

Abstract
Acyl CoA diacylglycerol acyltransferase (DGAT) is an integral membrane protein of the endoplasmic reticulum that catalyzes the synthesis of triacylglycerols. Two DGAT enzymes have been identified (DGAT1 and DGAT2) with unique roles in lipid metabolism. DGAT1 is a multifunctional acyl-transferase capable of synthesizing diacylglycerol, retinyl, and wax esters in addition to triacylglycerol. Here, we report the membrane topology for murine DGAT1 using protease protections assays and indirect immunofluorescence in conjunction with selective permeabilization of cellular membranes. Topology models based on prediction algorithms suggested that DGAT1 had eight transmembrane domains. In contrast, our data indicate that DGAT1 has three transmembrane domains with the N terminus oriented toward the cytosol. The C-terminal region of DGAT1, which accounts for ~50% of the protein, is present in the endoplasmic reticulum lumen and contains a highly conserved histidine residue (His-426) that may be part of the active site. Mutagenesis of His-426 to alanine impaired the ability of DGAT1 to synthesize triacylglycerols as well as retinyl and wax esters in an in vitro acyltransferase assay. Finally, we show that the N-terminal domain of DGAT1 is not required for the catalytic activity of DGAT1 but, instead, may be involved in regulating enzyme activity and dimer/tetramer formation.
Acyl coenzyme A:1,2-diacylglycerol acyltransferase (DGAT)2 is a membrane-bound enzyme that catalyzes the biosynthesis of triacylglycerols (TGs) (1). TGs are a class of neutral lipid that represents the major form of stored energy in eukaryotic organisms (2). However, excessive accumulation of TG in tissues can lead to obesity and insulin resistance, whereas increased TG in the blood is a risk factor for atherosclerosis (3–5).
DGAT catalyzes the formation of an ester bond between a long chain fatty acid (fatty acyl coenzyme A (CoA)) and the free hydroxyl group of diacylglycerol (DG), generating TG. Two DGAT genes have now been identified, Dgat1 and Dgat2, which share no sequence homology. DGAT1 belongs to a large family of membrane-bound O-acyltransferases (MBOAT) that includes acyl-CoA:cholesterol acyltransferase-1 and -2 (ACAT-1 and -2), which catalyze cholesterol ester biosynthesis (6–10). DGAT2 belongs to the DGAT2/acyl-CoA:monoacylglycerol acyltransferase gene family including monoacylglycerol acyl-CoA acyltransferases 1–3 and wax synthase (9, 11–16). Biochemical analyses have indicated that DGAT1 and DGAT2 have distinct roles in TG metabolism. When overexpressed in cells, DGAT2 yielded a much larger increase in intracellular triacylglycerol than DGAT1 (17). In in vitro assays, DGAT1, but not DGAT2, was capable of using a broad array of acyl acceptors to synthesize diacylglycerol, retinyl, and wax esters in addition to triacylglycerol (18).
In vivo experiments in mice have also provided strong evidence that DGAT1 and DGAT2 do not serve redundant roles in lipid metabolism. Dgat1-deficient (Dgat1−/−) mice were viable with modest reductions in TG content, were resistant to diet-induced obesity, and had improved glucose metabolism (19–22). More recently, overexpression of DGAT1 in mouse macrophages had a protective effect against the activation of inflammatory pathways induced by diet-induced obesity (23). In contrast, DGAT2 appears to be much more involved in promoting bulk TG synthesis. Dgat2-deficient (Dgat2−/−) mice had a ~90% reduction in carcass TG content and died within a few hours after birth due to skin abnormalities (17).
DGAT1 and DGAT2 are integral membrane proteins that reside at the endoplasmic reticulum (ER) (24, 25). Interestingly, DGAT2 also interacts with lipid droplets and mitochondria, whereas DGAT1 does not. Topological studies of DGAT2 demonstrated that this enzyme has two transmembrane domains, and the N and C termini are exposed to the cytosol (26). A highly conserved four-amino acid sequence (HPHG) present in all DGAT2 family members was required for the full catalytic activity of DGAT2 and is thought to be part of the active site. The HPHG sequence of DGAT2 faces the cytosol, which may be important for directing newly synthesized TG for their storage in lipid droplets.
The topology of DGAT1 has not been mapped experimentally, although it has been predicted to have 6–12 possible transmembrane domains (6). The membrane topologies of human ACAT1 and ACAT2, which share 15–25% identity with DGAT1, have been determined. ACAT1 appears to have 5–9 transmembrane domains with the N terminus facing the cytosol and the C terminus in the ER lumen (27, 28). Two different models have been proposed for human ACAT2. One model proposes that ACAT2 has two transmembrane domains, and both the N and C termini face the cytosol (29). The second model suggests that ACAT2 has five transmembrane domains, and the termini are on opposite sides of the ER membrane (28).
ACAT1 and ACAT2 are capable of forming homodimers and homotetramers (30, 31). For ACAT1, homotetramer formation is important for the in vitro catalytic efficiency of the enzyme (32). Deletion of part of the N terminus changes ACAT1 from a tetramer to dimer and increases the enzymatic activity of ACAT1. Like the ACAT enzymes, DGAT1 also can form dimers and tetramers (33, 34). The functional significance of this higher order structure for DGAT1 function has not been examined. It does appear that the ability to form a tetramer resides in the N-terminal domain and that the individual DGAT1 subunits of the tetramer catalyze TG synthesis independently of each other (33).
In this report we determined the membrane topology of murine DGAT1 using protease protection assays and immunofluorescence microscopy. We also identified a histidine residue that may be part of the active site of DGAT1. Lastly, we provide evidence that the N terminus of DGAT1 may be involved in regulating DGAT activity through tetramer formation.
EXPERIMENTAL PROCEDURES
Cell Culture and Transfection
HEK-293T and COS-7 cells (American Type Tissue Culture Collection) were cultured in Dulbecco’s modified Eagle’s medium (DMEM) with 10% fetal bovine serum in a 37 °C incubator with 5% CO2. For transfections, 20 μg of plasmid DNA was incubated with 430 μl of 0.15 M NaCl and 120 μl of 0.1% polyethyleneimine (pH 7.0) for 10 min at room temperature. The transfection mixture was then added dropwise to a 100-mm culture dish containing 10 ml of DMEM with10% fetal bovine serum and cells at ~50% confluence. After 4 h, the medium was removed, and cells were washed and re-fed with media. 48 h after transfection, cells were harvested and used for experiments.
Construction of DGAT1 Plasmids
N-terminal FLAG-tagged murine DGAT1 (FL-DGAT1) in the eukaryotic expression vector, pCDNA3.1, was used as a template for all mutagenesis reactions. The various mutations and insertion of Myc (EQKLI-SEEDL) and HA (YPYDVPDYA) epitope tags were generated using the primer pairs listed in supplemental Table 1 with the QuikChange II site-directed mutagenesis kit (Stratagene). All plasmids were sequenced to confirm the presence of the desired mutations (the cDNA for murine DGAT1 was a generous gift from Dr. Robert Farese, Jr.).
Protease Protection Assays
HEK-293T cells expressing the various DGAT1 mutants were washed twice with ice-cold PBS, harvested by scraping, and collected by centrifugation (1000 × g). Cell pellets were resuspended in PBS and disrupted by 10 passages through a 27-gauge needle. Cell debris and nuclei were pelleted by centrifugation at 1000 × g for 2 min. To isolate total cellular membranes, the supernatant was centrifuged at 100,000 × g for 30 min at 4 °C. The membrane pellet was resuspended in PBS. A typical 50-μl reaction contained 50 μg of total membrane protein and 20 μg/ml trypsin (Sigma) with or without 1% Triton X-100, which was incubated at 30 °C for 30 min. The reaction was terminated by the addition of soybean trypsin inhibitor (0.4 μg/μl final concentration) (Sigma). An equal volume of 2× SDS loading buffer (Bio-Rad) was added to the samples, which were then incubated at 37 °C for 20–30 min, separated by SDS-PAGE, and analyzed by immunoblotting.
