

Abstract
Hepatocyte apoptosis is intrinsically linked to chronic liver disease and hepatocarcinogenesis. Conversely, necroptosis of hepatocytes and other liver cell types and its relevance for liver disease is debated. Using liver parenchymal cell (LPC)-specific TGF-beta-activated kinase 1 (TAK1)-deficient (TAK1LPC-KO) mice, which exhibit spontaneous hepatocellular and biliary damage, hepatitis and early hepatocarcinogenesis, we have investigated the contribution of apoptosis and necroptosis in hepatocyte and cholangiocyte death and their impact on liver disease progression. Here, we provide in vivo evidence showing that TAK1-deficient cholangiocytes undergo spontaneous necroptosis induced primarily by TNFR1 and dependent on RIPK1 kinase activity, RIPK3 and NEMO. In contrast, TAK1-deficient hepatocytes die by FADD-dependent apoptosis, which is not significantly inhibited by LPC-specific RIPK1 deficiency, inhibition of RIPK1 kinase activity, RIPK3 deficiency or combined LPC-specific deletion of TNFR1, TRAILR and Fas. Accordingly, normal mouse cholangiocytes can undergo necroptosis, while primary hepatocytes are resistant to it and die exclusively by apoptosis upon treatment with cell death-inducing stimuli in vitro, likely due to the differential expression of RIPK3. Interestingly, the genetic modifications that conferred protection from biliary damage also prevented the spontaneous early lethality that was often observed in TAK1LPC-KO mice. In the presence of chronic hepatocyte apoptosis, preventing biliary damage delayed but did not avert hepatocarcinogenesis. On the contrary, inhibition of hepatocyte apoptosis fully prevented liver tumorigenesis even in mice with extensive biliary damage. Altogether, our results suggest that using RIPK1 kinase activity inhibitors could be therapeutically useful for cholestatic liver disease patients.
Keywords: apoptosis, necroptosis, liver disease, cholestasis, hepatocellular carcinoma
Introduction
Hepatocellular carcinoma (HCC) arises predominantly in a chronic setting of liver injury, hepatocyte proliferation, inflammation, and fibrosis. Liver cell death is a potent trigger of acute and chronic liver disease1–3. Different types of cell death are believed to induce stronger inflammatory responses than others. While apoptosis is believed to be less inflammatory due to the containment of the cellular content of dying cells within membrane-bound compartments, necrotic death, such as necroptosis or pyroptosis, is considered highly inflammatory due to uncontrolled release of cellular components that act as danger-associated molecular patterns4. Yet, a wealth of recent studies demonstrate that apoptosis is a key driver of chronic liver disease and HCC in mice and humans5–12. In contrast, the contribution of necroptosis in liver disease remains controversial13.
Receptor Interacting Protein Kinase 1 (RIPK1) has recently emerged as a key molecule that promotes both apoptosis and necroptosis through its kinase activity, but also cell survival via scaffolding functions. Although RIPK1 plays critical roles downstream of many receptors inducing inflammation, its functions are best characterized in Tumor Necrosis Factor receptor 1 (TNFR1) signaling3,14,15. Upon TNFR1 activation, RIPK1 forms Complex I together with TNFR1 associated death domain protein (TRADD), TNFR-associated factor 2 (TRAF2), cellular inhibitors of apoptosis 1 and 2 (cIAP1/2) and linear ubiquitin chain assembly complex (LUBAC). RIPK1 ubiquitination by cIAPs and LUBAC mediates the recruitment of transforming growth factor beta (TGFβ)-activated kinase 1 (TAK1)/TAK1-binding protein 2/3 (TAB2/3) complex, as well as the inhibitor of nuclear factor κB (IκB) kinase (IKK) complex, consisting of the IKK1/IKKα and IKK2/IKKβ kinases and the regulatory subunit NEMO/IKKγ. TAK1 and IKK complexes promote cell survival by activating nuclear factor κB (NF-κB) signaling but also by phosphorylating directly or indirectly RIPK110,14–16. In addition, RIPK1 phosphorylation by TBK1 and IKKε was recently shown to be required to prevent cell death induction following TNFR1 stimulation17,18. Upon TAK1, IKK1/2, TBK1/IKKε or cIAP inhibition, RIPK1 nucleates the formation of Complex IIb by recruiting Fas-associated protein with death domain (FADD) and Caspase-8, thereby triggering apoptosis. However, when Caspase-8 activity is inhibited, RIPK1 forms together with RIPK3 and Mixed Lineage Kinase-like (MLKL) the necrosome that induces necroptosis, although this event is cell-specific and depends on RIPK3 expression. Finally, FADD/Caspase-8 mediated apoptosis can also proceed through Complex IIa, the formation of which is RIPK1-independent but TRADD-dependent3.
Studies in mice lacking NEMO or TAK1 in liver parenchymal cells (LPCs) provided important insights into the role of cell death in chronic liver disease and cancer. Mice with LPC-specific ablation of NEMO (NEMOLPC-KO) develop spontaneous hepatocellular damage, steatohepatitis and ultimately HCC after 9-12 months. Liver disease in NEMOLPC-KO mice is driven by RIPK1 kinase activity/FADD/Caspase-8-mediated hepatocyte apoptosis5–8. TAK1LPC-KO mice also develop hepatocellular damage, but additionally show spontaneous biliary damage, lethal cholestasis, and early-onset hepatocarcinogenesis19. While tumors with HCC characteristics could be observed in TAK1LPC-KO mice as early as 16-20 weeks, only small dysplastic foci are detected in NEMOLPC-KO mice of similar age5,19. Considering that TAK1 and NEMO are both essential for NF-κB activation and pro-survival RIPK1 phosphorylation, the aggravated liver damage and early tumorigenesis in TAK1LPC-KO mice likely reflects additional TAK1 functions. Indeed, TAK1 regulates the activation of mitogen-activated protein kinase (MAPK) pathways, whereas it inhibits TGFβ-induced Smad2/3 activation that can induce hepatocyte death20,21. Furthermore, TAK1 was shown to regulate autophagy induction through AMP-activated protein kinase phosphorylation22.
TAK1LPC-KO mice are generated using Cre recombinase expression driven by albumin promoter and alpha-fetoprotein enhancer (Alfp-Cre) that leads to TAK1 deletion both in hepatocytes and cholangiocytes19,23. Interestingly, TAK1ΔHep mice, where TAK1 is deleted exclusively in hepatocytes using Albumin Cre, show chronic hepatocellular damage, compensatory proliferation, hepatitis and fibrosis, but have no biliary damage and develop HCC at the age of one year19,24. These differences have suggested that biliary damage promotes early tumorigenesis in TAK1LPC-KO mice.
In this study, we have used TAK1LPC-KO mice to dissect genetically the relative contribution of key molecules regulating apoptosis and necroptosis, such as FADD, RIPK1, RIPK3, NEMO and three death receptors (3DR; TNFR1, Fas and TRAILR), in hepatocellular and biliary damage and hepatocarcinogenesis. Our data suggest that cholangiocytes can undergo RIPK1 kinase activity-dependent necroptosis, whereas hepatocytes die exclusively by FADD-dependent apoptosis. Although not driving hepatocarcinogenesis, necroptosis-mediated biliary damage can act as “rheostat” regulating its aggressiveness.
