

Abstract
Signaling at NMDA receptors (NMDARs) is known to be important for memory reconsolidation, but while most studies show that NMDAR antagonists prevent memory restabilization and produce amnesia, others have shown that GluN2B-selective NMDAR antagonists prevent memory destabilization, protecting the memory. These apparently paradoxical, conflicting data provide an opportunity to define more precisely the requirement for different NMDAR subtypes in the mechanisms underlying memory reconsolidation, and to further understand the contribution of glutamatergic signaling to this process. Here, using rats with fully consolidated pavlovian auditory fear memories, we demonstrate a double dissociation in the requirement for GluN2B-containing and GluN2A-containing NMDARs within the basolateral amygdala in the memory destabilization and restabilization processes, respectively. We further show a double dissociation in the mechanisms underlying memory retrieval and memory destabilization, since AMPAR antagonism prevented memory retrieval while still allowing the destabilization process to occur. These data demonstrate that glutamatergic signaling mechanisms within the basolateral amygdala differentially and dissociably mediate the retrieval, destabilization and restabilization of previously consolidated fear memories.
Introduction
Memory reconsolidation is the process by which previously consolidated memories become destabilized at retrieval, and require restabilization in order to persist in the brain (Lewis, 1979; Nader, 2003). The neurochemical basis of reconsolidation has been intensively studied, particularly the requirement for signaling at the NMDA subtype of glutamate receptor (NMDAR). However, although NMDAR-mediated signaling is required for the reconsolidation (restabilization) of conditioned stimulus (CS)-drug (Sadler et al., 2007; Brown et al., 2008; Itzhak, 2008; Milton et al., 2008; Milton et al., 2012), spatial (Przybyslawski and Sara, 1997) and CS-fear (Pedreira et al., 2002; Lee et al., 2006) memories, antagonism at the GluN2B subtype of NMDAR has been shown to prevent the destabilization of CS-fear memories, thereby protecting them from the effects of amnestic agents (Ben Mamou et al., 2006). These paradoxical, apparently conflicting data provide the opportunity to better understand the contribution of signaling at glutamatergic receptors to the reconsolidation process.
NMDARs exist as tetramers, typically composed of two GluN1 and two GluN2 subunits (Dingledine et al., 1999). The GluN2 subunits consist of four different types (GluN2A-D), of which GluN2A and GluN2B have been most studied. In addition to differences between GluN2A-containing (GluN2A-NMDARs) and GluN2B-containing NMDARs (GluN2B-NMDARs) in their sensitivity to glutamate and their activation kinetics, these subtypes of receptor also couple to different proteins within the postsynaptic density, activating divergent intracellular signaling pathways (Kim et al., 2005; Ivanov et al., 2006; Zhang et al., 2008). For example, the C-terminal domain of GluN2B-NMDARs suppresses CREB and activates the ubiquitin-proteasome system (UPS), while GluN2A-NMDAR activation promotes CREB phosphorylation and is neuroprotective (Hardingham et al., 2002; Martel et al., 2012). These differences at the molecular level may have important functional implications; activation of GluN2B-NMDARs promotes long-term depression (LTD) while activation of GluN2A-NMDARs promotes long-term potentiation (LTP) in the hippocampus (Liu et al., 2004).
The basolateral amygdala (BLA) is required for both CS-fear memory consolidation (Campeau and Davis, 1995; Killcross et al., 1997; Koo et al., 2004) and reconsolidation (Nader et al., 2000). Furthermore, NMDARs within the BLA have been implicated in both memory destabilization (Ben Mamou et al., 2006) and restabilization (Milton et al., 2008) processes. Thus, we hypothesized that memory destabilization and restabilization may be mediated through the different subtypes of NMDAR within the BLA, GluN2B-NMDARs being required for destabilization, GluN2A-NMDARs being required for restabilization. Furthermore, since AMPARs are required for memory retrieval (Day et al., 2003; Bast et al., 2005; Winters and Bussey, 2005) and because memory reconsolidation can only occur when a memory is retrieved (Lewis, 1979; Nader, 2003), we further hypothesized that AMPARs would be necessary for the destabilization process. Finally, we investigated the effects of reducing presynaptic glutamate release by treatment with an agonist at metabotropic 2/3 glutamate receptors (mGlu2/3Rs) on the balance of these mnemonic processes. We hypothesized that the memory should neither be retrieved nor destabilized, and therefore restabilization of the memory would not be required for it to persist.
Materials and Methods
Subjects
Subjects were 93 male Lister-Hooded rats (Charles River) housed in pairs in a vivarium on a reversed light-dark cycle (lights on at 1900hrs). Subjects were food restricted, though not deprived, being fed 25 g per rat of lab chow after training or testing each day. Access to water was ad libitum except for when inside the conditioning chambers. All procedures were conducted in accordance with the UK Animals (Scientific Procedures) Act 1986.
Surgery
Rats were implanted with bilateral guide cannulae (16mm, 24 gauge; Coopers Needle Works Ltd) located just dorsal to the basolateral amygdala (Figure 1) as described previously (Milton et al., 2008). The co-ordinates for cannula implantation were AP - 2.6 mm and ML ± 4.5 mm (relative to bregma) and DV – 5.6 mm (relative to dura). A recovery period of 7 days was given before behavioral training and testing began.
Figure 1. Cannulae placements.

All cannulae placements were within the BLA. The placements for individual experiments are shown separately. For each figure, the white circles represent the vehicle/vehicle group; the white squares the vehicle/anisomycin group; the grey circles the drug/vehicle group, and; the grey squares the drug/anisomycin group, where the drugs were: (a) IFEN; (b) NVP-AAM077; (c) LY293558, and; (d) LY317206. Co-ordinates are given from bregma. This figure was modified, with permission, from Paxinos and Watson (2004).
