

Abstract
Previously, it has been shown that rat Schwann cells (SCs), but not olfactory ensheathing cells (OECs), form a boundary with astrocytes, due to a SC-specific secreted factor. Here, we identify highly sulfated heparan sulfates (HS), and fibroblast growth factors (FGF) 1 and FGF9, as possible determinants of boundary formation induced by rat SCs. Disaccharide analysis of HS in SC and rat OEC conditioned medium showed that SCs secrete more highly sulfated HS than OECs. The dependence of the boundary-forming activity on high levels of sulfation was confirmed using a panel of semi-synthetic modified heparins with variable levels of sulfation. Furthermore, extracellular HS 6-O-endosulfatase enzymes, Sulf 1 and Sulf 2, were expressed at a significantly lower level by SCs compared to OECs and siRNA reduction of Sulfs in OECs was, in itself, sufficient to induce boundary formation. This demonstrates a key role for remodelling (reduction) of HS 6-O-sulfation by OECs to suppress boundary formation, in comparison to SCs. Furthermore, specific anti-FGF1 and FGF9 antibodies disrupted SC/astrocyte boundary formation, supporting a role for an HS sulfation-dependent FGF signalling mechanism via FGF receptors (FGFR) on astrocytes. We propose a model in which FGF1 and FGF9 signalling is differentially modulated by patterns of glial cell HS sulfation, dependent on Sulf 1 and Sulf 2 expression, to control FGFR3-IIIb mediated astrocytic responses. Moreover, these data suggest manipulation of HS sulfation after CNS injury as a potential novel approach for therapeutic intervention in CNS repair.
INTRODUCTION
The adult mammalian central nervous system (CNS) has limited capacity for repair. Spinal cord injury usually results in formation of a glial scar and permanent loss of sensory, motor and autonomic function. A potential repair strategy is cell transplantation, for which glial cells or stem cells are popular candidates. Many researchers focus on glial cells such as Schwann cells (SCs) from the peripheral nervous system, or olfactory ensheathing cells (OECs) from the olfactory system, as they inherently support axon regeneration (Franklin and Barnett, 2000; Raisman, 2001; Barnett and Riddell, 2007). Previously, we have shown that there are some important differences between OECs and SCs that might influence their selection for transplantation. This difference, which has been detected not only in vitro (Lakatos et al., 2000; Fairless and Barnett, 2005), but also after transplantation in vivo, results in better integration of OECs than SCs with host astrocytes (Lakatos et al., 2003; Santos-Silva et al., 2007). This present work aims to determine differences in the cell biology of OECs compared to SCs that confer advantages for transplantation and may direct future efforts to improve transplantation of SCs.
It has been shown that OECs and SCs share many biological characteristics, including antigenic and morphological phenotypes, and the ability to myelinate axons with peripheral-type myelin (Franklin and Barnett, 2000; Chuah and West, 2002; Barnett and Riddell, 2007; Franssen et al., 2007). However, they interact differently with astrocytes, the main component of the glial scar (Fawcett and Asher, 1999), in that SCs induce a reactive astrocyte phenotype, through which axons cannot regenerate (Silver and Miller, 2004; Pekny and Nilsson, 2005), whereas OECs do not (Lakatos et al., 2000; Fairless and Barnett, 2005; Santos-Silva et al., 2007). This is in line with the native behaviour of OECs in the olfactory system where they intermix with astrocytes (Raisman, 1985; Li et al., 2005). Our previous studies demonstrated that SCs and astrocytes form a boundary on contact and occupy distinct, non-overlapping areas (Lakatos et al., 2000). In contrast, OECs and astrocytes freely intermingle, without inducing a reactive astrocyte phenotype, although the addition of SC-conditioned medium (SCM) or heparin can induce SC-like behaviour in OECs. Furthermore, SCs will mingle with astrocytes if treated with an FGF receptor (FGFR) inhibitor, suggesting an involvement of heparan sulfate (HS)-dependent FGF signalling (Santos-Silva et al., 2007).
HS proteoglycans are cell surface and matrix molecules, each composed of a core protein to which one or more HS polysaccharide chains are attached. These chains consist of repeating disaccharide units of N-acetylglucosamine and glucuronic acid that are modified enzymatically by N-deacetylation/N-sulfation, 2-O, 6-O and 3-O-sulfation and epimerisation of glucuronic acid to iduronic acid, to generate functionally specific saccharide sequences (Ori et al., 2008). Furthermore, HS chains are also remodelled extracellularly by 6-O-endosulfatase enzymes (Sulfs), which selectively remove 6-O-sulfates, and introduce a further level of structural sophistication (Dhoot et al., 2001; Morimoto-Tomita et al., 2002; Ai et al., 2006). The specific HS sequences synthesised modulate the cellular functions of a diverse array of protein ligands, including FGFs (Turnbull et al., 1992; Guimond et al., 1993; Maccarana et al., 1993; Brickman et al., 1998; Guimond and Turnbull, 1999; Kreuger et al., 2001; Ford-Perriss et al., 2002; Ashikari-Hada et al., 2004). Here, we demonstrate that OECs and SCs have distinct HS molecular phenotypes and implicate highly sulfated HS produced by SCs in the formation of a gliotic scar. We provide evidence that sulfation levels are differentially remodelled by OECs and SCs via Sulf 1 and Sulf 2, and identify FGF1 and FGF9 as possible growth factors involved in the induction process. We propose a model in which specific FGFs act in concert with regulated patterns of glial HS sulfation to control astrocytic responses.
MATERIALS AND METHODS
Generation of purified glial cells
All primary neural cultures were generated from Sprague-Dawley rat pups of either sex. As described previously (Noble and Murray, 1984; Lakatos et al., 2000), purified type 1 astrocytes were prepared by digesting cortices (dissected from 1-day old Sprague Dawley (SD) rats) in 1.33% (w/v) collagenase (Sigma-Aldrich, Gillingham, UK), seeding (~ 2×107 cells per 75 cm2 flask) and culturing the cells in poly-L-lysine (PLL, 13.3 μg/ml, Sigma-Aldrich, Gillingham, UK) coated 75 cm2 flasks for 10-12 days. The cells were maintained in DMEM (Invitrogen, Paisley, Scotland) supplemented with 10% (v/v) foetal bovine serum (FBS) (Sigma-Aldrich, Gillingham, UK) and L-glutamine (2 mM, Sigma-Aldrich, Gillingham, UK) (DMEM-FBS). Confluent flasks were shaken on a rotary platform overnight at 37°C to remove contaminating oligodendrocyte precursor cells. The remaining cells were 85-95% type 1 astrocytes as identified by labelling for glial fibrillary acidic protein (GFAP), an astrocyte cell specific marker.
OECs were isolated from the olfactory bulb of 7 day old SD rat pups and purified using magnetic nanoparticles (STEMCELL Technologies, UK) pre-bound with the p75NTR antibody (mouse IgG1, Abcam, Cambridge, UK) (Higginson and Barnett, 2010). The cells were grown in low glucose DMEM with 5% (v/v) FBS and 2 mM L-glutamine and further supplemented with DMEM-BS (Bottenstein, 1979), FGF2 (25 ng/ml, Peprotech, London UK), heregulin β-1 (50 ng/ml, R&D Systems, Oxon, UK), forskolin (5×10−7 M Sigma-Aldrich, Gillingham, UK) and astrocyte conditioned media (ACM) (1:5, fresh serum-free media taken after incubation with a confluent astrocyte monolayer for 48 h) (Noble and Murray, 1984; Alexander et al., 2002).
SCs were purified using a modification of the method described by Brockes and colleagues (Brockes et al., 1979). This modification involved treating the cultures with cytosine arabinoside (AraC, 10−5 M, Sigma-Aldrich, Gillingham, UK), followed by incubation with anti-Thy1.1 antibody (1:1 supernatant, Sigma-Aldrich, Gillingham, UK) and rabbit complement (1:6, Harlan Laboratories Ltd., UK) to reduce contamination by fibroblasts (Lakatos et al., 2000). All cell cultures were grown in PLL coated tissue culture flasks.
Collection of SC conditioned medium (SCM), OEC conditioned medium (OCM) and astrocyte conditioned medium (ACM)
Confluent cultures of purified SCs or OECs in 75 cm2 flasks (maintained in vitro for 2-6 weeks) were rinsed twice with phosphate buffered saline (PBS), pH 7.4 and 7 ml of DMEM-BS without growth factors added. Cultures were maintained for a further 2 days before medium collection. Collected medium was centrifuged to remove cellular debris and filtered through a 0.2 μm filter (Millipore, Hertfordshire, UK). The same procedure was used for generating ACM, except that confluent astrocyte cultures were maintained in 11 ml of DMEM-BS. Conditioned media was added to cell cultures at a 1:1 ratio with DMEM-FBS.