Results
FADD-dependent hepatocyte apoptosis drives hepatocarcinogenesis in TAK1LPC-KO mice
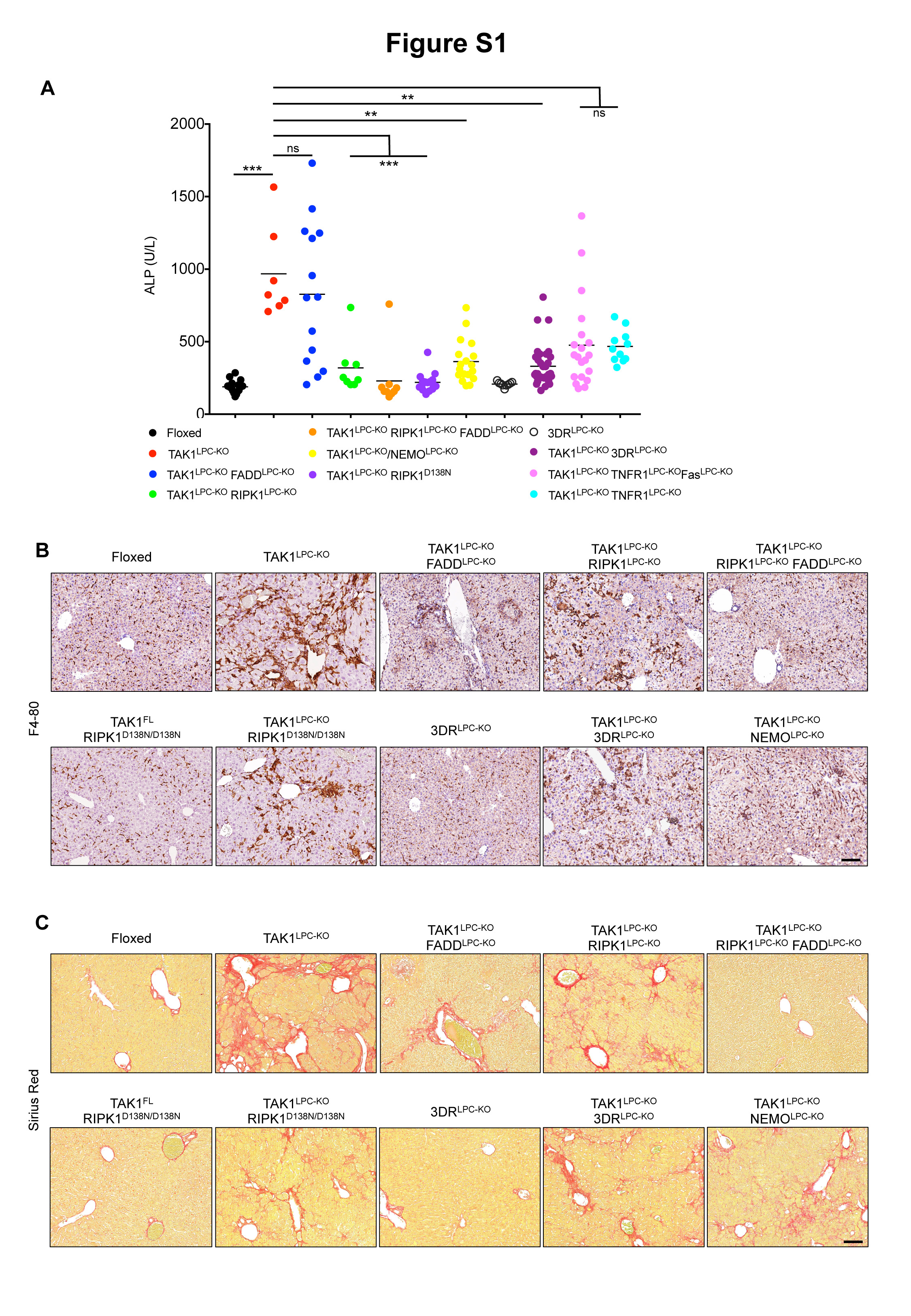
To investigate the role of apoptosis and necroptosis in chronic hepatocellular and biliary damage and hepatocarcinogenesis, we generated TAK1LPC-KO mice by crossing Tak1FL/FL 25 with Alfp-Cre mice23. As previously reported19, 6-week-old TAK1LPC-KO mice developed severe hepatobiliary disease characterized by increased hepatocyte apoptosis and biliary damage, as indicated by elevated serum ALT, total bilirubin and Alkaline Phosphatase (ALP) levels, increased cleaved Caspase-3 (CC3) and the presence of necrotic foci in periportal areas (Fig. 1A,B,D-E and S1A). Liver damage induced a strong regenerative response marked by increased number of Ki-67+ hepatocytes and cholangiocytes, expansion of Cytokeratin 19 (CK19)-positive cholangiocytes and hepatic progenitor cells (also known as ductular reaction) and elevated levels of Cyclin D1 and the oncofetal marker afp (Fig. 1D-F). In addition, we observed strong inflammation evidenced by increased Tnf mRNA expression and F4/80+ macrophage infiltration both at periportal areas and in the liver parenchyma, and increased fibrosis detected by Sirius Red staining and Tgfb1 mRNA expression (Fig. 1F and S1B-C). At this age, TAK1LPC-KO mice already exhibited an abnormal liver appearance with many macroscopically visible small nodules (Fig. 1C).
Fig. 1. FADD is required for hepatocellular but not biliary damage in TAK1LPC-KO mice.

(A) Serum ALT and total Bilirubin levels in 6-week-old mice. (B) Immunoblot analysis of total (C3) and cleaved Caspase-3 (CC3) in whole liver lysates from 6-week-old mice with the indicated genotypes. GAPDH was used as loading control. (C) Representative liver images from 6-week-old animals with the indicated genotypes. (D) Representative images of liver sections from 6-week-old mice with the indicated genotypes stained with H&E or immunostained for CC3, Ki-67 and cytokeratin 19 (CK19). Arrowheads point at intact bile ducts in periportal areas in floxed mice or necrotic foci observed in the same areas in TAK1LPC-KO and TAK1LPC-KO FADDLPC-KO livers. Arrows denote proliferating bile duct cells in the latter two genotypes. Asterisks indicate necrotic areas in the liver parenchyma only seen in TAK1LPC-KO FADDLPC-KO mice. (E) Quantification of CC3 and Ki-67 immunostaining shown in D. (F) qRT-PCR gene expression analysis in liver samples of mice with the indicated genotypes. Graphs show relative mRNA expression normalized to Tbp. The number of mice analyzed (n) is indicated in every graph. All graphs show mean values of the individual data points. *p <0.05, **p <0.01, ***p <0.005. Bars: (C) 1 cm; (D) 100 μm.
To assess the effect of FADD-dependent apoptosis on the liver damage of TAK1LPC-KO mice, we generated mice with combined deficiency of FADD and TAK1 in LPCs. Similar to TAK1LPC-KO Caspase-8LPC-KO mice9, TAK1LPC-KO FADDLPC-KO mice showed macroscopically smooth liver appearance and significant reduction in serum ALT levels, while the number of apoptotic and proliferating hepatocytes, inflammation, fibrosis and ductular reaction were nearly down to the levels of control mice (Fig. 1 and S1B-C). However, TAK1LPC-KO FADDLPC-KO mice still exhibited necrotic foci at the periportal areas, while additional scattered necrotic lesions were detected in the liver parenchyma (Fig. 1D), as previously reported for FADDLPC-KO and Caspase-8LPC-KO mice6,7. Despite the drastic decrease in hepatocellular damage, FADD ablation did not significantly reduce biliary damage, with only one third of TAK1LPC-KO FADDLPC-KO mice showing relatively low total bilirubin and ALP levels (Fig. 1A and S1A).
Many TAK1LPC-KO mice developed cachexia and died spontaneously prior to 30 weeks of age. The livers of TAK1LPC-KO mice surviving between 21-39 weeks exhibited macroscopically visible tumors (Fig. 2A and S2), and histopathological analysis revealed the presence of ceroid/bile-containing macrophages, marked portal inflammation and bile duct. Moreover, all TAK1LPC-KO mice displayed many dysplastic nodules with diameter up to 5 mm and some well-differentiated HCCs, both characterized by peritumoral fibrosis as illustrated by Masson’s trichrome staining (Fig. 2B-D). The tumor burden of TAK1LPC-KO mice was also evident by the increased liver-to-body weight (LW/BW) ratio (Fig. 2E). Strikingly, TAK1LPC-KO FADDLPC-KO mice did not show spontaneous mortality and lacked dysplastic lesions or HCC when aged to 28 weeks or 1 year (Fig. 2A-E, S2 and S3), although most of these mice exhibited as high bilirubinemia as TAK1LPC-KO mice, jaundice, and signs of liver fibrosis (Fig. 2B,F). These results show that FADD-dependent hepatocyte apoptosis is essential for driving hepatocarcinogenesis in TAK1LPC-KO mice even in the presence of strong biliary damage.
Fig. 2. LPC-specific deletion of FADD prevents hepatocarcinogenesis in TAK1LPC-KO mice.

(A) Representative liver images from 28-week-old mice with the indicated genotypes. (B) Representative images of Masson´s trichrome stained liver sections from 28-week-old mice with the indicated genotypes. HCC is outlined and marked with an asterisk. (C) Histopathological evaluation of hepatocarcinogenesis in samples of 21-to-28-week-old mice. (D) Quantification of dysplastic nodules/foci per lamella in liver sections of mice with the indicated genotype. (E-F) Liver:body weight (LW/BW) ratio (E) and serum ALT and total Bilirubin levels in 28-week-old mice with the indicated genotypes (F). The number of mice analyzed (n) is indicated in every graph. All graphs show mean values of the individual data points. *p <0.05, **p <0.01, ***p <0.005. Bars: (A) 1 cm; (B) 100 μm.
RIPK1-dependent necroptosis drives biliary damage in TAK1LPC-KO mice
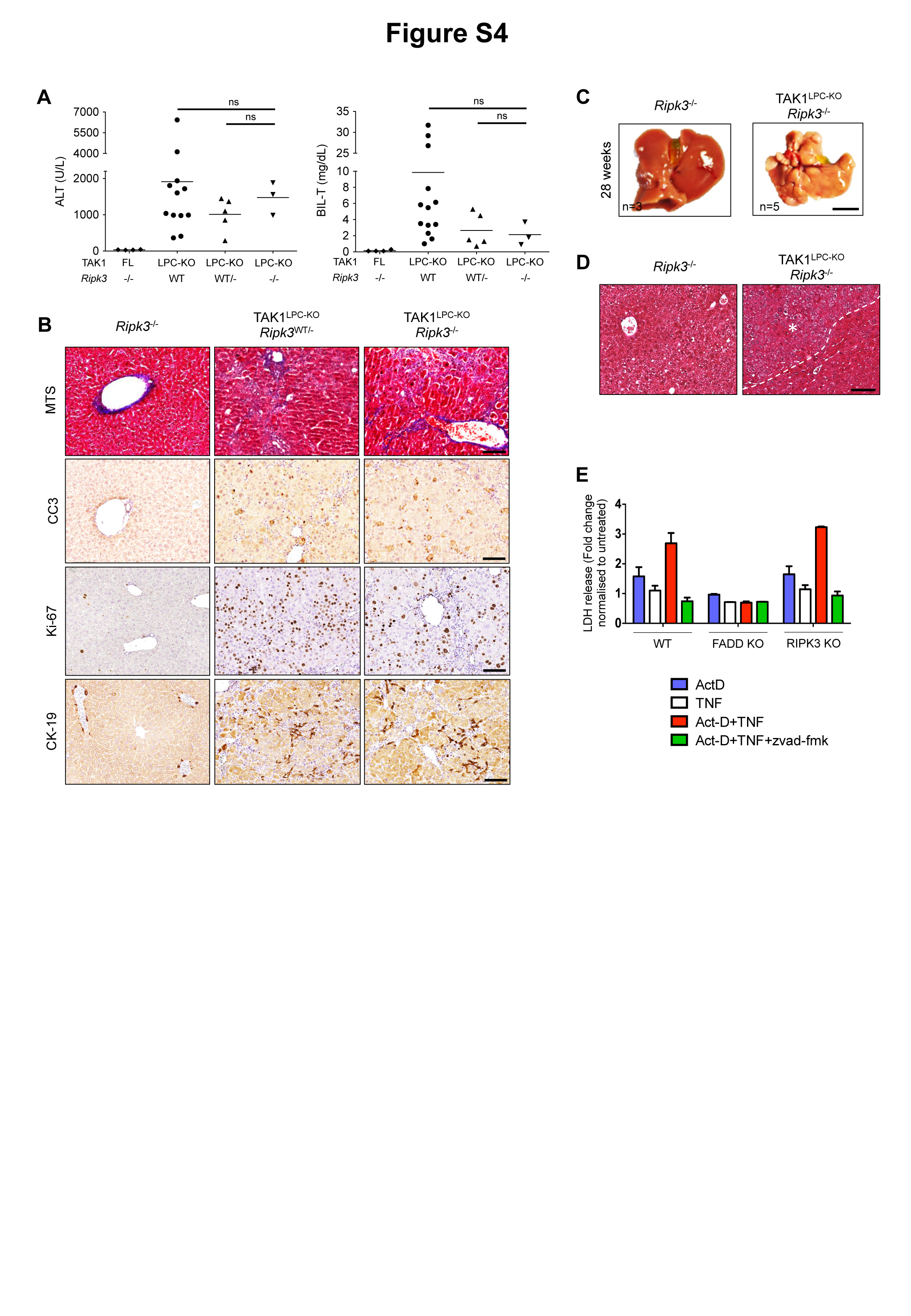
To examine the impact of inhibiting necroptosis in chronic liver damage, we generated TAK1LPC-KO Ripk3-/- mice. As shown previously9, systemic RIPK3 deficiency reduced biliary damage in 6-week-old mice but did not prevent hepatocyte death and the strong regenerative response elicited from it (Fig. S4A-B). Moreover, 28-week-old TAK1LPC-KO Ripk3-/- mice exhibited multiple dysplastic nodules (Fig. S4C-D).