Intracerebral drug administration
Infusions were carried out using a syringe pump (Harvard Apparatus) and 5 μl Hamilton syringes, connected to injectors (28 gauge, projecting 2 mm beyond the guide cannulae; Plastics One Inc.) by polyethylene tubing. The rats received two infusions; one immediately prior to the memory reactivation session, and one immediately afterwards. All infusions were begun 30 seconds after the insertion of the injectors and performed over 2 minutes at a rate of 0.25 μl min−1 (total volume of 0.5 μl side−1). One minute of waiting time was imposed from the end of the infusion to the removal of the injectors to allow diffusion of the solution away from the infusion site.
Drugs
Rats received either the protein synthesis inhibitor anisomycin or its vehicle as their second (post-reactivation) infusion. Anisomycin (Sigma-Aldrich; 125 μg μl−1) was dissolved in equimolar HCl and then pH-balanced to pH 7.4 with NaOH. This dose of anisomycin has previously been shown to disrupt memory reconsolidation (Ben Mamou et al., 2006).
Prior to memory reactivation, rats received infusions of drugs targeting the glutamatergic signaling system, or the appropriate vehicle. The GluN2B diheteromeric receptor-selective (Williams, 1993) NMDAR antagonist ifenprodil (Ascent Scientific) was dissolved in PBS at a concentration of 2 μg μl−1; this dose has previously been shown to disrupt memory destabilization (Ben Mamou et al., 2006). The GluN2A-preferring (Auberson et al., 2002) NMDAR antagonist NVP-AAM077 (Sigma-Aldrich) was dissolved in PBS at a concentration of 5 μg μl−1; this dose has been shown to reduce the expression of fear-potentiated startle (Walker and Davis, 2008). The mGlu2/3R agonist LY317206 (Doherty et al., 1999) and the AMPAR antagonist LY293558 (Ornstein et al., 1993) were both generous gifts from Eli Lilly. LY317206 was dissolved in PBS at a concentration of 2 μg μl−1, and LY293558 in ddH2O at a concentration of 1.33 μg μl−1. This dose of LY317206 has been shown to reduce fear-potentiated startle (Walker et al., 2002) and the dose of LY293558 is higher than ineffective doses used previously in the amygdala (Di Ciano and Everitt, 2004).
Behavioral procedures
Testing took place in four conditioning chambers (Med Associates Inc.) with the assignment of individual conditioning chambers counterbalanced across experimental groups within each experiment. Rats were first habituated to the context in a 2-h session in which neither the clicker CS nor the shock unconditioned stimulus (US) were presented. Following the end of this habituation session, they were returned to the home cage. Twenty-four hours later, they were placed back into the same experimental chamber for the fear conditioning session; during this time, they were first exposed to the context (with no CS or US) for 35 ± 1 minute, then exposed to a single CS-US pairing of a clicker (10 Hz, 80 dB, 60 s) CS and a 0.5 mA, 1 s scrambled footshock US. For the experiment investigating the requirement of AMPARs in memory reconsolidation, a different brand of experimental chamber (Paul Fray) was used, but these were set up in the same configuration as the Med Associates chambers other than the shock duration (0.5 s). To ensure comparability in the strength of learning, five CS-shock pairings were used in this experiment. All other aspects of the behavioral procedures remained the same.
Twenty-four hours following the end of training, the rats were returned to the same conditioning chambers for a memory reactivation session. After 1 minute of context exposure, the clicker CS was presented for 60s. The session was recorded through a CCTV system onto a DVD to allow for offline manual scoring of behavior. Two CS-fear memory tests were conducted following the memory reactivation session; these test sessions, conducted 24h and 8d following reactivation, had the same format as the memory reactivation session, except that no drug infusions were given.
Briefly, and as before (Ben Mamou et al., 2006), if memory destabilization was prevented by the pre-reactivation infusion, then anisomycin would not have an amnestic effect when it was infused subsequently, since the memory would not be in a destabilized and labile state when anisomycin was applied. If memory restabilization was prevented by the pre-reactivation infusion, then all experimental groups treated with the drug would be predicted to show amnesia at subsequent test, as would a group that had received an infusion of vehicle followed by anisomycin. If neither destabilization nor restabilization were prevented by the pre-reactivation infusion, then only the two groups that had received post-reactivation anisomycin would be predicted to show amnesia.
Histology
At the end of the experiments, the rats were killed with an overdose of anesthetic (Dolethal, Vétoquinol UK Ltd.) and transcardially perfused through the ascending aorta with 0.01 M PBS, followed by 4% paraformaldehyde (PFA). The brains were removed and stored in 4% PFA for at least 24 hours, before being transferred to 20% sucrose solution for cryoprotection prior to sectioning. The brains were sectioned at 60 μm and stained with Cresyl Violet. Cannulae placements (Figure 1) were verified using light microscopy (Leica).
Data collection and statistical analysis
Data for the reactivation and test sessions were scored offline and blind to treatment by A.L.M. Data from the conditioning session were not recorded, because there is no measure of conditioned freezing when animals receive only one pairing of the CS with shock. However, shock delivery was recorded by the experimenter observing the unconditioned response in the conditioning session. Freezing was defined as a cessation of movement apart from respiration, and was measured instantaneously at 5 s intervals. Freezing during the first minute of the session was assessed to provide a measure of fear to the context, and the second as a measure of fear to the CS. All data were converted to percentages before analysis. Data were analyzed by repeated measures ANOVA, with CS (Context vs. Cue) and Session (Reactivation vs. 24 h Test vs. 8 d Test) as within-subject factors, and Drug1 (VEH vs. drug) and Drug2 (VEH vs. ANI) as between-subjects factors. For clarity, these are reported in the text using the name of the drug used in the experimental group (e.g. Drug2 is shown as ‘ANI’). Where the data violated the assumption of sphericity as assessed using Mauchly’s test, a correction was applied; the Greenhouse-Geisser correction if ε < 0.75, and the Huynh-Feldt correction if ε > 0.75, as recommended by Cardinal & Aitken (2006). Where appropriate, further ANOVAs or pairwise comparisons were conducted; all pairwise comparisons were adjusted using the Sidák correction, which is a mathematically accurate form of the Bonferroni estimation (Cardinal and Aitken, 2006).
