

Abstract
Typical Rett syndrome (RTT) is a pediatric disorder caused by loss-of-function mutations in the MECP2 gene. The demonstrated reversibility of RTT-like phenotypes in mice suggests that MECP2 gene replacement is a potential therapeutic option in patients. We report improvements in survival and phenotypic severity in Mecp2-null male mice after neonatal intracranial delivery of a single-stranded (ss) AAV9/CBA-MECP2 vector. Median survival was 16.6 weeks for MECP2-treated versus 9.3 weeks for GFP-treated mice. ssAAV9/CBA-MECP2-treated mice also showed significant improvement in the phenotype severity score, in locomotor function and in exploratory activity, as well as a normalization of neuronal nuclear volume in transduced cells. Wild-type mice receiving neonatal injections of the same ssAAV9/CBA-MECP2 vector did not show any significant deficits, suggesting a tolerance for modest MeCP2 overexpression. To test a MECP2 gene replacement approach in a manner more relevant for human translation, a self-complementary AAV vector designed to drive MeCP2 expression from a fragment of the Mecp2 promoter was injected intravenously into juvenile (4-5 week-old) Mecp2-null mice. While the brain transduction efficiency in juvenile mice was low (~2-4% of neurons), modest improvements in survival were still observed. These results support the concept of MECP2 gene therapy for RTT.
Keywords: AAV, Gene Therapy, MeCP2, Rett Syndrome
Introduction
Most cases of Rett Syndrome (RTT), traditionally considered a neurodevelopmental disorder and mainly affecting girls [1, 2], involve loss-of-function mutations in the methyl-CpG binding protein 2 (MECP2) gene. MECP2 is X-linked, and in human males the complete absence of functional MeCP2 is normally fatal before, or soon after, birth. Due to random X chromosome-inactivation (XCI) females are mosaic at a cellular level for MeCP2-expression, and the absence of functional MeCP2 in approximately 50% of their cells leads to the typical RTT phenotype. RTT is characterized by a distinctive constellation of clinical features including cognitive and motor disabilities [1]. Knockout of Mecp2 in mice leads to a comparable phenotype [3, 4]. Currently, there are no direct, curative treatments for RTT, and the few interventions with demonstrated clinical efficacy are given to alleviate symptoms and improve signs. Understanding of the genotype-phenotype pathway remains limited and in the absence of a well-characterized set of downstream targets based on a known mechanism for MeCP2 action, a therapeutic strategy targeting the underlying causes of RTT more directly represents an attractive way forward. While there are multiple challenges facing its application in the clinical arena, gene-replacement therapy is a potential future treatment option for RTT patients, as postnatal activation of Mecp2 in mice has been shown to lead to an improved phenotype [5-7] thus demonstrating that aspects of the disorder may be reversible and potentially preventable if early treatment can be instigated [2, 8].
MECP2 is expressed widely throughout the body but at particularly high levels in post-mitotic central nervous system (CNS) neurons postnatally [9-11], and it has been demonstrated that activation of Mecp2 only in the brain of mice carrying a targeted gene results in restoration of a broadly normal phenotype [7, 12]. However, knockout of Mecp2 in adult mice produces RTT-like phenotypes, suggesting a persistent role for MeCP2 in the functioning of adult neurons [13]. It will thus be necessary for gene therapy approaches in RTT both to achieve widespread CNS delivery of a functional copy of MECP2 and to maintain long-term expression of the exogenously-derived MeCP2 protein [13]. Recombinant adeno-associated virus (AAV) vectors have been used in research and clinical gene-delivery studies primarily because they transduce non-dividing cells and confer long-term, stable gene expression without associated inflammation or toxicity [14-16]. Many AAV vectors are based on the genome of AAV serotype 2 (AAV2), whereas transduction of cells in the brain can be enhanced through use of the AAV9 capsid to package the recombinant construct [17], as AAV9 is known to be able to cross the blood-brain barrier. AAV has a 4.7 kb single-stranded (ss) DNA genome, and modern recombinant AAV vectors from which 4.4 kb of the viral genome has been removed can be packaged with a similar-sized piece of foreign DNA. A more recent advance in AAV vector technology has been the self-complementary (sc) vector, whose genome is comprised of complementary copies of the DNA insert linked in cis through a mutated AAV inverted terminal repeat. Sc AAV vectors have 10- to 100-fold higher transduction efficiency than traditional ss AAV vectors [18, 19], facilitating applications designed for global delivery of the virus particles, such as via intravenous (IV) or intra-cerebrospinal fluid (CSF) injection. However, a potential drawback of sc AAV vectors when the gene of interest is large is that the packaging capacity is cut in half, to approximately 2.2 kb of foreign DNA.
In the current study, we asked whether several aspects of the measurable phenotype of Mecp2-null mice could be improved by AAV2/9-mediated delivery of human MECP2 to Mecp2 knockout mice. We found that early or delayed postnatal delivery of a tagged MECP2 minigene resulted in an improvement in survival and overt RTT-like signs and that widespread overexpression of exogenous MeCP2 in cells already expressing a functional endogenous allele resulted in no obvious phenotype or overt toxicity effect.
Results
Neonatal CNS injection of AAV9/MECP2 results in widespread brain expression of MeCP2 at near physiological cellular levels
RTT is characterised by the onset of overt signs some months postnatally rather than at birth and a delayed onset is also observed in Mecp2-null mice [3, 4]. A previous study has shown that postnatal activation of a silenced Mecp2 gene can prevent the onset of RTT-like signs in a mouse model [6], suggesting that the introduction of a functional copy of Mecp2 may represent a viable therapeutic approach. To test this, we cloned the human MECP2_e1 isoform coding sequence carrying a C-terminal Myc tag under the chicken beta actin (CBA) promoter into an AAV2 vector backbone and produced AAV particles with capsid 9 (AAV2/9; this construct henceforth referred to as AAV9/CBA-MECP2; Fig. 1a). Direct brain injection of this vector bilaterally into male neonatal (P0-2) wild-type (WT) and Mecp2-null mice (Fig. 1b) resulted in the widespread expression of transgenic MeCP2 throughout the CNS (Fig. 1c). Transduction efficiency varied between brain areas, while expression endured throughout the course of the study (Fig. 1c-d; supplementary figure 1). Highest transduction efficiencies were observed in hypothalamus (41.5 ± 11.3% of all cells) and thalamus (37.2 ± 6.3% of cells) with lowest efficiency seen in the striatum (6.8 ± 2.3% of cells). Labelling was predominantly observed in NeuN-positive cells suggesting preferential transduction and/or expression in neuronal populations (Fig. 1d). In contrast, the proportion of transgene-expressing cells immunonegative for NeuN (presumed mostly to represent glia) was modest (Fig. 1d; range 0.5 – 6.0% of Myc-labelled nuclei).
Figure 1. Widespread expression of exogenous MeCP2 across the brain following neonatal delivery via an AAV2/9 vector.

a. MECP2_e1/Myc fusion and GFP (control) constructs were cloned into AAV2 backbones under a CBA promoter. b. Experimental plan in which male wild-type (WT) or Mecp2-/y mice were injected intracranially with AAV9/CBA-MECP2 at P0-2, scored for the progression of RTT-like signs from 3 weeks onwards and phenotyped quantitatively at weeks 8-10. c. Representative micrograph (whole brain, parasagittal section) showing the distribution of Myc-tagged MeCP2 expression in a 12 week old WT mouse following bilateral injection. White dashed arrow indicates approximate injection site. Insets are higher power micrographs corresponding to the box in the large image showing proportion of Myc +ve (transduced cells) relative to the NeuN-immunolabelled cell population. d. Quantification of transduction efficiency (as a proportion of DAPI-stained nuclei) revealed widespread, mainly neuronal, distribution across a range of CNS regions. e. Micrograph showing transduced and non-transduced cells in hippocampal area CA3 in a WT male mouse. At a cellular level, exogenous MeCP2 (revealed by Myc immunoreactivity) was localized to neuronal nuclei with a punctate distribution characteristic of proteins that colocalise with heterochromatin. f. Injection of AAV9/MECP2 into WT mice led to augmentation of MeCP2 levels (native + transgenic) in transduced cells and enabled quantification of relative transgenic MeCP2 abundance. Blue bars show native MeCP2 levels in non-transduced cells and red bars show levels of native + transgenic MeCP2 in transduced cells. Transduced cells exhibited mean levels of anti-MeCP2 immunofluorescence that were 105 - 124% higher than mean basal levels in untransduced nuclei. g. Distribution of MeCP2 level (immunofluorescence intensity) in transduced and non-transduced layer V pyramidal cells (primary motor cortex) from 4 injected brains. Analysis revealed native MeCP2 levels to be tightly regulated (narrow peak) while levels in transduced cells showed a broader and positively shifted distribution. Scale bars: c top, 1mm; c, bottom,100μm. Abbreviations: polyA, SV40 polyadenylation signal; CBA, chicken beta actin promoter; ITR, inverted terminal repeat; NeuN, ‘neuronal nuclei’ antigen (Rbfox3); AU, arbitrary units. Bars show mean ± SEM.
Maintaining the normal cellular level of MeCP2 has become considered to be crucial for normal function as transgenic overexpression of Mecp2 has a moderate phenotype in mice [7] and human chromosome Xq28 duplications involving MECP2 result in a pronounced clinical phenotype [20, 21]. Therefore, to quantify the cellular level of exogenously-derived MeCP2 and relate this to endogenous MeCP2 levels, WT mice were injected postnatally (P1) with AAV9/CBA-MECP2 and the resultant expression product assessed by quantitative immunofluorescence after 12 weeks. Analysis of principal cells within hippocampal area CA3 and layer V of primary motor cortex (Fig. 1e-f) revealed a 105 ± 0.4 to 124 ± 0.1 % increase in anti-MeCP2 immunofluorescence in transduced (Myc-positive) cells relative to untransduced cells, suggesting that the exogenously-derived MeCP2 was being expressed at approximately 1-1.25 times native levels. Distribution analysis of MeCP2 levels (measured as immunofluorescence intensity) in transduced and non-transduced cells from 4 brains revealed endogenous MeCP2 levels to be tightly regulated (narrow peak), while levels in transduced cells were higher and more variable (Fig. 1g).
