

Abstract
RNA molecules are essential for cellular information transfer and gene regulation, and RNAs have been implicated in many human diseases. Messenger and non-coding RNAs contain highly structured elements, and evidence suggests that many of these structures are important for function. Targeting these RNAs with small molecules offers opportunities to therapeutically modulate numerous cellular processes, including those linked to “undruggable” protein targets. Despite this promise, there is currently only a single class of human-designed small molecules that target RNA used clinically – the linezolid antibiotics. A growing number of small-molecule RNA ligands are being identified, however, leading to burgeoning interest in the field. Here, we discuss principles for discovering small-molecule drugs that target RNA and argue that the overarching challenge is to identify appropriate target structures – namely in disease-causing RNAs that have high information content, and consequently appropriate ligand binding pockets. If focus is placed on such druggable binding sites in RNA, extensive knowledge of the kinds of molecules that comprise conventional drugs could then enable small-molecule drug discovery for RNA targets to become (only) roughly as difficult as for protein targets.
Introduction
The vast majority of small-molecule therapeutics in clinical use target proteins. Successful protein-targeted drugs include small molecules that bind proteins expressed by humans (Fig. 1) or those expressed by bacteria, viruses, and other infectious organisms. If small-molecule therapeutics could be extended to modulate RNA, the landscape of targetable macromolecules would be expanded by more than an order of magnitude. For example, only roughly 1.5% of the human genome encodes proteins1,2. Within the roughly 20,000 expressed human proteins, 10–15% of these are thought to be disease-related3–5, meaning that disrupting or altering their activity is likely to have a therapeutically useful consequence. Within the group of disease-related proteins, many are termed “undruggable”, meaning that these molecules lack distinctive cleft-like motifs into which small molecules can bind with high specificity and affinity5. The net implication of this analysis is that de novo human-designed small-molecule drugs target a sliver (the 3.5% of drugged proteins) of a sliver (1.5% of the human genome that encodes proteins) of the human genome. Currently approved protein-targeted drugs interact with fewer than 700 gene products6, meaning that only 0.05% of the human genome has been drugged (Fig. 1).
Figure 1: The potential RNA-targeted druggable genome.
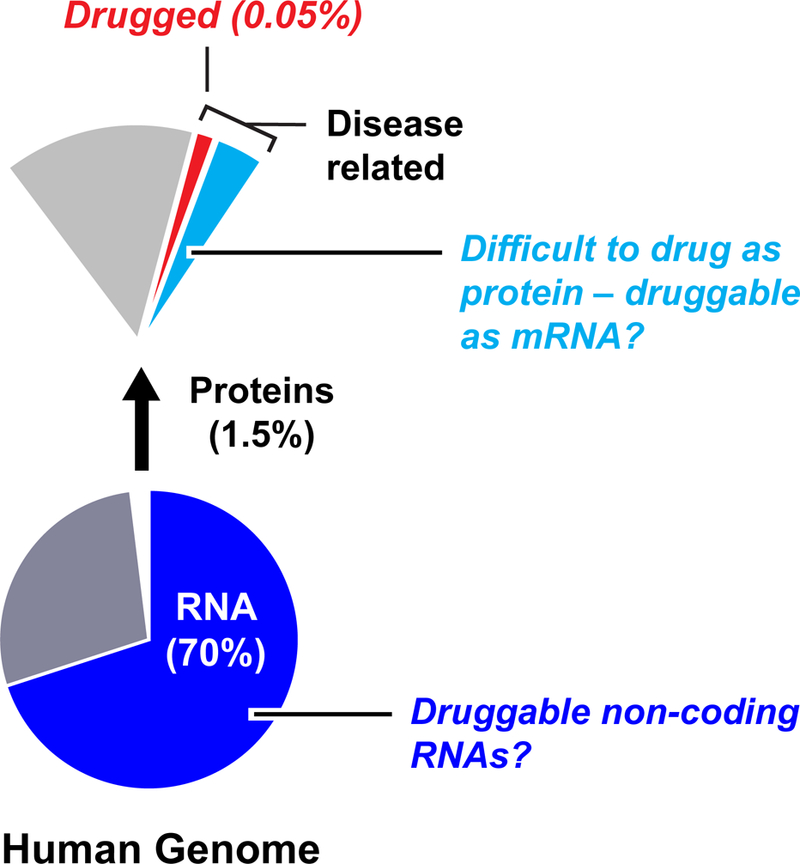
Only a small fraction of the human genome has been successfully drugged. Possible RNA targets include mRNAs that encode disease-related proteins characterized as undruggable or difficult to drug, plus those non-coding RNAs that influence disease.
RNA functions as a central conduit of information transfer in all biological systems7,8 and, in principle, there are numerous opportunities for creating small-molecule therapeutics targeting RNA. If mRNAs could be targeted, protein gene products could be modulated by up or down regulating translation efficiency or by altering mRNA abundance or stability. By directly targeting an mRNA, activities of proteins that are very difficult to drug or undruggable might be modulated, before or during their biogenesis. In addition, a large fraction of the human genome, roughly 70%, is transcribed into non-coding RNAs (RNAs that function directly as RNA)9 (Fig. 1). One class of non-coding RNAs, microRNAs, are now well-validated therapeutic targets10,11. Humans likely produce more than 15,000 long non-coding RNAs12 and a subset of these may eventually prove to be good drug targets. In summary, if a fraction of the tens of thousands of mRNAs and non-coding RNAs were targetable, the percentage of the “drugged” human genome could increase significantly.
Strong proof-of-principle for RNA-targeted drugs has been provided by experiments and clinical trials using antisense oligonucleotides (several of which have gained regulatory approval), as well as preclinical studies with synthetic RNAs that redirect the cellular RNAi machinery or that activate CRISPR-based systems10,11,13–16. Depending on the approach employed, it is possible to inhibit or to up-regulate expression13,14 of particular mRNAs and to inhibit non-coding RNA function by targeting specific RNA sequences. However, these nucleic acid-based approaches involve large, often highly charged, molecules and present significant delivery challenges. Given the choice between an oligonucleotide-based therapy versus a (currently hypothetical) small-molecule-based therapy that targets RNA, a small-molecule therapeutic would be preferred in many cases.
A number of small molecules that alter RNA function have been identified, providing encouraging evidence that RNA-targeted small-molecule therapeutics could be developed. Some of these studies are based on molecules identified in natural systems. For example, many RNA riboswitch regulatory elements have been characterized that bind diverse small-molecule metabolites and regulate gene expression17, and small-molecule antibiotics produced by numerous bacterial species bind ribosomal RNA and interfere with translation18. Furthermore, a growing number of synthetic mono- and multi-valent molecules that bind to secondary structure elements or repeat-containing RNAs have been shown to modulate biological function19. This progress is helping to address skepticism that RNA could be a viable small-molecule drug target, but key challenges remain to be addressed.
Any discussion of how to target RNA with small molecules needs to immediately acknowledge that no one currently knows how to create drug-like, RNA-targeted small molecules in a repeatable and scalable way. There is currently only a single human-designed and approved drug class that functions by binding RNA alone – the linezolid antibiotics, linezolid and tedizolid (Fig. 2). In our view, successful targeting of RNA in therapeutically useful ways with small molecules requires three distinct components: (1) identification of RNAs and RNA motifs with disease-related function, (2) use of screening approaches and libraries likely to identify drug-like molecules with appropriate pharmacological properties, and (3) identification of and focus on RNA motifs with sufficient structural sophistication that make it likely that high affinity and specificity in small-molecule binding can be achieved. To date, most work has focused on therapeutically well-validated targets (component 1), and there has been a notable recent focus on screening against RNA targets with drug-like lead compounds (component 2). However – and critically in our view – considerations of target complexity and achievable selectivity (component 3) have largely been neglected. With this in mind, after overviewing the current state of small-molecule ligand discovery for RNA targets and some instructional examples, we present a set of hypotheses regarding how to target RNA with small molecules and guidelines for achieving this goal. This article is intended to be provocative, and we look forward to the feedback and responses that our ideas will engender.
Figure 2: Instructional examples of bioactive small molecules that bind to RNA.
