

Abstract
Protein α-N-terminal methylation is catalyzed by protein N-terminal methyltransferases. Prevalent occurrence of this methylation in ribosome, myosin and histones implies its function in protein-protein interaction. Although its full spectrum of function is not outlined yet, recent discovery revealed the emerging roles of α-N-terminal methylation in protein-chromatin interaction, DNA damage repair, and chromosome segregation. Here, an overview of discovery of protein N-terminal methyltransferases and functions of α-N-terminal methylation is presented. In addition, substrate recognition, mechanism and inhibition of N-terminal methyltransferases are reviewed. Opportunities and gaps in protein α-N-terminal methylation are also discussed.
Keywords: protein methyltransferase, α-N-terminal methylation, substrate recognition, function, mechanism and inhibitors
Graphical Abstract
Protein α-N-terminal methylation mediates both protein-protein and protein-DNA interactions. Herein, we summarize the enzymes, cellular, and physiological consequences of α-N-terminal methylation. In addition, we overview the mechanism, substrate recognition, and inhibition of N-terminal methyltransferases. Opportunities and gaps in protein α-N-terminal methylation are also discussed.
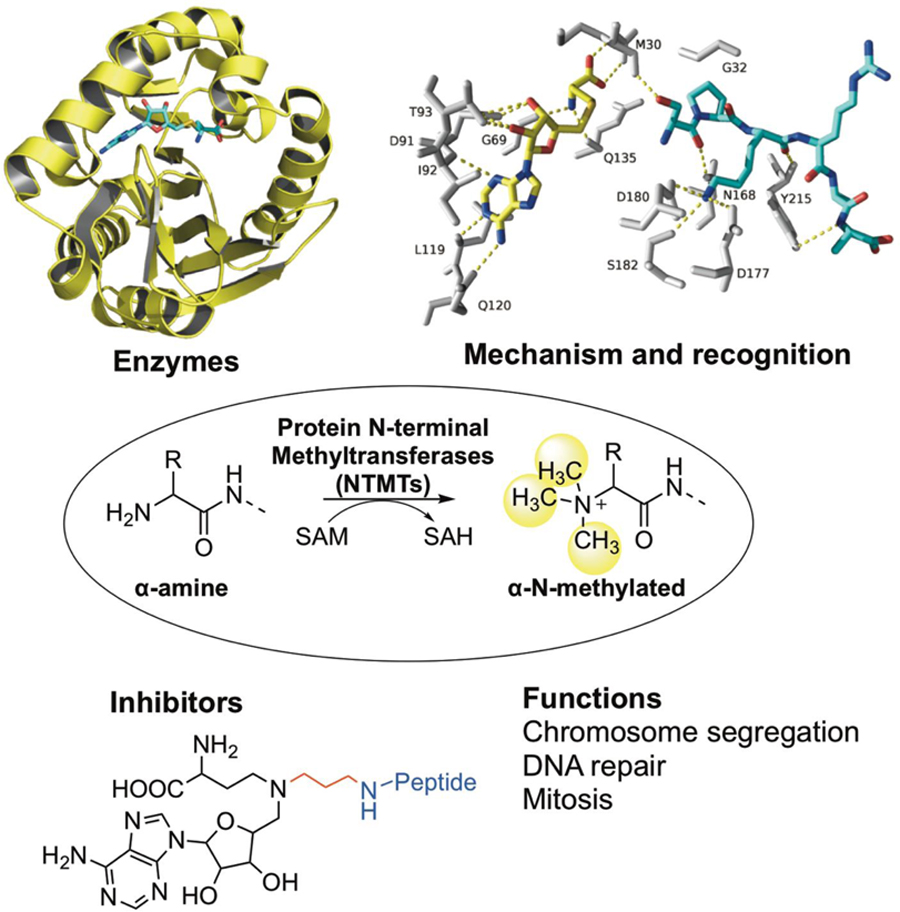
1. Early Discoveries of protein α-N-terminal methylation
Emerging epigenetic modulators have continued to reshape our understanding of eukaryotic gene expression and regulation. Lys and Arg methylation are two known epigenetic marks that play important roles in regulating chromatin dynamics and transcription activation. Protein lysine methyltransferases (PKMTs) and arginine methyltransferases (PRMTs) catalyze the covalent addition of a methyl group from S-adenosylmethionine (SAM) to the ε-amino group of Lys side chain and guanidino group of Arg side chain, respectively (Scheme 1). After the methyl group is transferred, SAM is transformed to S-adenosylhomocysteine (SAH). Unlike PKMTs and PRMTs that methylate the side chain, protein N-terminal methyltransferases (NTMTs) transfer a methyl group to the α-amino group at the protein N-terminus. Although protein α-N-terminal methylation is a known post-translational modification for over four decades, its emerging features have made it a relatively new addition to the list of players in these processes.
Scheme 1.
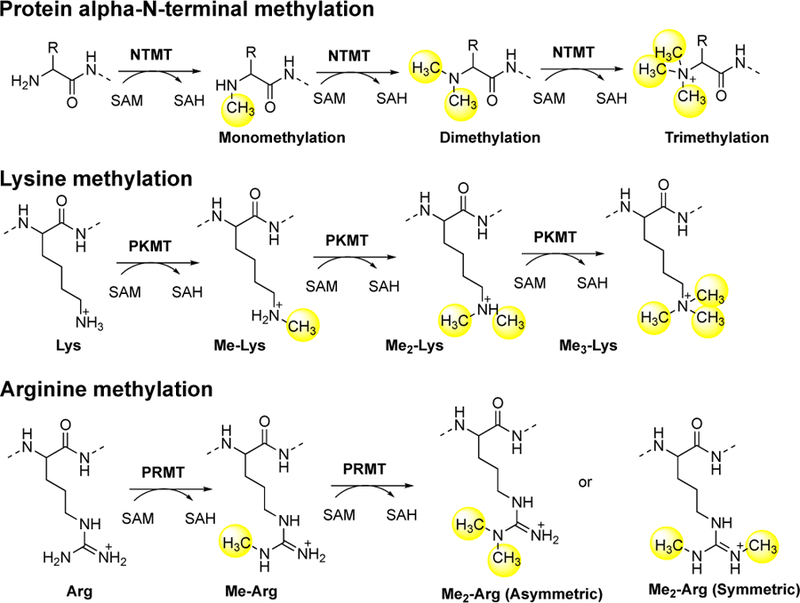
Schematic outline of protein methylation.