AAV9-mediated neonatal delivery of MECP2 improves the RTT-like phenotype and prolongs life span in Mecp2-null mice
To establish whether viral-mediated gene delivery of MECP2 can improve and/or prevent development of the RTT-like phenotype, Mecp2-null and corresponding WT littermate mice were injected intracerebrally at an early postnatal timepoint (P1/P2) with either AAV9/CBA-MECP2 or AAV9/GFP (as a non-therapeutic control) and monitored over a 37-week experimental period. AAV9/GFP-treated null mice showed markedly reduced survival compared to WT mice, as expected for this model [3] (Fig. 2a; median survival 9.3 weeks, range 5-19 weeks), whereas AAV9/CBA-MECP2-treated null mice showed significantly extended survival compared with the AAV9/GFP-treated null mice (median survival 16.6 weeks; range 7 to >37 weeks; p<0.0001, Gehan-Breslow-Wilcoxon test). In an AAV9/CBA-MECP2-treated WT cohort included to investigate the effect of overexpressing exogenously-derived MeCP2 in the brain no mortality was observed. Two of 12 AAV9/GFP-treated WT mice died suddenly before the end of the study. Weekly monitoring of mice for overt RTT-like features using an established observational scoring system [6, 12, 22] was carried out to investigate the trajectory of the Mecp2-null phenotype (Fig. 2b). AAV9/GFP control-injected Mecp2-null mice showed the expected phenotype with motor/activity and other deficits apparent at 3 weeks and a steep increase in phenotype severity score over the subsequent weeks. In contrast AAV9/CBA-MECP2-treated null mice exhibited slower progression of the phenotype, the severity of which reached a peak at around 14 weeks and then decreased towards WT values (supplementary videos 1-3). This decrease was in part due to sudden death in the most severely phenotypic mice, but also in part due to the lower and/or improving phenotype score in the longest-lived mice (supplementary figure 2a). There was no difference in severity score between WT mice injected with AAV9/CBA-MECP2 and those injected with AAV9/GFP and scores in these two groups did not change significantly over the course of the study. It should be noted that the downward trajectory of severity in the AAV9/GFP control-injected Mecp2-null mice at 12 weeks is largely a result of the death of the most severely affected animals reducing the mean severity in the survivors rather than a tendency for the phenotype severity to peak and then reduce.
Figure 2. Enhanced survival and reduced RTT-like signs following neonatal delivery of ssAAV9/CBA-MECP2.

a. Survival plot showing extended lifespan of Mecp2-/y mice injected with AAV9/CBA-MEPC2 (open circles, n=14) compared to AAV9/GFP injected controls (open boxes, n=11). The median survival period was significantly increased from 9.3 weeks to 16.6 weeks (P<0.0001 Gehan-Breslow-Wilcoxon test). The plot also shows absence of lethality associated with overexpression of MeCP2 in AAV9/MECP2-treated wild-type mice (filled circles, n=12). Filled boxes show data from AAV9/GFP-treated wild-type mice (n=12). b. Plot showing aggregate phenotype severity score in the mice used for survival analysis in a. The rate of phenotype progression in AAV9/MECP2-treated null mice was reduced compared to the aggressive phenotype trajectory seen in AAV9/GFP control-injected mice (P<0.05, Repeated measures ANOVA between weeks 3-12). In contrast, wild-type mice injected with AAV9/MECP2 and AAV9/GFP showed no difference between groups and no change in score over the study. c. Plot showing bodyweight changes over time in the same mice. There was a genotype effect (repeated measures ANOVA, P<0.05) but no significant treatment effect. Video camera symbols indicate time points at which movies were made (see supplemental files S1-3). # Indicates two AAV9/MECP2-treated Mecp2-null mice that survived to the end of the 30 week study (see supplementary figure 2). N Indicates that in the null untreated group, insufficient mice were still alive to plot the mean score after 12 weeks.
As growth deficits are commonly observed in RTT [1] and low bodyweight is a prominent characteristic of Mecp2-null mice, growth was also monitored weekly in these mice, but the difference in bodyweight between AAV9/GFP control-injected Mecp2-null mice and AAV9/CBA-MECP2-treated Mecp2-null mice was not significant (p > 0.05 repeated-measures ANOVA) despite a trend towards improved growth in the AAV9/CBA-MECP2-treated mice.
AAV9-mediated neonatal delivery of MECP2 improves motor phenotypes in Mecp2-null mice
Movement-related deficits constitute 3 of the 4 main criteria for RTT diagnosis [1] and represent a major feature of the syndrome in patients. Similarly, mobility and movement-dependent behaviour deficits constitute prominent characteristics of Mecp2-null mice [3, 4]. To explore the effect of AAV9/CBA-MECP2 treatment on movement-related phenotypes, mice were subjected to analysis of their locomotory behaviour at 8-9 weeks using a treadmill device and by open field test (Fig. 3a-e). Mice were challenged to perform on a treadmill at two different speeds. Challenged at the lower speed (10cm/s), there was no significant difference between AAV9/GFP-treated and AAV9/CBA-MECP2-treated Mecp2-null mice in their ability to perform to criteria (4/8 and 10/12 mice, respectively, could manage the task; Fisher’s exact test, p=0.14). When challenged using the more demanding, high-speed test (25cm/s), none of the AAV9/GFP-treated Mecp2-null mice could perform to criteria whereas 50% of AAV9/CBA-MECP2-treated Mecp2-null mice were still able to successfully complete the task, a significant difference in ability to perform in this test (0/8 vs 6/12 mice, respectively; Fisher’s exact test, p=0.024). All wild-type mice (both AAV9/CBA-MECP2 and AAV9/GFP treatment groups) could perform at both speeds.
Figure 3. Improved motor but not respiratory phenotypes following neonatal delivery of MECP2.
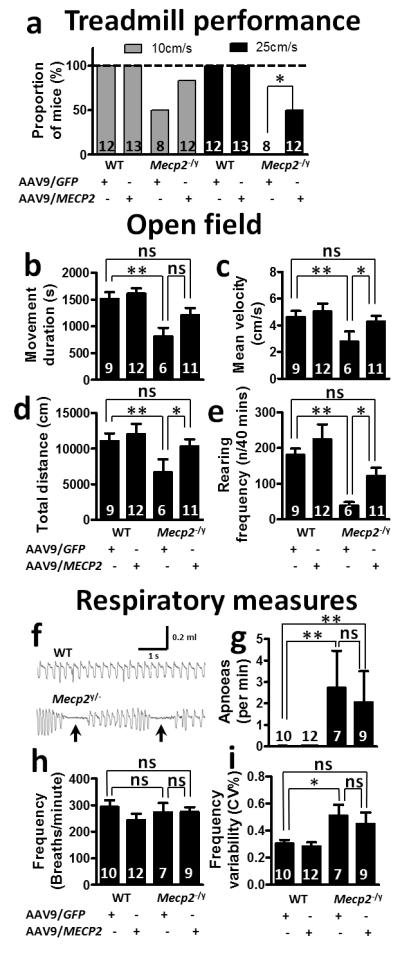
a. Proportion of mice able to perform at two different speeds on a motorized treadmill. There was a significant overall difference in performance between groups (10cm/s, P=0.0013; 25cm/s, P<0.0001; Fisher’s exact test). Post hoc pairwise comparison of AAV9/MECP2-treated and AAV9/GFP-treated mice showed a difference at 25cm/s (P=0.024), but not at 10cm/s. b-e. Open field tests performed at 9 weeks following AAV9 injection showing measures for movement duration (b), mean velocity (c) and total distance moved (d) together with rearing frequency (e). f. Representative whole-body plethysmograph traces showing regular and erratic breathing patterns/apnoeas (arrows) in WT and Mecp2-/y mice respectively. g. Apnoea frequency, h. baseline breathing frequency, i. breathing frequency variability. Both open field and respiratory measures were assessed by 2-way ANOVA with Tukey’s post hoc comparisons. *P<0.05, **P<0.01, ns=not significant. Numbers of animals per genotype/treatment group are shown within each bar.
AAV9/GFP-treated Mecp2-null mice (n=6) showed a significant deficit in locomotion-related parameters in the open field tests, including movement duration, total distance moved and mean velocity, compared to AAV9/GFP-treated WT controls (Figure 3b-d; n=9; all p<0.01). In contrast, AAV9/CBA-MECP2-treated Mecp2-null mice (n=11) were not significantly different from AAV9/GFP-treated WT controls (n=9), while post hoc comparisons revealed significantly higher mean velocity and total distance moved parameters in the AAV9/CBA-MECP2-treated Mecp2-null mice than in the AAV9/GFP-treated Mecp2-null cohort (both p<0.05). AAV9/CBA-MECP2-treated WT mice (n=12) demonstrated performance comparable to that of AAV9/GFP-treated WT mice (n=9; p>0.05). In addition to ambulatory movement, we tested exploration-related behaviour by assessing rearing frequency in the open field. AAV9/GFP-treated Mecp2-null mice showed a significantly reduced rearing frequency compared to AAV9/GFP-treated WT mice (p<0.01), while AAV9/CBA-MECP2-treated Mecp2-null mice showed increased rearing frequency relative to AAV9/GFP-treated Mecp2-null mice (p<0.05). As with ambulatory movements, there was no difference between the WT treatment groups.
Overt respiratory phenotypes persist following AAV9-mediated neonatal delivery of MECP2 to Mecp2-null mice
Apnoeas and abnormal breathing patterns were investigated using whole body plethysmography. At 9 weeks following neonatal delivery of AAV9/CBA-MECP2 or AAV9/GFP, Mecp2-null mice displayed a characteristic erratic breathing pattern with frequent apnoeas (Fig. 3f-g), in contrast to the regular baseline breathing pattern seen in both WT treatment groups. Overall, there was no difference in baseline respiratory frequency between genotypes or treatments (Fig. 3h; 2-way ANOVA, p>0.05). An increased incidence of apnoeas (Fig. 3g) was observed in the Mecp2-null mice compared to the WT mice (2-way ANOVA, genotype main effect p<0.05), but the AAV9/CBA-MECP2- and AAV9/GFP-treated mice did not differ. Similarly, higher breathing frequency variability (CV%; Fig. 3i) was observed in the Mecp2-null mice compared to the WT mice, but again the AAV9/CBA-MECP2- and AAV9/GFP-treated mice did not differ (2-way ANOVA, genotype main effect p<0.05, treatment main effect p>0.05, n=7-12 mice per group).
