

Abstract
The extracellular signal-regulated kinase (ERK) cascade comprised of the Raf, MEK, and ERK protein kinases constitutes a key effector cascade used by the Ras GTPases to relay signals regulating cell growth, survival, proliferation, and differentiation. Of the ERK cascade components, the regulation of the Raf kinases is by far the most complex, involving changes in subcellular localization, protein and lipid interactions, as well as alterations in the Raf phosphorylation state. The Raf kinases interact directly with active, membrane-localized Ras, and this interaction is often the first step in the Raf activation process, which ultimately results in ERK activation and the downstream phosphorylation of cellular targets that will specify a particular biological response. Here, we will examine our current understanding of how Ras promotes Raf activation, focusing on the molecular mechanisms that contribute to the Raf activation/inactivation cycle.
Members of the Raf serine/threonine kinase family are the initiating enzymes in the three-tiered extracellular signal-regulated kinase (ERK) cascade, and in mammalian cells, there are three Raf proteins, A-Raf, B-Raf, and C-Raf (also known as Raf-1) (Wellbrock et al. 2004). The Raf kinases were first discovered when the raf-1 gene was identified as the cellular counterpart of the murine retroviral oncogene, v-raf (Rapp et al. 1983). Later, this kinase family rose to prominence when they were found to be direct effectors of activated Ras, mediating signal transmission from Ras to the downstream kinases MEK and ERK (Marshall 1996). Not surprisingly, given their central position in such a major signaling pathway, mutations in the Raf kinases also contribute to human disease states. In particular, somatic mutations in B-Raf are important drivers of human cancer (Dhomen and Marais 2007), and germline mutations in either B-Raf or C-Raf can be causative for certain developmental disorders in the Rasopathies spectrum (Tartaglia et al. 2011).
Despite their critical function in cell signaling, determining how Raf activity is regulated has been a challenging task—resulting from the complexity of the Raf activation process. In this article, we will review the molecular mechanisms involved in Raf kinase regulation. Our discussion will include an examination of the presignaling inactive state of the Rafs as well as the events that contribute to Raf kinase activation, with a focus on the crucial role that the Ras GTPases play in promoting Raf activation under normal growth conditions as well as in human disease states. Whenever possible, we will also comment on the regulatory differences that have been identified for the individual Raf members, and we will conclude with a description of the mechanisms that attenuate Raf activity following signal transmission.
THE RAF PRESIGNALING INACTIVE STATE
All members of the Raf kinase family contain three conserved regions, CR1, CR2, and CR3, and each Raf protein can be divided into two functional domains—an amino-terminal regulatory domain and a carboxy-terminal catalytic domain (Fig. 1A) (Daum et al. 1994). Both the CR1, which consists of a Ras-binding domain (RBD) and a cysteine-rich domain (CRD), and the CR2, a region rich in serine/threonine residues, are found within the amino-terminal regulatory domain, whereas the CR3 comprises the carboxy-terminal kinase domain. The Raf proteins do not contain any intrinsic subcellular localization motifs, and in quiescent cells, they localize to the cytosol as monomers (Nan et al. 2013). Before pathway activation, the Raf monomers are maintained in a stable, presignaling inactive state through multiple regulatory mechanisms, including autoinhibition, phosphorylation of negative regulatory sites, and binding of inhibitory proteins (Figs. 1B and 2).
Figure 1.
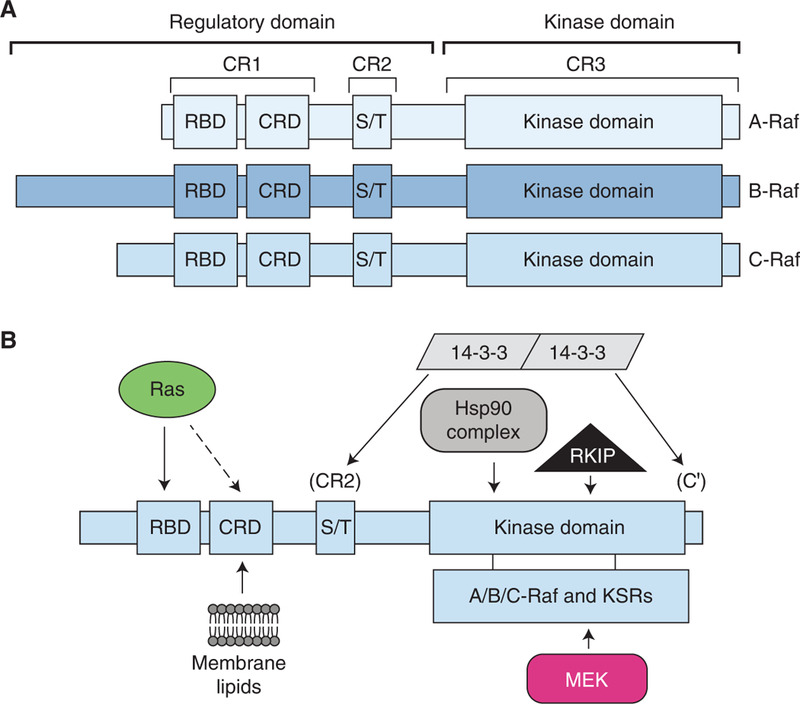
Raf domain structure and protein interactions. (A) The Raf kinases can be divided into two functional domains: an amino-terminal regulatory domain and a carboxy-terminal kinase domain and they contain three conserved regions (CRs): CR1, which contains a Ras-binding domain (RBD) and a cysteine-rich domain (CRD); CR2, an area rich in serine/threonine residues (S/T); and CR3, which contains the protein kinase domain. (B) Numerous Raf-binding partners have been identified.
Figure 2.

Regulation of the Raf kinases by phosphorylation. (Top) Raf is both positively and negatively regulated by phosphorylation on numerous sites. (Bottom) Shown are the amino acid sequence alignments of the sites of Raf phosphorylation, with the residues phosphorylated shown in red, and the relevant kinases indicated above the sequences. A glutamic acid residue that can act as a phosphomimetic to promote the active conformation of the activation segment (AS) is also shown in green.
Raf Autoinhibition
The first indication that the Raf amino-terminal domain might possess autoinhibitory activity came from the observation that this domain is absent in the oncogenic v-Raf protein (Rapp et al. 1983). Subsequently, it was shown that deletion of the amino-terminal domain could convert any of the mammalian Raf proteins into constitutively active kinases capable of inducing cellular transformation (Stanton et al. 1989; Heidecker et al. 1990). Further studies in which the amino-and carboxy-terminal domains of B-Raf or C-Raf were expressed as individual proteins revealed that the isolated amino-terminal domains of the Rafs could bind their respective kinase domains and block signal transmission to MEK and ERK (Cutler et al. 1998; Chong and Guan 2003; Tran and Frost 2003; Tran et al. 2005). Regions in the CR1, including the CRD and parts of the RBD, are critical for the amino-terminal autoinhibitory activity, and phosphorylation of sites in the carboxy-terminal kinase domain contribute prominently to the relief of autoinhibition, with phosphorylation of sites in the negative charge regulatory region (N-region) being particularly important (Cutler et al. 1998; Tran and Frost 2003; Tran et al. 2005).