Confrontation Assays
Confrontation assays were performed as described by Wilby et al. (1999) and Lakatos et al. (2000) with some modifications (Wilby et al., 1999; Lakatos et al., 2000). Briefly, 70 μl containing 10,000 OECs or SCs were seeded into one well of a silicon Ibidi culture insert on a PLL-coated glass coverslip (Ibidi GmbH, Munich, Germany). Into the opposing, parallel well, 10,000 astrocytes were seeded. Cells were allowed to attach for 1 h before careful removal of the insert followed by a wash with DMEM-FBS to remove unattached cells. Cultures were maintained in DMEM-FBS and allowed to grow towards each other over a period of 5-7 days, allowing time for cells to make contact and interact (Lakatos et al., 2000). In some experiments, tissue HS, modified heparins, blocking antibodies or conditioned medium were added to the cultures after the cells had contacted each other. Cultures were then immunolabelled using anti-GFAP for astrocytes (1:500; anti-rabbit (Dako, Ely, UK)) and anti-p75NTR for OECs and SCs (1:1; IgG1; hybridoma supernatant (Yan and Johnson, 1988)). Fluorescent images were captured using an Olympus BX51 fluorescent microscope and Image-Pro software. Using Adobe Photoshop Elements 7.0, a 300 μm line was drawn along the interface between astrocytes and either OECs or SCs. The numbers of OECs or SCs crossing the cell:cell boundary were counted and averaged over five randomly chosen fields. Experiments were repeated at least three times.
Treatments
Modified Heparins
Modified heparins (a gift from Dr EA Yates, University of Liverpool, UK) were produced semi-synthetically by chemical modification (selective desulfation) of heparin. These structurally distinct, model “HS-mimetic” polysaccharides (Yates et al., 1996) are useful tools for investigating structure-activity relationships of HS (Irie et al., 2002; Yates et al., 2004; Guimond et al., 2006; Patey et al., 2006). The disaccharide structures of the heparins are indicated in Fig. 3. Heparins were added to confrontation assays at 10 μg/ml at the stage when cells made contact (day 0) and treatment was repeated on day 2. Cultures were fixed and stained as described above on day 3.
Figure 3. HS sulfation is critical for boundary formation.

Confrontation assays of OECs and astrocytes were carried out in the presence of 10 μg/ml modified heparins (A-J). The disaccharide structures of the heparins are indicated. After 2 days of treatment, cells were fixed and stained for GFAP (red) and p75NTR (green). Assays were scored using a graded scoring system to assess the ability of each modified heparin to promote boundary formation, i.e., mingling (−), partial boundary (+/−) or complete boundary (++) (K). Control heparin effect was scored as ++. Modified heparins with the highest levels of sulfation (Hep1, Hep2, Hep3, Hep4 and Hep9) induced boundaries between OECs and astrocytes, whereas those with lower levels of sulfation (Hep5, Hep6, Hep7 and Hep8) did not. The images are representative of at least 3 independent experiments. Scale bar 50 μm.
HS from various tissue sources
Porcine mucosal HS (PMHS) was a gift from Organon (Oss, Netherlands), porcine liver and rat brain HS were purified using previously described methods (Lyon and Gallagher, 1991; Esko, 2001). Confrontation assays were treated with polysaccharides for 2 days (day 0 and day 2) at a final concentration of 30 μg/ml.
Heparinase and trypsin treatment of conditioned medium
Proteins in SCM were digested by the addition of trypsin at a final ratio of 1:50 trypsin:protein and incubated for 12 h at 37°C (amount of protein was estimated by measuring the UV absorbance at 280 nm). Trypsin was inactivated by addition of soybean trypsin inhibitor (1/10 of final trypsin concentration). HS was digested by the addition of 10 mU each of heparinase I (EC 4.2.2.7), II (EC number not assigned) and III (EC 4.2.2.8) (Ibex Technologies, Montreal, Canada) to 4 ml SCM, followed by incubation at 37°C for 6 h. A further 10 mU of each heparinase enzyme was added to SCM and the reaction incubated overnight at 37°C. Treatment of confrontation assays was carried out by replacing half of the media with untreated SCM, heparinase-treated SCM, trypsin-treated SCM or 50:50 heparinase:trypsin treated SCM. Confrontation assays were treated for 2 days.
FGF inhibition
Anti-FGF2 (Clone bFM-1) (Millipore, Hertfordshire, UK), anti-FGF9 (Clone 36912) and anti-FGF1 (both R&D Systems, Oxon, UK) neutralising antibodies were added to confrontation assays at a final concentration of 1 μg/mL, at the stage when cells made contact. Treatment was repeated for 2 days and then cultures were fixed and stained as described above on day 3.
HPLC analysis of HS disaccharides in SCM and OCM
SCM and OCM were collected from 75 cm2 flasks of confluent OECs or SCs, frozen and stored at -20°C until enough material was collected for detection (180 mL OCM and 90 mL SCM). OCM and SCM were rotated with 0.1 ml DEAE-sephacel (Sigma-Aldrich, Gillingham, UK) per 10 mL, overnight at 4 °C, and then centrifuged at 3382 x g for 5 min and unbound material in the supernatant removed. DEAE beads were washed three times with 10 column volumes of PBS, and then washed twice with 10 column volumes of 0.25 M NaCl in PBS. Bound material containing HS proteoglycans (HSPGs) was eluted with 10 column volumes of 2 M NaCl in PBS and desalted over two, in-line 5 mL HiTrap desalting columns (GE Healthcare UK Ltd, Buckinghamshire, UK) using an AKTA-FPLC system. Desalted material was then freeze-dried.
Heparinase digestion
Lyophilised material was taken up in heparinase buffer (100 mM sodium acetate, 0.1 mM calcium acetate, pH 7) and 2.5 mU each of heparinase I, II and III were added and incubated for 3 h at 37°C. After this time, a further 2.5 mU of heparinase I, II and III were added and the reaction incubated overnight at 37°C. Another 2.5 mU of heparinase I, II and III were then added and incubated for a further 3 h. As a control, 100 μg heparin was digested in the same way. Digested samples were then incubated at 95°C for 5 min to stop the reaction and taken up in 800 μl HPLC-grade water.
C18 HPLC
HS disaccharides were separated out from samples using a Discovery® C18 HPLC column (Supelco, Sigma-Aldrich, Gillingham, UK) (25 cm x 4.6 mm, 5 μm) on a Shimadzu SPD 10A HPLC system. Buffer A was HPLC-grade water and Buffer B was 80% (v/v) methanol. Elution profiles were monitored by UV absorbance at 232 nm. Samples were injected in buffer A and the void volume containing HS disaccharides was collected and freeze-dried for BODIPY labelling. Bound material (containing hydrophobic material, including HSPG core proteins), was eluted using a linear gradient of 0-50% buffer B over 30 min at a flow rate of 1 ml min−1.
BODIPY labelling of HS disaccharides
Freeze-dried HS disaccharides were labelled with BODIPY FL hydrazide (5 mg/ml; 4,4-difluoro-5, 7-dimethyl-4-bora-3a, 4a-diaza-s-indacene-3-propionic acid hydrazide; Molecular Probes) as previously described (Skidmore et al., 2006; Skidmore et al., 2009; Skidmore et al., 2010). Digested heparin control and HS disaccharide standards (Iduron, Manchester, UK) were labelled in the same way. Labelled samples were applied to silica gel thin layer chromatography (TLC) aluminium plates and free BODIPY tag separated from labelled disaccharides with butanol. Labelled HS disaccharides were removed from the TLC plates and solubilised in 800 μl HPLC-grade water.
SAX-HPLC
SAX separations were performed on a Propac PA1 column (25 cm x 9 mm, 5 μm) using a Shimadzu SPD 10A HPLC system. Elution profiles were monitored by UV absorbance at 232 nm and fluorescent detection using a Shimadzu RF10AXL spectrofluorometer. Buffer A was 150 mM NaOH and Buffer B was 150 mM NaOH, 2 M NaCl. Elution profiles were monitored by UV absorbance at 232 nm and fluorescent detection at λex = 488 nm λem = 520 nm. Samples were injected and the flow held at 2 ml min−1 in buffer A until all remaining free tag had been eluted. Fluorescently labelled disaccharides were then eluted using a linear gradient of 0-50% buffer B over 45 min at 2 ml min−1. The column was then washed with a 10 min elution in 300 mM NaOH, 2 M NaCl, before returning to 150 mM NaOH.