Protein α-N-terminal methylation was initially discovered in E. coli. on ribosomal subunits including methylalanine (MeAla) on α-N-terminus of L33 (MeAla-Lys-Gly) and S11 (MeAla-Lys-Ala), methylmethionine (MeMet) on L16 (MeMet-Leu-Gln) over four decades ago [1–3]. Since then, many cases of α-N-terminal methylation have been reported on various proteins, such as dimethylproline (Me2Pro) on Crithidia oncopelti cytochrome c and Asterias rubens histone H2B, trimethylalanine (Me3Ala) on E. coli ribosomal protein L11, Tetrahymena histone 2B, and all vertebrate striated muscle light chains[4–7]. Because the aforementioned protein substrates are components of macromolecular complexes, the α-N-terminal methylation has been inferred to mediate protein-protein interactions. In addition, eukaryotic N-terminal methylated proteins were postulated to be involved in protein degradation on the basis that methylation may interfere with N-terminal acetylation[8]. However, our knowledge about the physiological consequences of protein α-N-terminal methylation is still very limited. Recent identifications of eukaryotic protein α-N-terminal methyltransferases have prompted increasing discoveries of new protein substrates[9–13], supporting that α-N-terminal methylation is a widespread post-translational modification.
2. Discovery of protein N-terminal methyltransferases
2.1. Prokaryotic protein N-terminal methyltransferase
Protein L11 methyltransferase, PrmA is responsible for catalyzing the α-N-terminal methylation of the bacterial 50S ribosomal subunit protein L11[14,15]. It is conserved among bacteria but absent from archaea[16]. PrmA is a multifunctional methyltransferase because it is able to modify both the α-N-terminal amine and ε-amino groups of two different Lys residues[14,16]. PrmA consists of an N-terminal domain for substrate recognition, a C-terminal catalytic domain with a seven-β-strand structural fold, and a flexible linker helix (Figure 1A)[17]. Structural studies revealed a wide range of domain movements of PrmA, as exemplified by the structure of PrmA in bound with L11 in comparison with apo-form of PrmA (Figure 1B)[17]. Such conformational changes are necessary for recognition of multiple substrate sites. PrmA preferentially methylates free ribosomal protein L11 over assembled 50S ribosomal subunit, therefore, methylation of L11 may facilitate the assembly of the large subunit [16]. However, the role of L11 methylation remains a mystery since mutants and deletion of PrmA show no growth defects or any distinct phenotype in E. coli and T. thermophiles [15,16]. Investigation of those strains under different stress conditions may provide new insights on the function of N-terminal methylation of L11.
Figure 1.
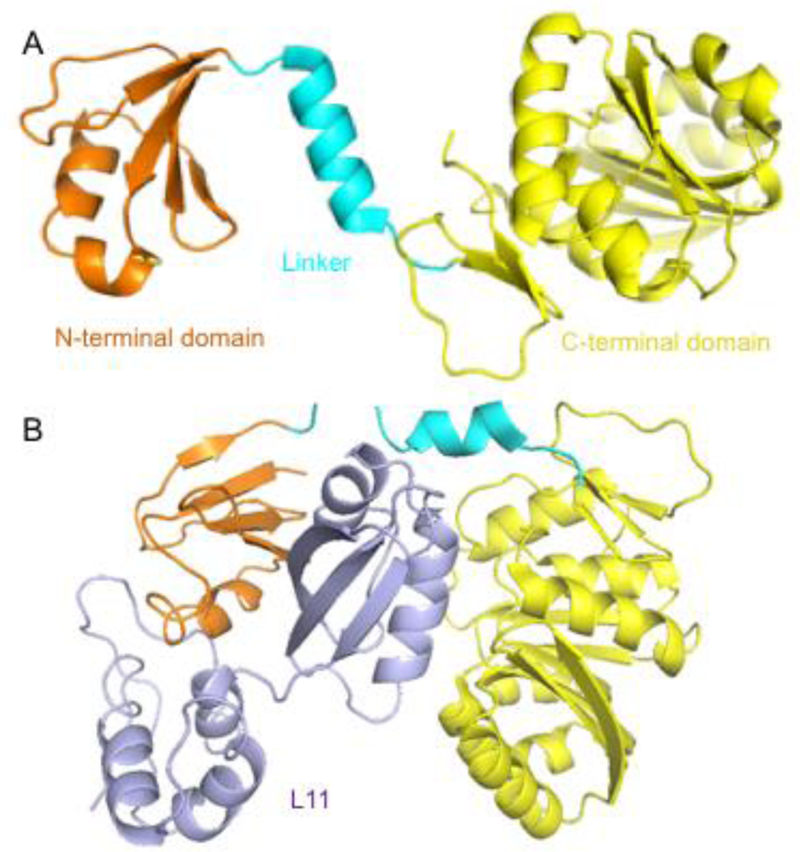
Representative crystal structures of PrmA. A) The N-terminal domain, linker, and C-terminal domain of Apo-PrmA (PDB ID:2NXC) are colored in orange, cyan, and yellow, respectively. B) PrmA-L11 complex structure (PDB ID: 2NXN) with L11 in purple.
2.2. Eukaryotic protein N-terminal methyltransferases that methylate an X-P-K/R motif
Yeast YBR261C/Tae1 protein was identified as the N-terminal methyltransferase Ntm1 in S. cerevisiae by Webb et. al. in 2010[11]. YBR261C recognizes the canonical X-P-K recognition motif and methylates ribosomal substrates, Rp112ab and Rps25a/Rps25b. Meanwhile, YBR261C is able to methylate 9-mer synthetic peptides including PPKQQLSKY that is derived from α-N-terminal Rps25a/b and A/S-PKQQLSKY with Ala or Ser replacing Pro[11]. Previous chemical genetic profile analysis indicated that deletion of YBR261C in yeast abolished N-terminal methylation, which consequently altered the ribosomal profile and led to defects in both translational efficiency and fidelity[11,18]. Overexpression of YBR261 validated its involvement in protein synthesis[18]. In addition, α-N-terminal methylation has been detected on the yeast Rpt1 (PPKEDW) subunit of the 19S regulatory particle of 26S proteasome[19]. When PK sequence at the second and third positions was deleted from Rpt1, N-terminal methylation of Rpt1 was abolished[19]. With this PK-deletion, yeast strains grow more slowly and are more sensitive to stress[19]. Despite of the implications of α-N-terminal methylation of Rpt1 in cell growth and stress tolerance in yeast[19], the molecular mechanism remains obscure. It is necessary to investigate how this methylation affects substrate recognition, ATPase activity, and the interactions of Rpt1 with other subunits of 26S proteasome, dNTMT (CG1675) is the enzyme for α-N-terminal methylation of H2B protein in Drosophila in 2012[20]. N-terminal methylation levels of H2B were increased during fly development and in the presence of cellular stress such as heat and proliferation stress[20]. dNTMT was mainly located in the nucleus where the majority of chromatin-bound H2B is methylated[19]. dNTMT recognizes the N-terminal sequence of Drosophila H2B (PPKTSG), which conforms to the canonical X-P-K recognition motif for its mammalian orthologs (X=A, P, or S). dNTMT methylation is not processive since monomethylated Pro was accumulated during the methylation reaction. A sequence search suggested about 36 proteins carrying a (M)-A/P/S-P-K recognition motif in the predicted proteome of D. melanogaster[20]. In addition, dART8, a PRMT for H3R2 methylation, negatively regulated H2B N-terminal methylation[20], suggesting a crosstalk between methylation on two histone tails.
