

INTRODUCTION
DICER1 syndrome1 predisposes to a variety of cancers, including pleuropulmonary blastoma (PPB),2 ovarian Sertoli-Leydig cell tumor (SLCT),3 embryonal rhabdomyosarcoma,4 and kidney tumors.5–8 In DICER1 syndrome, most patients bear a germline null mutation in DICER1, and the tumors uniformly bear a second-hit missense substitution at one of five hotspot positions (1705E, 1709D, 1809G, 1810D, and 1813E).
A wide range of clinical phenotypes can be seen in DICER1 syndrome5,9; some patients are asymptomatic, whereas others develop multiple tumors. In the classic cases, patients with a germ-line null mutation develop different somatic hotspot mutations in each tumor.7,10–16 However, in some patients, multiple tumors arise from germline mosaicism of the hotspot mutation, with subsequent somatic loss of the wild-type allele.17–19 Here we report a patient with DICER1 syndrome who developed four tumor types at six anatomic sites over the course of 12 years. These tumors harbor four distinct hotspot mutations, which is one of the highest numbers of distinct somatic DICER1 mutations reported in a single patient. By identifying these mutations, we show that the patient’s bilateral renal tumors and bilateral ovarian SLCTs each constituted a new primary tumor. Because we found no other mutations to explain her particularly severe clinical course, we speculate that her subsequent tumors were a product of the intense chemotherapy and radiation regimens she received.
METHODS
Informed consent was obtained from the patient and her guardian before collection of tumor specimens. All studies were conducted after approval by a local human investigations committee and in accord with an assurance filed with and approved by the Department of Health and Human Services.
Whole-exome sequencing of tumor and germ-line DNA has been described previously.20 For archival specimens, DNA was prepared using the QIAamp DNA FFPE kit (Qiagen, Santa Clarita, CA) and amplified using the REPLI-g FFPE kit (Qiagen). Polymerase chain reaction (PCR) primers spanning exons 21 to 26 of DICER1 are listed in Appendix Table A1. When needed, PCR products were subcloned into pCR2.1- Topo (Thermo-Fisher Scientific, Waltham, MA) before sequencing.
For TP53 sequencing, coding regions were amplified using the Accel-Amplicon Comprehensive TP53 Panel (Swift Biosciences, Ann Arbor, MI). Sequencing with the Illumina MiSeq Nano v2 kit (Illumina, San Diego, CA) generated 1.5 million paired-end reads. Reads were trimmed using trimmomatic21 in the Galaxy Project and were aligned to human genome GRCh38 using BWA-MEM22; 88% mapped to the TP53 locus, producing a mean depth of 13,367x in targeted regions. Variants were called using the FreeBayes algorithm v0.9.20 using frequency-based pooled calling instead of simple diploid calling, which allowed the algorithm to detect subclonal mutations.23
RESULTS
Clinical Presentation
The patient (CMCW11) presented originally at 5 years of age with a left kidney mass. She had no family history of cancer. She underwent left radical nephrectomy with lymph node resection. At the time, she was diagnosed with diffusely anaplastic Wilms tumor with extensive rhabdomyoblastic, osteoblastic, and chondroblastic differentiation (Fig 1A). This diagnosis was confirmed by central pathologic review. She had lymph node, pulmonary, and rib metastases. She was treated with 10 months of combination chemotherapy (vincristine, doxorubicin, cyclophosphamide, irinotecan, and etoposide) targeted at both anaplastic Wilms tumor and sarcoma, together with 2,100 cGy whole-abdomen irradiation given in 14 fractions over 19 days.
Fig 1.
