

Abstract
Prostate cancer is the second most common malignancy among men worldwide. Genome-wide association studies (GWAS) have identified 100 risk variants for prostate cancer, which can explain ~33% of the familial risk of the disease. We hypothesized that a comprehensive analysis of genetic variations found within the 3′ UTR of genes predicted to affect miRNA binding (miRSNPs) can identify additional prostate cancer risk variants. We investigated the association between 2,169 miRSNPs and prostate cancer risk in a large-scale analysis of 22,301 cases and 22,320 controls of European ancestry from 23 participating studies. Twenty-two miRSNPs were associated (p<2.3×10−5) with risk of prostate cancer, 10 of which were within the 7 genes previously not mapped by GWASs. Further, using miRNA mimics and reporter gene assays, we showed that miR-3162-5p has specific affinity for the KLK3 rs1058205 miRSNP T-allele whilst miR-370 has greater affinity for the VAMP8 rs1010 miRSNP A-allele, validating their functional role.
Introduction
Prostate cancer is the most common non-skin malignancy among men worldwide. In the US, an estimated 233,000 new cases and 29,480 deaths are expected in 2014 (1). Established risk factors for prostate cancer include advancing age, ethnicity, and a family history of the disease (2). Men with a family history of prostate cancer have a 2-fold increased risk of developing the disease and usually with an earlier age of onset (3). A significant role for genetic factors has been confirmed by genome-wide association studies (GWAS) and large scale replication studies, which have already identified 100 single nucleotide polymorphisms (SNP) associated with prostate cancer risk (4, 5). However, the identified SNPs account for only a small proportion of the (33%) excess familial risk suggesting additional SNPs remain to be identified (4).
MicroRNAs (miRNAs) are short ~19 – 24 nucleotide non-coding RNA molecules that post-transcriptionally regulate gene expression by cleaving or degrading mRNA and/or inhibiting its translation (6-8). Most miRNA binding has been observed within the 3′UTR of their target genes, although there are examples of binding within mRNA coding regions (9). As of March 2014, the miRBase database lists >2570 mature miRNAs for humans. miRNAs are expressed in a tissue and cell-specific manner with differential expression profiles in response to disease conditions, with many of these miRNA expression modulations contributing to disease progression (10-15). An impressive effort has been devoted to investigating miRNA dys-regulation profiles in prostate cancer. Hence, miRNAs have emerged as not only potential biomarkers for prostate cancer but also as potential therapeutic targets (15-17).
miRNAs negatively regulate their target mRNAs primarily through Watson-Crick base-pairing interactions (18, 19). The most critical region for mRNA binding and repression are miRNA nucleotides 2-8, referred to as the miRNA seed site. Experiments have shown that genetic variations within the seed site or in the target mRNA at sites complementary to miRNA seed sites, referred to as miRSNPs, may reduce effectiveness or abolish miRNA-mediated repression, having functional consequences for cancer risk (20, 21). For example, Liu et al recently reported that miRSNPs in ITGAv are associated with a decreased risk of prostate cancer (22). In another study assessing 61 putative miRSNPs in a Chinese population, three SNPs were associated with prostate cancer progression whilst four SNPs were associated with prostate cancer-specific mortality (23). However, all these studies have been conducted using small sample sizes and might not be reflective of true positive association.
To further explore the genetic association of miRSNPs and to derive more reliable risk estimates of previously identified prostate cancer risk miRSNPs, we investigated the association between 2,169 miRSNPs and prostate cancer risk and aggressiveness in 23 studies participating in the Prostate Cancer Association Group to Investigate Cancer Associated Alterations in the Genome (PRACTICAL) Consortium. This effort included 22,301 cases and 22,320 controls of European ancestry. We then validated the functional role of two prostate cancer risk miRSNPs, Kallikrein 3 (KLK3) rs1058205 (T>C) and Vesicle-associated membrane protein 8 (VAMP-8) rs1010 (A>G), as they were most strongly associated with disease aggressiveness. To our knowledge, this is the first large-scale investigation of the association between miRNA-related gene polymorphisms and prostate cancer risk.
Results
Patient characteristics
The characteristics of the study participants are presented in Table 1. The mean age at diagnosis for cases (64.8 years), was older than the age at interview for controls (60.6 years). Cases (22.1%) were more likely to have a family history of prostate cancer compared to controls (13.9%). As expected, the majority of cases were diagnosed with tumours with a low (≤ 7) Gleason score (85.5%) that were localized (72.8%) and non-aggressive (82.1%). Among the cases with data available on vital status, 14.6% died at a median follow up of 5 years with almost half (52.1%) of the deaths attributed to prostate cancer.
Table 1. Participant characteristics.
| Characteristic | Controls N=22,320 N (%) | Cases N=22,301 N (%) | p valuea |
|---|---|---|---|
| Age at diagnosis/interview mean± SD | 60.6±10.7 | 64.8± 8.0 | <.0001 |
| Family history of prostate cancer No Yes |
10992(86.1) 1779(13.9) |
10300(77.9) 2918(22.1) |
<.0001 |
| Gleason score 2-6 7 8-10 |
N/A |
8863(52.6) 5548(32.9) 2437(14.5) |
|
| SEER Stage Local Distant Regional Unknown |
N/A |
13246(72.8) 883(4.9) 3555(19.6) 503(2.8) |
|
| PSA at diagnosis (ng/ml) <100 ≥100 |
N/A |
12692(95.7) 565(4.3) |
|
| Aggressive diseaseb No Yes |
N/A |
17504(82.1) 3812(17.9) |
|
| Vital status Alive Prostate-specific death Other death |
4738(72.2) 0 1822(27.8) |
13794(85.4) 1233(7.6) 1134(7.0) |
<.0001 |
t-test for a continuous variable and chi-square test for a categorical variable
Aggressive disease is defined as a Gleason score of 8-10, PSA at diagnosis ≥100 ng/ml, distant stage or prostate cancer-specific death.
Association of miRSNPs with prostate cancer
Figure 1 and Supplementary Table 1 show the results of the association analyses for 2,169 putative miRSNPs with prostate cancer risk. Twenty five miRSNPs had a minor allele frequency <0.01 in control samples. A total of 22 SNPs (Table 2, Supplementary Figure 1) representing 16 genes were associated with risk of prostate cancer after correction for multiple testing (p<2.3×10−5). The most significant association was observed for rs1058205 located within the KLK3 3′UTR with an OR= 0.86(0.83-0.9), p = 1.7×10−14. This SNP was previously identified in a fine-mapping study (24). Similarly, MDM4 rs4245739 was recently reported in the primary iCOGS analysis (4). Ten SNPs - rs2450975, rs3103353, and rs3127593 (SLC22A2), rs1567669 (NKX3-1), rs1010 (VAMP8), rs1810126 (SLC22A3), rs2647257 (TET2), rs14082 and rs1043853 (PDLIM5), and rs17664 (ITGA6) were found in the genes/regions previously implicated by prostate cancer GWAS studies. Ten SNPs rs879161 (PHC3), rs7615039 (PHC3), rs12492606 (PHC3), rs311497 (GMEB2), rs1530865 (PDK1), rs2357637 (PDK1), rs12573077 (ARL3), rs7402 (MCAT), rs47340 (TTLL12), and rs4233979 (TMEM17) in seven genes (PHC3, GMEB2, PDK1, ARL3, MCAT, TTLL12, TMEM17) (Table 2) are at least 20 kb away from the previously reported index GWAS SNPs within the locus. Although GMEB2 and ARL3 have been previously reported, these seven genes have not been mapped by previous GWAS as per the NHGRI catalogue on Nov 2014(25).
Figure 1. miRSNP association with prostate cancer risk.
