

Abstract
Oncogene addiction postulates that the survival and growth of certain tumor cells is dependent upon the activity of one oncogene, despite their multiple genetic and epigenetic abnormalities. This phenomenon provides a foundation for molecular targeted therapy and a rationale for oncogene-based stratification. We have previously reported that the Promyelocytic Leukemia protein (PML) is upregulated in triple negative breast cancer (TNBC) and it regulates cancer-initiating cell function, thus suggesting that this protein can be therapeutically targeted in combination with PML-based stratification. However, the effects of PML perturbation on the bulk of tumor cells remained poorly understood. Here we demonstrate that TNBC cells are addicted to the expression of this nuclear protein. PML inhibition led to a remarkable growth arrest combined with features of senescence in vitro and in vivo. Mechanistically, the growth arrest and senescence were associated to a decrease in MYC and PIM1 kinase levels, with the subsequent accumulation of CDKN1B (p27), a trigger of senescence. In line with this notion, we found that PML is associated to the promoter regions of MYC and PIM1, consistent with their direct correlation in breast cancer specimens. Altogether, our results provide a feasible explanation for the functional similarities of MYC, PIM1 and PML in TNBC and encourage further study of PML targeting strategies for the treatment of this breast cancer subtype.
Keywords: PML, MYC, PIM1, p27, breast cancer, senescence, oncogene addition
Introduction
Breast cancer exemplifies the potential of gene expression profiling to classify the disease into molecular subtypes1, 2, 3. However, these classifications do not inform about the molecular mediators of tumor progression and metastasis in each subtype of breast cancer. To address this question, we and others have defined genes and pathways that are relevant to breast cancer progression, metastasis and resistance to therapy4, 5, 6. The Promyelocytic Leukemia protein (PML), the essential component of the PML nuclear bodies (PML-NBs), induces apoptosis and it inhibits angiogenesis and cell cycle progression in cancer, thus complying with the definition of a tumor suppressor7, 8. Paradoxically, PML exerts a pro-survival role conferring a selective advantage in chronic myeloid leukemia and specific solid tumors6, 9, 10, 11, 12, 13, 14, 15. In breast cancer, PML regulates aggressiveness and metastatic features through the control of the stem cell gene, SOX9, and the Hypoxia-inducible factor 1 alpha (HIF1α) signalling13, 15. Moreover, the regulation of cancer-initiating cell (CIC) and metastatic potential is restricted to PML high-expressing estrogen receptor-negative breast tumors, predominantly triple negative breast cancer (TNBC).
The concept that the perturbation of a driver cancer gene can exert an exacerbated tumor suppressive response in tumor cells is defined as “oncogene addiction” and provides a rationale for molecular targeted therapy16. Senescence is a stress response that involves a stable cell growth arrest as well as an adaptive process to reduce energy consumption for cell division or differentiation and therefore assure the survival and viability of the cell17, 18. Senescence is induced in vitro by different stimuli including DNA damage, oxidative stress, oncogene activation, mitochondrial dysfunction or chemotherapy17, 18. Cyclin-dependent kinase inhibitor family (CDKi) is a key regulator of the senescence response, predominantly through p53-p21 and/or p16-RB axes17, 18. To a lesser extent, CDKN1B (p27) has been reported to participate in the activation of the senescence response, in conditions where p21 and/or p16 are not active19, 20. Of note, PML is required for a fully functional senescence response upon oncogene activation in tumors where it functions as a tumor suppressor. In addition, the PML-NBs coordinate the activation of p53 and the formation of the Senescence-Associated Heterochromatin Foci (SAHF)21, 22, 23.
TNBC exhibits increased levels and activity of various oncogenes, including MYC and PIM124, 25, 26, 27. Importantly, these genes regulate metabolic and signaling activities in this breast tumor subtype, and they represent an attractive therapeutic vulnerability 24, 26, 27, 28. Whereas similarities exist among the reported activities of MYC, PIM1 and PML, their functional association remains obscure. In this study, we demonstrate that TNBC cells that express high PML levels are addicted to the nuclear protein, and its targeting elicits a growth suppressive response that encompasses MYC and PIM1 downregulation and the activation of p27-dependent senescence.
Results
PML silencing induces senescence and prevents tumor growth in vivo
The identification of PML as a novel target in aggressive breast cancer tumors13, 15 prompted us to investigate the molecular consequences of its inhibition in an established cell culture. To this end, we generated and validated three PML-targeting doxycycline-inducible and two constitutive short hairpin RNAs (shRNAs) (Fig. 1a and Supplementary Fig. 1a)15. PML silencing triggered a robust morphological change in PML-high expressing cells, MDA-MB-231 (Fig. 1b and Supplementary Fig. 1b), characterized by a significant increase in size (FSC-A) and granularity (SSC-A) analyzed by FACS (Fig. 1c and Supplementary Fig. 1c). These changes in morphology were indicative of a senescence response. Indeed, the evaluation of senescence-associated β-galactosidase (SA-β-gal) activity in both inducible and constitutive systems confirmed this notion (Fig. 1d-e and Supplementary Fig. 1d-e) in MDA-MB-231 cells. Senescence is defined as an irreversible cell cycle arrest. Indeed, we could confirm the cell cycle arrest upon PML genetic inhibition, by means of BrdU analysis (Fig. 1f) and crystal violet cell number assay (Supplementary Fig. 1f-g) and that it was not due to an increase in apoptosis (Supplementary Fig. 1h). Of note, arsenic trioxide (ATO) did not elicit a senescence phenotype (Supplementary Fig. 1i). This compound exerts a biphasic effect on PML; first favors the formation of the PML NBs and then the degradation of PML. Therefore, the inability of arsenic trioxide to recapitulate PML silencing could be due to its molecular mode of action.
Figure 1. PML silencing induces senescence.

(a-f) Effect of doxycycline-inducible (150 ng ml-1; 3+3 days) PML silencing (sh1, sh4, sh5) on PML protein expression (a, representative of at least 3 experiments), on the morphology (b, representative images, scale bar, 50μm), on cell size and granularity (c, FACS analysis, sh1 and sh4, n=4, sh5, n=5), on the number of senescent cells (d; n=3, representative images of SA-β-Galactosidase assay, scale bar 50μm (e)) and on the number of BrdU positive cells (f, n=4) in MDA-MB-231 cells. (g-j) Impact of doxycycline-inducible PML silencing (sh4) of established MDA-MB-231 xenografts on PML protein expression (g), on tumour growth rate represented as the growth rate of each tumour (h, sh4 no dox, n=10; sh4 dox, n=12; growth rate was inferred from the linear regression calculated for the progressive change in tumour volume of each individual tumour during the period depicted in Supplementary Fig. 1p) and on number of senescent cells measured by p-HP1γ staining (i, sh4 no dox, n=4; sh4 dox, n=4); representative images of p-HP1γ positive cells, scale bar 100μm (j) of the tumours. Error bars represent s.e.m. p, p-value (*p< 0.05, **p< 0.01, ***p< 0.001). One-tailed Student´s t-test was used for cell line data analysis (c, d, f) and one-tailed Mann-Whitney U-test for xenografts (h, i). sh1, sh4 and sh5: shRNA against PML, dox: doxycycline, SA-β-gal: Senescence-associated beta-galactosidase, BrdU: Bromodeoxyuridine, p-HP1γ: phospho-heterochromatin protein-1 gamma, molecular weight markers (kDa) are shown to the right.
We monitored additional features that were reported for certain types of oncogene-induced senescence29. On the one hand, proteomics analysis of the supernatant of these cells indicated that PML silencing resulted in a distinct secretome, without signs of a canonical SASP (senescence-associated secretory phenotype) (Supplementary Fig. 1j-l and Supplementary Table 1). On the other hand, we ascertained the formation of SAHF (senescence-associated heterochromatin foci). We could not confirm the existence of SAHF neither at the level of chromatin condensation nor the formation of macroH2A1.1 foci (Supplementary Fig. 1m). Lamin B1 loss is a senescence-associated biomarker30, 31. We demonstrated that in our system PML loss induced a decrease in Lamin B1 protein levels (Supplementary Fig. 1n-o).
Of note, PML regulates oxidative stress responses7, 32. We ruled out that reactive oxygen species (ROS) elevation drives senescence in our system since PML loss does not induce its accumulation (Supplementary Fig. 1p).