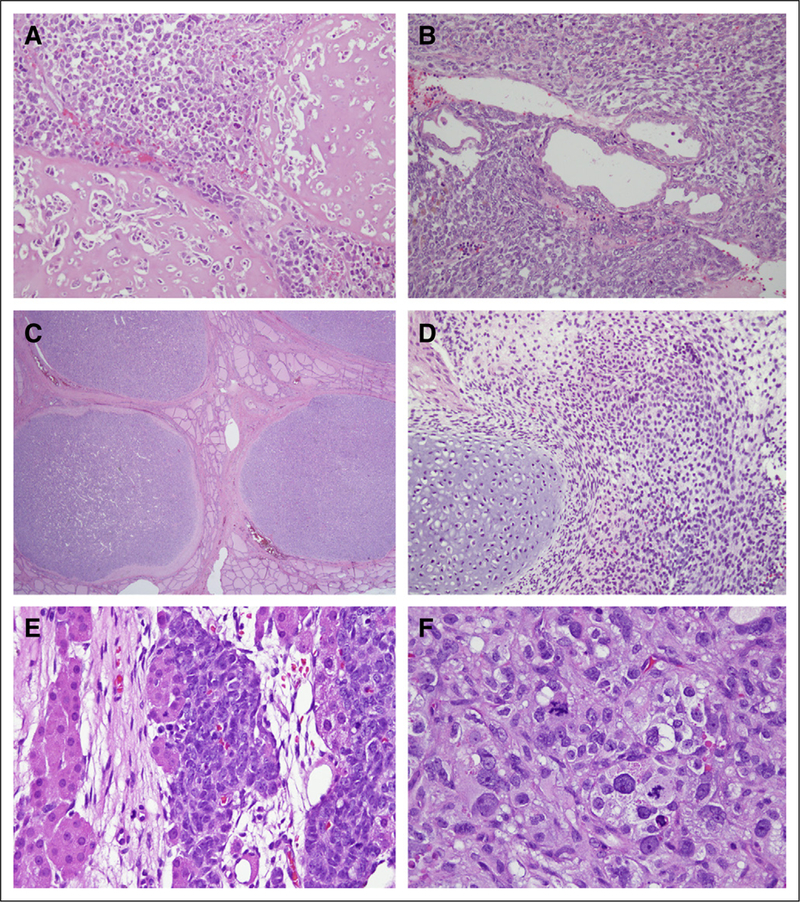
Histology of tumors. (A) Left kidney tumor (magnification, ×20). (B) Right kidney tumor (magnification, ×20). (C) Thyroid nodules (magnification, ×2). (D) Rhabdomyosarcoma arising from bladder (magnification, ×20). (E) Sertoli-Leydig cell tumor arising from right ovary (magnification, ×40). (F) Anaplastic elements of Sertoli-Leydig cell tumor arising from right ovary (magnification, ×40).
When the patient was 10 years old, a new mass arose in her remaining kidney, with a histology similar to her prior tumor, and she was diagnosed with relapsed Wilms tumor (Fig 1B). She initially received ifosfamide, carboplatin, and etoposide, but because of toxicity she was subsequently switched to vincristine, doxorubicin, cyclophosphamide, ifosfamide, and etoposide. On completion, she underwent right radical nephrectomy and started hemodialysis.
This patient’s kidney tumors were diagnosed initially as Wilms tumor, but in retrospect their unique histologic features are more consistent with anaplastic sarcoma of the kidney. This entity was first described in 2007, after our patient’s kidney tumor first arose.24 Biallelic DICER1 mutations have been seen in both Wilms tumor and anaplastic sarcoma of the kidney, which may represent neoplastic degeneration from cystic nephroma.6–8
At 12 years of age, the patient developed multiple nodules throughout her thyroid. Thyroidectomy revealed follicular adenomas (Fig 1C).
At 13 years of age, a mass arising from her bladder was diagnosed as embryonal rhabdomyosarcoma with focal cartilaginous differentiation (Fig 1D). She was treated with vincristine, dactinomycin, and cyclophosphamide and a radical cystectomy.