Results
GluN2B-NMDARs are required for memory destabilization, not restabilization
Administration of the GluN2B-selective NMDAR antagonist ifenprodil (IFEN) directly into the BLA prior to memory reactivation prevented the destabilization of the CS-fear memory. This was shown by the prevention of anisomycin-induced amnesia when IFEN was given before reactivation, but not when the vehicle was given prior to reactivation (Figure 2b). All groups had previously conditioned to the CS, as all rats showed greater freezing to the CS than the context in the reactivation and the test sessions [F1, 25 = 33.0, p < 0.001, η2 = 0.57]. While the VEH/ANI group showed less freezing than the VEH/VEH group at the 24 h test [F1, 14 = 5.8, p = 0.031, η2 = 0.29], the IFEN/ANI group froze comparably to the IFEN/VEH group [F < 1, p = 0.66]. Analysis of the 8d test was complicated by an overall reduction in conditioned freezing [CS × Session: F1, 25 = 4.7, p = 0.04, η2 = 0.16] most likely attributable to gradual extinction that occurred after the last non-reinforced retrieval test. However, while the VEH/VEH and VEH/ANI groups showed equivalent conditioned freezing at the 8d test [F1, 14 = 2.2, p = 0.16], so did the IFEN/VEH and IFEN/ANI groups [F1, 11 = 3.4, p = 0.09], consistent with the prevention of destabilization of the CS-fear memory by IFEN.
Figure 2. Effects of the GluN2B-selective NMDAR antagonist on CS-fear memory reconsolidation.
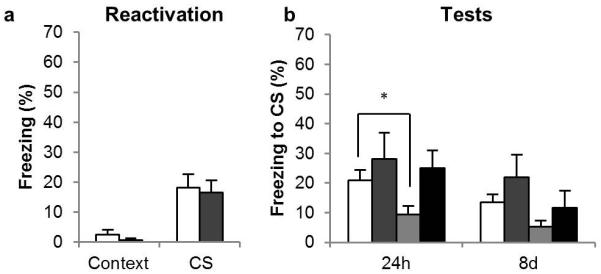
(a) Administration of the GluN2B-selective NMDAR antagonist had no effect on the retrieval of the CS-fear memory at reactivation, but (b) it did prevent the destabilization of the CS-fear memory. Data are presented as means ± s.e.m. Group sizes (and colors in b) were: VEH/VEH, n = 8 (white); IFEN/VEH, n = 8 (mid gray); VEH/ANI, n = 8 (pale gray); IFEN/ANI, n = 5 (dark gray). In (a) the groups are collapsed with the white bar representing the average of the VEH/VEH and VEH/ANI groups, and the gray bar the IFEN/VEH and IFEN/ANI groups. Asterisks denote p < 0.05.
IFEN did not acutely affect the retrieval of the conditioned fear memory (Figure 2a); collapsing across groups for the first infusion (VEH, n = 16; IFEN, n = 13) there was no difference in the level of conditioned freezing shown during the memory reactivation session [F < 1, p = 0.95]. These findings confirm the previous report (Ben Mamou et al., 2006) that GluN2B-NMDARs are required for memory destabilization.
GluN2A-NMDARs are required for memory restabilization, not destabilization
Administration of the GluN2A-preferring NMDAR antagonist NVP-AAM077 (NVP) prior to the memory reactivation session reduced conditioned freezing at subsequent tests conducted 24 hours and 8 days later (Figure 3b). All rats had conditioned to the CS, as all groups showed greater freezing to the CS than the context in the reactivation and the test sessions [F1, 15 = 12, p = 0.003, η2 = 0.45]. Administration of NVP reduced conditioned freezing in the subsequent test sessions [CS × Session × NVP: F2, 30 = 4.8, p = 0.016, η2 = 0.24], but not during the reactivation session. Analyses of individual sessions showed that there were no differences between experimental groups during reactivation [CS × NVP: F1, 15 = 2.3, p = 0.15; CS × ANI, F < 1, p = 0.52; CS × NVP × ANI: F < 1, p = 0.87] but that NVP-treated animals froze less than VEH-treated rats during the test at 24 h [CS × NVP: F1, 15 = 8.5, p = 0.011, η2 = 0.36]. The effect of NVP was persistent, in that there was no overall reduction in freezing between the 24h and the 8d test [F < 1], although there was a reduction in the VEH-treated groups [CS × Session × NVP: F1, 15 = 9.75, p = 0.007, η2 = 0.39; pairwise comparisons showed a reduction in conditioned freezing between the 24h and 8d test in the VEH-treated groups, p = 0.024, but not in the NVP-treated groups, p = 0.22]. This is consistent with extinction of the CS-US association in the VEH/VEH group, but not in the other experimental groups, which could not reduce freezing any further. Pairwise comparisons also revealed that the NVP-treated groups did not differ from the VEH/ANI group [all p’s > 0.41]. Thus, administration of NVP produced amnesia, regardless of whether anisomycin was also administered. Furthermore, the amnesia shown by the NVP/ANI group supports the view that NVP did not block destabilization, as if it had then this group would have shown intact memory.
Figure 3. Effects of the GluN2A-preferring NMDAR antagonist on CS-fear memory reconsolidation.