AAV9-mediated delivery of MECP2 rescues the Mecp2-null nuclear volume phenotype
A consistent feature of MeCP2 deficiency at the cellular level is a reduction in nucleus volume [5, 23]. In order to assess potential changes in this parameter, 3D nuclear volume measures were obtained for AAV9/CBA-MECP2-transduced and non-transduced granule cells of the dentate gyrus (Fig. 4). Consistent with previous reports, non-transduced neurons in Mecp2-null mice showed nuclear volumes that were on average 69% of volumes in age-matched WT mice (Fig. 4d). In Mecp2-null mice AAV9/CBA-MECP2- transduced cells displayed nuclear volumes that were significantly larger (by 29% on average) than those of neighbouring non-transduced cells in the same brains, and the AAV9/CBA-MECP2-transduced cells showed no significant difference from values observed in non-transduced WT mice. Interestingly, in WT mice, AAV9/CBA-MECP2-transduced cells showed a modest (approx. 1.2-fold on average) but significant increase in nuclear volume relative to neighbouring non-transduced cells (Figure 4c).
Figure 4. Increased nuclear volume in Mecp2-/y and wild-type mice after AAV9/CBA-MECP2 injection.
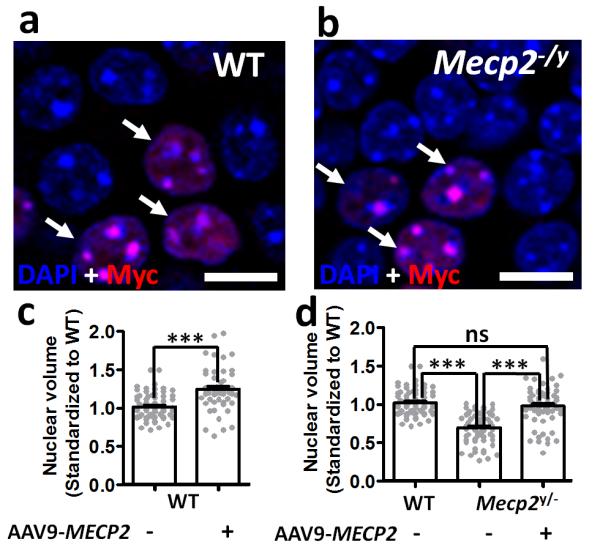
a-b. Representative image from the dentate gyrus of (a) wild-type and (b) Mecp2-null mouse at 12 weeks following AAV9/MECP2 injection. Sections show DAPI-stained nuclei within the stratum granulosum and transfected cells identified by anti-Myc immunostaining (arrows). c. Nuclear volume measurements from 3D-reconstructed DAPI-labelled nuclei in wild-type mice (n=4). d. Nuclear volume measurements from WT (n=122 datapoints in total from 4 mice) non-transfected cells and transfected and non-transfected cells from age-matched Mecp2-null mice (n=122 datapoints in total from from 3 mice). All data was normalized to the mean volume in untransfected wild-type cells. Bar plots show mean ± SEM with grey dots showing individual datapoints. Statistical analysis was carried out by 2-way ANOVA and Tukey’s post hoc pairwise comparison. ***= P<0.001. Scale bar = 10μm.
Intravenous delivery of MECP2 to the brains of juvenile mice using scAAV vectors is less efficient and of more limited benefit
Since widespread MECP2 gene delivery is likely to be a prerequisite for any successful RTT gene therapy approach, future translational studies of AAV-based therapeutic strategies will depend on the development of an appropriate scAAV/MECP2 vector. The MECP2 coding sequence spans nearly 1.5 kb, so packaging this gene in a sc form requires the use of a promoter and polyadenylation signal totaling ≤ 0.7 kb. A 229 bp truncated portion of the originally described 731 bp murine Mecp2 promoter containing the core transcriptional control elements was recently described as broadly recapitulating the overall pattern of MeCP2 expression [24]. Within the CNS, this promoter drives expression predominantly in neurons. We generated a scAAV9 vector packaging a construct consisting of Myc-tagged human MECP2 driven by the 229 bp Mecp2 promoter (MeP; Fig. 5a) and tested this vector in male Mecp2 knockout mice. We have previously shown that IV administration of scAAV9 vectors can lead to widespread and dose-responsive transduction of neurons and glia throughout the entire brain and spinal cord [25]. Four to five week-old mice received scAAV9/MeP-MECP2 by tail vein infusion. Following injection, mice were weighed weekly and assessed for survival, with a 20% drop from their peak weight marking the endpoint for sacrifice and thus scored as a death. To assess the transduction efficiency in the brain, sections were co-immunolabeled for NeuN and Myc-tagged MeCP2.
Figure 5. Enhanced survival following intravenous delivery of scAAV9/MeP-MECP2 virus to juvenile mice.

a. A MECP2_e1/Myc fusion construct was cloned into a scAAV2 ITR backbone under a truncated version of the murine Mecp2 promoter. b. Graph showing transduction efficiency in three brain areas of Mecp2-null mice (n=3) at endpoint following tail vein injection of scAAV9/MeP-MECP2. See suppl. figure 3 for representative images. Plots show mean ± SEM. c. Survival plot showing extended survival of Mecp2-/y mice injected with scAAV9/MeP-MEPC2 (purple open boxes, n=12) compared to untreated controls (green closed boxes, n=10). The median survival period in the treated mice was significantly higher than in the untreated group (15.4 vs 10.6 weeks, respectively, P<0.01 Gehan-Breslow-Wilcoxon test). Control WT mice showed no mortality over the experimental period (filled circles, n=8). Arrow indicates scAAV/MeP-MECP2 injection time. d. Micrographs showing GABA immunolabelling of transduced (MeCP2-positive, arrows) and non-transduced cells (arrowheads) in layer 2/3 of motor cortex of a Mecp2-null mouse. e. Bar plot showing elevated levels of anti-GABA immunofluorescence in transduced cells (***p<0.001, t-test, n=166 cells). mut ITR, mutated inverted terminal repeat; MeP, truncated murine Mecp2 promoter; BGHpolyA, boving growth hormone polyadenylation signal. Scale bar in d = 10μm.
Figure 5b and Suppl. Figure 3 show that even when a relatively high dose of 5×1011 vector genomes (vg) was administered, a relatively small proportion of cells were transduced in the motor cortex (2.54±1.04%), hippocampus (2.36±1.08%), and striatum (3.99±1.77%) (n=3 mice, >800 neurons counted per brain region across 5 sections per mouse; mean ± SEM). As expected from our previous studies on the tropism of AAV9 and the cell-type specificity of the Mecp2 promoter [24, 25], expression of MeCP2 was predominantly neuronal (Suppl. Figure 3). Despite the low transduction efficiency, median survival was significantly longer in the scAAV9/MECP2-treated Mecp2-null mice than in the untreated Mecp2-null mice used as controls (Fig. 5c; n=12 scAAV9/MeP-MECP2-treated and n=10 untreated mice; median survival was 15.4 weeks and 10.6 weeks in these groups, respectively, p<0.01 Gehan-Breslow-Wilcoxon test). There was no effect of scAAV9/MeP-MECP2 treatment on bodyweight (Suppl. Fig. 4b). These results indicate that the MeP-MECP2 construct provides sufficient MeCP2 expression to prolong the lifespan of juvenile male Mecp2 knockout mice. There was a trend towards a dosage effect (Suppl. Fig. 4a), with mice given the higher dose of scAAV9/MeP-MECP2 showing somewhat extended survival on average than those given the lower dose.
Given the low transduction efficiency in the brain following IV-injection at 4-5 weeks and the corresponding modest therapeutic effects on survival, an improvement in the phenotype severity score was not investigated, but a cellular phenotype was investigated instead. MeCP2 is required for the maintenance of synaptic function in mature neurons, and activation of Mecp2 in the postnatal brain has been shown to reverse at least a subset of synaptic defects in Mecp2 knockout mice [6]. Cell-type specific knockout of Mecp2 in GABAergic neurons by Cre-mediated homologous recombination reduces cellular GABA content by nearly 40% in the adult mouse cortex and similarly reduces the amplitude of inhibitory synaptic currents [26]. To determine whether postnatal scAAV9/MeP-MECP2-mediated MeCP2 expression is sufficient to rescue aspects of inhibitory neuron function in the Mecp2-null brain, we quantified GABA content in the soma of MeCP2-positive (transduced) and MeCP2-negative GABAergic interneurons in layer 2/3 of motor cortex (Fig. 5d-e). Mean GABA levels in exogenously-derived MeCP2-positive cells were 164.6±9.3% of levels in MeCP2-negative cells in the same fields (Fig. 5e; n=124 MeCP2-negative neurons, 42 MeCP2-positive neurons, p<0.001).
After intravenous delivery AAV9-mediated transduction is widespread in peripheral organs
The low transduction efficiency of brain cells after IV delivery of the scAAV9 vectors that we have described above led us to investigate the distribution of the packaged transgene (i.e. the vector biodistribution) in the periphery to assess the potential for MeCP2 overexpression-related toxicity. It is known that following IV injection of AAV9, the vector preferentially targets peripheral organs, while in the CNS a considerably smaller transduction rate is usually observed [25, 27].
Juvenile WT mice (4-5 weeks old) were injected IV with vehicle, with 1×1011 vg scAAV9/MeP-GFP, or with 1×1011 vg scAAV9/MeP-MECP2, then sacrificed after 1 or 3 weeks (i.e. at age 5-6 weeks or 7-8 weeks). As expected, the AAV9 vector preferentially targeted the liver and the spleen (Figure 6a), with between 1 and 10 copies of the vector present per cell on average (assuming that most liver cells are still diploid at the ages employed here). Levels of vector in heart and kidney were approximately 10-fold lower than this, and levels in the brain and spinal cord were nearly 100-fold lower, consistent with the transduction rates in brain reported above. No obvious differences in vector DNA levels were observed between the MECP2 and GFP vectors, or between the 1 and 3 week time points, for any organ. Within the limits of experimental sensitivity, no vector DNA signal could be detected in the vehicle-injected animals.
Figure 6. Vector biodistribution and gene expression after IV injection.
