

Abstract
Human liver pyruvate kinase (hLPYK) converts phosphoenolpyruvate to pyruvate in the final step of glycolysis. hLPYK is allosterically activated by fructose-1,6-bisphosphate (Fru-1,6-BP). The allosteric site, defined by previous structural studies, is located in domain C between the phosphate binding loop (residues 444–449) and the allosteric loop (residues 527–533). In this study, we solved the X-ray crystal structures of four hLPYK variants to make structural correlations with existing functional data. The variants are D499N, W527H, Δ529/S531G (called GGG herein), and S531E. Our results reveal a conformational toggle between the open and closed positions of the allosteric loop. In the absence of Fru-1,6-BP, the open position is stabilized, in part, by a cation-π bond between W527 and R538’ (from an adjacent monomer). In S531E, glutamate binds in place of the 6’-phosphate of Fru-1,6-BP in the allosteric site leading to partial allosteric activation. Finally, our D499N structure does not provide structural evidence for the previously observed allosteric activation of the D499N variant.
Keywords: Pyruvate Kinase, Allosterism, Human Liver Isozyme
1. Introduction
Human liver pyruvate kinase (hLPYK) catalyzes the conversion of phosphoenolpyruvate (PEP) to pyruvate through a phosphoryl transfer from PEP to ADP generating pyruvate and ATP. In human liver, this penultimate step of glycolysis is allosterically regulated by fructose-1,6-bisphosphate (Fru-1,6-BP), an earlier intermediate of glycolysis. Like many other allosteric systems, the structural changes that constitute the allosteric mechanism and give rise to altered function (i.e., a change in ligand affinity or catalysis(Carlson & Fenton, 2016)) are not well defined for hLPYK.
The binding position of Fru-1,6-BP in the pyruvate kinase allosteric site was first identified in yeast PYK (Jurica et al., 1998) and later in human erythrocyte PYK (Valentini et al., 2002). Previously, two hLPYK structures were solved (PDB: 4IMA and 4IP7), both with Fru-1,6-BP bound in the allosteric site (Holyoak et al., 2013). In a subsequent study (Ishwar et al., 2015), binding and allosteric activation were assayed for a Fru-1,6-BP effector analogue series and a mutational series that targeted residues surrounding the allosteric site including D499N, W527H, GGG and S531E (Figure 1). Analysis of these data led to several hypotheses. 1) D499 regulates allosteric coupling across a tetramerization interface between the adjacent C domain subunits (the C-C interface), possibly through interactions with W527 or R528 of the allosteric loop (residues 527–533) when Fru-1,6-BP is absent. 2) The increase in PEP affinity observed for D499N and W527H in the absence of Fru-1,6-BP is due to a disruption of allosteric coupling across the C-C interface. 3) The 6’-phosphate of Fru-1,6-BP contributes to binding by interacting with the phosphate binding loop (residues 444–449 between β 1of sheet D and α 22). A glutamate mutation to the allosteric loop serine (S531E) was found to mimic allosteric activation (i.e, increase PEP affinity) in the absence of Fru-1,6-BP (Ishwar et al., 2015). It was proposed that this gain-of-function was due to the S531E glutamate binding to the 444–449 loop in place of the 6’ phosphate of Fru-1,6-BP.
Figure 1. hLPYK Structural Overview.
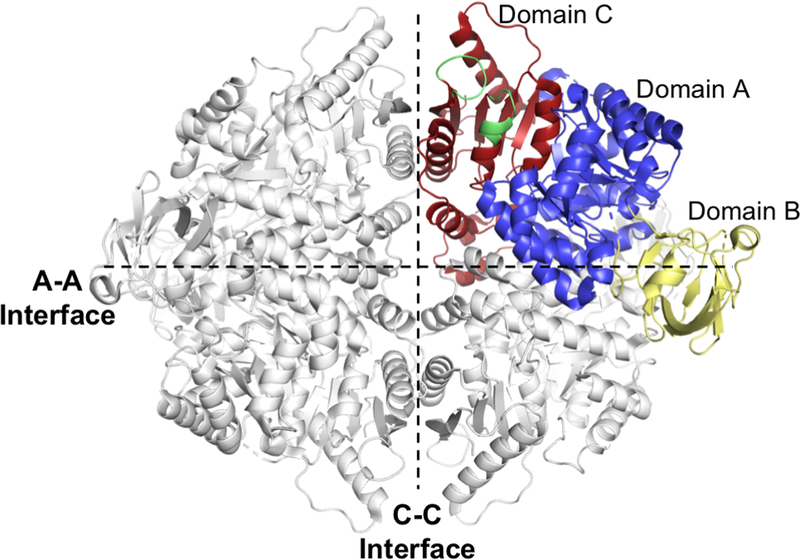
Model of hLPYK (PDB: 4IP7). Each monomer of the homotetramer is composed of three domains (A, B and C). The B domain interacts with the A domain of the same subunit. The A-A interface is formed between A domains from adjacent monomers. The C-C interface is formed by adjacent C domains. The allosteric sites are positioned along the C-C interface (shown in green for one monomer).