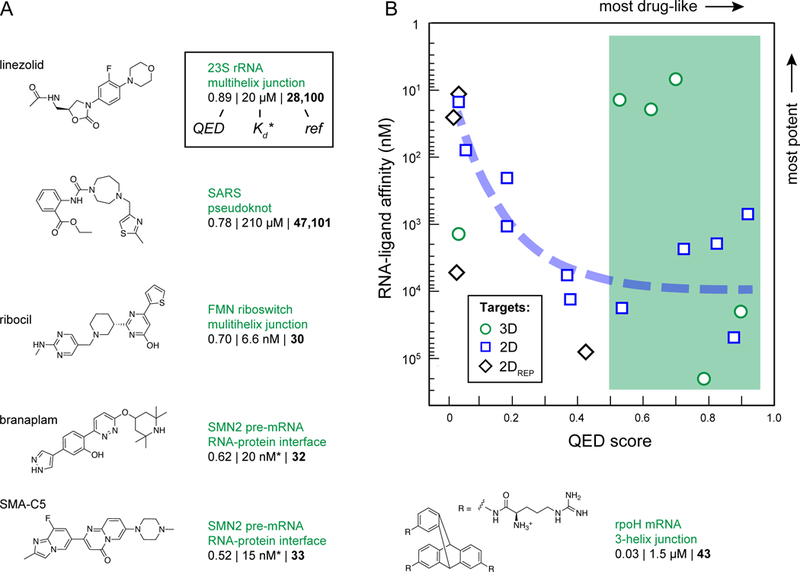
(A) Molecules that bind RNA tertiary structures. Molecules are listed by QED drug-likeness score (Box 1) with the most drug-like molecules first. Equilibrium dissociation constants (Kd) are listed for most molecules; if no Kd was available, the IC50 is provided instead (indicated by an asterisk). Molecular properties were calculated with SilicosIT. (B) Relationship between QED scores and RNA binding affinity. Molecules targeting tertiary structures (shown in this figure; 3D targets) and targeting secondary and repeat structures (from Fig. 3; 2D and 2DREP targets) are shown with circles, squares, and diamonds, respectively. The green box shows the regime where most (5/6) molecules targeting RNA tertiary structures fall; blue curve notes the trend that increasing potency correlates with low drug-likeness for molecules targeting RNA secondary structures and repeat sequences.
[H1] Current status of RNA-targeted ligands
Inspection of well-researched classic reviews on RNA-targeted small molecules reveals that, through 2012, efforts to find small-molecule ligands for RNAs mostly identified highly basic (and thus positively charged under physiological conditions) and planar molecules capable of intercalation between and stacking on RNA bases20–22. These molecules tend to bind to RNA with high affinity but with low selectivity. These kinds of molecules often have poor pharmacological properties including low cellular uptake and high toxicity. Thus, much early work was focused on molecules with decidedly undrug-like properties. The field of RNA-targeted ligand discovery is now changing rapidly, and many groups are now focusing on approaches that use strategies likely to identify molecules with plausible drug-like properties23–26 (Box 1).
Box 1: Drug-likeness.
Clinically useful small-molecule drugs have physicochemical properties that tend to fall within a narrow range of possible values. Compounds that have molecular features consistent with these properties are said to be drug-like. Emphasizing drug-likeness is important because this characteristic provides a useful shorthand for molecules that have properties that are desirable in a drug to be given orally to humans including solubility, cell and tissue permeability, metabolic stability, and lack of toxicity.
Lipinski’s Rule of Five is a well-known set of criteria used to evaluate the drug-likeness of a molecule. These rules were derived from retrospective analysis of known drugs and drug candidates96. Lipinski’s rules emphasize that molecules intended to become orally bioavailable drugs should not be too big (molecular mass less than 500), too polar, or too hydrophobic (fewer than five hydrogen-bond donors and logP, the octanol–water partition coefficient, less than 5). The emphasis on five serves as a mnemonic by which to remember the rules.
The idea of a privileged chemical space has recently been extended to quantify, in a single term, the overall drug-likeness for an orally bioavailable molecule. This approach allows some cutoffs (for example, one or more of Lipinski’s rules) to be “broken” if the overall molecular quality of a compound is high. In this work, we emphasize quantitative estimate of drug-likeness (QED) values27. QED values range from zero (all properties unfavorable) to one (all properties favorable). QED is a continuous function that allows the overall drug-likeness of a molecule to be summarized in a single term and that shows good discriminatory power and correlation with intuitive chemical aesthetics cultivated by medicinal chemists27,97. The median QED value for current orally bioavailable human drugs is approximately 0.65, and 75% of orally available drugs have QED values ≥ 0.5.
The concept of drug-likeness and the specific parameterization of QED is, of course, primarily based on small-molecule drugs that target proteins. Some successful drugs, especially antibiotics and viral protease inhibitors, have quite low QED scores27,97, so a low drug-likeness profile does not rule out the potential usefulness of a small-molecule drug candidate. It is also formally possible that RNA-targeted drugs will follow a different set of principles from those that target proteins. There is some evidence that molecules that bind RNA have different structural24 and kinetic98 properties, on average, than small molecules that bind proteins. Nonetheless, drug-likeness is strongly influenced by bioavailability and human physiology, which are the same for all drugs. Thus, we suggest that RNA-targeted drugs will ultimately follow roughly the same rules for drug-likeness as their currently better-characterized protein-targeting cousins.
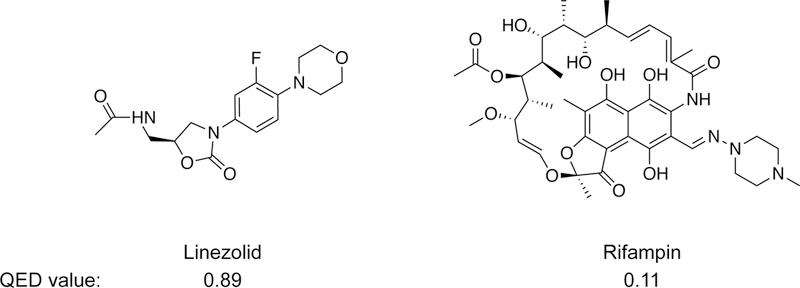
Bioactive molecules with high and low QED values.
We have compiled and analyzed a collection of instructional molecules that target RNA and are active in a cell culture or animal model or in humans, focusing on molecules with reasonable drug-like properties or high potencies or both (Fig. 2 & 3). We have de-emphasized aminoglycosides and molecules with likely non-specific intercalation behavior. In cases where a research group has developed a series of related compounds, we included a single representative example. Compounds are grouped, first, by the kind of RNA structure they target: tertiary structures involving multiple closely packed helices (Fig. 2A), irregular and usually bulge-containing secondary structures (Fig. 3A), or triplet repeats (Fig. 3B). Within each of these structure-based divisions, compounds are listed by their quantitative estimate of drug-likeness (QED) score27 (Box 1), with the most-drug-like first (reading left to right and then down the figure). Overall, there are a limited number of examples in this collection, and several of the molecules have the same targets, such as SMN2 pre-mRNA, the HIV TAR bulged helix and the DM1 CUG triplet repeat. Nonetheless, this collection represents the current state of the field and provides a reasonable starting point for thinking about principles for targeting RNA specifically and potently with small molecules.
Figure 3: Instructional examples of bioactive small molecules that bind RNA secondary structures.
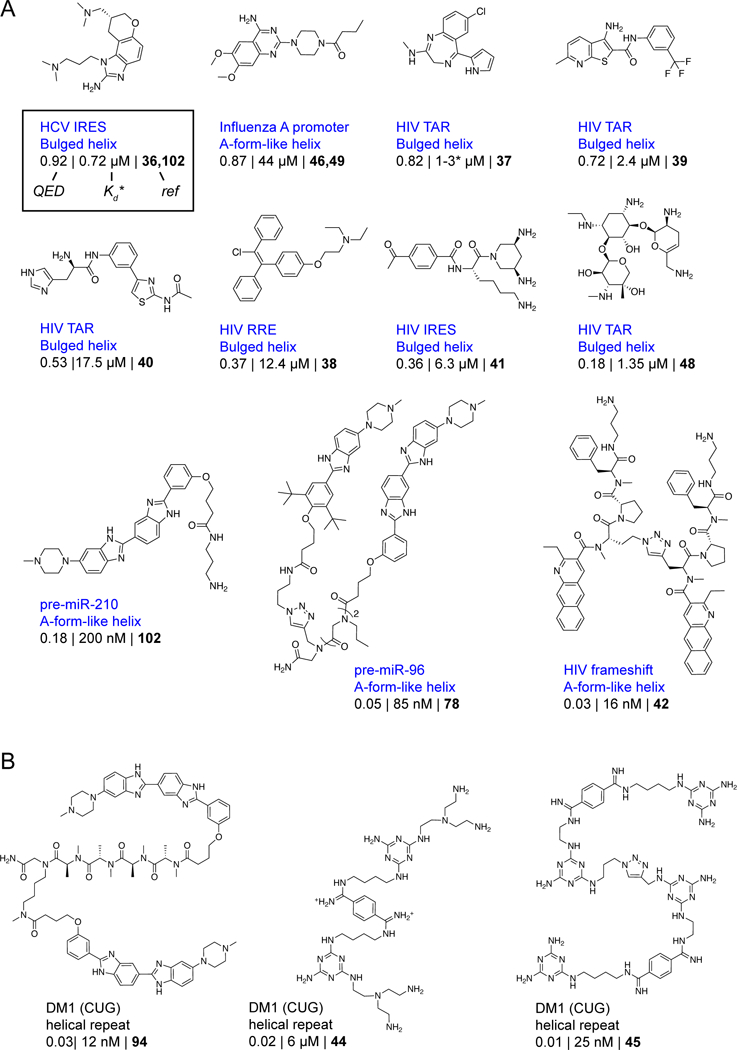
Molecules that bind RNA (A) secondary structures and (B) trinucleotide repeats. Molecules are listed by QED drug-likeness score with the most drug-like molecules first. Molecule attributes are reported as described in the legend to Fig. 2.