In Vitro Cross-linking
Total cellular membranes (1 μg/μl protein) were incubated with the cross-linker, disuccinimidyl suberate (DSS) (Pierce) at a final concentration of 1 mM in 10 mM Hepes (pH 7.4), 1 mM EDTA buffer for 50 min at room temperature. DSS was dissolved in DMSO (2.5% (final)). Reactions were terminated by the addition of 1/10 volume of 1 M Tris-Cl (pH 8.0). Samples were then separated by SDS-PAGE and analyzed by immunoblotting.
DGAT Activity Assays
Transfected HEK-293T cells were washed twice with ice-cold PBS, harvested by scraping, and collected by centrifugation (1000 × g). The cell pellet was resuspended in 200 μl of 50 mM Tris-HCl (pH 7.6), 250 mM sucrose and passed through a 27-gauge needle 10 times. Cell debris and nuclei were pelleted by centrifugation at 1000 × g for 2 min. Total cellular membranes (50 μg of protein) were used for in vitro DGAT activity assays (17). For some assays, the 1,2-diacylglycerol was replaced with either 0.2 mM all trans-retinol or 0.2 mM 1-hexadecanol to measure acyl-CoA:retinol acyltransferase (ARAT) and wax synthase activities, respectively (35).
Immunoblot Analyses
Samples were separated by SDS-PAGE on 10% polyacrylamide gels, transferred to PVDF membranes (Bio-Rad). Immunoblotting was performed by incubating PVDF membranes with antibodies at the following dilutions: mouse anti-FLAG-M2 (Sigma), 1:4000; rabbit anti-protein disulfide isomerase (PDI) (Stressgen), 1:1000; mouse anti-Myc (9E10 hybridoma supernatant), 1:10; mouse anti-HA (Sigma), 1:4000; anti-mouse IgG-HRP (Amersham Biosciences), 1:4000; anti-rabbit IgG-HRP (Bio-Rad), 1:4000. Protein-antibody complexes were visualized using the Supersignal West Pico kit (Pierce). Membranes were exposed to Hyblot Cl (Denville Scientific) film.
Immunofluorescence Microscopy
COS-7 cells were transiently transfected with FL-DGAT1-Myc. Twenty-four hours after transfection, cells were re-plated into 6-well dishes containing glass coverslips and allowed to adhere overnight. After washing three times with PBS, cells were fixed with 4% paraformaldehyde in PBS for 10 min at room temperature. Plasma membranes were selectively permeabilized with 10 μg/ml digitonin in 0.3 M sucrose, 5 mM MgCl2, 120 mM KCl, 0.15 mM CaCl2, 2 mM EGTA, and 25 mM Hepes/KOH (pH 7.6) for 10 min on ice. Alternatively, after fixation, total cellular membranes were permeabilized with 0.2% Triton X-100 for 2 min at room temperature. After permeabilization, cells were washed 3 times with PBS and incubated with 3% bovine serum albumin in PBS for 5 min to block non-specific antibody binding. Cells were incubated at room temperature for 1 h with either mouse anti-FLAG-M2 (1:500 dilution) and rabbit anti-PDI (1:200 dilution) or rabbit anti-FLAG (1:200 dilution) and mouse anti-Myc (clone (9E10) (1:10 dilution) and then with goat anti-rabbit Alexa Fluor 488 (Molecular Probes) (1:200 dilution) and donkey anti-mouse 594 (Molecular Probes) (1:200 dilution) secondary antibodies for 30 min at room temperature. Cells were washed three times with PBS, and the coverslips were mounted on glass slides with a drop of Immuno-Fluore mounting medium (ICN). Images were acquired using the appropriate filter sets with an Olympus Fluoview FV300 confocal microscope. Images were captured using the same wavelength, light intensity, and acquisition settings. Images shown are representative of results for the majority of transfected cells in each experiment.
RESULTS
Hydropathy Plot and Predicted Membrane Topology of DGAT1
A Kyte-Doolittle hydropathy plot (36) indicated that murine DGAT1 had several hydrophobic regions that could be potential transmembrane domains (Fig. 1A). Using several different algorithms that predict hydrophobic regions of a protein (Table 1), a consensus model of the membrane topology of murine DGAT1 was constructed (Fig. 1B). This model indicated that DGAT1 had eight potential transmembrane domains. No clear consensus was obtained using these algorithms to determine the orientation of the N and C termini. Some of the prediction algorithms suggested that the termini of DGAT1 were on the same side of the ER membrane, whereas others indicated that they were on opposite sides.
FIGURE 1. Hydropathy plot and predicted transmembrane domains of murine DGAT1.

A, a hydropathy plot of DGAT1 was generated by the method of Kyte and Doolittle using a window size of 19 amino acids (36). B, shown is a consensus model of the number and location of possible transmembrane domains of DGAT1 based on seven different prediction algorithms (Table 1). C, shown are DGAT1 expression constructs used to map the membrane topology of DGAT1. A Myc- (FL-DGAT1-Myc) and a HA- (FL-DGAT1-HA) epitope tag were placed at the C terminus of FL-DGAT1. Internal HA (H) or Myc (M) tags were then inserted into hydrophilic regions between the predicted transmembrane domains (black boxes) of FL-DGAT1-Myc or FL-DGAT1-HA, respectively.
TABLE 1.
Predicted transmembrane domains for murine DGAT1
| Analysis software | No. of TMDs |
|---|---|
| TMHMM | 9 |
| SOSUI | 6 |
| Phobius | 9 |
| TMAP | 8 |
| TMpred | 8 |
| TopPred | 9 |
| HMMTOP | 9 |
TMD, transmembrane domain.
To determine the orientation of the N and C termini of DGAT1, constructs were generated that contained a FLAG epitope tag at the N terminus and either an HA (FL-DGAT1-HA) or Myc (FL-DGAT1-Myc) epitope tag at the C terminus of murine DGAT1 (Fig. 1C). Expression of both Myc and HA C-terminal-tagged DGAT1 proteins in HEK-293T cells was confirmed by immunoblotting with anti-FLAG (Fig. 2A). FL-DGAT1-HA and FL-DGAT1-Myc migrated slightly slower by SDS-PAGE compared with FL-DGAT due to the presence of the additional epitope tag at the C terminus. The presence of a C-terminal epitope tag (Myc or HA) was not detrimental to DGAT1 function. In vitro DGAT activity in membranes isolated from cells expressing FL-DGAT1-HA and FL-DGAT1-Myc was similar to that of FL-DGAT1 and was ~20-fold higher than that of untransfected cells (Fig. 2B).
FIGURE 2. Expression of FLAG-tagged DGAT1 constructs in HEK-293T cells.

A, an immunoblot shows transient expression of the FL-DGAT1 constructs described in Fig. 1C in HEK-293T cells. Total cellular membranes (50 μg of protein) were immunoblotted with anti-FLAG and anti-calnexin antibodies. B, shown is in vitro DGAT activity in membranes from cells expressing FL-DGAT1 constructs. Membranes from cells were assayed for in vitro DGAT activity by measuring the formation of [14 C]TG from 1,2-diacylglycerol and [14C]oleoyl-CoA. The activities of the various mutants are normalized to that of FL-DGAT1. The activities for untransfected cells and cells transfected with FL-DGAT1 were 7.4 and 128 pmol/min/mg of protein, respectively. Data are from one experiment, performed in duplicate, which was repeated twice with similar results.