Quantitative real-time PCR
RNA from monocultures of OECs and SCs was extracted using a Qiagen RNeasy Mini Kit (Qiagen, West Sussex, UK) following manufacturer’s instructions and RNA quality and integrity were checked using the Nanodrop 1000 (Thermo Fisher Scientific Inc, IL, USA). Following RNA extraction, cDNA was synthesised from 1 μg RNA using the Quantitect Reverse Transcription kit (Qiagen, West Sussex, UK). Real-time PCR was performed with 100 ng cDNA in a 20 μl reaction volume using QuantiTect primer assays and the Quantifast SYBR-green PCR kit (Qiagen, West Sussex, UK). Experiments were performed in triplicate for each sample in 96-well plates using the Applied Biosystems 7500 real-time PCR system. PCR cycle settings were 95°C for 5 min, followed by 40 cycles of 95°C for 10 s, then 60°C for 30 s. Cycle threshold was calculated based on GAPDH (endogenous control), which was confirmed to be comparable in both cell types. Expression of all genes were expressed relative to GAPDH in each sample, derived using the comparative delta delta threshold change method (relative quantification, RQ). Three independent cell preparations were analysed.
siRNA Transfections
Purified OECs were seeded at a density of 5000 cells/100 μl into one chamber of an Ibidi culture insert (Ibidi GmbH, Munich, Germany) sealed onto a PLL coated coverslip in a well of a 24-well plate. Cells were cultured in defined OEC medium for 24 h, after which, the medium was replaced with low serum (2% (v/v) FBS) OEC medium containing 1 μM siRNA. siRNA sequences were obtained from Dharmacon/Thermo Scientific (Sulf 1: E-093746-00-0005; Sulf 2: E-093673-00-0005; non-targeting: D-001910-01-05; Thermo Fisher Scientific Inc, IL, USA). Sulf 1 and Sulf 2 siRNAs were added in combination. After 72 h, astrocytes were seeded into the opposing chamber of the culture insert and confrontation assays performed as previously described. The extent of gene knockdown was assessed by qPCR using RNA purified from siRNA treated cells and Sulf 1 and Sulf 2 specific primers.
RESULTS
The induction of astrocyte hypertrophy by SCs and the resulting formation of a cellular boundary remains a barrier to their use in cell transplantation therapies for the repair of spinal cord injury. Previous work has shown that SC induced boundary formation involves HS and FGFRs, since digestion of HS or chemical blockage of FGFR inhibits boundary formation and promotes cell mingling (Santos-Silva et al., 2007). Improved knowledge of the biological factors and signalling pathways underlying SC-induced boundary formation will aid the development of strategies to improve the incorporation of transplanted SCs into host CNS tissue and prevent activation of an astrocytic stress response by invading host SCs, to facilitate injury repair. Using a combination of cellular, biochemical and genetic approaches, we, therefore, investigated the role of HS and FGF signalling in the distinct interactions of SCs and OECs with astrocytes.
HS from different sources induces OEC/astrocyte boundaries
Heparin has been shown to induce a boundary between OECs and astrocytes (Santos-Silva et al., 2007). To determine if this activating effect of heparin on boundary formation could be induced by more physiologically relevant HS species, confrontation assays of OECs and astrocytes were treated with HS purified from different tissues. Whilst OECs and astrocytes mingled freely in control assays (Fig. 1A), strong boundaries were formed upon addition of HS purified from rat brain (Fig. 1B), rat liver (Fig. 1C) or porcine intestinal mucosa (Fig. 1D). Quantification of the extent of boundary formation, assessed by the number of OECs crossing into the astrocyte monolayer, showed that HS from all 3 sources had similar activity (Fig. 1E).
Figure 1. HS from a variety of tissue sources can induce a boundary in OEC:astrocyte confrontation assays.

30 ug/ml HS purified from brain (BHS, B), liver (LHS, C) and porcine intestinal mucosa (PMHS, D), were added to confrontation assays of OECs and astrocytes and compared to an untreated control (A). After 2 days of treatment, cells were fixed and stained for GFAP (red) and p75NTR (green). The numbers of OECs mingling with astrocytes across a 300 μm line were counted (E). HS from all 3 tissue sources prevented OECs from crossing into the astrocyte monolayer, resulting in the formation of a boundary. Error bars indicate ± SEM. Scale bar 50 μm. * p<0.05, *** p<0.001 versus control.
Induction of an OEC/astrocyte boundary by SCM requires both a protein and HS component
Initially, the relative requirements for protein and HS components in SCM for boundary formation were investigated. The active role of SCM HS in the induction of an OEC:astrocyte boundary was confirmed, since heparinase treatment of SCM negates the boundary forming effect (Fig. 2C). SCM treated separately with either heparinase or trypsin did not induce boundary formation (Fig. 2C, D, F), whilst combining the separately treated SCM samples reconstituted its biological activity and induced boundary formation (Fig. 2E, F). These data demonstrate that the activity of SCM in inducing boundary formation between cells requires both an HS and protein component, and that individually, these components are insufficient to induce a boundary.
Figure 2. Both HS and protein components of SCM are required for boundary formation.

Confrontation assays of OECs and astrocytes were carried out in the presence of; normal medium/untreated (A); SCM (B); heparinase treated SCM (C); trypsin treated SCM (D); 1:1 combination of heparinase treated and trypsin treated SCM (E). After 2 days of treatment, cells were fixed and stained for GFAP (red) and p75NTR (green). The number of cells mingling with astrocytes was counted across a 300 μm line (F). Heparinase or trypsin treated SCM did not induce boundary formation when added to assays individually, however, when combined, a boundary formed, suggesting that both an HS and protein component are required for activity. Error bars indicate ± SEM. Scale bar 50 μm. ** p<0.01 versus control.
HS sulfation determines the induction of OEC/astrocyte boundaries
In order to establish the effects of sulfation pattern on the inductive activity of HS, a panel of structurally defined, chemically modified (selectively desulfated) heparins were used to investigate the structural specificity of HS activity on boundary formation. After treatment with modified heparins (10 μg/ml) for 2 days, confrontation assays were stained for p75NTR and GFAP to visualise OECs and astrocytes, respectively. Results indicated that the most highly sulfated heparins (normal heparin (Hep1) or oversulfated heparin (Hep 9)) induced the strongest boundaries between OECs and astrocytes (Fig. 3 A, I), whilst lower sulfated heparin variants allowed OECs and astrocytes to mingle (Fig. 3 E-H). Interestingly, boundary formation was particularly dependent on O-sulfation, since N-acetylated heparin (in which N-sulfates are replaced with N-acetyl groups) induced significant boundary formation (Hep 2, Fig. 3 B), whereas either de-2-O-sulfated or de-6-O-sulfated heparins demonstrated lower boundary forming activity (Hep 3 and Hep 4, Fig. 3 C, D).
SCs secrete more highly sulfated HS than OECs
Since we have shown that HS in SCM plays an active role in boundary formation, we directly analysed the structure of HS synthesised by SCs and OECs and shed into their surrounding environment. The disaccharide composition of SCM and OCM HS was, therefore, analysed via separation by strong anion exchange (SAX)-HPLC (Fig. 4A, B) and the relative abundance of each of the eight standard HS disaccharides were calculated as a percentage of total HS (Fig. 4C). SCM contains approximately two fold more HS than OCM, and in addition, SCM HS is more highly sulfated, with an average of 1.02 sulfates per disaccharide, compared to 0.75 sulfates per disaccharide in OCM HS (Fig. 4D). SCM HS consists of a higher proportion of di- and tri-sulfated disaccharides (disaccharides 4, 5, 8 and 6) compared to OCM HS, and also a higher proportion of the singly 6-O-sulfated disaccharide (disaccharide 2). OCM HS, in contrast, contains a higher proportion of the unsulfated disaccharide (disaccharide 1). The proportion of singly N-sulfated (disaccharide 3) and singly 2-O-sulfated (disaccharide 7) disaccharides are similar in HS from both conditioned media.
Figure 4. SAX-HPLC analysis of fluorescently labelled HS disaccharides purified from OCM and SCM indicate that SCs and OECs secrete distinct HS structures.