In addition to the autoinhibition mediated by the amino-terminal domain, structural data indicate that the Raf kinase domain itself is held in an inactive conformation. Like all other protein kinases, the Raf kinase domain consists of an amino-terminal lobe (N-lobe) and a carboxy-terminal lobe (C-lobe) that are connected to one another through a flexible hinge region (Shaw et al. 2014). The N-lobe contains five antiparallel β-strands and the regulatory αC-helix, whereas the C-lobe is comprised primarily of α-helices and contains the activation segment (AS). For all kinases to assume the active/ON state conformation, spatially conserved hydrophobic res-idues spanning both lobes of the kinase domain must align to form two structural entities called the regulatory and catalytic spines (McClendon et al. 2014). The regulatory spine (R-spine) consists of four residues, with each residue derived from a key catalytic element in the kinase domain—a residue from the β4-strand, the αC-he-lix, the DFG motif, and the HRD sequence. In many inactive kinase structures, the R-spine is misconfigured and for the spine residues to align, both the αC-helix and the AS must change positions. Although the structure of an authentic OFF state Raf kinase has not been solved, the structure of a monomeric, inhibitor-bound B-Raf kinase domain has been determined that has features characteristic of other inactive protein kinases (Thevakumaran et al. 2015). In this monomeric B-Raf structure, the αC-helix is shifted to an outward position and residues adjacent to the DFG motif in the AS form a small helix called the AS-H1-helix (Fig. 3). Moreover, extensive interactions between the αC-helix, the AS-H1-helix and other residues in the active site, including residues in the conserved DFG and HRD motifs, stabilize the αC-helix in the inactive OUT position and inhibit the formation of an essential salt bridge that defines the active state (E501-K483 for B-Raf, E393-K375 for C-Raf, E354-K336 for A-Raf). A similarly config-ured AS-H1-helix has been observed in the inactive conformation of the epidermal growth factor receptor (EGFR) kinase (Zhang et al. 2006), and it is important to note that most activating B-Raf mutations identified in human cancer occur in residues comprising the AS-H1-helix (Lavoie and Therrien 2015). Thus, it is tempting to speculate that in the context of full-length Raf proteins, Raf monomers are held in an inactive state through intramolecular interactions occurring within the kinase domain as well as between the kinase domain and the amino-terminal regulatory domain.
Figure 3.
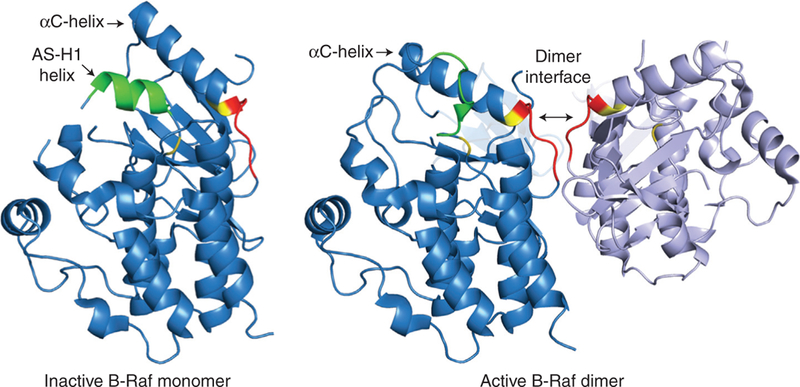
Regulation of the Raf kinase domain by dimerization. Shown are the crystal structures of a monomeric, B-Raf kinase domain in an inactive conformation (PDB, 4WO5) and of dimerized B-Raf kinase domains in an active conformation (taken from the B-Raf–MEK complex PDB, 4MNE). In the monomeric, inactive state, the αC-helix is shifted outward and residues adjacent to the DFG motif in the activation segment (AS) form a small helix known as the AS-H1-helix. When Raf kinase domains dimerize in a side-to-side manner, the αC-helix shifts inward to the active position and the AS-H1-helix is disrupted, allowing residues in the R-spine to align. The conserved Raf dimer interface motif RKTR is shown in red, the R-spine residues in the αC-helix (L505) and in the DFG motif (D595) are indicated in yellow, and the sequences comprising the AS-H1-helix are shown in green.
14-3-3 Binding to the CR2 Site
The autoinhibited Raf conformation is further stabilized by interactions with the 14-3-3 family of phosphoserine/phosphothreonine-binding proteins (Muslin et al. 1996). All Raf kinases contain two high-affinity, phosphorylation-dependent 14-3-3 binding sites, one in the CR2 (S365 of B-Raf, S259 of C-Raf, S214 of A-Raf) and one following the kinase domain (S729 of B-Raf, S621 of C-Raf, S582 of A-Raf), both of which are phosphorylated in the presignaling inactive state (Morrison et al. 1993; Dougherty et al. 2004; Ritt et al. 2010). 14-3-3 proteins form constitutive dimers with each protomer of the dimer containing an independent binding channel, and it has been proposed that a 14-3-3 dimer can interact with both the CR2 and carboxy-terminal sites simultaneously to stabilize the autoinhibited conformation (Tzivion et al. 1998). Moreover, binding of 14-3-3 to the CR2 site has a known negative regulatory function and appears to mask regions within the amino terminus, such as the CRD, which are required for Raf activation (Michaud et al. 1995; Clark et al. 1997; McPherson et al. 1999; Light et al. 2002). Mutation of the CR2 serine site or residues that comprise the 14-3-3 binding motif have been identified in tumors and Rasopathies as gain-of-function mutations (Lavoie and Therrien 2015), resulting in increased Ras/Raf binding, membrane recruitment, and Raf activation (Michaud et al. 1995; Dhillon et al. 2002a; Light et al. 2002; Pandit et al. 2007; Razzaque et al. 2007). Protein kinase A (PKA) and AKT have been identified as the primary cellular kinases that phosphorylate the CR2 site; however, the CR2 site can also be phosphorylated by LATS1 through signaling cross talk with the MST/Hippo pathway (Zimmermann and Moelling 1999; Dhillon et al. 2002b; Dumaz and Marais 2003; Romano et al. 2014).
Hsp90 Chaperone Complex
In their presignaling inactive state as well as in their active signaling state, all members of the Raf kinase family interact with Hsp90 complexes, which include Hsp90 proteins, the Cdc37 targeting subunit, and the peptidyl-prolyl cistrans isomerase FKBP5 (Stancato et al. 1994; Eisenhardt et al. 2016; Diedrich et al. 2017). The Hsp90 complex binds to the Raf kinase domain and this interaction is required for Raf protein stability (Schulte et al. 1995).
B-Raf–MEK1 Complex
Another protein that has been found to associate with inactive B-Raf complexes is MEK1 (Haling et al. 2014; Eisenhardt et al. 2016). In experiments investigating whether preformed Raf/ MEK complexes exist before Ras pathway activation, MEK1 was found to preferentially interact with B-Raf proteins found in the cytosol of quiescent cells or in cancer cell lines that contain low levels of active MEK (Haling et al. 2014). When cells were treated with growth factors to activate the pathway, the B-Raf–MEK complex was disrupted in a manner that correlated with the appearance of active phosphoMEK, but could reform when phosphoMEK levels re-turned to baseline, suggesting that phosphorylation and activation of MEK disrupts the interaction. Structural analysis of the B-Raf–MEK1 complex indicates that the two kinase domains interact in a face-to-face manner (Haling et al. 2014), with primary points of contact being the AS of both proteins and their respective αG-helices, a structural element implicated in kinase–substrate interactions. Although it is not clear why MEK1 preferentially binds inactive B-Raf, it is interesting to note that the RKIP inhibitory protein has been found to interact with the C-Raf kinase domain in a manner that is mutually exclusive with MEK binding (Yeung et al. 2000). Phosphorylation of RKIP on S153 by protein kinase C (PKC) disrupts the RKIP–C-Raf interaction and appears to account for the release of RKIP from C-Raf under signaling conditions (Corbit et al. 2003).
