

Abstract
Chemokine-directed leukocyte migration is a critical component of all innate and adaptive immune responses. The atypical chemokine receptor ACKR2 is expressed by lymphatic endothelial cells and scavenges pro-inflammatory CC chemokines to indirectly subdue leukocyte migration. This contributes to the resolution of acute inflammatory responses in vivo. ACKR2 is also universally expressed by innate-like B cells, suppressing their responsiveness to the non-ACKR2 ligand CXCL13, and controlling their distribution in vivo. The role of ACKR2 in autoimmunity remains relatively unexplored, although Ackr2 deficiency reportedly lessens the clinical symptoms of experimental autoimmune encephalomyelitis induced by immunization with encephalogenic peptide (MOG35-55). This was attributed to poor T cell priming stemming from the defective departure of dendritic cells from the site of immunization. However, we report here that Ackr2-deficient mice, on two separate genetic backgrounds, are not less susceptible to autoimmunity induced by immunization, and in some cases develop enhanced clinical symptoms. Moreover, ACKR2 deficiency does not suppress T cell priming in response to encephalogenic peptide (MOG35-55), and responses to protein antigen (collagen or MOG1-125) are characterized by elevated IL-17 production. Interestingly, after immunization with protein, but not peptide, antigen, Ackr2 deficiency was also associated with an increase in lymph node B cells expressing GM-CSF, a cytokine that enhances Th17 cell development and survival. Thus, Ackr2 deficiency does not suppress autoreactive T cell priming and autoimmune pathology, but can enhance T cell polarization toward Th17 cells and increase the abundance of GM-CSF+ B cells in lymph nodes draining the site of immunization.
Keywords: Autoimmunity, Animal models, Rheumatoid Arthritis, Multiple Sclerosis, Chemokines, Th17
INTRODUCTION
Chemokines play a major role in orchestrating innate and adaptive immune responses by controlling the migration of leukocytes using G-protein coupled chemokine receptors that decorate the surface of these cells1. Alongside the large chemokine receptor family is a small subfamily of ‘atypical’ chemokine receptors, members of which bind chemokines with high affinity and specificity but appear incapable of classical chemokine receptor behavior2. This subfamily is typified by ACKR2 (D6)3 a heptahelical membrane molecule structurally related to other chemokine receptors that binds a broad array of pro-inflammatory CC chemokines. In humans, ACKR2 is expressed by lymphatic endothelial cells (LEC), trophoblasts, and some leukocyte populations4-8. In mice, we have recently found that, amongst leukocytes, ACKR2 is highly restricted to innate-like B cells (IBC) (i.e. marginal zone (MZ) and B1 B cells), and is the best unifying marker of these cells9. IBC serve key roles during homeostasis, autoimmunity and infection, and new properties of these cells continue to be defined. For example, recent work has revealed that B1 B cells generate ‘innate response activator’ (IRA) B cells during inflammation that are dominant sources of the cytokine GM-CSF in secondary lymphoid tissue10.
What sets ACKR2 and other atypical chemokine receptors apart is their inability to couple to signaling pathways activated after classical chemokine receptor engagement. Neither ACKR2-transfected cell lines nor primary ACKR2-expressing leukocytes migrate towards ACKR2 ligands2,9. This, coupled with the ability of ACKR2 to continuously internalize chemokines11-15, supports the concept that the principal function of ACKR2 is to act as a ‘professional’ chemokine scavenger that indirectly modulates leukocyte migration through chemokine removal. This model is used to explain phenotypes in challenged Ackr2-deficient mice, which are often characterized by elevated chemokine abundance, exaggerated inflammation, and increased immunopathology2,5,6,16-21. However, Ackr2 deficiency also leads to cell-autonomous defects amongst IBC (e.g. increased responsiveness to the non-ACKR2 ligand CXCL139), that are not dependent on loss of chemokine scavenging and could be linked to the ability of ACKR2 to regulate the subcellular distribution of β-arrestins, key regulators of G protein-coupled receptors like CXCR514,15. B1 cell distribution in vivo is profoundly dependent on engagement of CXCR5 by its ligand CXCL1322, and Ackr2-deficient mice have fewer B1 B cells in their peritoneal cavity, omentum and spleen than WT animals9. It is not clear if this is due to the loss or redistribution of these cells, and the absence of suitable markers makes it difficult to distinguish B1 B cells from follicular B cells in other tissues, such as lymph nodes (LN).
Chemokines are capable of influencing all the key steps that lead to the development of pathology in mouse models of autoimmunity, such as inflammation at the site of immunization, delivery of antigen to draining LN, development of pathogenic lymphocytes and antibodies, and the orchestration of immunopathology in the target tissue23,24. Identifying how chemokine receptors contribute to the induction and maintenance of immunopathology has implications for the translation of chemokine receptor inhibition to the treatment of human disease25. Only one study has examined the impact of Ackr2 deficiency in a model of autoimmune disease26, specifically experimental autoimmune encephalomyelitis (EAE) induced by immunization with a short peptide from rat myelin oligodendrocyte glycoprotein (MOG), referred to hereafter as MOG35-55. This study reported that, in contrast to the exaggerated inflammation seen in the absence of Ackr2 in most other models, C57BL/6J Ackr2-deficient mice developed less brain inflammation and clinical symptoms of EAE than wild-type (WT) counterparts. This was attributed to the suppression of dendritic cell (DC) migration caused by excessive inflammation at the immunization site, and they reported a profound reduction in the proliferation of LN cells re-stimulated with MOG35-55 and reduced IFNγ release26. Indeed, we have subsequently shown that, during inflammation, the absence of Ackr2 is associated with the deposition of chemokines on skin LEC; peri-lymphatic accumulation of inflammatory leukocytes, including DCs; and concomitant ‘lymphatic congestion’27.
Here, using mice on two different genetic backgrounds, we report a detailed evaluation of the impact of Ackr2 deficiency in four models of autoimmune disease i.e. collagen-induced arthritis (CIA), collagen antibody-induced arthritis (CAIA) and EAE induced by immunization with MOG35-55 peptide or MOG1-125 protein. In none of these models did the absence of Ackr2 decrease the severity of disease, and in some cases Ackr2-deficient mice developed worse clinical symptoms than WT animals. Moreover, T cell priming was not diminished in Ackr2-deficient mice, and WT and Ackr2-deficient LN cells had comparable proliferation when re-stimulated with antigen. In fact, compared to WT, cells from the LN of Ackr2-deficient mice draining the site of immunization with protein (collagen or MOG1-125) but not peptide (MOG35-55) antigen showed an increased propensity to produce IL-17. Importantly, this was not an intrinsic T cell phenomenon as chimeric studies demonstrated that WT and Ackr2-deficient T cells differentiated equally into Th17 cells when in the same animal. Interestingly, enhanced Th17 responses were associated with an increase in the abundance of B cells producing GM-CSF, a cytokine known to enhance Th17 cell development and survival28. Thus, in the models we have tested, Ackr2-deficient mice are not less susceptible to autoimmunity and do not show suppressed T cell responses, but can develop enhanced Th17 responses and greater numbers of GM-CSF+ B cells after immunization with protein autoantigen.