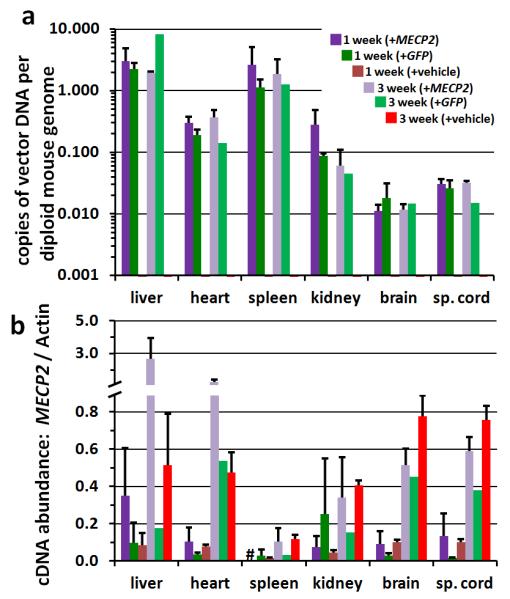
WT C57Bl/6 mice were injected IV at 4-5 weeks old with vehicle, 1×1011 vg scAAV/MeP-GFP (+GFP), or 1×1011 vg scAAV/MeP-MECP2 (+MECP2), then sacrificed at 1 or 3 weeks post-injection. a. qPCR biodistribution of GFP or MECP2 vector genomes to major organs. b. RT-qPCR of total MECP2 (mouse + human) relative to actin. Mean ± S.E.M. are plotted, n = 2-4, with the exception of the +GFP mice at 3 weeks (n =1). Legend for both panel a and b is provided in panel a. # = not determined.
MECP2 expression levels were also assessed by RT-qPCR in the same organs, using tissue from the same set of mice used for the biodistribution studies and expressing transcript abundance relative to that of actin transcripts in the same tissues (Figure 6b). We ascertained on the basis of a melting curve analysis of the PCR products (see Methods and Supplemental Table 1) that for the +MECP2 mice, in the organs with the highest vector biodistribution we could detect AAV9 vector-derived human MECP2 expression whereas only the endogenous mouse Mecp2 expression was detected in the GFP and vehicle-treated mice. At 3 weeks post-injection, relative levels of endogenous Mecp2 transcripts (ascertained from the vehicle-injected mice) were comparable with those observed previously at a comparable time point [10] except for spleen, in which transcript abundance was unexpectedly low. Although sample sizes prohibited detailed statistical analysis, we also observed that there was a trend towards increasing Mecp2 expression levels in all organs between the 1 week post-injection time point (i.e. 5-6 weeks of age) and the 3 week post-injection time point (i.e. 7-8 weeks of age), based on data from the vehicle-injected mice; such age-related changes have been reported before [28]. This pattern of transcript abundance change was also observed in the +MECP2 mice, and for kidney, brain and spinal cord, transcript levels were approximately the same as those in the vehicle-injected mice, consistent with the vector load (Fig. 6a). In the liver and heart, however, the transcript abundance was much higher at week 3 in the +MECP2 mice compared to the vehicle-injected mice, suggesting that the high vector load in these organs (Fig. 6a) led to high levels of human MECP2 expression in addition to the endogenous mouse transcript. The high vector DNA load in the spleen and the lack of additional MECP2 transcript in the +MECP2 mice are consistent with previous reports that the MeP promoter confers poor transgene expression in the spleen after intravenous AAV9-mediated transduction [24].
Where vector load is high and exogenous MECP2 is expressed at high levels from an efficient promoter, overexpression toxicity is a potential concern. We assessed a number of serum markers of organ dysfunction in the mice used for the biodistribution analysis (Supplemental Figure S5). There was no significant difference in the levels of these markers between the AAV9/GFP-treated and vehicle-injected mice. The difference between AAV9/MECP2-treated mice and vehicle-injected mice was, however, quite marked (p < 0.01) in the case of alanine amino transferase (ALT), a marker of acute liver damage, with increased levels observed in the AAV9/MECP2-treated mice. This may indicate that the high vector load and efficient transcription of MECP2 in liver cells has led to some liver dysfunction in the treated mice.
Discussion
Expression of MeCP2 as a result of AAV-mediated gene delivery to the Mecp2-knockout mouse CNS confers a survival benefit and phenotypic improvement
Incomplete understanding of the precise molecular actions of MeCP2 continues to impede the development of rational therapeutic strategies for Rett syndrome. In theory, delivery of a wild-type copy of the MECP2 gene to cells lacking functional MeCP2 represents a therapeutic approach worth considering. In this study, we provide evidence at the proof-of-concept level that exogenous delivery of the human MECP2 gene to the CNS of Mecp2-null male mice can result in levels of MeCP2 expression that lead to a reduction in phenotypic severity. We show that early postnatal delivery of exogenous MECP2 directly to the brain at transduction efficiencies in the range of ~7-42%, depending on brain region, results in a substantial prolongation of lifespan (~80% increase in median survival) and a less aggressive trajectory of RTT-like phenotype severity (Figure 2). Interestingly, a small number of treated mice survived to the end of the 37-week study period, well in excess of the median lifespan of the control Mecp2-null mice (lifespan was >3 fold greater for some AAV9/CBA-MECP2-treated mice). Furthermore, the longest-lived mice treated with the therapeutic vector also displayed a stabilization of the phenotype in terms of aggregate severity score and objective locomotor measures. Male mice lacking MeCP2 in all cells display the most severe phenotype observable in any mouse model of RTT and thus provide an especially challenging system for assessing the potential of novel therapeutic approaches. Nevertheless, these data show that treatment with the AAV9/CBA-MECP2 vector can influence phenotype progression and lifespan. In contrast to the observed beneficial impact on the RTT-like signs, AAV9/CBA-MECP2-treatment resulted in only a modest, non-significant increase in bodyweight of Mecp2-null mice compared to control-treated Mecp2-null mice, even for the most long-lived animals. Amongst several potential reasons for this observation are the possibility that exogenously-derived MeCP2 expression levels in relevant areas of the brain did not reach a threshold for affecting bodyweight or that reduced bodyweight in Mecp2-null mice is at least in part due to MeCP2 deficiency in peripheral organs rather than solely the brain. However, the relationship between animal ‘health’ and bodyweight is not straightforward, with other studies also showing little change in body weight despite a dramatic arrest of phenotype and prolongation of lifespan [29].
The effect of the AAV9/MECP2 treatment on the motor/locomotory phenotype of the Mecp2-null mice was especially notable and robust - treatment led to substantial improvements in both open field activity/locomotion and performance in a forced motor task. The impaired locomotor activity observed in the control vector-treated Mecp2-null mice in the open field experiments can be explained by genuine motor disability, by an anxiety phenotype or by a lack of motivation. The complete inability of AAV9/GFP-treated mice to perform in a forced motor challenge when tested at the high speed on the treadmill and their partial ability to perform at low speed suggest the presence of a genuine motor defect rather than inertia purely due to lack of motivation. This motor impairment was ameliorated by AAV9/CBA-MECP2 treatment, and the effect was mirrored in the open field test, in which the AAV9/CBA-MECP2-treated mice displayed significantly enhanced ambulatory activity. Locomotor improvement was accompanied by an increase in rearing frequency in AAV9/CBA-MECP2-treated mice, suggesting additional improvement in exploration-related behavior. Analysis of longitudinally-collected open field data from the long-term survivors suggests that the locomotor phenotypes are not progressive throughout life in AAV9/CBA-MECP2-treated null mice (supplementary figure 2b-d). Reports by other groups as to whether anxiety phenotypes that can be measured by open field test are visible in young Mecp2-null mice have not revealed consistent observations [12, 30, 31]. We found no genotype or treatment differences in the proportion of time spent in the central zone of the open field (data not shown) and therefore did not investigate this further.
The analysis of breathing was consistent with other reports that Mecp2-null mice exhibit a range of breathing phenotypes [32], mirroring the clinical picture where apnoeas and other breathing irregularities are characteristic and highly prevalent features of RTT [1]. Previous studies have shown that postnatal activation of Mecp2 globally [33] or in glial cells [12] can rescue abnormal breathing and this suggests that the respiratory phenotype is not solely the result of an earlier developmental deficit and is amenable to treatment at the level of causative gene function. The absence of any impact of AAV9/CBA-MECP2-treatment on breathing irregularity or on the incidence of apnoeas in the mutant mice in this study could be explained if, despite the widespread expression of exogenous MeCP2, the cells responsible for breathing rhythmogenesis or otherwise contributing to breathing regulation were transduced at levels below a threshold for improved functioning.
MeCP2-deficiency is known to be associated with a wide variety of morphological and neurochemical changes in the brain [2]. To examine the consequences of exogenous MECP2 delivery, simple measures of cellular phenotype, including neuronal nuclear volumes within the dentate gyrus(CBA-MECP2 construct) and the GABA content of GABAergic interneurons within the cortex (MeP-MECP2 construct), were investigated. The significant reduction in neuronal nuclear volume in MeCP2-deficient cells has been reported previously [5, 23] and our results show that this cell morphological measure can be effectively rendered normal by the expression of exogenously-derived MeCP2 at close to WT levels within the nucleus. Furthermore, the comparison between neighboring MeCP2-containing and MeCP2-deficient cells within individual brains suggests that these changes represent a cell-autonomous effect. This would be consistent with differences that we have observed in nuclear volume and soma size between neurons containing MeCP2 and those lacking MeCP2 in the heterozygous female (Mecp2+/−) mouse brain [34] (authors’ unpublished observations). The fact that nuclear volume becomes normal after expression of exogenously-derived MeCP2 suggests that the appearance of MeCP2 in a previously MeCP2-negative neuron leads to structural remodeling within the nucleus. Whether this remodeling also extends to changes in dendritic/axonal size and complexity, as shown in response to exogenously-derived MeCP2 in neuronal culture [35] and upon delayed activation of Mecp2 in mature brains [33], remains to be established.
The increases we observed in GABA content in cortical GABAergic interneurons upon MeCP2 re-expression in juvenile mice suggest one possible mechanism through which AAV-based gene therapy could lead to recovery of neuronal circuit function. A previous study has shown that cell type-specific deletion of Mecp2 in GABAergic neurons reduces GABA content (~40% in cortical interneurons) and leads to a variety of RTT-like phenotypes including motor deficits [26]. Our finding that exogenous delivery of MeCP2 elevates/restores GABA levels raises the possibility that improved GABAergic synaptic function in the scAAV9/MeP-MECP2-treated mice accounts at least in part for their milder motor phenotypes compared to control null mice.