From the above findings, the following model has emerged: In quiescent cells, members of the Raf kinase family exist as autoinhibited monomers, with the inactive conformation stabilized by the phosphorylation-dependent binding of 14-3-3 dimers as well as by the interaction with Hsp90 complexes. A pool of B-Raf and MEK1 exists in preformed complexes, whereas MEK may be precluded from C-Raf complexes as a result of RKIP binding. The presignaling, inactive Raf complexes localize to the cytosol, poised ready for recruitment in response to up-stream pathway activation (Fig. 4).
Figure 4.

The Raf activation–inactivation cycle. A model depicting the Raf kinases in a presignaling inactive state, a signaling active state, and a postsignaling inactive state is shown (see text for details). GDP, Guanosine diphosphate; GTP, guanosine triphosphate; RBD, Ras-binding domain; CRD, cysteine-rich domain; ERK, extracellular signal-regulated kinase.
THE RAF ACTIVATION PROCESS
For the Rafs to become active enzymes, auto-inhibition mediated by the amino-terminal domain must be relieved and the kinase domain must assume an active/ON state conformation. After more than 30 years of research, we now know that the cell accomplishes this task through an intricate series of events that include a change in Raf subcellular localization, protein and lipid interactions (Fig. 1B), as well as regulatory phosphorylation/dephosphorylationevents (Fig. 2).
Ras Binding
A major breakthrough in understanding Raf activation came with the discovery that all Raf kinases selectively interact with active guanosine triphosphate (GTP)-bound Ras (Van Aelst et al. 1993; Vojtek et al. 1993; Zhang et al. 1993). Binding to active Ras does not stimulate Raf enzymatic activity directly, but instead localizes the normally cytosolic Raf proteins to the plasma membrane. This change in localization is a critical step in the Raf activation process as artificially targeting Raf to the plasma membrane results in a kinase that is constitutively active in a Ras-independent manner (Leevers et al. 1994; Stokoe et al. 1994). The initial contact with Ras is made by the Raf-RBD, which adopts a conserved ubiquitin-like structure that binds with high affinity (18 nM) to the Ras effector domain in its GTP-bound state (Herrmann et al. 1995; Nassar et al. 1995). In addition to the RBD, the Raf CRD makes distinct contacts with lipid modified Ras. The Raf CRD is a zinc-coordinated structure of the atypical type (Mott et al. 1996), and as an isolated domain, the CRD shows constitutive low-affinity binding to Ras with no preference for its nucleotide state (Williams et al. 2000). However, in the context of the RBD, the CRD can further facilitate the membrane recruitment of the Rafs by selectively binding the farnesyl groups of processed Ras (Luo et al. 1997; Williams et al. 2000). Ras binding also allows the CRD zinc finger motifs to directly contact phosphatidyl serine in the plasma membrane (Ghosh et al. 1996; Hekman et al. 2002). Full activation of Raf requires that both the RBD and the CRD are engaged, and CRD interactions with Ras and phosphatidyl serine are critical for relief of autoinhibition as well as stably linking Raf to the plasma membrane (Roy et al. 1997; Cutler et al. 1998; Bondeva et al. 2002). Notably, more than 40% of activating B-Raf mutations identified in the Rasopathy cardio-facio-cutaneous (CFC) syndrome occur in the CRD, further demonstrating the biological importance of this domain (Sarkozy et al. 2009).
Displacement of 14-3-3 and Dephosphorylation of the CR2 Site
In addition to changing the subcellular localization of the Rafs, experimental evidence suggests that the interaction with Ras and membrane phospholipids displaces 14-3-3 from the CR2 site, thereby exposing the site to phosphatase activity (Rommel et al. 1996; McPherson et al. 1999). In response to epidermal growth factor signaling, a complex consisting of active M-Ras, Shoc2, and the catalytic subunit of protein phosphatase 1 (PP1) plays a prominent role in mediating the dephosphorylation of the CR2 site (Rodriguez-Viciana et al. 2006). However, the CR2 site is also a substrate for the heterotrimeric protein phosphatase PP2A (Abraham et al. 2000; Jaumot and Hancock 2001; Ory et al. 2003a). PP2A has been identified in active Raf complexes and may contribute to the dephos-phorylation of the CR2 site in specific cell types or under certain signaling conditions (Ory et al. 2003a; Eisenhardt et al. 2016).
Raf Dimerization and Activating Phosphorylation
The interaction with Ras also serves a critical function in promoting the phosphorylation of the Raf kinase domain on activating sites and is often a prerequisite for Raf dimerization. Phosphorylation sites that are particularly important for Raf activation are found in the negative charge regulatory region (N-region). The N-region of all Raf proteins contains either negatively charged amino acids or residues that become negatively charged as a result of phosphorylation (Mason et al. 1999). Negative charges in this region contribute to the relief of autoinhibition (Cutler et al. 1998; Tran and Frost 2003; Tran et al. 2005) and are needed to form intermolecular contacts that stabilize the Raf dimer structure (Baljuls et al. 2011; Hu et al. 2013). The N-regions of both C-Raf and A-Raf are comprised of phosphoacceptor residues that only become phosphorylated at the cell surface in response to signaling stimuli (Mason et al. 1999). In contrast, the N-region of B-Raf maintains a constant charge because of the presence of two aspartic acid residues and a serine site (S445) that is constitutively phosphorylated by the CK2 kinase (Mason et al. 1999; Ritt et al. 2007). The tyrosine residues in the N-region of C-Raf and A-Raf are phosphorylated primarily by members of the SRC family kinases, whereas the N-region serine site can be phosphorylated by members of the PAK and PKC families, or by CK2 if the tyrosine sites are phosphorylated (Fabian et al. 1993; Marais et al. 1995; Diaz et al. 1997; King et al. 1998; Chaudhary et al. 2000; Chiloeches et al. 2001; Hamilton et al. 2001; Ritt et al. 2007).
A second major breakthrough in understanding Raf activation came with the discovery that under most conditions, dimerization of the Raf kinases is required. Structural studies indicate that the Raf kinase domains form side-to-side dimers with interactions mediated by both the N-and C-lobes of the kinase domain (Raja-kulendran et al. 2009). Although it is unclear whether any contacts are made between the Raf aminoterminal domains, binding to Ras, concurrent with the membrane clustering of active Ras proteins, is thought to increase the local concentration of Raf at the cell surface and induce conformational changes that promote Raf dimerization (Plowman et al. 2005; Terai and Matsuda 2005; Tian et al. 2007; Hibino et al. 2009). In addition to the interaction with Ras, the retained binding of 14-3-3 to the Raf carboxy-terminal site is also required for Raf dimerization in cells (Rushworth et al. 2006). Whether the bound 14-3-3 dimers facilitate contact between the Raf kinase domains or act to stabilize the dimerized structure is currently un-known; however, Raf proteins that contain mutations in the carboxy-terminal serine site fail to dimerize and cannot be activated (Garnett et al. 2005; Rajakulendran et al. 2009; Ritt et al. 2010). The Raf kinases can homodimerize or heterodi-merize with any other Raf family member, and they can form side-to-side dimers with the closely related kinase suppressor of Ras (KSR) proteins (Hu et al. 2013). Although the factors that determine the dimerization preferences are poorly understood, it has been shown that B-Raf/C-Raf heterodimers predominate in Rasmediated signaling and appear to have the highest catalytic activity (Weber et al. 2001; Rushworth et al. 2006; Freeman et al. 2013).