Breast cancer-initiating cell (CIC) capacity is reduced upon PML knockdown in TNBC cells (with high PML expression), as we demonstrated in limiting dilution assays with MDA-MB-231 cells15. Here, we hypothesized that the activation of senescence would result in a tumor suppressive response in established tumors, where the contribution of CIC is negligible. To test this notion, MDA-MB-231 cells were injected in the flank of immunocompromised mice, and once the tumors were established (reaching a volume of 25-130 mm3) doxycycline was administered in the food pellets to induce PML silencing. In agreement with the response observed in vitro, xenograft growth was curbed upon PML knockdown (Fig. 1g-h and Supplementary Fig. 1q-r) and senescence increase was confirmed by means of p-HP1γ staining (Fig. 1i-j)33. Our data suggest that PML silencing in a PML-high expressing TNBC cell line triggers a senescence response with a partial presence of classical markers of this process.
p27 is the driver in PML loss-induced senescence
Senescence is executed and sustained at the molecular level through the activation of growth suppressors, including p53 and the cyclin-dependent kinase (CDKs) inhibitors p21, p16 and p2717, 20, 29. Since MDA-MB-231 cells harbor loss of p16 and p53 mutation34, 35, we proposed p27 as a candidate to drive PML silencing-induced senescence in our cell system. Importantly, p27 protein levels were increased upon both inducible (Fig. 2a and Supplementary Fig. 2a) and constitutive (Supplementary Fig. 2b) PML silencing with all the shRNA tested in MDA-MB-231 cells. Moreover, we observed that the induction of p27 protein levels occurred as soon as 2 days following PML inactivation and it was maintained up to 6 days of PML silencing (Fig. 2b-c and Supplementary Fig. 2c-f).
Figure 2. p27 is induced after PML silencing.

(a) Effect of doxycycline-inducible (150 ng ml-1; 3+3 days) PML silencing (sh4, sh5) on p27 and PML protein expression (representative of at least 3 experiments) on MDA-MB-231 cells. (b-c) Quantification of p27 and PML protein levels along 6 days of doxycycline-inducible PML silencing on MDA-MB-231 cells (n=3) with 2 different shRNAs. (d) Immunofluorescence quantification of nuclear p27 positive cells upon PML inducible silencing on MDA-MB-231 cells (n=4). (e-g) Immunofluorescence quantification of the correlation of p27 positive cells and PML levels in these cells (e-f) and representative images of p27 and PML staining (g) upon doxycycline-inducible PML silencing in MDA-MB-231 cells (n=4). (h) Effect of doxycycline-inducible (150 ng ml-1; 3+3 days) PML silencing (sh4) on RB phosphorylation (Ser780) (representative of 3 experiments) on MDA-MB-231 cells. (i) Expression of p27-related cell cycle genes upon PML inducible silencing in MDA-MB-231 cells (n=3). (j) Impact of doxycycline-inducible PML silencing (sh4) on p27 and PML protein expression on established MDA-MB-231 xenografts. Error bars represent s.e.m. p, p-value (*/$p< 0.05, **/$$p< 0.01, ***/$$$p< 0.001). One-tailed one-sample t-test (b-c, i) and one-tailed Student´s t-test were used for cell line data analysis (d-f). sh4 and sh5: shRNA against PML, dox: doxycycline. #: Unspecific band. Molecular weight markers (kDa) are shown to the right.
The function of p27 is controlled by changes in its levels along with its compartmentalization within the cell36. To confirm the functionality of accumulated p27 in PML-silenced cells, we quantified p27 nuclear localization by immunoflourescence. As predicted, PML silencing elicited an increase of nuclear p27 in cells with the three inducible shRNA tested (Fig. 2d and Supplementary Fig. 2g). Since the effect of shRNAs was analyzed in a pooled culture, it is plausible that there would be heterogeneity in PML levels across cells within the culture dish. We therefore evaluated whether the increase in nuclear p27 was ascribed to cells with a profound decrease in PML immunoreactivity. We established an immunofluorescence score based on previous studies6, 15 (PML low= 0-4 dots; PML high= more than 4 dots per cell nuclei). In line with our previous results, we observed a significant inverse association between PML immunoreactivity and p27 nuclear staining (Fig. 2e-g and Supplementary Fig. 2h). Moreover, the elevated activity of p27 upon PML silencing was consistent with the increase in its mRNA levels, the blockade of Retinoblastoma protein (Rb) phosphorylation and the reduced transcription of downstream regulated cell cycle-related genes (Fig. 2h-i and Supplementary Fig. 2i). In agreement with the results observed in vitro, p27 accumulation was recapitulated upon PML knockdown in vivo (Fig. 2j).
Our results reveal that PML silencing in TNBC cells with high expression of the nuclear protein triggers a senescence response associated to p27 accumulation. To ascertain the causal contribution of p27 to the execution of the senescence response, we silenced p27 in MDA-MB-231 cells concomitantly with PML silencing, using inducible (Fig. 3a-b) or constitutive (Supplementary Fig. 3a) shRNA systems. Preventing p27 accumulation upon PML loss hampered the induction of senescence in a dose-dependent manner according to the potency of the shRNA against p27 (Fig. 3c and Supplementary Fig. 3b). Our results demonstrate that PML loss elicits a senescence response mediated by the upregulation of p27 in PML high expressing TNBC cells.
Figure 3. p27 is the driver in PML loss-induced senescence.

(a) Experimental design for inducible p27 silencing (sh1p27 and sh2p27, B: blasticidin selection) alone or in combination with inducible PML silencing (sh4, P: puromycin selection) in MDA-MB-231 cells. (b) p27 and PML protein levels upon doxycycline inducible silencing of either p27 or PML or both in MDA-MB-231 cells (representative of 3 experiments). (c) Effect on the number of senescent cells (n=4) upon p27 and/or PML inducible silencing in MDA-MB-231 cells. (d) Effect on the relative cell number (n=4) upon p27 and/or PML inducible silencing in MDA-MB-231 cells. Error bars represent s.e.m. p, p-value (*p< 0.05, **p< 0.01, ***p< 0.001). One-tailed Student´s t-test (c) and one-tailed one-sample t-test (d) were used for cell line data analysis. shC: Scramble shRNA, Dox: doxycycline, SA-β-gal: Senescence-associated beta-galactosidase. Molecular weight markers (kDa) are shown to the right.
Although senescence is a major driver of growth arrest, we noticed that the amount of SA-β-Gal positivity upon PML silencing was not comparable to the extent of growth arrest detected (Fig. 1 and Supplementary Fig. 1). Taking advantage of our capacity to ablate senescence by silencing p27, we ascertained the contribution of this response to the cell number reduction. Importantly, the growth arrest caused by PML silencing was not recovered by blunting senescence (Fig. 3d and Supplementary Fig. 3c), thus suggesting that an additional mechanism may be involved.
MYC and PIM1 are regulated by PML in TNBC
Our data demonstrate that preventing p27 accumulation is not sufficient to rescue the growth arrest caused by PML loss. We reasoned that the mechanism through which PML was regulating growth arrest, p27 accumulation and senescence might depend on a larger growth-regulatory program. Interestingly, the oncogenic axis comprised of MYC and PIM1 kinase shares many similarities with PML concerning its activity in TNBC. MYC and PIM1 are upregulated in TNBC24, 26, 27 and inhibit p27 accumulation and function37. In addition, MYC regulates metabolic functions attributed to PML, including fatty acid β oxidation6, 10, 38, 39 and its inhibition induces cellular senescence in lymphoma, osteosarcoma, and hepatocellular carcinoma40. With this data in mind, we first evaluated the association of PML, MYC and PIM1 in breast cancer. We found a significant direct correlation in various breast cancer transcriptomics datasets. This association was evident in 2 out of 4 datasets for MYC-PML and 4 out of 4 for PIM1-PML, when accounting all breast cancer subtypes (Supplementary Fig. 4a). Since the effect of these genes is restricted to tumors that lack hormone receptors, we refined the analysis by focusing on ER negative tumors. In this scenario, the correlation was recapitulated in various datasets (Fig. 4a).
Figure 4. PML regulates MYC and PIM1 expression in TNBC.