In this study, we examine the structural basis for the above hypotheses, derived from the previously published allosteric activity data (Ishwar et al., 2015), by solving four X-ray crystal structures of hLPYK variants: D499N, W527H, S531E, and GGG (P529 deletion and S531G substitution resulting in glycine in each position from 530–532). This focus allows a direct comparison of different conformations of the allosteric site in a single PYK isozyme and offers a contrast to past studies that compared structures of different isozymes, including human muscle pyruvate kinase M2PYK and M1PYK (Dombrauckas et al., 2005, Morgan et al., 2013). Our functional characterizations show that D499N and W527H both served to mimic allosteric activation by Fru-1,6-BP and were included in this structural analysis to evaluate the previously proposed interaction between D499 and W527’ or R528’ (the (‘) symbol is used herein to designate a residue from an adjacent monomer). In contrast, the GGG variant demonstrated a lack of functional response to Fru-1,6-BP and GGG was generated for this structural study to test the effect of a shortened, more flexible allosteric loop with a reduced propensity to make hydrogen bonds with bound Fru-1,6-BP. The S531E mutation mimics of Fru-1,6-BP activation in functional assays. Here, the S531E structure was solved to test the hypothesis that glutamate, substituted at position 531, is able to bind within the allosteric site in place of Fru-1,6-BP, leading to partial activation. The results from this structural study build on the previous functional characterizations of mutants to reveal conformational changes that occur in the allosteric loop in the presence and absence of Fru-1,6-BP, confirm the proposed position of glutamate for the S531E variant and cause a revision of the proposed hypothesis regarding D499. The presence of a cation-π interaction between W527 and R538’ across the C-C interface was observed, but an interaction between D499’ and W527 or R528 was not.
2. Methods
2.1. Preparation of overexpression plasmids
The wild type human liver pyk gene (UniProt ID# P30613) was codon optimized for expression in Escherichia coli and purchased from Genscript in a pUC57 vector with a 5’ NdeI restriction site and a 3’ XhoI restriction site. The gene was digested and inserted into a p15TV-L vector (Addgene #26093) using NdeI and XhoI restriction enzymes. The resultant plasmid encodes the wild type human liver pyk gene with an N-terminal hexahistidine tag that can be cleaved with TEV protease. The hLPYK variants were produced by QuikChange mutagenesis (Agilent) using wild type hLPYK plasmid as the template with forward primers; S531E (5’ gttaccggttggcgtccgggtgaaggctataccaacatc), D499N (5’ gatctgggcggacgatgttaaccgtcgtgtg), GGG (5’ ggttaccggttggcgtggtggcggctata), and W527H (5’ tggtgatcgtggttaccggtcaccgtccgggtagc) and the complementary reverse primers. The variant plasmids were sequenced by ACGT Inc. and contained only the desired mutation. All plasmids were transformed into BL21 Star (DE3) E. coli cells (Invitrogen) for protein overexpression.
2.2. Protein overexpression and purification
BL21 Star (DE3) E. coli containing the hLPYK variant expression plasmid were grown in LB medium containing 200 μg/mL ampicillin at 37 °C with shaking (225 rpm) until an OD600 of ~1.0 was reached. The cultures were induced with 0.2 mM isopropyl β-D-thiogalactopyranoside and the temperature was reduced to 15 °C for 20 h. The cells were harvested by centrifugation (6000g, 10 min, 4 °C). The cell pellet was resuspended in 15 mL of 50 mM Tris-HCl pH 8.5, 500 mM NaCl, 50 mM imidazole, 10 mM KCl, 5 mM MgCl2 (buffer A) per liter of culture. Cells were disrupted by use of a French pressure cell (35 000 psi), and cellular debris was removed by centrifugation (12000g, 30 min, 4 °C). The supernatant was applied to a chelating Sepharose fast-flow column (Amersham Biosciences) charged with nickel chloride and pre-equilibrated in buffer A. hLPYK eluted at ~150 mM imidazole in a linear gradient of 50–500 mM imidazole in buffer A. The pooled fractions were applied to a Superdex 200 size-exclusion column (Amersham Biosciences) equilibrated with 50 mM 2-(N-morpholino) ethanesulfonic acid (MES) pH 6.8, 100 mM KCl, 10% glycerol. The fractions containing hLPYK were pooled and dithiothreitol (DTT) was added to a final concentration of 2 mM, concentrated by use of an Amicon stirred cell with a YM-30 membrane to ~5 mg/mL as determined by Bradford assay and stored at −80 °C.