To determine the orientation and number of the hydrophilic loops of DGAT1, internal HA or Myc epitopes were inserted into FL-DGAT1-Myc or FL-DGAT1-HA, respectively. FL-DGAT1-Myc constructs containing an internal HA epitope between amino acids 230 and 231 (230HA), 240 and 241 (240HA), and 250 and 251 (250HA) had DGAT activities at least equal to that of FL-DGAT1-Myc (Fig. 2B). Constructs with an internal HA epitope between amino acids 129 and 130 (129HA) and 196 and 197 (196HA) of FL-DGAT1-Myc and an internal Myc tag between amino acids 165 and 166 (165Myc) and 460 and 461 (460Myc) of FL-DGAT1-HA showed increased DGAT activity compared with untransfected cells but was lower than that of FL-DGAT1(Fig. 2B).
The insertion of an internal Myc epitope between amino acids 337 and 338 (337Myc) and 436 and 437 (436Myc) into FL-DGAT1-HA led to a significant decrease in DGAT activities, which were slightly above that of untransfected cells (Fig. 2B). Attempts to place epitope tags in adjacent areas were unsuccessful, indicating that these regions of DGAT1 are highly susceptible to perturbation (data not shown).
Orientation of the N and C Termini of DGAT1
To determine the orientation of the N terminus of DGAT1, protease protection assays using trypsin were performed with sealed membrane vesicles from HEK-293T cells expressing DGAT1 with an N-terminal FLAG tag (FL-DGAT1). Trypsin was chosen as it is a relatively weaker protease that cannot digest proteins within a membrane bilayer. Membrane vesicles were incubated with trypsin and then immunoblotted with anti-FLAG antibody. Under these conditions, a 55-kDa protein corresponding to DGAT1 was detected only in the absence of trypsin (Fig. 3A, lane 1). In the presence of the protease, with or without detergent (Fig. 3A, lanes 2 and 3), the FLAG epitope was destroyed, suggesting that the N terminus of DGAT1 was exposed to the cytosol. The integrity of membrane vesicles was confirmed by immunoblotting for the ER luminal protein, PDI. PDI could be detected both in the absence and presence of trypsin, indicating that the membrane vesicles were intact (Fig. 3A, lane 1 and 2). However, PDI was not detected when Triton X-100 was included in the reaction mix to solubilize membranes (Fig. 3A, lane 3).
FIGURE 3. The N terminus of DGAT1 has a cytosolic orientation, and the C terminus is exposed to the ER lumen as determined by protease protection analyses.

A, protease protection analysis is shown. Total membranes from HEK-293T cells expressing FL-DGAT1, FL-DGAT-HA, and FL-DGAT-Myc were prepared as described under “Experimental Procedures.” Aliquots of membranes (50 μg protein) were incubated in the absence or presence of 20 μg/ml of trypsin with or without 1% Triton X-100. Samples were immunoblotted with anti-FLAG, which recognizes the N terminus of DGAT1 (A) and anti-Myc (B) or anti-HA (C) that recognize the C terminus of DGAT1. The integrity of membrane vesicles was assessed by immunoblotting with an anti-PDI antibody. D, orientation of the N and C termini of DGAT1 by immunofluorescence microscopy is shown. COS-7 cells transiently expressing FL-DGAT1-Myc were fixed with 4% paraformaldehyde. Fixed cells were incubated with 0.2% Triton X-100 to permeabilize all cellular membranes or 10 μg/ml digitonin to selectively permeabilize the plasma membrane. Cells were exposed to anti-FLAG and anti-Myc antibodies and appropriate secondary antibodies to visualize the N-terminal FLAG (green) and the C-terminal Myc (red) epitopes of DGAT1, respectively (upper panel). Cells were exposed to anti-FLAG and anti-PDI antibodies and appropriate secondary antibodies to visualize the N-terminal FLAG (red) and PDI (green) in the ER lumen. Cells chosen for image comparison had similar levels of expression, as assessed by FLAG signal intensity (lower panel).
To determine the orientation of the C terminus of DGAT1, protease protection assays were performed using sealed membrane vesicles from HEK-293T cells expressing FL-DGAT1-HA and FL-DGAT1-Myc. In samples not treated with trypsin, the N terminus could be detected by immunoblotting with anti-FLAG (Fig. 3, B and C, lanes 1), and the C terminus was detected with anti-HA (Fig. 3B, lane 4) or anti-Myc (Fig. 3C, lane 4). When trypsin was included in the assay, DGAT1 could not be detected with anti-FLAG (Fig. 3, B and C, lanes 2 and 3). However, both anti-HA and anti-Myc detected a protected DGAT1 fragment that was 25 kDa (~50% of the protein) (Fig. 3, B and C, lanes 5), but not when detergent was added to the reaction (Fig. 3, B and C, lanes 6). Immunoblotting for PDI confirmed that the membrane vesicles were intact (Fig. 3, B and C). Protease protection assays were also performed on membrane vesicles from HEK-293T cells expressing FL-DGAT1-HA and FL-DGAT1-Myc using proteinase K with identical results of the orientation of the N and C termini of DGAT1 as trypsin (data not shown).
Indirect immunofluorescence was used to confirm the orientation of the N and C termini. In COS-7 cells transfected with FL-DGAT-Myc, the N-terminal FLAG epitope was detected when cellular membranes were completely permeabilized with detergent or when the plasma membrane was selectively permeabilized with digitonin (Fig. 3D, upper panel). In contrast, the C-terminal Myc epitope could only be detected when cellular membranes were permeabilized with detergent (Fig. 3D, upper panel). Integrity of ER membranes after permeabilization with digitonin was determined by probing for the ER luminal protein, PDI. PDI was only detected when cells were permeabilized with detergent and not with digitonin, indicating that the ER membranes were intact (Fig. 3D, lower panel). These results are consistent with the data from our protease protection assays and confirm that the N terminus of DGAT1 is exposed to the cytosol, whereas the C terminus is present in the ER lumen.
Determining the Location of the Transmembrane Domains of DGAT1 with Expression Plasmids Containing Internal Epitope Tags
The cytosolic orientation of the N terminus and luminal orientation of the C terminus suggested that DGAT1 has an odd number of transmembrane domains. Given the size of the C-terminal-protected fragment (25 kDa) observed in the protease protection assays, it appeared that DGAT1 also has fewer transmembrane domains than predicted in our consensus model (Fig. 1B), most likely only three. To determine the location of the transmembrane domains, protease protection assays were performed on DGAT1 constructs containing internal HA or Myc epitope tags (described in Fig. 1C and Fig. 2). Membrane vesicles prepared from HEK-293T cells expressing 129HA, 196HA, 240HA, and 165Myc were treated with trypsin with or without Triton X-100. DGAT1 in untreated samples was detected by immunoblotting with anti-FLAG, anti-HA, and anti-Myc antibodies (Fig. 4A, lanes 1, 4, 7, and 10). When treated with trypsin, DGAT1 could not be detected with anti-FLAG, but the 25-kDa protected fragment in the ER lumen that was observed in Fig. 3 was detected with anti-Myc for 129HA, 196HA, and 240HA (Fig. 4A, lanes 2, 5, and 8) and with anti-HA for 165Myc (Fig. 4A, lane 11). When trypsinized samples for 129HA and 196HA were immunoblotted with anti-HA to detect the orientation of the internal HA tags, a 16-kDa protected fragment was detected (Fig. 4A, lanes 2 and 5). The same 16-kDa fragment was seen when 165Myc was treated with trypsin and immunoblotted with anti-Myc (Fig. 4A, lane 11). These results indicate that the first transmembrane domain of DGAT1 precedes the HA epitope inserted between amino acids 129–130 and likely resides between amino acids 97 and 118.