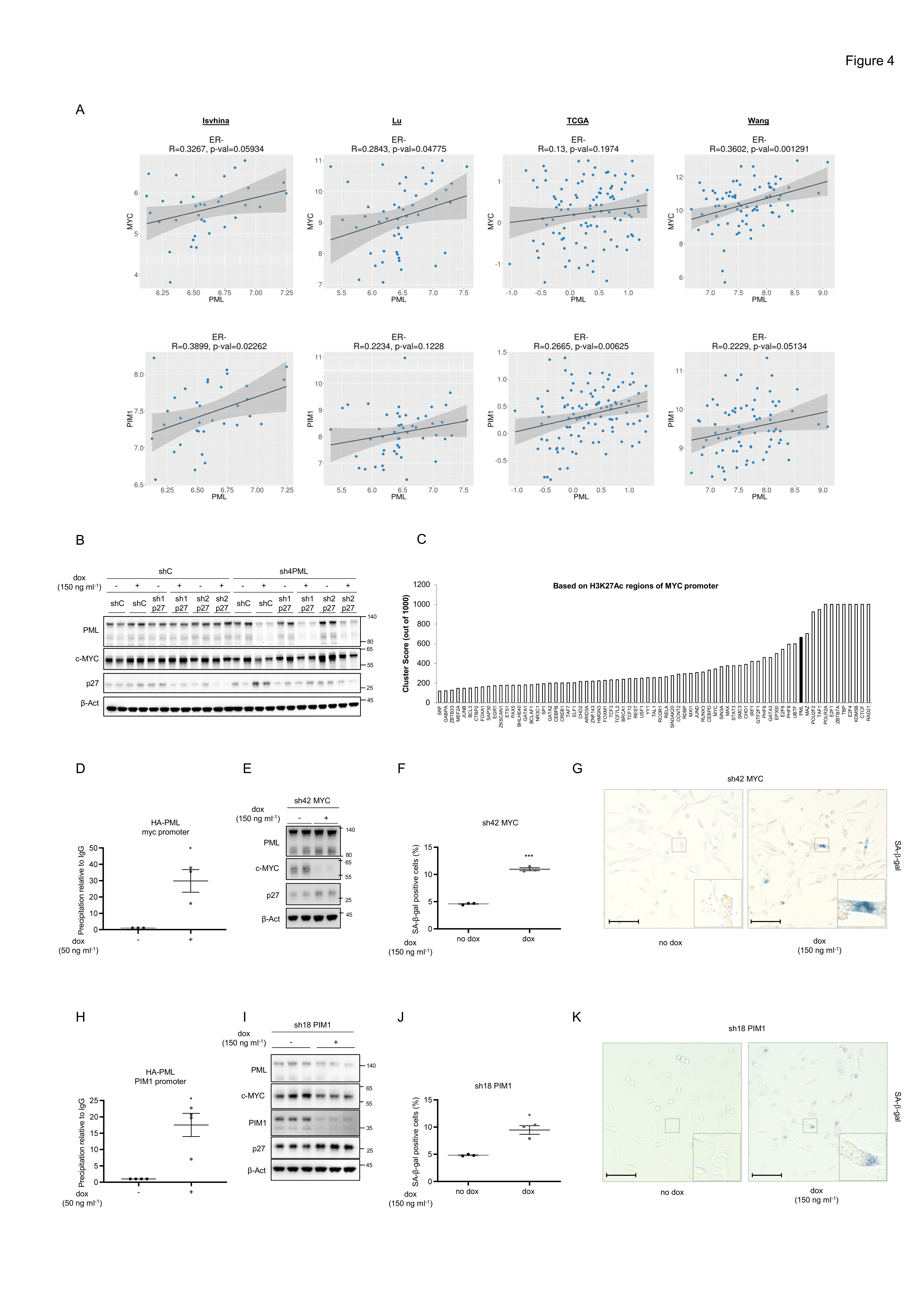
(a) Correlation analysis between PML and MYC (top panels) and between PML and PIM1 (bottom panels) mRNA levels in ER negative tumor specimens of the indicated breast cancer datasets. Sample sizes: Ivshina (n=34), Lu (n=49), TCGA (n=117) and Wang (n=77). (b) p27, MYC and PML protein levels upon doxycycline inducible silencing of either p27 or PML or both in MDA-MB-231 cells (representative of 3 experiments). (c) Cluster score of DNA-binding proteins in MYC promoter region using ENCODE database. (d) MYC promoter region abundance in chromatin immunoprecipitation (ChIP) of exogenous HA-PMLIV using HA-tag antibody in MDA-MB-231 cells after induction with 50 ng ml-1 doxycycline for 3 days (n=3). Data were normalized to IgG (negative-binding control). (e) p27, MYC and PML protein levels upon doxycycline inducible silencing of MYC (sh42) in MDA-MB-231 cells (representative of 3 experiments). (f-g) Effect on the number of senescent cells (n=3) (f) and representative images, scale bar 50μm, (g) upon MYC inducible silencing in MDA-MB-231 cells. (h) PIM1 promoter region abundance in chromatin immunoprecipitation (ChIP) of exogenous HA-PMLIV using HA-tag antibody in MDA-MB-231 cells after induction with 50 ng ml-1 doxycycline for 3 days (n=4). Data were normalized to IgG (negative-binding control). (i) p27, MYC, PIM1 and PML protein levels upon doxycycline inducible silencing of PIM1 (sh18) in MDA-MB-231 cells (representative of 3 experiments). (j-k) Effect on the number of senescent cells (n=3) (j) and representative images, scale bar 50μm, (k) upon PIM1 inducible silencing in MDA-MB-231 cells. Error bars represent s.e.m. p, p-value (*p< 0.05, ***p< 0.001). One-tailed one sample t-test (d, h) and One-tailed Student´s t-test (f, j) were used for cell line data analysis shC: Scramble shRNA, Dox: doxycycline, SA-β-gal: Senescence-associated beta-galactosidase. Molecular weight markers (kDa) are shown to the right.
We next aimed at deconstructing the molecular regulation of PML, MYC and PIM1. First, we monitored the impact of PML silencing on MYC abundance. As predicted, inducible PML shRNA activation resulted in a remarkable decrease in MYC protein and mRNA levels in vitro and in vivo (Supplementary Fig 4b-h) in two PML high expressing cells, MDA-MB-231 and MDA-MB-468. Of note, in line with our senescence results (Supplementary Fig. 1i), arsenic trioxide did not alter the abundance of p27 and MYC (Supplementary Fig 4i-j).
We next asked to which extent MYC downregulation was retained in cells devoid of p27-dependent senescence response. To address this question, we checked MYC expression upon PML/p27 double silencing in MDA-MB-231 cells. The decrease in MYC expression upon PML loss was not recovered with p27 silencing (Fig. 4b), thus providing a feasible explanation for the lack of rescue in growth capacity.
We have previously shown that PML can regulate gene expression in line with its association with discreet promoter regions15. Since PML silencing resulted in reduced MYC mRNA levels (Supplementary Fig. 4d, h), we interrogated MYC promoter in silico using ENCODE41. We found PML among the proteins with highest confidence DNA-binding score in MYC promoter region (Fig. 4c, cluster score: 527). We performed chromatin immunoprecipitation (ChIP) analysis of ectopically expressed PML and confirmed that PML is in the vicinity of MYC promoter (Fig. 4d). To ascertain if MYC silencing recapitulated the effect of PML inhibition, we used a validated shRNA targeting this oncogene (sh42)42, 43 and confirmed that MYC silencing resulted in increased p27 levels (Fig. 4e and Supplementary Fig. 4k), senescence (Fig. 4f-g) and growth arrest (Supplementary Fig. 4l).
In the last few years, an important body of work has demonstrated that PIM1 is an important partner of MYC function in prostate cancer and TNBC24, 26. Moreover, PIM1 can regulate MYC transcriptional signature and p2724, 26. We monitored the impact of PML on PIM1 expression and function. PML loss resulted in a decrease in PIM1 gene expression in two PML high expressing cells, MDA-MB-231 and MDA-MB-468 (Supplementary Fig. 4m-n). Importantly, we confirmed that PML is in close proximity to PIM1 promoter by ChIP analysis (Fig. 4h, cluster score: 383 in ENCODE). We hypothesized that loss of PIM1 would further impact on MYC function and recapitulate the aforementioned PML and MYC phenotype. We silenced PIM1 using a validated shRNA (sh18)24 and corroborated that the targeting of PIM1 led to decrease in MYC abundance, increase in p27 levels, senescence and growth arrest in MDA-MB-231 cells (Fig. 4i-k and Supplementary Fig. 4o-q). Altogether, our results provide strong support for the role of MYC-PIM1 axis supporting PML-elicited TNBC growth and preventing the accumulation of p27 and senescence.