2.3. Protein crystallization
All proteins were crystallized with their N-terminal hexahistidine-tag intact. All crystals were grown in hanging drops composed of 1.5 μl of protein and 1.5 μl of well solution (described below for each variant) at 24 °C. The protein buffer was 50 mM MES pH 6.8, 100 mM potassium chloride, 10% glycerol and 2 mM DTT. D499N, W527H and S531E crystals grew as rectangular prisms within two days and reached full size within two weeks, whereas GGG crystals grew as rectangular plates. Crystals were transferred into well solution supplemented with 15% (GGG), 20% (D499N) or 25% (W527H) ethylene glycol, or 24% PEG 3350 and 20% ethylene glycol (S531E) as a cryoprotectant and flash-cooled in liquid nitrogen prior to data collection.
D499N crystals were grown using 3.6 mg/ml purified protein preincubated with a final concentration of 10 mM pyruvate in a well solution of 0.2 M ammonium citrate dibasic and 20% PEG 3350. W527H crystals were grown using 6.0 mg/ml protein preincubated with a final concentration of 10 mM pyruvate in a well solution of 0.325 M ammonium citrate dibasic and 16% PEG 3350. GGG crystals were grown using 2.0 mg/ml protein in a well solution of 0.2 M ammonium citrate pH 6.0 and 24% PEG 3350. S531E crystals were grown using 2.0 mg/ml protein in a well solution of 0.2 M ammonium citrate pH 5.6 and 16% PEG 3350.
2.4. Data collection and structure determination
Diffraction data were collected remotely using BluIce (McPhillips et al., 2002) on beamlines 12–2 (D499N) and 9–2 (W527H, GGG and S531E) at the Stanford Synchrotron Radiation Lightsource (SSRL, Menlo Park, CA). All data sets were collected at a wavelength of 0.97946 Å with 0.15° oscillation and 0.2 s exposure at a temperature of 100 K. For D499N, 110° of data were collected at a detector distance of 416 mm and processed to 2.15 Å in XDS (Kabsch, 2010). A phasing solution was determined by molecular replacement in Phaser (McCoy et al., 2007) using the A and C domains of chain A of the S12D variant of hLPYK (PDB: 4IP7) as a rigid-body model with a resulting LLG of 21,178 and TFZ of 18.6. For W527H, 180° of data were collected at a detector distance of 350 mm and processed to 2.26 Å in AutoPROC (Vonrhein et al., 2011) using ellipsoidal resolution cut-off to account for the observed anisotropy. Ellipsoidal completeness in the outer shell exceeds 98% at 2.26 Å. Spherical completeness in the outer shell exceeds 98% at 2.55 Å. Following PDB guidelines we have deposited the merged intensities from Aimless (Evans & Murshudov, 2013) prior to resolution cut-off to allow unbiased reprocessing of the W527H data. Molecular replacement as described above resulted in an LLG of 17,337 and TFZ of 66.6. For GGG, which has a spacegroup of P1, 360° of data were collected at a detector distance of 385 mm and processed to 2.32 Å in XDS. The data set was 89.4% complete overall, but we have not been able to grow additional diffraction quality GGG crystals to improve data completeness. Molecular replacement resulted in an LLG of 30,905 and TFZ of 88.0. For S531E, 360° of data were collected at a detector distance of 430 mm and processed to 2.42 Å in XDS. Molecular replacement resulted in an LLG of 28,664 and TFZ of 82.8.
For each structure, rounds of model building and refinement were completed in Coot (Emsley et al., 2010)and phenix.refine (Adams et al., 2010) and waters were placed by phenix.refine, corrected manually and verified, using a 2mFo-DFc electron density map contoured at 1.5 σ, following a round of refinement. For the D499N variant, density for the allosteric site Fru-1,6-BP was visible following molecular replacement, but was not modeled until after refining the polypeptide backbone. Statistics for data collection and refinement are listed in Tables 3 and 4.
Table 3.
Structure solution and refinement – hLPYK-D499N
Values for the outer shell are given in parentheses.