RESULTS
Ackr2 is up-regulated in arthritic mouse joints and suppresses the severity of CIA in DBA1/j mice
By comparing healthy and arthritic knees from WT DBA1/j mice, we found that Ackr2 transcripts were significantly up-regulated in the target tissue of inflammatory arthritis (Figure 1A). We considered whether loss of the anti-inflammatory activity of ACKR2 at this site might have a more pronounced effect on the development of autoimmune disease than it is reported to have in the brain26. To explore this, we backcrossed Ackr2-deficient C57BL/6J mice onto DBA/1j and monitored development of arthritis in a large cohort of animals after immunization with bovine type II collagen, using WT DBA1/j littermate mice as controls. In contrast to previous observations in the EAE model26, the absence of Ackr2 resulted in a statistically significant increase in the clinical symptoms of arthritis (Figure 1B), and a substantial increase (p<0.05) in the cumulative clinical score of Ackr2-deficient (24.2 ± 3.6, mean ± SD) compared to WT mice (13.7 ± 2.6). There was, however, no change in the time at which these animals first developed symptoms (i.e. incidence of disease) (data not shown). Ackr2-deficient mice often have elevated levels of ACKR2-binding chemokines in inflamed tissues16,17,20. Consistent with this, 2-3-fold more ACKR2-binding chemokines CCL2, CCL3 and CCL4, but not non-ACKR2-binding chemokine CXCL10, were released from explants of patella and adjacent synovial tissue from arthritic Ackr2-deficient mice, compared with equivalent preparations from arthritic WT animals (Figure 1C). Furthermore, a corresponding increase in the release of TNFα was observed in the Ackr2-deficient patella and adjacent synovial tissue explants, whilst the levels of IL-10 were unchanged (Figure 1C). Histological analysis revealed a trend towards an increase in pathological changes in the inflamed joints in Ackr2-deficient mice, but this failed to achieve statistical significance (Figure 1D). Thus, ACKR2 is locally up-regulated in response to the development of inflammation in the mouse joint, controls the levels of inflammatory CC chemokines in the diseased tissue, and suppresses the clinical symptoms of arthritis.
Figure 1. ACKR2 regulates chemokine abundance in arthritic joints and suppresses disease severity in CIA.

(A) RNA was extracted from the knees of WT DBA/1j mice with established CIA (day 35) or age- and sex-matched naïve littermate control mice (n = 6). Q-RT-PCR was performed to evaluate the level of Ackr2 expression. Ackr2 gene expression was normalized to GAPDH and the fold-change in inflamed tissue calculated relative to naïve tissue (set to 1). (B-D) CIA was induced in wild-type littermate control (WT) and Ackr2-deficient (Ackr2−/−) DBA/1j mice with 100μg of Bovine CII in complete Freund’s adjuvant. (B) Clinical score, with each point representing the pooled mean ± SEM scores of three experiments using n = 29 mice of each genotype. Significance was determined by two-way repeated measures ANOVA with Bonferroni post-tests. *, p < 0.05 **, p < 0.01. (C) Knee patella and adjacent synovial tissue were harvested (n = 6-8), cultured ex vivo for 4h and the level of secreted chemokine and cytokines determined by ELISA. *, p <0.05; **, p<0.01 (Mann Whitney test). (D) Histological scores. H&E-stained tissue sections were scored in a blinded manner for hyperplasia, infiltration and erosion. Bars show mean ± SD of 5-6 per group.
Loss of ACKR2 does not alter anti-collagen antibody levels in CIA or the development of antibody-driven inflammatory arthritis
Loss of ACKR2 did not affect the level of subclass specific anti-CII antibodies produced during CIA (Figure 2A). However, we thought that the increased clinical disease in Ackr2-deficient mice in CIA might be the result of an altered innate response in the joint to these pathogenic antibodies. To investigate this directly, we examined the impact of Ackr2 deficiency on the development of anti-collagen antibody-induced arthritis. Pathology in this model relies heavily on innate effector mechanisms involving neutrophils, macrophages, Fc receptors, complement, and inflammatory chemokines and cytokines 29. However, loss of ACKR2 had no impact on the development or maintenance of arthritic disease in this model (Figure 2B). Thus, the enhanced disease observed in Ackr2-deficient mice in the CIA model was unlikely to be the result of exaggerated responses to anti-collagen antibodies.
Figure 2. Ackr2 deficiency has no impact on the generation of anti-collagen antibodies in CIA and or on antibody-induced arthritis.
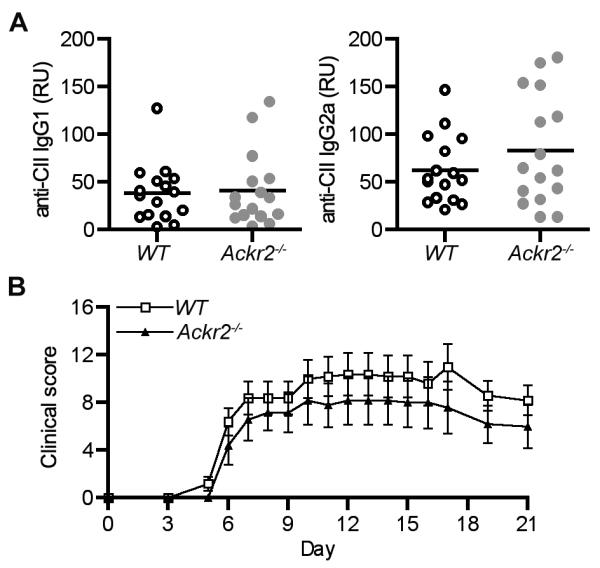
(A) Serum levels of anti-collagen mouse IgG1 and IgG2a in arthritic wild-type littermate control (WT) and Ackr2-deficient (Ackr2−/−) DBA/1j mice (n = 16) 35 days after the first collagen immunization. (B) CAIA was induced in wild-type (WT) and Ackr2-deficient (Ackr2−/−) DBA/1j mice with each animal assessed every 1-2 days for clinical symptoms of arthritis. Each point represents the mean ± SEM scores of n = 5 mice. Significance was determined by two-way repeated measures ANOVA.
Increased IL-17 production in LNs draining arthritic joints in Ackr2-deficient mice
Next, we examined whether there was any evidence of altered T cell responses in arthritic Ackr2-deficient mice by analyzing inguinal LNs draining the arthritic hind legs of WT and Ackr2-deficient mice 35 days after the induction of disease. Ackr2 deficiency had no impact on the cellularity of these LNs in resting mice (data not shown), but more cells were retrieved from the inflamed inguinal LNs of arthritic Ackr2-deficient mice than from WT counterparts (Figure 3A). There was no difference in the proliferative response of these cells to CII stimulation (Figure 3B), but, compared with WT, Ackr2-deficient cells released ~5-fold more IL-17 into the medium after CII stimulation (Figure 3C). In contrast, there was no difference in CII-induced TNFα or IL-10 production by LN cells harvested from WT and Ackr2-deficient mice (Figure 3D and E). However, Ackr2-deficient cells produced significantly more GM-CSF after stimulation with CII (Figure 3F), and interestingly, in the CII stimulated Ackr2-deficient cultures, the level of IL-17 significantly correlated with GM-CSF abundance (r2 = 0.72, p = 0.03) (Figure 3G). Moreover, compared to WT, the inguinal lymph nodes draining the inflamed joints of Ackr2-deficient mice showed a small, but statistically significant, increase in the proportion of CD4+ T cells capable of producing IL-17 after stimulation in vitro with PMA/ionomycin, indicative of an increased generation of Th17 cells in these mice (Figure 3H). This bias was restricted to CD4+ IL-17-producing cells as no significant differences were observed in the proportion of IL-17+ CD8+ or γδ T cells (data not shown and Supplemental Figure 1). Thus, Ackr2-deficient mice developed an effective T cell response to CII, and, by day 35, their CD4+ T cell population had a significantly greater Th17 component.
Figure 3. Arthritic Ackr2-deficient mice have enlarged draining lymph nodes capable of greater collagen-induced IL-17 production.

Draining lymph nodes (n = 6-10) were collected from wild-type (WT) and Ackr2-deficient (Ackr2−/−) DBA/1j mice 35 days after collagen immunization. (A) Number of lymph node cells retrieved. (B-G) Draining lymph node cells (3×105 cells/well) were stimulated ex vivo with or without antigen (CII, 60μg/ml). (B) Ag-induced proliferation (i.e. CII minus medium alone), as determined by incorporation of tritiated thymidine ([H3]-TdR). (C-F) Concentration of IL-17, TNFα, IL-10 and GM-CSF in the medium after stimulation with antigen (CII) or medium alone (m). Significance was determined by one-way ANOVA with Bonferroni multiple comparison post-tests. *, p < 0.05 **, p < 0.01. (G) Correlation between the concentration of GM-CSF and IL-17. Significance was determined by Spearman’s rho test. (H) Representative results from IL-17 intra-cellular flow cytometry and a bar graph showing the percentage of lymph node cells that were CD4+IL17+ cells. A repeat experiment yielded similar results. Significance was determined by t-test. *, p < 0.05.