Webb et. al. first identified the human METTL11A/NTMT1 that was encoded by METTL11A gene along with the discovery of YBR261C/TAE1[11]. Simultaneously, Schaner-Tooley et. al. isolated the same protein from HeLa cell soluble nuclear extracts and named it as N-terminal RCC1 methyltransferase (NRMT1) on account of its substrate Regulator of Chromosome Condensation 1 (RCC1)[10]. The discovery of NTMT1/NRMT1 has led to rising reports on N-terminal methylation existing in tumor suppressor retinoblastoma 1 (RB1), oncoprotein SET (also known as I2PP2A, TAF1alpha), damaged DNA-binding protein 2 (DDB2), poly(ADP-ribose) polymerase 3 (PARP3), and centromere proteins A&B (CENP-A&B) (Figure 2)[10,21–25]. Crystal structures of NTMT1 in complex with peptide substrates and SAH revealed the structural basis for preferred recognition motif X-P-K/R (X can be any amino acid except D or E)[26,27]. Substrate recognition will be discussed in more details in section 2.4. The NTMT1 gene is expressed in all tissues, and the protein is expressed in most tissues except spleen, liver and fallopian tube tissue according to the Human Protein Atlas (http://www.proteinatlas.org). Additionally, NTMT1 is overexpressed in patients’ tumor tissues including colorectal, melanoma, carcinoid, lung and liver according to ProteinAtlas (http://www.proteinatlas.org)[28]. Knockdown of NTMT1 results in mitotic defects and sensitizes etoposide and gamma irradiation in breast cancer cell lines like MCF-7 and LCC9, while NTMT1 knockout mice showed premature aging[29,30]. Both studies infer the function of NTMT1 in DNA damage repair.
Figure 2.
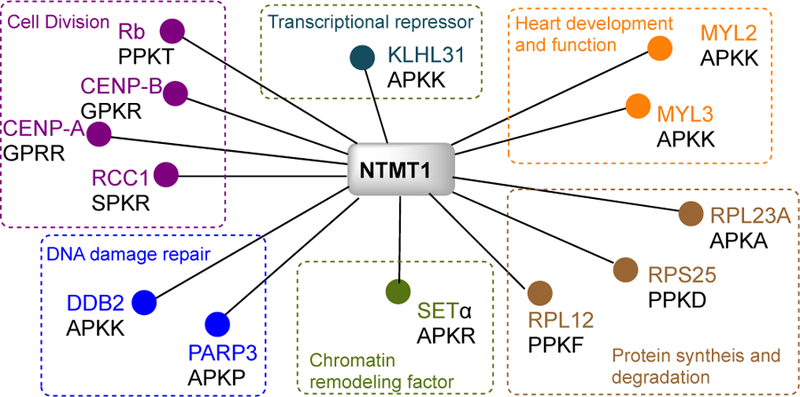
Validated substrates for NTMT1.
The NTMT1 homolog NTMT2/METTL11B shares over 50% sequence similarity with NTMT1 and was suggested as another N-terminal methyltransferase in 2010 (Figure 3A)[11]. Until 2013, NRMT2/NTMT2 was confirmed to be an N-terminal methyltransferase[9]. Both NTMT1 and 2 recognize a X-P-K/R consensus sequence, where X can be any amino acid except D or E[9–11,26,27,31]. While NTMT2 was originally proposed to be a monomethylase, recent studies indicate that it can also fully methylate both GPKRIA and PPKRIA peptides[9,31]. However, physiological substrate of NTMT2 remains to be uncovered. NTMT2 is predominantly expressed in heart and skeletal muscle tissues[28], which suggesting the possibility of its role in a tissue-specific context.
Figure 3.
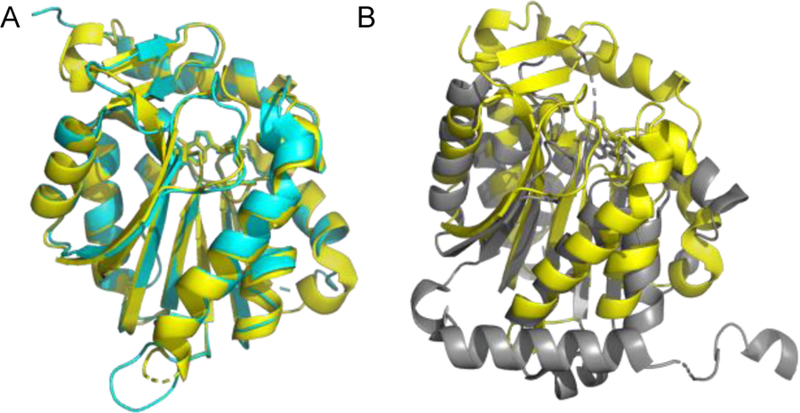
Structure alignment. A) Alignment of NTMT1 (yellow, PDB ID: 2EX4) with NTMT2 (cyan, PDB ID: 5UBB). B) Alignment of NTMT1 (yellow, PDB ID: 2EX4) with C-terminal domain of METTL13 (gray, PDB ID: 5WCJ)
YBR261C and NTMT1/2 are able to methylate 9-mer and 6-mer peptides[11,26,31–33], respectively. In addition, eukaryotic protein N-terminal methyltransferases share an X-P-K/R motif. Therefore, it is reasonable to speculate that an N-terminal linear sequence is sufficient for their recognition. Kinetic studies on protein substrates would shed lights on how other sequences contribute to recognition and methylation transfer.
2.3. Eukaryotic protein N-terminal methyltransferases that methylate elongation factor 1A
In addition to α-N-terminal methylation on the classical X-P-K/R motif in eukaryotic cells, a novel N-terminal methylation has been recently reported on eukaryotic elongation factor 1A (eEF1A) in both yeast and human[12,13]. YLR285W, also named as Elongation factor methyltransferase 7 (Efm7), is a dual methyltransferase that installs methyl groups at both N-terminal Gly1 and Lys2 residues of yeast eEF1A protein[12]. The Lys2 is methylated only after trimethylation of Gly1[12]. Yeast eEF1A starts with GKEKSHINV and is the only known substrate of Efm7, although there are 35 other yeast proteins with a G-K sequence at their N-termini[12]. Unlike NTMT1/2, Efm7 was not able to methylate the synthetic 10-mer peptide GKEKSHINVV derived from N-terminal of eEF1A[12]. Efm7 can methylate domain 1 (residues 1–238) of eEF1A but to a smaller degree of trimethylation[12]. In addition, methylation of eEF1A is increased in the presence of either GDP or GTP, which is known to bind to eEF1A and induce its conformational changes[12]. These data suggest that Efm7 substrate recognition may require the three-dimensional structure, which is different from the classic linear X-P-K/R motif recognition by other eukaryotic protein N-terminal methyltransferases.