HS purified from OCM (A) and SCM (B) was digested with heparinase enzymes to generate HS disaccharides, which were fluorescently labelled with BODIPY and separated by SAX-HPLC over a 45 minute, 0-1.5 M NaCl gradient, (A and B, black lines). HS disaccharide standards were separated over the same gradient as a reference guide (A and B, dashed lines). Relative abundances of the eight HS standard disaccharides in each conditioned medium sample were calculated as a percentage of total HS (C). A summary table detailing the composition of OCM HS and SCM HS is also presented (D). SCM HS was found to be more highly sulfated than OCM HS, with an average of 1.02 sulfates per disaccharide compared to 0.75 sulfates per disaccharide in OCM HS (D). SCM HS contained a higher proportion of di- and tri-sulfated disaccharides (disaccharides 4, 5, 8 and 6) and the singly 6-O-sulfated disaccharide (disaccharide 2) compared to OCM HS (C), which contained a higher proportion of the unsulfated disaccharide (disaccharide 1) (C, D).
(UA) uronic acid, (GlcN) glucosamine, (NAc) N-acetyl, (NS) N-sulfate, (6OS) 6-O-sulfate, (2OS) 2-O-sulfate.
SCs and OECs express different levels of HS biosynthetic enzymes
To determine if differences in the expression of HS biosynthetic enzymes could account for the higher sulfation of SCM HS, quantitative PCR was carried out using cDNA generated from monocultures of OECs and SCs. Whilst there appeared to be a trend towards differences in expression of several enzymes by SCs compared to OECs, these were not significant due to variability in biological replicates. For example, there were no significant differences in the expression of N-deacetylase/N-sulfotransferase (NDST1-4) enzymes or many of the sulfotransferase enzymes. However, HS 6-O-sulfotransferase 2 (HS6ST2) was expressed at a significantly higher level by OECs compared to SCs (Fig. 5B). In addition, HS 6-O-sulfotransferase 3 (HS6ST3) and HS 3-O-sulfotransferase 3a1 (HS3ST3a1) were found to be uniquely expressed by OECs and HS 3-O-sulfotransferase 6 (HS3ST6) was uniquely expressed by SCs (Fig. 5A). Interestingly, OECs express significantly higher levels of HS 6-O-sulfatase enzymes compared to SCs (Sulf 1 and Sulf 2; 8-fold and 4-fold respectively) (Fig. 5C), which would be expected to reduce 6-O-sulfation of OEC HS. These differences in HS biosynthetic and modification enzymes provide a potential explanation for the structural differences in HS synthesised and secreted by the two glial cell types. SCs and OECs express similar levels of the most common HSPG core proteins (Fig. 5D-E).
Figure 5. Expression profile of HS biosynthetic enzymes and HSPGs in OECs and SCs.

Using qRT-PCR, the expression of HS biosynthetic enzymes and HSPG core proteins by OECs and SCs were compared. Data are represented as relative quantification (RQ) of HS biosynthetic enzymes (A-C) and HSPGs (D-E) in SCs, normalised to the expression in OECs. HS6ST2, Sulf 1 and Sulf 2 were more highly expressed by OECs. HS3ST6 was expressed uniquely by SCs, whereas HS6ST3 and HS3ST3a1 were expressed uniquely by OECs. HS3ST2, GPC2 and GPC5 were not expressed by either cell type. Error bars indicate ± SEM. * p < 0.05, *** p < 0.001 versus control.
Reduction of Sulf 1 and Sulf 2 expression in OECs using RNAi promotes boundary formation with astrocytes
Consistent with increased levels of 6-O-sulfated HS in SCM, qPCR data indicated that SCs express lower levels of Sulf 1 and Sulf 2 6-O-endosulfatase enzymes compared to OECs. To determine if this was important for the ability of SCs to induce a boundary with astrocytes, we transfected OECs with siRNA targeted to Sulf 1 and Sulf 2 to see if the reduction in Sulf activity converted them to a more SC-like phenotype in confrontation assays. Prior to the addition of astrocytes, OECs were transfected with siRNA for 72 hours and the reduction of Sulf 1 and Sulf 2 mRNA was confirmed by qPCR (~73% and ~63% knockdown respectively). Once the cells had met in confrontation assays, the numbers of OECs mingling with astrocytes across a 300 μm line were counted. Significantly less Sulf siRNA-treated OECs crossed into the astrocyte monolayer than control siRNA-treated OECs (4.6 ± 1.3 Sulf siRNA cells compared with 15 ± 1.8 control SiRNA OECs. p <0.01) and a clear boundary was observed, whereas control OECs and astrocytes mingled freely (Fig. 6A-B). This suggests that an increase in HS 6-O-sulfation in OECs, via reduced Sulf expression, induces SC-like behaviour. The complimentary experiment to over-express Sulf 1 and Sulf 2 in SCs to determine if they adopted an OEC-like phenotype was attempted using pcDNA3.1/Myc-His(-)-MSulf-1 and pcDNA3.1/Myc-His(-)-MSulf-2 (Morimoto-Tomita et al., 2002). However, it was not possible to gain conclusive data using transfected cells in confrontation assays due to the low transfection efficiency of primary SCs (<3%, data not shown), which has also been reported by others (Kraus et al, 2010). We devised an alternative experiment based on the hypothesis that it may be possible to interfere competitively with endogenous highly sulfated HS in SC/astrocyte boundaries using an excess of low sulfated modified heparins that do not promote boundary formation (eg., Hep 6, 7 and 8 in Fig. 3). Confrontation assays of SC and astrocytes treated with each of the low sulfated modified heparins demonstrated significantly increased cell mingling (Fig. 7), confirming our hypothesis that these HS mimetics can inhibit boundary formation, presumably by competing for endogenous FGF ligands and reducing activation of astrocyte FGFR.
Figure 6. Knockdown of Sulf expression in OECs promotes boundary formation with astrocytes.
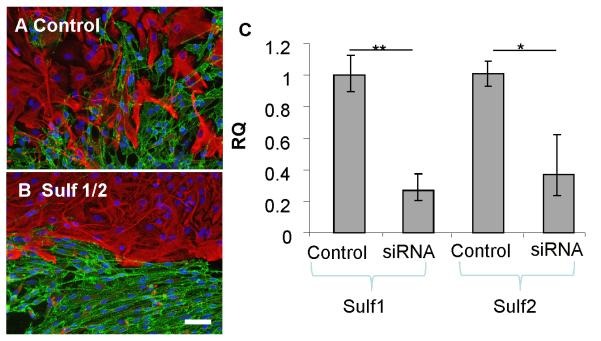
Confrontation assays were performed using astrocytes and either OECs transfected with non-targeting control siRNA (A) or OECs transfected with siRNAs targeted to Sulf 1 and Sulf 2 (B). After 2 days, cells were fixed and stained for GFAP (red) and p75NTR (green). OECs depleted of Sulf 1 and Sulf 2 expression formed a boundary on contact with astrocytes, whereas astrocytes and control treated OECs mingled. The extent of gene knockdown was assessed by qPCR using RNA purified from siRNA treated cells and Sulf 1 and Sulf 2 specific primers and was found to be ~ 70% and ~ 63% reduced respectively (C). Error bars indicate ± SEM. Scale bar 50 μm. * p < 0.05, ** p <0.01 versus control.
Figure 7. Interference of endogenous SC HS function using HS mimetics to inhibit boundary formation.

Confrontation assays of SCs and astrocytes were carried out in the absence (A) or presence (B-D) of 10 μg/ml selectively chemically modified heparins (Hep 6, 7, 8; see Figure 3 for structural details) with low levels of sulfation. After 2 days of treatment, cells were fixed and stained for GFAP (red) and p75NTR (green) and the numbers of SCs mingling with astrocytes across a 300 μm line were counted (E). The addition of low sulfated modified heparins (HS mimetics) to confrontation assays resulted in an increased number of SCs mingling with astrocytes (B-D) compared to in control cultures (A). Error bars indicate ± SEM. Scale bar 50 μm. * p < 0.05 versus control.
Inhibition of FGF1 or FGF9 disrupts SC boundary formation with astrocytes
It has long been established that HS is required for the proper function of FGF by supporting the binding of all members of the FGF family to their cognate FGFRs (Rapraeger et al., 1991; Yayon et al., 1991; Ornitz et al., 1992). The differential sulfation of HS is also known to regulate FGF activity (Guimond et al., 1993; Pye et al., 1998). Previously, it has been shown that FGFR inhibition disrupts SCM-induced boundary formation in OEC/astrocyte cultures (Santos-Silva et al., 2007), suggesting that a target of HS regulation of boundary formation is a member of the FGF family. We, therefore, investigated the role of particular FGF ligands in boundary formation. The FGFR inhibitor used in the aforementioned study (SU5402) was previously thought to specifically inhibit FGFR1, however, it has also been shown to effectively inhibit FGFR3 (Grand et al., 2004; Paterson et al., 2004). Astrocytes, but not OECs or SCs, express FGFR3-IIIb (Santos-Silva et al., 2007), suggesting that the response observed with the SU5402 inhibitor in confrontation assays may be due to inhibition of this receptor on astrocytes. FGF1 and FGF9 are the only known ligands for FGFR3-IIIb (Chellaiah et al., 1994; Hecht et al., 1995; Santos-Ocampo et al., 1996), therefore, the effect of blocking FGF1 or FGF9 using neutralising antibodies was investigated. Since FGFR3-IIIb does not bind FGF2 (Chellaiah et al., 1994), a neutralising antibody against FGF2 was used as a negative control. Inhibition of FGF1 or FGF9 in SC/astrocyte confrontation assays resulted in reduced boundary formation and increased cell mingling, whereas inhibition of FGF2 had no effect (Fig.8).