Structural studies have provided much insight regarding how dimerization promotes Raf activation. As stated above, for protein kinase domains to adopt an active/ON conformation, the hydrophobic residues comprising the regulatory R-spine must align, and for many kinases, this requires that the αC-helix in the N-lobe and the AS in the C-lobe shift to an active position. For the Raf kinases, the αC-helix and the dimer interface are allosterically linked in that the hydrophobic R-spine residue in the αC-helix (L505 for B-Raf, L397 for C-Raf, L358 for A-Raf) lies adjacent to the conserved RKTR motif in the dimer interface (Rajakulendran et al. 2009), and when dimerization occurs, contacts made at the dimer interface are thought to promote key conformational changes (Fig. 3). In particular, dimer interface contact is thought to shift the αC-helix into the active IN position and disrupt its interactions with the AS-H1-helix, which in turn may help destabilize the helix (Thevakumaran et al. 2015). Interestingly, a protomer does not need to possess cat-alytic activity to transactivate the second protomer as oncogenic mutations that abolish the kinase activity of B-Raf can promote tumorigenesis through the allosteric activation of C-Raf (Wan et al. 2004). B-Raf proteins that are inactivated by inhibitor treatment can also transactivate C-Raf in a Ras-dependent manner to promote paradoxical ERK cascade signaling (Hatzivassiliou et al. 2010; Heidorn et al. 2010; Poulikakos et al. 2010). Notably, the key determinant for whether a protomer in a dimer complex can function as an activator for the second protomer is the presence of charged and/or phosphorylated residues in the N-region (Hu et al. 2013).
Similar to many other kinases, it is also thought that phosphorylation of the AS is required for full Raf activity, functioning to expose the catalytic cleft and to promote the alignment of the R-spine residue in the DFG motif. Two conserved AS residues have been implicated, T598/S601 for B-Raf, T491/S494 for C-Raf, and T452/T455 for A-Raf, with substitution of phosphomimetic residues at these sites resulting in increased kinase activity (Zhang and Guan 2000; Chong et al. 2001). Given that these residues localize to the region of the AS that forms the AS-H1-helix, it has been proposed that phosphorylation of these sites may be required to disrupt the AS-H1-helix, which binds and stabilizes the αC-helix in an inactive OUT position (Thevakumaran et al. 2015). Through in vitro kinase assays, one study has provided evidence that the phosphorylation of these sites occurs in cis after Raf dimerization (Hu et al. 2013). Strikingly, however, detecting these phosphorylation events in cells has been particularly difficult. For example, these phosphorylation sites have not been identified in any large-scale phosphoproteomic study despite the frequent detection of other phosphorylation sites, including the Raf N-region sites (Lavoie and Therrien 2015). Thus, it is possible that phosphorylation of the AS sites occurs at a very low level or is highly transient, or based on the B-Raf/MEK1 structure may not always be required. In most Raf crystals the AS is largely unstructured; however, in the B-Raf–MEK1 complex, residue E611 in the B-Raf AS acts as a phosphomimetic to form an intramolecular salt bridge with R575 in the conserved HRD motif, allowing the AS to assume an active conformation (Haling et al. 2014). The E611 residue aligns with AS phosphorylation sites known to be required for the activation of other kinases, including PKCα, and given that residues analogous to E611 and R575 are conserved in all Raf members, it is possible that the AS of the Raf kinases can adopt an “active” conformation in the absence of phosphorylation. Thus, although phosphorylation of the AS sites would promote the active conformation, their phosphorylation may not be essential under all signaling conditions or for all Raf proteins.
Nonetheless, it is clear that the conformation of the AS is a critical determinant in Raf kinase activation, as many oncogenic B-Raf mutations occur in this segment, with V600E being the most prevalent. In the case of V600E, not only would this substitution act as a phosphomimetic to disrupt the AS-H1-helix, structural studies indicate that the negatively charged side chain of the glutamic acid can form an intramolecular salt bridge with K507, a component of the RKTR dimer interface motif that lies at the tip of the αC-helix. As a result of this interaction, both the αC-helix and the AS stably adopt an active conformation in the absence of dimerization (Haling et al. 2014; Thevakumaran et al. 2015). Moreover, recent studies indicate that mutations to the B-Raf V600 residue are among the few disease-associated Raf mutations that promote Raf activation in a dimerization-independent manner (Yao et al. 2015).
POSTSIGNALING INACTIVATION OF RAF
Once Raf has been activated and the signal transmitted to the downstream kinases MEK and ERK, it is critical that the Rafs return to an inactive state, as constitutive activation can result in tumorigenesis. Several mechanisms that contribute to Raf inactivation have been identified. As expected, activating phosphorylation sites must be dephosphorylated, and protein phosphatase 5 (PP5) has been reported to dephosphorylate the critical S338 N-region site of C-Raf (von Kriegsheim et al. 2006). Raf autophosphorylation has also been implicated as a mechanism for inactivating the Rafs. In particular, a report examining inhibitor-bound B-Raf dimers has identified autoinhibitory sites in the ATP-binding P-loop (Holderfield et al. 2013), and a phosphoproteomic study characterizing the V600E-B-Raf mutant has identified an autoinhibitory site in the AS (S614-B-Raf) (Dernayka et al. 2016). Further studies will be needed to determine the prevalence and contribution of these autophosphorylation events in the context of Ras-mediated signaling.
Another important mechanism that attenuates the activity of all Raf family members involves a negative feedback loop in which active ERK phosphorylates the Rafs on multiple S/TP sites (Brummer et al. 2003; Dougherty et al. 2004; Ritt et al. 2010). Phosphorylation of the S/TP sites disrupts the interaction with Ras as well as Raf dimerization, and results in Raf proteins that are signaling incompetent (Dougherty et al. 2004; Ritt et al. 2010). Of note, some of these S/TP sites are also phosphorylated by the c-Jun amino-terminal kinases (JNKs) to prevent Raf signaling in times of cellular stress, and this stress-mediated desensitization of the Rafs can be induced by certain cancer therapeutics, such as rigosertib and paclitaxel (Ritt et al. 2016).
Ultimately, the postsignaling hyperphosphorylated Raf proteins must be recycled to a presignaling inactive state that is competent to respond to a new round of activating signals. Dephosphorylation of the ERK target sites is mediated by PP2A in a manner that requires the Pin1 prolyl-isomerase (Dougherty et al. 2004). More specifically, the WW-domain of Pin1 binds to the pS/TP sites, allowing Pin1 to isomerize the pS/T–proline bond to a trans conformation that can then be dephosphorylated by PP2A. Whether PP2A also serves to dephosphorylate the sites of autophosphorylation is currently unknown; however, once dephosphorylated, the Raf proteins can then reestablish the intramolecular contacts that mediate the autoinhibited state. Finally, with the rephosphorylation of the CR2 site and the rebinding of 14-3-3 to this site, the Raf kinases are returned to their presignaling, inactive conformation.
OTHER DIMER PARTNERS: KSR
Members of the KSR family are close relatives of the Raf kinases, and like the mechanisms mediating Raf activation, the function of KSR in Ras signaling is complex. KSR was first discovered as a positive modulator of the Ras pathway through genetic studies performed in Drosophila and Caenorhabditis elegans (Kornfeld et al. 1995; Sundaram and Han 1995; Therrien et al. 1995) and two KSR proteins (KSR1 and KSR2) are present in mammalian cells. The KSR family shares significant sequence homology with the Rafs, and like the Rafs, have a carboxy-terminal kinase domain and an amino-terminal domain that contains a CRD followed by a region rich in serine/threonine residues (Therrien et al. 1995). However, the KSRs do not have an RBD, but instead contain a coiled-coil fused to a sterile-α motif (CC-SAM) domain and a region rich in proline residues (Therrien et al. 1995; Koveal et al. 2012). Although it was initially thought that the KSRs would function as typical protein kinases, the mammalian KSR proteins were found to lack a lysine residue that is normally required for the phosphotransferreaction (Therrien et al. 1995). Moreover, when residues known to be critical for enzyme catalysis were mutated in C. elegans or Drosophila KSR, little to no effect on the biological function of these KSR family members was observed (Stewart et al. 1999; Roy et al. 2002), raising the question of whether the KSRs indeed possess intrinsic kinase activity—a question that is still debated.