Potentially serious complications have been attributed to MeCP2 overexpression [7, 21, 36]. The present studies demonstrate the potential for achieving expression of exogenously-derived MeCP2 without overt overexpression-related toxicity within the CNS, at least with the vector designs utilized in this study and with the delivery efficiencies achieved. That said, in early pilot experiments (data not shown) we observed that introducing MeCP2 intracranially using scAAV9/MeP-MECP2 in neonatal mice achieved very high transduction efficiency and high levels of cellular expression, which resulted in a delayed-onset overexpression-related toxicity with similarities to that reported previously [7]. Moreover, we observed elevated levels of a liver damage marker (ALT) after peripheral delivery of the scAAV9/MECP2 vector to juvenile mice. Expression levels in these mice were consistent with a high vector load in the liver. These results suggest that the expression cassette provides a tolerable level of expression from one copy of the vector per cell, but overloading a particular cell with many copies of vector may lead to a deleterious overexpression phenotype. The lack of any phenotypic deficits in wild-type mice injected as neonates with the ssAAV9/CBA-MECP2 vector, however, which had approximately double normal levels of MeCP2 in transduced brain cells, suggests that in situations where vector load is modest (≤1 vector being present within each cell), this level of MeCP2 overexpression is tolerated. We cannot, of course, rule out the possibility of subtle or very delayed overexpression toxicity under these conditions [37], but the same dose in male Mecp2-null mice (conferring cellular MeCP2 levels in null cells that were 1- to 1.25-fold higher than levels in WT cells) provided a significant behavior and survival benefit. Moreover, none of the 4-5 week old male Mecp2-null mice in this study injected with the scAAV9/MeP-MeCP2 vector showed the mortality associated with up to 50% of mice upon rapid genetic reactivation of an inactive Mecp2 allele in 6 week-old male Mecp2-mutant mice [6]. The implications of these findings are particularly important when we consider this approach for RTT females (who have a mosaic pattern of cells expressing either the normal or mutant MECP2 allele). Here, there is a concern that overexpression toxicity might result from introduction of MeCP2 to cells expressing the endogenous WT allele while trying to achieve therapeutic levels of MeCP2 in the cells expressing the mutant allele. Our data shows that it may be possible to achieve a therapeutic outcome without the associated overexpression toxicity using an appropriate vector and promoter system.
Vector Design and Translational Implications
In the current study, delivery of MECP2 was initially conducted at an early postnatal age (P0-2) in order to achieve high neuronal transduction efficiency [14, 38, 39] and to test the effect of early introduction of exogenously-derived MeCP2 on the trajectory of the RTT-like phenotype. The clear influence that ssAAV9/CBA-MECP2 had on survival and motor phenotypes in this study contrasts with the failure of a similar vector to impact the survival of male Mecp2-null mice when injected intravenously at 4-8 weeks of age [12]. One interpretation of this difference is that later postnatal delivery of MECP2 might be ineffective in affording phenotypic benefit. However, in our study, delivery of MECP2 at an equivalent later time point using a self-complementary vector (Fig.5c) had a significant survival benefit, whilst reactivation studies by other groups in which Mecp2 silencing is reversed at late time points [6, 33] also suggest that the latency of MeCP2 restoration is not a critical issue. More important factors are likely to be the level of MeCP2 within each cell as well as the proportion of cells transduced. Indeed, reactivation studies have demonstrated a relationship between numbers of cells re-expressing MeCP2 and the degree of phenotypic rescue [33].
The increased transduction efficiency that comes from the use of scAAV vectors [18, 19, 25] is likely to be critical to the success of a translationally-relevant RTT gene therapy. The novel scAAV vector design we describe, in which MeCP2 expression is driven by a fragment of the Mecp2 promoter, was able to provide a modest survival benefit to Mecp2-null mice. However, the low transduction efficiency of neurons (2-4%) given the peripheral administration route, coupled with this modest therapeutic effect, suggest that CNS delivery efficiency remains the major factor limiting further efficacy with this approach. The high level of MeCP2 expression in the liver and associated evidence for liver damage after intravascular delivery of the vector suggest that a high concentration of vector in any particular cell population is something to be avoided. In formulating translational strategies, this is likely to be a critical issue. Any viable solution will require widespread distribution of vector across the CNS but minimal concentration of vector in peripheral tissues that are sensitive to MeCP2 overexpression. Whether the required distribution is more likely to come from direct brain injection, peripheral intravascular injection or injection into the cerebrospinal fluid (CSF) [25, 40-42] remains to be explored.
A further issue relates to whether we have reached a full understanding of the complement of cell types that underlie the RTT disease phenotype and the extent to which we have yet to explore which cell types, when rendered fully functional, will have the largest influence on amelioration of the phenotype after treatment. A recent report by Derecki and colleagues [29] demonstrated a robust arrest of the RTT-like phenotype after transplantation of WT microglial precursors from bone marrow into juvenile Mecp2-null mice. Application of this approach in patients is not straightforward, as it may involve first ablating the resident MeCP2-deficient population of microglia in the brain. If microglia do prove to be the cell type with the largest influence on the phenotype when corrective treatments are used, then a gene therapy approach in which microglia are targeted in preference to other cell types [12] could be used to circumvent this problem. The promoters used in our current study were not geared to achieving preferential expression in non-neuronal cells and this approach necessitates development of a new generation of appropriate vector constructs. Alternatively, however, we can envisage a future strategy in which vectors targeting microglial precursors in the periphery could be employed. Fixing both the genotype and phenotype of such cells could be successful if population of the brain by these peripheral precursors could be achieved. As an alternative, given the findings implicating various cell-autonomous defects in neurons lacking MeCP2, it is possible that a strategy involving both microglial cell transplantation and targeted delivery of MECP2 to neurons using viral vectors might be worth investigating for its potential to produce a synergistic outcome.
Whilst encouraging, the results of our study demonstrate only the first step in the development of a RTT gene therapy approach suitable for human translation. The results suggest that a more efficient and CNS-specific delivery system appropriate for the human scale is needed for RTT gene therapy. Alternative routes of administration and improvements in vector design offer possible solutions to this problem [40-42].
Concluding remarks
This work shows, at ‘proof-of-concept’ level, that exogenous MeCP2 can be delivered at a physiologically tolerable level via AAV2/9 vectors to the brain of Mecp2-null mice. Three main issues affecting the development of RTT gene therapy approaches have been addressed in this study. Firstly, we have shown that partial amelioration of the RTT-like phenotype in the null mouse model can be achieved via provision of exogenously-derived MeCP2. Secondly, we have shown that MeCP2 can be expressed at levels that are both efficacious in the null mouse and tolerated in WT mice, providing a broader-than-anticipated window of therapeutic MeCP2 expression. Thirdly, we have identified CNS delivery efficiency and overexpression in peripheral organs as limiting factors for any future translation approaches. Longer-term studies in heterozygous female mice, along with development of more sensitive mouse phenotyping tools and improvements in global CNS gene delivery are the next steps before any applied human application can be considered.
Material and methods
Animals
All investigations in Glasgow were carried out in accordance with the European Communities Council Directive (86/609/EEC) and with the terms of a project licence under the UK Scientific Procedures Act (1986). All experiments in North Carolina were approved by the University of North Carolina–Chapel Hill (UNC) Institutional Animal Care and Use Committee (IACUC). The Mecp2-null mice (B6.129P2(C)-Mecp2tm1.1Bird/J) used at UNC (treated at 4-5 weeks of age) were obtained from Jackson Laboratory (Bar Harbor, ME). The studies at Univ. of Glasgow (using mice that were treated as neonates) used Mecp2tm1.1Bird mice which were originally provided as a kind gift from Prof. Adrian Bird and maintained on a C57BL/6 / CBA mixed background. Animals were maintained on 12-hour light/dark cycles with free access to normal mouse food. Mice were genotyped as described previously [3].
Virus preparation
Recombinant AAV vector particles were generated using HEK293 cells grown in serum-free suspension conditions in shaker flasks, using proprietary methods developed at the UNC Gene Therapy Center Vector Core facility. In brief, single-stranded or self-complementary AAV particles (AAV2 ITR-flanked genomes packaged into AAV9 serotype capsids) were produced from suspension HEK293 cells (ATCC# CRL 1573) transfected using polyethyleneimine (Polysciences) with the following helper plasmids (pXX6-80 [43], pGSK2/9) plus one of three ITR-flanked transgene constructs (pTR-CBA-GFP, pTR-CBA-hMeCP2, pSJG-MeP229-hMeCP2, or pSJG-MeP229-GFP, respectively) to generate each respective single-stranded or self-complementary AAV vector. The CBA constructs utilized a CMV enhancer, modified chicken β-actin promoter, shortened SV40 intron, and SV40 polyadenylation sequence [44]. The MeP constructs used a truncated 229 bp murine Mecp2 promoter [44], along with the bovine growth hormone polyadenylation signal following MeCP2 or the SV40 polyadenylation signal for GFP [24]. All MeCP2-expressing constructs utilized the human MECP2 coding region (MECP2_e1 mRNA isoform) with a C-terminal Myc epitope tag. Forty-eight hours post-transfection, cell cultures were centrifuged and supernatant discarded. The cells were resuspended and lysed by sonication, as described [45]. DNase (550 U) was added to the lysate and incubated at 37°C for 45 minutes, followed by centrifugation at 9400 x g to pellet the cell debris and the clarified lysate was loaded onto a modified discontinuous iodixanol gradient followed by column chromatography, or purified by a step CsCl gradient centrifugation followed by a discontinuous CsCl gradient centrifugation. Both purification methods provide equivalent high levels of purity and in vivo transduction efficiency [46]. Purified vectors were dialyzed in 1x PBS with 5% D-sorbitol and a final NaCl concentration of 350 mM. AAV titer was obtained by both dot blot [45] and qPCR [25].