The four named molecules in Fig. 2A – linezolid, ribocil, branaplam and SMA-C5 – represent the current gold standards in biologically effective small molecules that target RNA. Each targets a complex RNA structural motif and each has a high QED score. But, notably, each is a special case. Moreover, each of these molecules was identified in a phenotypic screen, and only after the screen was completed was it discovered that these molecules function by interaction with RNA. Thus, none of these examples provide clear guidance on how to intentionally target RNA with small molecules.
Linezolid is a broad-spectrum antibacterial agent that binds to the large subunit RNA of the ribosome and appears to interfere with correct tRNA positioning28,29. Linezolid is a special case because ribosomes are highly abundant in cells and small molecules targeting the ribosome need only achieve modest binding affinity; the linezolid-ribosome Kd is approximately 20 µM. Merck’s ribocil binds to riboflavin riboswitches, which function as RNA-mediated regulators of gene expression in bacteria. Ribocil has many attractive features but is unlikely to be pursued further because bacteria rapidly develop resistance to this drug30,31. Ribocil bears no chemical similarity to the natural metabolite ligand and represents a major accomplishment in human-initiated RNA-targeted ligand development. Nonetheless, ribocil binding to a riboswitch is a special case because the drug binds in the same pocket as the natural flavin mononucleotide ligand and thus this RNA was predisposed to bind a small molecule. The binding sites for Novartis’ branaplam32 (originally called NVS-SM1) and PTC’s chemotypically similar SMA-C533 have been publicly shared in a qualitative way. These molecules both appear to bind at the RNA duplex formed between the U1 RNA and the target (SMN2) pre-mRNA and are likely stabilized either indirectly or directly by protein components of the U1 snRNP32–34. Compounds from these programs have been tested in clinical trials35. Discoveries of these molecules were impressive achievements but the target site pocket includes protein components, so neither is strictly an RNA-targeted drug.
The rest of the instructional compounds show distinct and interesting features (Fig. 2 & 3). Notably, these molecules were discovered using a wide variety of strategies that were intentionally focused on identifying RNA targets, and a subset are broadly drug-like, a feature emphasized by other recent reviewers24–26. Discovery strategies included high-throughput screening30,32,36–39, use of focused libraries40–42, structure-inspired design43–45, fragment-based approaches46, and computational modeling47,48. Both compact and multivalent molecules are represented. Together, the molecules in Figs. 2 and 3 strike us as reasonable validation of the potential of RNA as a small-molecule drug target. However, with the exceptions of the four named compounds in Fig. 2A, these molecules show deficiencies in one or more features of a conventional drug-like molecule.
The relationship between drug-likeness and potency for these instructional ligands has two intriguing characteristics. First, most (5 out of 6) of the molecules targeting higher-order tertiary structures are drug-like (as inferred by comparing their QED score with those of orally available drugs), independent of their potency (Fig. 2B, circles and green shading). This observation suggests that the ability to identify RNA-targeting small molecules with good drug-like properties is interrelated with the choice of RNA target. We explore this idea in depth below. Second, for compounds targeting RNA secondary structures or repeat sequences, it has been difficult to achieve good drug-likeness and high potency simultaneously. Indeed, for molecules targeting secondary structures, drug-likeness and RNA binding affinity are anti-correlated: high potencies are only achieved with the most undrug-like molecules (Fig. 2B, squares and diamonds, dashed line). Moreover, in the relatively few cases where authors have examined a significant number of molecules in a ligand discovery program, the observed structure-activity relationship (SAR) is often relatively flat39,40,46,49, indicating that it has proven difficult to improve the potency of a lead molecule. Thus, despite significant effort and creativity, most compounds identified to date do not have characteristics of known potent bioavailable small-molecule drugs and do not target complex RNA sites as do known high-specificity drugs.
We emphasize that we are focusing on targeting RNA with small molecules, which we think requires identifying a cleft- or pocket-containing RNA (see below). Our analysis does not rule out the possibility that low-complexity sites or large multi-valent ligands might prove therapeutically useful. In the protein-targeting field, these compounds would be roughly the analogs of those drugs that target protein–protein interfaces50 and constitute beyond rule-of-five (bRo5) molecules51,52, respectively. Targeting pathogenic repeat expansion sequences (for example, the CUG and CAG repeats characteristic of Myotonic Dystrophy Type 1 and Huntington’s, respectively) might represent an intriguing special case. Although trinucleotide repeat sequences have low complexity and do not appear to contain an obvious ligandable pocket (see below), it is possible that cooperative binding or in situ assembly of ligands driven by supra-secondary structure of the repeating sequence might afford critical specificity and selectivity19,53. Any of these strategies may prove fruitful but can generally be expected to have a notably increased degree of difficulty.
[H1] Hypotheses for RNA-targeted drugs
[H3] RNA-targeted drugs should probably look like conventional protein-targeted drugs.
One question that arises immediately when thinking about targeting RNA with small molecules is whether the structural differences between RNA and proteins render RNA less druggable than proteins. RNA is composed of only four primary nucleotide building blocks and is much more highly charged and more hydrophilic than a typical protein. We suggest that focusing on these global differences misses the point and that the key issue in drugging RNA is whether RNA can form specific binding sites capable of binding drug-like molecules. Current evidence suggests that bacterial17,54, viral55–59, and mammalian (both coding and non-coding)60–66 RNAs fold back on themselves to form complex structures. We will argue below that RNAs with complex structures also tend to contain pockets with sufficient structural sophistication to allow specific and high-affinity binding by small molecules.
That RNA binders can be “drug-like” is demonstrated by analysis of linezolid28,29 and ribocil30,31 (Fig. 2A). Linezolid and ribocil are both unremarkable molecules from the point of view of conventional medicinal chemistry. Both are consistent with Lipinski’s Rule of Five (Box 1), both have low total polar surface areas (tPSAs) consistent with good membrane permeability, and neither contains any red flags for toxicity. Most critically, both bind to structural “pockets” in their target RNAs — the large subunit bacterial ribosomal RNA and a bacterial flavin mononucleotide (FMN) riboswitch, respectively (Fig. 4). Both ligand binding sites are complex in the sense that they feature both ‘inner-sphere’ nucleobases that are in direct contact with the ligand and a set of ‘outer-sphere’ nucleotides that further rigidify and support the structure of the ligand binding site29–31,67. These factors – direct intimate contact with inner-sphere nucleotides and support by a complex three-dimensional network – make these binding sites similar to the many pockets in protein targets that bind therapeutically useful small molecules.
Figure 4. Drug-like molecules specifically targeting RNA.
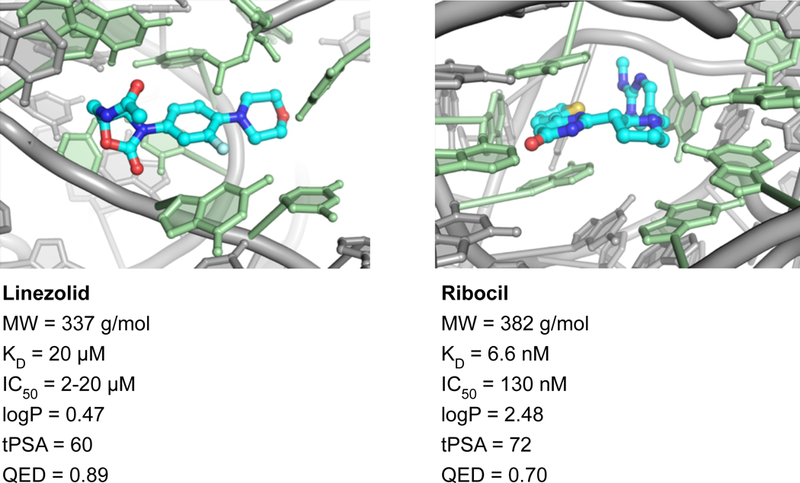
Both linezolid and ribocil have high QED drug-likeness scores27 and are bound within cleft-like sites29,30 composed of first-sphere nucleotides in contact with the ligand (green) supported by second-sphere nucleotides (gray). These motifs are high in information content.
Comparison of how a single ligand, riboflavin, can be recognized by both protein and RNA further supports the ability of RNA to recognize diverse physical features of a ligand (Fig. 5). Although riboflavin has a tPSA value that is high and other features that are moderate outliers relative to most drugs, it is, in fact, clinically useful68. Riboflavin contains three distinctive molecular entities: the nitrogen-rich pteridine-2,4-dione two-ring system, the hydrophobic dimethylbenzene ring, and the ribose group. Riboflavin kinase protein69 and a flavin RNA riboswitch70 recognize riboflavin in similar, but not identical, ways. Both protein and RNA form multiple hydrogen bonds with the pteridine ring system, both make multiple van der Waals contacts with the dimethylbenzene ring, both form hydrophobic or stacking interactions from above and below the plane of the three-ring system, and both form hydrogen bonds with a hydroxyl group in the ribose chain (Fig. 5). This analysis shows that, in principle, RNA is capable of making specific molecular interactions with a wide variety of functional groups and ligand surfaces. The most consistent feature of the examples discussed thus far is the ability of the RNA target to form a pocket into which a ligand fits intimately and forms multiple specific interactions.