To investigate the contribution of RIPK1 in TAK1LPC-KO mice, we generated mice lacking both TAK1 and RIPK1 in LPCs. TAK1LPC-KO RIPK1LPC-KO mice appeared healthy and did not show signs of cholestatic disease. At 6 weeks of age, these mice showed no significant reduction in ALT levels compared to TAK1LPC-KO mice. In stark contrast, total bilirubin and ALP were reduced to nearly normal levels (Fig. 3A and S1A). Accordingly, TAK1LPC-KO RIPK1LPC-KO mice had only slightly reduced number of CC3+ apoptotic hepatocytes compared to TAK1LPC-KO mice, but did not have substantial number of dying or Ki-67+ cholangiocytes or the characteristic periportal necrotic lesions seen in TAK1LPC-KO mice (Fig. 3B,D,E). The livers of 6-week-old TAK1LPC-KO RIPK1LPC-KO mice appeared macroscopically normal, while histological analysis revealed a moderate decrease in hepatocyte proliferation and ductular reaction (Fig. 3C-F). Furthermore, these mice showed only slight reduction in hepatitis and fibrosis indicators compared to TAK1LPC-KO mice (Fig.3F and S1B-C). Additional LPC-specific deletion of FADD normalized serum ALT levels and fully prevented inflammation, fibrosis and the remaining histopathological features of TAK1LPC-KO RIPK1LPC-KO mice (Fig. 3 and S1), suggesting that RIPK1-independent FADD/Caspase-8 signaling mediates apoptosis in TAK1-deficient hepatocytes.
Fig. 3. RIPK1 kinase activity drives cholestasis but is largely dispensable for hepatocellular damage in TAK1LPC-KO mice.

(A) Serum levels of ALT and total Bilirubin in 6-week-old mice with the indicated genotypes. (B) Immunoblot analysis of whole liver lysates from 6-week-old mice with the indicated genotypes. GAPDH was used as loading control. (C) Representative liver images from 6-week-old animals with the indicated genotypes. (D) Representative images of liver sections from 6-week-old mice with the indicated genotypes stained with H&E or immunostained for CC3, Ki-67 and CK19. (E) Quantification of CC3 and Ki-67 immunostaining shown in D. (F) qRT-PCR gene expression analysis in liver samples of mice with the indicated genotypes. Graphs show relative mRNA expression normalized to Tbp. The number of mice analyzed (n) is indicated in every graph. All graphs show mean values of the individual data points. *p <0.05, **p <0.01, ***p <0.005. Bars: (C) 1 cm; (D) 100 μm.
To address the role of RIPK1 kinase activity in TAK1LPC-KO mice, we crossed them to Ripk1D138N/D138N knock-in mice, which express kinase inactive RIPK1 26. Similar to complete lack of RIPK1, lack of RIPK1 kinase activity moderately decreased serum ALT levels but completely abrogated the bilirubinemia of TAK1LPC-KO mice (Fig. 3A). The livers of 6-week-old TAK1LPC-KO Ripk1D138N/D138N mice appeared macroscopically normal, lacked periportal necrotic lesions and showed reduced numbers of CC3+ apoptotic hepatocytes compared to TAK1LPC-KO mice. Accordingly, a modest decrease in most markers of hepatocyte proliferation, inflammation, fibrosis and ductular reaction was observed in TAK1LPC-KO Ripk1D138N/D138N compared to TAK1LPC-KO mice, although all these markers still remained significantly higher than in control mice (Fig. 3B-F and S1). Taken together, these genetic data suggest that the hepatocellular damage in TAK1LPC-KO mice is due to FADD/Caspase-8 mediated apoptosis, which is only partly driven by RIPK1 kinase activity and is largely independent of necroptosis. On the contrary, the biliary damage is dependent on RIPK1/RIPK3-mediated necroptosis.
Unlike hepatocytes, cholangiocytes can undergo necroptosis
The relevance of hepatocyte necroptosis in chronic liver disease mouse models has been debated13,27,28. Since Alfp Cre induces gene deletion both in hepatocytes and cholangiocytes23, we compared these two cell types for their ability to undergo necroptosis or apoptosis in vitro. Low passage, not transformed/immortalized, normal mouse cholangiocytes (NMCs) and primary hepatocytes were treated with TNF and the Smac mimetic (SM) Birinapant, a combination known to induce RIPK1-dependent cell death29. Indeed, TNF/SM-induced robust death in both cell types (Fig. 4A). Strikingly, while the pan-Caspase inhibitor zVAD-fmk fully prevented death of hepatocytes, it had no effect in NMCs. In contrast, the RIPK1 kinase inhibitor Nec1s inhibited almost completely TNF/SM-induced death in NMCs. Similar results were obtained in the presence of the TAK1 inhibitor 5Z-7-Oxozeaenol used to mimic TAK1 deficiency in LPCs (Fig. 4A). Additionally, we examined the susceptibility of these two cell types to TNF and actinomycin D (ActD), a combination inducing RIPK1-independent cell death29 (Fig. 4B). Again, TNF/ActD-induced death was fully rescued by zVAD-fmk in primary hepatocytes but as expected, Nec1s showed no protective effect. In NMCs, while zVAD-fmk or Nec1s treatment alone did not prevent cell death, combined treatment conferred full protection. Accordingly, FADD-deficient primary hepatocytes were resistant to TNF/ActD-induced cell death, whereas RIPK3-deficient hepatocytes were as sensitive as the wildtype ones (Fig. S4E). These results indicate that hepatocytes can exclusively undergo apoptosis, while cholangiocytes can undergo necroptosis when apoptosis is inhibited.
Fig. 4. Cholangiocytes, but not hepatocytes, can undergo necroptosis.

(A-B) Cell death expressed as released vs. total LDH ratio in normal mouse cholangiocytes and primary hepatocytes treated with TNF/Smac mimetic (A) or TNF/ActinomycinD (B) together with indicated inhibitors for 24 h (DMSO, C; T, TNF; S, Smac mimetic; Z, zVAD-fmk; N, Nec1s; A, Actinomycin D). Cells additionally pre-treated with 5Z-7-Oxozeaenol (TAK1-i) are shown in grey bars. The results of one representative experiment (n=3) are shown. Error bars represent SD of the technical triplicate. (C) Immunoblot analysis of apoptosis- and necroptosis-associated molecules in lysates from normal mouse cholangiocytes and primary hepatocytes treated with vehicle (PBS), TNF, Interferon α (IFNα) or IFNγ for 24 h. GAPDH was used as loading control. (D) Immunostaining for RIPK3 in liver sections from 6-week-old mice with the indicated genotypes. Boxed area on the right is shown magnified and the arrow points at high RIPK3-expressing bile epithelial cells levels observed in TAK1LPC-KO mice. Note the background RIPK3 staining in apoptotic hepatocytes (arrowheads) of TAK1LPC-KO Ripk3-/- mice. (E) Immunostaining for phospho-RIPK3 in liver sections from 6-week-old mice with the indicated genotypes. Note the occasional detection of pRIPK3+ cholangiocytes in bile ducts of TAK1LPC-KO and TAK1LPC-KO FADDLPC-KO mice. Bars: (D-E) 100 μm.
To investigate the reason for the differential behaviour between hepatocytes and cholangiocytes, we compared the protein levels of key apoptosis- and necroptosis-associated molecules by immunoblotting (Fig. 4C). The lack of RIPK3 expression in hepatocytes compared to its robust expression in NMCs was the most prominent difference observed. Interestingly, hepatocytes appear to express very high levels of FADD compared to NMCs. Since the expression of all other critical necroptosis-mediating molecules (MLKL, RIPK1, ZBP1) is similar between the two cell types, the absence of RIPK3 is likely responsible for hepatocyte resistance to necroptosis.