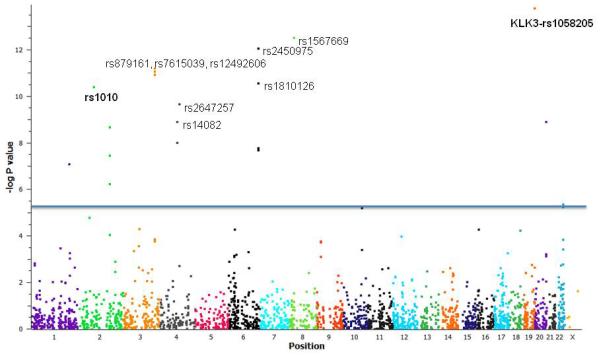
Manhattan Plot with −log p-values adjusted for study site and principal components. 2,169 miRSNPs were assessed for association with prostate cancer risk. 22 SNPs representing 16 genes were found to be associated with risk of prostate cancer after correction for multiple testing (p<2.3×10−5). (The 10 most significant SNPs are labelled). KLK3 rs1058205 and VAMP8 rs1010 (in bold) were selected for further functional validation studies. (OR = odds ratio).
Table 2. Risk estimates and predicted miRNAs for the 22 putative miRSNPs associated with prostate cancer risk.
| miRSNP | Gene | Transcript variante | Predicted miRNA(s)c | chr | position | Risk OR (95% CI)d | Risk p-value |
|---|---|---|---|---|---|---|---|
| rs4245739 | MDM4a | NM_002393, NM_001204171, NM_001204172 | miR-191-5p, miR-887, miR-3669 | 1 | 204518842 | 0.92 (0.89-0.95) | 7.81E-08 |
| rs4233979 | TMEM17 | NM_198276 | miR-299-5p | 2 | 62727902 | 1.08 (1.04-1.12) | 1.61E-05 |
| rs1010 | VAMP8b | NM_003761 | miR-370, miR-103 | 2 | 85808982 | 0.91 (0.89-0.94) | 3.79E-11 |
| rs17664 | ITGA6b | NM_001079818, NM_000210 | miR-548c-3p miR-548aj, miR-4691-3p | 2 | 173369231 | 0.93 (0.91-0.96) | 5.51E-07 |
| rs1530865 | PDK1 | NM_002610 | miR-877-5p, miR-3125, miR-3916 | 2 | 173461090 | 0.8 (0.75-0.86) | 2.28E-09 |
| rs2357637 | PDK1 | NM_002610 | miR-3916, miR-3125, miR-877-5p | 2 | 173463138 | 0.81 (0.76-0.88) | 3.66E-08 |
| rs7615039 | PHC3 | NM_024947 | miR-208a, miR-208b | 3 | 169806170 | 0.86 (0.83-0.9) | 8.72E-12 |
| rs12492606 | PHC3 | NM_024947 | miR-939, miR-362-5p | 3 | 169808354 | 0.86 (0.83-0.9) | 1.23E-11 |
| rs879161 | PHC3 | NM_024947 | miR-27a-5p, miR-220c, miR-3158-3p | 3 | 169812115 | 0.86 (0.83-0.9) | 6.57E-12 |
| rs14082 | PDLIM5b | NM_006457, NM_001011513, NM_001256425, NM_001256426, NM_001256428 | miR-128, miR-494 | 4 | 95586224 | 1.09 (1.06-1.12) | 1.27E-09 |
| rs1043853 | PDLIM5b | NM_006457, NM_001011513, NM_001256425, NM_001256426, NM_001256427, NM_001256428 | miR-567, miR-3120, miR-4310 | 4 | 95588274 | 1.08 (1.05-1.11) | 9.65E-09 |
| rs2647257 | TET2b | NM_001127208 | miR-301a, miR-301b, miR-4330 | 4 | 106199505 | 1.1 (1.06-1.13) | 2.05E-10 |
| rs2450975 | SLC22A2b | NM_003058, NM_003058 | miR-412, miR-4282 | 6 | 160637975 | 1.12 (1.09-1.16) | 9.36E-13 |
| rs3127593 | SLC22A2b | NM_003058 | miR-200a miR-302a miR-488-3p | 6 | 160638003 | 1.12 (1.08-1.17) | 2.22E-08 |
| rs3103353 | SLC22A2b | NM_003058 | miR-942, miR-4268 | 6 | 160638076 | 1.12 (1.08-1.17) | 1.75E-08 |
| rs1810126 | SLC22A3b | NM_021977 | miR-1205, miR-124-3p, miR-216b | 6 | 160872151 | 1.1 (1.07-1.13) | 2.81E-11 |
| rs1567669 | NKX3-1b | NM_006167, NM_001256339 | miR-637, miR-1275, miR-625 | 8 | 23538533 | 1.11 (1.08-1.15) | 3.15E-13 |
| rs12573077 | ARL3 | NM_004311 | miR-432, miR-1258, miR-1224-5p | 10 | 104434630 | 0.93 (0.91-0.96) | 6.43E-06 |
| rs1058205 | KLK3a | NM_001648, NM_001030047, NM_001030048 | miR-3162-5p, miR-219-1-3p, miR-4278 | 19 | 51363398 | 0.87 (0.83-0.9) | 1.73E-14 |
| rs311497 | GMEB2 | NM_012384 | miR-26c, miR-492, miR-619, miR-4648 | 20 | 62221249 | 0.92 (0.89-0.94) | 1.28E-09 |
| rs7402 | MCAT | NM_173467, NM_014507 | miR-616-3p | 22 | 43529029 | 0.93 (0.91-0.96) | 4.41E-06 |
| rs47340 | TTLL12 | NM_015140 | let-7f, let-7g, let-7i, miR-103, miR-107, miR-764 | 22 | 43562829 | 1.07 (1.04-1.1) | 5.79E-06 |
SNP previously identified in prostate cancer GWAS and fine-mapping studies.
Other SNPs within or around the genes for the listed SNPs have been identified in previous GWAS studies.
miRNAs were predicted using four algorithms. (See Materials & Methods).
adjusted for 6 principal components and study group.
miRSNPs are present within the 3′UTRs of the most the of the common splice variants of the miRNA target genes
In secondary analysis, seven SNPs showed significant differences in per-allele odds ratios between aggressive and non-aggressive disease (Supplementary Table 2). The most significant difference was observed for the KLK3 rs1058205 SNP, however this SNP was more strongly associated with nonaggressive disease, which is in line with previous reports on other Kallikrein SNPs (24). Interestingly, only two SNPs, rs1010 in VAMP8 and rs311497 in GMEB2, showed stronger association with aggressive disease. The rs1567669 SNP (NKX3-1) was associated with PSA levels in the patient cohort (Supplementary Table 3). Six SNPs (rs1043853, rs1058205, rs14082, rs2450975, rs3103353, rs3127593) including KLK3 rs1058205 were marginally associated with PSA levels in controls (Supplementary Table 3). Six SNPs showed a trend with respect to age at diagnosis including rs1058205, rs1043853, rs12492606, rs14082, rs7615039 and rs879161 (Supplementary Table 4).
Gene expression and eQTL analysis
Using Oncomine™ (Compendia Bioscience, Ann Arbor, MI) analysis tool, we compared the expression levels of the 16 genes harbouring 22 significant miRSNPs. Using the Grasso dataset (26) of 59 tumour and 28 non tumour samples, expression of 7 genes was found to be deregulated in prostate cancer vs matched benign tissue analysis (Figure 2). We found KLK3 and VAMP8 to be the second and seventh highest deregulated genes within the 16 genes analysed.
Figure 2. Expression levels of 16 genes harbouring 22 significant miRSNPs in cancerous and normal tissue from prostate cancer patients.