| D499N | W527H | GGG | S531E | |
|---|---|---|---|---|
| Resolution range (Å) | 39.44–2.15 (2.20–2.15) | 54.18–2.26 (2.28–2.26) | 39.06–2.32 (2.38–2.32) | 39.54–2.42 (2.48–2.42) |
| Completeness (%) | 98.5 | 90.4 (ellipsoidal) | 89.4 | 98.7 |
| No. of reflections, working set | 147251 (10216) | 84583 (1469) | 175816 (11360) | 173222 (11316) |
| No. of reflections, test set | 1982 (134) | 4187 (78) | 2000 (130) | 2006 (141) |
| Final Rcryst | 0.230 (0.3329) | 0.225 (0.256) | 0.191 (0.249) | 0.208 (0.267) |
| Final Rfree | 0.253 (0.4128) | 0.264 (0.336) | 0.251 (0.305) | 0.260 (0.361) |
| No. of non-H atoms | 13236 | 13016 | 29007 | 28375 |
| Protein | 12735 | 12811 | 28473 | 27957 |
| Ligand | 148 | 112 | 152 | 168 |
| Water | 353 | 195 | 382 | 250 |
| R.m.s. deviations | 1.142 | |||
| Bonds (Å) | 0.011 | 0.012 | 0.012 | 0.013 |
| Angles (°) | 1.142 | 1.268 | 1.252 | 1.318 |
| Average B factors (Å2) | ||||
| Protein | 40.6 | 36.7 | 44.76 | 55.42 |
| Ramachandran plot | ||||
| Most favoured (%) | 97.13 | 98.05 | 96.65 | 95.23 |
| Allowed (%) | 2.5 | 1.64 | 2.97 | 3.88 |
Table 4.
Model components
| Protein | PDB Code |
ASUa | Ordered Residues by Chain |
Waters | Ligandsb |
|---|---|---|---|---|---|
| D499N | CNN4 | 4 | A: 23–111, 117–130, 229–543 B: 24–110, 117–130, 231–543 C: 15–110, 117–130, 233–529, 532–543 D: 24–110, 117–133, 233–530, 532–543 |
353 | 4 fru-1,6-BP 4 phosphoenol pyruvate 7 ethylene glycol |
| W527H | CNN5 | 4 | A: 18–110, 117–129, 233–543 B: 23–110, 117–133, 232–543 C: 26–111, 116–129, 230–542 D: 23–111, 114–129, 229–543 |
195 | 10 ethylene glycol 12 glycerol |
| GGG | CNN7 | 8 | A: 16–137, 142–201, 205–527, 533–543 B: 26–132, 232–527, 533–543 C: 27–114, 117–543 D: 23–134, 142–148, 153–158, 163–176, 179–181, 186–195, 198–200, 211–224, 233–543 E: 25–543 F: 25–111, 116–131, 233–525, 534–543 G:15–111, 117–135, 145–202, 205–213, 218–224, 233–527, 533–543 H: 27–110, 113–129, 234–490, 495–525, 534–542 |
382 | 2 citrate 17 ethylene glycol 14 glycerol |
| S531E | CNN8 | 8 | A: 20–133, 144–155, 165–543 B: 23–138, 141–542 C: 18–135, 143–156, 166–200, 205–225, 233–543 D: 13–89, 92–129, 238–543 E: 23–137, 144–490, 494–529, 533–543 F: 22–110, 117–131, 230–529, 531–543 G: 20–89, 94–112, 116–130, 250–529, 533–543 H: 24–89, 92–110, 116–133, 236–243, 247–529, 531–543 |
250 | 42 ethylene glycol |
number of monomers in the asymmetric unit
ethylene glycol was used as a cryoprotectant, citrate was in the well solution and glycerol was in the protein buffer
2.5. Structure validation and model analysis
Summary data for the models, including residues and ligands built, are provided in Table 5. The PYK B domain (residues 130–230) interrupts the A domain. The B domain assumes multiple positions forming limited contact with the A domain, and thus resolution of the B domain is dependent on crystal packing. The B domain was modeled only if the residues connecting it to the A domain were largely continuous allowing accurate model building. As a result, unmodeled partial density exists for some B domains. The B domains were not modeled for D499N and W527H, though broken density is present, contributing to higher Rwork and Rfree values for these structures. Geometry analysis was performed by MolProbity (Chen et al., 2010). Residue T340, in the active site, is a consistent Ramachandran outlier that results from a hydrogen bond between the T340 carbonyl oxygen and the R306 side chain. T340 is also an outlier in the previously published hLPYK structures (Holyoak et al., 2013) and has excellent density in each variant structure presented here. Rotameric outliers were found at 2.4% (GGG), 1.5% (W527H), 1.43% (D499N) and 0.07% (S531E) of all residues.
Comparisons of structures and calculations of rmsd values were performed using PDBeFold (Krissinel & Henrick, 2004). Structure figures were generated in PyMOL (PyMOL Molecular Graphics System, version 2.0, Schrödinger, LLC). The atomic coordinates and structure factors have been deposited in the Protein Data Bank with accession codes 6NN4 (D499N), 6NN5 (W527H), 6NN7 (GGG) and 6NN8 (S531E).