Subsequently, reports emerged indicating that the KSR proteins may have scaffolding activities, interacting with the three core compo-nents of the ERK cascade (Morrison 2001). In quiescent cells, KSR is found in a multiprotein complex containing MEK, 14-3-3, and compo-nents of the Hsp90 complex (Xing et al. 1997; Denouel-Galy et al. 1998; Yu et al. 1998; Cacace et al. 1999; Stewart et al. 1999). Similar to the Rafs, binding of 14-3-3 to sites in the KSR amino-terminal domain sequesters the inactive KSR complex in the cytosol (Müller et al. 2001; Ory et al. 2003b), and as with B-Raf, the kinase domains of KSR and MEK interact in a face-to-face manner (Brennan et al. 2011). On signal activation, KSR rapidly translocates to the plasma membrane coincident with the dephosphorylation of one of the 14-3-3 binding sites (Ory et al. 2003b). The CRD and the CC-SAM domain are critical for membrane recruitment of KSR (Zhou et al. 2002; Koveal et al. 2012), and at the membrane, KSR serves to increase the local pool of MEK, thus facilitating MEK activation. More-over, KSR proteins can also form side-to-side dimers with the Rafs (Rajakulendran et al. 2009; Brennan et al. 2011), and if KSR is phosphorylated on residues analogous to the Raf N-region sites, it can function as an allosteric activator of Raf (Hu et al. 2013). Importantly, the KSR proteins also contain FxFP docking sites for activated ERK (Jacobs et al. 1999), and binding of ERK to the membrane-localized KSR complexes facilitates the phosphorylation of KSR and the Rafs on S/TP sites (McKay et al. 2009). Phosphorylation of these feedback sites disrupts the signaling complexes and promotes the release of KSR and the Rafs from the cell surface (McKay et al. 2009). Thus, KSR can modulate the dynamics of Ras pathway signaling by both potentiating and attenuating Raf activity and signal transmission to MEK and ERK.
CONCLUDING COMMENTS
The past decades have witnessed tremendous advances in our understanding of the Raf activation/inactivation process (Fig. 4). The early years of research were focused on elucidating the biochemical properties of the Rafs, determining their subcellular localization, phosphorylation state, and binding partners. Subsequently, the pace of discovery accelerated with the characterization of disease-associated mutations and the analysis of Raf crystallographic structures. Although much has been learned, outstanding questions still remain. In particular, it is unclear what factors determine Raf dimerization preferences and whether these preferences change depending on the signaling environment. Moreover, the structural features of a full-length Raf protein are unknown as are the structural changes induced by Ras binding and membrane interactions. The ultimate goal is that with further insight regarding the complexities of Raf regulation, new strategies for therapeutic intervention will emerge for targeting Ras-mediated Raf activation in human disease states.
REFERENCES
- Abraham D, Podar K, Pacher M, Kubicek M, Welzel N, Hemmings BA, Dilworth SM, Mischak H, Kolch W, Bac-carini M. 2000. Raf-1-associated protein phosphatase 2A as a positive regulator of kinase activation. J Biol Chem 275: 22300–22304. [DOI] [PubMed] [Google Scholar]
- Baljuls A, Mahr R, Schwarzenau I, Muller T, Polzien L, Hekman M, Rapp UR. 2011. Single substitution within the RKTR motif impairs kinase activity but promotes dimerization of RAF kinase. J Biol Chem 286: 16491–16503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondeva T, Balla A, Várnai P, Balla T. 2002. Structural determinants of Ras–Raf interaction analyzed in live cells. Mol Biol Cell 13: 2323–2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan DF, Dar AC, Hertz NT, Chao WC, Burlingame AL, Shokat KM, Barford D. 2011. A Raf-induced allosteric transition of KSR stimulates phosphorylation of MEK. Nature 472: 366–369. [DOI] [PubMed] [Google Scholar]
- Brummer T, Naegele H, Reth M, Misawa Y. 2003. Identification of novel ERK-mediated feedback phosphorylation sites at the C-terminus of B-Raf. Oncogene 22: 8823–8834. [DOI] [PubMed] [Google Scholar]
- Cacace AM, Michaud NR, Therrien M, Mathes K, Copeland T, Rubin GM, Morrison DK. 1999. Identification of constitutive and Ras-inducible phosphorylation sites of KSR: Implications for 14-3-3 binding, mitogen-activated protein kinase binding, and KSR overexpression. Mol Cell Biol 19: 229–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhary A, King WG, Mattaliano MD, Frost JA, Diaz B, Morrison DK, Cobb MH, Marshall MS, Brugge JS. 2000. Phosphatidylinositol 3-kinase regulates Raf1 through Pak phosphorylation of serine 338. Curr Biol 10: 551–554. [DOI] [PubMed] [Google Scholar]
- Chiloeches A, Mason CS, Marais R. 2001. S338 phosphorylation of Raf-1 is independent of phosphatidylinositol 3-kinase and Pak3. Mol Cell Biol 21: 2423–2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong H, Guan K-L. 2003. Regulation of Raf through phosphorylation and N-terminus–C terminus interaction. J Biol Chem 278: 36269–36276. [DOI] [PubMed] [Google Scholar]
- Chong H, Lee J, Guan KL. 2001. Positive and negative regulation of Raf kinase activity and function by phosphorylation. EMBO J 20: 3716–3727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark GJ, Drugan JK, Rossman KL, Carpenter JW, Rogers-Graham K, Fu H, Der CJ, Campbell SL. 1997. 14-3-3 ζ negatively regulates Raf-1 activity by interactions with the Raf-1 cysteine-rich domain. J Biol Chem 272: 20990–20993. [DOI] [PubMed] [Google Scholar]
- Corbit KC, Trakul N, Eves EM, Diaz B, Marshall M, Rosner MR. 2003. Activation of Raf-1 signaling by protein kinase C through a mechanism involving Raf kinase inhibitory protein. J Biol Chem 278: 13061–13068. [DOI] [PubMed] [Google Scholar]
- Cutler REJ, Stephens RM, Saracino MR, Morrison DK. 1998. Autoregulation of the Raf-1 serine/threonine kinase. Proc Natl Acad Sci 95: 9214–9219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daum G, Eisenmann-Tappe I, Fries HW, Troppmair J, Rapp UR. 1994. The ins and outs of Raf kinases. Trends Bio-chem Sci 19: 474–480. [DOI] [PubMed] [Google Scholar]
- Denouel-Galy A, Douville EM, Warne PH, Papin C, Laugier D, Calothy G, Downward J, Eychene A. 1998. Murine Ksr interacts with MEK and inhibits Ras-induced transfor-mation. Curr Biol 8: 46–55. [DOI] [PubMed] [Google Scholar]
- Dernayka L, Rauch N, Jarboui MA, Zebisch A, Texier Y, Horn N, Romano D, Gloeckner CJ, Kriegsheim A, Uef-fing M, et al. 2016. Autophosphorylation on S614 inhibits the activity and the transforming potential of BRAF. Cell Signal 28: 1432–1439. [DOI] [PubMed] [Google Scholar]