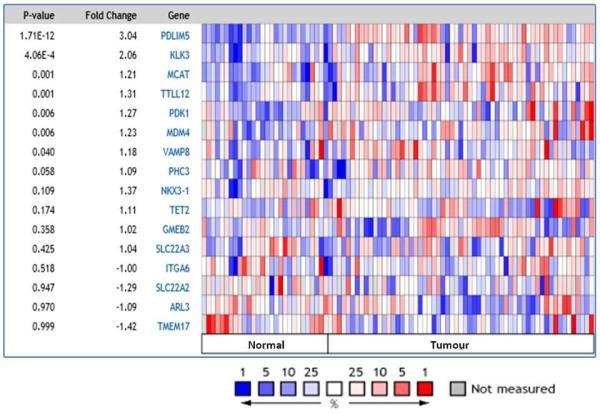
Oncomine analysis of the Grasso dataset (26) of 59 tumor and 28 non-tumor (normal) samples, shows the expression of 7 genes (PDLIM5 – VAMP8) to be deregulated (p<0.05) in prostate cancer.
We undertook a cis-eQTL analysis of the 22 prostate cancer associated miRSNPs using TCGA data. Three SNPs (rs1058205 (KLK3), rs1530865 and rs2357637 (PDK1)) were not covered by the TCGA genotyping platform (Affymetrix 6.0) and did not have an appropriate proxy SNP identified by SNAP(27), and thus could not be analysed. The remaining 19 SNPs (six index and 13 proxy SNPs identified by SNAP) were assessed for correlation with the respective gene expression harbouring these SNPs. SNP rs2450975 (indexed by rs316000, r2= 0.95) in the SLC22A2 gene was found to be associated with mRNA transcript expression (p = 1.76 × 10−5), while the SLC22A2 SNP, rs10945656 (indexing rs3103353 and rs3127593, r2 = 1.0) and VAMP8 rs1010 SNP showed a trend (p=0.09) towards genotype-transcript expression (Supplementary Table 5). None of the other SNPs were associated with transcript levels of the gene harbouring the SNP.
Functional validation of the KLK3 rs1058205 and VAMP8 rs1010 miRSNPs
Using a range of computational prediction algorithms, we identified three miRNAs predicted to have differences in binding affinity between the KLK3 rs1058205 SNP-alleles. SNPinfo (28) and mirsnpscore (29) predicted miR-219-1-3p to target the T-allele, MicroSNiPer (30) and mirsnpscore predicted miR-3162-5p also to target the T-allele and MicroSNiPer and mirsnpscore predicted miR-4278 to target the C-allele. Two miRNAs were predicted to have differences in binding affinity between the VAMP8 rs1010 SNP-alleles. SNPinfo predicted both miR-103 and miR-370-5p to target the A-allele (sense strand = T allele).
Reporter vector assays were then used to test the validity of these in silico predicted miRNA binding potential to their target gene/s with specific genotype. For KLK3 rs1058205 SNP, miRNA miR-3162-5p induced a ~29% (p = 0.048) decrease in luciferase levels for the T-allele compared to the C-allele suggesting that miR-3162-5p has specific affinity for the T allele (Figure 3A). No significant changes were observed for miR-219-1-3p or miR-4278 with either of the alleles for SNP rs1058205 (Supplementary Figure 2A-B). For VAMP8 rs1010 (A>G), though miR-370-5p induced a change in luciferase activity for both alleles, the decrease in luciferase levels for the A-allele was ~2 fold (p = 0.0067) stronger than for the G-allele (Figure 3B). Although miR-103 was found to regulate VAMP8 expression, it showed comparable results for both alleles (Supplementary Figure 2C).
Figure 3. miR-3162-5p directly targets the KLK3 rs1058205 SNP T allele and miR-370-5p targets the VAMP8 rs1010 SNP A allele with greater affinity.
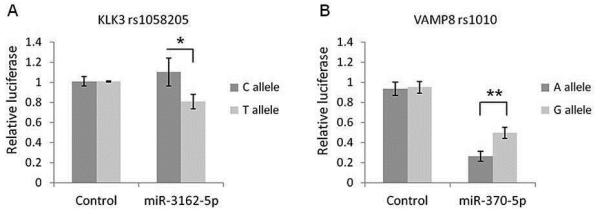
Following over-expression with miR-3162-5p, reporter vector assays demonstrated a ~29% decrease in luciferase levels (p = 0.048) for the KLK3 rs1058205 SNP T-allele compared to the C-allele (A). Over-expression of miR-370-5p resulted in a change in luciferase activity for both VAMP8 rs1010 SNP variants with the decrease for the A-allele ~2 fold (p = 0.0067) stronger than for the G-allele (B). Mean +/− SD, n = 3. (* = p < 0.05) (** = p < 0.01)
Expression of miR-3162-5p and miR-370-5p in prostate cancer
Though previously reported in melanoma, breast cancer and cervical cancer (with expression up-regulated in cervical cancer) (31-33), prostatic expression for miR-3162-5p has not been determined. However, the miR-3162-5p gene is located within intron seven of the Oxysterol binding protein gene, which is known to be expressed in the normal and cancerous prostate (31-34). Using qPCR, we specifically confirmed miR-3162-5p expression in a range of cancerous and non-cancerous prostatic cell lines (Supplementary Figure 3A) as well as in patient tissue samples (Supplementary Figure 3B). miR-3162-5p was detected in all the model cell lines and patient samples with varying expression levels. Prostatic expression for miR-370-5p has been reported previously to be up-regulated in cancer (17, 35).
Regulation of KLK3 mRNA and protein levels by miR-3162-5p
Given the importance of KLK3/PSA as a serum biomarker for prostate cancer, we further characterised the miR-3162-5p and KLK3 rs1058205 SNP interaction, to determine if miR-3162-5p was able to affect endogenous KLK3 mRNA and protein levels in cell lines using LNCaP cells homozygous TT for the rs1058205 SNP. With reference to the negative control miRNA mimic, over-expression of miR-3162-5p resulted in a 25% decrease in KLK3 mRNA (p = 0.016) as determined using qPCR analysis (Figure 4A).
Figure 4. miR-3162-5p induces a reduction in KLK3 mRNA and KLK3 protein expression in LNCaP cells homozygous for the rs1058205 T SNP-allele.
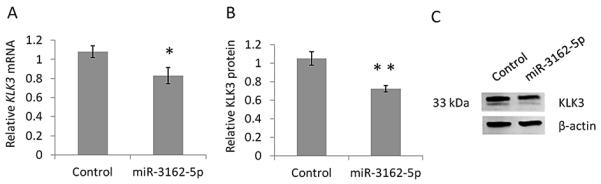
(A) qPCR analyses in LNCaP cells revealed a 25% decrease in KLK3 mRNA (p = 0.016) following over-expression of miR-3162-5p compared to the negative control miRNA mimic treatment. (B) Western blot analyses in LNCaP cells revealed a 32% decrease in cellular KLK3 protein (p = 0.007) following over-expression of miR-3162-5p compared to the negative control. (C) Representative Western blot. Mean +/− SD, n = 3. (* = p < 0.05) (** = p < 0.01)
We then assessed if miR-3162-5p was able to affect endogenous KLK3 protein levels. With reference to the negative control miRNA mimic, over-expression of miR-3162-5p resulted in a 32% decrease in cellular KLK3 protein (p = 0.007) as determined using Western blot analysis (Figure 4B-C).
Discussion
In this study we investigated the association between 2,169 putative miRSNPs and prostate cancer risk in a large sample including 22,301 cases and 22,320 controls of European ancestry. We identified 22 SNPs within the 3′ UTR of the 16 genes to be associated with risk of prostate cancer, seven of which although within the previously identified GWAS locus are not previously mapped by the GWAS studies. The most significant association is for the KLK3 rs1058205 SNP previously identified to be associated with prostate cancer risk in a recent study (24).