ACKR2 suppresses the development of Th17 cells during the initiation of the arthritogenic immune response
The resistance of Ackr2-deficient mice to EAE was ascribed to a defect in T cell priming 26, but our data showed that there was a plentiful supply of collagen-specific lymphocytes in arthritic Ackr2-deficient mice. This suggested that loss of ACKR2 might not suppress the priming of antigen-specific T cells in the CIA model. To specifically evaluate this, WT and Ackr2-deficient DBA1/j mice were immunized with CII in complete Freund’s adjuvant and draining LNs were harvested on day 8, the time point selected in previous work after immunization with MOG35-55 26. However, in marked contrast to the poor T cell responses reportedly elicited by MOG35-5526, proliferation and IFNγ secretion of antigen-stimulated lymphocytes were not suppressed in Ackr2-deficient LNs (Figure 4A-B). In fact, and in line with the data from the arthritic mice (Figure 3), a significant increase in the production of IL-17 was observed after stimulation of Ackr2-deficient lymphocytes with CII, and, strikingly, this cytokine was not detectably induced when WT lymphocytes were used (Figure 4C). Thus, loss of ACKR2 does not impair the ability to detect and respond to antigenic challenge in this model, and it actually enhances the initial Th17 response.
Figure 4. Ackr2-deficiency leads to an increased Th17 response during the induction of arthritis.
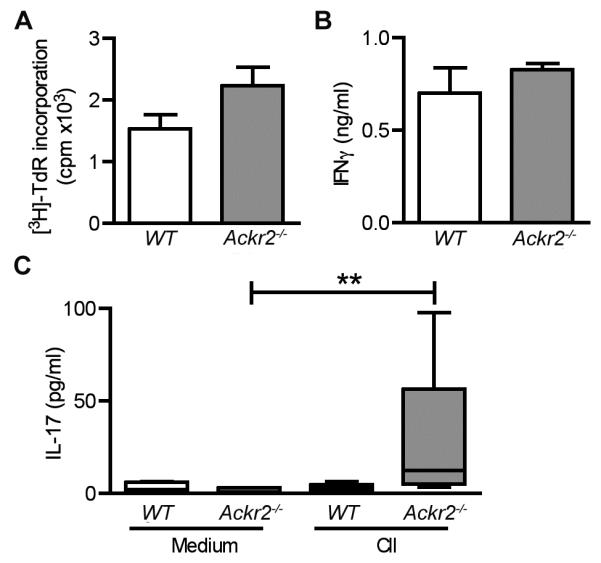
Eight days after challenge with CII, draining lymph nodes were harvested from WT littermate control (WT) and Ackr2-deficient (Ackr2−/−) DBA/1j mice (n = 5) and the recall immune response evaluated. Single cell suspensions of lymph node cells (3×105 cells/well) were stimulated with CII (60μg/ml) or medium alone and assessed for (A) specific proliferation (i.e. proliferation after CII stimulation minus proliferation in medium alone) or (B-C) the levels of IFNγ and IL-17 in the medium. **, p < 0.01 by a Kruskal-Wallis test with Dunn’s multiple comparison post-test. A repeat experiment yielded similar results.
MOG35-55-induced EAE is not suppressed by the absence of ACKR2
The absence of suppressed T cell priming after immunization with collagen led us to re-evaluate the role ACKR2 during EAE induction in C57BL/6J mice. In stark contrast to the previous work 26, we found that Ackr2 deficiency offered no protection against disease after immunization with MOG35-55 peptide (Figure 5A). We were also unable to observe a difference in the accumulation of DCs at the site of immunization in Ackr2-deficient mice 3 days after injection (Figure 5B), although in the draining lymph node at this time there was a significant decrease in the number of CD207+ EPCAM+ migratory DCs in Ackr2-deficient mice (Figure 5C), which was also evident at day 21 (data not shown). Nonetheless, no differences were detected in lymphocyte proliferation on day 11 or day 21 when the Ackr2-deficient and WT inguinal LN cells were stimulated ex vivo with MOG35-55 (Figure 5D and data not shown). Importantly, in this setting Ackr2-deficient cells did not produce more IL-17 than their WT counterparts (Figure 5E). Thus, although loss of ACKR2 did reduce the number of migratory DCs in the LN draining the site of MOG35-55 immunization, it did not hamper the ability of T cells to respond to antigenic challenge, and, in contrast to immunization with CII protein, there is no evidence of an enhanced Th17 response.
Figure 5. Ackr2 deficiency does not impair disease severity in the MOG35-55 peptide induced model of EAE or result in increased IL-17 production.

EAE was induced in WT littermate control (WT) and Ackr2-deficient C57BL/6 mice (Ackr2−/−) by immunization with MOG35-55 (100μg) in complete Freund’s adjuvant (4mg/ml Mycobacterium tuberculosis H37RA) and treatment on days 0 and 2 with 200ng of PTX. (A) All animals were assessed daily for the development of clinical symptoms of disease. Each point represents the mean ± SEM scores for n = 5 mice. A repeat experiment yielded similar results. (B) Injection site skin was harvested on day 3 and the number of CD207+ DCs in 10 high power fields (HPF) per mouse determined. (C-E) Draining lymph nodes were harvested on day 3 or day 11 and single cell suspensions were evaluated on for (C) the absolute number of CD207+EPCAM+ DCs (day 3) or (D-E) 3×105 cells/well stimulated with MOG35-55 (30μg/ml) (day 11) and assessed for (D) specific proliferation (i.e. proliferation after MOG35-55 stimulation minus proliferation in medium alone) or (E) the level of IL-17 in the medium. Significance was determined by t-test. *, p < 0.05.
MOG1-125-induced EAE is not suppressed by the absence of ACKR2 but Th17 responses are enhanced
Ackr2 deficiency did not affect the Th17 response in the peptide-driven C57BL/6 model of EAE, but enhanced this response in the DBA1/j collagen-driven model of CIA. This led us to question the role of ACKR2 and/or genetic background in protein-driven priming of T cell responses. As above, when EAE was induced in WT and Ackr2-deficient mice on the C57BL/6 or DBA1/j strain using MOG1-125 protein, Ackr2 deficiency did not offer any protection against disease (Figure 6A & B). In fact, there was a small, but statistically significant, increase in the clinical symptoms of disease in Ackr2-deficient DBA1/j mice 11 days after immunization (Figure 6B), although histological examination of the brains of these animals failed to reveal any change in leukocyte infiltrate (data not shown). The severe EAE induced in Ackr2-deficient DBA1/j mice by MOG1-125 meant that, under the terms of our licence, animals had to be euthanized on day 11. As in the other models, no differences were detected in lymphocyte proliferation when Ackr2-deficient and WT inguinal LN cells were stimulated ex vivo with MOG1-125, and IFNγ production was also unaffected (data not shown) but notably Ackr2-deficient cells from both C57BL/6 and DBA1/j mice produced significantly more IL-17 than their WT counterparts (Figure 6C & D). In further experiments, MOG1-125-immunized WT and Ackr2-deficient C57BL/6 mice were treated with PTX on day 0 and 2 (Supplemental Figure 2). As before, these animals developed comparable clinical symptoms and elevated Th17 responses. Thus, loss of ACKR2 does not suppress the pathogenicity of protein auto-antigens (collagen and MOG1-125) and is consistently associated with enhanced Th17 responses, regardless of genetic background.
Figure 6. Ackr2 deficiency leads to increased IL-17 production after the induction of EAE in C57BL/6 and DBA/1j mice, and has minimal effect on disease severity.