- Dhillon AS, Meikle S, Yazici Z, Eulitz M, Kolch W. 2002a. Regulation of Raf-1 activation and signalling by dephosphorylation. EMBO J 21: 64–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhillon AS, Pollock C, Steen H, Shaw PE, Mischak H, Kolch W. 2002b. Cyclic AMP-dependent kinase regulates Raf-1 kinase mainly by phosphorylation of serine 259. Mol Cell Biol 22: 3237–3246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhomen N, Marais R. 2007. New insight into BRAF mutations in cancer. Curr Opin Genet Dev 17: 31–39. [DOI] [PubMed] [Google Scholar]
- Diaz B, Barnard D, Filson A, MacDonald S, King A, Marshall M. 1997. Phosphorylation of Raf-1 serine 338-serine 339 is an essential regulatory event for Ras-dependent activa-tion and biological signaling. Mol Cell Biol 17: 4509–4516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diedrich B, Rigbolt KT, Roring M, Herr R, Kaeser-Pebernard S, Gretzmeier C, Murphy RF, Brummer T, Dengjel J. 2017. Discrete cytosolic macromolecular BRAF com-plexes exhibit distinct activities and composition. EMBO J 36: 646–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougherty MK, Müller J, Ritt DA, Zhou M, Zhou XZ, Copeland TD, Conrads TP, Veenstra TD, Lu KP, Morrison DK. 2004. Regulation of Raf-1 by direct feedback phosphory-lation. Mol Cell 17: 215–224. [DOI] [PubMed] [Google Scholar]
- Dumaz N, Marais R. 2003. Protein kinase A blocks Raf-1 activity by stimulating 14-3-3 binding and blocking Raf-1 interaction with Ras. J Biol Chem 278: 29819–29823. [DOI] [PubMed] [Google Scholar]
- Eisenhardt AE, Sprenger A, Roring M, Herr R, Weinberg F, Kohler M, Braun S, Orth J, Diedrich B, Lanner U, et al. 2016. Phosphoproteomic analyses of B-Raf protein complexes reveal new regulatory principles. Oncotarget 7:26628–26652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabian JR, Daar IO, Morrison DK. 1993. Critical tyrosine residues regulate the enzymatic and biological activity of Raf-1 kinase. Mol Cell Biol 13: 7170–7179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman AK, Ritt DA, Morrison DK. 2013. The importance of Raf dimerization in cell signaling. Small GTPases 4: 180–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garnett MJ, Rana S, Paterson H, Barford D, Marais R. 2005. Wild-type and mutant B-RAF activate C-RAF through distinct mechanisms involving heterodimerization. Mol Cell 20: 963–969. [DOI] [PubMed] [Google Scholar]
- Ghosh S, Strum JC, Sciorra VA, Daniel L, Bell RM. 1996. Raf1 kinase possesses distinct binding domains for phosphatidylserine and phosphatidic acid. J Biol Chem 271: 8472–8480. [DOI] [PubMed] [Google Scholar]
- Haling JR, Sudhamsu J, Yen I, Sideris S, Sandoval W, Phung W, Bravo BJ, Giannetti AM, Peck A, Masselot A, et al. 2014. Structure of the BRAF–MEK complex reveals a kinase activity independent role for BRAF in MAPK signaling. Cancer Cell 26: 402–413. [DOI] [PubMed] [Google Scholar]
- Hamilton M, Liao J, Cathcart MK, Wolfman A. 2001. Constitutive association of c-N-Ras with c-Raf-1 and protein kinase Cɛ in latent signaling modules. J Biol Chem 276: 29079–29090. [DOI] [PubMed] [Google Scholar]
- Hatzivassiliou G, Song K, Yen I, Brandhuber BJ, Anderson DJ, Alvarado R, Ludlam MJ, Stokoe D, Gloor SL, Vigers G, et al. 2010. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature 464: 431–435. [DOI] [PubMed] [Google Scholar]
- Heidecker G, Huleihel M, Cleveland JL, Kolch W, Beck TW, Lloyd P, Pawson T, Rapp UR. 1990. Mutational activation of c-raf-1 and definition of the minimal transforming sequence. Mol Cell Biol 10: 2503–2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidorn SJ, Milagre C, Whittaker S, Nourry A, Niculescu-Duvas I, Dhomen N, Hussain J, Reis-Filho JS, Springer CJ, Pritchard C, et al. 2010. Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell 140: 209–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hekman M, Hamm H, Villar AV, Bader B, Kuhlmann J, Nickel J, Rapp UR. 2002. Associations of B-and C-Raf with cholesterol, phosphatidylserine, and lipid second messengers. J Biol Chem 277: 24090–24102. [DOI] [PubMed] [Google Scholar]
- Herrmann C, Martin GA, Wittinghofer A. 1995. Quantitative analysis of the complex between p21ras and the Ras-binding domain of the human Raf-1 protein kinase. J Biol Chem 270: 2901–2905. [DOI] [PubMed] [Google Scholar]
- Hibino K, Shibata T, Yanagida T, Sako Y. 2009. A RasGTP-induced conformational change in C-RAF is essential for accurate molecular recognition. Biophys J 97: 1277–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holderfield M, Merritt H, Chan J, Wallroth M, Tandeske L, Zhai H, Tellew J, Hardy S, Hekmat-Nejad M, Stuart DD, et al. 2013. RAF inhibitors activate the MAPK pathway by relieving inhibitory autophosphorylation. Cancer Cell 23: 594–602. [DOI] [PubMed] [Google Scholar]
- Hu J, Stites EC, Yu H, Germino EA, Meharena HS, Stork PJ, Kornev AP, Taylor SS, Shaw AS. 2013. Allosteric activation of functionally asymmetric RAF kinase dimers. Cell 154: 1036–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs D, Glossip D, Xing H, Muslin AJ, Kornfeld K. 1999. Multiple docking sites on substrate proteins form a modular system that mediates recognition by ERK MAP kinase. Genes Dev 13: 163–175. [PMC free article] [PubMed] [Google Scholar]
- Jaumot M, Hancock JF. 2001. Protein phosphatases 1 and 2A promote Raf-1 activation by regulating 14-3-3 interactions. Oncogene 20: 3949–3958. [DOI] [PubMed] [Google Scholar]
- King AJ, Sun H, Diaz B, Barnard D, Miao W, Bagrodia S, Marshall MS. 1998. The protein kinase Pak3 positively regulates Raf-1 activity through phosphorylation of serine 338. Nature 396: 180–183. [DOI] [PubMed] [Google Scholar]
- Kornfeld K, Hom DB, Horvitz HR. 1995. The ksr-1 gene encodes a novel protein kinase involved in Ras-mediated signaling in C. elegans. Cell 83: 903–913. [DOI] [PubMed] [Google Scholar]
- Koveal D, Schuh-Nuhfer N, Ritt D, Page R, Morrison DK, Peti W. 2012. A CC-SAM, for coiled coil-sterile α motif, domain targets the scaffold KSR-1 to specific sites in the plasma membrane. Sci Signal 5: ra94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavoie H, Therrien M. 2015. Regulation of RAF protein kinases in ERK signalling. Nature Rev Mol Cell Biol 16: 281–298. [DOI] [PubMed] [Google Scholar]
- Leevers SJ, Paterson HF, Marshall CJ. 1994. Requirement for Ras in Raf activation is overcome by targeting Raf to the plasma membrane. Nature 369: 411–414. [DOI] [PubMed] [Google Scholar]