Seven of these SNPs including KLK3 (rs1058205) and VAMP8 (rs1010), a gene for which little is known about its prostatic function, showed significant differences between aggressive and non-aggressive disease. As expected, these results were not as robust due to the small sample size in the aggressive disease sub cohort. Thus, it was not surprising that, as has been the case for many previously GWAS identified SNPs (36), we could identify only two SNPs to be more significantly associated with aggressive disease.
Using Oncomine, we compared the expression levels of these 16 genes in a dataset consisting of 59 tumor and 28 non-tumor samples, revealing the expression of seven of these genes to be deregulated in prostate cancer. KLK3 was the second most deregulated gene. We then chose to validate the functional role of the KLK3 rs1058205 SNP, it being the most significant miRSNP identified. We demonstrated that miR-3162-5p has specific affinity for the KLK3 rs1058205 T-allele. Our results support the emerging “miRNA network” that contributes to prostate cancer by regulating kallikrein and non-kallikrein genes (37).
As prostatic expression for miR-3162-5p had not been determined previously, we confirmed its expression in a range of cancerous and non-cancerous prostatic cell lines and in patient tissue samples. It is interesting to note that prostate cancer PC3 cell lines with the highest miR-3162-5p expression do not produce any endogenous KLK3. In previous studies this differential expression has been attributed to absence of an androgen receptor in the PC3 cell lines since KLK3 expression is androgen dependent in other prostate cancer cell lines. However, its regulation by miR-3162-5p could be an alternative regulatory mechanism and rationale for no KLK3 expression in these cells.
Kallikrein-3 (KLK3) is also referred to as Prostate-specific antigen (PSA), as it is expressed at orders of magnitude higher in the prostate compared to other tissues (38). As KLK3 serum levels are often elevated in prostate cancer, largely due to leakage associated with a loss of tissue architecture, KLK3 is thus utilized as the major serum biomarker for this disease (38, 39). Given the potential importance of KLK3 for prostate cancer diagnosis we further characterized the miRNA-KLK3 rs1058205 interaction demonstrating that miR-3162-5p is able to cause a decrease in KLK3 mRNA and KLK3 protein expression in LNCaP cells homozygous for the T-allele. Interestingly, the KLK3 rs1058205 SNP was associated with PSA levels in the control population (Supplementary Table 3), which may reflect some effects of strong regulatory factors (such as miRNA) exerting genotype-specific effects for this locus. The rs1058205 SNP, in addition to other PSA associated SNPs, may therefore have implications for PSA-based diagnoses, hence requiring adjustments to PSA ranges for specific genotype. Furthermore, as KLK3 belongs to a family of 15 homologous genes, it would be important to consider potential additional effects of miR-3162-5p miRNA on other kallikrein and non-kallikrein targets in future studies.
The KLK3 rs1058205 T allele was previously shown to be associated with increased prostate cancer risk (24). Here we demonstrate that decreased KLK3 expression induced by miR-3162-5p targeting of the T-allele represents a mechanism by which the rs1058205 T-allele may be associated with increased prostate cancer risk. Interestingly, it has been shown that more aggressive prostate tumors have lower tissue levels of KLK3 (40). One mode via which KLK3 may act in a protective capacity in prostate cancer is through inhibition of angiogenesis (41, 42). Although the full mechanism is unclear, the anti-angiogenic effect of KLK3 has been attributed to its proteolytic function on various angiogenic and anti-angiogenic proteins. (43, 44). However, due to its additional proteolytic function and its subsequent potential to target components of cell-cell adhesion and the extra cellular matrix, high levels of KLK3 have also been proposed as a risk for prostate cancer. It is therefore possible that the effects of KLK3 on tumor development are stage-specific, with low KLK3 contributing to increased localised tumor growth (as observed in genetic risk analysis) whilst high KLK3 poses a risk at later metastatic stages of tumor development.
The VAMP8 rs1010 SNP was also selected for functional validation due to its significant association with aggressive prostate cancer, where miR-370-5p was found to have greater affinity for the VAMP8 rs1010 A-allele versus the minor G-allele. Interestingly, prostatic expression for miR-370-5p has been reported previously to be up-regulated in cancer (17, 35).
To our knowledge, this is the first study to report an association and mechanism of action between a VAMP8 miRSNP and prostate cancer risk. VAMP8 is an integral membrane protein that is involved in the fusion of synaptic vesicles with the pre-synaptic membrane. It also plays a complex role in the control of granule secretion, transport vesicle trafficking, phagocytosis and endocytosis (45, 46). Loss of VAMP8 has been shown to affect glucose metabolism, energy expenditure and insulin sensitivity in mice (47). Though a direct role of VAMP8 in cancer is unknown, its ability to influence glucose metabolism and energy expenditure makes it a potential candidate in carcinogenesis, in relation to the shift in cellular metabolism from oxidative phosphorylation to glycolysis (the Warburg effect) that occurs in cells undergoing malignant transformation (47, 48). Hence, the role of VAMP8 may be important for prostate cancer. The VAMP8 rs1010 SNP was previously associated with risk for early onset myocardial infarction (49) and is in high LD (r2=0.98) with an intergenic SNP (rs10187424) identified in a previous GWAS for prostate cancer risk (OR=0.92, 95% CI=0.89-0.94, P=2.1×10−9) (36). No functional relevance has been assigned to rs10187424 SNP. In the current study, we demonstrated that miR-370-5p has greater affinity for the VAMP8 rs1010 A-allele, thus identifying the likely causal variant behind the GWAS marker SNP. Nevertheless, the possibility of another functional variant in LD with rs1010 or any other putative functional miRSNPs (including KLK3) identified in our study cannot be ruled out. Larger sample sizes are now required to provide additional power to assess true independence and/or the effect of these SNPs as modifiers of the unknown functional variants and/or top risk GWAS SNPs using conditional regression and/or haplotype analysis. Furthermore, it should be noted that the size effects of these variants are very small though comparable to previous GWAS studies. Thus, once independent causal variants or haplotypes at each of the known GWAS loci are identified, it would be interesting to undertake risk score calculations to assess the additive effects of all GWAS identified SNPs including the miRSNPs identified in our study.
Although our analysis has identified several miRSNPs previously not reported by the GWAS analysis, functional validation of these variants is required. Our eQTL analysis did not yield any significant results for genotype - mRNA expression correlation except for a SNP in SLC22A2, which is not surprising given that the miRNA machinery might not affect the mRNA levels in situ but will only inhibit the translation of these genes. Future studies are warranted to correlate genotypes with protein expression using immunohistochemistry and/or western blot analysis. Additional functional studies may further clarify the role of these novel miRSNPs in prostate cancer aetiology.
In conclusion, our study has identified putative functional SNPs associated with prostate cancer risk in several genes that further show differential expression in tumor vs normal tissue from prostate cancer patients. The functional validation for the rs1058205 and rs1010 miRSNPs herein provides increasing evidence that miRSNPs may be associated with prostate cancer risk.
