

Abstract
To identify previously unknown genetic loci associated with fasting glucose concentrations, we examined the leading association signals in ten genome-wide association scans involving a total of 36,610 individuals of European descent. Variants in the gene encoding melatonin receptor 1B (MTNR1B) were consistently associated with fasting glucose across all ten studies. The strongest signal was observed at rs10830963, where each G allele (frequency 0.30 in HapMap CEU) was associated with an increase of 0.07 (95% CI = 0.06-0.08) mmol/l in fasting glucose levels (P = 3.2 = × 10−50) and reduced beta-cell function as measured by homeostasis model assessment (HOMA-B, P = 1.1 × 10−15). The same allele was associated with an increased risk of type 2 diabetes (odds ratio = 1.09 (1.05-1.12), per G allele P = 3.3 × 10−7) in a meta-analysis of 13 case-control studies totaling 18,236 cases and 64,453 controls. Our analyses also confirm previous associations of fasting glucose with variants at the G6PC2 (rs560887, P = 1.1 × 10−57) and GCK (rs4607517, P = 1.0 × 10−25) loci.
Blood and plasma fasting glucose levels are tightly regulated within a narrow physiologic range by a feedback mechanism that targets a particular fasting glucose set point for each individual1,2. Disruption of normal glucose homeostasis and substantial elevations of fasting glucose are hallmarks of type 2 diabetes (T2D) and typically result from sustained reduction in pancreatic beta-cell function and insulin secretion.
However, even within healthy, nondiabetic populations there is substantial variation in fasting glucose levels. Approximately one-third of this variation is genetic3, but little of this heritability has been explained. There is growing evidence to suggest that common variants contributing to variation in fasting glucose are largely distinct from those associated with major disruptions of beta-cell function that predispose to T2D. Common sequence variants in the GCK (gluco-kinase) promoter4-6, and around genes encoding the islet-specific glucose-6-phosphatase (G6PC2)5,6 and the glucokinase regulatory protein (GCKR)7-9, have each been associated with individual variation in fasting glucose levels, but have, at best, weak effects on T2D risk8,10. Furthermore, although there are now over 15 genetic loci strongly associated with the risk of T2D7,10-14, none shows compelling evidence for association with fasting glucose in the two genome-wide association scans (GWAS) so far reported5,6.
MAGIC (the Meta-Analyses of Glucose and Insulin-related traits Consortium) represents a collaborative effort to combine data from multiple GWAS to identify additional loci that affect glycemic and metabolic traits. Our genetic studies of fasting glucose levels were originally organized as four distinct consortia: (i) European Network for Genetic and Genomic Epidemiology (ENGAGE), combining data from deCODE, Northern Finland Birth Cohort 1966 (NFBC1966), Netherlands Twins Register/Netherlands Study of Depression and Anxiety (NTR/NESDA) and the Rotterdam Study; (ii) Genetics of Energy Metabolism (GEM), a meta-analysis of the Lausanne (CoLaus) and TwinsUK scans; (iii) DFS, involving the Diabetes Genetics Initiative (DGI), Finland-United States Investigation of NIDDM Genetics (FUSION) and SardiNIA scans; and (iv) the Framingham Heart Study (FHS). Details of the ten component studies (n = 1,233-6,479) are provided in Supplementary Table 1 online.
As a prelude to more extensive data-sharing, the four consortia initially exchanged the identities of between 10 and 20 SNPs prominently associated with fasting glucose in their individual, interim, meta-analyses (n = 6,479-12,389; Supplementary Table 2 online). Comparison of these signals revealed three loci with consistent effects on fasting glucose detected in multiple studies. Two of these represented the previously reported signals in G6PC2 and GCK. In addition, all four groups independently generated evidence for an association between fasting glucose and SNPs around the MTNR1B(melatonin receptor 1B) locus (ENGAGE: rs1387153, P = 2.2 10−17; GEM: rs10830963, P = 7.4 × 10−11; DFS: rs10830963, P = 2.5 × 10−7 FHS: rs11020107, P = 5.8 × 10−4, for the most strongly associated SNP exchanged from each analysis). The association signals at all three loci were confirmed on formal meta-analysis including results from all ten studies, after exclusion of individuals with known diabetes (rs560887 (G6PC2), P = 1.1 × 10−57; rs4607517), (GCK), P = 1.0 × 1.0−25; rs10830963 (MTNR1B), P = P 3.2 × 10−50; Table 1, Fig. 1, Supplementary Fig. 1, Supplementary Table 3 and Supplementary Methods online). Subsequent efforts to harmonize additional aspects of data analysis strategies (including the additional exclusion, where necessary, of individuals with fasting glucose measures ≥7mmol/l) had only a marginal impact on estimates of significance and effect size (Supplementary Table 4 online).