PML loss-elicited growth suppression in breast cancer is selective of high PML expressing TNBC
The inactivation of a single oncogene can compromise the development and survival of tumor cells despite their genetic or epigenetic abnormalities16. We have previously reported that high PML levels in TNBC are required for adequate CIC function15. Here, the data presented support the notion that the bulk of tumor cells in a TNBC with elevated PML is “addicted” to the expression of the protein. In turn, acute depletion of the nuclear protein results in growth arrest and senescence. To ascertain whether the “addiction” was restricted to TNBC cells, we took advantage of various breast cancer cell lines belonging to distinct subtypes with differing levels of PML. A second TNBC cell line (MDA-MB-468) which presented high levels of PML protein was compared to estrogen receptor positive (ER+) cells15 (Fig. 5a). Silencing of PML in MDA-MB-468 cells elicited a remarkable growth arrest, which was not detected in the ER+ cells Cama-1 and MCF7 (Fig. 5b and Supplementary Fig. 5a-c). In line with this notion, Cama-1 and MCF7 cells did not exhibit neither the reduction in MYC expression nor the induction of p27-dependent senescence, as compared to MDA-MB-468 cells (Fig. 5c-d and Supplementary Fig. 5d-g). Moreover, the morphological changes induced by the loss of PML were only present in TNBC cells (Supplementary Fig. 5h). Since PML silencing resulted in a distinct secretory phenotype (albeit not a canonical SASP), we monitored the secretome of the ER+ cell line Cama-1. In agreement with our prior data, unsupervised clustering based on the secretome was ineffective in segregating experimental conditions according to PML status. Similarly, principal component analysis and hierarchical clustering reinforced the notion that Cama-1 are refractory to PML level perturbation (Supplementary Fig. 5i-k and Supplementary Table 2).
Figure 5. The antiproliferative program elicited by PML loss is restricted to PML-high expressing TNBC cells.

(a-d) Effect of doxycycline-inducible (150 ng ml-1; 3+3 days) PML silencing (sh4) on PML and p27 protein expression (a, representative of 3 experiments), on cell number (b, MDA-MB-468 and Cama-1, n=3, MCF7, n=4) and on the number of senescent cells (c, MDA-MB-468 and Cama-1, n=3, MCF7, n=4; representative images of SA-β-galactosidase positive cells (d)) in MDA-MB-468, MCF-7 and Cama-1 cells, scale bar 50μm. Error bars represent s.e.m. p, p-value (*p< 0.05, **p< 0.01, ***p< 0.001, ns: not significant). One-tailed one sample t-test (b) and one-tailed Student´s t-test were used for cell line data analysis (c). Dox: doxycycline, SA-β-gal: Senescence-associated beta-galactosidase. Molecular weight markers (kDa) are shown to the right.
Discussion
PML has been a paradigmatic tumor suppressor since its discovery7, 8, 44. A variety of molecular activities directly support its reported capacity to prevent many of the hallmarks of cancer, including the induction of apoptosis and the inhibition of proliferation or angiogenesis7. Molecular partners such as p53 have reinforced this notion. However, the discovery of tumoral contexts where the presence of PML is required has broadened the picture of the roles of this nuclear protein in disease. Depletion of PML impairs the self-renewal activity in the leukemic stem cell from chronic myeloid leukemia9, 10. This phenotype is the consequence of both cell autonomous (the hyper-activation of mTOR complex1 and the reduction in PPARδ-fatty acid oxidation activity9, 10) and non-cell autonomous activities (regulation of the mesenchymal stem cell in the leukemic niche45), which trigger symmetric commitment and the loss of the CIC compartment.
The regulation of CIC activity was recently translated to solid tumors. To date, glioma and a subset of breast cancers exhibit PML-dependent self-renewal activity14, 15, whereas other tumors, such as ovarian cancers or some experimental models of hepatocarcinoma development, exhibit a broad tumor suppressive response upon PML inhibition11, 12. PML expression is selectively exacerbated in a subset of breast tumors (TNBC)6, 13, 15. Yet, we lack basic understanding around the impact of PML on the function of this cell subtype. In this study, we demonstrate that PML depletion in the bulk of TNBC cells in culture and in vivo triggers a tumor suppressive response consisting on growth arrest and the activation of senescence.
PML has been previously related to the regulation of the senescence response21. However, the majority of studies associate PML expression to the execution of this growth suppressive response upon the activation of oncogenes or replicative stress11. Experimentally, ectopic PML expression triggers senescence, and, conversely, PML deletion bypasses the senescence response elicited by the oncogenic form of RAS, thus enabling transformation46, 47, 48. Mechanistically, PML supports p53 activity and participates in the formation of SAHF21. In turn, PML loss bypasses the senescence response. Paradoxically, our results indicate that, in cancer cells with high dependence on PML expression, its inhibition also triggers a senescence response that lacks canonical SASP and SAHF. This phenomenon might have been overlooked in prior studies due to the lack of data at the time on the role of PML favoring cancer cell function in specific tumor subsets. Of note, the activation of senescence in non-transformed fibroblasts upon PML depletion adds complexity to already extensive portfolio of PML activities49.
Oncogene addiction16 is perceived as an attractive opportunity in the era of targeted therapies. Our results are consistent with PML addiction in TNBC cells, even if this protein cannot be formally considered an oncogene. The data produced by us and others6, 13, 15 argue in favor of a molecular make up in this subtype of breast cancer that requires the presence of PML in high doses, as opposed to estrogen receptor-positive tumor cells. In this regard, the control of MYC and PIM1 expression by PML provides a feasible explanation for the accumulation of p27 and the induction of senescence when PML is silenced. To which extent the relationship between PML, MYC and PIM1 is operative in other tumor types becomes now an exciting question to address. Since PML lacks a dedicated domain to recognize and bind discreet DNA sequences, the existence of yet unidentified PML-interacting transcription factors that enable this regulatory mode warrants further research.
The results obtained in this study represent a conceptual leap in how we perceive the role of PML in TNBC, and suggest that targeting this nuclear protein can be beneficial at multiple levels, including impairing the CIC function15, blunting hypoxia signaling13 and triggering a senescence response. The quantification of the relative relevance of each PML effector pathway in the overall activity of PML could open new opportunities to apply the biology of PML-regulated TNBC function for breast cancer treatment.
Materials and methods
Cell culture
MDA-MB-231, MDA-MB-468, MCF-7 and Cama-1 cell lines were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA) or from Leibniz-Institut - Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH (DMSZ) who provided an authentication certificate. None of the cell lines used in this study was found in the database of commonly misidentified cell lines maintained by ICLAC and NCBI Biosample. Cell lines were routinely monitored for mycoplasma contamination and quarantined while treated if positive. MDA-MB-231 and MCF-7 cells were maintained in DMEM media, MDA-MB-468 were maintained in RPMI media and Cama-1 were maintained in DMEM-F12 media, all supplemented with 10% (v/v) foetal bovine serum (FBS) and 1% (v/v) penicillin-streptomycin.
Generation of stable cell lines
293FT cells were used for lentiviral production. Lentiviral vectors expressing shRNAs against human PML and p27 from the Mission® shRNA Library were purchased from Sigma‐Aldrich. Cells were transfected with lentiviral vectors following standard procedures, and viral supernatant was used to infect cells. Selection was done using puromycin (2 μg ml-1; P8833, Sigma) for 48 h or blasticidin (10 μg ml-1; Cat. 15205, Sigma) for 5 days. As a control, a lentivirus with scrambled shRNA (shC) was used. Short hairpins sequence: shC: CCGGCAACAAGATGAAGAGCACCAACTCGAGTTGGTGCTCTTCATCTTGTTGTTTTT; sh1PML (TRCN0000003865): CCGGCAATACAACGACAGCCCAGAACTCGAGTTCTGGGCTGTCGTTGTATTGTTTTT; sh4PML (TRCN 0000003867): CCGGGCCAGTGTACGCCTTCTCCATCTCGAGATGGAGAAGGCGTACACTGGCTTTTT; sh5PML (TRCN 0000003868): CCGGGTGTACCGGCAGATTGTGGATCTCGAGATCCACAATCTGCCGGTACACTTTTT; sh1p27 (TRCN 0000039928): CCGGGTAGGATAAGTGAAATGGATACTCGAGTATCCATTTCACTTATCCTACTTTTTG; sh2p27 (TRCN 0000039930): CCGGGCGCAAGTGGAATTTCGATTTCTCGAGAAATCGAAATTCCACTTGCGCTTTTTG. sh42MYC (TRCN0000039642): CCGGCCTGAGACAGATCAGCAACAACTCGAGTTGTTGCTGATCTGTCTCAGGTTTTTG. sh18PIM1 (TRCN0000010118): CCGGACATCCTTATCGACCTCAATCCTCGAGGATTGAGGTCGATAAGGATGTTTTTT.