- Light Y, Paterson H, Marais R. 2002. 14-3-3 Antagonizes Ras-mediated Raf-1 recruitment to the plasma membrane to maintain signaling fidelity. Mol Cell Biol 22: 4984–4996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Z, Diaz B, Marshall MS, Avruch J. 1997. An intact Raf zinc finger is required for optimal binding to processed Ras and for Ras-dependent Raf activation in situ. Mol Cell Biol 17: 46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marais R, Light Y, Paterson HF, Marshall CJ. 1995. Ras recruits Raf-1 to the plasma membrane for activation by tyrosine phosphorylation. EMBO J 14: 3136–3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall CJ. 1996. Ras effectors. Curr Opin Cell Biol 8: 197–204. [DOI] [PubMed] [Google Scholar]
- Mason CS, Springer CJ, Cooper RG, Superti-Furga G, Mar-shall CJ, Marais R. 1999. Serine and tyrosine phosphorylations cooperate in Raf-1, but not B-Raf activation. EMBO J 18: 2137–2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClendon CL, Kornev AP, Gilson MK, Taylor SS. 2014. Dynamic architecture of a protein kinase. Proc Natl Acad Sci 111: E4623–E4631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKay MM, Ritt DA, Morrison DK. 2009. Signaling dynam-ics of the KSR1 scaffold complex. Proc Natl Acad Sci 106: 11022–11027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPherson RA, Harding A, Roy S, Lane A, Hancock JF. 1999. Interactions of c-Raf-1 with phosphatidylserine and 14-3-3. Oncogene 18: 3862–3869. [DOI] [PubMed] [Google Scholar]
- Michaud NR, Fabian JR, Mathes KD, Morrison DK. 1995. 14-3-3 is not essential for Raf-1 function: Identification of Raf-1 proteins that are biologically activated in a 14-3-3 and Ras-independent manner. Mol Cell Biol 15: 3390–3397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison DK. 2001. KSR: A MAPK scaffold of the Ras path-way? J Cell Sci 114: 1609–1612. [DOI] [PubMed] [Google Scholar]
- Morrison DK, Heidecker G, Rapp UR, Copeland TD. 1993. Identification of the major phosphorylation sites of the Raf-1 kinase. J Biol Chem 268: 17309–17316. [PubMed] [Google Scholar]
- Mott HR, Carpenter JW, Zhong S, Ghosh S, Bell RM, Campbell SL. 1996. The solution structure of the Raf-1 cysteine-rich domain: A novel ras and phospholipid binding site. Proc Natl Acad Sci 93: 8312–8317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller J, Ory S, Copeland T, Piwnica-Worms H, Morrison DK. 2001. C-TAK1 regulates Ras signaling by phosphorylating the MAPK scaffold, KSR1. Mol Cell 8: 983–993. [DOI] [PubMed] [Google Scholar]
- Muslin AJ, Tanner JW, Allen PM, Shaw AS. 1996. Interaction of 14-3-3 with signaling proteins is mediated by the recognition of phosphoserine. Cell 84: 889–897. [DOI] [PubMed] [Google Scholar]
- Nan X, Collisson EA, Lewis S, Huang J, Tamguney TM, Liphardt JT, McCormick F, Gray JW, Chu S. 2013. Single-molecule superresolution imaging allows quantitative analysis of RAF multimer formation and signaling. Proc Natl Acad Sci 110: 18519–18524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nassar N, Horn G, Herrmann C, Scherer A, McCormick F, Wittinghofer A. 1995. The 2.2 Å crystal structure of the Ras-binding domain of the serine/threonine kinase c-Raf1 in complex with Rap1A and a GTP analogue. Nature 375: 554–560. [DOI] [PubMed] [Google Scholar]
- Ory S, Zhou M, Conrads TP, Veenstra TD, Morrison DK. 2003a. Protein phosphatase 2A positively regulates Ras signaling by dephosphorylating KSR1 and Raf-1 on critical 14-3-3 binding sites. Curr Biol 13: 1356–1364. [DOI] [PubMed] [Google Scholar]
- Ory S, Zhou M, Conrads TP, Veenstra TD, Morrison DK. 2003b. Protein phosphatase 2A positively regulates Ras signaling by dephosphorylating KSR1 and Raf-1 on critical 14-3-3 binding sites. Curr Biol 13: 1356–1364. [DOI] [PubMed] [Google Scholar]
- Pandit B, Sarkozy A, Pennacchio LA, Carta C, Oishi K, Martinelli S, Pogna EA, Schackwitz W, Ustaszewska A, Landstrom A, et al. 2007. Gain-of-function RAF1 mutations cause Noonan and LEOPARD syndromes with hypertrophic cardiomyopathy. Nat Genet 39: 1007–1012. [DOI] [PubMed] [Google Scholar]
- Plowman SJ, Muncke C, Parton RG, Hancock JF. 2005. H-Ras, K-Ras, and inner plasma membrane raft proteins operate in nanoclusters with differential dependence on the actin cytoskeleton. Proc Natl Acad Sci 102: 15500–15505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N. 2010. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature 464: 427–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajakulendran T, Sahmi M, Lefrancois M, Sicheri F, Therrien M. 2009. A dimerization-dependent mechanism drives RAF catalytic activation. Nature 461: 542–545. [DOI] [PubMed] [Google Scholar]
- Rapp UR, Goldsborough MD, Mark GE, Bonner TI, Groffen J, Reynolds J, Stephenson FH Jr. 1983. Structure and biological activity of v-raf, a unique oncogene transduced by a retrovirus. Proc Natl Acad Sci 80: 4218–4222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razzaque MA, Nishizawa T, Komoike Y, Yagi H, Furutani M, Amo R, Kamisago M, Momma K, Katayama H, Nakagawa M, et al. 2007. Germline gain-of-function mutations in RAF1 cause Noonan syndrome. Nat Genet 39: 1013–1017. [DOI] [PubMed] [Google Scholar]
- Ritt DA, Zhou M, Conrads TP, Veenstra TD, Copeland TD, Morrison DK. 2007. CK2 Is a component of the KSR1 scaffold complex that contributes to Raf kinase activation. Curr Biol 17: 179–184. [DOI] [PubMed] [Google Scholar]
- Ritt DA, Monson DM, Specht SI, Morrison DK. 2010. Impact of feedback phosphorylation and Raf heterodimerization on normal and mutant B-Raf signaling. Mol Cell Biol 30: 806–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritt DA, Abreu-Blanco MT, Bindu L, Durrant DE, Zhou M, Specht SI, Stephen AG, Holderfield M, Morrison DK. 2016. Inhibition of Ras/Raf/MEK/ERK pathway signaling by a stress-induced phospho-regulatory circuit. Mol Cell 64: 875–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Viciana P, Oses-Prieto J, Burlingame A, Fried M, McCormick F. 2006. A phosphatase holoenzyme comprised of Shoc2/Sur8 and the catalytic subunit of PP1 functions as an M-Ras effector to modulate Raf activity. Mol Cell 22: 217–230. [DOI] [PubMed] [Google Scholar]
- Romano D, Nguyen LK, Matallanas D, Halasz M, Doherty C, Kholodenko BN, Kolch W. 2014. Protein interaction switches coordinate Raf-1 and MST2/Hippo signalling. Nat Cell Biol 16: 673–684. [DOI] [PubMed] [Google Scholar]
- Rommel C, Radziwill G, Lovric J, Noeldeke J, Heinicke T, Jones D, Aitken A, Moelling K. 1996. Activated Ras displaces 14-3-3 protein from the amino terminus of c-Raf-1. Oncogene 12: 609–619. [PubMed] [Google Scholar]
- Roy S, Lane A, Yan J, McPherson R, Hancock JF. 1997. Activity of plasma membrane-recruited Raf-1 is regulated by Ras via the Raf zinc finger. J Biol Chem 272: 20139–20145. [DOI] [PubMed] [Google Scholar]