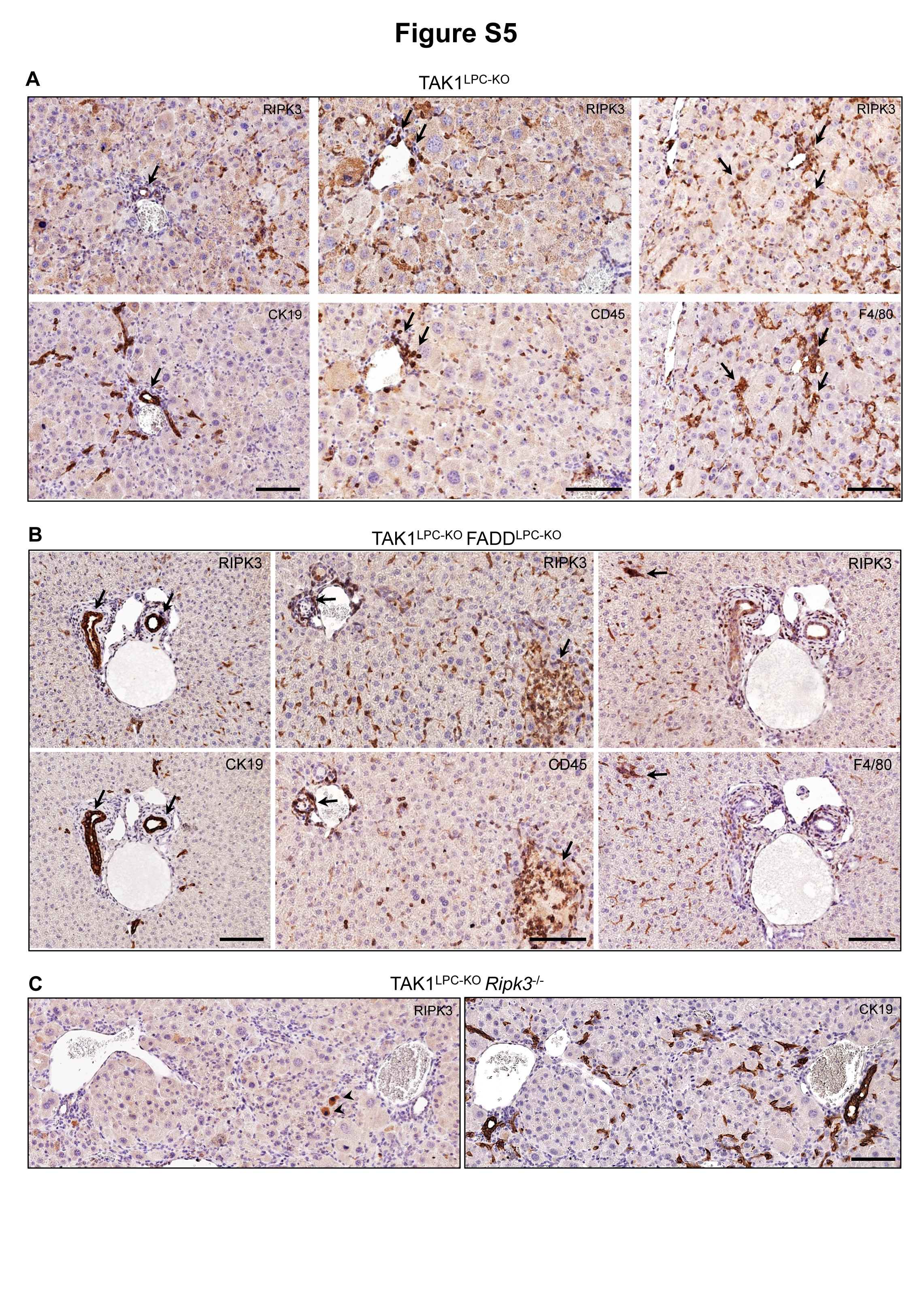
Supporting these data, immunostaining with a highly specific anti-RIPK3 antibody30 showed that RIPK3 was predominantly expressed in non-parenchymal liver cells, while some non-specific staining was observed in hepatocytes undergoing apoptosis (Fig. 4D). Immunohistochemistry on serial liver sections showed that RIPK3-expressing cells represented mainly F4/80+ macrophages and other CD45+ leukocytes (Fig. S5). Interestingly, bile ducts in TAK1LPC-KO and TAK1LPC-KO FADDLPC-KO mice often expressed high levels of RIPK3, although this was rarely observed in wildtype mice (Fig. 4D and S5). Moreover, immunohistochemical detection of phospho-RIPK3 was not observed in hepatocytes, but mostly in liver leukocytes and to a lower extent in cholangiocytes of TAK1LPC-KO and TAK1LPC-KO FADDLPC-KO mice (Fig. 4E). The low frequency of necroptotic cholangiocytes detected is somewhat expected as dying cells are rapidly shed into the bile duct lumen. Altogether, these results are in agreement with previous studies reporting that RIPK3-dependent necroptosis does not contribute to hepatocellular injury8,10,11,30–32, while they support a role for necroptosis in biliary injury33.
RIPK1 regulates HCC development in TAK1LPC-KO mice
To assess the role of RIPK1 scaffolding properties and kinase activity in hepatocarcinogenesis in TAK1LPC-KO mice, we followed mice up to 52 weeks of age. In contrast to TAK1LPC-KO mice, both TAK1LPC-KO RIPK1LPC-KO and TAK1LPC-KO Ripk1D138N/D138N mice survived well beyond the age of 28 weeks without developing lethal cholestasis, had normal serum bilirubin levels, and showed moderately reduced ALT levels at 28 weeks of age (Fig. 5A). Moreover, 28-week-old TAK1LPC-KO RIPK1LPC-KO and TAK1LPC-KO Ripk1D138N/D138N mice exhibited aberrant liver architecture but showed almost no macroscopically visible tumors bigger than 2 mm in diameter (Fig. 5B and S2). Histological analysis revealed that the majority of the examined livers had many but predominantly small clear cell foci/nodules, while no high-grade dysplastic nodules or HCC were identified (Fig. 5C-E). Accordingly, both mouse strains had a LW/BW ratio similar to control mice and significantly lower than TAK1LPC-KO mice (Fig. 5F). Upon ageing to one year, TAK1LPC-KO RIPK1LPC-KO and TAK1LPC-KO Ripk1D138N/D138N mice eventually developed liver tumors with premalignant or HCC features, although in TAK1LPC-KO Ripk1D138N/D138N mice, there was a clear tendency for delayed hepatocarcinogenesis (Fig. S3). Finally, no tumors or dysplastic lesions were observed in TAK1LPC-KO RIPK1LPC-KO FADDLPC-KO mice (Fig. 5, S2 and S3). Therefore, lack of RIPK1 or inhibition of its kinase activity significantly ameliorated early hepatocarcinogenesis but could not prevent HCC development in aged TAK1LPC-KO mice.
Fig. 5. RIPK1 promotes early hepatocarcinogenesis in TAK1LPC-KO mice through its kinase activity.

(A) Serum levels of ALT and total Bilirubin in 28-week-old mice with the indicated genotypes. (B) Representative liver images from 28-week-old mice with the indicated genotypes. (C) Representative images of Masson´s trichrome stained liver sections from 28-week-old mice with the indicated genotypes. Dysplastic nodules are outlined and marked with an asterisk. (D-E) Histopathological evaluation of hepatocarcinogenesis (D) and quantification of dysplastic nodules/foci per lamella (E) in liver sections of 28-week-old mice with the indicated genotype. (F) LW/BW ratio in 28-week-old mice with the indicated genotypes. The number of mice analyzed (n) is indicated in every graph. All graphs show mean values of the individual data points. *p <0.05, **p <0.01, ***p <0.005. Bars: (B) 1 cm; (C) 100 μm.
NEMO mediates biliary damage but is dispensable for liver tumorigenesis in TAK1LPC-KO mice
TAK1 acts sequentially with the IKK complex to promote cell survival both by activating NF-κB signaling and through NF-κB-independent mechanisms3,14,15. Surprisingly, a former study showed that lack of NEMO in LPCs prevented cholestasis and hepatocarcinogenesis in 21-week-old TAK1LPC-KO mice, although it did not reduce hepatocyte death19. Considering reports suggesting that NEMO can have a scaffolding pro-necroptosis role under certain experimental conditions34,35, we decided to re-investigate these intriguing findings by generating and analyzing TAK1LPC-KO NEMOLPC-KO mice.
Indeed, we confirmed that TAK1LPC-KO NEMOLPC-KO mice did not show spontaneous lethality or signs of jaundice. Accordingly, at 6 and 12 weeks of age, serum ALT levels of TAK1LPC-KO NEMOLPC-KO mice were comparable to the single knockout mice, while total bilirubin was in most animals similar to wildtype controls (Fig. 6A-B). However, in contrast to the study by Bettermann et al. 19, our macroscopic and histopathological evaluation revealed that 12-week-old TAK1LPC-KO NEMOLPC-KO mice already exhibited many dysplastic foci and nodules resembling more the phenotype of TAK1LPC-KO rather than NEMOLPC-KO mice (Fig. 6C). All analyzed livers from 28- and 52-week-old mice presented macroscopically visible tumors, and almost all of them exhibited high-grade dysplastic nodules or HCC (Fig. 6D-F), stages that could be detected only in 52-week-old NEMOLPC-KO mice5,8. Interestingly, most one-year-old TAK1LPC-KO NEMOLPC-KO mice did eventually develop high bilirubinemia and had a higher LW/BW ratio indicative of increased tumor load (Fig. 6B,G). These results suggest that NEMO promotes biliary damage during the early stages of the disease development but only marginally delays early hepatocarcinogenesis in TAK1LPC-KO mice.