(A-D) EAE was induced in WT littermate control (WT) and Ackr2-deficient (Ackr2−/−) C57BL/6 (A & C) or DBA/1j (B & D) mice by immunization with MOG1-125 protein (50μg in 3mg/ml Mycobacterium tuberculosis H37RA CFA). (A and B) Animals evaluated daily for clinical symptoms of disease. In A, each point represents the means ± SEM (n = 5 mice per genotype). A repeat experiment generated a similar result. In B, the pooled mean ± SEM scores of two experiments are shown (n = 10-11 mice per genotype). Significance was determined by two-way repeated measures ANOVA with Bonferroni post-tests. ***, p < 0.001. (C and D) 21 (n = 5) (C) or 11 days (n = 10-11) (D) after immunization, 3×105 cells/well from lymph nodes draining the site of immunization were stimulated ex vivo with MOG1-125 (60μg/ml) or medium alone and the concentration of IL-17 in the medium was determined. *, p < 0.05 by a Mann Whitney test. (E-F) Mixed bone marrow chimeric mice (WT:Ackr2−/−) were immunized with MOG1-125 protein (50μg in 3mg/ml Mycobacterium tuberculosis H37RA CFA), draining inguinal LNs were harvested on day 15, and the number of CD4+TCRβ+CD45.1+ (WT) or CD4+TCRβ+CD45.1− (Ackr2−/−) IL-17A+ and IFNγ+ T cells determined. (E) Bar graphs showing the percentage of CD4+TCRβ+ cells that were IL17+ or IFNγ+. (F) Representative flow cytometry plots pre-gated on live singlet CD4+TCRβ+CD45.1+ (WT) or CD4+TCRβ+CD45.1− (Ackr2−/−).
Enhanced Th17 responses in Ackr2-deficient mice are not due to intrinsic T cell defects
Although there is no evidence that T cells express ACKR2 protein in mice9, transcripts encoding ACKR2 can be detected in mouse T cells activated in culture, and in samples of mouse secondary lymphoid tissues that have been enriched for T cells8. It was possible therefore that Ackr2 expression by T cells played a role in regulating the generation of Th17 cells. To examine this, we generated chimeric mice carrying both WT and Ackr2-deficient hematopoietic cells to allow us to evaluate whether WT and Ackr2-deficient T cells activated in the same host differed in their ability to generate Th17 cells. These animals were immunized with MOG1-125 protein, and Th1 and Th17 cells identified in the WT and Ackr2-deficient CD4+ T cell populations in the inguinal LN by using intracellular cytokine staining to identify T cells capable of producing IFNγ or IL-17, respectively. Under these circumstances, there was no difference in the percentage of WT and Ackr2-deficient T cells that could be classified as Th1 or Th17 cells (Figure 6E-F). Thus, ACKR2 in T cells does not detectably influence their ability to develop into Th17 cells.
Enhanced Th17 responses in Ackr2-deficient mice are associated with increased numbers of GM-CSF+ B cells in draining LN
ACKR2 is expressed by IBCs, including rare B1 cells in LN, and the B1 B cell compartment is perturbed in resting Ackr2-deficient mice9. B1a B cells have been shown to give rise to a population of cells in the spleen during inflammation that are a dominant source of GM-CSF10. GM-CSF, via IL-6 and IL-23, enhances Th17 cell development and survival28. Thus, we considered whether during the initiation of an immune response, GM-CSF+ B cells were present in draining LN, and whether their abundance was affected by Ackr2 deficiency. LN were harvested 8 days after immunization of WT and Ackr2-deficient C57BL/6 mice with MOG35-55 or MOG 1-125, and GM-CSF expression assessed by intracellular cytokine staining (Figure 7 and data not shown). Small numbers of GM-CSF+ B cells were detected in the LN of WT mice, and the number retrieved was similar irrespective of whether they had been immunized with peptide (66107 ± 27981 GM-CSF+ B cells; mean ±SD) or protein (79406 ±18994 GM-CSF+ B cells; mean ±SD). Interestingly, however, draining LN from Ackr2-deficient mice immunized with MOG1-125 protein contained significantly more GM-CSF+ B cells than their WT counterparts, while no increase was observed when MOG35-55 peptide was used. No differences were detected in the levels of GM-CSF+ non-B cell populations in Ackr2-deficient and WT counterparts (Figure 7). Surface immunophenotyping revealed that these cells, whether WT or Ackr2-deficient, were mostly CD138− CD43− IgMhi IgDhi, although approximately a quarter had much lower levels of surface IgM (data not shown). Thus, enhanced Th17 responses of Ackr2-deficient mice are associated with an increase in the number of B cells making GM-CSF, a cytokine that controls the development and survival of Th17 cells.
Figure 7. Ackr2 deficiency leads to increased numbers of GM-CSF+ B cells in draining lymph nodes after immunization with MOG1-125 protein.
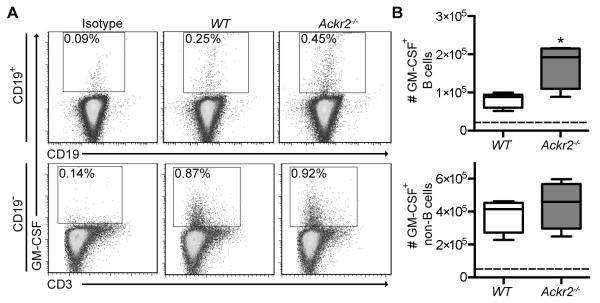
EAE was induced in WT littermate control (WT) and Ackr2-deficient (Ackr2−/−) C57BL/6 mice (n = 4) by immunization with MOG1-125 protein (50μg in 3mg/ml Mycobacterium tuberculosis H37RA CFA), draining lymph nodes harvested on day 8, and the number of GM-CSF+ CD19+ B cells determined. (A) Representative flow cytometry plots, which were pre-gated on live IgD+IgM+ singlets (top panels) and live IgD−IgM−CD19− singlets (bottom panels), and the percentage of GM-CSF+ CD19+ B cells and GM-CSF+ CD19− non-B cells respectively are shown. (B) Box & whisker plots, where the boxes represent the 25th to 75th percentiles, the lines within the boxes represent the median, and the lines outside the boxes represent the 5th and 95th percentiles, show the absolute number of GM-CSF+ B cells (top plot) and GM-CSF+ non-B cells (bottom plot) in the draining LN. The dotted line represents the isotype control level. A repeat experiment yielded similar results. *, p < 0.05 by a Mann Whitney test.
DISCUSSION
In the models we have examined, and using mice on two different genetic backgrounds, we find that deletion of Ackr2 does not suppress T cell priming or provide protection against autoimmunity. It can however enhance Th17 responses and the abundance of GM-CSF+ B cells in LN after immunization with protein, but not peptide, autoantigen.
The responses we observed in WT and Ackr2-deficient C57BL/6j mice after immunization with MOG35-55 clearly differ from those reported by Liu and colleagues26, even though very similar immunization strategies were used. In their study, the reduced clinical and histopathological signs of disease seen in Ackr2-deficient mice from day 14 after immunization with MOG35-55 were attributed to dysregulated inflammation at the site of immunization causing defective DC migration from the skin26. We have found that Ackr2 deficiency can lead to ‘lymphatic congestion’ and the impaired movement of antigen-presenting cells from the skin27, and shown here that DC migration to draining LNs is attenuated in Ackr2-deficient mice after immunization with MOG35-55. Thus, ACKR2 does allow optimal DC trafficking to LN from inflamed skin. Where our studies differ is in the impact of Ackr2 deficiency on T cell priming and disease course. Liu and colleagues reported that LN cells draining the site of immunization in Ackr2-deficient mice were virtually unable to proliferate after re-stimulation with antigen ex vivo, in stark contrast to the strong responses seen when LN cells from immunized WT mice were used. We saw robust proliferative responses in Ackr2-deficient mice after immunization with MOG35-55 (or, indeed, collagen or MOG1-125) and these responses were no weaker than those seen in WT controls. While EAE was successfully induced in both studies, we failed to see the divergence in the clinical scores of WT and Ackr2-deficient C57BL/6j mice 14 days after immunization with MOG35-55 that was reported in the previous work. This was also the case after immunization of WT and Ackr2-deficient C57BL/6j mice with MOG1-125. There was in fact a small increase in EAE score when Ackr2-deficient mice on a DBA/1j background were compared to WT DBA/1j mice, and Ackr2-deficient DBA/1j mice also developed worse arthritis than WT animals during CIA. We have yet to resolve the differences between our study and those of Liu and colleagues, but our findings clearly challenge the notion that ACKR2 facilitates the development of pathogenic T cells in mouse models of autoimmune disease.