Recently, human methyltransferase (MTase)-like protein 13 (METTL13) was identified as a dual methyltransferase for both the N-terminal Gly1 and Lys55 of human eEF1A[13]. Human eEF1A contains a very similar N-terminal sequence GKEKTHIN, with Thr substituted at the fifth position instead of Ser as seen in yeast[12]. METTL13 has two distinct MTase domains: N-terminal domain and C-terminal domain that appear to have different recognition preferences. Specifically, the C-terminal domain is able to methylate peptides derived from the first 15 amino acids of eEF1A, while the N-terminal domain is sufficient for methylation of Lys 55[13]. Structural alignment of C-terminal domain of METTL13 with NTMT1 displayed striking differences (Figure 3B). So far, eEF1A is the only reported substrate for METTL13 although 49 potential substrates have been suggested for METTL13[13]. METTL13 is also called FEAT (faint expression in normal tissues, aberrant overexpression in tumors). As the meaning of its alternative name, METTL13/FEAT is implicated in tumorigenesis in vivo by suppressing apoptosis[34]. FEAT was observed in the cytoplasm, mitochondria and nucleus of HeLa cells, as well as in the blood of cancer patients[35]. On the other hand, METTL13/FEAT protein was also implied as a tumor suppressor in bladder carcinoma by negatively regulating cell proliferation, migration and invasion in bladder cancer cells[36].
2.4. Substrate recognition
The α-N-terminal methylation has been reported on various N-terminal sequences in prokaryotic proteins. However, all identified N-terminal methylation on eukaryotic proteins is either a conserved X-P-K/R motif or specific to eEF1A (Table 1). We will focus on our discussion of substrate recognition and structure on three human N-terminal methyltransferases here.
Table 1.
Protein N-terminal Methyltransferases.
| Protein | Organism | Substrate recognition | PDB ID |
|---|---|---|---|
| PrmA | E. coli., T. thermophilus | A-K-A/G/K M-L/M/K-G/Q |
3CJT, 3CJS, 3CJR, 3EGV, 2NXC, 2NXN |
| dNTMT (CG1675) | Drosophila | X-P-K | |
| YBR261C/TAE1 | S. cerevisiae | X-P-K | |
| NTMT1/NRMT1/METTL11A | Homo sapiens | X-P-K/R | 2EX4, 5E1B, 5E1M, 5E1O, 5E1D, 5E2B, 5E2A, 5CVD, 5CVE |
| NTMT2/NRMT2/METTL11B | Homo sapiens | X-P-K/R | 5UBB, 6DUB |
| Efm7/YLR285W | S. cerevisiae | GKEKSH | |
| METTL13/FEAT | Homo sapiens | GKEKTH | 5WCJ |
Crystal structures of NTMT1-SAH-peptide substrates ternary complexes revealed that NTMT1 contains a seven-strand β sheet and five α helixes, which is a Rossmann-fold methyltransferase (Figure 3)[26,27]. In addition, NTMT1 has two unique structural components: an N-terminal extension containing three helixes and a pair of β hairpins[26]. Peptide substrates bind in a similar manner into a negatively charged channel[26]. The first three residues (X-P-K/R) inserts into a defined binding pocket, which explains the unique specificity of NTMT1 in methylation of the α-N-terminal amine versus the ε-amine of Lys[26].
Proteins starting with S/P/A/G-P-K/R have been confirmed to be methylated in vivo. Furthermore, NTMT1 is able to methylate X-P-K/R peptides (X is any amino acid except D or E) in vitro.[26] This expanded consensus implies that there are about 300 possible substrates for NTMT1. Among them, centromere H3 variants (CENP-A/B), damaged DNA-binding protein 2 (DDB2), and poly (ADP-ribose) polymerase 3 have subsequently been confirmed (Figure 2). The tolerance for the first position of the substrate is because of its backbone hydrogen bond with a Asn168 residue of NTMT1 and a spacious binding pocket surrounding the side chain of first residue X[10,11,26]. The importance of Pro2 is revealed by its stacking interaction with Trp136[11,26]. Petkowski et. al. reported that the Pro2 can be replaced by other residues including Ala, Glu, Met, Asn, Gln, Gly and Ser[37]. However, Dong et. al. demonstrated that substitution of Pro2 with Ile, Gln, Glu or Ser abolished its interaction with NTMT1[26]. Basic residues including Lys3 or Arg3 are preferred for the peptide substrate, where electrostatic interactions are formed with key residues Asp177 and Asp180 of NTMT1 (Figure 4)[10,26,37]. Mutation of D180 to lysine had abolished enzyme activity[26] Co-crystal structures also indicated that NTMT1 has no significant preference for nonmethylated or monomethylated substrates as they have the same orientation to interact with NTMT1[26]. This is consistent with the comparable kinetic parameters of NTMT1 methylation with both substrates, as well as the distributive mechanism of NTMT1 methylation[26,32,33].
Figure 4.