Fig. 6. NEMO deletion strongly prevents biliary damage but leads to only a marginal delay in HCC progression in TAK1LPC-KO mice.

(A-B) Serum levels of ALT (A) and total Bilirubin (B) in 6-, 12- and 52-week-old mice with the indicated genotypes. (C-E) Representative liver photos and images of liver sections stained for Masson’s trichrome from 12- (C), 28- (D), and 52-week-old mice (E) with the indicated genotypes. Dysplastic nodules or HCC are outlined and marked with an asterisk in D and E. (F) Histopathological evaluation of hepatocarcinogenesis in samples of liver samples from mice with the indicated age. (G) LW/BW ratio in 52-week-old mice with the indicated genotypes. The number of mice analyzed (n) is indicated in every graph. All graphs show mean values of the individual data points. *p <0.05, **p <0.01, ***p <0.005. Bars: (C-E) upper, 1 cm; lower, 100 μm.
TNFR1/DR signaling triggers biliary but not hepatocellular damage in in TAK1Δhep mice
TNFR1 signaling inhibition was previously shown to ameliorate liver damage, hepatitis and fibrosis in TAK1Δhep mice, which do not show significant biliary damage24. To address the role of DR signaling in mice that show both hepatocellular and biliary damage, we have generated TAK1LPC-KO mice that also lack 3 death receptors (3DR; TNFR1, Fas and TRAILR) in LPCs7. Strikingly, TAK1LPC-KO 3DRLPC-KO mice had nearly normal serum bilirubin and ALP levels but not significantly reduced ALT levels compared to TAK1LPC-KO mice (Fig. 7A and S1A). A major contribution of TRAILR in biliary damage was excluded, since TAK1LPC-KO mice only lacking TNFR1 and Fas showed comparable decrease in total bilirubin levels. Moreover, TAK1LPC-KO TNFR1LPC-KO mice were almost equally protected from biliary damage as TAK1LPC-KO 3DRLPC-KO mice (Fig. 7A), suggesting that TNFR1 signaling is the main driver of cholangiocyte death. Similar to TAK1LPC-KO RIPK1LPC-KO and TAK1LPC-KO Ripk1D138N/D138N mice that did not develop cholestasis, 6-week-old TAK1LPC-KO 3DRLPC-KO mice did not show macroscopically visible liver nodules, although they showed elevated numbers of CC3+ hepatocytes, fibrosis, hepatocytes proliferation and ductular reaction (Fig. 7B-C and S1B-C).
Fig. 7. Death receptor signaling drives biliary but not hepatocellular damage, and significantly delays hepatocarcinogenesis in TAK1LPC-KO mice.

(A) Serum levels of ALT and total Bilirubin in 6-week-old mice with the indicated genotypes. (B) Representative liver images from 6-week-old animals with the indicated genotypes. (C) Representative images of liver sections from 6-week-old mice with the indicated genotypes stained with H&E or immunostained for CC3, Ki-67 and CK19. (D-E) Representative liver photos (D) and images of liver sections stained for Masson’s trichrome (E) from 28-week-old mice with the indicated genotypes. Dysplastic nodules are outlined and marked with an asterisk. (F-G) Histopathological evaluation of hepatocarcinogenesis (F) and LW/BW ratio (G) in 28-week-old mice with the indicated genotypes. The number of mice analyzed (n) is indicated in every graph. All graphs show mean values of the individual data points. *p <0.05, **p <0.01, ***p <0.005. Bars: (B,D) 1 cm; (C,E) 100 μm.
TAK1LPC-KO 3DRLPC-KO mice were aged to 28 weeks without showing spontaneous lethality. Unlike TAK1LPC-KO mice, only 4 out 16 TAK1LPC-KO 3DRLPC-KO mice developed one or more liver tumors larger than 2 mm, although all livers displayed aberrant architecture (Fig. 7D). Accordingly, histological analysis revealed the presence of multiple dysplastic foci/nodules but not HCC in TAK1LPC-KO 3DRLPC-KO mice, while even LPC-specific TNFR1 deficiency alone conferred significant delay in hepatocarcinogenesis of TAK1LPC-KO mice (Fig. 7E-F). The reduced tumor burden in these mice was also reflected in the decreased LW/BW ratio compared to TAK1LPC-KO mice (Fig. 7G). These results suggest that the spontaneous death of TAK1-deficient cholangiocytes observed in vivo is triggered predominantly by TNFR1 activation, while apoptosis of TAK1-deficient hepatocytes is 3DR-independent.
Discussion
Chronically elevated hepatocyte apoptosis has been recognized as a prominent driver of liver disease pathogenesis5–12. In contrast, the role of necroptosis in acute and chronic liver injury models remains debatable due to non-conclusive or contradicting data13. Here, we show that in TAK1LPC-KO mice both apoptotic and necroptotic cell death pathways with distinct molecular requirements contribute to liver pathology, albeit in different cell types (Fig. 8). TAK1-deficient hepatocytes are prone to FADD/Caspase-8-mediated apoptosis, but this pathway is independent of TNFR1, FAS and TRAILR (3DR), and only partially dependent on RIPK1 kinase activity. On the contrary, TAK1-deficient bile epithelial cells are susceptible to RIPK1/RIPK3-dependent necroptosis triggered by DRs and primarily TNFR1.
Fig. 8. Model for the role of cell death in TAK1-deficient LPCs.

Proposed model for the contribution of hepatocyte apoptosis and bile epithelial cell necroptosis in the development of chronic liver inflammation, regeneration and hepatocarcinogenesis in TAK1LPC-KO mice. The observed cholestasis aggravates inflammation likely by inducing more hepatocyte apoptosis through released bile acid toxicity. Although the role of necroptosis per se is currently unclear, it could regulate the aggressiveness of hepatocarcinogenesis by shaping the inflammatory microenvironment.
Hepatocytes are considered refractory to necroptosis due to very low RIPK3 expression (Ref 30 and this study). Nevertheless, upregulation of RIPK3 expression in the liver is reported in liver injury models of alcoholic and non-alcoholic steatohepatitis27,28. Our immunostaining data support previous studies showing that in healthy liver, RIPK3 is mainly expressed in resident immune cells, such as Kupffer cells30,36. In chronically injured liver of TAK1LPC-KO mice, RIPK3 expression was higher but this reflected strong inflammatory cell infiltration, as well as an increased RIPK3 expression in cholangiocytes rather than in hepatocytes. Accordingly, phospho-RIPK3, which is used as marker of an activated necroptotic pathway, was observed mainly in immune cells and a small fraction of cholangiocytes but not in hepatocytes. Of note, RIPK3 has also necroptosis-independent, pro-inflammatory functions, particularly in immune cells, which could affect liver disease development37. However, in TAK1LPC-KO mice, the strong amelioration in biliary damage by RIPK3 deficiency is likely due to necroptosis inhibition in cholangiocytes, as LPC-specific deletion of RIPK1 conferred similar protection.
RIPK1 kinase activity is dispensable for proinflammatory and prosurvival signaling but it can promote both apoptosis and necroptosis3. The in vivo relevance of this pro-apoptotic function in hepatocytes was recently exemplified in NEMOLPC-KO mice or mice treated with TNF and the IKK1/2 inhibitor TPCA-18,16. The present study illustrates the existence of a RIPK1 kinase-dependent necroptotic pathway that can be specifically activated in cholangiocytes and may be relevant for biliary disease. In line with the pro-necroptotic role of RIPK1’s scaffolding properties4, necroptosis also prevented when RIPK1 was completely absent from TAK1-deficient cholangiocytes. In contrast, lack of RIPK1 from TAK1-, NEMO- or IKK1/2-deficient hepatocytes did not significantly reduce hepatocellular damage (Refs 8,10 and this study). In NEMOLPC-KO mice, this was due to the presence of an alternative, TRADD-mediated apoptotic pathway 8, and a recent study confirmed that TRADD-dependent apoptosis is activated in several embryonic tissues in RIPK1-deficient mice38. Furthermore, expression of kinase-inactive RIPK1 conferred only moderate protection in TAK1LPC-KO mice from hepatocyte apoptosis, suggesting that RIPK1 kinase-independent pathways are also involved. These could include in addition to TRADD-dependent apoptosis, death induced by TGFβ/Smad activation or inhibition of autophagy, which have been reported in TAK1-deficient hepatocytes22,24. Whichever of these alternative pathways is activated, it should converge to FADD/Caspase-8 activation, as hepatocyte apoptosis was fully rescued in TAK1LPC-KO RIPK1LPC-KO FADDLPC-KO mice.
Our study confirmed the previously reported role of NEMO in driving necroptosis-mediated biliary damage in the absence of TAK119. A necroptosis-promoting role of NEMO has been described in mouse embryonic fibroblasts either by acting as scaffold nucleating RIPK1/RIPK3 necrosome formation or by inducing mitochondria permeabilization34,35. It is, however, whether NEMO facilitates cholangiocyte necroptosis or drives biliary damage through other mechanisms, and whether this pathway operates in other RIPK3-competent cell types. In contrast, our results did not confirm the reported significant inhibition of early hepatocarcinogenesis in TAK1LPC-KO NEMOLPC-KO mice19, since high-grade dysplastic nodules or HCC could be identified in the livers of 28- and 52-week-old animals. The reason for this discrepancy is not clear. A possible explanation could be the fact that Bettermann and colleagues used mice bred on a mixed C57/BL6-SV129Ola genetic background, while our mice were bred on pure C57/BL6 background, which could result in different genetic susceptibility to HCC development.