FIGURE 4. Determination of the orientation of the hydrophilic loops of DGAT1 by trypsin digestion.

Total membranes were prepared from HEK-293T cells expressing FL-DGAT1-Myc with internal HA tags (A) or FL-DGAT1-HA (B) with internal Myc tags. Protease protection assays were performed as described in Fig. 3. Samples were analyzed by immunoblotting with anti-FLAG, anti-Myc, anti-HA, or anti-PDI antibodies.
In contrast, the internal HA epitope of 240HA was not detected in the presence of trypsin (Fig. 4A, lane 8), indicating that this region of DGAT1 had a cytosolic orientation and was not part of the 16- or 25-kDa protected fragments. Immunoblotting for PDI confirmed membrane vesicles were intact in the absence or presence of trypsin (Fig. 4A). When membrane vesicles were disrupted with Triton X-100, PDI immunoreactivity was lost. These data suggest that DGAT1 has two hydrophilic loops present in the ER lumen; that is, a 16-kDa fragment near the N terminus and a second 25-kDa fragment containing the C-terminal half of DGAT1.
The identification of the large 25-kDa C-terminal protected fragment in the ER lumen of DGAT1 indicated that this region likely did not contain any transmembrane domains, although our model predicted that there could be four, between amino acids 292–312, 343–364, 439–459, and 467–484 (Fig. 1B). We generated three DGAT1 constructs containing a C-terminal HA epitope with internal Myc epitopes between each of the predicted transmembrane domains (337Myc, 436Myc, and 460Myc) (Fig. 1C). When expressed in HEK-293T cells, DGAT activity of 460Myc was similar to that of FL-DGAT (Fig. 2B). However, 337Myc and 436Myc were essentially devoid of DGAT activity, and the internal Myc epitope could not be detected for 337Myc (Fig. 2B). Attempts to place the internal tag in these three constructs in adjacent areas and retain significant activity were unsuccessful. 337Myc was not used further. We did continue to use 436Myc in protease protection assays because we could detect the internal Myc epitope.
In protease protection assays, 436Myc and 460Myc were both observed with anti-FLAG in the absence but not presence of trypsin (Fig. 4B). When samples were immunoblotted after digestion of microsomes with trypsin, both anti-HA and anti-Myc antibodies detected a 25-kDa protected fragment, indicating that the C-terminal region of DGAT1 resided in the ER lumen and did not contain any of the predicted transmembrane domains (Fig. 4B, lanes 2 and 5). Epitope tags at the N and C termini, and internal tags of 436Myc and 460Myc could not be detected when samples were incubated with detergent and trypsin (Fig. 4B, lanes 3 and 6).
436Myc and 460Myc had identical protease protection patterns, suggesting that the insertion of a Myc tag between amino acids 436 and 437 likely disrupted the active site of the enzyme rather than by altering its structure. The active site of DGAT has been predicted to be in this region of DGAT1 (2).
The HA epitope of 240HA could not be detected in the presence of protease, suggesting that this region of DGAT1 was orientated toward the cytosol and was flanked on either side by the second and third transmembrane domains. Because the HA epitope tag of 196HA was found in the 16-kDa protected region, we surmised that the second transmembrane domain of DGAT1 is likely between amino acids 196 and 240, whereas the third trans-membrane is likely between amino acids 240 and 337. To test this, we introduced an HA tag between amino acids 230 and 231 (230HA) and 250 and 251 (250HA) of FL-DGAT1-Myc (Figs. 1C and 2). Membrane vesicles were prepared from HEK-293T cells expressing 230HA and 250HA and used in protease protection assays. Both 230HA and 250HA were detected by immunoblotting with anti-FLAG, anti-HA, and anti-Myc in the absence of trypsin (Fig. 5, lanes 1 and 4). For both 230HA and 250HA, a 25-kDa protected C-terminal fragment was observed in the presence of trypsin using anti-Myc. However, immunoblotting with anti-HA identified a 16-kDa protected fragment for 230HA and a 25-kDa protected fragment for 250HA (Fig. 5, lanes 2 and 5). These results indicate that the second transmembrane domain is between amino acids 230 and 240, and the third transmembrane domain is between amino acids 240 and 250. A model of the membrane topology of murine DGAT1 based on our experimental data is shown in Fig. 6.
FIGURE 5. Orientation of amino acids 230 –250 of DGAT1.

Total membranes were prepared from HEK-293T cells expressing FL-DGAT1-Myc with internal HA tags at amino acids position 230, 240, or 250 of DGAT1. Protease protection assays were performed as described in Fig. 3. Samples were analyzed by immunoblotting with anti-FLAG, anti-Myc, anti-HA or anti-PDI antibodies.
FIGURE 6. Proposed model of the membrane topology of murine DGAT1.
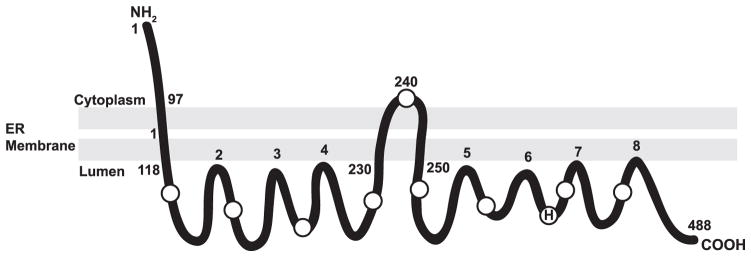
Our topology studies demonstrate that DGAT1 contains three potential transmembrane domains with the N terminus oriented toward the cytosol and the C terminus oriented toward the ER lumen. An alternative model, also consistent with the current data, would show that amino acids 230 –250 do not span the lipid bilayer but are instead embedded within the membrane. H indicates the putative active site histidine at position 426. Circles represent the location of internal Myc or HA epitope tags. Amino acid numbers of regions exposed to the cytosol or ER lumen are indicated.
His-426 Is Important for DGAT Activity
A highly conserved histidine residue has been determined to be part of the active site for human ACAT1 (His-460) and ACAT2 (His-438) (37, 38). Mutation of this amino acid in ACAT1 or ACAT2 resulted in the complete loss of in vitro ACAT activity. Alignment of ACAT/DGAT1 family members shows that this histidine is not only present in ACAT enzymes but is also highly conserved in DGAT1 across species (histidine 426 of murine DGAT1) (Fig. 7A). We sought to determine whether His-426 of murine DGAT1, which our model suggests is present in the ER lumen, was important for DGAT activity by mutating it to alanine (H426A). FL-DGAT1 and the H426A mutant were expressed in HEK-293T cells at almost equal levels as assessed by immunoblotting with anti-FLAG (Fig. 7B). Although the in vitro activity of FL-DGAT1 was 15-fold higher than that of untransfected cells, the H426A mutant had no detectible enzymatic activity (Fig. 7B).
FIGURE 7. A highly conserved histidine (His-426) of the ACAT/DGAT1 family is important for the catalytic activity of murine DGAT1.