3. Results
Functional characterizations of all variants used in the current structural study have previously been reported (Ishwar et al., 2015). X-ray diffraction data were collected to determine the crystal structures for the D499N, W527H, GGG and S531E variants of human liver pyruvate kinase to evaluate structural determinants for allosteric loop motions in the presence and absence of Fru-1,6-BP. Each variant was generated through site-directed mutagenesis of the wild type hLPYK sequence. D499N, W527H and S531E are single position substitutions that functionally mimic the activation (i.e., increased PEP affinity) caused by Fru-1,6-BP (Ishwar et al., 2015). GGG contains both a deletion of proline at position 529 and a substitution of serine with glycine at position 531, thus shortening the allosteric loop by one residue. In the above structures, differences are evident only for the region surrounding the Fru-1,6-BP allosteric site and primarily involve the allosteric loop (residues 527–533).
3.1. Overall structure
D499N and W527H crystallized in P21212 with 4 monomers in the asymmetric unit arranged as a tetramer. GGG and S531E crystallized in P1 and P21, respectively, each with 8 monomers, forming two tetramers, within the asymmetric unit. For the tetramers, the calculated surface areas for the interface between adjacent A domains (A-A interface) range from 2502 to 2749 Å2 and interface surface areas between adjacent C domains (C-C interface) range from 1421 to 1691 Å2 across the four structures, as calculated by PDBePISA (Krissinel & Henrick, 2007). This is consistent with previously published hLPYK variants C436M (PDB: 4IMA) and S12D (PDB: 4IP7) (Figure 1, Figure S1). PDBeFold (Krissinel & Henrick, 2004) was used for comparisons of overall structural homology. Alignments between structures were made for each combination of monomer of the variants presented here and with C436M (Table S1). All structural alignments show high homology. Rmsd values ranged from 0.27 to 0.69 Å for the AC domains. For comparisons that included the B domains for GGG (Chains A, C, D, E, G) and S531E (Chains A, B, C, E) rmsd values ranged as high as 2.23 Å. This was due to the varied conformations of the B domains. For example, the 2.23 Å rmsd value is from a comparison between GGG and C436M in which the GGG B domain is rotated 26° relative to the C436M B domain according to DynDom (Girdlestone & Hayward, 2016). When the B domains are removed from the calculation, AC domain alignments drop to below 0.6 Å rmsd. Alignments of the tetrameric form of each variant were performed by aligning Cα carbons across the A and C domains, but not the B domain (residue 130–230), with the combinatorial extension algorithm in PyMOL. These alignments also have low rmsd values ranging from 0.47–1.73 Å. The higher rmsd values (> 1.0 Å) are found between variants crystallizing in different space groups (Figure S1, Table S2). Thus, the structures of all variants generated share very similar global conformations with the exception of the B domains that are conformationally flexible except when stabilized by crystallographic contacts. This high homology leads us to conclude that the global structural changes that are likely contributors to the allosteric mechanism are not represented in these structures, likely due to the low pH of the crystallization conditions (Fenton & Alontaga, 2009). Attempts were made to find a crystallization condition at a pH value above 7, but without success and efforts are ongoing. Thus, we restrict our attention to the observed local changes in the allosteric loop within the allosteric site.
3.2. The allosteric site of D499N
The D499N structure was determined to evaluate a structural basis for the recently proposed mechanism for the increased PEP affinity observed for D499N. It was hypothesized that in the absence of Fru-1,6-BP, D499 interacts across the C-C interface with W527’ and possibly R528’ (Ishwar and Fenton, 2015). This hypothesis was supported by the M2PYK structure (PDB: 4FXJ) in the absence of Fru-1,6-BP in which W515 forms a 2.8 Å hydrogen bond with D487’ across the C-C interface (structurally equivalent to W527 and D499 in hLPYK) (Morgan et al., 2013). Thus, the D499N variant could prevent this interaction, such that the allosteric loop becomes less constrained in the absence of Fru-1,6-BP when compared to the wild type protein.
D499N was purified and crystallized in the absence of added Fru-1,6-BP; however, clear density corresponding to Fru-1,6-BP in the allosteric site for all four chains was visible following molecular replacement. The position of Fru-1,6-BP matches that found in both S12D and C436M structures (mutations that were included in previous structures in attempts to evaluate how the N-terminus of hLPYK interacts with the main body of the protein (Holyoak et al., 2013)) that were co-crystallized with added Fru-1,6-BP. The binding site is located between two loops, the phosphate binding loop (residues 444–449) and the allosteric loop (residues 527–533). The Fru-1,6-BP 6’-phosphate forms hydrogen bonds with the hydroxyl groups of T444, T446 and S449 from the phosphate binding loop as well as S531 from the allosteric loop. The 1’-phosphate forms an electrostatic interaction with R501. Finally, the C3, C4 and C5 hydroxyl groups of Fru-1,6-BP hydrogen bond with backbone amide and carbonyl groups of residues R528, G530 and Y533 in the allosteric loop (Figure 2). To form these interactions, the allosteric loop must adopt a closed position to bring the residues in proximity to Fru-1,6-BP. In this position, the Y533, R528 and W527 side chains pack roughly parallel to each other and are directed out into solvent (Figure 3A). We find that D499N is in a position very similar to D499 in the S12D and C436M structures. It is ~13 Å distant from R528’ in the allosteric loop and 4.3 Å, on average, from R538 within the same subunit (Figure 3A). R538 forms a cation-π stacking interaction with the open form of the allosteric loop as discussed below.