Figure 8. Neutralisation of FGF1 and FGF9 disrupts boundary formation between SCs and astrocytes.

SC and astrocyte confrontation assays were treated with 1 μg/ml FGF1 (B), FGF2 (C) or FGF9 (D) blocking antibodies for 2 days after the cell populations had met. Cells were fixed and stained for GFAP (red) and p75NTR (green) and the numbers of SCs mingling with astrocytes across a 300 μm line were counted (E). Neutralisation of FGF1 and FGF9, but not FGF2, resulted in an increase in mingling between SCs and astrocytes compared to control cultures (A). Error bars indicate ± SEM. Scale bar 50 μm. * p < 0.05, *** p < 0.001 versus control.
DISCUSSION
Glial cell transplantation is a promising strategy for the repair of damaged CNS following injury or disease, with OECs and SCs as potential candidates. OECs may be the preferred candidate due to their ability to evoke less of a stress response in astrocytes (Lakatos et al., 2000; Fairless and Barnett, 2005; Santos-Silva et al., 2007). However, since SCs often invade the CNS after injury when the blood brain barrier is breached (Bruce et al., 2000), it is important to understand the mechanisms by which they induce an astrocytic stress response, in order to devise strategies to prevent it. Previously, it has been shown that addition of heparin to OEC/astrocyte co-cultures induces boundary formation and removal of endogenous HS or inhibition of HS sulfation in SC/astrocyte cultures results in cell mingling (Santos-Silva et al., 2007). In this study, we have demonstrated that the sulfation of HS synthesised by SCs and OECs is a crucial feature of their molecular phenotype influencing their activity on contacting astrocytes. The ability of HS to induce a boundary between SCs and astrocytes is shown to be dependent upon the level and pattern of HS sulfation, is modulated by the extracellular sulfatases, Sulf 1 and Sulf 2, and is likely to be mediated via FGF1 and/or FGF9 activation of astrocyte FGFR3-IIIb signalling. This information provides new targets for the development of strategies to enhance post-transplantation integration of SCs into CNS tissue.
Using chemically modified heparins, which are model HS compounds with different levels of sulfation, it was possible to correlate HS sulfation with the extent of mingling/boundary formation induced in OEC/astrocyte confrontation assays. The more highly sulfated structures induced a stronger OEC:astrocyte boundary compared to the less sulfated structures, with a particular dependence on O-sulfation (Fig. 3). In support of this, HS isolated from SCM, which induces a boundary in OEC/astrocyte cultures, was 27% more sulfated than OCM HS, with a particular enhancement of 6-O-sulfated and 2-O-sulfated disaccharides (Fig. 4). Differences in the structure of HS synthesised by the glial cells will reflect a difference in biological activity and function, since HS:protein interactions are mediated by specific HS structures. For example, FGF family members show variations in the optimal HS sugar motifs required for binding, e.g., FGF1 has been shown to bind IdoA(2S)-GlcNS containing oligosaccharides that are additionally 6-O-sulfated, whereas binding of FGF2 to HS does not appear to require 6-O-sulfation (Turnbull et al., 1992; Maccarana et al., 1993; Kreuger et al., 1999; Kreuger et al., 2001; Ashikari-Hada et al., 2004). However, 6-O-sulfated HS is necessary for FGF2 dependent signalling, suggesting that, although not required for the initial FGF:HS interaction, 6-O-sulfated HS structures participate in HS:FGF:FGFR interactions and the formation of active signalling complexes (Guimond et al., 1993; Pye et al., 1998). The requirement of distinct HS structures for protein binding and variations in HS:protein binding kinetics (Powell et al., 2002) has been suggested to influence the assembly of particular ternary complexes and their frequency of formation, e.g., specific FGF:FGFR pairs, thus, regulating growth factor signalling dynamics (Kan et al., 1999; Powell et al., 2002; Allen and Rapraeger, 2003). Variations in the fine structure of HS synthesised by SCs and OECs are, therefore, anticipated to result in differences in biological activity via altered interactions with protein binding partners, such as FGFs (Fig. 9).
Figure 9. A model of HS function in SC/astrocyte boundary formation.

SCs secrete a highly sulfated HS (green chains), due to low level expression of Sulf enzymes. This HS promotes FGF1 and/or FGF9 ligand (black circles) binding to and/or activation of FGFR3-IIIb receptor (blue) on astrocytes, activating intracellular signalling pathways and resulting in altered cell phenotype (blocking of mingling) and the formation of a boundary. OECs secrete a lower sulfated HS (red chains), due to higher expression of Sulf enzymes (yellow stars), which selectively remove 6-O-sulfates. These lower sulfated HS chains cannot promote FGF1 or FGF9 binding to and/or activation of FGFR3-IIIb on astrocytes, and so fail to activate the required signalling pathways that lead to boundary formation, thus, enabling OECs to mingle with astrocytes.
To investigate possible mechanisms underlying the differences in HS structure synthesised by SCs and OECs, the expression of HS biosynthetic enzymes by the two glial cell types were analysed. The majority of enzymes were expressed at comparable levels by SCs and OECs, although some differences were observed, with OECs expressing a number of sulfotransferase enzymes at a higher level than SCs (Fig. 5). However, it is noteworthy that enzyme mRNA levels do not necessarily directly reflect the resulting HS structure that is synthesised and the mechanisms regulating the HS biosynthetic machinery remain incompletely understood. Furthermore, a number of HS biosynthetic enzymes may be chiefly regulated at the translational level through the presence of structured 5′-untranslated regions and internal ribosomal entry sites, allowing cells to regulate the level of translation, which is particularly relevant for regulatory proteins (Grobe and Esko, 2002). In addition, HS biosynthetic enzymes have been shown to interact and form complexes, further regulating their activities in vivo (Esko and Selleck, 2002; Presto et al., 2008).
Of more direct interest, HS 6-O-sulfatase enzymes, Sulf 1 and Sulf 2, were found to be expressed at relatively lower levels by SCs compared to OECs (Fig. 5). These enzymes act extracellularly on sulfated HS chains post-synthesis, to selectively remove 6-O-sulfates and ‘fine-tune’ the final HS structure (Dhoot et al., 2001; Morimoto-Tomita et al., 2002; Ai et al., 2006). Lower expression of Sulfs by SCs is, therefore, anticipated to result in changes to 6-O-sulfation in the mature HS chains synthesised, which was confirmed by our HPLC data. Differences in other sulfate moieties were also observed in our HPLC disaccharide analysis, for example the 2-O-/6-O-sulfated disaccharide. These differences can also be attributed to distinct levels of Sulf expression, since feedback mechanisms have been identified between Sulfs and other members of HS biosynthetic machinery, such that Sulf expression levels influence not only 6-O-sulfation, but also to some extent, N- and 2-O-sulfation (Lamanna et al., 2008).
Further evidence for a role of Sulf modified HS in boundary formation was obtained by siRNA mediated knock down of Sulf 1 and Sulf 2 expression in OECs (Fig 6). Cell behaviour was altered dramatically, resulting in a phenotypic switch towards a SC-like response on contact with astrocytes and formation of a boundary. This strongly suggests that Sulf enzymes expressed by OECs play a vital role in modifying HS fine structure to determine their biological function. Removal of specific 6-O-sulfates from HS by Sulf enzymes will affect HS:protein interactions and, therefore, subsequent signalling pathways involved in boundary formation, allowing the cells to mingle with astrocytes. Reduction in Sulf enzyme expression diminishes this extracellular control of HS 6-O-sulfation, resulting in the synthesis of OEC HS that is more highly sulfated and SC-like and, thus, able to activate the signalling cascades that underlie boundary formation. Conversely, by saturating SC/astrocyte confrontation assays with low sulfated HS mimetics, we demonstrated that it is possible to inhibit SC-induced boundary formation (Fig. 7), potentially by out-competing SC HS for the activating FGF ligands. This is consistent with the notion that highly sulfated HS produced by SCs is essential for activation of boundary forming signalling pathways.