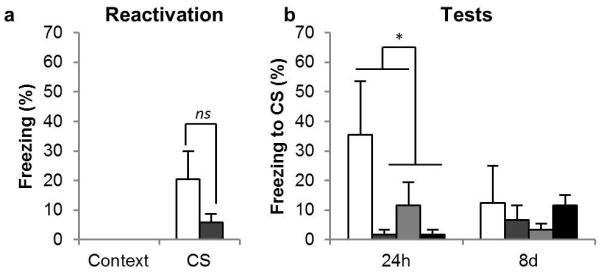
(a) Administration of the GluN2A-preferring NMDAR antagonist had no effect on the retrieval of the CS-fear memory at reactivation, but (b) it prevented the restabilization of the CS-fear memory. Data are presented as means ± s.e.m. Group sizes (and colors in b) were: VEH/VEH, n = 4 (white); NVP/VEH, n = 5 (mid gray); VEH/ANI, n = 5 (pale gray); NVP/ANI, n = 5 (dark gray). In (a) the groups are collapsed with the white bar representing the average of the VEH/VEH and VEH/ANI groups, and the gray bar the NVP/VEH and NVP/ANI groups. Asterisks denote p < 0.05; ns denotes non-significant differences.
NVP did not acutely affect the retrieval of the conditioned fear memory during the reactivation session (Figure 3a); collapsing across groups for the first infusion (VEH, n = 9; NVP, n = 10), despite the apparent numerical reduction in freezing in the NVP-treated group, there was no statistical difference between freezing levels in the two experimental groups [F1, 17 = 2.4, p = 0.14]. However, the NVP-treated group showed levels of freezing that did not significantly differ from zero [p = 0.34] consistent with the previous observation that this dose of NVP reduces the expression of fear-potentiated startle (Walker and Davis, 2008). This result supports our hypothesis that GluN2A-NMDARs are required for memory restabilization, while not being required for memory destabilization.
AMPARs are not required for destabilization or restabilization
Administration of the AMPAR antagonist LY293558 prior to memory reactivation did not reduce conditioned freezing during the test sessions, and did not prevent the post-reactivation anisomycin infusion from inducing amnesia (Figure 4b). All rats had conditioned to the CS, as they showed greater freezing to the CS than the context following training [F1, 21 = 37.7, p < 0.001, η2 = 0.64]. LY293558 did not affect freezing in the 24h test session [CS × LY293558: F < 1, p = 0.45], but anisomycin infusion resulted in amnesia [CS × ANI: F1, 21 = 16, p = 0.001, η2 = 0.44]. Indeed, the group that received anisomycin following LY293558 froze less than the group that received vehicle prior to reactivation [CS × ANI: F1, 9 = 6.0, p = 0.037, η2 = 0.40] demonstrating that LY293558 did not prevent the destabilization of the memory. This anisomycin-induced amnesia was also observed at the 8d test [F1, 21 = 7.82, p = 0.011, η2 = 0.27], even though there was extinction of conditioned freezing at the 8d test compared to the 24h test [F1, 21 = 10.1, p = 0.005, η2 = 0.32]. This is consistent with a previous report, in which the AMPA/kainate receptor antagonist CNQX was shown to leave memory destabilization and restabilization intact when given prior to memory reactivation (Ben Mamou et al., 2006).
Figure 4. Effects of the AMPAR antagonist on CS-fear memory reconsolidation.
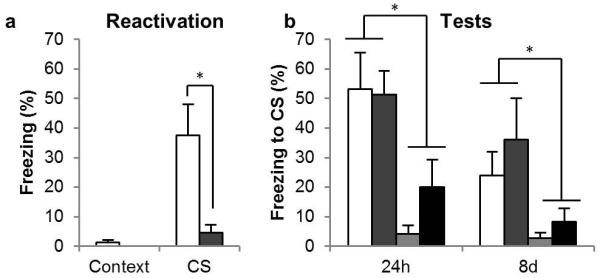
(a) The AMPAR antagonist LY293558 acutely impaired the retrieval of the CS-fear memory at reactivation, but (b) it affected neither the destabilization nor the restabilization of the CS-fear memory. Group sizes (and colors in b) were: VEH/VEH, n = 8 (white); LY293558/VEH, n = 6 (mid gray); VEH/ANI, n = 6 (pale gray); LY293558/ANI, n = 5 (dark gray). In (a) the groups are collapsed with the white bar representing the average of the VEH/VEH and VEH/ANI groups, and the gray bar the LY293558/VEH and LY293558/ANI groups. Asterisks denote p < 0.05.
LY293558 acutely reduced conditioned freezing during the memory reactivation session (Figure 4a); collapsing across groups for the first infusion (VEH, n = 14; LY293558, n = 11), the groups given LY293558 prior to the memory reactivation froze to the CS less than groups that had received the infusion of vehicle [CS × LY293558: F1, 23 = 6.8, p = 0.016, η2 = 0.23]. Thus AMPARs are required for memory retrieval, but not memory destabilization.
Blockade of Glu release left destabilization and restabilization intact
LY317206, an agonist at presynaptic class II/III metabotropic glutamate receptors (mGlu2/3Rs), affected neither the destabilization nor the restabilization of the CS-fear memory (Figure 5b). All groups had conditioned to the CS during training, as shown by increased freezing during the CS compared to the context [F1, 16 = 29, p < 0.001, η2 = 0.64]. However, although anisomycin produced amnesia as assessed at the 24 h test [CS × ANI: F1, 16 = 7.2, p = 0.016, η2 = 0.31], there was no effect of LY317206 on conditioned freezing [CS × LY317206: F < 1, p = 0.74], indicating that it did not prevent the restabilization of the CS-fear memory. Furthermore, as the LY317206/ANI group showed less freezing to the CS than the LY317206/VEH group at the 24 h test [CS × ANI: F1, 10 = 11, p = 0.008, η2 = 0.52], the memory destabilized during the reactivation session, and this process was not prevented by agonism at mGlu2/3Rs. The anisomycin-induced amnesia observed at 24h persisted at the trend level in the 8d test [F1, 16 = 4.14, p = 0.059, η2 = 0.21], although the data from the 8d test were compromised by the extinction of conditioned freezing in the VEH/VEH group [pairwise comparisons revealed reduced conditioned freezing at the 8d test in this group, p = 0.035, but no differences in the other groups, all p’s > 0.49].