Figure 2. D499N allosteric site.
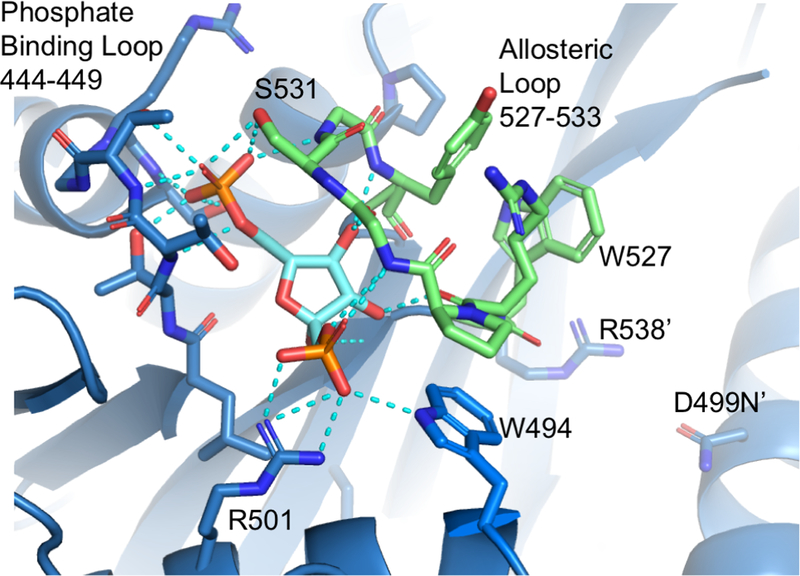
Allosteric loop colored green. Fructose-1,6-bisphosphate colored cyan. R538’ and D499N’ are located on the adjacent C domain across the C-C interface.
Figure 3. Allosteric loop comparison.
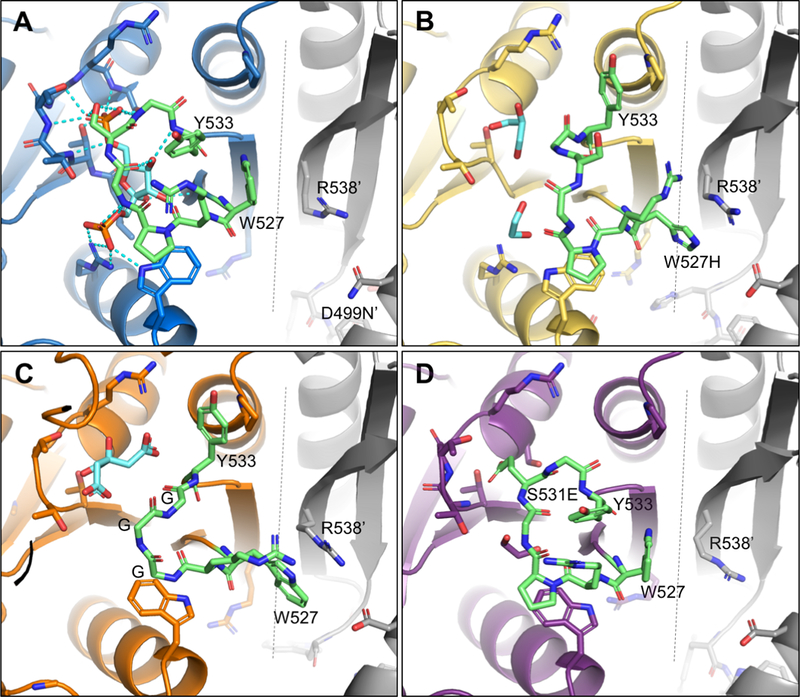
Allosteric loop colored green. Ligands colored cyan. Dashed line marks the C-C interface. Adjacent monomers are colored gray. A) D499N. D499N has a closed allosteric loop with fructose-1,6-bisphosphate (Fru-1,6-BP) bound. B) W527H. Allosteric loop in an open position. W527H exhibits a π-cation interaction with R538 across the C-C interface stabilizing the conformation. C) GGG with an open allosteric loop. W527 forms a π stacking interaction with R538. D) S531E with a closed allosteric loop. The E531 carboxylate is bound in the position of the 6’ phosphate of Fru-1,6-BP stabilizing the closed conformation.