Table 1. Association of rs10830963 (MTNR1B) with fasting glucose levels in ten studies within MAGIC and meta-analysis of best SNPs across all ten studies for three loci associated with fasting glucose (MTNR1B, G6PC2 and GCK).
| Mean mmol/l fasting glucosea per genotype (s.d.) |
|||||||
|---|---|---|---|---|---|---|---|
| Study sample | N | G allele frequency | CC | CG | GG | Per-allele effect, mmol/l (s.e.m.) | P value |
| CoLaus | 5,000 | 0.32 | 5.36 (0.71) | 5.46 (0.80) | 5.54 (0.81) | 0.094 (0.016) | 1.9 × 10−9 |
| deCODE | 6,240 | 0.27 | 5.29 (0.71) | 5.39 (0.71) | 5.44 (0.71) | 0.086 (0.016) | 9.2 × 10−8 |
| DGI | 1,455 | 0.31 | 5.29 (0.54) | 5.32 (0.53) | 5.39 (0.60) | 0.042 (0.022) | 0.054 |
| Framinghamb | 6,479 | 0.28 | 5.16 (0.48) | 5.21 (0.48) | 5.26 (0.46) | 0.050 (0.012) | 2.2 × 10−13 |
| FUSION | 1,233 | 0.33 | 5.28 (0.49) | 5.33 (0.47) | 5.40 (0.44) | 0.057 (0.016) | 5.8 × 10−4 |
| NFBC1966 | 4,245 | 0.34 | 5.63 (0.46) | 5.70 (0.49) | 5.80 (0.46) | 0.079 (0.012) | 1.7 × 10−11 |
| NTR/NESDA | 3,166 | 0.27 | 5.22 (0.64) | 5.26 (0.62) | 5.38 (0.63) | 0.062 (0.019) | 1.2 × 10−3 |
| Rotterdam | 2,058 | 0.28 | 5.58 (0.81) | 5.75 (0.91) | 5.83 (1.03) | 0.145 (0.029) | 7.9 × 10−7 |
| Sardinia | 4,108 | 0.20 | 5.62 (0.89) | 5.68 (0.89) | 5.76 (0.89) | 0.070 (0.019) | 3.2 × 10−4 |
| TwinsUKc | 1,828 | 0.30 | 4.58 (0.65) | 4.67 (0.50) | 4.74 (0.57) | 0.084 (0.032) | 7.9 × 10−3 |
| rs10830963 (MTNR1B) | Meta-analysis | 0.072 (0.005) | 3.2 × 10−50 | ||||
| rs560887 (G6PC2) | Meta-analysis | 0.064 (0.004) | 1.1 × 10−57 | ||||
| rs4607517 (GCK) | Meta-analysis | 0.062 (0.007) | 1.0 × 10−25 | ||||
Fasting glucose levels (mmol/l) are reported untransformed and unadjusted for covariates. Effect of the risk allele and s.e.m. were calculated using untransformed fasting glucose values. P values are reported for the additive genetic model with study-specific transformation of fasting glucose values, adjusted for sex and age.
Fasting glucose levels in NFBC1966 and SardiNIA were measured in whole blood; in other samples measures were conducted on plasma samples. For these two studies, values in the table are corrected to plasma fasting glucose using a correction factor of 1.13.