Figure 5. Effects of the mGlu2/3R agonist on CS-fear memory reconsolidation.
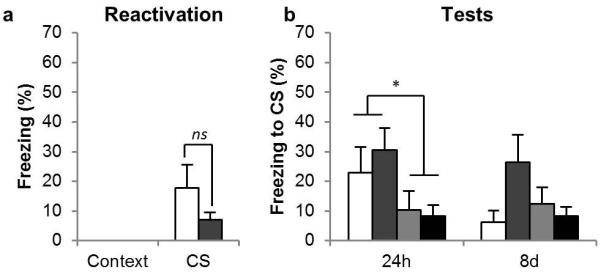
(a) The mGlu2/3R agonist LY317206 did not prevent retrieval of the CS-fear memory at reactivation, and (b) it prevented neither destabilization nor restabilization of the CS-fear memory. Group sizes (and colors in b) were: VEH/VEH, n = 4 (white); LY317206/VEH, n = 6 (mid gray); VEH/ANI, n = 4 (pale gray); LY317206/ANI, n = 6 (dark gray). In (a) the groups are collapsed with the white bar representing the average of the VEH/VEH and VEH/ANI groups, and the gray bar the LY317206/VEH and LY317206/ANI groups. Asterisks denote p < 0.05; ns denotes non-significant differences.
LY317206 did not acutely affect the retrieval of the conditioned fear memory during the reactivation session (Figure 5a); collapsing across groups for the first infusion (VEH, n = 8; LY317206, n = 12) there was no statistically significant difference between freezing levels in the two groups [F1, 18 = 2.4, p = 0.14] despite the numerical reduction in conditioned freezing. Thus, agonism at presynaptic mGlu2/3Rs did not affect the destabilization or restabilization, and did not produce a statistically significant reduction in the retrieval, of the CS-fear memory.
Discussion
The present data support the view that GluN2B-NMDARs and GluN2A-NMDARs within the BLA are required for memory destabilization and restabilization respectively. This is the first demonstration of a double dissociation in the function of these two subtypes of NMDAR in memory reconsolidation. Furthermore, we also demonstrate a double dissociation between memory retrieval (dependent upon AMPARs) and the induction of memory lability (dependent upon GluN2B-NMDARs).
The doubly dissociable involvement of different NMDAR subtypes in destabilization and restabilization enables resolution of the apparent discrepant findings in the literature that antagonism at GluN2B-NMDARs prevents memory destabilization (Ben Mamou et al., 2006), but that non-selective NMDAR antagonism with D-APV (Milton et al., 2008) or MK-801 (Przybyslawski and Sara, 1997; Pedreira et al., 2002; Sadler et al., 2007; Brown et al., 2008; Itzhak, 2008; Lee and Everitt, 2008; von der Goltz et al., 2009) prevents the restabilization of memories. We hypothesize (Figure 6) that the differential effects of non-subtype selective NMDAR antagonists on these mnemonic processes depend upon the balance between signaling at GluN2B-NMDARs and GluN2A-NMDARs, since only after the brief destabilization process is there a requirement for the longer-lasting restabilization process to be engaged. The differences in duration of the two processes, subserved by different subpopulations of NMDAR expressed within BLA neurons (Müller et al., 2009), would suggest that the non-subtype selective NMDAR antagonists tested in previous studies appear to be sufficient to prevent GluN2A-dependent signaling, but fail significantly to affect GluN2B-dependent signaling.
Figure 6. Hypothesized glutamatergic mechanisms underlying memory stability.
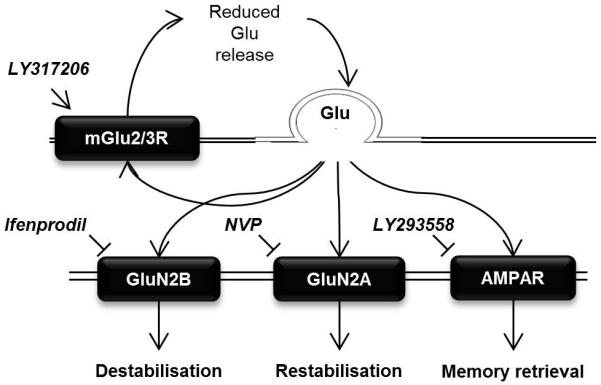
GluN2B-containing NMDARs are required for memory destabilization, and IFEN prevents this process. GluN2A-containing NMDARs are required for memory restabilization, which is prevented by NVP-AAM077. The AMPAR antagonist LY293558 reduces memory retrieval, but has no effect on destabilization or restabilization. The presynaptic mGlu2/3R agonist LY317206 reduces glutamate release, reducing memory retrieval via AMPARs, but leaving the balance of GluN2A and GluN2B activity intact (so having no effect on destabilization or restabilization).
Thus, we hypothesize that non-selective NMDAR antagonists exert their amnestic effects primarily through GluN2A-containing NMDARs. Consistent with this view are the findings that GluN2A-preferring NMDAR antagonists prevent the acquisition of spatial memory (Hu et al., 2009) and conditioned fear (Dalton et al., 2012), as do non-subtype-selective NMDAR antagonists (Morris et al., 1986; Davis et al., 1992; Fanselow and Kim, 1994). Furthermore, the effects of non-selective NMDAR antagonists on other processes, such as cortical oscillations, are more similar to the effects of GluN2A-preferring NMDAR antagonists than GluN2B-selective NMDAR antagonists (Kocsis, 2012). Thus the glutamate transmission-dependent destabilization and restabilization processes would be predicted to engage parallel, independent molecular mechanisms mediated by the two subtypes of NMDAR. This perspective can be integrated with already known mechanisms underlying memory destabilization, for example GluN2B-containing NMDARs recruit the ubiquitin-proteasome system via CaMKII (Bingol et al., 2010), thus allowing the protein degradation that is required for the induction of memory lability (Lee et al., 2008).