Materials and Methods
Study populations
The Collaborative Oncological Gene-environment Study (COGS) is a large collaborative effort among different consortia, including PRACTICAL, to evaluate genetic variants for associations with the risk of prostate, ovarian and breast cancers. Details of the study have been reported previously (4). Briefly, 32 studies participating in the PRACTICAL consortium contributed samples from 25,074 prostate cancer cases and 24,272 controls to COGS. The majority of studies were nested, population-based or hospital-based case-control studies. Individuals were excluded from the study based on strict quality control criteria including: overall genotype call rate <95%, genotypically non-European origin, samples that were XX or XXY and therefore not genotypically males or samples not concordant with previous genotyping within PRACTICAL. The present analysis included 44,621 samples (22,301 cases and 22,320 controls) of European ancestry. Demographic and clinical information on study participants including age at diagnosis, Gleason score, stage of disease, prostate-specific antigen (PSA) and cause of death were obtained through in-person interviews or medical or death records. Aggressive disease was defined as Gleason score ≥ 8, prostate-specific antigen (PSA) >100 ng/ml, disease stage of ‘distant’ (outside the pelvis) or prostate cancer associated death. Study was approved by each institutional review board (IRB) and informed consent was obtained from each participant. Patient studies were conducted in accordance with the Declaration of Helsinki.
miRSNP selection and genotyping
A total of 2,169 miRSNPs within the 3′ UTRs of the cancer associated genes were selected for genotyping. A SNP was selected if differential miRNA binding potential for the alternative alleles was predicted by at least two of four algorithms: 1. Mirsnpscore (29); 2. Miranda and 3. Sanger (both available through SNPinfo) (28); and 4. MicroSNiPer (30). Genotyping was performed using a custom Illumina Infinium array that included 211,115 SNPs (the iCOGS chip) (4). Genotypes were called using Illumina’s proprietary GenCall algorithm. SNPs were excluded from further analysis if the call rate was <95%, deviated from Hardy-Weinberg Equilibrium (HWE) in controls at P<10−7, or if genotypes were discrepant in more than 2% of duplicate samples.
cis-eQTL analysis
For each index miRSNP, we retrieved all the correlated (r2 ≥ 0.8) variants in EUR populations from 1000 Genomes using SNAP (27). The pre-processed (Level 2) germline genotypes of the index or correlated SNPs were downloaded from the TCGA data portal and the expression levels of genes harbouring these SNPs were obtained via the cBio Portal for Cancer Genomics. Using standard QC analysis, 6 samples were removed either due to discordant sex information (X-chromosome homozygosity rate between 0.2 and 0.8) or due to heterozygosity rate >3 standard deviations from the mean. An additional 45 individuals were removed due to ethnic heterogeneity as calculated using principal component analysis. Data from 178 Caucasian individuals was used for the final genotype expression correlation analysis by Kruskal-Wallis test using IBM SPSS Statistics (version 22).
miRNA target reporter vector assays
To assess validity of in silico predictions for miRNA-mRNA affinity, miRNA target luciferase reporter vector assays were performed. Reporter vectors were constructed for the major and minor SNP allele variants for both KLK3 and VAMP-8 using the pmirGLO Dual-Luciferase miRNA Target Expression Vector (Promega) for KLK3 constructs, and the pMIR REPORT vector (Ambion) for VAMP-8 constructs. (Portions of KLK3 and VAMP-8 pertaining to predicted miRNA binding regions were synthesised by Integrated DNA Technologies). LNCaP cells were co-transfected with vector and mirVana miRNA Mimics (Life Technologies) using FuGENE transfection reagent (Promega) then analysed 24 hours later using the Dual-Luciferase Reporter Assay System (Promega) according to the manufacturer’s instructions. Luciferase levels were normalised against Renilla co-expressed from the same vector or against the β-galactosidase derived from co-expression with a second vector measured using Galacto-Light (Tropix) for VAMP8. For KLK3, on the day of transfection cells were cultured in 1% charcoal stripped serum for the remainder of the experiment to induce lower levels of endogenous KLK3 to minimise the impact of miRNA mimic-endogenous KLK3 binding on reporter vector assay sensitivity. A negative control mirVana miRNA Mimic - Negative Control #1 (Life Technologies) was used for analysis alongside candidate miRNAs. A single experiment consisted of each miRNA/vector treatment cultured in triplicate. Three independent experiments were conducted in total.
RT-qPCR analysis to assess miRNA expression
miRNA was extracted as total RNA from cell lines (LNCaP, LAPC4, DUCAP, DU145, PC3, 22Rv1, RWPE1 and BPH1 obtained from ATCC unless otherwise stated (see Acknowledgements) using TRIzol reagent (Life Technologies). Cell line authentication (STR profiling) was performed by either the Queensland Institute of Medical Research (Brisbane, Queensland, Australia) or DDC Medical (Fairfield, Ohio, U.S.A.).
Formalin fixed and paraffin-embedded (FFPE) blocks from prostate tumours and their adjacent non-cancer prostate were obtained from the Australian Prostate Cancer Bio-Resource tumour bank. Tissue blocks containing the tumour cells were serially sectioned (20 μm sections) and transferred to glass slides. Slides were stained with methyl green and the tumour areas were marked by a pathologist. Marked areas were then manually micro-dissected under a microscope using a sterile injection needle (size 0.65 × 25 mm). RNA was extracted using the miRNeasy FFPE kit (Qiagen, Chadstone, Australia).
To assess expression, reverse transcription and qPCR was performed using the TaqMan MicroRNA Reverse Transcription Kit and TaqMan MicroRNA Assays (Life Technologies). The small nuclear RNA - RNU24 was used as an endogenous quantitative normalization control (16). Relative expression levels were calculated using the Comparative Ct method. As TaqMan MicroRNA Assays were not commercially available for miR-3162-5p, we designed in-house assays for miR-3162-5p (Supplementary Methods 1) following the TaqMan methodology (50).
RT-qPCR and Western blot analysis to assess KLK3 mRNA and protein expression
LNCaP cells were plated at 150 000 cells per well on a 6 well plate overnight, then cultured in 1% charcoal stripped serum (for the remaining duration of the experiment) and transiently transfected with 30 nM of mirVana miRNA Mimics using Lipofectamine RNAiMAX transfection reagent (Life Technologies) followed by a treatment with 10 nM dihydrotestosterone (DHT) to stimulate KLK3 expression via the androgen receptor pathway. Total RNA was isolated after 24 hours incubation post DHT stimulation using the RNAeasy Mini Kit (Qiagen) and assessed for quality and yield using a Nanodrop ND-1000 spectrophotometer. RNA was reversed transcribed using oligo dT primers and 500 ng of total RNA. qPCR for KLK3 was then performed using the SYBR Green PCR Master Mix (Life Technologies) for each sample in triplicate with Beta-actin used as an endogenous quantitative normalisation control. Relative expression levels were calculated using the Comparative Ct method. Primers were synthesised by Integrated DNA Technologies. Primers sequences for KLK3 were: forward primer 5′- agtgcgagaagcattcccaacc -3′, reverse primer 5′- ccagcaagatcacgcttttgttcct -3′. Primers sequences for Beta-actin were: forward primer 5′- gcgttacaccctttcttgacaaaacct -3′, reverse primer 5′- gctgtcaccttcaccgttcca -3′.
Total protein was isolated using SDS lysis buffer (1% SDS, 5% glycerol, 10 nM Tris, Roche Complete protease inhibitor); concentration was assessed via the BCA method and 10 ug of total protein was run using standard techniques on a 12% resolving poly-acrylamide gel. Western blotting was performed using standard techniques with primary antibodies including Rb anti KLK3 (Dako - A0562) and Rb anti Beta-actin (Abcam - ab25894). Western blots were imaged on an Odyssey Imaging System (LI-COR Biosciences) using fluorescently labelled secondary antibodies (Alexa Fluor 680 & 790 - Invitrogen) with protein band intensities analysed via densitometry using Odyssey Imaging System software.
Statistical analysis
Demographic, clinical and mortality information was summarised by mean (SD) and number (%). Ethnic groups were defined based on a subset of 37,000 uncorrelated markers that passed quality control (including ~1,000 selected as ancestry informative markers). The COGS data was combined with the three Hapmap2 populations and multi-dimensional scaling was used to identify and exclude ethnic outliers (4). After exclusion of ethnic outliers, principal component analyses were carried out for Europeans. The first six principal components were used to control for population substructure as additional principal components did not reduce inflation further (4). Associations between individual SNPs and prostate cancer risk or aggressive disease were evaluated using logistic regression models to estimate per minor allele odds ratios (OR) and 95% confidence intervals (95% CI). Associations between individual SNPs and prostate cancer risk were also evaluated in a similar manner for different age categories. The associations between SNP genotypes and PSA level were assessed using linear regression, after log-transformation of PSA level to correct for skewness. Analyses were performed using SPSS and R. All models included study site and principal components as covariates.