In Framingham study, mean fasting glucose values for the imputed SNPs are reported for proxies: rs560887 (proxy rs573225, r2 = 0.96); rs4607517 (proxy rs1799884, r2 = 1); rs10830963 (proxy rs7936247, r2 = 0.59).
In the TwinsUK study, mean fasting glucose values per genotype are estimated for a subset of unrelated individuals only.
Figure 1.
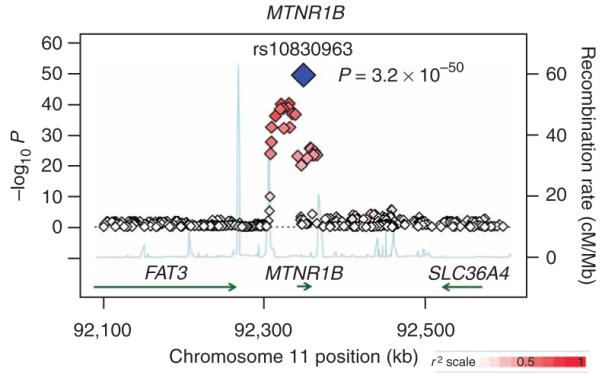
Regional plot of fasting glucose association results for the MTNR1B locus across ten MAGIC GWAS. Meta-analysis −log10 P values are plotted as a function of genomic position (NCBI build 35). The SNP with the strongest signal (rs10830963) is denoted by a blue diamond. Estimated recombination rates (from HapMap) are plotted to reflect the local linkage disequilibrium structure around associated SNPs and proxies (according to a white-to-red scale from r2 = 0 to r2 = 1 and based on pairwise r2 values from HapMap CEU). Gene annotations were taken from the University of California Santa Cruz genome browser.
We attempted to refine the location of the MTNR1B association signal by extending the meta-analysis to all SNPs (genotyped and imputed from the HapMap) within the 1-Mb region flanking the gene (n = 35,812; 981 SNPs). In all, 30 genotyped and imputed SNPs showed compelling evidence for association with fasting glucose (P < 10−8). The strongest signal was detected at rs10830963: the minor (G) allele (frequency 0.30 in HapMap CEU15) at this SNP was associated with a per-allele increase of 0.07 (95% CI = 0.06-0.08) evidence for mmol/l in fasting glucose (P = 3.2 × 10−50). Consistent association at rs10830963 was observed in all ten component GWAS, irrespective of whether this SNP was genotyped or imputed, and of the genotyping platform (Table 1 and Supplementary Table 1). Repeat meta-analysis within the region after conditioning on rs10830963 revealed no additional independent signals of association (Supplementary Note online).
The strength of the association between rs10830963 and fasting glucose was unchanged after adjustment for body mass index (Supplementary Table 4). Analyses of fasting insulin levels as well as indices of beta-cell function (HOMA-B) and insulin sensitivity (HOMA-IR) estimated by the homeostasis model assessment16 were possible in ~24,000 participants from the ten studies. These established that the glucose-raising allele at rs10830963 was associated with reduced beta-cell function (P = 1.1 × 10−15), with no appreciable effect on fasting insulin or insulin sensitivity (Supplementary Table 5 and Supplementary Note online).
To determine the impact of variants within MTNR1B on T2D risk, we carried out a large-scale meta-analysis of 13 T2D case-control samples (18,236 T2D cases, 64,453 controls; corresponding to an effective sample size of 21,179 unrelated cases and 21,179 unrelated controls). We combined data from the deCODE13, Rotterdam17, KORA18, FUSION stage 2 (ref. 11) and METSIM10 studies and from several case-control samples from the UK10 with publicly available data from the DIAGRAM consortium (which itself aggregates GWA data from the WTCCC, DGI and FUSION scans)10 (Supplementary Note). We found strong evidence that the minor G allele of rs10830963 was associated with increased risk of T2D (odds ratio = 1.09 (1.05-1.12), P = 3.3 × 10−7; Fig. 2 and Supplementary Table 6 online). The possibility that the fasting glucose association might reflect the inclusion within the cross-sectional study samples of subjects with undiagnosed T2D can be discounted given that exclusion of those with either known diabetes, or a fasting glucose ≥7mmol/l had little impact on the strength of the association signal (Table 1 and Supplementary Table 4). Although the association with T2D does not, despite large-scale replication efforts, reach the 5 × 10−8 threshold consistent with ‘genome-wide significance’15, it seems highly probable, given the strong impact of this variant on beta-cell function (Supplementary Table 5), that this is a genuine effect.