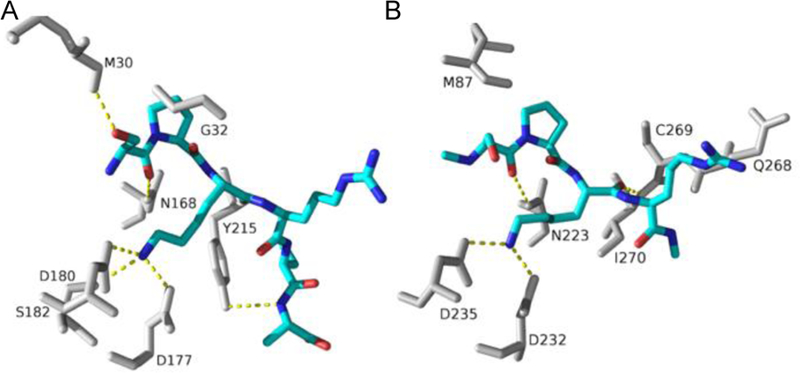
Substrate binding (SPKR, blue) in the active site with NTMT1 (A, grey, PDB ID: 5E1B) and NTMT2 (B, grey, PDB ID: 6DUB)
NTMT2 shares a comparable substrate recognition mode as NTMT1. A structural comparison of the substrate-binding region of NTMT2 with that of NTMT1 revealed a similar set of residues (Figure 4)[31]. In NTMT2, key substrate-engaging residues are Asn223, Trp191, Asp232 and Asp235, which are in agreement with Asn168, Trp136, Asp177 and Asp180 in NTMT1[26,31]. NTMT2 is able to methylate XPKRIA hexapeptides[31], although its physiological substrate has yet to be identified. However, NTMT2 exhibits a different product specificity from NTMT1. NTMT2 is able to fully methylate G/P-PKRIA, but it predominantly installs one methyl group for other X-PKRIA peptides[31]. The residue Asn89 of NTMT2 serves as a gatekeeper to govern the product specificity[31]. Besides those residues that involve with substrate binding, multiple mutations have been reported in human cancer samples (Cosmic Catalogue of Somatic Mutations in cancer; https://cancer.sanger.ac.uk/cosmic/). Among them, the N209I endometrial and P211S lung cancer mutants decreased trimethylation level of RCC1, while the Q144H lung cancer mutant increased trimethylation level of RCC1 with minimal levels of mono- and di-methylated RCC1[38]. For NTMT2, the V224L breast cancer mutant showed marginal methylation activity for methylation states[38]. Those data infer that methylation levels may play different roles in different cancers. Meanwhile, it would be worth to investigate how those mutations affect kinetic parameters of the enzyme.
The C-terminal domain of dual methyltransferase METTL13 is responsible for the α-N-terminal methylation of eEF1A[13]. Although yeast Efm7 is not able to methylation the 10-mer peptide that is derived from yeast N-terminal eEF1A, METTL13 can methylate the 15-mer peptide derived from human N-terminal eEF1A. Peptide array study suggested an N-terminal consensus sequence [G/A/P]-[K/RF/Y/Q/H]-E-[K/R/Q/I/H/L] for METTL13, implying 49 candidate substrates in human proteome[13]. However, only eEF1A was validated to be the substrate for METTL13[13]. Crystal structure of its core methyltransferase domain (residues 470–699, PDB ID 5WCJ) in complex with SAH reveals a Rossmann fold-like structure[13]. Docking study of the 6-mer peptide GKEKTH suggested a hydrogen bond between the carboxylate O atom of Gly1 and side chain of Asn647[13]. Such interaction is conserved in both NTMT1 and 2[13,26,31]. However, unique interaction of Asp577 with the α-amino group of Gly1 is required for enzymatic activity since the activity of D577A mutant decreased about half[13]. It is imperative to obtain a co-crystal structure of METTL13 in complex of eEF1A substrate to elucidate its substrate recognition and high specificity. In addition, structural information of full-length METTL13 would shed lights on how both N-terminal and C-terminal domains orient to favor the methylation on α-N-terminus and Lys55 of eEF1A, respectively. A wide range of domain movements is speculated to occur to induce conformational changes of METTL13.
3. Functions of α-N-terminal Methylation
The α-N-terminal amines (pKa = 6–8) are less basic in comparison with the side chain aliphatic amines (pKa ~10.5). Consequently, methylation of α-N-terminus alters not only the hydrophobicity and steric hindrance, but also charge state under the physiological condition. Early reported N-terminal methylated proteins such as myosin light chain LC-1, histone H2B, and cytochrome c-557 are involved in large macromolecular complexes[39]. Therefore, protein N-terminal methylation has been proposed to regulate protein-protein interactions in complex macromolecular structures. N-terminal methylation was also suggested to be involved in protein stability since methylation interplays with another predominant acetylation at the N-terminus. Additionally, Hershko et. al. demonstrated that a free α-N-amine group is required for ubiquitin-mediated protein degradation in comparison with chemical methylated protein[40]. The relevance of α-N-terminal methylation in protein-DNA interactions has been unveiled in interactions of RCC1 and CENP-A/B with chromatin, DDB2 with DNA damage foci[21,23,24,41]. In addition, the level of α-N-terminal methylation increases in response to a variety of extracellular stimuli, including increased cell density, heat shock, and arsenite treatment[20,42], suggesting its potential as a new epigenetic mark.
3.1. Functions of methylated Pro
Dimethylproline (Me2Pro) was present in the relatively free-moving N-terminal region of Crithidia oncopelti cytochrome c557 as a novel N-terminal protein modification[43]. The observation of only one CαH resonance of Me2Pro at 4.03 ppm in nuclear magnetic resonance (NMR) spectroscopy suggested that dimethylation may limit the rate of the interconversion between cis and trans conformations[44]. Thus, relatively rigid Me2Pro may yield specific folding for the interaction with other partners including proteins and DNA. The occurrence of Me2Pro was also found in starfish histone H2B. N-terminal methylation of yeast 26S proteasome subunit Rpt1 (starts with Pro-Pro-Lys) is involved in cell growth or stress tolerance to oxidant and canavanine stress[19]. Heat shock and arsenite treatments induced a rapid increase of Me2Pro of histone H2B and a shift of methylation sites of H3 in Drosophila, which correlated with chromatin remodeling and gene inactivation[45]. The N-terminal end of H2B was inferred to interact preferentially with DNA rather than histone[45], suggesting that this methylation can regulate both protein-protein and protein-DNA interactions.
3.2. Functions of methylated Ala
Accumulating evidence revealed a widespread occurrence of the α-N-trimethylalanine (Me3Ala) in a highly mobile N-terminal region (about 40 residues) of myosin alkali 1 (A1) and LC2 light chains in vertebrate striated skeletal and cardiac muscles[6]. The disappearance of the NMR signal for N-methyl protons of Me3Ala in the presence of actin implied its role in the interaction of myosin light chain with the C-terminal region of actin[6,44]. Such interaction is weakened by increased ionic strength[6,44], suggesting the involvement of electrostatic interactions in this case. Hayashibara et. al. demonstrated that the positively charged α-N-Me3Ala is critical for the actin-A1 interaction, as it lowers both the Vmax and KM of the actin-activated ATPase activity of globular head of myosin A1[46]. Such interaction results in a higher binding affinity for myosin to actin and a slower turnover rate of actin-activated ATPase activity[46]. Hence, trimethylation of Ala1 may have a suppressive effect on the mobility between actin and myosin filaments.
Meanwhile, trimethylation of Ala1 can have a positive effect on DNA damage repair. DDB2 possesses an N-terminal sequence of Ala-Pro-Lys, which undergoes α-N-methylation with trimethylation being the predominant form[23]. DDB2 recruits DDB1-CUL4A-based E3 ligase to initiate DNA repair process. The α-helical N-terminal domain (102 residue) of DDB2 is important to mediate interactions with DDB1 and damaged DNA[47]. α-N-methylated DDB2 enhances its nuclear localization, recruitment to cyclobutene pyrimidine dimers, and ATM activation, indicating the function of N-terminal methylation in UV-damaged DNA repair[23].