Unless otherwise stated, for all other analyses three independent experiments were conducted with results presented as mean +/− standard deviation, and analysed using a Student’s t test with a p-value of <0.05 considered statistically significant for the functional studies
Supplementary Material
Significance.
Findings from this large association study suggest that a focus on miRSNPs, including functional evaluation, can identify candidate risk loci below currently accepted statistical levels of genome-wide significance. Studies of miRNAs and their interactions with SNPs could provide further insights into the mechanisms of prostate cancer risk.
Acknowledgements
J. Batra is supported by an NHMRC Career Development Fellowship. A.B. Spurdle is an NHMRC Senior Research Fellow; J.A. Clements is an NHMRC Principal Research Fellow. This work is supported by Cure Cancer Australia Foundation and Cancer Australia PdCCRS grant 1068321 and NHMRC grant 1050742 (PI: Batra), DOD grant W81XWH-12-1-0113(PI: Park) and NCI R01CA128813 (PI: Park). E.K. Amankwah was supported by a Cancer prevention fellowship from the NCI (R25T CA147832) during the conduct of this study. The APCB funding was obtained from the National Health and Medical Research Council (Enabling Grant 614296) and an infrastructure grant from the Prostate Cancer Foundation of Australia. The authors would also like to thank John Lai, Farhana Matin and Leire Moya for technical support and to Charles Sawyers for providing the prostate cancer cell line LAPC4 and Matthias Nees for providing the prostate cancer cell line DuCAP. Additionally, the results published here are based partly on data generated by The Cancer Genome Atlas (TCGA), established by the National Cancer Institute and the National Human Genome Research Institute, and we are grateful to the specimen donors and relevant research groups associated with this project.
Funding for the CRUK study and PRACTICAL consortium:
This work was supported by the Canadian Institutes of Health Research, European Commission’s Seventh Framework Programme grant agreement n° 223175 (HEALTH-F2-2009-223175), Cancer Research UK Grants C5047/A7357, C1287/A10118, C5047/A3354, C5047/A10692, C16913/A6135, and The National Institute of Health (NIH) Cancer Post-Cancer GWAS initiative grant: No. 1 U19 CA 148537-01 (the GAME-ON initiative). We acknowledge support from the NIHR to the Biomedical Research Centre at The Institute of Cancer Research and Royal Marsden NHS Foundation Trust.
COGS acknowledgement:
This study would not have been possible without the contributions of the following: Per Hall (COGS); Douglas F. Easton, Paul Pharoah, Kyriaki Michailidou, Manjeet K. Bolla, Qin Wang (BCAC), Andrew Berchuck (OCAC), Rosalind A. Eeles, Douglas F. Easton, Ali Amin Al Olama, Zsofia Kote-Jarai, Sara Benlloch (PRACTICAL), Georgia Chenevix-Trench, Antonis Antoniou, Lesley McGuffog, Fergus Couch and Ken Offit (CIMBA), Joe Dennis, Alison M. Dunning, Andrew Lee, and Ed Dicks, Craig Luccarini and the staff of the Centre for Genetic Epidemiology Laboratory, Javier Benitez, Anna Gonzalez-Neira and the staff of the CNIO genotyping unit, Jacques Simard and Daniel C. Tessier, Francois Bacot, Daniel Vincent, Sylvie LaBoissière and Frederic Robidoux and the staff of the McGill University and Génome Québec Innovation Centre, Stig E. Bojesen, Sune F. Nielsen, Borge G. Nordestgaard, and the staff of the Copenhagen DNA laboratory, and Julie M. Cunningham, Sharon A. Windebank, Christopher A. Hilker, Jeffrey Meyer and the staff of Mayo Clinic Genotyping Core Facility.
Funding for the iCOGS infrastructure came from: the European Community’s Seventh Framework Programme under grant agreement n° 223175 (HEALTH-F2-2009-223175) (COGS), Cancer Research UK (C1287/A10118, C1287/A 10710, C12292/A11174, C1281/A12014, C5047/A8384, C5047/A15007, C5047/A10692), the National Institutes of Health (CA128978) and Post-Cancer GWAS initiative (1U19 CA148537, 1U19 CA148065 and 1U19 CA148112 - the GAME-ON initiative), the Department of Defence (W81XWH-10-1-0341), the Canadian Institutes of Health Research (CIHR) for the CIHR Team in Familial Risks of Breast Cancer, Komen Foundation for the Cure, the Breast Cancer Research Foundation, and the Ovarian Cancer Research Fund.
The PRACTICAL Consortium- In addition to those named in the author list
| Authors | Affiliations |
|---|---|
| Margaret Cook | PRACTICAL coordination |
| Angela Morgan, Artitaya Lophatananon, Cyril Fisher, Daniel Leongamornlert, Edward J. Saunders, Emma J. Sawyer, Koveela Govindasami, Malgorzata Tymrakiewicz, Michelle Guy, Naomi Livni, Rosemary Wilkinson, Sara Jugurnauth-Little, Steve Hazel, Tokhir Dadaev | UKGPCS |
| John Pedersen, John L Hopper and Melissa C Southey | MCCS |
| Ami Karlsson, Carin Cavalli-Bjoerkman, Jan-Erik Johansson, Jan Adolfson, Markus Aly, Michael Broms, Paer Stattin | CAPS & STHM1 |
| Brian E. Henderson, Fredrick Schumacher | MEC |
| Anssi Auvinen, Kimmo.Taari Kimmo.Taari, Liisa Maeaettaenen, Paula Kujala, Teemu Murtola, Teuvo LJ Tammela, Tiina Wahlfors | TAMPERE |
| Andreas Roder, Peter Iversen, Peter Klarskov, Sune F. Nielsen, Maren Weischer | CPCS1 & CPCS2 |
| Tim J. Key, Hans Wallinder, Sven Gustafsson | EPIC |
| Jenny L. Donovan, Freddie Hamdy, Anne George, Athene Lane, Gemma Marsden, Michael Davis, Paul Brown | ProtecT & ProMPT |
| Nora Pashayan | SEARCH |
| Sarah Holt | FHCRC |
| Lisa B. Signorello, Wei Zheng | SCCS |
| Liang Wang, Lori Tillmans, Shaun Riska | MAYO |
| Antje Rinckleb, Kathleen Herkommer, Manuel Luedeke, Walther Vogel Poland Dominika Wokozorczyk, Jan Lubiski, Wojciech Kluzniak | ULM |
| Aida k. Dieffenbach, Christa Stegmaier, Volker Arndt | ESTHER |
| Babu Zachariah, Hyun Park, Julio Pow-Sang, Maria Rincon, Selina Radlein, | MOFFITT |
| Aleksandrina Vlahova, Atanaska Mitkova, Chavdar Slavov, Darina Kachakova, Elenko Popov, Svetlana Christova, Tihomir Dikov, Vanio Mitev | PCMUS |
| Felicity Lose, APCB* | QLD |
| Joana Santos, Joao Barros-Silva, Paula Paulo, Pedro Pinto, Rui Henrique, Sofia Maia | IPO-Porto |
The Australian Prostate Cancer BioResource is contributed by following author other than those named in the author list Prof Gail Risbridger1,2, Dr Renea Taylor1,2, Prof Wayne Tilley3,4, A/Prof Lisa Butler3,4, A/Prof Lisa Horvath5,6,7, Dr Trina Yeadon8,9,10, Ms Allison Eckert8,9,10, Dr Glen Wood11, Dr Peter Heathcote11, Dr Greg Malone11, Dr Kris Kerr12, Dr Megan Turner12, Dr Angus Collins12
1. Prostate Cancer Research Group, Monash University, Clayton, VIC, 2. Australian Prostate Cancer BioResource, Melbourne, VIC, 3. Adelaide Prostate Cancer Research Centre, Adelaide, SA, 4. Australian Prostate Cancer BioResource, Adelaide, SA, 5. Garvan Institute of Medical Research/The Kinghorn Cancer Centre, Darlinghurst, NSW, 6. Chris O’Brien Lifehouse, Camperdown, NSW, 7. Australian Prostate Cancer BioResource, Sydney, NSW, 8. Cancer Program, Institute of Health and Biomedical Innovation, Queensland University of Technology, Brisbane, QLD, 9. Australian Prostate Cancer Research Centre-Queensland at the Princess Alexandra Hospital, Queensland University of Technology, Brisbane, QLD, 10. Australian Prostate Cancer BioResource, Brisbane, QLD, 11. The Brisbane Urology Clinic Central Queensland Urology Clinic, QLD, 12. Sullivan and Nicolaides Pathology, Brisbane, QLD.