Sub-cloning of shC, sh1PML, sh4PML, sh5PML and sh42myc into pLKO-Tet-On-Puromycin vector was done introducing AgeI and EcoRI in the 5´end of top and bottom shRNA oligos respectively (following the strategy provided by Dr. Dmitri Wiederschain50, Addgene plasmid: 21915). Sub-cloning of shC, sh1p27, sh2p27 and sh18PIM1 into pLKO-Tet-On- Blasticidin was done following the same procedure. Puromycin resistance cassette was replaced by Blasticidin cassette following Gibson assembly strategy.
Reagents
Doxycycline (Cat. D9891, Sigma) was used at 150 ng ml-1 to induce the expression of shRNA from pLKO-Tet-On vectors. Doxycycline-mediated inducible shRNA expression was performed by treating cell cultures for 72 h with the antibiotic (150 ng ml−1) and then seeding for cellular or molecular assays in the presence of doxycycline for 3 more days (unless otherwise specified). Arsenic trioxide (ATO; Cat. A1010, Sigma-Aldrich) was prepared at a concentration of 100 mM in NaOH 1N and subsequently diluted to 0.1 mM in PBS for a working solution. ATO was used at 150 nM for 6 days as indicated in figure legends.
Cell growth analysis and size measurement by FACS
Cell number quantification was done with crystal violet as reported5. For FACS analysis MDA-MB-231 cells were trypsinized and resuspended in PBS to be analysed based on their size (FSC) and granularity (SSC) using a BD FACSCanto™ II (BD Biosciences) flow cytometer upon PML doxycycline-inducible silencing. Data represented in Fig.1c correspond to the sum of Q1+Q2+Q3 populations selected as in Supplementary Fig. 1c. Data were analysed using the FlowJo software; cell populations were selected for each shRNA (no dox condition) and differences quantified for increasing size and granularity.
Senescence associated-β-galactosidase detection
To quantify the number of senescent cells, constitutive or inducible PML (and/or p27) silencing cells was performed as described previously and cells were seeded in 24-well plates in duplicate. An overnight incubation with the senescence detection kit (QIA117, Calbiochem) was performed and SA-β-Gal activity was revealed and quantified (3 areas per well, more than 200 cells per condition). The number of senescent cells in each area was relativized to the number of total cells counted per area. Cells were seeded in plates or glass cover slips to acquire images with EVOS® cell imaging station (20x magnification objective).
Western blotting, immunofluorescence and BrdU
Western blot analysis was carried out as previously described5. Briefly, cells were seeded on 6-well plates. Cell lysates were prepared with RIPA buffer (50 mM TrisHCl pH 7.5, 150 mM NaCl, 1 mM EDTA, 0.1% SDS, 1% Nonidet P40, 1% sodium deoxycholate, 1 mM Sodium Fluoride, 1 mM sodium orthovanadate, 1 mM beta-glycerophosphate and protease inhibitor cocktail; Roche). The following antibodies were used for Western blotting: rabbit polyclonal anti-PML, 1:1000 dilution (Cat: A301-167A, Bethyl laboratories), mouse monoclonal anti-p27[Kip1], 1:1000 dilution (Cat: 610242, BD Biosciences), mouse monoclonal anti-beta-ACTIN, 1:2000 dilution (Cat: 3700, Cell Signaling), rabbit polyclonal Hsp90, 1:2000 dilution (Cat: 4874, Cell Signaling), rabbit polyclonal c-Myc, 1:1000 dilution (Cat: 13987, Cell Signaling), rabbit polyclonal PIM1, 1:1000 dilution (ab75776, Abcam), rabbit polyclonal Lamin B1 (ab133741, Abcam), rabbit monoclonal anti-cleaved PARP (Asp214), 1:1000 dilution (Cat: 5625, Cell Signaling), rabbit polyclonal anti-cleaved caspase 3 (Asp175), 1:1000 dilution (Cat: 9661, Cell Signaling), mouse monoclonal anti-α-Tubulin (66031-1-Ig, Proteintech), 1:2500 dilution, rabbit monoclonal anti-phospho-Rb (Ser780) 1:1000 dilution (Cat: 9307, Cell Signaling). After standard SDS-PAGE and Western blotting techniques, proteins were visualized using the ECL on iBright™ CL1000 Imaging System (Cat: A32749, Invitrogen). Densitometry-based quantification was performed using ImageJ software. Uncropped scans are provided in Supplementary Fig. 6.
For immunofluorescence, cells were seeded on glass cover slips in 24-well plates, cells were fixed with 4% para-formaldehyde (15 min), PBS (3 times wash), 1% Triton X-100 (5 min), PBS (3 times wash), 10% goat serum (1 h) and anti-PML antibody 1:100 dilution (catalog A301-167A; Bethyl laboratories), anti-p27[Kip1] antibody 1:100 dilution (Cat: 610242, BD Biosciences) and anti-macroH2A1.1 antibody 1:100 (Cat: 12455, Cell Signaling) were added ON (4ºC) in goat serum. Cover slips were washed with PBS 3 times and incubated with secondary antibodies (anti-rabbit Alexa488, anti-rabbit Alexa594, anti-mouse Alexa488 and anti-mouse Alexa594; Invitrogen-Molecular Probes) for 1 h (room temperature). Cover slips were washed with PBS 3 times, and DAPI added to stain nuclei (10 min), followed by mounting with ProLong™ Gold Antifade Mountant (Cat: P36930, Invitrogen). Immunofluorescence images were obtained with AxioImager D1 microscope (Zeiss) or with a confocal microscopy (Leica SP8) with 63x objectives. At least three different areas per cover slip were quantified.
For BrdU analysis cells were seeded as for immunofluoresce. Prior to fixing, cells were incubated in the presence of BrdU (3 ug ml-1). Cells were fixed with 4% para-formaldehyde (15 min), PBS (3 times wash) and DNA exposed with 2M HCl (5 min), PBS (3 times wash) and 0,1M sodium borate. After that, PBS (3 times wash), 1% Triton X-100 (5 min), PBS (3 times wash), 10% goat serum (1 h) and monoclonal anti-BrdU (MoBU-1) antibody 1:100 dilution (Cat: B35128, Invitrogen) was added ON (4ºC) in goat serum. Cover slips were washed three times with PBS and incubated with secondary antibodies (anti-mouse Alexa594; Invitrogen-Molecular Probes) for 1 h (room temperature). Cover slips were washed three times with PBS and DAPI added to stain nuclei (10 min), followed by mounting with ProLong™ Gold Antifade Mountant (Cat: P36930, Invitrogen). Images were obtained with an AxioImager D1 microscope (Zeiss). At least three different areas per cover slip were quantified.
Quantitative real-time PCR
Cells were seeded as for western blot. Total RNA was extracted from cells using NucleoSpin RNA isolation kit from Macherey-Nagel (ref: 740955.250). Complementary DNA was produced from 1 μg of RNA using Maxima™ H Minus cDNA Synthesis Master Mix (Cat# M1682, Invitrogen). Taqman probes were obtained from Applied Biosystems. Amplifications were run in a Viia7 or QS6 Real-Time PCR Systems (Applied Biosystems) using the following probes: PML (Hs00971694_m1, cat: 4331182). For p27 (CDKN1B), MYC, PIM1, CDK2, CDK4, E2F3, AURKA and CDC25A amplification, Universal Probe Library (Roche) primers and probes were employed (p27, For: ccctagagggcaagtacgagt, Rev: agtagaactcgggcaagctg, probe: 39; MYC, For: gctgcttagacgctggattt, Rev: taacgttgaggggcatcg, probe: 66; PIM1, For: atcaggggccaggttttc, Rev: gggccaagcaccatctaat, probe: 13; CDK2, For: aaagccagaaacaagttgacg, Rev: gtactgggcacaccctcagt, probe 77; CDK4, For: gtgcagtcggtggtacctg, Rev: aggcagagattcgcttgtgt, probe 25; E2F3, For: ggtttcggaaatgcccttac, Rev: gatgaccgctttctcctagc, probe 40; AURKA, For: gcagattttgggtggtcagt, Rev: tccgaccttcaatcatttca, probe 79; CDC25A, For: cgtcatgagaactacaaaccttga, Rev: tctggtctcttcaacactgacc, probe 67). All quantitative PCR with reverse transcription data presented were normalized using GAPDH (Hs02758991_g1, cat: 4331182) from Applied Biosystems as housekeeping.