Figure 5: RNA and protein recognition of the same riboflavin ligand.
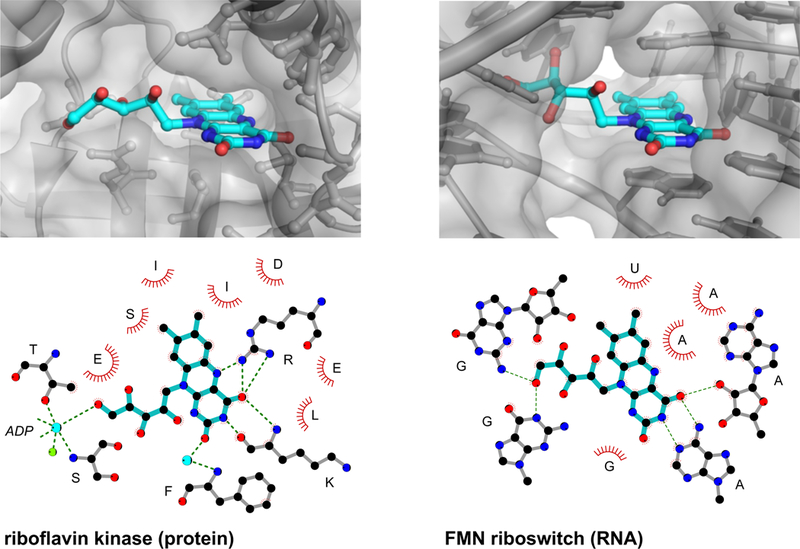
Molecular interactions of riboflavin (blue) with riboflavin kinase protein (PDB inb9)69 (left) and FMN riboswitch RNA (3f4g)70 (right). Hydrogen bonds are shown with green dashed lines and van der Waals and stacking interactions are emphasized with red arcs. Water molecules and metal ions are shown as cyan and green spheres, respectively. Riboflavin has molecular weight = 376 g/mol; logP = – 0.76; tPSA = 162; and QED = 0.33.
[H3] Good RNA targets should have high information content.
An overarching concern in targeting RNA is how to achieve both high binding affinity and also high selectivity in the cellular context in which there are many similar RNA motifs. To date, a significant fraction of the effort devoted to targeting RNA has focused on simple RNA structures consisting of base-paired helices or irregular and bulge-containing duplexes (Fig. 3). In contrast, the RNAs that interact with the drug-like RNA-targeting molecules such as linezolid, ribocil, and riboflavin bind to complex structures and are encapsulated in cleft-like sites reminiscent of ligand-binding sites in proteins (Figs. 2,4,5).
The complexity of an RNA can be characterized in terms of its information content, measured in bits71–73 (Box 2). One bit is the amount of information required to distinguish between two possibilities. RNA secondary structures can now be modeled with high accuracy based on sequence covariation information (when many alignable sequences are available)74 and using chemical probing strategies, especially SHAPE, for individual RNAs75,76, enabling a useful estimation of information content. The number of bits in an RNA target is approximated by summing the information in individual structure elements (Box 2). Analysis of information content is especially attractive for RNA, as this metric is relatively easy to calculate, given a secondary structure model and modest additional biochemical information, and provides an impartial way by which to evaluate current and future targets. A generic six base-pair RNA helix has an information content of roughly 9 bits, whereas the linezolid, ribocil, and branaplam binding sites likely exceed 50 bits. The information content of a ligand binding site reflects both inner- and outer-sphere interactions (see Fig. 4), such that nucleotides do not necessarily have to touch an RNA ligand directly to affect the information content of a site31,67,77.
Box 2: Information content of RNA targets.
The amount of information required to specify an RNA target can be defined by its structural complexity, measured in bits71,72. One bit corresponds to the information required to distinguish between two possibilities, for example, between the purine and pyrimidine bases. Two bits are therefore required to define an invariant or absolutely required nucleotide in an RNA element. Nucleotide positions with no sequence constraints correspond to zero bits. The information required to define a conserved base pair can be subtle because a degree of mismatched or wobble pairing is often allowed73. A canonical Watson-Crick base pair, allowing G-U, corresponds to roughly 2 bits. For many RNA motifs, a short helix is required to stabilize a given structure, but the specific sequence, length, and degree of allowed mismatches can vary. We estimate that a generic helix requires the equivalent of six base pairs, half of which could be wobble or mismatch pairs, for (3 × 2) + (3 × 1) = 9 bits.
The total information content for a given RNA motif or drug target is given by summing the information content for every position in the target element (see image). RNA structural motifs can be modeled with good accuracy based on chemical probing data, especially using the SHAPE strategy. SHAPE experiments use reagents that form covalent adducts with the 2’-hydroxyl group in RNA, such that the degree of reactivity reports local nucleotide flexibility55,75,76. These SHAPE reactivities can then be interpreted using RNA folding software to define the secondary structure of an RNA target, where regions with low versus high reactivities tend to be base paired or single stranded, respectively. The total number of bits in an RNA target site can be estimated from this secondary structure, taken together with additional biochemical data including comparative sequence analysis71,73, biochemical experiments72, or inspection of interactions observed in high resolution studies (Fig. 6, 7). Applying these rules to recognition of the HIV-1 TAR RNA element by short Tat-derived peptides99, scored based on the information content of structural elements recognized by the peptides, reveals that this interaction site contains about 22 ± 5 bits of information. Boxed nucleotides show significant sequence specificity; brackets indicate elements where structure, but not sequence, is the primary constraint.
There are a few helpful reference points that help calibrate the distinctiveness of an RNA motif with a given amount of information content. For example, 10 bits is equivalent to an expected occurrence of 1 in 1024; whereas 30 bits corresponds to a motif that will occur roughly once in a billion nucleotides. Thus, 30 bits are required to be reasonably confident that an RNA target is unique in the human transcriptome.
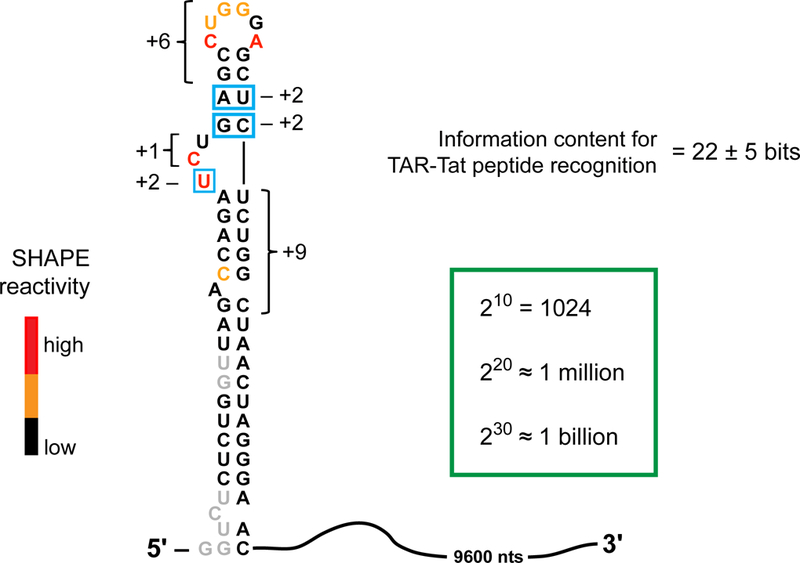
Information content for recognition of the HIV TAR RNA.
Perhaps the most instructive illustration so far of interrelationships between RNA complexity and ligand binding comes from prior studies focusing on examining RNA sequences that have been identified by in vitro selection to bind guanosine triphosphate73. Interactions between GTP and RNA likely involve primarily the guanosine entity77 (MW = 283; QED score = 0.40). GTP binding affinity shows a strong correlation with the complexity and information content of the RNA target (Fig. 6, circles). For GTP, increasing the information content of the target RNA by 10 bits has been experimentally observed to yield a roughly 10-fold increase in binding affinity73, which we term the ‘10→10’ relationship’ for brevity. This 10→10 relationship also holds for another ligand for which affinity versus target-site complexity data are available, Targaprimir-96 (Fig. 6, diamonds; data from ref. 78). The Targaprimir-96 line is offset from the GTP line because Targaprimir-96 has a higher affinity for a given information content relative to GTP, likely due to its larger size.
Figure 6: Relationship between information content and specificity and binding affinity.