3.3. The allosteric site of W527H
Similar to our reasoning for D499N, the W527H structure was determined to examine the structural basis of the allosteric activation observed for the W527H variant (Ishwar et al., 2015). W527H was purified and crystallized in the absence of added Fru-1,6-BP. Weak density corresponding to solvent molecules such as glycerol and ethylene glycol was evident in the positions occupied by the 6’- and 1’-phosphates of Fru-1,6-BP in the D499N structure, but density suggesting bound Fru-1,6-BP was not evident. The D499N and W527H variants have similar functional activity, making the presence of Fru-1,6-BP in the D499N structure and absence in the W527H structure difficult to explain.
In the absence of Fru-1,6-BP (i.e, the W527H structure), the allosteric loop position is quite different from that found in the D499N structure (Figure 3B). Y533 is rotated 90° and packs parallel to P420. H527 is rotated 150° to form a cation- interaction across the C-C interface with R538 from the adjacent monomer. Residues 529–532 are not positioned to form hydrogen bonds with Fru-1,6-BP in this open conformation. With knowledge of the open conformation of the allosteric loop, we looked again at the D499N structure. Indeed, chain C of the D499N structure has weak density for the open allosteric loop, observed as the presence of density for Y533 parallel to P420 and W527 density parallel to R538’. Because the D499N structure retains an unmodified allosteric loop, this suggests that the open position is sampled by the native loop and is not merely due to the W527H mutation. Residue D499, in the W527H variant, is much closer to residue R538 and residue W527H’ (4.3 Å on average), but still beyond hydrogen bonding distance.
3.4. The allosteric site of GGG
GGG, with the deletion of P529 and substitution of S531G, is not activated by Fru-1,6-BP (Ishwar and Fenton, 2015). The original design of the GGG mutation was based on the idea that the shortened loop would not allow formation of hydrogen bonds between the allosteric loop and Fru-1,6-BP. Fru-1,6-BP was not included in the crystallization condition and no Fru-1,6-BP was present in the allosteric site of the GGG structure. The allosteric loop adopts an open conformation that is fully resolved in 3 of the 8 chains (Figure 3C). Again, only solvent molecules, citrate or glycerol, could be modeled in the allosteric site and attempts at co-crystallization with Fru-1,6-BP did not yield diffracting crystals. In chains C, D and E, Y533 opposes P420 and W527 stacks parallel to R538’, forming a cation-π interaction analogous to that seen in the W527H structure. Consistent with the original design of this variant, the GGG residues 530–532 are held open and are not in position to hydrogen bond with Fru-1,6-BP. In the remaining chains the allosteric loop had incomplete electron density that could not be modeled.
3.5. The allosteric site of S531E
The S531E variant has a higher affinity for PEP than wild type hLPYK in the absence of Fru-1,6-BP (Ishwar et al., 2015). Addition of Fru-1,6-BP causes only a very small additional increase in PEP affinity. Therefore, the S531E mutation serves as a mutational mimic of the allosteric activation caused by Fru-1,6-BP. It was hypothesized that the introduced negative charge of glutamate in S531E would bind in the position of the Fru-1,6-BP 6’-phosphate (i.e, to the phosphate binding loop), thus stabilizing the allosteric loop in a closed position and leading to activation of the enzyme. The S531E structure, with 8 chains, captures a variety of allosteric loop positions (closed, disordered and open) (Figure S2). The allosteric loop is modeled in the closed position in 5 chains. In chain C, high quality and complete electron density is present for the closed position as seen in Figure 3D. In chain B the allosteric loop is modeled in the open position with partial occupancy. Density is incomplete for 530–532 in chains E and G. Overall the density across the eight allosteric loops suggests a sampling of both open and closed conformations with higher occupancy for the closed position. From the mosaic of conformations in the S531E structure, we conclude that S531E is able to bind in the 6’-phosphate binding site; however, the loop is in flux leading to partial allosteric activation, consistent with previously collected kinetic data (Ishwar et al., 2015).
4. Discussion
The hLPYK variant crystal structures presented here have high global structural homology and appear to be in the same conformational state as the C436M and S12D variants previously published. Other pyruvate kinase enzymes, such as Leishmania mexicana PYK and human M2PYK, have been shown to adopt alternative conformational states in the presence and absence of effector (Morgan et al., 2010, Morgan et al., 2013). We propose that the reason the structures included in this study show local, but not global changes is due to a pH ranging below 6.0 in the crystallization drops for each of the variants. Allosteric coupling between PEP affinity and Fru-1,6-BP binding is known to decrease for hLPYK at low pH (Fenton & Alontaga, 2009). Thus, our structures may represent a fixed conformation that is determined primarily by pH and not the presence or absence of effector.