Figure 2.
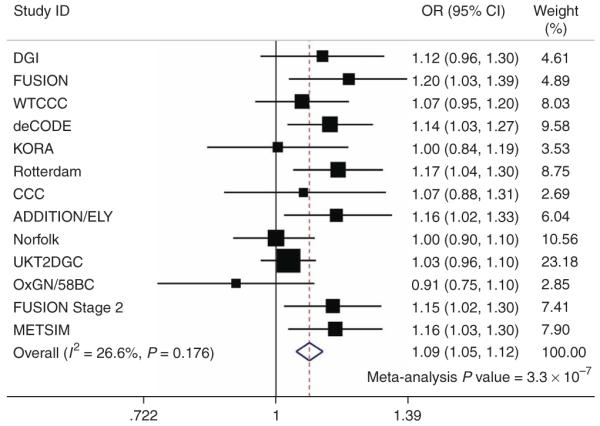
Association of rs10830963 with type 2 diabetes (T2D) in 13 case-control studies.
The analyses we performed interrogate only a fraction of common sequence variants in a given region—it is likely that the causal variant for this locus is yet to be identified. The SNP with the strongest statistical evidence so far, rs10830963, maps within the single 11.5-kb intron of MTNR1B but does not seem to disrupt consensus transcription factor binding or cryptic alternative splice sites. The association signal is bounded by recombination hot spots defining a ~60-kb interval within which all our strongly associated SNPs lie and the causal variant is likely to reside. This interval contains the entire coding region of MTNR1B. The only other nearby genes (the coding regions of which lie well outside this 60-kb region) are SLC36A4 and FAT3, neither of which are compelling candidates. SLC36A4 encodes a proton/amino acid transmembrane transporter moderately similar to Rattus norvegicus lysosomal amino acid transporter 1, and FAT3 encodes a cadherin family member which is the human homolog of the Drosophila melanogaster FAT tumor suppressor gene. Ultimately, detailed fine mapping and functional analyses will be required to define the causal allele(s) and to confirm that this effect is mediated through altered function or expression of MTNR1B.
The size of the MAGIC dataset also allowed us to examine the G6PC2 and GCK regions in greater detail than had previously been possible. In the G6PC2 region, rs560887, within intron 3 of the gene, remained the strongest signal whether or not imputed data were included (P = 1.1 × 10−57 across all ten studies; Supplementary Fig. 1 online). This is the same SNP reported in one recent paper5 and is in substantial linkage disequilibrium (LD; r2 = 0.72 in HapMap CEU) with the lead SNP (rs563694) identified in another6. In the GCK region, rs4607517, which lies 6.6-kb upstream of the gene, was the most strongly associated SNP (P = 1.0 × 10−25; Supplementary Fig. 1 and Table 1). This SNP is also in strong LD (r2 1 in HapMap CEU) with the GCK promoter SNP (rs1799884) that was featured in previous reports4. Repeat meta-analysis after conditioning on the respective lead SNPs revealed no additional independent association signals at either locus (Supplementary Note).
As with the variant in MTNR1B, the magnitude of the fasting glucose associations for both these signals was unchanged after adjustment for BMI (Supplementary Table 4). Glucose-raising alleles at GCK and G6PC2 were associated with reduced beta-cell function (rs4607517[A], P = 9.8 × 10−6; rs560887[C], P = 1.2 × 10−26; Supplementary Table 5 and Supplementary Note). However, in line with previous reports4,9, neither signal was strongly associated with T2D in the large-scale meta-analysis: in fact, the glucose-raising allele at G6PC2 was weakly associated with reduced T2D risk (rs4607517[A], per-allele OR = 1.05 (1.00-1.10), P = 0.031; rs560887[C], 0.93 (0.89-0.97), P = 0.0017; Supplementary Table 6).