Surprisingly, we found a further dissociation in the mechanisms underlying memory retrieval (dependent upon AMPARs) and destabilization (dependent upon GluN2B-NMDARs). These data refute our original hypothesis concerning AMPARs, although they are consistent with previous findings that signaling via AMPARs is necessary for memory expression and retrieval (Day et al., 2003; Bast et al., 2005; Winters and Bussey, 2005). It is perhaps surprising, from a theoretical perspective, that memory retrieval and destabilization might be disrupted independently, since reconsolidation theories maintain that memories must be retrieved in order to become once again susceptible to disruption with amnestic agents (Lewis, 1979; Nader, 2003). We therefore propose that the processes of memory retrieval and memory destabilization are doubly dissociable, but that behavioral procedures used to induce memory destabilization often induce memory retrieval as well. This hypothesis clearly warrants further investigation.
As predicted, the LY317206-induced reduction in glutamate release (Doherty et al., 1999) resulted in decreased activity at AMPARs, GluN2B-NMDARs and GluN2A-NMDARs and hence no observable effect on memory retrieval, destabilization or restabilization. Although this is consistent with the mechanisms shown in Figure 6, these data also indicate that such drugs are unlikely to have utility in the treatment of maladaptive memories that characterize some neuropsychiatric disorders. There has been interest in indirectly modulating signaling at NMDARs by reducing glutamate release presynaptically, since as has been noted previously (Milton and Everitt, 2010), systemic NMDAR antagonists are unlikely to be used clinically because of their problematic psychotomimetic side effects. However, our data do support the view that selectively targeting GluN2A-NMDARs may provide a useful therapeutic strategy; the psychotomimetic effects of systemic NMDAR antagonism are likely mediated through GluN2B-NMDARs (De Vry and Jentzsch, 2003) and so it may be possible to develop GluN2A-NMDAR selective therapies that are appropriate for clinical use.
The results of these experiments reveal the complexity of the glutamatergic mechanisms underlying CS-fear memory reconsolidation within the BLA. In summary, GluN2A-containing and GluN2B-containing NMDARs have dissociable roles in memory restabilization and destabilization, respectively. While AMPARs are required for memory retrieval, they do not appear necessary for memory destabilization, suggesting that these two processes are independently regulated. Furthermore, agonism at presynaptic mGlu2/3Rs has no overall effect on the strength of a CS-fear memory. In addition to further elucidating the glutamatergic mechanisms underlying the reconsolidation of fear memories, these data also further support the possible utility of modulating specific glutamate receptors in the clinical treatment of anxiety disorders in order to disrupt persistent maladaptive and intrusive memories.
Acknowledgements
This work was supported by a UK Medical Research Council Programme Grant (no. 9536855) to B.J.E. and was conducted in the Behavioural and Clinical Neuroscience Institute (BCNI), an initiative jointly funded by the MRC and the Wellcome Trust. A.L.M. was supported by a BCNI lectureship, E.M. was supported by a Royal Society Newton International Fellowship, and B.L.G. by a Prior Scholarship from St. John’s College, Cambridge. The authors would like to thank D. Theobald and A. Lyon for technical assistance.
Footnotes
Conflicts of Interest: The authors declare no competing financial interests.
References
- Auberson YP, Allgeier H, Bischoff S, Lingenhoehl K, Moretti R, Schmutz M. 5-phosphonomethylquinoxalinediones as competitive NMDA receptor antagonists with a preference for the human 1A/2A, rather than 1A/2B receptor composition. Bioorg Med Chem Lett. 2002;12:1099–1102. doi: 10.1016/s0960-894x(02)00074-4. [DOI] [PubMed] [Google Scholar]
- Bast T, da Silva BM, Morris RGM. Distinct contributions of hippocampal NMDA and AMPA receptors to encoding and retrieval of one-trial place memory. The Journal of Neuroscience. 2005;25:5845–5856. doi: 10.1523/JNEUROSCI.0698-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben Mamou C, Gamache K, Nader K. NMDA receptors are critical for unleashing consolidated auditory fear memories. Nat Neurosci. 2006;9:1237–1239. doi: 10.1038/nn1778. [DOI] [PubMed] [Google Scholar]
- Bingol B, Wang C-F, Arnott D, Cheng D, Peng J, Sheng M. Autophosphorylated CaMKIIα acts as a scaffold to recruit proteasomes to dendritic spines. Cell. 2010;140:567–578. doi: 10.1016/j.cell.2010.01.024. [DOI] [PubMed] [Google Scholar]
- Brown TE, Lee BR, Sorg BA. The NMDA antagonist MK-801 disrupts reconsolidation of a cocaine-associated memory for conditioned place preference but not for self-administration in rats. Learn Mem. 2008;15:857–865. doi: 10.1101/lm.1152808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campeau S, Davis M. Involvement of the central nucleus and basolateral complex of the amygdala in fear conditioning measured with fear-potentiated startle in rats trained concurrently with auditory and visual conditioned stimuli. The Journal of Neuroscience. 1995;15:2301–2311. doi: 10.1523/JNEUROSCI.15-03-02301.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardinal RN, Aitken MRF. ANOVA for the Behavioural Sciences Researcher. Lawrence Erlbaum Associates, Inc.; London: 2006. [Google Scholar]