Mice
Xenograft experiments were carried out following the ethical guidelines established by the Biosafety and Welfare Committee at CIC bioGUNE. The procedures employed were carried out following the recommendations from AAALAC. Xenograft experiments were performed as previously described5, injecting 3.106 cells per tumour, two injections per mouse, one per flank. All mice (female Hsd:Athymic Nude-Foxn1 nu/nu) were inoculated at 8-12 weeks of age. 19 days post-injection, once tumours were stablished (25-130mm3), mice were fed with chow or doxycycline diet (Research diets, D12100402) until the experimental endpoint.
p-HP1γ immunohistochemistry
After sacrifice, formalin-fixed paraffin embedded xenograft tissues were stained for p-HP1γ. Tissues were deparaffinized using the standard procedure and unmasking/antigen retrieval was performed using pH 6.0 solution for 20 min at 98°C in water bath. Tissue sections were stained for p-HP1γ using primary antibody Phospho-HP1γ (Ser83) (Cat. No: 2600, Cell Signaling technologies, 1:200) and secondary antibody Biotinylated antibody Anti-Rabbit (BP-9100, Vector Laboratories, 1:200). This was followed by Vectastain ABC solution incubation (PK-6100, Vector laboratories, 1:150) and DAB staining (SK-4105, Vector laboratories) as per the manufacturer’s protocol. Stained slides were scanned using Leica Aperio AT2 slide scanner. The criteria for senescent staining used for quantification was a very prominent nuclear staining in which the nucleus was bigger in size and its staining was darker brown than the other cells.
ChIP
Chromatin Immunoprecipitation (ChIP) was performed using the SimpleChIP® Enzymatic Chromatin IP Kit (Cat #9003, Cell Signaling Technology, Inc) as reported15. DNA quantification was carried out using a Viia7 Real-Time PCR System (Applied Biosystems) with SybrGreen reagents and primers that amplify the predicted PML binding region to MYC promoter (chr8:128748295-128748695) as follows: left primer: CCGGCTAGGGTGGAAGAG, right primer: GCTGCTATGGGCAAAGTTTC and PIM1 promoter (chr6:37137097-37137612) as follows: left primer: ACTCCCTCCGTGACTCATGT, right primer: ACGAGGGTGGTCTTTCTGTG.
Secretome analysis
Secretomes were prepared as previously described51. MDA-MB-231 sh4 PML tet on and Cama-1 sh4 PML tet on cells were pre-induced with doxycycline (150 ng ml-1) for three days. Three 150 cm2 plates where seeded per condition: 4x106 cells per plate of non-induced cells and 5x106 cells per plate of doxycycline induced cells. After two days, cell supernatants were removed and cells were washed 5 times: the first two washes were performed with PBS and the last three were made with serum-depleted DMEM. Cells were left to grow for 24h in serum-depleted DMEM. Doxycicline was maintained (150 ng ml-1). Two biological replicates, each with three technical replicates were processed.
After 24 hours supernatant was collected and one dish per condition was trypsinized and counted to check cell number and PML expression. The supernatant was first spun at 1000 rpm for 5 minutes followed by filtration through 0.2 μm filtering bottles. After this, it was concentrated using 10 kDa Amicons; first, 15 mL Amicons (Ref. UCF901024, Merck) were used, followed by 0.5 mL Amicons (Ref. UCF501069, Merck) to get final volumes close to 80 μL. The concentrated secretome was frozen at -20ºC until proteomics analysis. Protein concentration was determined with a Pierce BCA protein assay kit (Thermo Scientific). All samples were digested with trypsin in-solution prior to analysis by liquid chromatography−mass spectrometry (LC−MS). Tryptic digests were analysed by shotgun proteomics using an LTQ Velos-Orbitrap mass spectrometer (Thermo Fisher Scientific, Bremen, Germany). The RAW files of each MS run were processed using Proteome Discoverer (Thermo Fisher Scientific), and MS/MS spectra were searched against the human database of Swiss-Prot using the MASCOT (Matrix Science, London, U.K) algorithm. The results files generated from MASCOT (.DAT files) were then loaded into Scaffold (Proteome Software, Portland, OR), resulting in a nonredundant list of identified proteins per sample achieving a protein false discovery rate (FDR) under 1.0%, as estimated by a search against a decoy database.
Secretome statistical analysis
Relative spectral counting-based protein quantification analysis was performed on the different samples analyzed using Scaffold. Files containing all spectral counts for each sample and its replicates were generated and then exported to R software for normalization and statistical analysis52. All statistical computations were done using the open-source statistical package R. The data were assembled in a matrix of spectral counts, where the different conditions are represented by the columns and the identified proteins are represented by the rows. An unsupervised exploratory data analysis (EDA) by means of principal components analysis and hierarchical clustering of the samples on the SpC matrix was first performed. Then, the GLM model based on the Poisson distribution was used as a significance test52. Finally the Benjamini and Hochberg multitest correction was used to adjust the p values with control on the FDR.
Full information regarding the proteins detected in the secretome analysis can be found in Supplementary Tables 1 and 2.
Reactive Oxygen Species analysis
MDA-MB-231 cells with inducible shRNA against PML (sh4) were pre-induced with doxycycline (150 ng ml-1) for three days. Then, cells were seeded in a 6 well plate in triplicate (1.5 x 105 cells / well) maintaining the doxycycline concentration. Two additional wells with non-induced cells were used for positive and negative ROS controls respectively.
After 72 hours, 10 μM of 2′, 7′-Dichlorofluorescin diacetate (DCF-DA) (Sigma-Aldrich Ref: 35845) reactive was added to each well and cells were incubated for 30 minutes. In the last 5 minutes of the incubation time, 1 M hydrogen peroxide (H2O2) was added to the positive control well.
Subsequently, cells were washed with PBS and raised from plates employing 500 μL of trypLE reactive (Gibco™ ref: 12563-011). After that, cells were washed twice with abundant PBS to eliminate the excess of DCF-DA reactive and pellets were re-suspended in 500 μL PBS for FACS analysis. Samples were analyzed in FACS CANTO II for green fluorescence.
Datasets
Database normalization
all the datasets used for the data mining analysis were downloaded from GEO and TCGA, and subjected to background correction, log2 transformation and quartile normalization. In the case of using a pre-processed dataset, this normalization was reviewed and corrected if required.
Correlation analysis
Pearson correlation test was applied to analyse the relationship between paired genes. From this analysis, Pearson coefficient (R) indicates the existing linear correlation (dependence) between two variables X and Y, giving a value between +1 and −1 (both included), where 1 is total positive correlation, 0 is no correlation, and −1 is total negative correlation. The p-value indicates the significance of this R coefficient.
Statistical analysis
No statistical method was used to predetermine sample size. The experiments were not randomized. The investigators were not blinded to allocation during experiments and outcome assessment. Data analysed by parametric tests are represented by the mean ± s.e.m. of pooled experiments unless otherwise stated. n values represent the number of independent experiments performed or the number of individual mice. For each in vitro independent experiment, technical replicates were used and a minimum number of three experiments were performed to ensure adequate statistical power. In the in vitro experiments, normal distribution was assumed and one sample t-test was applied for one component comparisons with control and Student´s t test for two component comparisons. For in vivo experiments, a non-parametric Mann-Whitney U-test was used. Two-tailed statistical analysis was applied for experimental design without predicted result, and one-tail for validation or hypothesis-driven experiments. The confidence level used for all the statistical analyses was of 0.95 (alpha value=0.05).