Injection and phenotyping
For the studies employing AAV delivery neonatally, male littermates were sexed at birth and at P0-3 received, unanaesthetised, direct bilateral injections of virus (3μl per site; 8×1012 vg/ml titer; ie. 4.8×1010 vg/mouse) into the neuropil (Fig. 1B) via a 33g needle / 10μl Hamilton syringe over a 10-20 second period. The needle was then removed slowly after a delay of 30-60 seconds. The injection sites were over the temporal cortex, approximately 1-2mm lateral from the midline and ~2 mm anterior to lambda and at an approximate depth of 2-4mm [47].
The injected pups were returned to the home cage containing their uninjected littermates. Genotyping was carried out at 3 weeks, at which time phenotyping also commenced. Mice were observed and scored (blind to genotype and treatment) on a weekly basis according to an established phenotype scoring system [6].
Open field test
Motor function and anxiety were evaluated at 9-11 weeks of age using the well-established open field test. Mice were placed in the centre of a 60cm diameter arena and allowed to ambulate freely for 40 minutes during which time the experimenter left the room. The arena was filmed using an overhead digital camera and the digital tracks for each mouse were analysed for various motor and anxiety parameters using Ethovision 3.1 tracking software (Noldus Inc,. Leesburg, VA).
Whole body plethysmograph
Respiratory phenotype was determined at 9-11 weeks of age in conscious and unrestrained animals using whole body plethysmography (EMMS, Bordon, U.K.). Animals were placed inside a plexiglass chamber for 20 minutes to become adapted to the environment after which their breathing was monitored for 30 minutes. A continuous bias airflow supply allowed the animal to be kept in the chamber for extended periods of time. Pressure changes caused by alterations in the temperature and humidity of the air as it enters and leaves the subjects’ lungs were detected by a pressure transducer and signals amplified and converted to a digital display using the custom software (EMMS, Bordon, U.K.) to produce a waveform representing the breathing pattern of the animal. This waveform was then exported and analysed using pClamp 10.2 (Molecular Devices inc., California, USA). Pressure changes were calibrated by injecting 1ml of air via syringe. Respiratory waveforms were analysed for frequency, frequency variability and the presence of abnormal breaths and apnoeas (expiratory pauses lasting >3 times the mean breath-length for that animal) [33].
Treadmill motor challenge test
A treadmill challenge test was carried out using the DigiGait imaging system (Mouse Specifics, Boston, MA) as described [48]. Digital videos were captured as mice ran at varying speeds (10 and 25cm/s) on a transparent treadmill. Mice were contained within a plexiglass chamber, with front and rear bumpers, that was placed on top of the treadmill to ensure the animals remained visible to the camera at all times. A run was considered as successful if the animals could run at the set speed for 10 consecutive steps (~ 2 seconds) without sliding back and hitting the rear bumper with their hindquarters.
AAV injection experiments using juvenile mice
To investigate the effects of juvenile delivery of scAAV9/MeP-MECP2, 100μl of vector of the indicated titers (either low dose: 1×1011 vg; or high dose: 5×1011 vg) were injected into the tail vein of 4-5 week-old male Mecp2-null mice. Mice were weighed at the time of injection and weekly thereafter. Mice were euthanized as the study endpoint when their body weight decreased to 80% of its peak level.
Vector biodistribution analysis utilized tail vein injections (100 μl, as above) into 4-5 week-old WT C57BL/6 mice. Mice were injected with vehicle (350 mM PBS containing 5% sorbitol), 1×1011 vg scAAV9/MeP-MECP2, or 1×1011 vg of scAAV9/MeP-GFP. Mice were sacrificed after 1 or 3 weeks. At sacrifice, blood was collected transcardially and the following organs were harvested for DNA and RNA purification: brain, lumbar spinal cord, liver, heart, spleen, and kidney. A portion of the liver was retained for immunohistochemical detection of GFP or MeCP2.
Immunohistochemistry
To validate the expression of exogenously-derived MeCP2 in mice injected neonatally, mice were deeply anaesthetized with pentobarbitone (50 mg, I.P) and transcardially perfused with 4% paraformaldehyde (PFA; 0.1M PBS). A vibrating microtome (Leica VT1200, Leica UK) was used to obtain 50μm sections of brain, spinal cord and a variety of different peripheral organs. Sections were washed three times in 0.3M PBS followed by blocking using 15% normal goat serum in 0.3M PBS with 0.3% triton X-100 for one hour at room temperature. Samples then were incubated for 48 hours on a shaker at 4°C with the following primary antibodies: rabbit anti-Myc (Abcam product code ab9106; 1/500 dilution); mouse anti-NeuN (Millipore; 1/500), mouse anti-MeCP2 (Sigma; 1/500). The primary antibodies were then washed off (3 × 0.3M PBS) and secondary antibodies applied to the sections overnight at 4°C: Alexa fluor 488 goat anti-mouse/rabbit (Invitrogen; 1/500), Alexa fluor 546 goat anti-mouse/rabbit (Invitrogen; 1/500). Finally, sections were incubated with DAPI nuclear stain (Sigma, UK; 1/1000) for 30 minutes at room temperature before mounting with Vectashield (Vector labs, UK).
For immunofluorescence studies of mice injected at 4-5 weeks old, mice were transcardially perfused with PBS, then after 48 hours of fixation in freshly made PBS containing 4% PFA, the entire brain and liver was sectioned at 40 μm using a Leica vibrating microtome. Sections were incubated for 1 hour at room temperature in blocking solution (10% goat serum, 0.1% triton X-100, 1X PBS), then incubated 48-72 hours at 4° C in primary antibody solution (3% goat serum, 0.1% triton X-100, 1X PBS, rabbit anti-Myc [Abcam ab9106; 1:500] alone, rabbit anti-Myc plus mouse anti-NeuN [Millipore; 1:500], or mouse anti-MeCP2 [Abcam, 1:1000] plus rabbit anti-GABA [Sigma 1:100]). After washing 3 times with PBS, the sections were incubated for 1 hour at room temperature in secondary antibody solution (3% goat serum, 0.1% triton X-100, 1X PBS, secondary antibodies), then washed 3 more times in PBS. Secondary antibodies were as appropriate, each at 1:1000 dilution: goat anti-rabbit Alexa 488 (Invitrogen #A11008), goat anti-mouse Alexa 594 (Invitrogen #A11032).
Image analysis
Analysis of expression patterns, transduction efficiency and quantification of exogenously-derived MeCP2 levels within nuclei was carried out on image stacks captured using a Zeiss LSM710 or Zeiss Axiovert LSM510 laser confocal microscope. Z-series were taken at 0.6-1.3 μm intervals through the section of interest using a 20x objective.
To estimate transduction efficiency in neonatally-injected mice, images were captured as above and the ratio of Myc-immunopositive nuclei to DAPI-stained nuclei was calculated for random fields (n=18 from 4 mice) from sections of hippocampus (CA3), layer 5 of primary motor cortex, thalamus, hypothalamus, brainstem and striatum. For mice injected as juveniles, hippocampus, motor cortex and striatum were analysed similarly. To assess the percentage of transduced cells that were neurons, the ratio of NeuN-immunopositive cells to Myc-immunopositive cells was determined. To quantify levels of exogenously-derived MeCP2 per nucleus in WT mice, injections of ssAAV9/CBA-MECP2 were carried out at P0-2 and brains perfused at 12 weeks. Sections were stained with anti-Myc and anti-MeCP2 and confocal stacks obtained as above. ImageJ software (http://rsbweb.nih.gov/ij/) was used to determine mean MeCP2-channel fluorescence intensity within transduced (Myc +ve) and non-transduced (Myc −ve) cells within a 15 μm image stack.
Nuclear volume estimation was conducted by serial reconstruction of nuclei in a Z-series (0.6μm) using ImageJ. Fluorescence in the DAPI channel was used to define the nuclear boundary and thus to calculate nuclear volumes, while the transduction status of cells was ascertained by the presence or absence of anti-Myc labelling in another channel.
For quantitative immunofluorescence of GABA immunoreactivity, we captured images of neurons in layer 2/3 of motor cortex that had been double-stained with antibodies against MeCP2 and GABA. Fields containing MeCP2-expressing cells were identified, and cellular colocalisation of MeCP2-expression and GABA subsequently evaluated. Images of these cells were captured with a 40x oil lens on a Leica DMI4000 inverted fluorescence microscope using a Cascade 512B camera and digital images were analysed using MetaMorph 7.0 Image Analysis software (Molecular Devices, Sunnyvale, CA), using the ‘Count Nuclei’ module to evaluate the integrated intensity of the GABA immunoreactivity in the sentinel MeCP2-expressing neuron. GABA content was then quantified in the three closest Mecp2-null GABA-expressing neurons in the same field of view.
scAAV vector biodistribution and MECP2 transcript quantification
Genomic DNA was recovered from tissues using the DNeasy Blood and Tissue kit (Qiagen), with automated purifications done on a Qiacube (Qiagen). RNA was recovered from tissues using the RNA Mini Plus kit (Qiagen), tissue lysis performed with the TissueLyser (Qiagen) and automated purification carried out on a Qiacube (Qiagen). cDNA was synthesized from the purified RNA using the Transcriptor First Strand cDNA Synthesis kit (Roche), following the manufacturer’s instructions, then used directly as template for RT-qPCR. Genomic qPCR and RT-qPCR reactions and analysis were carried out on a Roche Lightcycler 480, following the manufacturer’s instructions and as described [49].
For the quantification of vector biodistribution, the amount of vector genome present in each sample was standardized against an amplicon from a single copy mouse gene, laminB2, amplified from genomic DNA. For the quantification of GFP, mouse Mecp2, and human MECP2 cDNAs, standardization was achieved by comparison against standard curves generated by amplification from plasmid constructs specific for each of these transcripts. This enabled estimation of absolute numbers of transcripts in each sample, and absolute numbers of a reference housekeeping gene transcript, actin, were estimated using a standard curve generated by amplification from mouse genomic DNA. Vector biodistribution data is reported as the number of double-stranded vector DNA (GFP or human MECP2) molecules per 2 copies of the mouse LaminB2 locus, i.e. the number of vector DNA copies per diploid mouse genome. Transcript quantification is reported as the absolute number of human MECP2 + mouse Mecp2 transcripts/μl (using ‘universal’ primers that amplify both human and mouse transcripts with equal efficiency) divided by the absolute number of actin transcripts/μl in the sample. Primers for human MECP2 were as follows: FORWARD: 5′-caccacgagacccaaggcggcca-3′; REVERSE: 5′-ggtctcctgcacagatcggata-3′. Universal MECP2/Mecp2 primers were as follows: FORWARD: 5′-ccggggacctatgtatgatg-3′; REVERSE: 5′-aagcttttccctggggatt-3′. Primers for mouse actin were as follows: FORWARD: 5′-tggcaccacaccttctacaat-3′; REVERSE: 5′-aggcatacagggacagcaca-3′. Primers for GFP and LaminB2 were as described [49]. A melting curve analysis was performed at the end of the RT-qPCR run, following the manufacturer’s instructions, to distinguish and quantify the mouse Mecp2 and human MECP2 transcript amplification products. The mouse product melted at 81.9°C and the human melted at 83.5°C. Only the MECP2-treated mice showed evidence for human MECP2 expression (Supplemental Table 1).