We found no influence of the noncoding lead SNPs rs10830963, rs560887 or rs4607517 on gene expression of MTNR1B, SLC36A4, FAT3, G6PC2 or GCK in genome-wide expression QTL datasets from lymphocyte-derived cell lines19,20, cerebral cortex21 or liver22, and no evidence for epistatic effects among the three lead SNPs was observed (P for two-way interactions >0.19 in each of the seven studies including only unrelated individuals; interactions were not examined in the other three studies).
MTNR1B encodes one of two known human melatonin receptors23. Although this is the first study to implicate genetic variation in MTNR1B in the regulation of fasting glucose levels and predisposition to T2D, this relationship is biologically credible. As well as being highly expressed in the brain, retina and elsewhere24, MTNR1B is transcribed in human islets and rodent insulinoma cell lines25, and the translated receptor is thought to mediate the inhibitory effect of melatonin on insulin secretion26. Melatonin release is characterized by marked circadian variability and these inhibitory effects on insulin secretion may contribute to the entrainment of circadian patterns of insulin release27. There is substantial evidence in human and rodent studies linking disturbances of circadian rhythmicity to metabolic conditions including diabetes28,29, and overexpression of melatonin receptors has been observed in islets from individuals with T2D as compared to nondiabetic controls30. Taken together, these findings suggest that the association with raised fasting glucose and T2D may be driven by variants that augment expression and/or activity of islet melatonin receptors.
Our findings bring the number of common variant loci influencing fasting glucose levels to four, three of which were detected in the present study. Variants in GCKR have a smaller effect size than the others7,9, and the present study design (based on exchange of a limited number of prominent signals between component groups) was not well-powered to detect these. However, subsequent meta-analysis of GCKR variants across all ten study samples confirms the association with fasting glucose (rs780094, P = 8.5 10−9; Supplementary Table 4). The total variance in fasting glucose now attributable to these four signals is 1.5%, indicating that additional loci remain to be found3. In comparison with GCK and G6PC2, variants in MTNR1B seem to have a more marked effect on risk of T2D, the effect size being comparable in magnitude (OR = 1.09 (1.05-1.12)) to several other T2D-susceptibility genes recently identified in GWAS10. Thus, although the physiological regulation of fasting glucose set point and the pathological decline in beta-cell function that characterizes common forms of T2D generally seem to involve different processes, the MTNR1B finding suggests that this is not always the case. Not only can the study of diabetes-related quantitative traits provide an important path to the identification of additional T2D susceptibility loci, but there may also be opportunities for useful therapeutic overlap.
Supplementary Material
ACKNOWLEDGMENTS
The authors would like to thank the many colleagues who contributed to collection and phenotypic characterization of the clinical samples, as well as genotyping and analysis of the GWA data. They would also like to acknowledge those who agreed to participate in these studies. Major funding for the work described in this paper comes from Academy of Finland (124243); the Administration of Lanusei, Ilbono, Arzana and Elini (Sardinia, Italy); American Diabetes Association (1-05-RA-140); the Center for Inherited Disease Research; Clinical Research Institute (HUCH); Diabetes UK; the European Bioinformatics Institute; the European Commission (contracts LSHM-CT-2006-037197, LSHM-CT-2003-503041, QLK6-CT-2002-02629, QLG2-CT-2002-01254, HEALTH-F4-2007-201413, LSHG-CT-2004-512066, QLRT-2001-01254, LSHG-CT-2004-518153); the Faculty of Biology and Medicine of Lausanne; Finnish Diabetes Research Foundation; Folkhalsan Research Foundation; Foundation of the NIH (GAIN initiative); German Federal Ministry of Education and Research; German Federal Ministry of Health and Social Security; German National Genome Research Network; GlaxoSmithKline; GSF-National Research Center for Environment and Health; LMUinnovativ; Ministry of Science and Research of the State North-Rhine Westphalia; Municipality of Rotterdam; US National Institutes of Health (HG-02651, HL-084729, HL-087679, HC-25195, N02-HL-6-4278, DK-078616, DK-080140, DK-065978, RR-163736, MH059160, DK069922, DA-021519, DK-062370, DK-072193, US National Human Genome Research Institute intramural project HG-000024; and the Intramural Program of the National Institute on Aging); the UK National Institute for Health Research (Oxford Biomedical Research Centre and Guys and St. Thomas’ Biomedical Research Centre); the Netherlands Ministry of Education, Culture and Science; the Netherlands Ministry of Health, Welfare and Sports; Novartis; NWO (904-61-090, 904-61-193, 480-04-004, 400-05-717); NWOGenomics; NWOInvestments; Research Institute for Diseases in the Elderly (RIDE); Sigrid Juselius Foundation; Spinozapremie; Swedish Research Council (349-2006-237); UK Medical Research Council (G0500539, G0000649, G016121); UK National Health Services Research and Development; the Wellcome Trust (including intramural support for the Wellcome Trust Sanger Institute, GR069224, Strategic Awards 076113 and 083948, Biomedical Collections Grant GR072960); and ZonMw (10-000-1002).