TNFR1 has been partly implicated in triggering hepatocellular damage in TAK1ΔHep mice24. However, our data support the involvement of DRs, and particularly of TNFR1, primarily in inducing cholangiocyte necroptosis but not hepatocyte apoptosis in TAK1LPC-KO mice. Accordingly, DRs were also shown to be dispensable for spontaneous hepatocyte apoptosis in NEMOLPC-KO and HOIP/LUBACΔHep mice7,8,39. The marginal reduction in ALT levels observed in TAK1LPC-KO 3DRLPC-KO or TAK1LPC-KO RIPK1LPC-KO mice is probably an indirect effect of preventing bile duct destruction. Conversely, TNFR1 is clearly involved as inducer of hepatocyte apoptosis in RIPK1LPC-KO, NEMOLPC-KO, and TAK1LPC-KO mice upon acute liver injury models, triggered by LPS, TNF, Concanavalin A or α-galactosyl ceramide injection, which cause release of proinflammatory cytokines by activated immune cells5,8,11,19,31,32,40,41. Yet, what drives spontaneous hepatocyte apoptosis in the aforementioned mice remains unresolved. The role of DR3, DR6 and TLR3 pathways or DR-independent formation of Complex IIb induced by replicative/genotoxic stress during liver organogenesis the first weeks after birth, should be subject of future investigation3,42,43.
In agreement with a recent study12, our results support the decisive role of hepatocyte apoptosis in driving hepatitis and HCC in TAK1LPC-KO mice. The combined strong hepatocellular and biliary damage observed in these mice leads to severe hepatitis and tissue regeneration. While most TAK1LPC-KO mice succumb to death due to cholestasis, the survivors develop accelerated HCC as early as 20-28 weeks of age (Fig. 8). Inhibition of hepatocyte apoptosis alone could effectively prevent HCC development even in the presence of strong biliary damage, moderate inflammation and fibrosis. When necroptosis of cholangiocytes was inhibited without a concomitant substantial reduction in hepatocyte apoptosis, lethal cholestasis was prevented but hepatocarcinogenesis still progressed albeit slower. It is noteworthy that out of those genetic modifications that prevented biliary damage in TAK1LPC-KO mice, expression of kinase-inactive or lack of RIPK1 led to the strongest delay in hepatocarcinogenesis. This effect is compatible with the proposed cell death-independent role of RIPK1 kinase activity in promoting DNA damage12 or an additional role of RIPK1 in promoting proliferation of transformed hepatocytes similar to that described in melanoma cells44.
In summary, our study in TAK1LPC-KO mice demonstrates the ability of hepatocytes to die exclusively through FADD/Caspase-8 mediated apoptosis, whereas cholangiocytes can undergo necroptosis. This striking difference between two cell types, which originate from the same hepatic progenitor cells, is likely due to RIPK3 availability. Intriguingly, a recent study has suggested that hepatocyte necroptosis generates a cytokine microenvironment that promotes the preferential development of intrahepatic cholangiocarcinomas from oncogenically transformed hepatocytes45. Considering that mature hepatocytes can be reprogrammed to cholangiocytes following liver injury46, it remains to be seen whether the reported necroptotically-dying cells are hepatocytes or cholangiocytes. Finally, an emerging question is whether RIPK1/RIPK3-necroptosis of cholangiocytes is relevant in human cholangiopathies. Interestingly, a recent study reported increased RIPK3 and MLKL levels in primary biliary cirrhosis patients and a partial protection of RIPK3-/- mice from biliary damage induced by bile duct ligation33, without however addressing the role of RIPK1 kinase activity. If confirmed as a universal mechanism, our findings suggest that preventing necroptosis using RIPK1 kinase inhibitors could be effective in treating patients with biliary injury, while it could prevent cholangiocarcinoma development.
Materials and methods
Animals
The following mouse lines were used in this study: Alfp-Cre 23, Tak1FL 25, NemoFL 47, FaddFL 48, Ripk1FL 49, Ripk1D138N 26, Ripk3-/- 50, Tnfr1FL 51, FasFL 52 and TrailrFL 53. All alleles were maintained on a C57BL/6 genetic background. Littermates carrying the floxed alleles but not the Alfp-Cre transgene served as controls. Animals were housed in individually ventilated cages in a specific pathogen-free (SPF) mouse facility at the Institute for Genetics, University of Cologne, kept under a 12-hr light cycle, and given a regular chow diet (Harlan diet no. 2918 or Prolab Isopro RMH3000 5P76) and water ad libitum. All animal procedures were conducted in accordance with European, national and institutional guidelines and protocols and were approved by local government authorities (Landesamt für Natur, Umwelt und Verbraucherschutz Nordrhein-Westfalen, Germany).
Biochemical serum analysis
Alanine aminotransferase (ALT), Alkaline Phosphatase (ALP) and Total Bilirubin (BIL-T) were measured in blood serum using standard assays in a Cobas C111 biochemical analyzer (Roche, Mannheim, Germany).
Histology, Immunohistochemistry (IHC), and quantification
Tissues were fixed overnight in 4% paraformaldehyde, embedded in paraffin, and cut into 5 μm sections. H&E, Masson’s trichrome. Sirius Red and IHC stainings were performed using standard procedures that have previously been described8,32. The following antibodies were used: cleaved Caspase-3 (R&D systems; clone AF835), Ki-67 (Dako, M724901), F4/80 (AbD Serotec, clone A3-1), CK19 (Developmental Studies Hybridoma Bank, TROMA-III), CD45 (ebioscience, 14-0451), p-RIPK3 (Cell Signaling, #57220) and RIP3 (gift from Genentech). Biotinylated secondary antibodies were purchased from Perkin Elmer, DAKO and Vector Laboratories. Stainings were visualized with ABC Kit Vectastain Elite (Vector Laboratories, PK6100) and DAB substrate (Dako, K3466). Stained sections were scanned using a Leica SCN400 slidescanner (Imaging facility, CECAD, Cologne) and representative snapshots were subsequently selected. For optimal visualization, levels and brightness/contrast adjustments of the pictures were equally applied using Adobe Photoshop.
IHC quantification was performed on 3-5 randomly selected high power fields (HPF) snapshots. Liver sections from 4-8 mice per genotype were analyzed. Quantification of Ki-67+ and active Caspase-3+ apoptotic hepatocytes cells was performed using Image J software (Version 1.48; http://rsbweb.nih.gov/ij/). For Ki-67 quantification, selected images were spectrally unmixed and the nuclear size was defined to match the manual counts of hepatocytes per field under the microscope. The images were then analyzed using the measure function by running a macro in batch mode. Data is represented as number of proliferating hepatocytes per HPF. For cleaved Caspase 3 evaluation appropriate pixel threshold intensity was equally applied on all selected pictures and the total brown area was determined using measure function. Data is represented as % of stained area per HPF.
Macroscopic and histopathological evaluation of liver tumors
Livers of sacrificed mice were excised, digitally photographed with their dorsal side exposed, and weighed to calculate the liver/body weight ratio. Histopathological evaluation was performed on H&E stained liver sections, where the number, size (diameter) and stage of tumors were assessed. For the hepatocarcinogenesis quantification, the following stages were used in ascending order of severity: No severe pathology, ≤ 5 or >5 dysplastic foci/nodules per liver lobe cross-section, and high-grade dysplastic nodules and early or well-differentiated HCC. In the corresponding graphs, each bar represents the % of livers per genotype in which the indicated stage was identified as the most advanced hepatocarcinogenesis stage.
Hepatocytes, cholangiocytes and cell death assay
Primary hepatocytes were isolated from 4-week-old mice by retrograde liver perfusion with a collagenase solution (15mg Collagenase D and 2mg Trypsin inhibitor in EBSS) through the caval vein, as previously described8. Hepatocytes were cultured in DMEM (Sigma-Aldrich) containing 2% FCS, penicillin and streptomycin in collagen-coated plates. Normal mouse cholangiocytes (NMCs) are not immortalized/transformed cells that were isolated and cultured as previously described54. The cells maintain their strong biliary phenotype (expression of markers such as AE2, CFTR and AQP1, and characteristic primary cilium in the apical membrane) for at least 15 cell passages before going into senescence. NMCs from passages 4 to 8 were used for our described in vitro experiments.
For cell death assays, hepatocytes and NMCs were treated with the following reagents: 20 ng/ml TNF (VIB Protein Service Facility, Ghent), 10 μM Birinapant (Smac mimetic, Biovision), 20ng/ml Actinomycin D (transcription inhibitor, Sigma-Aldrich), 20 μM zVAD-fmk (Pan-Caspase inhibitor, Enzo Life Sciences), 20 μM Necrostatin-1s (RIPK1 kinase inhibitor, Biovision) and 2 (in NMCs) or 10 μM (in primary hepatocytes) 5Z-7-Oxozeaenol (TAK1 inhibitor, Sigma-Aldrich) for 24 hr. Cell death was estimated by measuring LDH release CytoTox 96® Nonradioactive Cytotoxicity Assay kit (Promega, G1780) according to the manufacturer’s instructions. The experiments were performed in triplicates using primary hepatocytes that were isolated from at least 3 different mice per indicated genotype. At least 3 independent cell death experiments were performed in NMCs.