3.3. Functions of methylated Ser
RCC1 is pivotal in regulating nuclear transport, mitotic spindle formation, and nuclear envelope assembly through its dynamic association with chromatin[48]. Chen et. al. identified that an α-N-terminal methylation of the first Ser residue is important for RCC1 interacting with chromosomes, which is critical for mitotic spindle assembly and function[21,49]. In addition, RCC1 mutant K3Q or knockdown of NTMT1 abolished N-terminal methylation of RCC1 and led to multipolar spindle formation and mitotic defects[21]. Further studies by Hao et. al. manifested that α-N-methylation may enhance the association of RCC1 with chromatin through electrostatic interaction with DNA[50]. Hitakomate et. al. provided strong support for the importance of alpha-N-methylated tail for stable association of RCC1 with interphase chromatin[51].
3.4. Functions of methylated Gly
Human centromere plays an essential role in chromosome segregation to ensure genome stability. Trimethylation of Gly has a positive effect on the functions of both CENP-A and B proteins. CENP-A, a centromere-specific histone H3 variant, contains a N-terminal motif Gly-Pro-Arg, which is subject to α-N-methylation in cells[25]. This methylation is independent of other posttranslational modifications of CENP-A tail[25]. CENP-A replaces H3 in centromeric nucleosomes and is indispensable for recruitment and assembly of some components to the centromere and kinetochore[52]. The N-terminal tail of CENP-A was proposed to direct proper deposition of CENP-B at centromeres and stabilize its binding to centromeres through direct interaction[53]. The α-N-trimethylation has implied to be essential for CENP-A in maintaining chromosome segregation fidelity since methylation of CENP-A is required for cell survival, localization of CENP-T and CENP-I, and formation of bipolar spindle[41]. Loss of CENP-A methylation caused defects in chromosome segregation and cell death in presence of p53. Methylation in CENP-A demonstrated different effects in the absence of p53[41], suggesting the link between p53 and α-N-methylation.
CENP-B contains a N-terminal DNA-binding domain that binds specifically to a 17-bp CENP-B box in centromeric α-satellite DNA, and a C-terminal dimerization domain. Interaction of CENP-B box with DNA supports faithful chromosome segregation through direct interaction with CENP-A and CENP-C[54]. CENP-B possesses a Gly-Pro-Lys sequence at its N-terminus and it is primarily trimethylated in cells under stressed conditions. The α-N-trimethylation level of chromatin-bound CENP-B is greater and increases the binding of CENP-B to the centromeric DNA[24].
4. Mechanism and Inhibitors
Protein methyltransferases generally promote a nucleophilic substitution reaction to transfer a methyl group from cofactor SAM to their substrates. NTMT1 was proposed to follow a common SN2 reaction mechanism[26,27]. NTMT1-catalyzed methylation follows a random sequential Bi Bi mechanism, which involves a formation of a ternary complex with either substrate binding to NTMT1 first [32]. Two highly conserved Asp180 and His140 act as general bases to facilitate the deprotonation of the α-amino group of the N-terminus to attack SAM to transfer the methyl group [26]. Mutant H140A lost the catalytic activity, but retained binding affinity to the peptide substrate [26]. NTMT1 is known to be a trimethylase that catalyzes the mono-, di-, and trimethylation[32,33]. During the process of multiple methylations, the substrate can be released and rebind to NTMT1, which proceeds a distributive mechanism for multiple methylations.[32,33]
The biological significances of NTMT1 in cell mitosis, chromatin segregation, damaged DNA repair, along with its implications in cancer and aging, have stimulated the interest in discovering NTMT1 inhibitors. Potent and specific inhibitors are critical to probe the function of each individual protein. Bisubstrate analogs that simultaneously target both binding sites have been proven to be an effective strategy to obtain potent and selective inhibitors for many enzymes with two binding sites[55–57]. Because NTMT1 forms a ternary complex during its catalysis, a bisubstrate strategy has been applied to design and synthesize bisubstrate inhibitors by covalently linking a SAM analogue with a peptide substrate to mimic the transition state[58,59]. NTMT1 bisubstrate inhibitors contain three components: an N-adenosyl-L-methionine (NAM) that replaces the sulfonium ion of SAM with a nitrogen atom, a hexapeptide derived from the N-terminal sequence of NTMT1 substrate, and a linker (Scheme 2). Compound NAM-TZ-SPKRIA (IC50=0.81±0.13 μM) contains a triazole linker and incorporated an SPKRIA peptide derived from N-terminus of RCC1, while NAM-C3-GPRRRS (IC50=0.94±0.16 μM) links a GPKRRS peptide derived from CENP-A through a propylene group [58, 59]. NAM-TZ-SPKRIA showed less than 50% inhibition against protein lysine methyltransferase G9a and protein arginine methyltransferase 1 (PRMT1) at 50 μM[58]. NAM-C3-GPRRRS showed no significant inhibition at 30 μM against either G9a nor PRMT1[59]. Kinetic analysis exhibited that inhibitor NAM-TZ-SPKRIA engages with both substrate binding sites[58]. Potency of such bisubstrate inhibitors corroborate the Bi Bi mechanism of NTMT1 methylation. Despite the stability and cellular permeability of the above bisubstrate inhibitors, they lay a foundation to discover small molecule inhibitors for NTMT1.
Scheme 2.
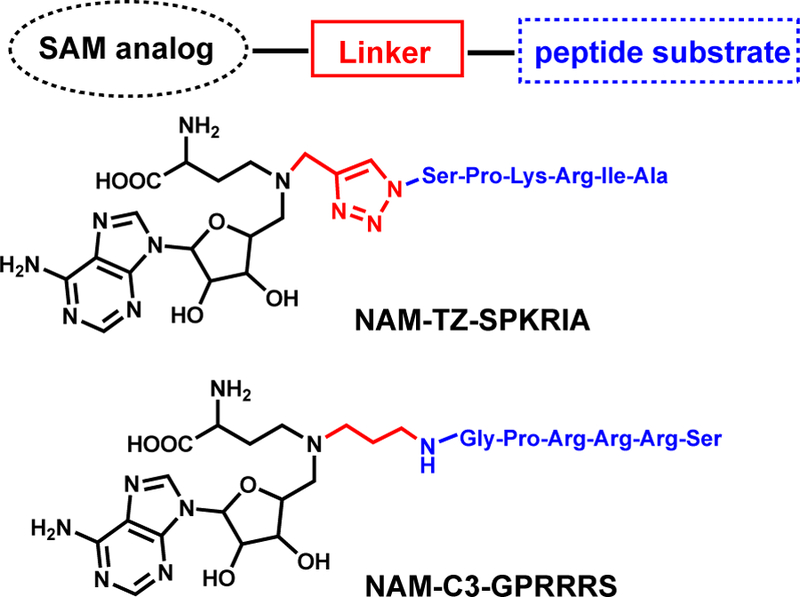
NTMT1 inhibitors.