In addition to the requirement for an HS component in SCM to induce an OEC:astrocyte boundary, a protein factor in SCM is also needed, since treatment of SCM with trypsin abolishes SCM activity. Reconstitution of separately heparinase- or trypsin-treated SCM samples restores SCM activity, further demonstrating the necessity of both an HS and protein component for activity (Fig. 2). In our previous work, FGFs were implicated, since inhibition of FGFR in SC:astrocyte cultures and OEC:astrocyte cultures treated with SCM reduced astrocyte hypertrophy and boundary formation (Santos-Silva et al., 2007). Notably, in the same study, the expression of FGFRs in SCs, OECs and cortical astrocytes were analysed, and FGFR3-IIIb was found to be uniquely expressed by astrocytes. FGF1 and FGF9 are the only known ligands for FGFR3-IIIb (Chellaiah et al., 1994; Hecht et al., 1995; Santos-Ocampo et al., 1996). In the current study, inhibition of FGF1 and FGF9 activity in confrontation assays resulted in mingling of SCs and astrocytes, whilst inhibition of FGF2 had no effect (Fig. 8). Interestingly, FGF9, also known as glial activating factor (Santos-Ocampo et al., 1996), has previously been reported to stimulate GFAP expression in human glioma cells (Miyagi et al., 1999), a process that also occurs during boundary formation (Lakatos et al., 2000). This data is consistent with the notion that signalling through FGFR3-IIIb is crucial for astrocyte boundary formation.
Figure 9 presents a model in which FGF1 and/or FGF9 can only bind to and activate FGFR3-IIIb if a more highly sulfated HS is present in the conditioned medium via direct secretion of an extracellular matrix HSPG such as perlecan or by shedding of cell surface HSPGs such as syndecans (Asundi et al., 2003; Ramani et al., 2012) or glypicans (Kreuger et al., 2004; Traister et al., 2008). This leads to activation of astrocytes and subsequent boundary formation with SCs. Lower sulfated HS synthesised by OECs, formed as a result of higher Sulf expression, cannot support FGF induced FGFR3-IIIb signalling, resulting in mingling between astrocytes and OECs. However, it cannot be discounted that FGFR1, which is expressed by all three glial cell types, may be differentially activated and also play a role in boundary formation.
It has been demonstrated in previous work that N-cadherin influences boundary formation and differentially regulates SC and OEC adhesion and migration responses upon contact with astrocytes (Fairless et al., 2005). The N-cadherin dependent pathway involved in astrocytosis and boundary formation may be independent of the FGF/HS dependent pathway or, alternatively, the two may be cooperatively linked, since interactions between FGFR and N-cadherins have been demonstrated (Utton et al., 2001; Williams et al., 2001; Suyama et al., 2002; Sanchez-Heras et al., 2006). Other reports have demonstrated a role for ephrins in SC:astrocyte boundary formation, since blocking the EphA receptors reduces boundary formation (Afshari et al., 2010a). Whilst there may not be a direct link between HS/FGF and ephrin signalling, they may be working in combination to induce a boundary following cell-cell contact. A function for chondroitin sulfate proteoglycans (CSPGs) in SC migration on astrocytes has also been demonstrated via modulation of integrin function (Afshari et al., 2010b). However, the evidence gained in the present study suggests that HSPGs are functioning via a separate mechanism to CSPGs.
The findings of this study afford the prospect for significant advancements in combinatorial approaches for CNS repair after injury and in neurodegenerative diseases in which astrocytes become reactive. For example, a novel strategy could be to transiently modify HS structure by modulating the expression of HS sulfatase enzymes in transplanted SCs to enhance engraftment. It may also be possible to manipulate HS sulfation levels at the CNS injury site, possibly by direct addition of Sulf enzymes into the lesion, or alternatively, to interfere with endogenous HS activities using HS mimetics. Further work is necessary to determine the validity of these approaches as novel strategies for CNS repair.
Acknowledgements
JRH and SMT were funded by a grant from The Wellcome Trust to JET, SCB and SEG.
Footnotes
The authors declare no competing financial interests.
REFERENCES
- Afshari FT, Kwok JC, Fawcett JW. Astrocyte-produced ephrins inhibit schwann cell migration via VAV2 signaling. J Neurosci. 2010a;30:4246–4255. doi: 10.1523/JNEUROSCI.3351-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afshari FT, Kwok JC, White L, Fawcett JW. Schwann cell migration is integrin-dependent and inhibited by astrocyte-produced aggrecan. Glia. 2010b;58:857–869. doi: 10.1002/glia.20970. [DOI] [PubMed] [Google Scholar]
- Ai X, Do AT, Kusche-Gullberg M, Lindahl U, Lu K, Emerson CP., Jr Substrate specificity and domain functions of extracellular heparan sulfate 6-O-endosulfatases, QSulf1 and QSulf2. J Biol Chem. 2006;281:4969–4976. doi: 10.1074/jbc.M511902200. [DOI] [PubMed] [Google Scholar]
- Alexander CL, Fitzgerald UF, Barnett SC. Identification of growth factors that promote long-term proliferation of olfactory ensheathing cells and modulate their antigenic phenotype. Glia. 2002;37:349–364. [PubMed] [Google Scholar]
- Allen BL, Rapraeger AC. Spatial and temporal expression of heparan sulfate in mouse development regulates FGF and FGF receptor assembly. J Cell Biol. 2003;163:637–648. doi: 10.1083/jcb.200307053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashikari-Hada S, Habuchi H, Kariya Y, Itoh N, Reddi AH, Kimata K. Characterization of growth factor-binding structures in heparin/heparan sulfate using an octasaccharide library. J Biol Chem. 2004;279:12346–12354. doi: 10.1074/jbc.M313523200. [DOI] [PubMed] [Google Scholar]
- Asundi VK, Erdman R, Stahl RC, Carey DJ. Matrix metalloproteinase-dependent shedding of syndecan-3, a transmembrane heparan sulfate proteoglycan, in Schwann cells. J Neurosci Res. 2003;73:593–602. doi: 10.1002/jnr.10699. [DOI] [PubMed] [Google Scholar]
- Barnett SC, Riddell JS. Olfactory ensheathing cell transplantation as a strategy for spinal cord repair--what can it achieve? Nat Clin Pract Neurol. 2007;3:152–161. doi: 10.1038/ncpneuro0447. [DOI] [PubMed] [Google Scholar]
- Brickman YG, Ford MD, Gallagher JT, Nurcombe V, Bartlett PF, Turnbull JE. Structural modification of fibroblast growth factor-binding heparan sulfate at a determinative stage of neural development. J Biol Chem. 1998;273:4350–4359. doi: 10.1074/jbc.273.8.4350. [DOI] [PubMed] [Google Scholar]
- Brockes JP, Fields KL, Raff MC. Studies on cultured rat Schwann cells. I. Establishment of purified populations from cultures of peripheral nerve. Brain Res. 1979;165:105–118. doi: 10.1016/0006-8993(79)90048-9. [DOI] [PubMed] [Google Scholar]
- Bruce JH, Norenberg MD, Kraydieh S, Puckett W, Marcillo A, Dietrich D. Schwannosis: role of gliosis and proteoglycan in human spinal cord injury. J Neurotrauma. 2000;17:781–788. doi: 10.1089/neu.2000.17.781. [DOI] [PubMed] [Google Scholar]
- Chellaiah AT, McEwen DG, Werner S, Xu J, Ornitz DM. Fibroblast growth factor receptor (FGFR) 3. Alternative splicing in immunoglobulin-like domain III creates a receptor highly specific for acidic FGF/FGF-1. J Biol Chem. 1994;269:11620–11627. [PubMed] [Google Scholar]
- Chuah MI, West AK. Cellular and molecular biology of ensheathing cells. Microsc Res Tech. 2002;58:216–227. doi: 10.1002/jemt.10151. [DOI] [PubMed] [Google Scholar]