Ackr2 expression was up-regulated in the knees or CNS of DBA1/j mice with CIA or EAE, respectively (Figure 1 and data not shown), perhaps in an effort to counteract ongoing inflammation. The source of this Ackr2 is not clear and there are no effective anti-ACKR2 antibodies available with which to identify and localize ACKR2-expressing cells by immunohistochemistry in mouse tissues. However, since ACKR2 is found in subsets of leukocytes, LECs, and epithelial cells in resting and inflamed human tissues, including those affected by autoimmune disease4,8,30,31, it seems likely that these cell types are responsible for Ackr2 expression in inflamed tissues in mice. However, it remains unclear whether the enhanced Ackr2 expression seen during autoimmune inflammation is due to either its presence on infiltrating leukocytes or its induction on resident cells by signals that are still to be defined. Synovial explants from arthritic Ackr2-deficient DBA1/j mice released more ACKR2-binding chemokine (CCL2, 3 and 4) than WT counterparts, while levels of the non-ACKR2 ligand CXCL10 were unaffected. These data suggest that loss of ACKR2-mediated chemokine scavenging in the target tissue might contribute to the enhanced clinical symptoms seen in Ackr2-deficient DBA1/j mice in CIA. However, the enhanced Th17 responses alone could be responsible, and this is supported by the fact that clinical symptoms of disease are unaffected by Ackr2 deficiency in models that exclusively examine the effector arm of disease development (i.e. anti-collagen antibody induced arthritis in DBA1/j mice; EAE in C57BL/6J mice driven by transfer of MOG35-55-primed T cells26). Thus, it is unlikely that ACKR2 in the joint or CNS plays any significant role in regulating the inflammation that develops in these tissues in the models that we have used. The amount of ACKR2 expressed at these sites may be insufficient to scavenge enough chemokine to exert any inhibitory effect on disease progression in the presence of autoreactive T cells and autoantibodies, and joints and brain are relatively poor sources of ACKR2 compared to skin, lung, gut and placenta where the effects of Ackr2 deficiency are most apparent2,5,6,16-21.
One consistent feature of our study was the enhanced Th17 responses seen in Ackr2-deficient mice after immunization with protein (collagen or MOG1-125) but not peptide (MOG35-55). The data from the analysis of mixed bone marrow chimeras, in which WT and Ackr2-deficient T cells developed equally well into Th17 cells in response to protein antigen, suggests that this was not due to an intrinsic T cell defect arising from loss of Ackr2. It may instead be due to differences in antigen presentation, and interestingly, peptide and protein can be presented by distinct APCs32,33. In general terms, peptides are most efficiently presented to naïve T cells by DCs, but proteins can be processed and presented by other cells, most notably B cells. Indeed, a dependence on B cell presentation for T cell priming has been demonstrated in protein-induced models of both arthritis and EAE34,35. Moreover, B cell depletion leads to diametrically opposed outcomes in the two modes of immunization; exacerbating peptide-driven EAE but suppressing protein-induced disease35. In the protein model, high titers of pathogenic MOG-specific antibody are generated and B cells are capable of efficiently priming encephalogenic Th1 and Th17 cells. In the peptide model, MOG-specific antibody is less important, DCs are thought to dominate T cell priming, and IL-10-producing regulatory B cells can suppress disease 36. Thus, in Ackr2-deficient mice immunized with MOG1-125 or collagen, it appears likely that B cell-mediated T cell priming overcomes any ‘lymphatic congestion’ or reduction in DC-mediated T cell priming that might conceivably be occurring in these animals.
An increased dependence on B cells for T cell priming in Ackr2-deficient mice might be responsible for the increase in IL-17-producing cells that we see in LN after their immunization with collagen or MOG1-125 protein. In this regard, it is intriguing that IBC (i.e. MZ and B1 B cells) are the principal leukocytes that express ACKR2 in mice9. These cells exhibit a number of properties that might contribute to the generation of Th17 responses. For example, compared to follicular B cells, antigen-loaded IBC are particularly adept at priming naïve T cells37, and B1 B cells are known to bias T cell differentiation towards a Th17 phenotype ex vivo38. Moreover, B1 B cells can give rise to GM-CSF+ B cells in the spleen10, and during inflammation these cells appear to be a dominant leukocytic source of this cytokine, which is known to enhance Th17 cell development and survival28. Our work now demonstrates that GM-CSF+ B cells are present in LNs draining immunized skin, and their abundance is regulated by ACKR2 after immunization with protein autoantigens. The precise identity and origin of these cells remains uncertain, and their surface immunophenotype is clearly distinct from the GM-CSF+ B cells that can be generated in the spleen to provide protection against microbial sepsis, which express CD43 and CD138, amongst other markers10. Nonetheless, we speculate that GM-CSF+ B cells contribute, at least in part, to the enhanced Th17 responses seen in Ackr2-deficient animals and experiments are underway to explore this possibility. Mechanistically, their increased abundance in inflamed Ackr2-deficient LN may be linked to cell-autonomous changes in the migratory properties of these cells, or their precursors. Ackr2 deficiency enhances the migration of B1 B cells towards the non-ACKR2 ligand CXCL139, which is the principal chemokine involved in the localization of B cells, including IBCs22. Through its receptor CXCR5, it recruits B cells into follicles in secondary lymphoid tissues and regulates their intrafollicular motility. It is conceivable enhanced CXCL13 responsiveness alters the recruitment or positioning of certain subsets of B cells in Ackr2-deficient mice before or during inflammation, and that this favors the development of GM-CSF+ B cells. These possibilities are currently being addressed in our laboratory.
In conclusion, ACKR2 can regulate T cell priming and GM-CSF-producing B cells during the induction of arthritic and neuropathic autoimmunity in mice, and its deletion leads to subtle changes in the development of disease. The precise nature of this regulation depends on whether peptide or protein antigen is used to induce the disease, clearly demonstrating the importance of using multiple models when assessing the role of specific genes in autoimmunity. Given the regulatory role of ACKR2 during inflammation and T cell priming, it is important that studies are now undertaken to further investigate the expression and regulation of ACKR2 in the inflamed tissues and draining lymph nodes of patients with autoimmune disease, and it remains to be seen whether ACKR2-mediated chemokine scavenging can influence disease progression in humans. A greater understanding of these aspects of ACKR2 biology could potentially lead to the development of new treatments in which the artificial elevation of ACKR2 in chronically inflamed tissues could be used to broadly suppress chemokine-driven inflammation.
METHODS
Mice
Ackr2-deficient mice (DBA1/j (F10) and C57BL/6J (F11)) and WT counterparts were housed under specific pathogen-free conditions at Glasgow University’s Central Research Facility. Animal studies were approved by the University of Glasgow Ethical Review Process and licensed by the UK Home Office.
Induction and assessment of arthritis
CIA was induced in DBA/1j mice with 100μg of bovine type-II collagen (CII) emulsified in complete Freund’s adjuvant (MD Biosciences) at the base of the tail on day 0, and boosted on day 21 with an i.p. injection of CII in PBS, as previously described 39. Collagen antibody-induced arthritis (CAIA) was induced by intravenous injection of 2mg of collagen antibody cocktail (Chondrex). Three days later LPS (50μg/mouse, Chondrex) was injected i.p.. Mice were scored by a ‘genotype-blinded’ observer for clinical signs of disease, as previously described 39. Hind paws were histologically scored for inflammation and joint damage, as previously described 40. In brief, 0 = healthy, 1 = mild, 2 = moderate, 3 = severe. A genotype-blinded observer scored 3 sections per knee and the mean score per group was calculated.
Quantitative PCR
RNA was extracted using RNeasy columns with DNAse treatment (Qiagen), cDNA generated (AffinityScript (Stratagene)) and QPCR done using Ackr2-specific Taqman assay (mm0044555_m1) (Applied Biosystems) on a Prism 7900HT (Applied Biosystems). GAPDH-specific probes were used to normalise Ackr2 expression. Analysis used the relative quantitation ΔΔ−2 CT method to give an RQ (fold change) value, with naïve tissue as calibrators set to 1.