At 15 years of age, she developed ovarian masses and underwent sequential salpingooophorectomies. Both ovaries harbored SLCTs with intermediate to poor differentiation (Figs 1E and 1F). One of the tumors had anaplastic sarcomatoid foci. Since that time, she has been observed for > 2 years, with no further disease.
DICER1 Mutations
We performed whole-exome sequencing on the patient’s first kidney tumor and germline DNA as part of a retrospective Wilms tumor sequencing project.20 Her germline harbored a frameshift deletion (c.3307_3311delGACAG, p.Ile1102fs; Fig 2). Her tumor bore an additional somatic hotspot mutation (c.5425G>A; p.G1809R; Fig 2 and Table 1).
Fig 2.
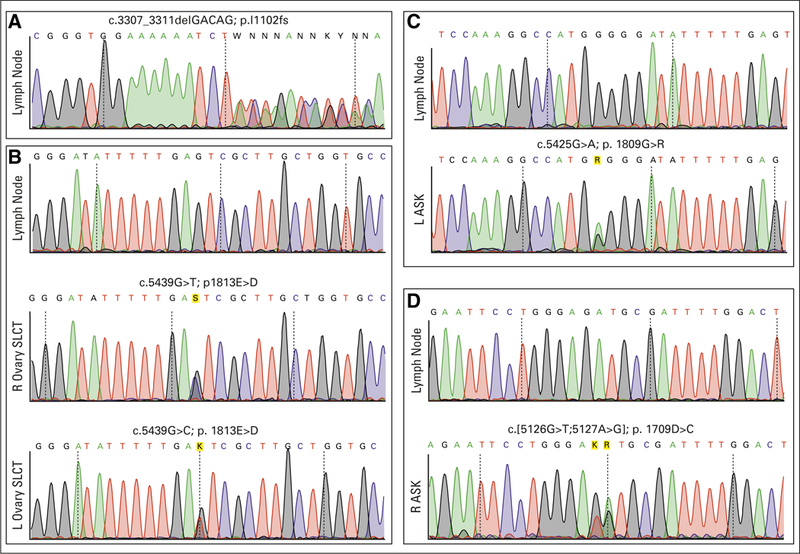
Sanger sequencing chromatograms demonstrating the patient’s (A) DICER1 germline frameshift mutation and (B) RNase Illb hotspot mutations in her ovarian Sertoli-Leydig cell tumors (SLCTs); (C) left (L) kidney tumor; and (D) right (R) kidney tumor. ASK, anaplastic sarcoma of the kidney.
Table 1.
RNase IIIb Hotspot Mutations Identified in Each Tumor Sample
| Tissue of Origin | cDNA Mutation | Amino Acid Change |
|---|---|---|
| Left kidney tumor | c.5425G>A | p.1809G>R |
| Right kidney tumor | c.5125_5126delinsTG | p.1709D>C |
| Thyroid adenoma | c.5439G>T | p.1813E>D |
| Bladder rhabdomyosarcoma | c.5439G>T | p.1813E>D |
| Left ovary SLCT | c.5439G>T | p.1813E>D |
| Right ovary SLCT | c.5439G>C | p.1813E>D |
NOTE. cDNA mutation notations are based on ENST00000526495.5; amino acid notations are based on ENSP00000437256.1.
Abbreviations: cDNA, complementary DNA; SLCT, Sertoli-Leydig cell tumor.
We then sequenced the RNase III domains of DICER1 in archival specimens from her other tumors. Her contralateral renal tumor exhibited a different hotspot mutation, c.5125_5126-delinsTG; p.D1709C (Fig 2 and Table 1). Because this change spanned two nucleotides, it was possible that the two nucleotide variants (c.G5125T and c.A5126G) occurred on separate strands. To sequence strands separately, we subcloned the PCR amplicon spanning this mutation and found that both single-nucleotide variants occurred on the same strand, making the tumor heterozygous for a single somatic hotspot mutation (Appendix Fig A1). Contrary to our clinical suspicion at the time, her contralateral kidney tumor was, in fact, a second primary.