- Dhoot GK, Gustafsson MK, Ai X, Sun W, Standiford DM, Emerson CP., Jr Regulation of Wnt signaling and embryo patterning by an extracellular sulfatase. Science. 2001;293:1663–1666. doi: 10.1126/science.293.5535.1663. [DOI] [PubMed] [Google Scholar]
- Esko JD. Special considerations for proteoglycans and glycosaminoglycans and their purification. Curr Protoc Mol Biol. 2001 doi: 10.1002/0471142727.mb1702s22. Chapter 17:Unit17 12. [DOI] [PubMed] [Google Scholar]
- Esko JD, Selleck SB. Order out of chaos: assembly of ligand binding sites in heparan sulfate. Annu Rev Biochem. 2002;71:435–471. doi: 10.1146/annurev.biochem.71.110601.135458. [DOI] [PubMed] [Google Scholar]
- Fairless R, Barnett SC. Olfactory ensheathing cells: their role in central nervous system repair. Int J Biochem Cell Biol. 2005;37:693–699. doi: 10.1016/j.biocel.2004.10.010. [DOI] [PubMed] [Google Scholar]
- Fairless R, Frame MC, Barnett SC. N-cadherin differentially determines Schwann cell and olfactory ensheathing cell adhesion and migration responses upon contact with astrocytes. Mol Cell Neurosci. 2005;28:253–263. doi: 10.1016/j.mcn.2004.09.009. [DOI] [PubMed] [Google Scholar]
- Fawcett JW, Asher RA. The glial scar and central nervous system repair. Brain Res Bull. 1999;49:377–391. doi: 10.1016/s0361-9230(99)00072-6. [DOI] [PubMed] [Google Scholar]
- Ford-Perriss M, Guimond SE, Greferath U, Kita M, Grobe K, Habuchi H, Kimata K, Esko JD, Murphy M, Turnbull JE. Variant heparan sulfates synthesized in developing mouse brain differentially regulate FGF signaling. Glycobiology. 2002;12:721–727. doi: 10.1093/glycob/cwf072. [DOI] [PubMed] [Google Scholar]
- Franklin RJ, Barnett SC. Olfactory ensheathing cells and CNS regeneration: the sweet smell of success? Neuron. 2000;28:15–18. doi: 10.1016/s0896-6273(00)00080-5. [DOI] [PubMed] [Google Scholar]
- Franssen EH, de Bree FM, Verhaagen J. Olfactory ensheathing glia: their contribution to primary olfactory nervous system regeneration and their regenerative potential following transplantation into the injured spinal cord. Brain Res Rev. 2007;56:236–258. doi: 10.1016/j.brainresrev.2007.07.013. [DOI] [PubMed] [Google Scholar]
- Grand EK, Chase AJ, Heath C, Rahemtulla A, Cross NC. Targeting FGFR3 in multiple myeloma: inhibition of t(4;14)-positive cells by SU5402 and PD173074. Leukemia. 2004;18:962–966. doi: 10.1038/sj.leu.2403347. [DOI] [PubMed] [Google Scholar]
- Grobe K, Esko JD. Regulated translation of heparan sulfate N-acetylglucosamine N-deacetylase/n-sulfotransferase isozymes by structured 5′-untranslated regions and internal ribosome entry sites. J Biol Chem. 2002;277:30699–30706. doi: 10.1074/jbc.M111904200. [DOI] [PubMed] [Google Scholar]
- Guimond S, Maccarana M, Olwin BB, Lindahl U, Rapraeger AC. Activating and inhibitory heparin sequences for FGF-2 (basic FGF). Distinct requirements for FGF-1, FGF-2, and FGF-4. J Biol Chem. 1993;268:23906–23914. [PubMed] [Google Scholar]
- Guimond SE, Turnbull JE. Fibroblast growth factor receptor signalling is dictated by specific heparan sulphate saccharides. Curr Biol. 1999;9:1343–1346. doi: 10.1016/s0960-9822(00)80060-3. [DOI] [PubMed] [Google Scholar]
- Guimond SE, Turnbull JE, Yates EA. Engineered bio-active polysaccharides from heparin. Macromol Biosci. 2006;6:681–686. doi: 10.1002/mabi.200600042. [DOI] [PubMed] [Google Scholar]
- Hecht D, Zimmerman N, Bedford M, Avivi A, Yayon A. Identification of fibroblast growth factor 9 (FGF9) as a high affinity, heparin dependent ligand for FGF receptors 3 and 2 but not for FGF receptors 1 and 4. Growth Factors. 1995;12:223–233. doi: 10.3109/08977199509036882. [DOI] [PubMed] [Google Scholar]
- Higginson JR, Barnett SC. The culture of olfactory ensheathing cells (OECs)-a distinct glial cell type. Exp Neurol. 2010;229:2–9. doi: 10.1016/j.expneurol.2010.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irie A, Yates EA, Turnbull JE, Holt CE. Specific heparan sulfate structures involved in retinal axon targeting. Development. 2002;129:61–70. doi: 10.1242/dev.129.1.61. [DOI] [PubMed] [Google Scholar]
- Kan M, Wu X, Wang F, McKeehan WL. Specificity for fibroblast growth factors determined by heparan sulfate in a binary complex with the receptor kinase. J Biol Chem. 1999;274:15947–15952. doi: 10.1074/jbc.274.22.15947. [DOI] [PubMed] [Google Scholar]
- Kreuger J, Perez L, Giraldez AJ, Cohen SM. Opposing activities of Dally-like glypican at high and low levels of Wingless morphogen activity. Dev Cell. 2004;7:503–512. doi: 10.1016/j.devcel.2004.08.005. [DOI] [PubMed] [Google Scholar]
- Kreuger J, Prydz K, Pettersson RF, Lindahl U, Salmivirta M. Characterization of fibroblast growth factor 1 binding heparan sulfate domain. Glycobiology. 1999;9:723–729. doi: 10.1093/glycob/9.7.723. [DOI] [PubMed] [Google Scholar]
- Kreuger J, Salmivirta M, Sturiale L, Gimenez-Gallego G, Lindahl U. Sequence analysis of heparan sulfate epitopes with graded affinities for fibroblast growth factors 1 and 2. J Biol Chem. 2001;276:30744–30752. doi: 10.1074/jbc.M102628200. [DOI] [PubMed] [Google Scholar]
- Lakatos A, Franklin RJ, Barnett SC. Olfactory ensheathing cells and Schwann cells differ in their in vitro interactions with astrocytes. Glia. 2000;32:214–225. doi: 10.1002/1098-1136(200012)32:3<214::aid-glia20>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- Lakatos A, Barnett SC, Franklin RJ. Olfactory ensheathing cells induce less host astrocyte response and chondroitin sulphate proteoglycan expression than Schwann cells following transplantation into adult CNS white matter. Exp Neurol. 2003;184:237–246. doi: 10.1016/s0014-4886(03)00270-x. [DOI] [PubMed] [Google Scholar]
- Lamanna WC, Frese MA, Balleininger M, Dierks T. Sulf loss influences N-, 2-O-, and 6-O-sulfation of multiple heparan sulfate proteoglycans and modulates fibroblast growth factor signaling. J Biol Chem. 2008;283:27724–27735. doi: 10.1074/jbc.M802130200. [DOI] [PubMed] [Google Scholar]
- Li Y, Field PM, Raisman G. Olfactory ensheathing cells and olfactory nerve fibroblasts maintain continuous open channels for regrowth of olfactory nerve fibres. Glia. 2005;52:245–251. doi: 10.1002/glia.20241. [DOI] [PubMed] [Google Scholar]
- Lyon M, Gallagher JT. Purification and partial characterization of the major cell-associated heparan sulphate proteoglycan of rat liver. Biochem J. 1991;273(Pt 2):415–422. doi: 10.1042/bj2730415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maccarana M, Casu B, Lindahl U. Minimal sequence in heparin/heparan sulfate required for binding of basic fibroblast growth factor. J Biol Chem. 1993;268:23898–23905. [PubMed] [Google Scholar]
- Miyagi N, Kato S, Terasaki M, Aoki T, Sugita Y, Yamaguchi M, Shigemori M, Morimatsu M. Fibroblast growth factor-9 (glia-activating factor) stimulates proliferation and production of glial fibrillary acidic protein in human gliomas either in the presence or in the absence of the endogenous growth factor expression. Oncol Rep. 1999;6:87–92. [PubMed] [Google Scholar]
- Morimoto-Tomita M, Uchimura K, Werb Z, Hemmerich S, Rosen SD. Cloning and characterization of two extracellular heparin-degrading endosulfatases in mice and humans. J Biol Chem. 2002;277:49175–49185. doi: 10.1074/jbc.M205131200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble M, Murray K. Purified astrocytes promote the in vitro division of a bipotential glial progenitor cell. EMBO J. 1984;3:2243–2247. doi: 10.1002/j.1460-2075.1984.tb02122.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ori A, Wilkinson MC, Fernig DG. The heparanome and regulation of cell function: structures, functions and challenges. Front Biosci. 2008;13:4309–4338. doi: 10.2741/3007. [DOI] [PubMed] [Google Scholar]