Immunoblotting
Protein extracts were prepared by homogenizing liver tissue, primary hepatocytes, or NMCs in lysis buffer (150 mM NaCl, 1% NP-40, 0.1% SDS in a 50 mM Tris buffer at pH 7.5) supplemented with phosSTOP phosphatase inhibitors and complete protease inhibitors (Roche). Lysates were separated by SDS-polyacrylamide gel electrophoresis (PAGE), transferred to Immobilon-P PVDF membranes (Millipore), and analyzed by immunoblotting. The following primary antibodies were used: cleaved Caspase-3 (Cell Signaling, #9661), Caspase 3 (Cell Signaling, #9662), RIPK1 (BD Biosciences, 610459), FADD (Millipore, 05-486), RIPK3 (Enzo Life Sciences, ADI-905-242-100), MLKL (Millipore, MABC604), Caspase-8 (Enzo Life Sciences, ALX-804-447-C100), ZBP1 (Adipogen, AG-20B-0010), CK-19 (Developmental Studies Hybridoma Bank, TROMA-III), Albumin (Santa Cruz, sc-46291), and GAPDH (Imgenex, IMG-5019A-1). Membranes were incubated with secondary, HRP-coupled antibodies (GE Healthcare and Jackson ImmunoResearch), followed by detection with ECL-Prime reagent (RPN 2106, GE Healthcare).
Quantitative real-time polymerase chain reaction (qRT-PCR)
Isolation of total RNA from frozen whole liver tissue and cDNA synthesis was performed as previously described8. qRT-PCR was performed using gene-specific TaqMan assays (Life technologies). mRNA expression of each gene was normalized to the expression of the housekeeping gene Tbp.
Statistical Analysis
For comparisons between 2 datasets, unpaired Student’s t-test or Mann-Whitney U test was used depending on whether the datasets fulfilled the D’Agostino & Pearson normality test criteria or not. For comparisons of multiple datasets that followed Gaussian distribution or not, 1-way ANOVA with a post-hoc Tukey’s test or a Kruskal-Wallis test with a post-hoc Dunn’s test was performed, respectively. In all dot plots, horizontal lines represent the mean values, while column graphs represent the mean ±SD. A P value of less than 0.05 was considered significant (*P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.005). Statistical analysis was performed with Prism version 6.0 (GraphPad).
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We thank V. Dixit and Genentech for anti-RIPK3 antibody and Ripk3-/- mice. We thank J. Kuth, C. Uthoff-Hachenberg, E. Gareus, B. Kühnel and E. Stade for technical assistance. This work was supported by grants from Worldwide Cancer Research (award no. 15-0228) and the European Research Council (grant agreement no. 323040 to MP). VK was supported by a Marie Curie Career Development Fellowship (FP7-PEOPLE-2010-IEF; proposal no. 275767).
Footnotes
Authors’ contributions
S.K-S. performed genetic crosses, tissue sampling, IHC, immunoblotting, qRT-PCR, and data interpretation and drafted the manuscript. V.K. conducted genetic crosses, tissue sampling, IHC analysis, in vitro experiments in NMCs and hepatocytes and data interpretation. S.S, K.H. and P.S. performed histopathological analysis of mouse livers. M.A. and G.K. provided Tak1FL and H.W. TrailrFL mice. J.M.B. provided NMCs. V.K. and M.P. coordinated the project and wrote the manuscript.
Conflict of Interest
The authors declare no conflict of interest.
References
- 1.Guicciardi ME, Malhi H, Mott JL, Gores GJ. Apoptosis and necrosis in the liver. Comprehensive Physiology. 2013;3(2):977–1010. doi: 10.1002/cphy.c120020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schwabe RF, Luedde T. Apoptosis and necroptosis in the liver: a matter of life and death. Nat Rev Gastroenterol Hepatol. 2018;15(12):738–52. doi: 10.1038/s41575-018-0065-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kondylis V, Pasparakis M. RIP Kinases in Liver Cell Death, Inflammation and Cancer. Trends Mol Med. 2019;25(1):47–63. doi: 10.1016/j.molmed.2018.10.007. [DOI] [PubMed] [Google Scholar]
- 4.Pasparakis M, Vandenabeele P. Necroptosis and its role in inflammation. Nature. 2015;517(7534):311–20. doi: 10.1038/nature14191. [DOI] [PubMed] [Google Scholar]
- 5.Luedde T, Beraza N, Kotsikoris V, van Loo G, Nenci A, De Vos R, et al. Deletion of NEMO/IKKgamma in liver parenchymal cells causes steatohepatitis and hepatocellular carcinoma. Cancer Cell. 2007;11(2):119–32. doi: 10.1016/j.ccr.2006.12.016. [DOI] [PubMed] [Google Scholar]
- 6.Liedtke C, Bangen JM, Freimuth J, Beraza N, Lambertz D, Cubero FJ, et al. Loss of caspase-8 protects mice against inflammation-related hepatocarcinogenesis but induces non-apoptotic liver injury. Gastroenterology. 2011;141(6):2176–87. doi: 10.1053/j.gastro.2011.08.037. [DOI] [PubMed] [Google Scholar]
- 7.Ehlken H, Krishna-Subramanian S, Ochoa-Callejero L, Kondylis V, Nadi NE, Straub BK, et al. Death receptor-independent FADD signalling triggers hepatitis and hepatocellular carcinoma in mice with liver parenchymal cell-specific NEMO knockout. Cell death and differentiation. 2014;21(11):1721–32. doi: 10.1038/cdd.2014.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kondylis V, Polykratis A, Ehlken H, Ochoa-Callejero L, Straub BK, Krishna-Subramanian S, et al. NEMO Prevents Steatohepatitis and Hepatocellular Carcinoma by Inhibiting RIPK1 Kinase Activity-Mediated Hepatocyte Apoptosis. Cancer Cell. 2015;28(5):582–98. doi: 10.1016/j.ccell.2015.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vucur M, Reisinger F, Gautheron J, Janssen J, Roderburg C, Cardenas DV, et al. RIP3 inhibits inflammatory hepatocarcinogenesis but promotes cholestasis by controlling caspase-8- and JNK-dependent compensatory cell proliferation. Cell reports. 2013;4(4):776–90. doi: 10.1016/j.celrep.2013.07.035. [DOI] [PubMed] [Google Scholar]
- 10.Koppe C, Verheugd P, Gautheron J, Reisinger F, Kreggenwinkel K, Roderburg C, et al. I kappa B Kinase alpha/beta Control Biliary Homeostasis and Hepatocarcinogenesis in Mice by Phosphorylating the Cell-Death Mediator Receptor-Interacting Protein Kinase 1. Hepatology. 2016;64(4):1217–31. doi: 10.1002/hep.28723. [DOI] [PubMed] [Google Scholar]
- 11.Schneider AT, Gautheron J, Feoktistova M, Roderburg C, Loosen SH, Roy S, et al. RIPK1 Suppresses a TRAF2-Dependent Pathway to Liver Cancer. Cancer Cell. 2017;31(1):94–109. doi: 10.1016/j.ccell.2016.11.009. [DOI] [PubMed] [Google Scholar]
- 12.Boege Y, Malehmir M, Healy ME, Bettermann K, Lorentzen A, Vucur M, et al. A Dual Role of Caspase-8 in Triggering and Sensing Proliferation-Associated DNA Damage, a Key Determinant of Liver Cancer Development. Cancer Cell. 2017;32(3):342–59 e10. doi: 10.1016/j.ccell.2017.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dara L, Liu ZX, Kaplowitz N. Questions and controversies: the role of necroptosis in liver disease. Cell Death Discov. 2016;2 doi: 10.1038/cddiscovery.2016.89. 16089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ting AT, Bertrand MJ. More to Life than NF-kappaB in TNFR1 Signaling. Trends Immunol. 2016;37(8):535–45. doi: 10.1016/j.it.2016.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Annibaldi A, Meier P. Checkpoints in TNF-Induced Cell Death: Implications in Inflammation and Cancer. Trends Mol Med. 2018;24(1):49–65. doi: 10.1016/j.molmed.2017.11.002. [DOI] [PubMed] [Google Scholar]
- 16.Dondelinger Y, Jouan-Lanhouet S, Divert T, Theatre E, Bertin J, Gough PJ, et al. NF-kappa B-Independent Role of IKK alpha/IKK beta in Preventing RIPK1 Kinase-Dependent Apoptotic and Necroptotic Cell Death during TNF Signaling. Mol Cell. 2015;60(1):63–76. doi: 10.1016/j.molcel.2015.07.032. [DOI] [PubMed] [Google Scholar]
- 17.Lafont E, Draber P, Rieser E, Reichert M, Kupka S, de Miguel D, et al. TBK1 and IKKepsilon prevent TNF-induced cell death by RIPK1 phosphorylation. Nat Cell Biol. 2018;20(12):1389–99. doi: 10.1038/s41556-018-0229-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xu D, Jin T, Zhu H, Chen H, Ofengeim D, Zou C, et al. TBK1 Suppresses RIPK1-Driven Apoptosis and Inflammation during Development and in Aging. Cell. 2018;174(6):1477–91 e19. doi: 10.1016/j.cell.2018.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bettermann K, Vucur M, Haybaeck J, Koppe C, Janssen J, Heymann F, et al. TAK1 Suppresses a NEMO-Dependent but NF-kappa B-Independent Pathway to Liver Cancer. Cancer Cell. 2010;17(5):481–96. doi: 10.1016/j.ccr.2010.03.021. [DOI] [PubMed] [Google Scholar]