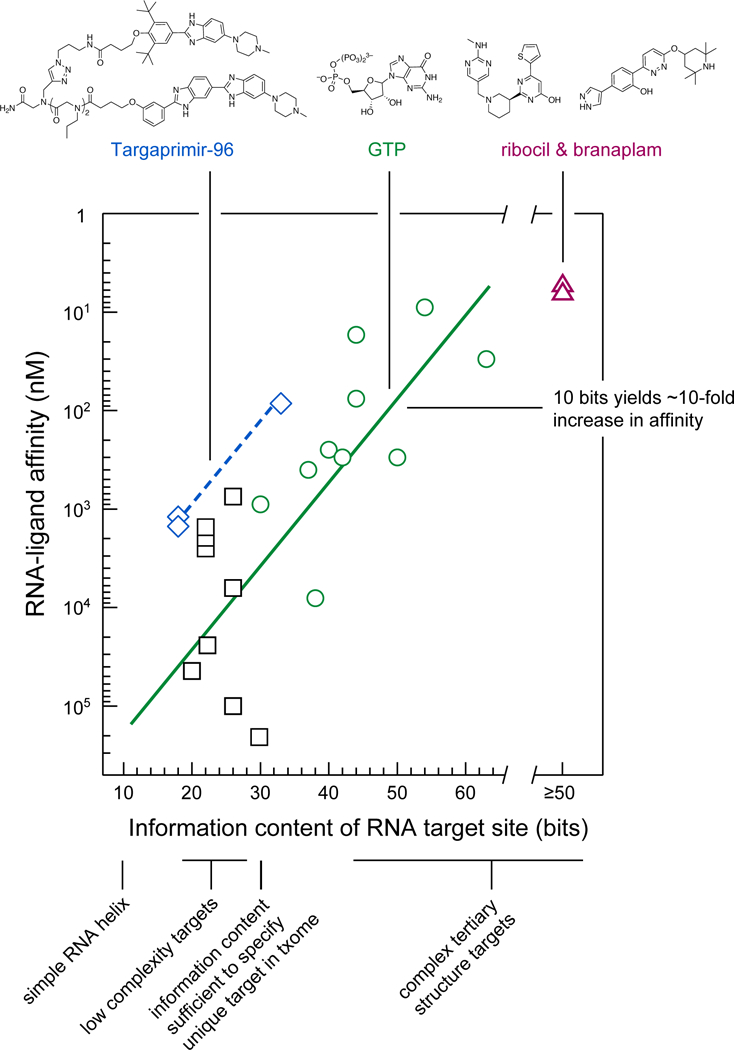
Solid and dashed lines show the relationship between RNA target information content and binding affinity for GTP (circles) and Targaprimir-96 (diamonds) (based on data from refs. 73,78). Simple helices are assigned 9 ± 2 bits of information. Ribocil30 and branaplam32 are estimated to have ≥50 bits of information and are placed on a separate scale. Binding data for medium-complexity targets (squares) are from the following: influenza A promoter stem-loop (20 bits)49, TAR (22 bits) 37,39,40,48 HCV IRES bulged-stem (26 bits)36,41,102, and SARS pseudoknot (30 bits)47,101.
The approximate 10→10 relationship identified in prior studies73 provides a framework for interpreting recent efforts to target RNA with small molecules. For example, ribocil and branaplam both have low-nanomolar affinities and bind to RNA and RNA–protein sites, respectively, with high-information contents (Fig. 6, triangles). Similarly, diverse efforts focused on identifying ligands against targets with low-to-moderate information content – including a stem loop (20 bits), the TAR hairpin (22 bits), and an asymmetric loop in the HCV IRES element (26 bits) – have resulted in molecules with affinities in the 1 to 100 µM range. These micromolar binding affinities for low-molecular-mass ligands agree closely with expectations based on the 10→10 relationship (Fig. 6, squares). All of these low-to-moderate affinity target sites fall below the 30-bit threshold, which is roughly the amount of information required to uniquely specify an RNA target in the context of the human transcriptome (Box 2).
The critical conclusion from this analysis of information content is that current efforts at targeting RNA with drug-like molecules are doing about as well as expected. Efforts to target relatively simple structures have achieved modest potency and selectivity. Thus, it is not necessarily the methods used or the small-molecule molecular frameworks that are limiting the RNA-targeted drug discovery field. We argue that the field has been limited by the choice of targets without sufficient complexity and information content.
[H3] Relationship between RNA information content and quality of the ligand pocket.
Our hypothesis is that focusing on RNA target sites with high information content is important both in order to identify sites that can be targeted with the roughly one in a billion (30 bits) selectivity necessary to identify a site uniquely in a complex transcriptome and in order to achieve high potency. Here we discuss a third critical advantage of targeting complex sites: Complex sites contain high-quality pockets into which ligands and drugs can bind.
We examined the ligandablity of representative targets in terms of their pocket quality. Pocket prediction algorithms use a combination of sequence, structure, and ligand information to provide estimations of potential binding sites within a target79,80. We used an algorithm, PocketFinder81, to estimate the quality of potential ligand-binding pockets formed from a representative set of RNAs. The pocket-finding algorithm defines a ligand-binding envelope based on the transformation of the Lennard–Jones potential determined from the target surface and does not require pre-existing knowledge about a putative ligand. This pocket-finding strategy predicts the location of a pocket and estimates its shape and size (Fig. 7). For example, the pockets in riboflavin kinase and the FMN riboswitch are distinguished by both their volumes (>160 Å3) and their ‘buriedness’ (>0.75; where 0.5 indicates the surface is flat and 1.0 corresponds to a completely buried pocket), values that indicate large cleft-like regions (Fig. 7A). Similarly, the pocket in the 23S ribosomal RNA into which linezolid binds is large and highly buried (Fig. 7B).
Figure 7: Pocket analysis for current and aspirational RNA targets.
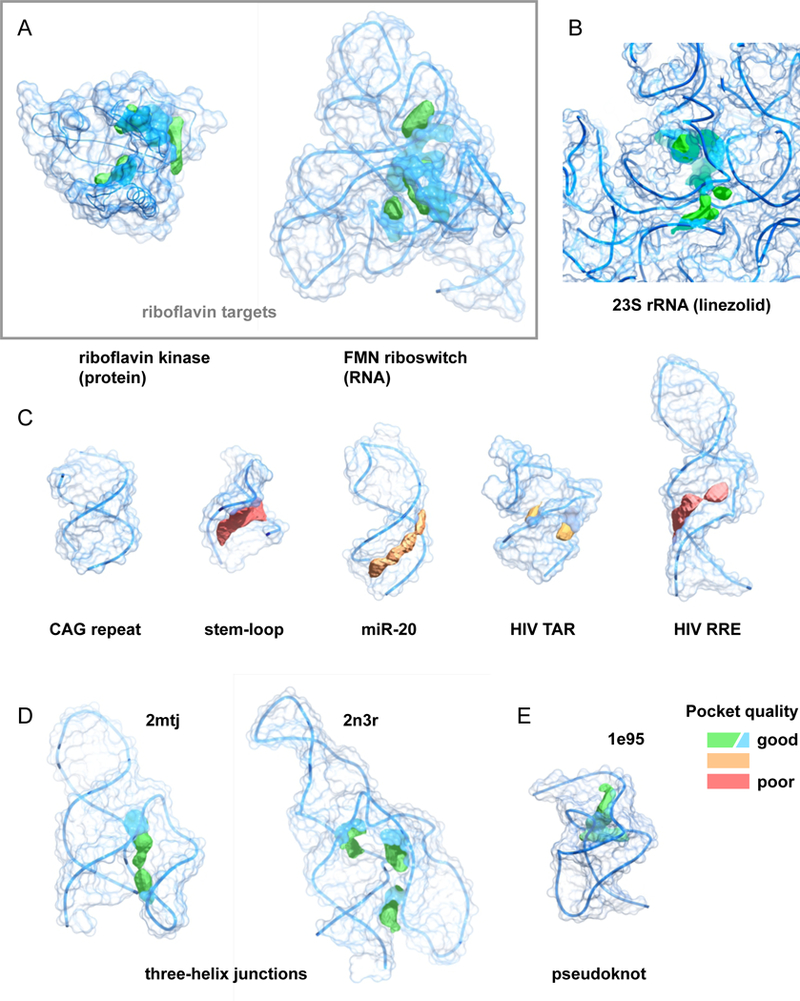
Structures are colored by pocket quality, with larger and more buried pockets ranking higher. (A) Riboflavin binding pockets for protein (1nb9)69 and RNA (3f4g)70 targets. Macromolecular targets are the same as shown in Fig. 5. (B) Pocket for linezolid in the E. coli 23S rRNA (3dll)29. (C) Ligand binding pockets in representative low-to-medium complexity targets including a CAG helical repeat (PDB 4j50)104, stem-loop (1oq0)105, microRNA (2n7x)106, and HIV TAR (1qd3)107 and RRE (1i9f) 108. (D, E) Potential ligand binding pockets in high complexity sites. Three helix junctions (2mtj, 2n3r) 82,83 and the SRV-1 pseudoknot (1e95)85. Pocket qualities were calculated using Pocket-Finder81, as part of the MolSoft package.