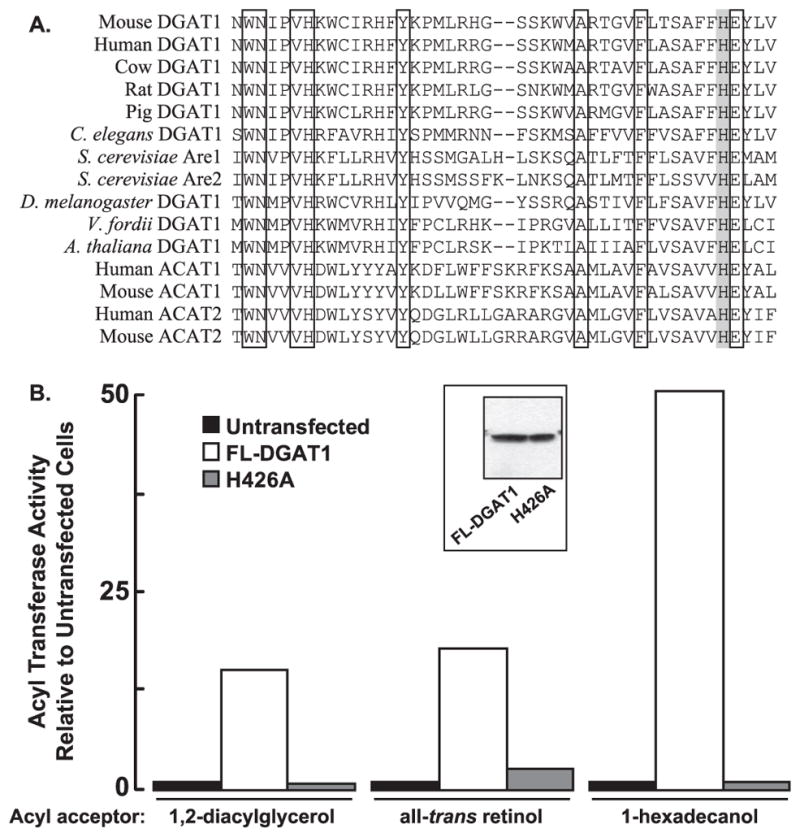
A, shown is alignment of partial sequences of ACAT/DGAT1 family members (amino acids 387– 430 of murine DGAT1). Identical amino acids are indicated by a white box, and a highly conserved active site histidine residue (His-426) is indicated by a gray box. B, in vitro acyltransferase assays were performed on total cellular membranes from untransfected HEK-293T cells or from HEK-293T cells expressing FL-DGAT1 or H426A using [14C]oleoyl CoA as the acyl donor and 2 mM 1,2-DG, all-trans retinol or 1-hexadecanol as acyl group acceptors. The acyltransferase activities of cells transfected with FL-DGAT1 were 2241 (1,2-diacylglycerol), 234 (all-trans retinol), and 86 (1-hexadecanol) pmol/min/mg of protein, respectively. Data shown are from one representative experiment that was repeated twice with similar results. Expression of DGAT1 and a H426A mutant in HEK-293T cells was confirmed by immunoblotting with anti-FLAG (inset).
We also determined if His-426 was required for the ARAT and wax synthase activities of DGAT1. When 1,2-diacylglycerol (acyl acceptor) was replaced with either all-trans retinol and 1-hexadecanol in our acyltransferase assay, ARAT and wax synthase activities of FL-DGAT1 were 18- and 50-fold higher than that of untransfected cells, respectively, but were essentially undetectable in the H426A mutant (Fig. 7B).
Role of the N Terminus of DGAT1
Our topology experiments have indicated that the majority of DGAT1, including a potential active site histidine, is orientated toward the ER lumen. However, the N terminus of DGAT1, consisting of approximately the first 100 amino acids, along with a small internal loop near the middle of the protein were exposed to the cytosol. We sought to determine whether the N terminus of DGAT1 was important for the catalytic activity of DGAT1. Total membranes from untransfected HEK-293T cells or cells expressing FL-DGAT1 and FL-DGAT1-HA were treated with or without trypsin, and then in vitro DGAT activity was measured. Immunoblotting with anti-FLAG demonstrated that protease treatment was effective in destroying the N termini of FL-DGAT1 and FL-DGAT1-HA exposed to the cytosol (Fig. 8A). We were also able to detect the 25-kDa C-terminal protected fragment of FL-DGAT1-HA when immunoblotting with anti-HA, indicating that DGAT1 had not been completely degraded by protease. The integrity of membrane vesicles was confirmed by immunoblotting for PDI, which could be detected both in the absence and presence of trypsin. In vitro DGAT activity in membranes from HEK-293T cells expressing FL-DGAT1 or FL-DGAT1-HA was ~9-fold higher than that of untransfected cells (Fig. 8B). Surprisingly, in the presence of trypsin, DGAT activity in untransfected cells and in cells expressing both FL-DGAT1 and FL-DGAT-HA was increased 3–4-fold compared with samples not exposed to trypsin.
FIGURE 8. Stimulation of DGAT1 in vitro activity in the absence of its N terminus.

Total membranes from untransfected HEK-293T cells and HEK-293T cells expressing FL-DGAT1 and FL-DGAT-HA were prepared as described under “Experimental Procedures.” Aliquots of membranes (50 μg protein) were incubated in the absence or presence of20 μg/ml of trypsin (as described in Fig. 3). After the addition of soybean trypsin inhibitor, samples were immunoblotted with anti-FLAG, anti-HA, and anti-PDI antibodies (A), and in vitro DGAT activity was determined (B). C, an immunoblot shows transient expression of FL-DGAT1, DGAT1/Δ2–37, and FL-DGAT1/Δ2– 84 in HEK-293T cells. Total cellular membranes (50 μg of protein) were immunoblotted with anti-FLAG. D, shown is in vitro DGAT activity in membranes from cells expressing FL-DGAT1, DGAT1/Δ2–37, and FL-DGAT1/Δ2– 84. Membranes from cells were assayed for in vitro DGAT activity as described in Fig. 2. The activities of the various mutants were normalized to the amount of FL-DGAT1 proteins that were quantified using a fluorescently labeled secondary antibody. The fluorescence signal was obtained with a VersaDoc 4000 imaging system (Bio-Rad) and quantified with Quantity One imaging software. The activities for untransfected cells and cells transfected with FL-DGAT1 were 64.2 and 690.2 pmol/min/mg of protein, respectively. Data are from one experiment, performed in duplicate, which was repeated once with similar results.
Because the increase in DGAT activity in the presence of trypsin could also be due to cleavage of the short cytosolic loop or other nonspecific effects, we generated two DGAT1 N-terminal deletion mutants. DGAT1 mutants lacking amino acids 2–37 (FL-DGAT1/Δ2–37) and 2–84 (FL-DGAT1/Δ2–84) along with FL-DGAT1 were expressed in HEK-293T cells as confirmed by immunoblotting (Fig. 8C). The amount of FL-DGAT1/Δ2–84 protein was consistently less abundant than either FL-DGAT1 or FL-DGAT1/Δ2–37. DGAT activity in membranes from cells expressing FL-DGAT1/Δ2–34 was comparable with that of FL-DGAT1 (Fig. 8D). However, consistent with our results in Fig. 8B, DGAT activity of the FL-DGAT1/Δ2–84 mutant was ~14-fold higher than that of FL-DGAT1 when normalized to the amount of DGAT1 protein. Taken together, these results suggest that the cytosolic N terminus, in particular the region between amino acids 38–84, may play a role in regulating DGAT1 activity.