Footnotes
Conflict of interest
No author has any competing financial interests or conflicts of interest in respect of this work and its publication
References
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA: a cancer journal for clinicians. 2013;63:11–30. doi: 10.3322/caac.21166. [DOI] [PubMed] [Google Scholar]
- 2.Hsing AW, Chokkalingam AP. Prostate cancer epidemiology. Frontiers in bioscience : a journal and virtual library. 2006;11:1388–413. doi: 10.2741/1891. [DOI] [PubMed] [Google Scholar]
- 3.Bratt O. What should a urologist know about hereditary predisposition to prostate cancer? BJU international. 2007;99:743–7. doi: 10.1111/j.1464-410X.2006.06666.x. discussion 7-8. [DOI] [PubMed] [Google Scholar]
- 4.Eeles RA, Olama AA, Benlloch S, Saunders EJ, Leongamornlert DA, Tymrakiewicz M, et al. Identification of 23 new prostate cancer susceptibility loci using the iCOGS custom genotyping array. Nature genetics. 2013;45:385–91. 91e1–2. doi: 10.1038/ng.2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Al Olama AA, Kote-Jarai Z, Berndt SI, Conti DV, Schumacher F, Han Y, et al. A meta-analysis of 87,040 individuals identifies 23 new susceptibility loci for prostate cancer. Nature genetics. 2014 doi: 10.1038/ng.3094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kiriakidou M, Tan GS, Lamprinaki S, De Planell-Saguer M, Nelson PT, Mourelatos Z. An mRNA m7G cap binding-like motif within human Ago2 represses translation. Cell. 2007;129:1141–51. doi: 10.1016/j.cell.2007.05.016. [DOI] [PubMed] [Google Scholar]
- 7.Pillai RS, Bhattacharyya SN, Artus CG, Zoller T, Cougot N, Basyuk E, et al. Inhibition of translational initiation by Let-7 MicroRNA in human cells. Science. 2005;309:1573–6. doi: 10.1126/science.1115079. [DOI] [PubMed] [Google Scholar]
- 8.Zinovyev A, Morozova N, Nonne N, Barillot E, Harel-Bellan A, Gorban AN. Dynamical modeling of microRNA action on the protein translation process. BMC systems biology. 2010;4:13. doi: 10.1186/1752-0509-4-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang S, Wu S, Ding J, Lin J, Wei L, Gu J, et al. MicroRNA-181a modulates gene expression of zinc finger family members by directly targeting their coding regions. Nucleic acids research. 2010;38:7211–8. doi: 10.1093/nar/gkq564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Greve TS, Judson RL, Blelloch R. microRNA Control of Mouse and Human Pluripotent Stem Cell Behavior. Annual review of cell and developmental biology. 2013;29:213–39. doi: 10.1146/annurev-cellbio-101512-122343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lemons D, Maurya MR, Subramaniam S, Mercola M. Developing microRNA screening as a functional genomics tool for disease research. Frontiers in physiology. 2013;4:223. doi: 10.3389/fphys.2013.00223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cimmino A, Calin GA, Fabbri M, Iorio MV, Ferracin M, Shimizu M, et al. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:13944–9. doi: 10.1073/pnas.0506654102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fonseca-Sanchez MA, Perez-Plasencia C, Fernandez-Retana J, Arechaga-Ocampo E, Marchat LA, Rodriguez-Cuevas S, et al. microRNA-18b is upregulated in breast cancer and modulates genes involved in cell migration. Oncology reports. 2013;30:2399–410. doi: 10.3892/or.2013.2691. [DOI] [PubMed] [Google Scholar]
- 14.Pinho FG, Frampton AE, Nunes J, Krell J, Alshaker H, Jacob J, et al. Downregulation of microRNA-515-5p by the Estrogen Receptor Modulates Sphingosine Kinase 1 and Breast Cancer Cell Proliferation. Cancer research. 2013;73:5936–48. doi: 10.1158/0008-5472.CAN-13-0158. [DOI] [PubMed] [Google Scholar]
- 15.Ventura A, Jacks T. MicroRNAs and cancer: short RNAs go a long way. Cell. 2009;136:586–91. doi: 10.1016/j.cell.2009.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carlsson J, Helenius G, Karlsson M, Lubovac Z, Andren O, Olsson B, et al. Validation of suitable endogenous control genes for expression studies of miRNA in prostate cancer tissues. Cancer genetics and cytogenetics. 2010;202:71–5. doi: 10.1016/j.cancergencyto.2010.06.009. [DOI] [PubMed] [Google Scholar]
- 17.Porkka KP, Pfeiffer MJ, Waltering KK, Vessella RL, Tammela TL, Visakorpi T. MicroRNA expression profiling in prostate cancer. Cancer research. 2007;67:6130–5. doi: 10.1158/0008-5472.CAN-07-0533. [DOI] [PubMed] [Google Scholar]
- 18.Carthew RW, Sontheimer EJ. Origins and Mechanisms of miRNAs and siRNAs. Cell. 2009;136:642–55. doi: 10.1016/j.cell.2009.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Muckstein U, Tafer H, Hackermuller J, Bernhart SH, Stadler PF, Hofacker IL. Thermodynamics of RNA-RNA binding. Bioinformatics. 2006;22:1177–82. doi: 10.1093/bioinformatics/btl024. [DOI] [PubMed] [Google Scholar]
- 20.Pelletier C, Weidhaas JB. MicroRNA binding site polymorphisms as biomarkers of cancer risk. Expert review of molecular diagnostics. 2010;10:817–29. doi: 10.1586/erm.10.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liang D, Meyer L, Chang DW, Lin J, Pu X, Ye Y, et al. Genetic variants in MicroRNA biosynthesis pathways and binding sites modify ovarian cancer risk, survival, and treatment response. Cancer research. 2010;70:9765–76. doi: 10.1158/0008-5472.CAN-10-0130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu J, Huang J, He Y, Liu J, Liao B, Liao G. Genetic variants in the integrin gene predicted microRNA-binding sites were associated with the risk of prostate cancer. Molecular carcinogenesis. 2014;53:280–5. doi: 10.1002/mc.21973. [DOI] [PubMed] [Google Scholar]
- 23.Bao BY, Pao JB, Huang CN, Pu YS, Chang TY, Lan YH, et al. Polymorphisms inside microRNAs and microRNA target sites predict clinical outcomes in prostate cancer patients receiving androgen-deprivation therapy. Clinical cancer research: an official journal of the American Association for Cancer Research. 2011;17:928–36. doi: 10.1158/1078-0432.CCR-10-2648. [DOI] [PubMed] [Google Scholar]