In strong contrast, many RNA motifs targeted by small molecules to date do not contain what would be considered either intuitively or quantitatively notable pockets. For example, helices containing CAG or CUG trinucleotide repeats (and almost certainly other repeat sequences) do not contain features that meet common definitions of a ligand-binding pocket. RNA motifs comprised primarily of secondary structures, including regulatory stem-loops, microRNAs, and the HIV TAR and RRE stem-loops, contain small or shallow pockets (Fig. 7C). It is possible that these kinds of motifs can be targeted, but doing so will likely prove challenging in much the same way as has targeting the shallow grooves that characterize many protein–protein interactions51.
It is straightforward to identify RNAs that adopt high-quality, likely targetable, structures. The simplest motifs that have complex, high-information-content structures are multi-helix junctions. Analysis of the structures of three-helix junctions82,83 immediately reveals that these motifs can have information contents exceeding 30 bits and contain high-quality pockets (Fig. 7D). These pockets are simply consequences of having a complex structure. Multi-helix junctions occur widely in large RNAs, many are known to overlap or occur within functional motifs, and a subset form well-defined, stable structural elements. Pseudoknots are a simple motif formed when unpaired nucleotides in a loop pair with a region outside the stem that closes the loop84. Many pseudoknots contain high-quality pockets, as illustrated by the structure of the SRV-1 pseudoknot85, which forms the common H-type pseudoknot (Fig. 7E). Pseudoknots are thought to be relatively rare in large RNAs, but appear to be overrepresented in functionally important regions86.
[H1] Provocative guidelines
We first reiterate that no one currently knows how to target RNA with drug-like small molecules in a scalable and reproducible way. Proof-of-principle examples exist (Fig. 2,3), but the most drug-like of these molecules were discovered based on phenotypic assays and only later found to target RNA. The molecules discovered to date via strategies intentionally designed to target RNA generally suffer from tradeoffs in drug-likeness and potency due to low information content and low pocket quality. Nonetheless, current examples are supportive of the fundamental target-ability of RNA. We suspect that a wide variety of screening strategies – including phenotypic assays, high-throughput screening, fragment-based screening, small-molecule microarrays, other biophysical partitioning approaches, and structure-inspired and computationally-assisted design – will eventually prove useful. In this light, we would like to propose the following guidelines for future efforts directed at RNA-targeted drug discovery:
Focus on RNA motifs of sufficient complexity that they will be unique, able to form high-quality pockets, and capable of binding small-molecule ligands with high affinity. We think that good RNA targets should have “30 bits and a pocket”. Such motifs are likely to be common in large RNAs. We posit that few good targetable pockets exist in the absence of high information content.
Be cautious of early attempts to define specialized privileged chemical scaffolds for targeting RNA. These might exist, but a lot more information is likely to be required before promising scaffolds can be defined. Instead, emphasize rules derived from decades of development of protein-targeted drugs. Most are likely to apply to RNA-targeted drugs as well.
De-emphasize “lessons” from the ribosome. Due to their uniquely high concentration in cells, the ribosomal RNAs are a special case. The only engagement expected for a ligand with micromolar affinity is a target that is present at micromolar concentration. Drugs targeting ribosomal RNAs thus achieve a unique kind of specificity by binding relatively weakly to the ribosome.
Be cautious in interpreting the results of studies focused on compounds that are highly basic, intercalating, strongly stacking, and/or highly hydrophobic. These kinds of compounds may bind RNA with high affinity but are unlikely to show strong discrimination between target and off-target sites and are likely to be difficult to improve in a medicinal chemistry campaign.
Target RNA–protein interactions or RNA elements stabilized by protein binding, as branaplam and SMA-C5 appear to do32,33. It is easy to imagine that recognition of a target-specific RNA element in conjunction with a general protein component of cellular metabolism or regulatory network might be a highly productive strategy for achieving both specificity (via a selective RNA-targeted component) and affinity (due to the high information content of a protein interface).
Use methods that enable rapid identification of RNA motifs with high information content structures and high-quality pockets. A clear advantage of RNA over proteins is that tools such as quantitative chemical probing approaches55,75,87 already exist that make it possible to identify high information content72,73 and structurally well-determined (low entropy) target sites54,55, to examine the influence of the cellular environment on a specific RNA structure, and to ensure that simplified screening constructs recapitulate the native RNA motif.
Develop additional tools and new refinements of current tools, including advanced structural biology and modeling strategies for RNAs with complex structures88–90. These technologies can then be leveraged to identify and rapidly characterize the subset of complex RNA structures with high-quality ligand-binding pockets and to rapidly assess the impact of ligand binding on the functions of these sites.
Implement reproducible strategies for connecting ligand binding to complex targets with clear therapeutic mechanisms. There is modest-to-good evidence that disrupting or stabilizing RNA structures near translation initiation sites can modulate gene expression in therapeutically useful ways13,64,91. There is some evidence that binding of small molecules to mRNAs can modulate the efficiency of translation92,93. Both disrupting and stabilizing RNA– protein interactions are therapeutically promising. Covalent drugs are gaining acceptance in the protein-targeting field; analogously, ligands that can form covalent linkages or induce cleavage at an RNA target site44,94 also hold promise.
[H1] Perspective
Three key components are necessary to enable effective, repeatable, and scalable RNA-targeted drug discovery: (1) a therapeutically compelling RNA target, (2) a screening approach that will identify drug-like lead molecules with appropriate pharmacological properties, and (3) identification of RNA motifs with sufficient information content that high-specificity and high-potency binding to a high-quality pocket is achieved. We are arguing for a broad change in strategy for creating RNA-targeted drugs. Much greater emphasis needs to be placed on the quality of the RNA target – component 3. Biologically well-validated RNA targets are important, of course, but much greater effort needs to be placed on ensuring the ligand-ability of these targets. Ultimately, as we learn more about what constitutes a high-quality RNA target, RNA might prove to be no more difficult to “drug” than proteins. Indeed, given that targetable RNA motifs appear to be ubiquitous (refs. 17,54–66 and Fig. 7D, E) and that the principles of RNA structure and folding are simpler than those for proteins95, we wonder if RNA might eventually prove to be more generically targetable than proteins. Success of RNA-targeted therapeutic discovery efforts will open up vast opportunities (Fig. 1) for modulating the functions of currently undruggable protein-mediated pathways and the non-coding transcriptome.
Acknowledgement
Work on understanding RNA structure-function interrelationships in the laboratory of K.M.W. is supported by the NIH (R35 GM122532 and R01 AI068462).
Footnotes
Disclosure
K.M.W. is an advisor to and holds equity in Ribometrix. K.D.W. and C.E.H. are employees of Ribometrix.