Identification of the Oligomerization Domains of Murine DGAT1
Human DGAT1, ACAT1, and ACAT2 have been demonstrated to exist in oligomeric forms (30–34). Because murine DGAT1 lacks the dimer forming motif present in the N terminus of ACAT1 (32), we sought to identify the region of murine DGAT1 that interacts to form oligomers. Total membranes from HEK-293T cells expressing DGAT1 were cross-linked with the membrane permeable cross-linker, DSS. After the cross-linking reaction was quenched, samples were analyzed by SDS-PAGE and immunoblotted with anti-FLAG. In the absence of cross-linker, a predominant 55-kDa band was observed that represented the monomeric form of DGAT1 (Fig. 9A, lane 1). When DSS was included in the reaction, two immunoreactive bands were observed of ~110 and 220 kDa, which represented a dimer (2×) and tetramer (4×) of DGAT1, respectively (Fig. 9A, lane 2).
FIGURE 9. The N terminus of DGAT1 has a role in oligomer formation.

Total membranes isolated from HEK-293T cells expressing (A) FL-DGAT1, FL-DGAT1(Δ2–34), and FL-DGAT1(Δ2–95) were incubated in the (−) absence or (+) presence of 1 mM DSS. After the cross-linking reactions were terminated, samples were subjected to SDS-PAGE and immunoblotted with anti-FLAG. 1×, 2×, and 4× refer to DGAT1 monomer, dimer, and tetramer, respectively. B, after cross-linking with 1 mM DSS, samples were treated with or without trypsin and immunoblotted with anti-FLAG, anti-Myc, and anti HA antibodies. Oligomers of the cross-linked 16-kDa protected fragment recognized by anti-HA (*). An oligomer of unknown composition is indicated by ?.
To determine whether a region in the N terminus of DGAT1 was responsible for dimer/tetramer formation, FL-DGAT1/Δ2–34 and FL-DGAT1/Δ2–95 were expressed in HEK-293T cells, and cross-linking was performed on cellular membranes as described previously. As expected, in the absence of DSS the monomeric forms of both DGAT1 mutants were the predominant bands (Fig. 9A, lanes 3 and 5). When exposed to DSS, FL-DGAT1/Δ2–37 formed dimers and tetramers of the expected size (Fig. 9A, lane 4). In contrast, DGAT1 lacking amino acids 2–84 was only capable of forming a dimer when cross-linked with DSS (Fig. 9A, lane 6). These results suggest that a region between amino acids 37 and 84 is partially responsible for tetramer formation of DGAT1.
To determine whether other regions of DGAT1 contributed to dimer/tetramer formation, we performed cross-linking reactions on 230HA with DSS in conjunction with protease protection assays. After cross-linking with DSS, membrane vesicles from HEK-293T cells expressing 230HA were incubated in the presence or absence of trypsin. In samples cross-linked with DSS, the dimer and tetramer of DGAT1 could be seen when immunoblotted with anti-FLAG, whereas only the monomer of DGAT1 was seen in untreated samples (Fig. 9B, lanes 1 and 3). Trypsin treatment of both non-cross-linked and cross-linked samples destroyed the N-terminal FLAG epitope, and DGAT1 could not be detected with anti-FLAG (Fig. 9B, lanes 2 and 4). Similar results were seen when immunoblotting with anti-Myc, which recognizes the C terminus of DGAT1 (Fig. 9B).
When samples were probed with anti-HA, a 16-kDa protected fragment of DGAT1 from amino acids 119–230 was seen in samples treated only with trypsin (Fig. 5 and 9B, lane 2). In lane 2 we also observed a faint 32-kDa band that likely represented incompletely denatured DGAT1. When first cross-linked with DSS and then exposed to trypsin, anti-HA recognized additional protected bands representing oligomers of the 16-kDa fragment (Fig. 9B, lane 4). These oligomers were ~32 and ~48 kDa and were likely a dimer and trimer of the 16-kDa fragment, respectively. A third oligomer of ~50 kDa of unknown composition was also present. These data suggest that the 16-kDa fragment of DGAT1 present in the ER lumen may also be involved in dimer/tetramer formation.
DISCUSSION
In this study we determined the membrane topology of murine DGAT1, identified a highly conserved histidine residue (His-426) that is essential for DGAT1 function, and demonstrated that the N terminus of DGAT1 is not required for DGAT activity but instead may be required for tetramer formation.
Using protease protection assays and indirect immunofluorescence microscopy in conjunction with epitope tagging, we provide evidence showing that DGAT1 has three transmembrane domains, with its N terminus exposed to the cytosol and the C terminus in ER lumen. This is in stark contrast to the eight transmembrane domains that were originally predicted.
The first transmembrane domain of DGAT1 is located near the N terminus, approximately between amino acids 97 and 118. The second and third transmembrane domains are more centrally located within DGAT1 and appear to reside between amino acids 230 and 240 and between 240 and 250, respectively. These two transmembrane domains were not identified in our original consensus model. However, both of these regions have a significant number of nonpolar amino acids that could potentially form α-helices. An alternative model to what has been proposed is that the second and third transmembrane domains do not completely cross the ER membrane and are instead imbedded within the lipid bilayer. The addition of the HA epitope to this region of DGAT1 (i.e. 240HA) may have forced the tag through the ER membrane where it then became accessible to digestion by trypsin. However, in our protease protection assays, we were still able to detect the 16- and 25-kDa protected fragments in DGAT1 constructs lacking internal tags after digestion with protease. Therefore, a portion of DGAT1 between amino acids 230–250 must completely span the ER membrane and be accessible to protease.
Murine DGAT1 shares ~55% similarity and 27% identity to both human ACAT1 and ACAT2. However, the membrane topology of DGAT1 resembles that of neither ACAT enzyme. ACAT1 has nine transmembrane domains, with its N terminus facing the cytosol and the C terminus in the ER lumen (37, 39). ACAT2 has only two transmembrane domains, with both the N and C termini facing the cytosol (29). A second study reported that both ACAT1 and ACAT2 have five transmembrane domains, with their N and C termini on opposite sides of the ER membrane (28). It has been speculated that the different membrane topologies observed for ACAT1 and ACAT2 may be due to significant differences in the amino acid sequences within the putative transmembrane domains (29). This may apply to DGAT1 as well.
DGAT1 also has a different membrane topology than that of a plant DGAT1 orthologue from Vernicia fordii, which has both its N and C termini exposed to the cytosol (40). Alignment of the amino acid sequences of ACAT/DGAT1 family members showed that DGAT1 orthologues from several plant species actually shared more homology with mammalian ACAT1 and ACAT2 than with DGAT1. Therefore, it is possible that the plant orthologues of DGAT1 are more structurally similar to ACATs than DGAT1.
Our model suggests that the active site of DGAT1 resides in the ER lumen. All DGAT1/ACAT family members have a highly conserved histidine residue (His-426 of murine DGAT1), which has been shown to be essential for both ACAT1 and ACAT2 function and is likely part of the active site of these enzymes (10, 29, 37). Our own studies demonstrated that His-426 of DGAT1 is also essential for DGAT activity. Mutation of His-426 of murine DGAT1 resulted in a non-functional enzyme that lacked the ability to synthesize TG, retinyl, and wax esters. Histidine residues have now been shown to be important for the in vitro activities of both DGAT1 (His-426) and DGAT2 (HPHG motif) (26). However, it is not clear if these two histidines have a similar role in catalyzing TG synthesis.
A pathway for the synthesis of TG in the ER lumen would require the transport of fatty acyl-CoA substrates from the cytosol across the ER membrane. A carnitine acyltransferase system capable of transporting fatty acids from the cytosol across membranes via a fatty acylcarnitine intermediate has been identified at the ER (41–43). Carnitine acyltransferase enzymes have previously been demonstrated to have a role in TG synthesis in the ER lumen both in vivo in rat small intestine and with a cell-free system, providing evidence for a mechanism that could deliver fatty acid substrates across the ER membrane from the cytosol to the active site of DGAT1 in the ER lumen (44, 45).
