaj SK, Tse YC, Srivastava LK, Wong TP. Opposing alterations in excitation and inhibition of layer 5 medial prefrontal cortex pyramidal neurons following neonatal ventral hippocampal lesion. Cereb Cortex. 2013;23:1198–1207. doi: 10.1093/cercor/bhs111. [DOI] [PubMed] [Google Scholar]
- Santini E, Quirk GJ, Porter JT. Fear conditioning and extinction differentially modify the intrinsic excitability of infralimbic neurons. J Neurosci. 2008;28:4028–4036. doi: 10.1523/JNEUROSCI.2623-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheiderer CL, Dobrunz LE, McMahon LL. Novel form of long-term synaptic depression in rat hippocampus induced by activation of alpha 1 adrenergic receptors. J Neurophysiol. 2004;91:1071–1077. doi: 10.1152/jn.00420.2003. [DOI] [PubMed] [Google Scholar]
- Sirvio J, MacDonald E. Central alpha1-adrenoceptors: their role in the modulation of attention and memory formation. Pharmacol Ther. 1999;83:49–65. doi: 10.1016/s0163-7258(99)00017-0. [DOI] [PubMed] [Google Scholar]
- Sparta DR, Hovelso N, Mason AO, Kantak PA, Ung RL, Decot HK, et al. Activation of prefrontal cortical parvalbumin interneurons facilitates extinction of reward-seeking behavior. J Neurosci. 2014;34:3699–3705. doi: 10.1523/JNEUROSCI.0235-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thierry AM, Gioanni Y, Degenetais EG, Glowinski J. Hippocampo-prefrontal cortex pathway: Anatomical and electrophysiological characteristics. Hippocampus. 2000;10:411–419. doi: 10.1002/1098-1063(2000)10:4<411::AID-HIPO7>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- Thomson AM, Deuchars J. Synaptic interactions in neocortical local circuits: dual intracellular recordings in vitro. Cereb Cortex. 1997;7:510–522. doi: 10.1093/cercor/7.6.510. [DOI] [PubMed] [Google Scholar]
- Tseng KY, Chambers RA, Lipska BK. The neonatal ventral hippocampal lesion as a heuristic neurodevelopmental model of schizophrenia. Behav Brain Res. 2009;204:295–305. doi: 10.1016/j.bbr.2008.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng KY, Lewis BL, Hashimoto T, Sesack SR, Kloc M, Lewis DA, et al. A neonatal ventral hippocampal lesion causes functional deficits in adult prefrontal cortical interneurons. J Neurosci. 2008;28:12691–12699. doi: 10.1523/JNEUROSCI.4166-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van den Oever MC, Goriounova NA, Li KW, Van der Schors RC, Binnekade R, Schoffelmeer AN, et al. Prefrontal cortex AMPA receptor plasticity is crucial for cue-induced relapse to heroin-seeking. Nat Neurosci. 2008;11:1053–1058. doi: 10.1038/nn.2165. [DOI] [PubMed] [Google Scholar]
- Volk DW, Lewis DA. Prefrontal cortical circuits in schizophrenia. Curr Top Behav Neurosci. 2010;4:485–508. doi: 10.1007/7854_2010_44. [DOI] [PubMed] [Google Scholar]
- Wood GK, Quirion R, Srivastava LK. Early environment contributes to developmental disruption of MPFC after neonatal ventral hippocampal lesions in rats. Synapse. 2003;50:223–232. doi: 10.1002/syn.10265. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.