5. Gaps and opportunities in protein α-N-terminal methylation
Evolutionary conservation of N-methylation and the recognition motif of N-terminal methyltransferase are low. It remains a puzzle whether cellular N-terminal methylation plays differing roles in different organisms or if similar functions are exerted through different pathways. Recognition motifs of centromere related human substrates of NTMT1 including RCC1 and CENP-A/B are retained in mammals but are missing in lower organisms [27]. The recognition sequence of histone 2B protein for N-terminal methylation is conserved from protozoans to insects, but not in mammals[27]. For example, human H2B is eight residues longer than Tetrahymena H2B, which masking the APKK motif.
Although an X-P-K/R motif search suggested about 300 proteins that may undergo α-N-terminal methylation, profiling a full spectrum of physiological protein substrates would shed light pathways mediated by α-N-terminal methylation. Despite increased number of reports on α-N-terminal methylation roles, functions of N-terminal methylation are still under-explored for those validated NTMT1 protein substrates including the tumor suppressor RB1 and oncoprotein SET. Meanwhile, no recognition motif has been identified to read protein N-terminal methylation. Revealing the molecular basis for recognizing α-N-terminal methylation could benefit the discovery of α-N-terminal methylation functions, as well as potential therapeutic applications involving α-N-terminal methylation in cancer and aging.
The reversibility of α-N-methylation is still under debate. Lys methylation used to be believed to be irreversible, but the discovery of histone demethylases including lysine specific demethylase 1 and jmjc domain-containing enzymes demonstrate the dynamic character of histone methylation. Therefore, it is rational to hypothesize that α-N-terminal methylation may be reversible as well. Although predominant N-terminal acetylation has led to a belief of interplay between N-terminal methylation and acetylation, the rare acetylation of X-P-K/R motif with Pro at the second position favors the likelihood of reversibility. Future studies will need to determine if α-N-terminal methylation is reversible.
Besides the specific X-P-K/R recognition motif of α-N-terminal methylation in eukaryotes, the new discovery of N-terminal methylation on eEF1A demonstrates a completely different recognition preference. This shifts our curiosity to question if there exist any extra α-N-terminal methylation sites and/or writers. In summary, identification of possible new writers, readers, and erasers would be important to illuminate the comprehensive function and regulation of the α-N-terminal methylation network. Finally, cell-potent and druglike inhibitors are in need for N-terminal methyltransferase family to enable the research community to reveal their physiological and pharmacological functions.
Acknowledgements
This work was funded by the National Institutes of Health R01GM117275 (R.H.), U01CA214649 (R.H.), and Purdue University. The author acknowledges Andy Hudmon for critical comments and Huang laboratory members for helpful feedback.
Biography

Rong Huang obtained her PhD in bioorganic chemistry with Prof. Richard F. Borch at Purdue University in 2006. After a postdoctoral training with Prof. Philip A. Cole at Johns Hopkins University, she started her independent career as an Assistant Professor of Medicinal Chemistry at Virginia Commonwealth University in 2011. Since 2017, she has been an Associate Professor of Medicinal Chemistry and Molecular Pharmacology at Purdue University. Research in Huang Lab focuses on mechanism, recognition, and inhibition of protein α-N-terminal methylation and acetylation to identify and validate novel therapeutic targets
Footnotes
Conflict of Interest
The author declares no conflict of interest.
References
- [1].Brosius J, Chen R, FEBS Lett 1976, 68, 105–109. [DOI] [PubMed] [Google Scholar]
- [2].Wittmann-Liebold B, Pannenbecker R, FEBS Lett 1976, 68, 115–118. [DOI] [PubMed] [Google Scholar]
- [3].Chen R, Brosius J, Wittmann-Liebold B, J. Mol. Biol 1977, 111, 173–181. [DOI] [PubMed] [Google Scholar]
- [4].Martinage A, Briand G, Van Dorsselaer A, Turner CH, Sautiere P, Eur. J. Biochem 1985, 147, 351–359. [DOI] [PubMed] [Google Scholar]
- [5].Nomoto M, Kyogoku Y, Iwai K. J. Biochem 1982, 92, 1675–1678. [DOI] [PubMed] [Google Scholar]
- [6].Henry GD, Trayer IP, Brewer S, Levine BA, Eur. J. Biochem 1985, 148, 75–82. [DOI] [PubMed] [Google Scholar]
- [7].Trayer IP, Trayer HR, Levine BA, Eur. J. Biochem 1987, 164, 259–266. [DOI] [PubMed] [Google Scholar]
- [8].Van Damme P, Arnesen T, Gevaert K, FEBS J 2011, 278, 3822–3834. [DOI] [PubMed] [Google Scholar]
- [9].Petkowski JJ, Bonsignore LA, Tooley JG, Wilkey DW, Merchant ML, Macara IG, Schaner Tooley CE, Biochem. J 2013, 456, 453–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Schaner Tooley CE, Petkowski JJ, Muratore-Schroeder TL, Balsbaugh JL, Shabanowitz J, Sabat M, Minor W, Hunt DF, Macara IG, Nature 2010, 466, 1125–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Webb KJ, Lipson RS, Al-Hadid Q, Whitelegge JP, Clarke SG, Biochemistry 2010, 49, 5225–5235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Hamey JJ, Winter DL, Yagoub D, Overall CM, Hart-Smith G, Wilkins MR, Mol. Cell. Proteomics 2016, 15, 164–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Jakobsson ME, Małecki JM, Halabelian L, Nilges BS, Pinto R, Kudithipudi S, Munk S, Davydova E, Zuhairi FR, Arrowsmith CH, et al. , Nat. Commun 2018, 9, 3411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Demirci H, Gregory ST, Dahlberg AE, Jogl G, Structure 2008, 16, 1059–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Vanet A, Plumberidge JA, Guerin MF, Alix JH, Mol. Microbiol 1994, 14, 947–958. [DOI] [PubMed] [Google Scholar]