The presence of its active site in the ER lumen suggests that DGAT1 could produce a pool of TG readily accessible to the cellular machinery involved in the formation of chylomicrons and very low density lipoproteins (VLDL). The luminal orientation of the DGAT1 active site would provide an efficient way to channel TG to nascent lipoproteins. Other studies have provided additional evidence that TG synthesis can occur in the ER lumen. A latent DGAT activity was identified in rat liver microsomes after sealed microsomal vesicles were disrupted with permeabilizing agents (44, 46–48). In a separate study mice overexpressing DGAT1, but not DGAT2, had increased latent DGAT activity with a corresponding increase in VLDL secretion (49). Overexpression of DGAT1 was also able to stimulate secretion of TG-rich VLDL from rat hepatoma (McArdle RH7777) cells (50). Taken together, DGAT1 appears to have a role in lipoprotein formation, whereas DGAT2 promotes bulk TG synthesis that accumulates in large cytosolic lipid droplets (17).
The luminal localization of the active site of DGAT1 may also have a role in dietary retinol absorption, which occurs in a very similar manner to that of TG (51). Dietary retinyl esters are hydrolyzed by lipases in the intestinal lumen. Free retinol is transported across the intestinal brush border where it is re-esterified to retinyl esters and incorporated into chylomicrons that are released into the circulation. The re-esterification of retinol in the small intestine has recently been shown to be at least partially dependent on the activity of DGAT1. Chylomicrons isolated from DGAT1-deficient mice had less retinyl esters compared with those of wild-type mice, establishing an in vivo role for DGAT1 in intestinal retinol metabolism (52).
We investigated the role of the cytosolic N terminus of DGAT1 and found that this region of the enzyme was not required for the catalytic activity of the enzyme. Similar observations were made for both ACAT1 and DGAT2 (24, 32). However, in the case of DGAT2, the N terminus contained a mitochondrial targeting sequence. Like ACAT1, the N-terminal domain of DGAT1 had a role in tetramer formation and may be involved in regulating DGAT1 activity. Using deletion mutants, we found that DGAT1 tetramer formation required amino acids 37–84. When these amino acids were deleted, only a dimer of DGAT1 was observed. These findings are consistent with a previous study in which a truncated form of DGAT1 lacking the last 101 amino acids was isolated from human adipose tissue (33). Although catalytically inactive, this DGAT1mutant was still capable of forming tetramers, suggesting that sequences required for tetramer formation reside in the N terminus. The inability of DGAT1 to form a tetramer also corresponded to a severalfold increase in in vitro enzyme activity, indicating that the functional catalytic unit of DGAT1 may be a dimer. We speculate that DGAT1 activity may be regulated by switching between an active dimer to an inactive tetramer.
A second region of DGAT1 was identified that may be involved in the quaternary organization of the enzyme. When DGAT1 was cross-linked before digestion with trypsin, an antibody specific for the 16-kDa loop present in the ER lumen detected material that was ~32 and ~48 kDa, possibly a dimer and trimer, respectively. In addition to the N terminus, oligomerization of DGAT1 may also require the interaction of a domain present in the ER lumen. Further mutagenesis experiments will be required to provide direct evidence for such a role of this luminal region in the structural organization of DGAT1.
Our findings in this study provide valuable insight into the structural features of DGAT1 and how this might relate to TG synthesis, storage, and secretion. The data from this study will provide the basis for future experiments examining the importance of dimer/tetramer formation in regulating DGAT1 activity.
Supplementary Material
Footnotes
The on-line version of this article (available at http://www.jbc.org) contains supplemental Table 1.
The abbreviations used are: DGAT, acyl-CoA:diacylglycerol acyltransferase; ACAT, acyl-CoA:cholesterol acyltransferase; ARAT, acyl-CoA:retinol acyl-transferase; DG, diacylglycerol; DSS, disuccinimidyl suberate; ER, endoplasmic reticulum; PDI, protein disulfide isomerase; TG, triacylglycerol.
References
- 1.Kennedy EP. Ann Rev Biochem. 1957;26:119–148. doi: 10.1146/annurev.bi.26.070157.001003. [DOI] [PubMed] [Google Scholar]
- 2.Yen CL, Stone SJ, Koliwad S, Harris C, Farese RV., Jr J Lipid Res. 2008;49:2283–2301. doi: 10.1194/jlr.R800018-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Unger RH. Annu Rev Med. 2002;53:319–336. doi: 10.1146/annurev.med.53.082901.104057. [DOI] [PubMed] [Google Scholar]
- 4.Friedman J. Nature. 2002;415:268–269. doi: 10.1038/415268a. [DOI] [PubMed] [Google Scholar]
- 5.Rosenbaum M, Leibel RL, Hirsch J. N Engl J Med. 1997;337:396–407. doi: 10.1056/NEJM199708073370606. [DOI] [PubMed] [Google Scholar]
- 6.Cases S, Smith SJ, Zheng YW, Myers HM, Lear SR, Sande E, Novak S, Collins C, Welch CB, Lusis AJ, Erickson SK, Farese RV., Jr Proc Natl Acad Sci USA. 1998;95:13018–13023. doi: 10.1073/pnas.95.22.13018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Farese RV, Jr, Cases S, Smith SJ. Curr Opin Lipidol. 2000;11:229–234. doi: 10.1097/00041433-200006000-00002. [DOI] [PubMed] [Google Scholar]
- 8.Buhman KK, Chen HC, Farese RV., Jr J Biol Chem. 2001;276:40369–40372. doi: 10.1074/jbc.R100050200. [DOI] [PubMed] [Google Scholar]
- 9.Turkish A, Sturley SL. Am J Physiol Gastrointest Liver Physiol. 2007;292:G953–G957. doi: 10.1152/ajpgi.00509.2006. [DOI] [PubMed] [Google Scholar]
- 10.Hofmann K. Trends Biochem Sci. 2000;25:111–112. doi: 10.1016/s0968-0004(99)01539-x. [DOI] [PubMed] [Google Scholar]
- 11.Cases S, Stone SJ, Zhou P, Yen E, Tow B, Lardizabal KD, Voelker T, Farese RV., Jr J Biol Chem. 2001;276:38870–38876. doi: 10.1074/jbc.M106219200. [DOI] [PubMed] [Google Scholar]
- 12.Yen CL, Farese RV., Jr J Biol Chem. 2003;278:18532–18537. doi: 10.1074/jbc.M301633200. [DOI] [PubMed] [Google Scholar]
- 13.Yen CL, Stone SJ, Cases S, Zhou P, Farese RV., Jr Proc Natl Acad Sci USA. 2002;99:8512–8517. doi: 10.1073/pnas.132274899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cao J, Burn P, Shi Y. J Biol Chem. 2003;278:25657–25663. doi: 10.1074/jbc.M302835200. [DOI] [PubMed] [Google Scholar]