- Dalton GL, Wu DC, Wang YT, Floresco SB, Phillips AG. NMDA GluN2A and GluN2B receptors play separate roles in teh induction of LTP and LTD in the amygdala and in the acquisition and extinction of conditioned fear. Neuropharmacology. 2012;62:797–806. doi: 10.1016/j.neuropharm.2011.09.001. [DOI] [PubMed] [Google Scholar]
- Davis S, Butcher SP, Morris RG. The NMDA antagonist D-2-amino-5-phosphonopentanoate (D-AP5) impairs spatial learning and LTP in vivo at intracerebral concentrations comparable to those that block LTP in vitro. The Journal of Neuroscience. 1992;12:21–34. doi: 10.1523/JNEUROSCI.12-01-00021.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day M, Langston R, Morris RGM. Glutamate-receptor-mediated encoding and retrieval of paired-associate learning. Nature. 2003;424:205–209. doi: 10.1038/nature01769. [DOI] [PubMed] [Google Scholar]
- De Vry J, Jentzsch KR. Role of the NMDA receptor NR2B subunit in the discriminative stimulus effects of ketamine. Behavioural Pharmacology. 2003;14:229–235. doi: 10.1097/00008877-200305000-00007. [DOI] [PubMed] [Google Scholar]
- Di Ciano P, Everitt BJ. Direct interactions between the basolateral amygdala and nucleus accumbens core underlie cocaine-seeking behavior by rats. The Journal of Neuroscience. 2004;24:7167–7173. doi: 10.1523/JNEUROSCI.1581-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dingledine R, Borges K, Bowie D, Traynelis SF. The glutamate receptor ion channels. Pharmacol Rev. 1999;51:7–61. [PubMed] [Google Scholar]
- Doherty AJ, Coutinho V, Collingridge GL, Henley JM. Rapid internalization and surface expression of a functional, fluorescently tagged G-protein-coupled glutamate receptor. Biochem J. 1999;341:415–422. [PMC free article] [PubMed] [Google Scholar]
- Fanselow MS, Kim JJ. Acquisition of contextual pavlovian fear conditioning is blocked by application of an NMDA receptor antagonist D,L-2-amino-5-phosphonovaleric acid to the basolateral amygdala. Behav Neurosci. 1994;108:210–212. doi: 10.1037//0735-7044.108.1.210. [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Fukunaga Y, Bading H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat Neurosci. 2002;5:405–414. doi: 10.1038/nn835. [DOI] [PubMed] [Google Scholar]
- Hu M, Sun Y-J, Zhou Q-G, Auberson YP, Chen L, Hu Y, Luo C-X, Wu J-Y, Zhu D-Y, Li L-X. Reduced spatial learning in mice treated with NVP-AAM077 through down-regulating neurogenesis. Eur J Pharmacol. 2009;622:37–44. doi: 10.1016/j.ejphar.2009.09.031. [DOI] [PubMed] [Google Scholar]
- Itzhak Y. Role of the NMDA receptor and nitric oxide in memory reconsolidation of cocaine-induced conditioned place preference in mice. Ann N Y Acad Sci. 2008;1139:350–357. doi: 10.1196/annals.1432.051. [DOI] [PubMed] [Google Scholar]
- Ivanov A, Pellegrino C, Rama S, Dumalska I, Salyha Y, Ben-Ari Y, Medina I. Opposing role of synaptic and extrasynaptic NMDA receptors in regulation of the extracellular signal-related kinases (ERK) activity in cultured rat hippocampal neurons. The Journal of Physiology. 2006;572:789–798. doi: 10.1113/jphysiol.2006.105510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Killcross S, Robbins TW, Everitt BJ. Different types of fear-conditioned behaviour mediated by separate nuclei within amygdala. Nature. 1997;388:377–380. doi: 10.1038/41097. [DOI] [PubMed] [Google Scholar]
- Kim MJ, Dunah AW, Wang YT, Sheng M. Differential roles of NR2A- and NR2B-containing NMDA receptors in Ras-ERK signaling and AMPA receptor trafficking. Neuron. 2005;46:745–760. doi: 10.1016/j.neuron.2005.04.031. [DOI] [PubMed] [Google Scholar]
- Kocsis B. Differential role of NR2A and NR2B subunits in N-methyl-D-aspartate receptor antagonist-induced aberrant cortical gamma oscillations. Biological Psychiatry. 2012;71:987–995. doi: 10.1016/j.biopsych.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo JW, Han JS, Kim JJ. Selective neurotoxic lesions of basolateral and central nuclei of the amygdala produce differential effects on fear conditioning. The Journal of Neuroscience. 2004;24:7654–7662. doi: 10.1523/JNEUROSCI.1644-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JLC, Everitt BJ. Appetitive memory reconsolidation depends upon NMDA receptor-mediated neurotransmission. Neurobiol Learn Mem. 2008;90:147–154. doi: 10.1016/j.nlm.2008.02.004. [DOI] [PubMed] [Google Scholar]
- Lee JLC, Milton AL, Everitt BJ. Reconsolidation and extinction of conditioned fear: inhibition and potentiation. The Journal of Neuroscience. 2006;26:10051–10056. doi: 10.1523/JNEUROSCI.2466-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, Choi JH, Lee N, Lee HR, Kim JI, Yu NK, Choi SL, Kim H, Kaang BK. Synaptic protein degradation underlies destabilization of retrieved fear memory. Science. 2008;319:1253–1256. doi: 10.1126/science.1150541. [DOI] [PubMed] [Google Scholar]
- Lewis DJ. Psychobiology of active and inactive memory. Psychological Bulletin. 1979;86:1054–1083. [PubMed] [Google Scholar]
- Liu L, Wong TP, Pozza MF, Lingenhoehl K, Wang Y, Sheng M, Auberson YP, Wang YT. Role of NMDA receptor subtypes in governing the direction of hippocampal synaptic plasticity. Science. 2004;304:1021–1024. doi: 10.1126/science.1096615. [DOI] [PubMed] [Google Scholar]