Footnotes
COMPETING INTERESTS STATEMENT
The authors declare competing financial interests: details accompany the full-text HTML version of the paper at http://www.nature.com/naturegenetics/.
Note: Supplementary information is available on the Nature Genetics website.
Published online at http://www.nature.com/naturegenetics/
Reprints and permissions information is available online at Published online at http://npg.nature.com/reprintsandpermissions/
References
- 1.Xiang AH, et al. Coordinate changes in plasma glucose and pancreatic beta-cell function in Latino women at high risk for type 2 diabetes. Diabetes. 2006;55:1074–1079. doi: 10.2337/diabetes.55.04.06.db05-1109. [DOI] [PubMed] [Google Scholar]
- 2.Mason CC, Hanson RL, Knowler WC. Progression to type 2 diabetes characterized by moderate then rapid glucose increases. Diabetes. 2007;56:2054–2061. doi: 10.2337/db07-0053. [DOI] [PubMed] [Google Scholar]
- 3.Watanabe RM, et al. Finland-United States Investigation of NIDDM Genetics (FUSION) Study investigators Familiality of quantitative metabolic traits in Finnish families with non-insulin-dependent diabetes mellitus. Hum. Hered. 1999;49:159–168. doi: 10.1159/000022865. [DOI] [PubMed] [Google Scholar]
- 4.Weedon MN, et al. A common haplotype of the glucokinase gene alters fasting glucose and birth weight: association in six studies and population-genetics analyses. Am. J. Hum. Genet. 2006;79:991–1001. doi: 10.1086/509517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bouatia-Naji N, et al. A polymorphism within the G6PC2 gene is associated with fasting plasma glucose levels. Science. 2008;320:1085–1088. doi: 10.1126/science.1156849. [DOI] [PubMed] [Google Scholar]
- 6.Chen WM, et al. Variations in the G6PC2/ABCB11 genomic region are associated with fasting glucose levels. J. Clin. Invest. 2008;118:2620–2628. doi: 10.1172/JCI34566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Saxena R, et al. Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science. 2007;316:1331–1336. doi: 10.1126/science.1142358. [DOI] [PubMed] [Google Scholar]
- 8.Vaxillaire M, et al. Impact of common type 2 diabetes risk polymorphisms in the DESIR prospective study. Diabetes. 2008;57:244–254. doi: 10.2337/db07-0615. [DOI] [PubMed] [Google Scholar]
- 9.Orho-Melander M, et al. A common missense variant in the glucokinase regulatory protein gene (GCKR) is associated with increased plasma triglyceride and C-reactive protein but lower fasting glucose concentrations. Diabetes. 2008;57:3112–3121. doi: 10.2337/db08-0516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zeggini E, et al. Meta-analysis of genome-wide association data and large-scale replication identifies additional susceptibility loci for type 2 diabetes. Nat. Genet. 2008;40:638–645. doi: 10.1038/ng.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Scott LJ, et al. A genome-wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science. 2007;316:1341–1345. doi: 10.1126/science.1142382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sladek R, et al. A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature. 2007;445:881–885. doi: 10.1038/nature05616. [DOI] [PubMed] [Google Scholar]
- 13.Steinthorsdottir V, et al. A variant in CDKAL1 influences insulin response and risk of type 2 diabetes. Nat. Genet. 2007;39:770–775. doi: 10.1038/ng2043. [DOI] [PubMed] [Google Scholar]