- 15.Cao J, Lockwood J, Burn P, Shi Y. J Biol Chem. 2003;278:13860–13866. doi: 10.1074/jbc.M300139200. [DOI] [PubMed] [Google Scholar]
- 16.Cheng D, Nelson TC, Chen J, Walker SG, Wardwell-Swanson J, Meegalla R, Taub R, Billheimer JT, Ramaker M, Feder JN. J Biol Chem. 2003;278:13611–13614. doi: 10.1074/jbc.C300042200. [DOI] [PubMed] [Google Scholar]
- 17.Stone SJ, Myers HM, Watkins SM, Brown BE, Feingold KR, Elias PM, Farese RV., Jr J Biol Chem. 2004;279:11767–11776. doi: 10.1074/jbc.M311000200. [DOI] [PubMed] [Google Scholar]
- 18.Yen CL, Brown CH, 4th, Monetti M, Farese RV., Jr J Lipid Res. 2005;46:2388–2397. doi: 10.1194/jlr.M500168-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang SJ, Cornick C, O’Dowd J, Cawthorne MA, Arch JR. Lipids Health Dis. 2007;6:2. doi: 10.1186/1476-511X-6-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen HC, Jensen DR, Myers HM, Eckel RH, Farese RV., Jr J Clin Invest. 2003;111:1715–1722. doi: 10.1172/JCI15859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen HC, Smith SJ, Ladha Z, Jensen DR, Ferreira LD, Pulawa LK, McGuire JG, Pitas RE, Eckel RH, Farese RV., Jr J Clin Invest. 2002;109:1049–1055. doi: 10.1172/JCI14672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Smith SJ, Cases S, Jensen DR, Chen HC, Sande E, Tow B, Sanan DA, Raber J, Eckel RH, Farese RV., Jr Nat Genet. 2000;25:87–90. doi: 10.1038/75651. [DOI] [PubMed] [Google Scholar]
- 23.Koliwad SK, Streeper RS, Monetti M, Cornelissen I, Chan L, Terayama K, Naylor S, Rao M, Hubbard B, Farese RV., Jr J Clin Invest. 2010;120:756–767. doi: 10.1172/JCI36066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stone SJ, Levin MC, Zhou P, Han J, Walther TC, Farese RV., Jr J Biol Chem. 2009;284:5352–5361. doi: 10.1074/jbc.M805768200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kuerschner L, Moessinger C, Thiele C. Traffic. 2008;9:338–352. doi: 10.1111/j.1600-0854.2007.00689.x. [DOI] [PubMed] [Google Scholar]
- 26.Stone SJ, Levin MC, Farese RV., Jr J Biol Chem. 2006;281:40273–40282. doi: 10.1074/jbc.M607986200. [DOI] [PubMed] [Google Scholar]
- 27.Guo ZY, Chang CC, Chang TY. Biochemistry. 2007;46:10063–10071. doi: 10.1021/bi7011367. [DOI] [PubMed] [Google Scholar]
- 28.Joyce CW, Shelness GS, Davis MA, Lee RG, Skinner K, Anderson RA, Rudel LL. Mol Biol Cell. 2000;11:3675–3687. doi: 10.1091/mbc.11.11.3675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lin S, Lu X, Chang CC, Chang TY. Mol Biol Cell. 2003;14:2447–2460. doi: 10.1091/mbc.E02-11-0725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cho KH, An S, Lee WS, Paik YK, Kim YK, Jeong TS. BBRC. 2003;309:864–872. doi: 10.1016/j.bbrc.2003.08.077. [DOI] [PubMed] [Google Scholar]
- 31.Yu C, Chen J, Lin S, Liu J, Chang CC, Chang TY. J Biol Chem. 1999;274:36139–36145. doi: 10.1074/jbc.274.51.36139. [DOI] [PubMed] [Google Scholar]
- 32.Yu C, Zhang Y, Lu X, Chen J, Chang CC, Chang TY. Biochemistry. 2002;41:3762–3769. doi: 10.1021/bi0120188. [DOI] [PubMed] [Google Scholar]
- 33.Cheng D, Meegalla RL, He B, Cromley DA, Billheimer JT, Young PR. Biochem J. 2001;359:707–714. doi: 10.1042/0264-6021:3590707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weselake RJ, Madhavji M, Szarka SJ, Patterson NA, Wiehler WB, Nykiforuk CL, Burton TL, Boora PS, Mosimann SC, Foroud NA, Thibault BJ, Moloney MM, Laroche A, Furukawa-Stoffer TL. BMC Biochemistry. 2006;7:24. doi: 10.1186/1471-2091-7-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yen CL, Monetti M, Burri BJ, Farese RV., Jr J Lipid Res. 2005;46:1502–1511. doi: 10.1194/jlr.M500036-JLR200. [DOI] [PubMed] [Google Scholar]
- 36.Kyte J, Doolittle RF. J Mol Biol. 1982;157:105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- 37.Guo ZY, Lin S, Heinen JA, Chang CC, Chang TY. J Biol Chem. 2005;280:37814–37826. doi: 10.1074/jbc.M508384200. [DOI] [PubMed] [Google Scholar]
- 38.Das A, Davis MA, Rudel LL. J Lipid Res. 2008;49:1770–1781. doi: 10.1194/jlr.M800131-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lin S, Cheng D, Liu MS, Chen J, Chang TY. J Biol Chem. 1999;274:23276–23285. doi: 10.1074/jbc.274.33.23276. [DOI] [PubMed] [Google Scholar]
- 40.Shockey JM, Gidda SK, Chapital DC, Kuan JC, Dhanoa PK, Bland JM, Rothstein SJ, Mullen RT, Dyer JM. Plant Cell. 2006;18:2294–2313. doi: 10.1105/tpc.106.043695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Broadway NM, Saggerson ED. Biochem J. 1995;310:989–995. doi: 10.1042/bj3100989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lilly K, Bugaisky GE, Umeda PK, Bieber LL. Arch Biochem Biophys. 1990;280:167–174. doi: 10.1016/0003-9861(90)90532-4. [DOI] [PubMed] [Google Scholar]
- 43.Markwell MA, Tolbert NE, Bieber LL. Arch Biochem Biophys. 1976;176:479–488. doi: 10.1016/0003-9861(76)90191-0. [DOI] [PubMed] [Google Scholar]
- 44.Abo-Hashema KA, Cake MH, Power GW, Clarke D. J Biol Chem. 1999;274:35577–35582. doi: 10.1074/jbc.274.50.35577. [DOI] [PubMed] [Google Scholar]
- 45.Washington L, Cook GA, Mansbach CM., 2nd J Lipid Res. 2003;44:1395–1403. doi: 10.1194/jlr.M300123-JLR200. [DOI] [PubMed] [Google Scholar]
- 46.Owen MR, Corstorphine CC, Zammit VA. Biochem J. 1997;323:17–21. doi: 10.1042/bj3230017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Waterman IJ, Price NT, Zammit VA. J Lipid Res. 2002;43:1555–1562. doi: 10.1194/jlr.m200051-jlr200. [DOI] [PubMed] [Google Scholar]
- 48.Waterman IJ, Zammit VA. Int J Obes Relat Metab Disord. 2002;26:742–743. doi: 10.1038/sj.ijo.0801981. [DOI] [PubMed] [Google Scholar]
- 49.Yamazaki T, Sasaki E, Kakinuma C, Yano T, Miura S, Ezaki O. J Biol Chem. 2005;280:21506–21514. doi: 10.1074/jbc.M412989200. [DOI] [PubMed] [Google Scholar]
- 50.Liang JJ, Oelkers P, Guo C, Chu PC, Dixon JL, Ginsberg HN, Sturley SL. J Biol Chem. 2004;279:44938–44944. doi: 10.1074/jbc.M408507200. [DOI] [PubMed] [Google Scholar]
- 51.Harrison EH, Hussain MM. J Nutr. 2001;131:1405–1408. doi: 10.1093/jn/131.5.1405. [DOI] [PubMed] [Google Scholar]
- 52.Wongsiriroj N, Piantedosi R, Palczewski K, Goldberg IJ, Johnston TP, Li E, Blaner WS. J Biol Chem. 2008;283:13510–13519. doi: 10.1074/jbc.M800777200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.