- Ornitz DM, Yayon A, Flanagan JG, Svahn CM, Levi E, Leder P. Heparin is required for cell-free binding of basic fibroblast growth factor to a soluble receptor and for mitogenesis in whole cells. Mol Cell Biol. 1992;12:240–247. doi: 10.1128/mcb.12.1.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paterson JL, Li Z, Wen XY, Masih-Khan E, Chang H, Pollett JB, Trudel S, Stewart AK. Preclinical studies of fibroblast growth factor receptor 3 as a therapeutic target in multiple myeloma. Br J Haematol. 2004;124:595–603. doi: 10.1111/j.1365-2141.2004.04814.x. [DOI] [PubMed] [Google Scholar]
- Patey SJ, Edwards EA, Yates EA, Turnbull JE. Heparin derivatives as inhibitors of BACE-1, the Alzheimer’s beta-secretase, with reduced activity against factor Xa and other proteases. J Med Chem. 2006;49:6129–6132. doi: 10.1021/jm051221o. [DOI] [PubMed] [Google Scholar]
- Pekny M, Nilsson M. Astrocyte activation and reactive gliosis. Glia. 2005;50:427–434. doi: 10.1002/glia.20207. [DOI] [PubMed] [Google Scholar]
- Powell AK, Fernig DG, Turnbull JE. Fibroblast growth factor receptors 1 and 2 interact differently with heparin/heparan sulfate. Implications for dynamic assembly of a ternary signaling complex. J Biol Chem. 2002;277:28554–28563. doi: 10.1074/jbc.M111754200. [DOI] [PubMed] [Google Scholar]
- Presto J, Thuveson M, Carlsson P, Busse M, Wilen M, Eriksson I, Kusche-Gullberg M, Kjellen L. Heparan sulfate biosynthesis enzymes EXT1 and EXT2 affect NDST1 expression and heparan sulfate sulfation. Proc Natl Acad Sci U S A. 2008;105:4751–4756. doi: 10.1073/pnas.0705807105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pye DA, Vives RR, Turnbull JE, Hyde P, Gallagher JT. Heparan sulfate oligosaccharides require 6-O-sulfation for promotion of basic fibroblast growth factor mitogenic activity. J Biol Chem. 1998;273:22936–22942. doi: 10.1074/jbc.273.36.22936. [DOI] [PubMed] [Google Scholar]
- Raisman G. Specialized neuroglial arrangement may explain the capacity of vomeronasal axons to reinnervate central neurons. Neuroscience. 1985;14:237–254. doi: 10.1016/0306-4522(85)90176-9. [DOI] [PubMed] [Google Scholar]
- Raisman G. Olfactory ensheathing cells - another miracle cure for spinal cord injury? Nat Rev Neurosci. 2001;2:369–375. doi: 10.1038/35072576. [DOI] [PubMed] [Google Scholar]
- Ramani VC, Pruett PS, Thompson CA, Delucas LD, Sanderson RD. Heparan sulfate chains of syndecan-1 regulate ectodomain shedding. J Biol Chem. 2012;287:9952–61. doi: 10.1074/jbc.M111.330803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapraeger AC, Krufka A, Olwin BB. Requirement of heparan sulfate for bFGF-mediated fibroblast growth and myoblast differentiation. Science. 1991;252:1705–1708. doi: 10.1126/science.1646484. [DOI] [PubMed] [Google Scholar]
- Sanchez-Heras E, Howell FV, Williams G, Doherty P. The fibroblast growth factor receptor acid box is essential for interactions with N-cadherin and all of the major isoforms of neural cell adhesion molecule. J Biol Chem. 2006;281:35208–35216. doi: 10.1074/jbc.M608655200. [DOI] [PubMed] [Google Scholar]
- Santos-Ocampo S, Colvin JS, Chellaiah A, Ornitz DM. Expression and biological activity of mouse fibroblast growth factor-9. J Biol Chem. 1996;271:1726–1731. doi: 10.1074/jbc.271.3.1726. [DOI] [PubMed] [Google Scholar]
- Santos-Silva A, Fairless R, Frame MC, Montague P, Smith GM, Toft A, Riddell JS, Barnett SC. FGF/heparin differentially regulates Schwann cell and olfactory ensheathing cell interactions with astrocytes: a role in astrocytosis. J Neurosci. 2007;27:7154–7167. doi: 10.1523/JNEUROSCI.1184-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver J, Miller JH. Regeneration beyond the glial scar. Nat Rev Neurosci. 2004;5:146–156. doi: 10.1038/nrn1326. [DOI] [PubMed] [Google Scholar]
- Skidmore M, Atrih A, Yates E, Turnbull JE. Labelling heparan sulphate saccharides with chromophore, fluorescence and mass tags for HPLC and MS separations. Methods Mol Biol. 2009;534:157–169. doi: 10.1007/978-1-59745-022-5_12. [DOI] [PubMed] [Google Scholar]
- Skidmore MA, Guimond SE, Dumax-Vorzet AF, Yates EA, Turnbull JE. Disaccharide compositional analysis of heparan sulfate and heparin polysaccharides using UV or high-sensitivity fluorescence (BODIPY) detection. Nat Protoc. 2010;5:1983–1992. doi: 10.1038/nprot.2010.145. [DOI] [PubMed] [Google Scholar]
- Skidmore MA, Guimond SE, Dumax-Vorzet AF, Atrih A, Yates EA, Turnbull JE. High sensitivity separation and detection of heparan sulfate disaccharides. J Chromatogr A. 2006;1135:52–56. doi: 10.1016/j.chroma.2006.09.064. [DOI] [PubMed] [Google Scholar]
- Suyama K, Shapiro I, Guttman M, Hazan RB. A signaling pathway leading to metastasis is controlled by N-cadherin and the FGF receptor. Cancer Cell. 2002;2:301–314. doi: 10.1016/s1535-6108(02)00150-2. [DOI] [PubMed] [Google Scholar]
- Traister A, Shi W, Filmus J. Mammalian Notum induces the release of glypicans and other GPI-anchored proteins from the cell surface. Biochem J. 2008;410:503–511. doi: 10.1042/BJ20070511. [DOI] [PubMed] [Google Scholar]
- Turnbull JE, Fernig DG, Ke Y, Wilkinson MC, Gallagher JT. Identification of the basic fibroblast growth factor binding sequence in fibroblast heparan sulfate. J Biol Chem. 1992;267:10337–10341. [PubMed] [Google Scholar]
- Utton MA, Eickholt B, Howell FV, Wallis J, Doherty P. Soluble N-cadherin stimulates fibroblast growth factor receptor dependent neurite outgrowth and N-cadherin and the fibroblast growth factor receptor co-cluster in cells. J Neurochem. 2001;76:1421–1430. doi: 10.1046/j.1471-4159.2001.00140.x. [DOI] [PubMed] [Google Scholar]
- Wilby MJ, Muir EM, Fok-Seang J, Gour BJ, Blaschuk OW, Fawcett JW. N-Cadherin inhibits Schwann cell migration on astrocytes. Mol Cell Neurosci. 1999;14:66–84. doi: 10.1006/mcne.1999.0766. [DOI] [PubMed] [Google Scholar]
- Williams EJ, Williams G, Howell FV, Skaper SD, Walsh FS, Doherty P. Identification of an N-cadherin motif that can interact with the fibroblast growth factor receptor and is required for axonal growth. J Biol Chem. 2001;276:43879–43886. doi: 10.1074/jbc.M105876200. [DOI] [PubMed] [Google Scholar]
- Yan Q, Johnson EM., Jr An immunohistochemical study of the nerve growth factor receptor in developing rats. J Neurosci. 1988;8:3481–3498. doi: 10.1523/JNEUROSCI.08-09-03481.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates EA, Guimond SE, Turnbull JE. Highly diverse heparan sulfate analogue libraries: providing access to expanded areas of sequence space for bioactivity screening. J Med Chem. 2004;47:277–280. doi: 10.1021/jm0309755. [DOI] [PubMed] [Google Scholar]
- Yates EA, Santini F, Guerrini M, Naggi A, Torri G, Casu B. 1H and 13C NMR spectral assignments of the major sequences of twelve systematically modified heparin derivatives. Carbohydr Res. 1996;294:15–27. doi: 10.1016/s0008-6215(96)90611-4. [DOI] [PubMed] [Google Scholar]
- Yayon A, Klagsbrun M, Esko JD, Leder P, Ornitz DM. Cell surface, heparin-like molecules are required for binding of basic fibroblast growth factor to its high affinity receptor. Cell. 1991;64:841–848. doi: 10.1016/0092-8674(91)90512-w. [DOI] [PubMed] [Google Scholar]