Synovial tissue explants
Patella and adjacent synovium were dissected and immediately incubated in complete RPMI for 4h at 37°C. The supernatant was harvested and assayed for chemokines, as described below.
Restimulation of LN cells
Inguinal LN cells were harvested and single cell suspensions prepared by enzymatic digestion using 1mg/ml collagenase D (Roche) in HBSS without calcium and magnesium. Cells were cultured in triplicate in 96-well round-bottomed plates at 3×105 cells/well in complete DMEM. Cells were re-stimulated with medium, 60μg/ml of bovine tracheal cartilage (Sigma) (in CIA model) or 30μg/ml of the MOG35-55 peptide or MOG1-125 protein (in EAE model). Proliferation was analyzed at 88h by [3H]Thymidine (GE healthcare) incorporation during the last 16h of culture.
Luminex and ELISA
Cytokines, chemokines and anti-collagen Ab levels were quantified by Luminex or ELISA using appropriately diluted sera or culture supernatants. Reagents for the quantification of mouse chemokines, IFNγ, IL-17 and TNFα were from Biosource, and assays were performed according to the manufacturer’s instructions. Anti-collagen Ab titers of individual sera were evaluated using ELISA-grade collagen (Chondrex) and detected with HRP-conjugated anti-mouse IgG1 or IgG2a (Southern Biotech). Total IgG was determined using an unlabeled anti-mouse IgG capture antibody and detected with HRP-conjugated anti-mouse IgG1 or IgG2a. Antibody ELISAs were developed using o-phenylenediamine dihydrochloride substrate (Sigma).
Flow cytometry
LN cells were harvested and single cell suspensions prepared by enzymatic digestion using 1mg/ml collagenase D (Roche) in HBSS without calcium and magnesium. These cells were re-suspended in FACS buffer (PBS, 1% FCS, 0.02% sodium azide, 5mM EDTA). 1-3×106 cells per well of a 96 well round bottomed plate were incubated with 50μl of a 5μg/ml solution of FC block (BD Biosciences) for 15 minutes on ice, washed twice with FACS buffer and stained with fluorescently-labelled Abs (various concentrations) and Viaprobe (BD Biosciences) or fixable viability dye eFluor® 506 or 780 (eBioscience) (to exclude dead cells). FACS analysis of blood samples was performed similarly with an additional blood cell lysis step performed using ammonium chloride solution (Stemcell Technologies) according to manufacturer’s instructions immediately prior to staining. Abs against the following surface markers were used, with clone names and suppliers given in parentheses: TCRβ (H57-597), CD3 (145-2C11), CD4 (H129.19), CD11b (M1/70), GMCSF (MP1-22E9), IL-17 (TC11-18H10), IFNγ (XMG1.2) (BD biosciences), CD5 (53-7.3), CD11c (N418), CD207 (Langerin) (EbioL31), MHCII (I-A/I-E) (M5/114.15.2), (eBioscience), CD19 (6D5), EpCAM-1 (2E7) (Biolegend) with a variety of conjugated fluorochromes); appropriate isotype controls were purchased from BD Biosciences or eBioscience. For intracellular cytokine staining the BD Biosciences Cytofix/Cytoperm™ Fixation/Permeabilization Solution Kit with BD GolgiPlug™ was used according to manufacturer’s instructions. Briefly, for assessment of cytokine secretion in T cells 1×106 isolated LN cells were cultured for 5 hours at 37°C in complete RPMI in the presence of 500ng/ml of ionomycin, 50ng/ml PMA and Golgistop™. To detect GM-CSF production by B cells, LN cells were cultured as described above but in the absence PMA and ionomycin. Cultured cells were subsequently stained for surface antigens as above and dead cells were excluded using ethidium monoazide or fixable viability dye eFluor® 506 or 780 (eBioscience). Cells were fixed and permeabilised, and stained for intracellular cytokines. The Cytofix/Cytoperm™ Fixation/Permeabilization Solution Kit was used to stain for CD207. In this case the 5h culture step was excluded; otherwise the methodology remained the same. Positive populations were defined on the basis of size (to exclude doublet populations), viability (i.e. viability dye negative), and ‘fluorescence minus one’ isotype controls. Data were analysed using FlowJo software (Treestar Inc).
Induction and assessment of EAE
EAE was induced in DBA/1j and C57BL/6 mice with either 50μg of recombinant rat MOG1-125 41 emulsified in incomplete Freund’s adjuvant (Sigma) supplemented with 3mg/ml of heat-inactivated Mycobacterium tuberculosis H37RA (Difco Laboratories) or in C57BL/6 mice with 100μg of MOG35-55 peptide (AnaSpec) emulsified in incomplete Freund’s adjuvant (Sigma) supplemented with 4mg/ml of heat-inactivated Mycobacterium tuberculosis H37RA (Difco Laboratories). The emulsion was injected intra-dermally at the base of the tail. In some experiments (as indicated in the figure legends), animals were treated with 200ng of pertussis toxin (PTX) (Enzo Life Sciences) via i.p. injection on day 0 and 2. Clinical assessment was performed daily by an observer ‘blinded’ to mouse genotype and according to the following criteria: 0, no disease; 1, decreased tail tone; 2, abnormal gait (ataxia) and/or impaired righting re-flex (hind limb weakness or partial paralysis); 3, partial hind limb paralysis; 4, complete hind limb paralysis; 5, hind limb paralysis with partial fore limp paralysis; and 6, moribund or dead.
Generation of mixed chimeric mice
Mice with a mixed WT and ACKR2-deficient hematopoietic compartment were generated using the mixed bone marrow chimera system. WT (CD45.1) mice were irradiated with 5.5Gy followed by 2hours rest period and then a second dose of 5.5Gy. The irradiated mice were reconstituted immediately with a mixed inoculum of bone marrow (50% CD45.1/.2 WT and 50% CD45.2 ACKR2-deficient). The hematopoietic compartment was left for 2 months to repopulate. Chimerism was confirmed by FACS analysis of blood retrieved from the lateral tail vein as described previously using antibodies against mouse CD45.1 (A20) and CD45.2 (104) (E-bioscience).
Immunohistochemistry
Skin was harvested from the injection site immediately proximal to the base of the tail as well as uninvolved skin from the back. The skin samples were embedded in Cryomatrix (Thermo scientific) and frozen in a bath of isopentane chilled with dry ice. 8μm frozen tissue sections were cut on a Shandon Crotome FSE and stored at −80°C until required. Tissue sections were fixed in acetone at −20°C for 20 minutes. Unless stated otherwise all subsequent steps were performed at room temperature. Tissue sections were rehydrated with PBS for 5 minutes and blocked with 10% normal goat serum 3% Bovine Serum Albumin fraction V (SIGMA) for 1h. Sections were stained with unlabeled 5μg/ml CD207 (Langerin) (EbioL31) overnight at 4°C. The sections were then washed twice for 5 minutes with PBS tween 0.05% and once for 5 minutes with PBS alone before staining with 10μg/ml goat anti-rat IgG H+L Alexa Fluor® 647 (Invitrogen). Wash steps were repeated as previous. Finally sections were stained with a 0.2ug/ml DAPI (Invitrogen) solution in PBS for 30 minutes before mounting using Vectashield (Vector). Images were taken on a Zeiss imager M2 microscope and Axiovision 4.8 software. The numbers of CD207-positive cells per 10 high power fields per samples was determined.
Statistical analysis
GraphPad Prism was used for all statistical analyses using (as appropriate and as indicated in the Figure Legends) t-test; Mann Whitney test; Kruskal-Wallis test with Dunn’s multiple comparison post-test; One-way ANOVA with Bonferroni’s Multiple comparison post-tests; or Two-way ANOVAs with repeated measured. To determine correlations, Spearman’s rho test was used. Data are presented as mean ± SD, except for clinical scores, which are mean ± SEM. p ≤0.05 was considered statistically significant and all tests were two-sided.