- 24.Kote-Jarai Z, Amin Al Olama A, Leongamornlert D, Tymrakiewicz M, Saunders E, Guy M, et al. Identification of a novel prostate cancer susceptibility variant in the KLK3 gene transcript. Human genetics. 2011;129:687–94. doi: 10.1007/s00439-011-0981-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Welter D, MacArthur J, Morales J, Burdett T, Hall P, Junkins H, et al. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic acids research. 2014;42:D1001–6. doi: 10.1093/nar/gkt1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012;487:239–43. doi: 10.1038/nature11125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Johnson AD, Handsaker RE, Pulit SL, Nizzari MM, O’Donnell CJ, de Bakker PI. SNAP: a web-based tool for identification and annotation of proxy SNPs using HapMap. Bioinformatics. 2008;24:2938–9. doi: 10.1093/bioinformatics/btn564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xu Z, Taylor JA. SNPinfo: integrating GWAS and candidate gene information into functional SNP selection for genetic association studies. Nucleic acids research. 2009;37:W600–5. doi: 10.1093/nar/gkp290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thomas LF, Saito T, Saetrom P. Inferring causative variants in microRNA target sites. Nucleic acids research. 2011;39:e109. doi: 10.1093/nar/gkr414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Barenboim M, Zoltick BJ, Guo Y, Weinberger DR. MicroSNiPer: a web tool for prediction of SNP effects on putative microRNA targets. Hum Mutat. 2010;31:1223–32. doi: 10.1002/humu.21349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stark MS, Tyagi S, Nancarrow DJ, Boyle GM, Cook AL, Whiteman DC, et al. Characterization of the Melanoma miRNAome by Deep Sequencing. PloS one. 2010;5:e9685. doi: 10.1371/journal.pone.0009685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Persson H, Kvist A, Rego N, Staaf J, Vallon-Christersson J, Luts L, et al. Identification of new microRNAs in paired normal and tumor breast tissue suggests a dual role for the ERBB2/Her2 gene. Cancer research. 2011;71:78–86. doi: 10.1158/0008-5472.CAN-10-1869. [DOI] [PubMed] [Google Scholar]
- 33.Chen J, Yao D, Li Y, Chen H, He C, Ding N, et al. Serum microRNA expression levels can predict lymph node metastasis in patients with early-stage cervical squamous cell carcinoma. International journal of molecular medicine. 2013;32:557–67. doi: 10.3892/ijmm.2013.1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thorsen K, Schepeler T, Oster B, Rasmussen MH, Vang S, Wang K, et al. Tumor-specific usage of alternative transcription start sites in colorectal cancer identified by genome-wide exon array analysis. BMC genomics. 2011;12:505. doi: 10.1186/1471-2164-12-505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ambs S, Prueitt RL, Yi M, Hudson RS, Howe TM, Petrocca F, et al. Genomic profiling of microRNA and messenger RNA reveals deregulated microRNA expression in prostate cancer. Cancer research. 2008;68:6162–70. doi: 10.1158/0008-5472.CAN-08-0144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kote-Jarai Z, Olama AA, Giles GG, Severi G, Schleutker J, Weischer M, et al. Seven prostate cancer susceptibility loci identified by a multi-stage genome-wide association study. Nat Genet. 2011;43:785–91. doi: 10.1038/ng.882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Samaan S, Lichner Z, Ding Q, Saleh C, Samuel J, Streutker C, et al. Kallikreins are involved in an miRNA network that contributes to prostate cancer progression. Biol Chem. 2014;395:991–1001. doi: 10.1515/hsz-2013-0288. [DOI] [PubMed] [Google Scholar]
- 38.Lawrence MG, Lai J, Clements JA. Kallikreins on steroids: structure, function, and hormonal regulation of prostate-specific antigen and the extended kallikrein locus. Endocrine reviews. 2010;31:407–46. doi: 10.1210/er.2009-0034. [DOI] [PubMed] [Google Scholar]
- 39.Thorek DL, Evans MJ, Carlsson SV, Ulmert D, Lilja H. Prostate-specific kallikrein-related peptidases and their relation to prostate cancer biology and detection. Established relevance and emerging roles. Thrombosis and haemostasis. 2013;110:484–92. doi: 10.1160/TH13-04-0275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stege R, Grande M, Carlstrom K, Tribukait B, Pousette A. Prognostic significance of tissue prostate-specific antigen in endocrine-treated prostate carcinomas. Clinical cancer research: an official journal of the American Association for Cancer Research. 2000;6:160–5. [PubMed] [Google Scholar]
- 41.Fortier AH, Nelson BJ, Grella DK, Holaday JW. Antiangiogenic activity of prostate-specific antigen. Journal of the National Cancer Institute. 1999;91:1635–40. doi: 10.1093/jnci/91.19.1635. [DOI] [PubMed] [Google Scholar]
- 42.Fortier AH, Holaday JW, Liang H, Dey C, Grella DK, Holland-Linn J, et al. Recombinant prostate specific antigen inhibits angiogenesis in vitro and in vivo. The Prostate. 2003;56:212–9. doi: 10.1002/pros.10256. [DOI] [PubMed] [Google Scholar]
- 43.Mattsson JM, Valmu L, Laakkonen P, Stenman UH, Koistinen H. Structural characterization and anti-angiogenic properties of prostate-specific antigen isoforms in seminal fluid. The Prostate. 2008;68:945–54. doi: 10.1002/pros.20751. [DOI] [PubMed] [Google Scholar]
- 44.Mattsson JM, Narvanen A, Stenman UH, Koistinen H. Peptides binding to prostate-specific antigen enhance its antiangiogenic activity. The Prostate. 2012;72:1588–94. doi: 10.1002/pros.22512. [DOI] [PubMed] [Google Scholar]
- 45.Ho YH, Cai DT, Huang D, Wang CC, Wong SH. Caspases regulate VAMP-8 expression and phagocytosis in dendritic cells. Biochemical and biophysical research communications. 2009;387:371–5. doi: 10.1016/j.bbrc.2009.07.028. [DOI] [PubMed] [Google Scholar]
- 46.Behrendorff N, Dolai S, Hong W, Gaisano HY, Thorn P. Vesicle-associated membrane protein 8 (VAMP8) is a SNARE (soluble N-ethylmaleimide-sensitive factor attachment protein receptor) selectively required for sequential granule-to-granule fusion. The Journal of biological chemistry. 2011;286:29627–34. doi: 10.1074/jbc.M111.265199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zong H, Wang CC, Vaitheesvaran B, Kurland IJ, Hong W, Pessin JE. Enhanced energy expenditure, glucose utilization, and insulin sensitivity in VAMP8 null mice. Diabetes. 2011;60:30–8. doi: 10.2337/db10-0231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhu D, Zhang Y, Lam PP, Dolai S, Liu Y, Cai EP, et al. Dual role of VAMP8 in regulating insulin exocytosis and islet beta cell growth. Cell metabolism. 2012;16:238–49. doi: 10.1016/j.cmet.2012.07.001. [DOI] [PubMed] [Google Scholar]
- 49.Shiffman D, Rowland CM, Louie JZ, Luke MM, Bare LA, Bolonick JI, et al. Gene variants of VAMP8 and HNRPUL1 are associated with early-onset myocardial infarction. Arteriosclerosis, thrombosis, and vascular biology. 2006;26:1613–8. doi: 10.1161/01.ATV.0000226543.77214.e4. [DOI] [PubMed] [Google Scholar]
- 50.Chen C, Ridzon DA, Broomer AJ, Zhou Z, Lee DH, Nguyen JT, et al. Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic acids research. 2005;33:e179. doi: 10.1093/nar/gni178. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.