Supplementary Material
Acknowledgements
Apologies to those whose related publications were not cited due to space limitations. LA, AS, MPu, AA-A and LV-J were supported by the Basque Government of education. IH was supported by the Program “Juan de la Cierva” from MINECO. FC acknowledges the Proteomics Unit at VHIO is a member ProteoRed, PRB3 (Grant IPT17/0019 - ISCIII-SGEFI / ERDF. RB acknowledges projects BFU2017-84653-P, Consolider BFU2014-57703-REDC and SAF2017-90900-REDT (MINECO/FEDER, EU). The work of VT is founded by Fundación Vasca de Innovación e Investigación Sanitarias, BIOEF (BIO15/CA/052), the AECC J.P. Bizkaia, the Basque Department of Health (2016111109) and the MINECO (RTI2018-097267-B-I00 (MCIU/AEI/FEDER, UE)). AA was supported by ERC consolidator (683136) and Swiss Cancer League (KFS4267-08-2017) grant, Dr. Josef Steiner Foundation, Swiss Card-Onco-Grant of Alfred and Annemarie von Sick grant, Helmut Horten Foundation, SNSF (310030_176045) and PCUK (RIA15-ST2-018). NM-M was supported by the Spanish Association Against Cancer (AECC), AECC JP Vizcaya and CIBERONC. The work of A. Carracedo is supported by the Basque Department of Industry, Tourism and Trade (Elkartek) and the department of education (IKERTALDE IT1106-16), the BBVA foundation, the MINECO (SAF2016-79381-R (FEDER/EU); Severo Ochoa Excellence Accreditation SEV-2016-0644; Excellence Networks SAF2016-81975-REDT), European Training Networks Project (H2020-MSCA-ITN-308 2016 721532), the AECC (IDEAS175CARR, GCTRA18006CARR), La Caixa Foundation (HR17-00094), the AstraZeneca Oncology prize and the European Research Council (Starting Grant 336343, PoC 754627). CIBERONC was co-funded with FEDER funds and funded by ISCIII.
Footnotes
Authors´ contributions
LA, NM-M and MP performed the majority of the in vitro and in vivo experiments, unless specified otherwise. SF-R, JRC, AA-A, LV-J, PZ-G and IH contributed to specific in vitro analyses. MPu and AZ-L performed ROS analyses. AS and VT coordinated or performed secretome preparation. AR performed the histochemical staining and quantification of p-HP1γ staining. IA contributed to the sub-cloning of shC, sh1p27 and sh2p27 into pLKO-Tet-On- Blasticidin. ARC performed the bioinformatic and biostatistical analysis. JV and FC performed or coordinated the secretome analysis. LB-B provided support with imaging analyses. RB and JDS provided technical advice. AA supervised the histochemical staining and quantification of p-HP1γ staining. AC and NM-M directed the project, contributed to data analysis and wrote the manuscript.
The authors declare no conflict of interest
References
- 1.Sorlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A. 2001;98(19):10869–10874. doi: 10.1073/pnas.191367098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.van de Vijver MJ, He YD, van't Veer LJ, Dai H, Hart AA, Voskuil DW, et al. A gene-expression signature as a predictor of survival in breast cancer. N Engl J Med. 2002;347(25):1999–2009. doi: 10.1056/NEJMoa021967. [DOI] [PubMed] [Google Scholar]
- 3.Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, Dunning MJ, et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012;486(7403):346–352. doi: 10.1038/nature10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bos PD, Zhang XH, Nadal C, Shu W, Gomis RR, Nguyen DX, et al. Genes that mediate breast cancer metastasis to the brain. Nature. 2009;459(7249):1005–1009. doi: 10.1038/nature08021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carracedo A, Ma L, Teruya-Feldstein J, Rojo F, Salmena L, Alimonti A, et al. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J Clin Invest. 2008;118(9):3065–3074. doi: 10.1172/JCI34739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carracedo A, Weiss D, Leliaert AK, Bhasin M, de Boer VC, Laurent G, et al. A metabolic prosurvival role for PML in breast cancer. The Journal of clinical investigation. 2012;122(9):3088–3100. doi: 10.1172/JCI62129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tessier S, Martin-Martin N, de The H, Carracedo A, Lallemand-Breitenbach V. Promyelocytic Leukemia Protein, a Protein at the Crossroad of Oxidative Stress and Metabolism. Antioxid Redox Signal. 2017;26(9):432–444. doi: 10.1089/ars.2016.6898. [DOI] [PubMed] [Google Scholar]
- 8.Bernardi R, Pandolfi PP. Structure, dynamics and functions of promyelocytic leukaemia nuclear bodies. Nat Rev Mol Cell Biol. 2007;8(12):1006–1016. doi: 10.1038/nrm2277. [DOI] [PubMed] [Google Scholar]
- 9.Ito K, Bernardi R, Morotti A, Matsuoka S, Saglio G, Ikeda Y, et al. PML targeting eradicates quiescent leukaemia-initiating cells. Nature. 2008;453(7198):1072–1078. doi: 10.1038/nature07016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ito K, Carracedo A, Weiss D, Arai F, Ala U, Avigan DE, et al. A PML–PPAR-δ pathway for fatty acid oxidation regulates hematopoietic stem cell maintenance. Nat Med. 2012;18(9):1350–1358. doi: 10.1038/nm.2882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chung YL, Wu ML. Dual oncogenic and tumor suppressor roles of the promyelocytic leukemia gene in hepatocarcinogenesis associated with hepatitis B virus surface antigen. Oncotarget. 2016;7(19):28393–28407. doi: 10.18632/oncotarget.8613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu SB, Shen ZF, Guo YJ, Cao LX, Xu Y. PML silencing inhibits cell proliferation and induces DNA damage in cultured ovarian cancer cells. Biomed Rep. 2017;7(1):29–35. doi: 10.3892/br.2017.919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ponente M, Campanini L, Cuttano R, Piunti A, Delledonne GA, Coltella N, et al. PML promotes metastasis of triple-negative breast cancer through transcriptional regulation of HIF1A target genes. JCI Insight. 2017;2(4):e87380. doi: 10.1172/jci.insight.87380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhou W, Cheng L, Shi Y, Ke SQ, Huang Z, Fang X, et al. Arsenic trioxide disrupts glioma stem cells via promoting PML degradation to inhibit tumor growth. Oncotarget. 2015;6(35):37300–37315. doi: 10.18632/oncotarget.5836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Martin-Martin N, Piva M, Urosevic J, Aldaz P, Sutherland JD, Fernandez-Ruiz S, et al. Stratification and therapeutic potential of PML in metastatic breast cancer. Nat Commun. 2016;7 doi: 10.1038/ncomms12595. 12595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weinstein IB, Joe A. Oncogene addiction. Cancer Res. 2008;68(9):3077–3080. doi: 10.1158/0008-5472.CAN-07-3293. discussion 3080. [DOI] [PubMed] [Google Scholar]
- 17.Childs BG, Durik M, Baker DJ, van Deursen JM. Cellular senescence in aging and age-related disease: from mechanisms to therapy. Nat Med. 2015;21(12):1424–1435. doi: 10.1038/nm.4000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nardella C, Clohessy JG, Alimonti A, Pandolfi PP. Pro-senescence therapy for cancer treatment. Nat Rev Cancer. 2011;11(7):503–511. doi: 10.1038/nrc3057. [DOI] [PubMed] [Google Scholar]
- 19.Lin HK, Chen Z, Wang G, Nardella C, Lee SW, Chan CH, et al. Skp2 targeting suppresses tumorigenesis by Arf-p53-independent cellular senescence. Nature. 2010;464(7287):374–379. doi: 10.1038/nature08815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Majumder PK, Grisanzio C, O'Connell F, Barry M, Brito JM, Xu Q, et al. A prostatic intraepithelial neoplasia-dependent p27 Kip1 checkpoint induces senescence and inhibits cell proliferation and cancer progression. Cancer Cell. 2008;14(2):146–155. doi: 10.1016/j.ccr.2008.06.00. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ivanschitz L, De The H, Le Bras M. PML, SUMOylation, and Senescence. Front Oncol. 2013;3:171. doi: 10.3389/fonc.2013.00171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang R, Poustovoitov MV, Ye X, Santos HA, Chen W, Daganzo SM, et al. Formation of MacroH2A-containing senescence-associated heterochromatin foci and senescence driven by ASF1a and HIRA. Dev Cell. 2005;8(1):19–30. doi: 10.1016/j.devcel.2004.10.019. [DOI] [PubMed] [Google Scholar]