Supplementary Material
Supplemental Figure 1. The number of IL-17+ γδ T cells is unaffected by the absence of Ackr2. Draining lymph nodes (n = 6-10) were collected from wild-type (WT) and Ackr2-deficient (Ackr2−/−) DBA/1j mice 35 days after collagen immunization. Representative results from IL-17 intra-cellular flow cytometry and a bar graph showing the percentage of lymph node cells that were IL17+ γδ+ T cells.
Supplemental Figure 2. EAE disease severity in the absence of Ackr2 is unaffected by PTX. (A) EAE was induced in WT littermate control (WT) and Ackr2-deficient C57BL/6 mice (Ackr2−/−) by immunization with MOG1-125 (50μg in 3mg/ml Mycobacterium tuberculosis H37RA CFA) and treatment on days 0 and 2 with 200ng of PTX. All animals were assessed daily for the development of clinical symptoms of disease. Each point represents the mean ± SEM scores for n = 5 mice. (B) On day 21, single cell suspensions of lymph nodes (3×105 cells/well) were stimulated with MOG1-125 (60μg/ml) and the concentration of IL-17 was determined.
ACKNOWLEDGEMENTS
This work was supported by the Biotechnology and Biological Sciences Research Council (CAHH, EA, CSG, RJBN); Arthritis Research UK (CAHH, IBM, RJBN, CSG); a Glasgow University Medical Faculty Scholarship (RSO, RJBN); and Medical Research Council studentship (JD, CSG). Work in RJBNs lab is funded, in part, by a Medical Research Council programme grant. CSG was supported by an Arthritis Research UK fellowship (grants 17653 & 19701). RJBN acknowledges support provided by Dr A Wilson.
Non-standard abbreviations
- CII
type II collagen
- CAIA
Collagen antibody-induced arthritis
- CIA
Collagen-induced arthritis
- DC
dendritic cells
- EAE
Experimental autoimmune encephalomyelitis
- IBC
innate-like B cell
- LEC
lymphatic endothelial cell
- LN
lymph node
- MOG
myelin oligodendrocyte glycoprotein
- MZ
marginal zone
- PTX
pertussis toxin
- WT
wild-type
Footnotes
The authors declare that no conflict of interest exists.
REFERENCES
- 1.Rot A, Andrian von UH. Chemokines in innate and adaptive host defense: basic chemokinese grammar for immune cells. Annu Rev Immunol. 2004;22:891–928. doi: 10.1146/annurev.immunol.22.012703.104543. [DOI] [PubMed] [Google Scholar]
- 2.Nibbs RJB, Graham GJ. Immune regulation by atypical chemokine receptors. Nat Rev Immunol. 2013;13(11):815–29. doi: 10.1038/nri3544. [DOI] [PubMed] [Google Scholar]
- 3.Bachelerie F, Ben-Baruch A, Burkhardt AM, Combadiere C, Farber JM, Graham GJ, et al. International Union of Basic and Clinical Pharmacology. LXXXIX. Update on the extended family of chemokine receptors and introducing a new nomenclature for atypical chemokine receptors. Pharmacol Rev. 2014;66(1):1–79. doi: 10.1124/pr.113.007724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nibbs RJ, Kriehuber E, Ponath PD, Parent D, Qin S, Campbell JD, et al. The beta-chemokine receptor D6 is expressed by lymphatic endothelium and a subset of vascular tumors. Am J Pathol. 2001;158(3):867–77. doi: 10.1016/s0002-9440(10)64035-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Madigan J, Freeman DJ, Menzies F, Forrow S, Nelson SM, Young A, et al. Chemokine scavenger D6 is expressed by trophoblasts and aids the survival of mouse embryos transferred into allogeneic recipients. J Immunol. 2010;184(6):3202–12. doi: 10.4049/jimmunol.0902118. [DOI] [PubMed] [Google Scholar]
- 6.Martinez de la Torre Y, Buracchi C, Borroni EM, Dupor J, Bonecchi R, Nebuloni M, et al. Protection against inflammation- and autoantibody-caused fetal loss by the chemokine decoy receptor D6. Proc Natl Acad Sci USA. 2007;104(7):2319–24. doi: 10.1073/pnas.0607514104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nibbs RJ, Wylie SM, Yang J, Landau NR, Graham GJ. Cloning and characterization of a novel promiscuous human beta-chemokine receptor D6. J Biol Chem. 1997;272(51):32078–83. doi: 10.1074/jbc.272.51.32078. [DOI] [PubMed] [Google Scholar]
- 8.McKimmie CS, Fraser AR, Hansell C, Gutiérrez L, Philipsen S, Connell L, et al. Hemopoietic cell expression of the chemokine decoy receptor D6 is dynamic and regulated by GATA1. J Immunol. 2008;181(5):3353–63. doi: 10.4049/jimmunol.181.5.3353. [DOI] [PubMed] [Google Scholar]
- 9.Hansell CAH, Schiering C, Kinstrie R, Ford L, Bordon Y, McInnes IB, et al. Universal expression and dual function of the atypical chemokine receptor D6 on innate-like B cells in mice. Blood. 2011;117(20):5413–24. doi: 10.1182/blood-2010-11-317115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rauch PJ, Chudnovskiy A, Robbins CS, Weber GF, Etzrodt M, Hilgendorf I, et al. Innate response activator B cells protect against microbial sepsis. Science. 2012;335(6068):597–601. doi: 10.1126/science.1215173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bonecchi R, Locati M, Galliera E. Differential recognition and scavenging of native and truncated macrophage-derived chemokine (macrophage-derived chemokine/CC chemokine ligand 22) by the D6 decoy receptor. J Immunol. 2004;172(8):4972–6. doi: 10.4049/jimmunol.172.8.4972. [DOI] [PubMed] [Google Scholar]
- 12.Weber M, Blair E, Simpson CV, O’Hara M, Blackburn PE, Rot A, et al. The chemokine receptor D6 constitutively traffics to and from the cell surface to internalize and degrade chemokines. Mol Biol Cell. 2004;15(5):2492–508. doi: 10.1091/mbc.E03-09-0634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fra AM, Locati M, Otero K, Sironi M, Signorelli P, Massardi ML, et al. Cutting edge: scavenging of inflammatory CC chemokines by the promiscuous putatively silent chemokine receptor D6. J Immunol. 2003;170(5):2279–82. doi: 10.4049/jimmunol.170.5.2279. [DOI] [PubMed] [Google Scholar]
- 14.Galliera E, Jala VR, Trent JO, Bonecchi R, Signorelli P, Lefkowitz RJ, et al. beta-Arrestin-dependent constitutive internalization of the human chemokine decoy receptor D6. J Biol Chem. 2004;279(24):25590–7. doi: 10.1074/jbc.M400363200. [DOI] [PubMed] [Google Scholar]
- 15.McCulloch CV, Morrow V, Milasta S, Comerford I, Milligan G, Graham GJ, et al. Multiple roles for the C-terminal tail of the chemokine scavenger D6. J Biol Chem. 2008;283(12):7972–82. doi: 10.1074/jbc.M710128200. [DOI] [PubMed] [Google Scholar]
- 16.Di Liberto D, Locati M, Caccamo N, Vecchi A, Meraviglia S, Salerno A, et al. Role of the chemokine decoy receptor D6 in balancing inflammation, immune activation, and antimicrobial resistance in Mycobacterium tuberculosis infection. J Exp Med. 2008;205(9):2075–84. doi: 10.1084/jem.20070608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jamieson T, Cook DN, Nibbs RJB, Rot A, Nixon C, McLean P, et al. The chemokine receptor D6 limits the inflammatory response in vivo. Nat Immunol. 2005;6(4):403–11. doi: 10.1038/ni1182. [DOI] [PubMed] [Google Scholar]
- 18.Martinez de la Torre Y, Locati M, Buracchi C, Dupor J, Cook DN, Bonecchi R, et al. Increased inflammation in mice deficient for the chemokine decoy receptor D6. Eur J Immunol. 2005;35(5):1342–6. doi: 10.1002/eji.200526114. [DOI] [PubMed] [Google Scholar]