We also sequenced DICER1 from her thyroid follicular adenoma and bladder rhabdomyosarcoma.
Both showed c.G5439T (p.E1813D) mutations (Table 1). DICER1-associated ovarian and cervical embryonal rhabdomyosarcomas frequently exhibit cartilaginous differentiation,11,25 and her bladder tumor may be a related tumor. Bladder rhabdomyosarcomas have been associated previously with DICER1 syndrome, although specific discussion of their histologic features was not reported.4
Her two ovarian tumors were detected at the same time, and it was unclear whether they represented distinct primary tumors or metastatic spread. The ovarian tumors harbored different mutations at the same position: c.G5439T and c.G5439C (Fig 2 and Table 1), indicating that they arose independently.
TP53 Status
In many tumors, DICER1 mutations are associated with TP53 inactivation,20,26,27 but clinical sequencing of our patient’s ovarian tumor did not detect any mutations in TP53. Because her phenotype was so severe, we performed targeted TP53 sequencing in her other tumor samples (Appendix Table A2). Her initial renal tumor bore a p.V274L variant, as we described previously.20 However, her remaining tumors had no additional somatic TP53 variants and did not undergo loss of heterozygosity at TP53.
DISCUSSION
Clinically, the discovery of a second tumor with similar histology can raise the question of whether it is a recurrence or a new primary tumor. The answer has important therapeutic implications; new primary tumors are treated with front-line therapy to which recurrent tumors rarely respond. In this patient, the bilateral renal and SLCTs were suspected to represent metastatic spread. The differing DICER1 mutations were identified retrospectively. Had we known that her second kidney tumor was, in fact, metachronous, she might have avoided the increased toxicity of chemotherapy regimens designed for relapsed or refractory tumors. Similarly, when she was diagnosed with bilateral ovarian tumors, we were concerned that she might need aggressive therapy for eradicating hematogenous metastases. In fact, the cartilaginous elements of her bladder tumor led us to question whether it could instead represent a distant recurrence of her renal sarcoma, although its distinct DICER1 mutation proved its independence.
Thus, this case highlights a powerful but often overlooked application of clinical tumor sequencing: better understanding of the biology behind tumor formation. We used sequencing here to show that the contralateral tumors arose independently. As sequencing costs fall, clinicians should be cognizant that tumor sequencing can help distinguish between new primary tumors and recurrences.
In this case, sequencing at initial diagnosis would have also had the added benefit of alerting clinicians to her potential DICER1 status. In the case of DICER1 syndrome, because second-hit mutations occur at such stereotyped positions, sequencing separate tumors could even occur by simple Sanger sequencing rather than by next-generation sequencing. In patients with DICER1 syndrome, dedicated tests may be developed for identifying these stereotyped mutations, perhaps including circulating tumor DNA assays as a noninvasive screening method.
This is one of the most severe cases in the literature of multiple distinct somatic DICER1 mutations arising in a patient. Germline whole-exome sequencing did not detect any other known cancer-predisposing mutations. Although we cannot rule out other secondary mutations, targeted TP53 sequencing did not reveal TP53 inactivation in her subsequent tumors. We speculate that the particular severity of her clinical course could have been related to the intense chemo- and radiotherapy she received at her initial cancer diagnoses. A few case reports have described patients with PPB/ DICER1 syndrome who developed one or more cancers after alkylating chemotherapy (Table 2). However, these cases constitute a small sample size and are likely the most extreme cases, highlighted because of publication bias. Ongoing prospective studies such as the International PPB Registry are necessary to study the long-term effects of chemotherapy and radiation in patients with DICER1.
Table 2.