REFERENCES
- 1.Clamp M et al. Distinguishing protein-coding and noncoding genes in the human genome. Proc Natl Acad Sci U S A 104, 19428–19433 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ezkurdia I et al. Multiple evidence strands suggest that there may be as few as 19,000 human protein-coding genes. Hum. Mol. Genet 23, 5866–5878 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hopkins AL & Groom CR The druggable genome. Nat Rev Drug Discov 1, 727–730 (2002). [DOI] [PubMed] [Google Scholar]
- 4.Overington JP, Al-Lazikani B & Hopkins AL How many drug targets are there? Nat Rev Drug Discov 5, 993–996 (2006). [DOI] [PubMed] [Google Scholar]
- 5.Dixon SJ & Stockwell BR Identifying druggable disease-modifying gene products. Curr Opin Chem Biol 13, 549–555 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Santos R et al. A comprehensive map of molecular drug targets. Nat Rev Drug Discov 16, 19–34 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sharp PA The centrality of RNA. Cell 136, 577–580 (2009). [DOI] [PubMed] [Google Scholar]
- 8.Cech TR & Steitz JA The noncoding RNA revolution-trashing old rules to forge new ones. Cell 157, 77–94 (2014). [DOI] [PubMed] [Google Scholar]
- 9.Djebali S et al. Landscape of transcription in human cells. Nature 489, 101–108 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Matsui M & Corey DR Non-coding RNAs as drug targets. Nat Rev Drug Discov 16, 167–179 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Adams BD, Parsons C, Walker L, Zhang WC & Slack FJ Targeting noncoding RNAs in disease. J. Clin. Invest 127, 761–771 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harrow J et al. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res 22, 1760–1774 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liang X-H et al. Translation efficiency of mRNAs is increased by antisense oligonucleotides targeting upstream open reading frames. Nature Biotechnol 34, 875–880 (2016). [DOI] [PubMed] [Google Scholar]
- 14.Liang X-H et al. Antisense oligonucleotides targeting translation inhibitory elements in 5’ UTRs can selectively increase protein levels. Nucleic Acids Res 45, 9528–9546 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Crooke ST, Witztum JL, Bennett CF & Baker BF RNA-Targeted Therapeutics. Cell Metab 27, 714–739 (2018). [DOI] [PubMed] [Google Scholar]
- 16.Fellmann C, Gowen BG, Lin P-C, Doudna JA & Corn JE Cornerstones of CRISPR-Cas in drug discovery and therapy. Nat Rev Drug Discov 16, 89–100 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McCown PJ, Corbino KA, Stav S, Sherlock ME & Breaker RR Riboswitch diversity and distribution. RNA 23, 995–1011 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wilson DN Ribosome-targeting antibiotics and mechanisms of bacterial resistance. Nat Rev Microbiol 12, 35–48 (2014). [DOI] [PubMed] [Google Scholar]
- 19.Childs-Disney JL & Disney MD Approaches to Validate and Manipulate RNA Targets with Small Molecules in Cells. Annu. Rev. Pharmacol. Toxicol 56, 123–140 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thomas JR & Hergenrother PJ Targeting RNA with small molecules. Chem. Rev 108, 1171–1224 (2008). [DOI] [PubMed] [Google Scholar]
- 21.Aboul-ela F Strategies for the design of RNA-binding small molecules. Future Med Chem 2, 93–119 (2010). [DOI] [PubMed] [Google Scholar]
- 22.Guan L & Disney MD Recent advances in developing small molecules targeting RNA. ACS Chem Biol 7, 73–86 (2012). [DOI] [PubMed] [Google Scholar]
- 23.Connelly CM, Moon MH & Schneekloth JS The Emerging Role of RNA as a Therapeutic Target for Small Molecules. Cell Chem Biol 23, 1077–1090 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Morgan BS, Forte JE, Culver RN, Zhang Y & Hargrove AE Discovery of Key Physicochemical, Structural, and Spatial Properties of RNA-Targeted Bioactive Ligands. Angew. Chem. Int. Ed. Engl 56, 13498–13502 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hermann T Small molecules targeting viral RNA. Wiley Interdiscip Rev RNA 7, 726–743 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rizvi NF & Smith GF RNA as a small molecule druggable target. Bioorg. Med. Chem. Lett 27, 5083–5088 (2017). [DOI] [PubMed] [Google Scholar]
- 27.Bickerton GR, Paolini GV, Besnard J, Muresan S & Hopkins AL Quantifying the chemical beauty of drugs. Nat Chem 4, 90–98 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moellering RC Linezolid: the first oxazolidinone antimicrobial. Ann. Intern. Med 138, 135–142 (2003). [DOI] [PubMed] [Google Scholar]
- 29.Wilson DN et al. The oxazolidinone antibiotics perturb the ribosomal peptidyl-transferase center and effect tRNA positioning. Proc Natl Acad Sci U S A 105, 13339–13344 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Howe JA et al. Selective small-molecule inhibition of an RNA structural element. Nature 526, 672–677 (2015). [DOI] [PubMed] [Google Scholar]
- 31.Howe JA et al. Atomic resolution mechanistic studies of ribocil: A highly selective unnatural ligand mimic of the E. coli FMN riboswitch. RNA Biol 13, 946–954 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Palacino J et al. SMN2 splice modulators enhance U1-pre-mRNA association and rescue SMA mice. Nat. Chem. Biol 11, 511–517 (2015). [DOI] [PubMed] [Google Scholar]
- 33.Sivaramakrishnan M et al. Binding to SMN2 pre-mRNA-protein complex elicits specificity for small molecule splicing modifiers. Nat Commun 8, 1476 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kondo Y, Oubridge C, van Roon A-MM & Nagai K Crystal structure of human U1 snRNP, a small nuclear ribonucleoprotein particle, reveals the mechanism of 5’ splice site recognition. Elife 4, 360 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Calder AN, Androphy EJ & Hodgetts KJ Small Molecules in Development for the Treatment of Spinal Muscular Atrophy. J. Med. Chem 59, 10067–10083 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Seth PP et al. SAR by MS: discovery of a new class of RNA-binding small molecules for the hepatitis C virus: internal ribosome entry site IIA subdomain. J. Med. Chem 48, 7099–7102 (2005). [DOI] [PubMed] [Google Scholar]
- 37.Mei HY et al. Discovery of selective, small-molecule inhibitors of RNA complexes--I. The Tat protein/TAR RNA complexes required for HIV-1 transcription. Bioorg. Med. Chem 5, 1173–1184 (1997). [DOI] [PubMed] [Google Scholar]
- 38.Prado S et al. Bioavailable inhibitors of HIV-1 RNA biogenesis identified through a Rev-based screen. Biochem. Pharmacol 107, 14–28 (2016). [DOI] [PubMed] [Google Scholar]
- 39.Sztuba-Solinska J et al. Identification of biologically active, HIV TAR RNA-binding small molecules using small molecule microarrays. J Am Chem Soc 136, 8402–8410 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Joly J-P et al. Artificial nucleobase-amino acid conjugates: a new class of TAR RNA binding agents. Chemistry 20, 2071–2079 (2014). [DOI] [PubMed] [Google Scholar]
- 41.Carnevali M, Parsons J, Wyles DL & Hermann T A modular approach to synthetic RNA binders of the hepatitis C virus internal ribosome entry site. Chembiochem 11, 1364–1367 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hilimire TA et al. HIV-1 Frameshift RNA-Targeted Triazoles Inhibit Propagation of Replication-Competent and Multi-Drug-Resistant HIV in Human Cells. ACS Chem Biol 12, 1674–1682 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Barros SA, Yoon I & Chenoweth DM Modulation of the E. coli rpoH Temperature Sensor with Triptycene-Based Small Molecules. Angew. Chem. Int. Ed. Engl 55, 8258–8261 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nguyen L et al. Rationally designed small molecules that target both the DNA and RNA causing myotonic dystrophy type 1. J Am Chem Soc 137, 14180–14189 (2015). [DOI] [PubMed] [Google Scholar]
- 45.Luu LM et al. A Potent Inhibitor of Protein Sequestration by Expanded Triplet (CUG) Repeats that Shows Phenotypic Improvements in a Drosophila Model of Myotonic Dystrophy. ChemMedChem 11, 1428–1435 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee M-K et al. A novel small-molecule binds to the influenza A virus RNA promoter and inhibits viral replication. Chem. Commun. (Camb.) 50, 368–370 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Park S-J, Kim Y-G & Park H-J Identification of RNA pseudoknot-binding ligand that inhibits the −1 ribosomal frameshifting of SARS-coronavirus by structure-based virtual screening. J Am Chem Soc 133, 10094–10100 (2011). [DOI] [PubMed] [Google Scholar]
- 48.Stelzer AC et al. Discovery of selective bioactive small molecules by targeting an RNA dynamic ensemble. Nat. Chem. Biol 7, 553–559 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bottini A et al. Targeting Influenza A Virus RNA Promoter. Chem Biol Drug Des 86, 663–673 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jin L, Wang W & Fang G Targeting protein-protein interaction by small molecules. Annu. Rev. Pharmacol. Toxicol 54, 435–456 (2014). [DOI] [PubMed] [Google Scholar]
- 51.Doak BC, Over B, Giordanetto F & Kihlberg J Oral druggable space beyond the rule of 5: insights from drugs and clinical candidates. Chem Biol 21, 1115–1142 (2014). [DOI] [PubMed] [Google Scholar]
- 52.Matsson P, Doak BC, Over B & Kihlberg J Cell permeability beyond the rule of 5. Adv. Drug Deliv. Rev 101, 42–61 (2016). [DOI] [PubMed] [Google Scholar]
- 53.Zimmerman SC A journey in bioinspired supramolecular chemistry: from molecular tweezers to small molecules that target myotonic dystrophy. Beilstein J Org Chem 12, 125–138 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mustoe AM et al. Pervasive Regulatory Functions of mRNA Structure Revealed by High-Resolution SHAPE Probing. Cell 173, 181–195.e18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Siegfried NA, Busan S, Rice GM, Nelson JAE & Weeks KM RNA motif discovery by SHAPE and mutational profiling (SHAPE-MaP). Nature Methods 11, 959–965 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mauger DM et al. Functionally conserved architecture of hepatitis C virus RNA genomes. Proc Natl Acad Sci U S A 112, 3692–3697 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pirakitikulr N, Kohlway A, Lindenbach BD & Pyle AM The Coding Region of the HCV Genome Contains a Network of Regulatory RNA Structures. Mol. Cell 62, 111–120 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Villordo SM, Carballeda JM, Filomatori CV & Gamarnik AV RNA Structure Duplications and Flavivirus Host Adaptation. Trends Microbiol 24, 270–283 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kutchko KM et al. Structural divergence creates new functional features in alphavirus genomes. Nucleic Acids Res 58, 491–3670 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cencig S et al. Mapping and characterization of the minimal internal ribosome entry segment in the human c-myc mRNA 5’ untranslated region. Oncogene 23, 267–277 (2004). [DOI] [PubMed] [Google Scholar]
- 61.Chakraborty S, Mehtab S, Patwardhan A & Krishnan Y Pri-miR-17–92a transcript folds into a tertiary structure and autoregulates its processing. RNA 18, 1014–1028 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Smola MJ et al. SHAPE reveals transcript-wide interactions, complex structural domains, and protein interactions across the Xist lncRNA in living cells. Proc Natl Acad Sci U S A 113, 10322–10327 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ball CB, Solem AC, Meganck RM, Laederach A & Ramos SBV Impact of RNA structure on ZFP36L2 interaction with luteinizing hormone receptor mRNA. RNA 23, 1209–1223 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Corley M et al. An RNA structure-mediated, posttranscriptional model of human α−1-antitrypsin expression. Proc Natl Acad Sci U S A 114, E10244–E10253 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lee ASY, Kranzusch PJ & Cate JHD eIF3 targets cell-proliferation messenger RNAs for translational activation or repression. Nature 522, 111–114 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Xue Z et al. A G-Rich Motif in the lncRNA Braveheart Interacts with a Zinc-Finger Transcription Factor to Specify the Cardiovascular Lineage. Mol. Cell 64, 37–50 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Long KS & Vester B Resistance to linezolid caused by modifications at its binding site on the ribosome. Antimicrob. Agents Chemother 56, 603–612 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.WHO Model Lists of Essential Medicines, March 2017.