Serum chemistry
Blood recovered from mice was allowed to clot for 2 hours at room temperature, and was then centrifuged at 1000 x g for 10 minutes. The clear upper phase (serum) was collected and preserved at −80°C. Serum analysis was conducted by the University of North Carolina Animal Clinical Laboratory Core Facility.
Statistical analysis
Differences between treatment groups were carried out using 2-way ANOVA, repeated-measures ANOVA, Fisher’s exact test, Student’s t-test, and Gehan-Breslow-Wilcoxon test (survival curves), as appropriate. p<0.05 was used to define statistical significance.
Supplementary Material
a. Low power micrograph showing whole-brain transduction efficiency at 26 weeks following P1 neonatal brain injection as revealed by anti-Myc labelling (red). b-e Higher power images of areas denoted by boxes in a and showing relative neuronal tranduction efficiency (red = anti-Myc; green = anti-NeuN immunolabelling). f. Representative images from lumbar region of the spinal cord showing transduction in ChAT-immunopositive lower motor neurons. g. Micrograph of liver showing peripheral expression of the transgene. Scale bars: a, 1mm; b-e, 100μm; f-g, 10μm. Abbrevations: ChAT; Choline Acetyl Transferase; NeuN; ‘neuronal nuclei’ antigen (Rbfox3).
a. Plot showing individual aggregate phenotype severity scores for the longest-surviving mice (Nos. 15, 23 and 30). Numbers 15 and 23 survived to the end of the study (30 weeks; ie. 12 weeks beyond the longest surviving AAV9/GFP –treated Mecp2-/y mouse), whilst number 30 died suddenly at 22 weeks. Black arrow indicates the peak mean severity score (around 14 weeks of age). b-d. plots showing the performance of these three mice on the open field test at two time points (11 and 22 weeks). Plots show data as mean ± SEM. Statistical analysis was carried out by paired t test.
These representative sample images illustrate the neuronal transduction efficiency data shown in figure 5b. Green (left column) is NeuN, red (middle column) is myc-MeCP2, and the right column is the merged imaged. Arrows show examples of non-neuronal MeCP2-positive cells that do not co-localize with NeuN. Scale bar: 50μm
a. Survival plot showing extended lifespan of Mecp2-/y mice injected with a low dose (1×1011 vg, open boxes, n=7; p=0.03, Gehan-Breslow-Wilcoxon test) or high dose (5×1011 vg, open circles, n=5; p=0.08, Gehan-Breslow-Wilcoxon test) scAAV9/MeP-MEPC2 compared to untreated Mecp2-/y mice (closed boxes, n=10). Untreated WT mice (black circles, n=8). Numbers indicate median survival (weeks). b. Plot showing bodyweight changes over time (symbols as in a). There was a significant genotype effect (lower bodyweight in mutant mice) but no effect of treatment (p>0.05, ANOVA).
The mice indicated in figure 6 had blood drawn at the time of termination. Serum was analyzed from mice sacrificed at 1 week or 3 weeks post-injection. Asterisks indicate a significant difference between AAV9/MECP2- and vehicle-injected mice at the same termination time by t-test (* = p<0.05, ** = p<0.01). Mean ± S.E.M. are plotted, n = 2-4 with the exception of GFP-treated mice at 3 weeks, n =1. ALT = alanine amino transferase, ALK = alkaline phosphatase, BUN = blood urea nitrogen, HDL = high density lipoprotein.
Representative video showing AAV9/CBA-GFP treated Mecp2-/y mouse at 7 weeks
Representative video showing AAV9/CBA-Mecp2 treated Mecp2-/y mouse at 7 weeks
Representative video showing AAV9/CBA-Mecp2 treated Mecp2-/y mouse at 16 weeks
ACKNOWLEDGEMENTS
This work was supported by grants from the International Rett Syndrome Foundation (SJG), from the Medical Research Council (SRC; grant no. G0800401) and from the Rett Syndrome Association Scotland (SRC/MESB). KKG was funded by an Egyptian Government Scholarship. We thank the UNC Animal Studies Core for help with injections, along with Swati Yadav and Hung-Jui “Sophia” Shih (UNC) for assistance with qPCR.
Footnotes
CONFLICTS OF INTEREST The authors declare no conflicts of interest.
REFERENCES
- 1.Neul JL, et al. Rett syndrome: revised diagnostic criteria and nomenclature. Ann Neurol. 2010;68:944–950. doi: 10.1002/ana.22124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gadalla KK, Bailey ME, Cobb SR. MeCP2 and Rett syndrome: reversibility and potential avenues for therapy. Biochem J. 2011;439:1–14. doi: 10.1042/BJ20110648. [DOI] [PubMed] [Google Scholar]
- 3.Guy J, Hendrich B, Holmes M, Martin JE, Bird A. A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat Genet. 2001;27:322–326. doi: 10.1038/85899. [DOI] [PubMed] [Google Scholar]
- 4.Chen RZ, Akbarian S, Tudor M, Jaenisch R. Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat Genet. 2001;27:327–331. doi: 10.1038/85906. [DOI] [PubMed] [Google Scholar]
- 5.Giacometti E, Luikenhuis S, Beard C, Jaenisch R. Partial rescue of MeCP2 deficiency by postnatal activation of MeCP2. Proc Natl Acad Sci U S A. 2007;104:1931–1936. doi: 10.1073/pnas.0610593104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guy J, Gan J, Selfridge J, Cobb S, Bird A. Reversal of neurological defects in a mouse model of Rett syndrome. Science. 2007;315:1143–1147. doi: 10.1126/science.1138389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Luikenhuis S, Giacometti E, Beard CF, Jaenisch R. Expression of MeCP2 in postmitotic neurons rescues Rett syndrome in mice. Proc Natl Acad Sci U S A. 2004;101:6033–6038. doi: 10.1073/pnas.0401626101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cobb S, Guy J, Bird A. Reversibility of functional deficits in experimental models of Rett syndrome. Biochem Soc Trans. 2010;38:498–506. doi: 10.1042/BST0380498. [DOI] [PubMed] [Google Scholar]
- 9.LaSalle JM, Goldstine J, Balmer D, Greco CM. Quantitative localization of heterogeneous methyl-CpG-binding protein 2 (MeCP2) expression phenotypes in normal and Rett syndrome brain by laser scanning cytometry. Hum Mol Genet. 2001;10:1729–1740. doi: 10.1093/hmg/10.17.1729. [DOI] [PubMed] [Google Scholar]
- 10.Shahbazian MD, Antalffy B, Armstrong DL, Zoghbi HY. Insight into Rett syndrome: MeCP2 levels display tissue- and cell-specific differences and correlate with neuronal maturation. Hum Mol Genet. 2002;11:115–124. doi: 10.1093/hmg/11.2.115. [DOI] [PubMed] [Google Scholar]
- 11.Zhou Z, et al. Brain-specific phosphorylation of MeCP2 regulates activity-dependent Bdnf transcription, dendritic growth, and spine maturation. Neuron. 2006;52:255–269. doi: 10.1016/j.neuron.2006.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lioy DT, et al. A role for glia in the progression of Rett’s syndrome. Nature. 2011;475:497–500. doi: 10.1038/nature10214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McGraw CM, Samaco RC, Zoghbi HY. Adult Neural Function Requires MeCP2. Science. 2011 doi: 10.1126/science.1206593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Foust KD, Nurre E, Montgomery CL, Hernandez A, Chan CM, Kaspar BK. Intravascular AAV9 preferentially targets neonatal neurons and adult astrocytes. Nat Biotechnol. 2009;27:59–65. doi: 10.1038/nbt.1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goncalves MA. Adeno-associated virus: from defective virus to effective vector. Virol J. 2005;2:43. doi: 10.1186/1743-422X-2-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jiang H, et al. Evidence of multiyear factor IX expression by AAV-mediated gene transfer to skeletal muscle in an individual with severe hemophilia B. Mol Ther. 2006;14:452–455. doi: 10.1016/j.ymthe.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 17.Foust KD, Nurre E, Montgomery CL, Hernandez A, Chan CM, Kaspar BK. Intravascular AAV9 preferentially targets neonatal neurons and adult astrocytes. Nat Biotechnol. 2009;27:59–65. doi: 10.1038/nbt.1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McCarty DM, Fu H, Monahan PE, Toulson CE, Naik P, Samulski RJ. Adeno-associated virus terminal repeat (TR) mutant generates self-complementary vectors to overcome the rate-limiting step to transduction in vivo. Gene Ther. 2003;10:2112–2118. doi: 10.1038/sj.gt.3302134. [DOI] [PubMed] [Google Scholar]
- 19.McCarty DM, Monahan PE, Samulski RJ. Self-complementary recombinant adeno-associated virus (scAAV) vectors promote efficient transduction independently of DNA synthesis. Gene Ther. 2001;8:1248–1254. doi: 10.1038/sj.gt.3301514. [DOI] [PubMed] [Google Scholar]
- 20.Meins M, et al. Submicroscopic duplication in Xq28 causes increased expression of the MECP2 gene in a boy with severe mental retardation and features of Rett syndrome. J Med Genet. 2005;42:e12. doi: 10.1136/jmg.2004.023804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Van Esch H, et al. Duplication of the MECP2 region is a frequent cause of severe mental retardation and progressive neurological symptoms in males. Am J Hum Genet. 2005;77:442–453. doi: 10.1086/444549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weng SM, McLeod F, Bailey ME, Cobb SR. Synaptic plasticity deficits in an experimental model of rett syndrome: long-term potentiation saturation and its pharmacological reversal. Neuroscience. 2011;180:314–321. doi: 10.1016/j.neuroscience.2011.01.061. [DOI] [PubMed] [Google Scholar]