Other Patients With PPB/DICER1 Syndrome Who Developed Tumors After Alkylating Chemotherapy or Radiation
| Age (years) | Tumor Type | Therapy | Somatic Mutation | First Author |
|---|---|---|---|---|
| 3 | PPB | Vincristine, pirarnbicin, cyclophosphamide, cisplatin; tandem autoBMT (ifosfamide, melphalan; then busulfan, thiotepa) | Oue28 | |
| 6 | DTC | |||
| 3 | PPB | Ifosfamide, vincristine, actinomycin, carboplatin, teniposide; autoBMT | Rome29 | |
| 7 | Cervical embryonal RMS | Ifosfamide, vincristine, actinomycin, cyclophosphamide | ||
| 12 | Bladder RMS | vincristine, actinomycin, cyclophosphamide | ||
| 16 | MNG and follicular carcinoma | |||
| 2 | PPB | Vincristine, actinomycin, cyclophosphamide, ifosfamide, etoposide, cisplatin, doxorubicin | Shin30; de Kock31 | |
| 4 | PPB relapse in muscle | Ifosfamide, carboplatin, etoposide; autoBMT (melphalan, carboplatin, etoposide) | ||
| 7 | MNG | |||
| 9 | DTC | p.1813E>D | ||
| 1 | PPB | Unspecified chemotherapy | de Kock31 | |
| 6 | Ciliary body medulloepithelioma | |||
| 7 | DTC | p.1813E>G | ||
| 2 | PPB, CN | Unspecified chemotherapy | de Kock31 | |
| 11 | DTC | p.1705E>K | ||
| 5 | Type II PPB | Vincristine, actinomycin, cyclophosphamide, doxorubicin, cisplatin | p.1810D>Y | Schultz10 |
| 8 | DTC | p.1813E>V | ||
| 13 | Nasal chondromesenchymal hamartoma | p.1813E>D | ||
| 15 | SLCT | p.1813E>D | ||
| 6 | Ovarian embryonal RMS | Vincristine, actinomycin, cyclophosphamide, ifosfamide, etoposide, XRT | p.1809G>R | de Kock11 |
| 12 | Cystic nephroma | p.1813E>D | ||
| 13 | MNG | |||
| 16 | Breast fibroadenoma | |||
| 10 | Ovarian undifferentiated sarcoma | Vincristine, actinomycin, cyclophosphamide | p.1709D>N | Schultz14 |
| 14 | SLCT | Bleomycin, etoposide, cisplatin | p.1813E>D |
Abbreviations: autoBMT, autologous bone marrow transplant; CN, cystic nephroma; DTC, differentiated thyroid carcinoma; MNG, multinodular goiter; PPB, pleuropulmonary blastoma; RMS, rhabdomyosarcoma; SLCT, Sertoli-Leydig cell tumor; XRT, radiation therapy.
Supplementary Material
Acknowledgments
Support
Supported by Grants No. P50CA196516 (J.F.A.) and 1K08CA207849 (K.S.C.) from the National Institutes of Health, by a grant from Curing Kids Cancer (J.F.A.), and by a grant from Alex’s Lemonade Stand Foundation (K.S.C.).
Appendix.
Fig A1.
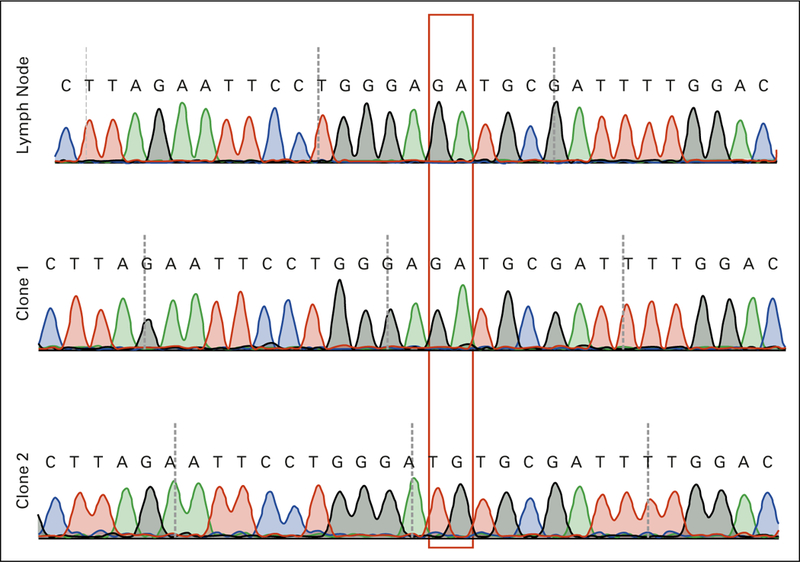
Sequence of the D1709 region in normal lymph node and in representative clones of the amplicon from the right kidney tumor, demonstrating that the c.G5126T and c.A5127G mutations are on the same DNA strand.