- [16].Cameron DM, Gregory ST, Thompson J, Suh MJ, Limbach PA, Dahlberg AE, J. Bacteriol 2004, 186, 5819–5825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Demirci H, Gregory ST, Dahlberg AE, Jogl G, EMBO J 2007, 26, 567–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Alamgir M, Eroukova V, Jessulat M, Xu J, Golshani A, BMC Genomics 2008, 9, 583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Kimura Y, Kurata Y, Ishikawa A, Okayama A, Kamita M, Hirano H, Proteomics 2013, 13, 3167–3174. [DOI] [PubMed] [Google Scholar]
- [20].Villar-Garea A, Forne I, Vetter I, Kremmer E, Thomae A, Imhof A, Nucleic Acids Res 2012, 40, 1536–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Chen T, Muratore TL, Schaner-Tooley CE, Shabanowitz J, Hunt DF, Macara IG, Nat. Cell Biol 2007, 9, 596–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Dai X, Rulten SL, You C, Caldecott KW, Wang Y, J Proteome Res 2015, 14, 2575–2582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Cai Q, Fu L, Wang Z, Gan N, Dai X, Wang Y, J. Biol. Chem 2014, 289, 16046–16056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Dai X, Otake K, You C, Cai Q, Wang Z, Masumoto H, Wang Y, J. Proteome Res 2013, 12, 4167–4175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Bailey AO, Panchenko T, Sathyan KM, Petkowski JJ, Pai P-J, Bai DL, Russell DH, Macara IG, Shabanowitz J, Hunt DF, et al. , Proc. Natl. Acad. Sci. U. S. A 2013, 110, 11827–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Dong C, Mao Y, Tempel W, Qin S, Li L, Loppnau P, Huang R, Min J, Genes Dev 2015, 29, DOI 10.1101/gad.270611.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Wu R, Yue Y, Zheng X, Li H, Genes Dev 2015, 29, 2337–2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Uhlen M, Bjo E, Agaton C, Szigyarto CA, Amini B, Andersen E, Andersson A, Angelidou P, Asplund A, Asplund C, et al. , Mol. Cell. Proteomics 2005, 4, 1920–1932. [DOI] [PubMed] [Google Scholar]
- [29].Bonsignore LA, Butler JS, Klinge CM, Schaner Tooley CE, Oncotarget 2015, 6, 12248–12263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Bonsignore LA, Tooley JG, Van Hoose PM, Wang E, Cheng A, Cole MP, Schaner Tooley CE, Mech. Ageing Dev 2015, 146, 42–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Dong C, Dong G, Li L, Zhu L, Tempel W, Liu Y, Huang R, Min J, Commun. Biol 2018, 1, 183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Richardson SL, Mao Y, Zhang G, Hanjra P, Peterson DL, Huang R, J. Biol. Chem 2015, 290, 11601–11610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Richardson SL, Hanjra P, Zhang G, Mackie BD, Peterson DL, Huang R, Anal. Biochem 2015, 478, 59–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Liang H, Fu Z, Jiang X, Wang N, Wang F, Wang X, Zhang S, Wang Y, Yan X, Guan W, et al. , BMC Cancer 2015, 15, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Li Y, Kobayashi K, Mona MM, Satomi C, Okano S, Inoue H, Tani K, Takahashi A, J. Transl. Med 2016, 14, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Zhang Z, Zhang G, Kong C, Zhan B, Dong X, Man X, Sci. Rep 2016, 6, 19261. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [37].Petkowski JJ, Schaner Tooley CE, Anderson LC, a Shumilin I, Balsbaugh JL, Hunt DF, Minor W, Macara IG, Biochemistry 2012, 51, 5942–5950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Shields KM, Tooley JG, Petkowski JJ, Wilkey DW, Garbett NC, Merchant ML, Cheng A, Schaner Tooley CE, Protein Sci 2017, 26, 1639–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Stock A, Clarke S, Clarke C, Stock J, FEBS Lett 1987, 220, 8–14. [DOI] [PubMed] [Google Scholar]
- [40].Hershko A, Heller H, Eytan E, Kaklij G, Rose IA, Proc. Natl. Acad. Sci. U. S. A 1984, 81, 7021–7025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Sathyan KM, Fachinetti D, Foltz DR, Nat. Commun 2017, 8, 14678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Dai X, Otake K, You C, Cai Q, Wang Z, Masumoto H, Wang Y, J. Proteome Res 2013, 12, 4167–4175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Pettigrew GW, Smith GM, Nature 1977, 265, 661–662. [DOI] [PubMed] [Google Scholar]
- [44].Smith GM, Pettigrew GW, Eur. J. Biochem 1980, 110, 123–130. [DOI] [PubMed] [Google Scholar]
- [45].Desrosiers R, Tanguay RM, J. Biol. Chem 1988, 263, 4686–4692. [PubMed] [Google Scholar]
- [46].Hayashibara T, Miyanishi T, Biochemistry 1994, 33, 12821–12827. [DOI] [PubMed] [Google Scholar]
- [47].Yeh JI, Levine AS, Du S, Chinte U, Ghodke H, Wang H, Shi H, Hsieh CL, Conway JF, Van Houten B, et al. , Proc. Natl. Acad. Sci 2012, 109, E2737–E2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Renault L, Kuhlmann J, Henkel A, Wittinghofer A, Cell 2001, 105, 245–255. [DOI] [PubMed] [Google Scholar]
- [49].Clarke PR, Nat. Cell Biol 2007, 9, 485–487. [DOI] [PubMed] [Google Scholar]
- [50].Hao Y, Macara IG, J. Cell Biol 2008, 182, 827–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Hitakomate E, Hood FE, Sanderson HS, Clarke PR, BMC Cell Biol 2010, 11, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Shuaib M, Ouararhni K, Dimitrov S, Hamiche A, Proc. Natl. Acad. Sci 2010, 107, 1349–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Fachinetti D, Diego Folco H, Nechemia-Arbely Y, Valente LP, Nguyen K, Wong AJ, Zhu Q, Holland AJ, Desai A, Jansen LET, et al. , Nat. Cell Biol 2013, 15, 1056–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Fachinetti D, Han JS, McMahon MA, Ly P, Abdullah A, Wong AJ, Cleveland DW, Dev. Cell 2015, 33, 314–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Dowden J, Hong W, V Parry R, Pike RA, Ward SG, Bioorg. Med. Chem. Lett 2010, 20, 2103–2105. [DOI] [PubMed] [Google Scholar]
- [56].Osborne T, Weller Roska RL, Rajski SR, Thompson PR, J. Am. Chem. Soc 2008, 130, 4574–4575. [DOI] [PubMed] [Google Scholar]
- [57].Foyn H, Jones JE, Lewallen D, Narawane R, Varhaug JE, Thompson PR, Arnesen T, ACS Chem. Biol 2013, 8, 1121–1127. [DOI] [PubMed] [Google Scholar]
- [58].Zhang G, Richardson SL, Mao Y, Huang R, Org. Biomol. Chem 2015, 13, 4149–4154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Zhang G, Huang R, RSC Adv 2016, 6, 6768–6771. [DOI] [PMC free article] [PubMed] [Google Scholar]