- 23.Johnson RA, et al. 7,8-dihydroxyflavone (7,8-DHF) exhibits therapeutic efficacy in a mouse model of Rett syndrome. J Appl Physiol. 2011 doi: 10.1152/japplphysiol.01361.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gray SJ, et al. Optimizing Promoters for Recombinant Adeno-Associated Virus-Mediated Gene Expression in the Peripheral and Central Nervous System Using Self-Complementary Vectors. Hum Gene Ther. 2011 doi: 10.1089/hum.2010.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gray SJ, Matagne V, Bachaboina L, Yadav S, Ojeda SR, Samulski RJ. Preclinical differences of intravascular AAV9 delivery to neurons and glia: a comparative study of adult mice and nonhuman primates. Mol Ther. 2011;19:1058–1069. doi: 10.1038/mt.2011.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chao HT, et al. Dysfunction in GABA signalling mediates autism-like stereotypies and Rett syndrome phenotypes. Nature. 2010;468:263–269. doi: 10.1038/nature09582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zincarelli C, Soltys S, Rengo G, Rabinowitz JE. Analysis of AAV serotypes 1-9 mediated gene expression and tropism in mice after systemic injection. Mol Ther. 2008;16:1073–1080. doi: 10.1038/mt.2008.76. [DOI] [PubMed] [Google Scholar]
- 28.Mullaney BC, Johnston MV, Blue ME. Developmental expression of methyl-CpG binding protein 2 is dynamically regulated in the rodent brain. Neuroscience. 2004;123:939–949. doi: 10.1016/j.neuroscience.2003.11.025. [DOI] [PubMed] [Google Scholar]
- 29.Derecki NC, et al. Wild-type microglia arrest pathology in a mouse model of Rett syndrome. Nature. 2012;484:105–109. doi: 10.1038/nature10907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stearns NA, Schaevitz LR, Bowling H, Nag N, Berger UV, Berger-Sweeney J. Behavioral and anatomical abnormalities in Mecp2 mutant mice: a model for Rett syndrome. Neuroscience. 2007;146:907–921. doi: 10.1016/j.neuroscience.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 31.Pelka GJ, et al. Mecp2 deficiency is associated with learning and cognitive deficits and altered gene activity in the hippocampal region of mice. Brain. 2006;129:887–898. doi: 10.1093/brain/awl022. [DOI] [PubMed] [Google Scholar]
- 32.Viemari JC, et al. Mecp2 deficiency disrupts norepinephrine and respiratory systems in mice. J Neurosci. 2005;25:11521–11530. doi: 10.1523/JNEUROSCI.4373-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Robinson L, et al. Morphological and functional reversal of phenotypes in a mouse model of Rett syndrome. Brain. 2012;135:2699–710. doi: 10.1093/brain/aws096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Belichenko NP, Belichenko PV, Mobley WC. Evidence for both neuronal cell autonomous and nonautonomous effects of methyl-CpG-binding protein 2 in the cerebral cortex of female mice with Mecp2 mutation. Neurobiol Dis. 2009;34:71–77. doi: 10.1016/j.nbd.2008.12.016. [DOI] [PubMed] [Google Scholar]
- 35.Rastegar M, et al. MECP2 isoform-specific vectors with regulated expression for Rett syndrome gene therapy. PLoS One. 2009;4:e6810. doi: 10.1371/journal.pone.0006810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Friez MJ, et al. Recurrent infections, hypotonia, and mental retardation caused by duplication of MECP2 and adjacent region in Xq28. Pediatrics. 2006;118:e1687–1695. doi: 10.1542/peds.2006-0395. [DOI] [PubMed] [Google Scholar]
- 37.Collins AL, et al. Mild overexpression of MeCP2 causes a progressive neurological disorder in mice. Hum Mol Genet. 2004;13:2679–2689. doi: 10.1093/hmg/ddh282. [DOI] [PubMed] [Google Scholar]
- 38.Rahim AA, et al. Intravenous administration of AAV2/9 to the fetal and neonatal mouse leads to differential targeting of CNS cell types and extensive transduction of the nervous system. The FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2011;25:3505–3518. doi: 10.1096/fj.11-182311. [DOI] [PubMed] [Google Scholar]
- 39.Miyake N, Miyake K, Yamamoto M, Hirai Y, Shimada T. Global gene transfer into the CNS across the BBB after neonatal systemic delivery of single-stranded AAV vectors. Brain Res. 2011;1389:19–26. doi: 10.1016/j.brainres.2011.03.014. [DOI] [PubMed] [Google Scholar]
- 40.Federici T, et al. Robust spinal motor neuron transduction following intrathecal delivery of AAV9 in pigs. Gene Ther. 2011 doi: 10.1038/gt.2011.130. [DOI] [PubMed] [Google Scholar]
- 41.Samaranch L, et al. AAV9 Transduction In The Central Nervous System Of Non-Human Primates. Human gene therapy. 2011 doi: 10.1089/hum.2011.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bevan AK, et al. Systemic gene delivery in large species for targeting spinal cord, brain, and peripheral tissues for pediatric disorders. Mol Ther. 2011;19:1971–1980. doi: 10.1038/mt.2011.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xiao X, Li J, Samulski RJ. Production of high-titer recombinant adeno-associated virus vectors in the absence of helper adenovirus. J Virol. 1998;72:2224–2232. doi: 10.1128/jvi.72.3.2224-2232.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gray SJ, et al. Optimizing promoters for recombinant adeno-associated virus-mediated gene expression in the peripheral and central nervous system using self-complementary vectors. Human gene therapy. 2011;22:1143–1153. doi: 10.1089/hum.2010.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Grieger JC, Choi VW, Samulski RJ. Production and characterization of adeno-associated viral vectors. Nat Protoc. 2006;1:1412–1428. doi: 10.1038/nprot.2006.207. [DOI] [PubMed] [Google Scholar]
- 46.Gray SJ, Matagne V, Bachaboina L, Yadav S, Ojeda SR, Samulski RJ. Preclinical differences of intravascular AAV9 delivery to neurons and glia: a comparative study of adult mice and nonhuman primates. Mol Ther. 2011;19:1058–1069. doi: 10.1038/mt.2011.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pilpel N, Landeck N, Klugmann M, Seeburg PH, Schwarz MK. Rapid, reproducible transduction of select forebrain regions by targeted recombinant virus injection into the neonatal mouse brain. Journal of neuroscience methods. 2009;182:55–63. doi: 10.1016/j.jneumeth.2009.05.020. [DOI] [PubMed] [Google Scholar]
- 48.Kale A, Amende I, Meyer GP, Crabbe JC, Hampton TG. Ethanol’s effects on gait dynamics in mice investigated by ventral plane videography. Alcohol Clin Exp Res. 2004;28:1839–1848. doi: 10.1097/01.alc.0000148103.09378.81. [DOI] [PubMed] [Google Scholar]
- 49.Gray SJ, Blake BL, Criswell HE, Nicolson SC, Samulski RJ, McCown TJ. Directed evolution of a novel adeno-associated virus (AAV) vector that crosses the seizure-compromised blood-brain barrier (BBB) Mol Ther. 2010;18:570–578. doi: 10.1038/mt.2009.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
a. Low power micrograph showing whole-brain transduction efficiency at 26 weeks following P1 neonatal brain injection as revealed by anti-Myc labelling (red). b-e Higher power images of areas denoted by boxes in a and showing relative neuronal tranduction efficiency (red = anti-Myc; green = anti-NeuN immunolabelling). f. Representative images from lumbar region of the spinal cord showing transduction in ChAT-immunopositive lower motor neurons. g. Micrograph of liver showing peripheral expression of the transgene. Scale bars: a, 1mm; b-e, 100μm; f-g, 10μm. Abbrevations: ChAT; Choline Acetyl Transferase; NeuN; ‘neuronal nuclei’ antigen (Rbfox3).
a. Plot showing individual aggregate phenotype severity scores for the longest-surviving mice (Nos. 15, 23 and 30). Numbers 15 and 23 survived to the end of the study (30 weeks; ie. 12 weeks beyond the longest surviving AAV9/GFP –treated Mecp2-/y mouse), whilst number 30 died suddenly at 22 weeks. Black arrow indicates the peak mean severity score (around 14 weeks of age). b-d. plots showing the performance of these three mice on the open field test at two time points (11 and 22 weeks). Plots show data as mean ± SEM. Statistical analysis was carried out by paired t test.
These representative sample images illustrate the neuronal transduction efficiency data shown in figure 5b. Green (left column) is NeuN, red (middle column) is myc-MeCP2, and the right column is the merged imaged. Arrows show examples of non-neuronal MeCP2-positive cells that do not co-localize with NeuN. Scale bar: 50μm
a. Survival plot showing extended lifespan of Mecp2-/y mice injected with a low dose (1×1011 vg, open boxes, n=7; p=0.03, Gehan-Breslow-Wilcoxon test) or high dose (5×1011 vg, open circles, n=5; p=0.08, Gehan-Breslow-Wilcoxon test) scAAV9/MeP-MEPC2 compared to untreated Mecp2-/y mice (closed boxes, n=10). Untreated WT mice (black circles, n=8). Numbers indicate median survival (weeks). b. Plot showing bodyweight changes over time (symbols as in a). There was a significant genotype effect (lower bodyweight in mutant mice) but no effect of treatment (p>0.05, ANOVA).
The mice indicated in figure 6 had blood drawn at the time of termination. Serum was analyzed from mice sacrificed at 1 week or 3 weeks post-injection. Asterisks indicate a significant difference between AAV9/MECP2- and vehicle-injected mice at the same termination time by t-test (* = p<0.05, ** = p<0.01). Mean ± S.E.M. are plotted, n = 2-4 with the exception of GFP-treated mice at 3 weeks, n =1. ALT = alanine amino transferase, ALK = alkaline phosphatase, BUN = blood urea nitrogen, HDL = high density lipoprotein.
Representative video showing AAV9/CBA-GFP treated Mecp2-/y mouse at 7 weeks
Representative video showing AAV9/CBA-Mecp2 treated Mecp2-/y mouse at 7 weeks
Representative video showing AAV9/CBA-Mecp2 treated Mecp2-/y mouse at 16 weeks