Appendix.
Table A1.
Primer Sequences Used For Validation
| Exon | Forward Primer | Reverse Primer |
|---|---|---|
| 21 | GATGGCCGTAATGCCTGGTA | CTTGGCCGGTGTAGCAATTT |
| 22 | ATGCCTTGAATGAATTCCAGCAGTG | TTTGGCTCACCGAAAAGTAAATCC |
| 23 | ACCTTTCAGTGAGCTGTGCT | ACAAGGCCAACACGATGAGAT |
| 24 | AAGCTTACGGTTCCACTTCG | CCTGCTGTCCCTTTAGACCA |
| 25 | CTACATCTGTGGACTGCCTGT | CAATTCTCGCACAGGGGAAC |
| 26 | TGTACTATCCCATGATGCGGC | TGACAACAGCACACCACAGT |
Appendix.
Table A2.
TP53 Variants Detected by Amplicon Sequencing
| Position (hg38) | Reference | Alternate | Affected Region |
AA Change | Variant Call (No. reference/alternate reads) | ||||
|---|---|---|---|---|---|---|---|---|---|
| Germline | Left Kidney | Right Kidney | Thyroid | Bladder | |||||
| chrl7:7673294 | T | A | UTR3 | Het (26,088/6,788) | Het (12,234/4,390) |
Het (31,962/7,128) | Het (21,770/6,568) |
Het (23,234/7,806) |
|
| chrl7:7673800 | C | G | Exonic | p.274V>L | Horn ref (25,104/0) | Het (4,214/18,666) |
Horn ref (13,554/0) |
Horn ref (27,700/0) |
Horn ref (22,112/0) |
| chrl 7:7676153 | G | C | Exonic (common SNP rsl 042 522 ) |
p.72P>R | Horn alt (0/6,310) | Horn alt (0/5,598) |
Horn alt (0/4,484) | Horn alt (14/15,398) |
Horn alt (8/7,880) |
Abbreviations: AA, amino acid; alt, alternate; het, heterozygous; hom, homozygous; ref, reference.
Footnotes
Prior presentation
Presented in part at the Spring 2014 meeting of the Society for Pediatric Pathology, Phoenix, AZ, February 28 to March 3, 2014.
AUTHORS’ DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO’s conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Kenneth S. Chen
No relationship to disclose
Sarai H. Stuart
No relationship to disclose
Emily K. Stroup
No relationship to disclose
Abhay S. Shukla
No relationship to disclose
Jason Wang
No relationship to disclose
Veena Rajaram
No relationship to disclose
Gordan Vujanic
No relationship to disclose
Tamra Slone
No relationship to disclose
Dinesh Rakheja
No relationship to disclose
James F. Amatruda
No relationship to disclose
Contributor Information
Kenneth S. Chen, University of Texas Southwestern Medical Center Children’s Health Children’s Medical Center, Dallas, TX.
Sarai H. Stuart, University of Texas Southwestern Medical Center
Emily K. Stroup, University of Texas Southwestern Medical Center
Abhay S. Shukla, University of Texas Southwestern Medical Center
Jason Wang, University of Texas Southwestern Medical Center.