- Martel M-A, Ryan TJ, Bell KFS, Fowler JH, McMahon A, Al-Mubarak B, Komiyama NH, Horsburgh K, Kind PC, Grant SGN, Wyllie DJA, Hardingham GE. The subtype of GluN2 C-terminal domain determines the response to excitotoxic insults. Neuron. 2012;74:543–556. doi: 10.1016/j.neuron.2012.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milton AL, Everitt BJ. The psychological and neurochemical mechanisms of drug memory reconsolidation: implications for the treatment of addiction. Eur J Neurosci. 2010;31:2308–2319. doi: 10.1111/j.1460-9568.2010.07249.x. [DOI] [PubMed] [Google Scholar]
- Milton AL, Lee JLC, Butler VJ, Gardner RJ, Everitt BJ. Intra-amygdala and systemic antagonism of NMDA receptors prevents the reconsolidation of drug-associated memory and impairs subsequently both novel and previously acquired drug-seeking behaviors. The Journal of Neuroscience. 2008;28:8230–8237. doi: 10.1523/JNEUROSCI.1723-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milton AL, Schramm MJW, Wawrzynski J, Gore F, Oikonomou-Mpegeti F, Wang NQ, Samuel D, Economidou D, Everitt BJ. Antagonism at NMDA receptors, but not β-adrenergic receptors, disrupts the reconsolidation of pavlovian conditioned approach and instrumental transfer for ethanol-associated conditioned stimuli. Psychopharmacology. 2012;219:751–761. doi: 10.1007/s00213-011-2399-9. [DOI] [PubMed] [Google Scholar]
- Morris RG, Anderson E, Lynch GS, Baudry M. Selective impairment of learning and blockade of long-term potentiation by an N -methyl-D-aspartate receptor antagonist, AP5. Nature. 1986;319:774–776. doi: 10.1038/319774a0. [DOI] [PubMed] [Google Scholar]
- Müller T, Albrecht D, Gebhardt C. Both NR2A and NR2B subunits of the NMDA receptor are critical for long-term potentiation and long-term depression in the lateral amygdala of horizontal slices of adult mice. Learn Mem. 2009;16:395–405. doi: 10.1101/lm.1398709. [DOI] [PubMed] [Google Scholar]
- Nader K. Memory traces unbound. Trends Neurosci. 2003;26:65–72. doi: 10.1016/S0166-2236(02)00042-5. [DOI] [PubMed] [Google Scholar]
- Nader K, Schafe GE, Le Doux JE. Fear memories require protein synthesis in the amygdala for reconsolidation after retrieval. Nature. 2000;406:722–726. doi: 10.1038/35021052. [DOI] [PubMed] [Google Scholar]
- Ornstein PL, Arnold MB, Augenstein NK, Lodge D, Leander JD, Schoepp DD. (3SR,4aRS,6RS,8aRS)-6-[2-(1H-tetrazol-5-yl)ethyl]decahydroisoquinoline-3-carboxylic acid: a structurally novel, systemically active, competitive AMPA receptor antagonist. J Med Chem. 1993;36:2046–2048. doi: 10.1021/jm00066a016. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The Rat Brain in Stereotaxic Co-ordinates. 4th Ed. Academic Press; New York: 2004. [Google Scholar]
- Pedreira ME, Pérez-Cuesta L, Maldonado H. Reactivation and reconsolidation of long-term memory in the crab Chasmagnathus: protein synthesis requirement and mediation by NMDA-type glutamatergic receptors. The Journal of Neuroscience. 2002;22:8305–8311. doi: 10.1523/JNEUROSCI.22-18-08305.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Przybyslawski J, Sara SJ. Reconsolidation of memory after its reactivation. Behav Brain Res. 1997;84:241–246. doi: 10.1016/s0166-4328(96)00153-2. [DOI] [PubMed] [Google Scholar]
- Sadler R, Herzig V, Schmidt WJ. Repeated treatment with the NMDA antagonist MK-801 disrupts reconsolidation of memory for amphetamine-conditioned place preference. Behavioural Pharmacology. 2007;18:699–703. doi: 10.1097/FBP.0b013e3282effb81. [DOI] [PubMed] [Google Scholar]
- von der Goltz C, Vengeliene V, Bilbao A, Perreau-Lenz S, Pawlak CR, Kiefer F, Spanagel R. Cue-induced alcohol seeking behaviour is reduced by disrupting the reconsolidation of alcohol-related memories. Psychopharmacology. 2009;205:389–397. doi: 10.1007/s00213-009-1544-1. [DOI] [PubMed] [Google Scholar]
- Walker DL, Davis M. Amygdala infusions of an NR2B-selective or an NR2A-preferring NMDA receptor antagonist differentially influence fear conditioning and expression in the fear-potentiated startle test. Learn Mem. 2008;15:67–74. doi: 10.1101/lm.798908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker DL, Rattiner LM, Davis M. Group II metabotropic glutamate receptors within the amygdala regulate fear as assessed with potentiated startle in rats. Behav Neurosci. 2002;116:1075–1083. doi: 10.1037//0735-7044.116.6.1075. [DOI] [PubMed] [Google Scholar]
- Williams K. Ifenprodil discriminates subtypes of the N-methyl-D-aspartate receptor: selectivity and mechanisms at recombinant heteromeric receptors. Mol Pharmacol. 1993;44:851–859. [PubMed] [Google Scholar]
- Winters BD, Bussey TJ. Glutamate receptors in perirhinal cortex mediate encoding, retrieval and consolidation of object recognition memory. The Journal of Neuroscience. 2005;25:4243–4251. doi: 10.1523/JNEUROSCI.0480-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X-H, Liu F, Chen Q, Zhang C-L, Zhuo M, Xiong Z-Q, Li B-M. Conditioning-strength dependent involvement of NMDA NR2B subtype receptor in the basolateral nucleus of amygdala in acquisition of auditory fear memory. Neuropharmacology. 2008;55:238–246. doi: 10.1016/j.neuropharm.2008.05.030. [DOI] [PubMed] [Google Scholar]