- 23.Lo Re O, Vinciguerra M. Histone MacroH2A1: A Chromatin Point of Intersection between Fasting, Senescence and Cellular Regeneration. Genes (Basel) 2017;8(12) doi: 10.3390/genes8120367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Braso-Maristany F, Filosto S, Catchpole S, Marlow R, Quist J, Francesch-Domenech E, et al. PIM1 kinase regulates cell death, tumor growth and chemotherapy response in triple-negative breast cancer. Nat Med. 2016;22(11):1303–1313. doi: 10.1038/nm.4198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cancer Genome Atlas N. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490(7418):61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Horiuchi D, Camarda R, Zhou AY, Yau C, Momcilovic O, Balakrishnan S, et al. PIM1 kinase inhibition as a targeted therapy against triple-negative breast tumors with elevated MYC expression. Nat Med. 2016;22(11):1321–1329. doi: 10.1038/nm.4213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Horiuchi D, Kusdra L, Huskey NE, Chandriani S, Lenburg ME, Gonzalez-Angulo AM, et al. MYC pathway activation in triple-negative breast cancer is synthetic lethal with CDK inhibition. J Exp Med. 2012;209(4):679–696. doi: 10.1084/jem.20111512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhao W, Qiu R, Li P, Yang J. PIM1: a promising target in patients with triple-negative breast cancer. Med Oncol. 2017;34(8):142. doi: 10.1007/s12032-017-0998-y. [DOI] [PubMed] [Google Scholar]
- 29.Hernandez-Segura A, Nehme J, Demaria M. Hallmarks of Cellular Senescence. Trends Cell Biol. 2018;28(6):436–453. doi: 10.1016/j.tcb.2018.02.001. [DOI] [PubMed] [Google Scholar]
- 30.Dreesen O, Chojnowski A, Ong PF, Zhao TY, Common JE, Lunny D, et al. Lamin B1 fluctuations have differential effects on cellular proliferation and senescence. J Cell Biol. 2013;200(5):605–617. doi: 10.1083/jcb.201206121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Freund A, Laberge RM, Demaria M, Campisi J. Lamin B1 loss is a senescence-associated biomarker. Mol Biol Cell. 2012;23(11):2066–2075. doi: 10.1091/mbc.E11-10-0884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Niwa-Kawakita M, Ferhi O, Soilihi H, Le Bras M, Lallemand-Breitenbach V, de The H. PML is a ROS sensor activating p53 upon oxidative stress. J Exp Med. 2017;214(11):3197–3206. doi: 10.1084/jem.20160301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Di Mitri D, Toso A, Chen JJ, Sarti M, Pinton S, Jost TR, et al. Tumour-infiltrating Gr-1+ myeloid cells antagonize senescence in cancer. Nature. 2014;515(7525):134–137. doi: 10.1038/nature13638. [DOI] [PubMed] [Google Scholar]
- 34.Bazarov AV, Van Sluis M, Hines WC, Bassett E, Beliveau A, Campeau E, et al. p16(INK4a) -mediated suppression of telomerase in normal and malignant human breast cells. Aging Cell. 2010;9(5):736–746. doi: 10.1111/j.1474-9726.2010.00599.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bykov VJ, Issaeva N, Shilov A, Hultcrantz M, Pugacheva E, Chumakov P, et al. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat Med. 2002;8(3):282–288. doi: 10.1038/nm0302-282. [DOI] [PubMed] [Google Scholar]
- 36.Rodier G, Montagnoli A, Di Marcotullio L, Coulombe P, Draetta GF, Pagano M, et al. p27 cytoplasmic localization is regulated by phosphorylation on Ser10 and is not a prerequisite for its proteolysis. EMBO J. 2001;20(23):6672–6682. doi: 10.1093/emboj/20.23.6672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Morishita D, Katayama R, Sekimizu K, Tsuruo T, Fujita N. Pim kinases promote cell cycle progression by phosphorylating and down-regulating p27Kip1 at the transcriptional and posttranscriptional levels. Cancer Res. 2008;68(13):5076–5085. doi: 10.1158/0008-5472.CAN-08-0634. [DOI] [PubMed] [Google Scholar]
- 38.Carracedo A, Cantley LC, Pandolfi PP. Cancer metabolism: fatty acid oxidation in the limelight. Nat Rev Cancer. 2013;13(4):227–232. doi: 10.1038/nrc3483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Camarda R, Zhou AY, Kohnz RA, Balakrishnan S, Mahieu C, Anderton B, et al. Inhibition of fatty acid oxidation as a therapy for MYC-overexpressing triple-negative breast cancer. Nat Med. 2016;22(4):427–432. doi: 10.1038/nm.4055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wu CH, van Riggelen J, Yetil A, Fan AC, Bachireddy P, Felsher DW. Cellular senescence is an important mechanism of tumor regression upon c-Myc inactivation. Proc Natl Acad Sci U S A. 2007;104(32):13028–13033. doi: 10.1073/pnas.0701953104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Consortium EP. The ENCODE (ENCyclopedia Of DNA Elements) Project. Science. 2004;306(5696):636–640. doi: 10.1126/science.1105136. [DOI] [PubMed] [Google Scholar]
- 42.Liu R, Zhang T, Zhu G, Xing M. Regulation of mutant TERT by BRAF V600E/MAP kinase pathway through FOS/GABP in human cancer. Nat Commun. 2018;9(1):579. doi: 10.1038/s41467-018-03033-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nakano T, Kanai Y, Amano Y, Yoshimoto T, Matsubara D, Shibano T, et al. Establishment of highly metastatic KRAS mutant lung cancer cell sublines in long-term three-dimensional low attachment cultures. PLoS One. 2017;12(8):e0181342. doi: 10.1371/journal.pone.0181342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Martin-Martin N, Sutherland JD, Carracedo A. PML: Not all about Tumor Suppression. Front Oncol. 2013;3:200. doi: 10.3389/fonc.2013.00200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Guarnerio J, Mendez LM, Asada N, Menon AV, Fung J, Berry K, et al. A non-cell-autonomous role for Pml in the maintenance of leukemia from the niche. Nat Commun. 2018;9(1) doi: 10.1038/s41467-017-02427-x. 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bischof O, Kirsh O, Pearson M, Itahana K, Pelicci PG, Dejean A. Deconstructing PML-induced premature senescence. EMBO J. 2002;21(13):3358–3369. doi: 10.1093/emboj/cdf341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ferbeyre G, de Stanchina E, Querido E, Baptiste N, Prives C, Lowe SW. PML is induced by oncogenic ras and promotes premature senescence. Genes Dev. 2000;14(16):2015–2027. [PMC free article] [PubMed] [Google Scholar]
- 48.Pearson M, Carbone R, Sebastiani C, Cioce M, Fagioli M, Saito S, et al. PML regulates p53 acetylation and premature senescence induced by oncogenic Ras. Nature. 2000;406(6792):207–210. doi: 10.1038/35018127. [DOI] [PubMed] [Google Scholar]
- 49.Marchesini M, Matocci R, Tasselli L, Cambiaghi V, Orleth A, Furia L, et al. PML is required for telomere stability in non-neoplastic human cells. Oncogene. 2016;35(14):1811–1821. doi: 10.1038/onc.2015.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wiederschain D, Wee S, Chen L, Loo A, Yang G, Huang A, et al. Single-vector inducible lentiviral RNAi system for oncology target validation. Cell Cycle. 2009;8(3):498–504. doi: 10.4161/cc.8.3.7701. [DOI] [PubMed] [Google Scholar]
- 51.Gregori J, Villarreal L, Sanchez A, Baselga J, Villanueva J. An effect size filter improves the reproducibility in spectral counting-based comparative proteomics. Journal of proteomics. 2013;95:55–65. doi: 10.1016/j.jprot.2013.05.030. [DOI] [PubMed] [Google Scholar]
- 52.Villarreal L, Mendez O, Salvans C, Gregori J, Baselga J, Villanueva J. Unconventional secretion is a major contributor of cancer cell line secretomes. Mol Cell Proteomics. 2013;12(5):1046–1060. doi: 10.1074/mcp.M112.021618. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.