Veena Rajaram, University of Texas Southwestern Medical Center.
Gordan M. Vujanic, Sidra Medical and Research Center, Doha, Qatar.
Tamra Slone, University of Texas Southwestern Medical Center; Children’s Health Children’s Medical Center, Dallas, TX.
Dinesh Rakheja, University of Texas Southwestern Medical Center.
James F. Amatruda, University of Texas Southwestern Medical Center Children’s Health Children’s Medical Center, Dallas, TX.
REFERENCES
- 1.Slade I, Bacchelli C, Davies H, et al. : DICER1 syndrome: Clarifying the diagnosis, clinical features and management implications of a pleiotropic tumour predisposition syndrome. J Med Genet 48:273–278, 2011 [DOI] [PubMed] [Google Scholar]
- 2.de Kock L, Plourde F, Carter MT, et al. : Germ-line and somatic DICER1 mutations in a pleuropulmonary blastoma. Pediatr Blood Cancer 60:2091–2092, 2013 [DOI] [PubMed] [Google Scholar]
- 3.Heravi-Moussavi A, Anglesio MS, Cheng SW, et al. : Recurrent somatic DICER1 mutations in nonepithelial ovarian cancers. N Engl J Med 366:234–242, 2012 [DOI] [PubMed] [Google Scholar]
- 4.Doros L, Yang J, Dehner L, et al. : DICER1 mutations in embryonal rhabdomyosarcomas from children with and without familial PPB-tumor predisposition syndrome. Pediatr Blood Cancer 59:558–560, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Foulkes WD, Bahubeshi A, Hamel N, et al. : Extending the phenotypes associated with DICER1 mutations. Hum Mutat 32:1381–1384, 2011 [DOI] [PubMed] [Google Scholar]
- 6.Wu MK, Sabbaghian N, Xu B, et al. : Biallelic DICER1 mutations occur in Wilms tumours. J Pathol 230:154–164, 2013 [DOI] [PubMed] [Google Scholar]
- 7.Wu MK, Goudie C, Druker H, et al. : Evolution of renal cysts to anaplastic sarcoma of kidney in a child with DICER1 syndrome. Pediatr Blood Cancer 63:1272–1275, 2016 [DOI] [PubMed] [Google Scholar]
- 8.Wu MK, Vujanic GM, Fahiminiya S, et al. : Anaplastic sarcomas of the kidney are characterized by DICER1 mutations. Mod Pathol 31:169–178, 2018 [DOI] [PubMed] [Google Scholar]
- 9.Schultz KA, Pacheco MC, Yang J, et al. : Ovarian sex cord-stromal tumors, pleuropulmonary blastoma and DICER1 mutations: A report from the International Pleuropulmonary Blastoma Registry. Gynecol Oncol 122:246–250, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schultz KA, Yang J, Doros L, et al. : DICERl-pleuropulmonary blastoma familial tumor predisposition syndrome: A unique constellation of neoplastic conditions. Pathol Case Rev 19:90–100, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.de Kock L, Druker H, Weber E, et al. : Ovarian embryonal rhabdomyosarcoma is a rare manifestation of the DICER1 syndrome. Hum Pathol 46:917–922, 2015 [DOI] [PubMed] [Google Scholar]
- 12.Wu MK, de Kock L, Conwell LS, et al. : Functional characterization of multiple DICER1 mutations in an adolescent. Endocr Relat Cancer 23:L1–L5, 2015 [DOI] [PubMed] [Google Scholar]
- 13.de Kock L, Bah I, Wu Y, et al. : Germline and somatic DICER1 mutations in a well-differentiated fetal adenocarcinoma of the lung. J Thorac Oncol 11:e31–e33, 2016 [DOI] [PubMed] [Google Scholar]