- 69.Karthikeyan S et al. Crystal structure of human riboflavin kinase reveals a beta barrel fold and a novel active site arch. Structure 11, 265–273 (2003). [DOI] [PubMed] [Google Scholar]
- 70.Serganov A, Huang L & Patel DJ Coenzyme recognition and gene regulation by a flavin mononucleotide riboswitch. Nature 458, 233–237 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schneider TD, Stormo GD, Gold L & Ehrenfeucht A Information content of binding sites on nucleotide sequences. J. Mol. Biol 188, 415–431 (1986). [DOI] [PubMed] [Google Scholar]
- 72.Witherell GW & Uhlenbeck OC Specific RNA binding by Q beta coat protein. Biochemistry 28, 71–76 (1989). [DOI] [PubMed] [Google Scholar]
- 73.Carothers JM, Oestreich SC, Davis JH & Szostak JW Informational complexity and functional activity of RNA structures. J Am Chem Soc 126, 5130–5137 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Shang L, Xu W, Ozer S & Gutell RR Structural constraints identified with covariation analysis in ribosomal RNA. PLoS ONE 7, e39383 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Weeks KM & Mauger DM Exploring RNA structural codes with SHAPE chemistry. Acc Chem Res 44, 1280–1291 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Smola MJ, Rice GM, Busan S, Siegfried NA & Weeks KM Selective 2’-hydroxyl acylation analyzed by primer extension and mutational profiling (SHAPE-MaP) for direct, versatile and accurate RNA structure analysis. Nat Protoc 10, 1643–1669 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Carothers JM, Davis JH, Chou JJ & Szostak JW Solution structure of an informationally complex high-affinity RNA aptamer to GTP. RNA 12, 567–579 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Velagapudi SP et al. Design of a small molecule against an oncogenic noncoding RNA. Proc Natl Acad Sci U S A 113, 5898–5903 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pérot S, Sperandio O, Miteva MA, Camproux A-C & Villoutreix BO Druggable pockets and binding site centric chemical space: a paradigm shift in drug discovery. Drug Discov. Today 15, 656–667 (2010). [DOI] [PubMed] [Google Scholar]
- 80.Fauman EB, Rai BK & Huang ES Structure-based druggability assessment--identifying suitable targets for small molecule therapeutics. Curr Opin Chem Biol 15, 463–468 (2011). [DOI] [PubMed] [Google Scholar]
- 81.An J, Totrov M & Abagyan R Pocketome via comprehensive identification and classification of ligand binding envelopes. Mol. Cell Proteomics 4, 752–761 (2005). [DOI] [PubMed] [Google Scholar]
- 82.Bonneau E & Legault P Nuclear magnetic resonance structure of the III-IV-V three-way junction from the Varkud satellite ribozyme and identification of magnesium-binding sites using paramagnetic relaxation enhancement. Biochemistry 53, 6264–6275 (2014). [DOI] [PubMed] [Google Scholar]
- 83.Bonneau E, Girard N, Lemieux S & Legault P The NMR structure of the II-III-VI three-way junction from the Neurospora VS ribozyme reveals a critical tertiary interaction and provides new insights into the global ribozyme structure. RNA 21, 1621–1632 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Staple DW & Butcher SE Pseudoknots: RNA structures with diverse functions. PLoS Biol 3, e213 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Michiels PJ et al. Solution structure of the pseudoknot of SRV-1 RNA, involved in ribosomal frameshifting. J. Mol. Biol 310, 1109–1123 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hajdin CE et al. Accurate SHAPE-directed RNA secondary structure modeling, including pseudoknots. Proc Natl Acad Sci U S A 110, 5498–5503 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Smola MJ, Calabrese JM & Weeks KM Detection of RNA-Protein Interactions in Living Cells with SHAPE. Biochemistry 54, 6867–6875 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gilbert SD, Reyes FE, Edwards AL & Batey RT Adaptive ligand binding by the purine riboswitch in the recognition of guanine and adenine analogs. Structure 17, 857–868 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Warner KD et al. Validating fragment-based drug discovery for biological RNAs: lead fragments bind and remodel the TPP riboswitch specifically. Chem Biol 21, 591–595 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Miao Z & Westhof E RNA Structure: Advances and Assessment of 3D Structure Prediction. Annu Rev Biophys 46, 483–503 (2017). [DOI] [PubMed] [Google Scholar]
- 91.Kozak M Regulation of translation via mRNA structure in prokaryotes and eukaryotes. Gene 361, 13–37 (2005). [DOI] [PubMed] [Google Scholar]
- 92.Harvey I, Garneau P & Pelletier J Inhibition of translation by RNA-small molecule interactions. RNA 8, 452–463 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bandyopadhyay S et al. Novel 5’ untranslated region directed blockers of iron-regulatory protein-1 dependent amyloid precursor protein translation: implications for down syndrome and Alzheimer’s disease. PLoS ONE 8, e65978 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Rzuczek SG et al. Precise small-molecule recognition of a toxic CUG RNA repeat expansion. Nat. Chem. Biol 13, 188–193 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Tinoco I & Bustamante C How RNA folds. J. Mol. Biol 293, 271–281 (1999). [DOI] [PubMed] [Google Scholar]
- 96.Lipinski CA Lead- and drug-like compounds: the rule-of-five revolution. Drug Discov Today Technol 1, 337–341 (2004). [DOI] [PubMed] [Google Scholar]
- 97.Ritchie TJ & Macdonald SJF How drug-like are ‘ugly’ drugs: do drug-likeness metrics predict ADME behaviour in humans? Drug Discov. Today 19, 489–495 (2014). [DOI] [PubMed] [Google Scholar]
- 98.Gleitsman KR, Sengupta RN & Herschlag D Slow molecular recognition by RNA. RNA 23, 1745–1753 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Weeks KM & Crothers DM RNA recognition by Tat-derived peptides: interaction in the major groove? Cell 66, 577–588 (1991). [DOI] [PubMed] [Google Scholar]
- 100.Lin AH, Murray RW, Vidmar TJ & Marotti KR The oxazolidinone eperezolid binds to the 50S ribosomal subunit and competes with binding of chloramphenicol and lincomycin. Antimicrob. Agents Chemother 41, 2127–2131 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ritchie DB, Soong J, Sikkema WKA & Woodside MT Anti-frameshifting ligand reduces the conformational plasticity of the SARS virus pseudoknot. J Am Chem Soc 136, 2196–2199 (2014). [DOI] [PubMed] [Google Scholar]
- 102.Parsons J et al. Conformational inhibition of the hepatitis C virus internal ribosome entry site RNA. Nat. Chem. Biol 5, 823–825 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Costales MG et al. Small Molecule Inhibition of microRNA-210 Reprograms an Oncogenic Hypoxic Circuit. J Am Chem Soc 139, 3446–3455 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Yildirim I, Park H, Disney MD & Schatz GC A dynamic structural model of expanded RNA CAG repeats: a refined X-ray structure and computational investigations using molecular dynamics and umbrella sampling simulations. J Am Chem Soc 135, 3528–3538 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Leeper T, Leulliot N & Varani G The solution structure of an essential stem-loop of human telomerase RNA. Nucleic Acids Res 31, 2614–2621 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Chen Y et al. Rbfox proteins regulate microRNA biogenesis by sequence-specific binding to their precursors and target downstream Dicer. Nucleic Acids Res 44, 4381–4395 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Faber C, Sticht H, Schweimer K & Rösch P Structural rearrangements of HIV-1 Tat-responsive RNA upon binding of neomycin B. J. Biol. Chem 275, 20660–20666 (2000). [DOI] [PubMed] [Google Scholar]
- 108.Zhang Q, Harada K, Cho HS, Frankel AD & Wemmer DE Structural characterization of the complex of the Rev response element RNA with a selected peptide. Chem Biol 8, 511–520 (2001). [DOI] [PubMed] [Google Scholar]