- Roy F, Laberge G, Douziech M, Ferland-McCollough D, Therrien M. 2002. KSR is a scaffold required for activation of the ERK/MAPK module. Genes Dev 16: 427–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rushworth LK, Hindley AD, O’Neill E, Kolch W. 2006. Regulation and role of Raf-1/B-Raf heterodimerization. Mol Cell Biol 26: 2262–2272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkozy A, Carta C, Moretti S, Zampino G, Digilio MC, Pantaleoni F, Scioletti AP, Esposito G, Cordeddu V, Lepri F, et al. 2009. Germline BRAF mutations in Noonan, LEOPARD, and cardiofaciocutaneous syndromes: Molecular diversity and associated phenotypic spectrum. Hum Mutat 30: 695–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulte TW, Blagosklonny MV, Ingui C, Neckers L. 1995. Disruption of the Raf-1-Hsp90 molecular complex results in destabilization of Raf-1 and loss of Raf-1-Ras association. J Biol Chem 270: 24585–24588. [DOI] [PubMed] [Google Scholar]
- Shaw AS, Kornev AP, Hu J, Ahuja LG, Taylor SS. 2014. Kinases and pseudokinases: Lessons from RAF. Mol Cell Biol 34: 1538–1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stancato LF, Chow YH, Owens-Grillo JK, Yem AW, Deibel MR Jr, Jove R, Pratt WB. 1994. The native v-Raf.hsp90. p50 heterocomplex contains a novel immunophilin of the FK506 binding class. J Biol Chem 269: 22157–22161. [PubMed] [Google Scholar]
- Stanton VP, Nichols DW, Laudano AP, Cooper GM. 1989. Definition of the human raf amino-terminal regulatory region by deletion mutagenesis. Mol Cell Biol 9: 639–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart S, Sundaram M, Zhang Y, Lee J, Han M, Guan KL. 1999. Kinase suppressor of Ras forms a multiprotein signaling complex and modulates MEK localization. Mol Cell Biol 19: 5523–5534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stokoe D, Macdonald SG, Cadwallader K, Symons M, Han-cock JF. 1994. Activation of Raf as a result of recruitment to the plasma membrane. Science 264: 1463–1467. [DOI] [PubMed] [Google Scholar]
- Sundaram M, Han M. 1995. The C. elegans ksr-1 gene encodes a novel Raf-related kinase involved in Ras-mediated signal transduction. Cell 83: 889–901. [DOI] [PubMed] [Google Scholar]
- Tartaglia M, Gelb BD, Zenker M. 2011. Noonan syndrome and clinically related disorders. Best Pract Res Clin Endo-crinol Metab 25: 161–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terai K, Matsuda M. 2005. Ras binding opens c-Raf to expose the docking site for mitogen-activated protein kinase kinase. EMBO Rep 6: 251–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Therrien M, Chang HC, Solomon NM, Karim FD, Wassarman DA, Rubin GM. 1995. KSR, a novel protein kinase required for RAS signal transduction. Cell 83: 879–888. [DOI] [PubMed] [Google Scholar]
- Thevakumaran N, Lavoie H, Critton DA, Tebben A, Marinier A, Sicheri F, Therrien M. 2015. Crystal structure of a BRAF kinase domain monomer explains basis for alloste-ric regulation. Nat Struct Mol Biol 22: 37–43. [DOI] [PubMed] [Google Scholar]
- Tian T, Harding A, Inder K, Plowman S, Parton RG, Hancock JF. 2007. Plasma membrane nanoswitches generate high-fidelity Ras signal transduction. Nat Cell Biol 9: 905–914. [DOI] [PubMed] [Google Scholar]
- Tran NH, Frost JA. 2003. Phosphorylation of Raf-1 by p21-activated kinase 1 and Src regulates Raf-1 autoinhibition. J Biol Chem 278: 11221–11226. [DOI] [PubMed] [Google Scholar]
- Tran NH, Wu X, Frost JA. 2005. B-Raf and Raf-1 are regu-lated by distinct autoregulatory mechanisms. J Biol Chem 280: 16244–16253. [DOI] [PubMed] [Google Scholar]
- Tzivion G, Luo Z, Avruch J. 1998. A dimeric 14-3-3 protein is an essential cofactor for Raf kinase activity. Nature 394: 88–92. [DOI] [PubMed] [Google Scholar]
- Van Aelst L, Barr M, Marcus S, Polverino A, Wigler M. 1993. Complex formation between Ras and Raf and other protein kinases. Proc Natl Acad Sci 90: 6213–6217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vojtek AB, Hollenberg SM, Cooper JA. 1993. Mammalian Ras interacts directly with the serine/threonine kinase Raf. Cell 74: 205–214. [DOI] [PubMed] [Google Scholar]
- von Kriegsheim A, Pitt A, Grindlay GJ, Kolch W, Dhillon AS. 2006. Regulation of the Raf-MEK-ERK pathway by protein phosphatase 5. Nat Cell Biol 8: 1011–1016. [DOI] [PubMed] [Google Scholar]
- Wan PT, Garnett MJ, Roe SM, Lee S, Niculescu-Duvaz D, Good VM, Jones CM, Marshall CJ, Springer CJ, Barford D, et al. 2004. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell 116: 855–867. [DOI] [PubMed] [Google Scholar]
- Weber CK, Slupsky JR, Kalmes HA, Rapp UR. 2001. Active Ras induces heterodimerization of cRaf and BRaf. Cancer Res 61: 3595–3598. [PubMed] [Google Scholar]
- Wellbrock C, Karasarides M, Marais R. 2004. The RAF proteins take centre stage. Nat Rev Mol Cell Biol 5: 875–885. [DOI] [PubMed] [Google Scholar]
- Williams JG, Drugan JK, Yi GS, Clark GJ, Der CJ, Campbell SL. 2000. Elucidation of binding determinants and functional consequences of Ras/Raf-cysteine-rich domain interactions. J Biol Chem 275: 22172–22179. [DOI] [PubMed] [Google Scholar]
- Xing H, Kornfeld K, Muslin AJ. 1997. The protein kinase KSR interacts with 14-3-3 protein and Raf. Curr Biol 7: 294–300. [DOI] [PubMed] [Google Scholar]
- Yao Z, Torres NM, Tao A, Gao Y, Luo L, Li Q, de Stanchina E, Abdel-Wahab O, Solit DB, Poulikakos PI, et al. 2015. BRAF mutants evade ERK-dependent feedback by different mechanisms that determine their sensitivity to pharmacologic inhibition. Cancer Cell 28: 370–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung K, Janosch P, McFerran B, Rose DW, Mischak H, Sedivy JM, Kolch W. 2000. Mechanism of suppression of the Raf/MEK/extracellular signal-regulated kinase pathway by the Raf kinase inhibitor protein. Mol Cell Biol 20: 3079–3085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu W, Fantl WJ, Harrowe G, Williams LT. 1998. Regulation of the MAP kinase pathway by mammalian KSR through direct interaction with MEK and ERK. Curr Biol 8: 56–64. [DOI] [PubMed] [Google Scholar]
- Zhang BH, Guan KL. 2000. Activation of B-Raf kinase requires phosphorylation of the conserved residues Thr598 and Ser601. EMBO J 19: 5429–5439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XF, Settleman J, Kyriakis JM, Takeuchi-Suzuki E, Elledge SJ, Marshall MS, Bruder JT, Rapp UR, Avruch J. 1993. Normal and oncogenic p21ras proteins bind to the amino-terminal regulatory domain of c-Raf-1. Nature 364: 308–313. [DOI] [PubMed] [Google Scholar]
- Zhang X, Gureasko J, Shen K, Cole PA, Kuriyan J. 2006. An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell 125: 1137–1149. [DOI] [PubMed] [Google Scholar]
- Zhou M, Horita DA, Waugh DS, Byrd RA, Morrison DK. 2002. Solution structure and functional analysis of the cysteine-rich C1 domain of kinase suppressor of Ras (KSR). J Mol Biol 315: 435–446. [DOI] [PubMed] [Google Scholar]
- Zimmermann S, Moelling K. 1999. Phosphorylation and regulation of Raf by Akt ( protein kinase B). Science 286: 1741–1744. [DOI] [PubMed] [Google Scholar]