- 14.Schultz KA, Harris A, Messinger Y, et al. : Ovarian tumors related to intronic mutations in DICER1: A report from the international ovarian and testicular stromal tumor registry. Fam Cancer 15:105–110, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rutter MM, Jha P, Schultz KA, et al. : DICER1 mutations and differentiated thyroid carcinoma: Evidence of a direct association. J Clin Endocrinol Metab 101:1–5, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Durieux E, Descotes F, Mauduit C, et al. : The co-occurrence of an ovarian Sertoli-Leydig cell tumor with a thyroid carcinoma is highly suggestive of a DICER1 syndrome. Virchows Arch 468:631–636, 2016 [DOI] [PubMed] [Google Scholar]
- 17.Klein S, Lee H, Ghahremani S, et al. : Expanding the phenotype of mutations in DICER1: Mosaic missense mutations in the RNase IIIb domain of DICER1 cause GLOW syndrome. J Med Genet 51:294–302, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.de Kock L, Wang YC, Revil T, et al. : High-sensitivity sequencing reveals multi-organ somatic mosaicism causing DICER1 syndrome. J Med Genet 53:43–52, 2015 [DOI] [PubMed] [Google Scholar]
- 19.Brenneman M, Field A, Yang J, et al. : Temporal order of RNase IIIb and loss-of-function mutations during development determines phenotype in DICER1 syndrome: A unique variant of the two-hit tumor suppression model. F1000 Res 4:214, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rakheja D, Chen KS, Liu Y, et al. : Somatic mutations in DROSHA and DICER1 impair microRNA biogenesis through distinct mechanisms in Wilms tumours. Nat Commun 2:4802, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bolger AM, Lohse M, Usadel B: Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li H, Durbin R: Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26:589–595, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Garrison E, Marth G: Haplotype-based variant detection from short-read sequencing. arXiv:1207.3907v2, 2012 [Google Scholar]
- 24.Vujanić GM, Kelsey A, Perlman EJ, et al. : Anaplastic sarcoma of the kidney: A clinicopathologic study of 20 cases of a new entity with polyphenotypic features. Am J Surg Pathol 31:1459–1468, 2007 [DOI] [PubMed] [Google Scholar]
- 25.Dehner LP, Jarzembowski JA, Hill DA: Embryonal rhabdomyosarcoma of the uterine cervix: A report of 14 cases and a discussion of its unusual clinicopathological associations. Mod Pathol 25:602–614, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Seki M, Yoshida K, Shiraishi Y, et al. : Biallelic DICER1 mutations in sporadic pleuropulmonary blastoma. Cancer Res 74:2742–2749, 2014 [DOI] [PubMed] [Google Scholar]
- 27.Pugh TJ, Yu W, Yang J, et al. : Exome sequencing of pleuropulmonary blastoma reveals frequent biallelic loss of TP53 and two hits in DICER1 resulting in retention of 5p-derived miRNA hairpin loop sequences. Oncogene 33:5295–5302, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Oue T, Inoue M, Kubota A, et al. : Pediatric thyroid cancer arising after treatment for pleuropulmonary blastoma. Pediatr Blood Cancer 50:901–902, 2008 [DOI] [PubMed] [Google Scholar]
- 29.Rome A, Gentet JC, Coze C, et al. : Pediatric thyroid cancer arising as a fourth cancer in a child with pleuropulmonary blastoma. Pediatr Blood Cancer 50:1081, 2008 [DOI] [PubMed] [Google Scholar]
- 30.Shin SH, Yoon JH, Son MH, et al. : Follicular thyroid carcinoma arising after hematopoietic stem cell transplantation in a child with pleuropulmonary blastoma. Thyroid 22:547–551, 2012 [DOI] [PubMed] [Google Scholar]
- 31.de Kock L, Sabbaghian N, Soglio DB, et al. : Exploring the association Between DICER1 mutations and differentiated thyroid carcinoma. J Clin Endocrinol Metab 99:E1072–E1077, 2014 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.