- 20.Roh YS, Song J, Seki E. TAK1 regulates hepatic cell survival and carcinogenesis. J Gastroenterol. 2014;49(2):185–94. doi: 10.1007/s00535-013-0931-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mihaly SR, Ninomiya-Tsuji J, Morioka S. TAK1 control of cell death. Cell death and differentiation. 2014;21(11):1667–76. doi: 10.1038/cdd.2014.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Inokuchi-Shimizu S, Park EJ, Roh YS, Yang L, Zhang B, Song J, et al. TAK1-mediated autophagy and fatty acid oxidation prevent hepatosteatosis and tumorigenesis. J Clin Invest. 2014;124(8):3566–78. doi: 10.1172/JCI74068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kellendonk C, Opherk C, Anlag K, Schutz G, Tronche F. Hepatocyte-specific expression of Cre recombinase. Genesis. 2000;26(2):151–3. doi: 10.1002/(sici)1526-968x(200002)26:2<151::aid-gene17>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 24.Inokuchi S, Aoyama T, Miura K, Osterreicher CH, Kodama Y, Miyai K, et al. Disruption of TAK1 in hepatocytes causes hepatic injury, inflammation, fibrosis, and carcinogenesis. Proc Natl Acad Sci U S A. 2010;107(2):844–9. doi: 10.1073/pnas.0909781107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Eftychi C, Karagianni N, Alexiou M, Apostolaki M, Kollias G. Myeloid TAKI [corrected] acts as a negative regulator of the LPS response and mediates resistance to endotoxemia. PLoS One. 2012;7(2):e31550. doi: 10.1371/journal.pone.0031550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Polykratis A, Hermance N, Zelic M, Roderick J, Kim C, Van TM, et al. Cutting edge: RIPK1 Kinase inactive mice are viable and protected from TNF-induced necroptosis in vivo. Journal of immunology. 2014;193(4):1539–43. doi: 10.4049/jimmunol.1400590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gautheron J, Vucur M, Reisinger F, Cardenas DV, Roderburg C, Koppe C, et al. A positive feedback loop between RIP3 and JNK controls non-alcoholic steatohepatitis. Embo Molecular Medicine. 2014;6(8):1062–74. doi: 10.15252/emmm.201403856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Roychowdhury S, McMullen MR, Pisano SG, Liu X, Nagy LE. Absence of receptor interacting protein kinase 3 prevents ethanol-induced liver injury. Hepatology. 2013;57(5):1773–83. doi: 10.1002/hep.26200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang L, Du F, Wang X. TNF-alpha induces two distinct caspase-8 activation pathways. Cell. 2008;133(4):693–703. doi: 10.1016/j.cell.2008.03.036. [DOI] [PubMed] [Google Scholar]
- 30.Dara L, Johnson H, Suda J, Win S, Gaarde W, Han D, et al. Receptor Interacting Protein Kinase 1 Mediates Murine Acetaminophen Toxicity Independent of the Necrosome and Not Through Necroptosis. Hepatology. 2015;62(6):1847–57. doi: 10.1002/hep.27939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Suda J, Dara L, Yang L, Aghajan M, Song Y, Kaplowitz N, et al. Knockdown of RIPK1 Markedly Exacerbates Murine Immune-Mediated Liver Injury through Massive Apoptosis of Hepatocytes, Independent of Necroptosis and Inhibition of NF-kappaB. Journal of immunology. 2016;197(8):3120–9. doi: 10.4049/jimmunol.1600690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Van TM, Polykratis A, Straub BK, Kondylis V, Papadopoulou N, Pasparakis M. Kinase-independent functions of RIPK1 regulate hepatocyte survival and liver carcinogenesis. J Clin Invest. 2017;127(7):2662–77. doi: 10.1172/JCI92508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Afonso MB, Rodrigues PM, Simao AL, Ofengeim D, Carvalho T, Amaral JD, et al. Activation of necroptosis in human and experimental cholestasis. Cell Death Dis. 2016;7(9):e2390. doi: 10.1038/cddis.2016.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pescatore A, Esposito E, Draber P, Walczak H, Ursini MV. NEMO regulates a cell death switch in TNF signaling by inhibiting recruitment of RIPK3 to the cell death-inducing complex II. Cell Death Dis. 2016;7(8):e2346. doi: 10.1038/cddis.2016.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Irrinki KM, Mallilankaraman K, Thapa RJ, Chandramoorthy HC, Smith FJ, Jog NR, et al. Requirement of FADD, NEMO, and BAX/BAK for aberrant mitochondrial function in tumor necrosis factor alpha-induced necrosis. Mol Cell Biol. 2011;31(18):3745–58. doi: 10.1128/MCB.05303-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gunther C, He GW, Kremer AE, Murphy JM, Petrie EJ, Amann K, et al. The pseudokinase MLKL mediates programmed hepatocellular necrosis independently of RIPK3 during hepatitis. J Clin Invest. 2016;126(11):4346–60. doi: 10.1172/JCI87545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moriwaki K, Chan FK, et al. Necroptosis-independent signaling by the RIP kinases in inflammation. Cell Mol Life Sci. 2016;73(11–12):2325–34. doi: 10.1007/s00018-016-2203-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Anderton H, Bandala-Sanchez E, Simpson DS, Rickard JA, Ng AP, Di Rago L, et al. RIPK1 prevents TRADD-driven, but TNFR1 independent, apoptosis during development. Cell death and differentiation. 2018 doi: 10.1038/s41418-018-0166-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shimizu Y, Peltzer N, Sevko A, Lafont E, Sarr A, Draberova H, et al. The linear ubiquitin chain assembly complex acts as a liver tumor suppressor and inhibits hepatocyte apoptosis and hepatitis. Hepatology. 2017 doi: 10.1002/hep.29074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Filliol A, Piquet-Pellorce C, Le Seyec J, Farooq M, Genet V, Lucas-Clerc C, et al. RIPK1 protects from TNF-alpha-mediated liver damage during hepatitis. Cell death & disease. 2016;7(11):e2462. doi: 10.1038/cddis.2016.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Filliol A, Piquet-Pellorce C, Raguenes-Nicol C, Dion S, Farooq M, Lucas-Clerc C, et al. RIPK1 protects hepatocytes from Kupffer cells-mediated TNF-induced apoptosis in mouse models of PAMP-induced hepatitis. Journal of hepatology. 2017 doi: 10.1016/j.jhep.2017.01.005. [DOI] [PubMed] [Google Scholar]
- 42.Zinngrebe J, Rieser E, Taraborrelli L, Peltzer N, Hartwig T, Ren H, et al. --LUBAC deficiency perturbs TLR3 signaling to cause immunodeficiency and autoinflammation. J Exp Med. 2016;213(12):2671–89. doi: 10.1084/jem.20160041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tenev T, Bianchi K, Darding M, Broemer M, Langlais C, Wallberg F, et al. The Ripoptosome, a signaling platform that assembles in response to genotoxic stress and loss of IAPs. Mol Cell. 2011;43(3):432–48. doi: 10.1016/j.molcel.2011.06.006. [DOI] [PubMed] [Google Scholar]
- 44.Liu XY, Lai F, Yan XG, Jiang CC, Guo ST, Wang CY, et al. RIP1 Kinase Is an Oncogenic Driver in Melanoma. Cancer Res. 2015;75(8):1736–48. doi: 10.1158/0008-5472.CAN-14-2199. [DOI] [PubMed] [Google Scholar]
- 45.Seehawer M, Heinzmann F, D'Artista L, Harbig J, Roux PF, Hoenicke L, et al. Necroptosis microenvironment directs lineage commitment in liver cancer. Nature. 2018;562(7725):69–75. doi: 10.1038/s41586-018-0519-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yanger K, Zong Y, Maggs LR, Shapira SN, Maddipati R, Aiello NM, et al. Robust cellular reprogramming occurs spontaneously during liver regeneration. Genes Dev. 2013;27(7):719–24. doi: 10.1101/gad.207803.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schmidt-Supprian M, Bloch W, Courtois G, Addicks K, Israel A, Rajewsky K, et al. NEMO/IKK gamma-deficient mice model incontinentia pigmenti. Mol Cell. 2000;5(6):981–92. doi: 10.1016/s1097-2765(00)80263-4. [DOI] [PubMed] [Google Scholar]
- 48.Mc Guire C, Volckaert T, Wolke U, Sze M, de Rycke R, Waisman A, et al. Oligodendrocyte-specific FADD deletion protects mice from autoimmune-mediated demyelination. J Immunol. 2010;185(12):7646–53. doi: 10.4049/jimmunol.1000930. [DOI] [PubMed] [Google Scholar]
- 49.Dannappel M, Vlantis K, Kumari S, Polykratis A, Kim C, Wachsmuth L, et al. RIPK1 maintains epithelial homeostasis by inhibiting apoptosis and necroptosis. Nature. 2014;513(7516):90–4. doi: 10.1038/nature13608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Newton K, Sun X, Dixit VM. Kinase RIP3 is dispensable for normal NF-kappa Bs, signaling by the B-cell and T-cell receptors, tumor necrosis factor receptor 1, and Toll-like receptors 2 and 4. Mol Cell Biol. 2004;24(4):1464–9. doi: 10.1128/MCB.24.4.1464-1469.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Van Hauwermeiren F, Armaka M, Karagianni N, Kranidioti K, Vandenbroucke RE, Loges S, et al. Safe TNF-based antitumor therapy following p55TNFR reduction in intestinal epithelium. J Clin Invest. 2013;123(6):2590–603. doi: 10.1172/JCI65624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hao Z, Hampel B, Yagita H, Rajewsky K. T cell-specific ablation of Fas leads to Fas ligand-mediated lymphocyte depletion and inflammatory pulmonary fibrosis. J Exp Med. 2004;199(10):1355–65. doi: 10.1084/jem.20032196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Grosse-Wilde A, Voloshanenko O, Bailey SL, Longton GM, Schaefer U, Csernok AI, et al. TRAIL-R deficiency in mice enhances lymph node metastasis without affecting primary tumor development. J Clin Invest. 2008;118(1):100–10. doi: 10.1172/JCI33061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Uriarte I, Banales JM, Saez E, Arenas F, Oude Elferink RP, Prieto J, et al. Bicarbonate secretion of mouse cholangiocytes involves Na(+)-HCO(3)(-) cotransport in addition to Na(+)-independent Cl(-)/HCO(3)(-) exchange. Hepatology. 2010;51(3):891–902. doi: 10.1002/hep.23403. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.