- 19.Berres M-L, Trautwein C, Zaldivar MM, Schmitz P, Pauels K, Lira SA, et al. The chemokine scavenging receptor D6 limits acute toxic liver injury in vivo. Biol Chem. 2009;390(10):1039–45. doi: 10.1515/BC.2009.119. [DOI] [PubMed] [Google Scholar]
- 20.Nibbs RJB, Gilchrist DS, King V, Ferra A, Forrow S, Hunter KD, et al. The atypical chemokine receptor D6 suppresses the development of chemically induced skin tumors. J Clin Invest. 2007;117(7):1884–92. doi: 10.1172/JCI30068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Whitehead GS, Wang T, DeGraff LM, Card JW, Lira SA, Graham GJ, et al. The chemokine receptor D6 has opposing effects on allergic inflammation and airway reactivity. Am J Respir Crit Care Med. 2007;175(3):243–9. doi: 10.1164/rccm.200606-839OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ansel KM, Harris RBS, Cyster JG. CXCL13 is required for B1 cell homing, natural antibody production, and body cavity immunity. Immunity. 2002;16(1):67–76. doi: 10.1016/s1074-7613(01)00257-6. [DOI] [PubMed] [Google Scholar]
- 23.Szekanecz Z, Vegvari A, Szabo Z, Koch AE. Chemokines and chemokine receptors in arthritis. Front Biosci. 2010;2:153–67. doi: 10.2741/s53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Proudfoot AEI, de Souza ALS, Muzio V. The use of chemokine antagonists in EAE models. J Neuroimmunol. 2008;198(1-2):27–30. doi: 10.1016/j.jneuroim.2008.04.007. [DOI] [PubMed] [Google Scholar]
- 25.Schall TJ, Proudfoot AEI. Overcoming hurdles in developing successful drugs targeting chemokine receptors. Nat Rev Immunol. 2011;11(5):355–63. doi: 10.1038/nri2972. [DOI] [PubMed] [Google Scholar]
- 26.Liu L, Graham GJ, Damodaran A, Hu T, Lira SA, Sasse M, et al. Cutting edge: the silent chemokine receptor D6 is required for generating T cell responses that mediate experimental autoimmune encephalomyelitis. J Immunol. 2006;177(1):17–21. doi: 10.4049/jimmunol.177.1.17. [DOI] [PubMed] [Google Scholar]
- 27.Lee KM, McKimmie CS, Gilchrist DS, Pallas KJ, Nibbs RJ, Garside P, et al. D6 facilitates cellular migration and fluid flow to lymph nodes by suppressing lymphatic congestion. Blood. 2011;118(23):6220–9. doi: 10.1182/blood-2011-03-344044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sonderegger I, Iezzi G, Maier R, Schmitz N, Kurrer M, Kopf M. GM-CSF mediates autoimmunity by enhancing IL-6-dependent Th17 cell development and survival. J Exp Med. 2008;205(10):2281–94. doi: 10.1084/jem.20071119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nandakumar K, Holmdahl R. Antibody-induced arthritis: disease mechanisms and genes involved at the effector phase of arthritis. Arthritis Res Ther. 2006;8(6):223. doi: 10.1186/ar2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vetrano S, Borroni EM, Sarukhan A, Savino B, Bonecchi R, Correale C, et al. The lymphatic system controls intestinal inflammation and inflammation-associated Colon Cancer through the chemokine decoy receptor D6. Gut. 2010;59(2):197–206. doi: 10.1136/gut.2009.183772. [DOI] [PubMed] [Google Scholar]
- 31.Singh MD, King V, Baldwin H, Burden D, Thorrat A, Holmes S, et al. Elevated expression of the chemokine-scavenging receptor D6 is associated with impaired lesion development in psoriasis. Am J Pathol. 2012;181(4):1158–64. doi: 10.1016/j.ajpath.2012.06.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Constant S, Sant’Angelo D, Pasqualini T, Taylor T, Levin D, Flavell R, et al. Peptide and protein antigens require distinct antigen-presenting cell subsets for the priming of CD4+ T cells. J Immunol. 1995;154(10):4915–23. [PubMed] [Google Scholar]
- 33.Constant S, Schweitzer N, West J, Ranney P, Bottomly K. B lymphocytes can be competent antigen-presenting cells for priming CD4+ T cells to protein antigens in vivo. J Immunol. 1995;155(8):3734–41. [PubMed] [Google Scholar]
- 34.O’Neill SK, Shlomchik MJ, Glant TT, Cao Y, Doodes PD, Finnegan A. Antigen-specific B cells are required as APCs and autoantibody-producing cells for induction of severe autoimmune arthritis. J Immunol. 2005;174(6):3781–8. doi: 10.4049/jimmunol.174.6.3781. [DOI] [PubMed] [Google Scholar]
- 35.Weber MS, Prod’homme T, Patarroyo JC, Molnarfi N, Karnezis T, Lehmann-Horn K, et al. B-cell activation influences T-cell polarization and outcome of anti-CD20 B-cell depletion in central nervous system autoimmunity. Ann Neurol. 2010;68(3):369–83. doi: 10.1002/ana.22081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Matsushita T, Yanaba K, Bouaziz J-D, Fujimoto M, Tedder TF. Regulatory B cells inhibit EAE initiation in mice while other B cells promote disease progression. J Clin Invest. 2008;118(10):3420–30. doi: 10.1172/JCI36030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Attanavanich K, Kearney JF. Marginal zone, but not follicular B cells, are potent activators of naive CD4 T cells. J Immunol. 2004;172(2):803–11. doi: 10.4049/jimmunol.172.2.803. [DOI] [PubMed] [Google Scholar]
- 38.Zhong X, Gao W, Degauque N, Bai C, Lu Y, Kenny J, et al. Reciprocal generation of Th1/Th17 and T(reg) cells by B1 and B2 B cells. Eur J Immunol. 2007;37(9):2400–4. doi: 10.1002/eji.200737296. [DOI] [PubMed] [Google Scholar]
- 39.MacLellan LM, Montgomery J, Sugiyama F, Kitson SM, Thummler K, Silverman GJ, et al. Co-opting endogenous immunoglobulin for the regulation of inflammation and osteoclastogenesis. Arthritis Rheum. 2011;63(12):3897–907. doi: 10.1002/art.30629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Joosten LA, Lubberts E, Durez P, Helsen MM, Jacobs MJ, Goldman M, et al. Role of interleukin-4 and interleukin-10 in murine collagen-induced arthritis. Protective effect of interleukin-4 and interleukin-10 treatment on cartilage destruction. Arthritis Rheum. 1997;40(2):249–60. doi: 10.1002/art.1780400209. [DOI] [PubMed] [Google Scholar]
- 41.Adelmann M, Wood J, Benzel I, Fiori P, Lassmann H, Matthieu JM, et al. The N-terminal domain of the myelin oligodendrocyte glycoprotein (MOG) induces acute demyelinating experimental autoimmune encephalomyelitis in the Lewis rat. J Neuroimmunol. 1995;63(1):17–27. doi: 10.1016/0165-5728(95)00124-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. The number of IL-17+ γδ T cells is unaffected by the absence of Ackr2. Draining lymph nodes (n = 6-10) were collected from wild-type (WT) and Ackr2-deficient (Ackr2−/−) DBA/1j mice 35 days after collagen immunization. Representative results from IL-17 intra-cellular flow cytometry and a bar graph showing the percentage of lymph node cells that were IL17+ γδ+ T cells.
Supplemental Figure 2. EAE disease severity in the absence of Ackr2 is unaffected by PTX. (A) EAE was induced in WT littermate control (WT) and Ackr2-deficient C57BL/6 mice (Ackr2−/−) by immunization with MOG1-125 (50μg in 3mg/ml Mycobacterium tuberculosis H37RA CFA) and treatment on days 0 and 2 with 200ng of PTX. All animals were assessed daily for the development of clinical symptoms of disease. Each point represents the mean ± SEM scores for n = 5 mice. (B) On day 21, single cell suspensions of lymph nodes (3×105 cells/well) were stimulated with MOG1-125 (60μg/ml) and the concentration of IL-17 was determined.