- 14.Zeggini E, et al. Replication of genome-wide association signals in UK samples reveals risk loci for type 2 diabetes. Science. 2007;316:1336–1341. doi: 10.1126/science.1142364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.The International HapMap Consortium A haplotype map of the human genome. Nature. 2005;437:1299–1320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Matthews DR, et al. Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia. 1985;28:412–419. doi: 10.1007/BF00280883. [DOI] [PubMed] [Google Scholar]
- 17.Hofman A, et al. The Rotterdam Study: objectives and design update. Eur. J. Epidemiol. 2007;22:819–829. doi: 10.1007/s10654-007-9199-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Herder C, et al. Variants of the PPARG, IGF2BP2, CDKAL1, HHEX, and TCF7L2 genes confer risk of type 2 diabetes independently of BMI in the German KORA Studies. Horm. Metab. Res. 2008;40:722–726. doi: 10.1055/s-2008-1078730. [DOI] [PubMed] [Google Scholar]
- 19.Dixon AL, et al. A genome-wide association study of global gene expression. Nat. Genet. 2007;39:1202–1207. doi: 10.1038/ng2109. [DOI] [PubMed] [Google Scholar]
- 20.Stranger BE, et al. Population genomics of human gene expression. Nat. Genet. 2007;39:1217–1224. doi: 10.1038/ng2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Myers AJ, et al. A survey of genetic human cortical gene expression. Nat. Genet. 2007;39:1494–1499. doi: 10.1038/ng.2007.16. [DOI] [PubMed] [Google Scholar]
- 22.Schadt EE, et al. Mapping the genetic architecture of gene expression in human liver. PLoS Biol. 2008;6:e107. doi: 10.1371/journal.pbio.0060107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reppert SM, et al. Molecular characterization of a second melatonin receptor expressed in human retina and brain: the Mel1b melatonin receptor. Proc. Natl. Acad. Sci. USA. 1995;92:8734–8738. doi: 10.1073/pnas.92.19.8734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Su AI, et al. Large-scale analysis of the human and mouse transcriptomes. Proc. Natl. Acad. Sci. USA. 2002;99:4465–4470. doi: 10.1073/pnas.012025199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ramracheya RD, et al. Function and expression of melatonin receptors on human pancreatic islets. J. Pineal Res. 2008;44:273–279. doi: 10.1111/j.1600-079X.2007.00523.x. [DOI] [PubMed] [Google Scholar]
- 26.Stumpf I, Muhlbauer E, Peschke E. Involvement of the cGMP pathway in mediating the insulin-inhibitory effect of melatonin in pancreatic beta-cells. J. Pineal Res. 2008;45:318–327. doi: 10.1111/j.1600-079X.2008.00593.x. [DOI] [PubMed] [Google Scholar]
- 27.Boden G, Ruiz J, Urbain JL, Chen X. Evidence for a circadian rhythm of insulin secretion. Am. J. Physiol. 1996;271:E246–E252. doi: 10.1152/ajpendo.1996.271.2.E246. [DOI] [PubMed] [Google Scholar]
- 28.Spiegel K, Leproult R, Van CE. Impact of sleep debt on metabolic and endocrine function. Lancet. 1999;354:1435–1439. doi: 10.1016/S0140-6736(99)01376-8. [DOI] [PubMed] [Google Scholar]
- 29.Turek FW, et al. Obesity and metabolic syndrome in circadian Clock mutant mice. Science. 2005;308:1043–1045. doi: 10.1126/science.1108750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Peschke E, et al. Melatonin and type 2 diabetes - a possible link? J. Pineal Res. 2007;42:350–358. doi: 10.1111/j.1600-079X.2007.00426.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.