Based on past structural comparisons of M1PYK and M2PYK (Dombrauckas et al., 2005, Morgan et al., 2013) and outcomes from mutational studies of hLPYK, it was proposed that an interaction between R528 and W527 from one subunit and D499’ from the neighboring subunit was responsible for stabilizing the open form of the allosteric loop. Indeed, the D499N mutation and a series of substitutions at the 527 position all caused increased PEP affinity, even in the absence of Fru-1,6-BP (Ishwar et al., 2015). The position of D499N is very similar to that of D499 in the S12D and C436M structures (Figure S3). Due to the increased PEP affinity caused by the D499N position, we speculate that D499 interacts with W527’ and R538 in the wild type protein stabilizing the open allosteric loop, as homologous residues do in M2PYK (Morgan et. al.). However, this would require closing a distance greater than 4 Å between D499 and W527H’ and R538. Again, it is possible that the low pH crystallization conditions are preventing conformational changes that would otherwise allow interaction between D499 and W527’. It is also possible that hLPYK undergoes far more subtle conformational changes to link effector binding with increased PEP affinity. Nevertheless, D499N does not appear to be in position to interact with W527 or R528 and so a reason for the ability of D499N to mimic the Fru-1,6-BP allosteric effect remains unresolved. Given that Fru-1,6-BP must have originated from the cell and remained throughout purification, we suggest that Fru-1,6-BP binds tightly to the D499N variant. It is possible that the D499N protein included in the previous functional study also had Fru-1,6-BP present and the presence of that activator caused increased PEP affinity.
These four hLPYK structures suggest that the allosteric loop toggles between a closed and open position. In the absence of Fru-1,6-BP, the loop is stabilized in the open position by interactions across the C-C interface between R538 and W527’. Once Fru-1,6-BP binds, the closed conformation is favored due to the hydrogen bonding network between the allosteric loop and Fru-1,6-BP. Of the changes in the allosteric loop, Y533 and W527 undergo the most significant repositioning. In the closed form, with Fru-1,6-BP bound, Y533 and W527 pack proximal to each other and are exposed to solvent. In the open form, Y533 packs parallel to P420 and W527H forms a cation-π interaction with R538’ across the C-C interface. Further structural studies will be necessary to determine how conformational changes of the allosteric loop of hLPYK lead to altered PEP affinity in the active site.
Supplementary Material
Table 1.
Crystallization
| D499N | W527H | GGG | S531E | |
|---|---|---|---|---|
| Method | All crystals were grown by hanging drop vapor diffusion at 298K in VDX 24-well plates | |||
| Protein concentration | 3.6 mg/mL | 6.0 mg/mL | 2.0 mg/mL | 2.0 mg/mL |
| Buffer composition of protein solution | 50 mM MES pH 6.8, 100 mM KCl, 10% glycerol, 2 mM DTT | 50 mM MES pH 6.8, 100 mM KCl, 10% glycerol, 2 mM DTT | 50 mM MES pH 6.8, 100 mM KCl, 10% glycerol, 2 mM DTT | 50 mM MES pH 6.8, 100 mM KCl, 10% glycerol, 2 mM DTT |
| Composition of reservoir solution | 200 mM ammonium citrate dibasic, 20% PEG 3350 | 325 mM ammonium citrate dibasic, 16% PEG 3350 | 200 mM ammonium citrate pH 6.0, 24% PEG 3350 | 200 mM ammonium citrate pH 5.6, 16% PEG 3350 |
| Volume and ratio of drop | 3.0 μL, 1:1 | 3.0 μL, 1:1 | 3.0 μL, 1:1 | 3.0 μL, 1:1 |
| Volume of reservoir | 1000 μL | 1000 μL | 1000 μL | 1000 μL |
Table 2.
Data collection and processing – hLPYK-D499N
Values for the outer shell are given in parentheses.
| D499N | W527H | GGG | S531E | |
|---|---|---|---|---|
| Diffraction source | SSRL 12–2 | SSRL 9–2 | SSRL 9–2 | SSRL 9–2 |
| Wavelength (Å) | 0.97946 | 0.97946 | 0.97946 | 0.97946 |
| Temperature (K) | 100 | 100 | 100 | 100 |
| Detector | Pilatus 6M | Pilatus 6M | Pilatus 6M | Pilatus 6M |
| Crystal-detector distance (mm) | 416 | 350 | 385 | 430 |
| Rotation range per image (°) | 0.15 | 0.15 | 0.15 | 0.15 |
| Total rotation range (°) | 110 | 180 | 360 | 360 |
| Exposure time per image (s) | 0.2 | 0.2 | 0.2 | 0.2 |
| Space group | P21212 | P21212 | P1 | P21 |
| a, b, c (Å) | 120.57, 204.66, 112.33 | 120.0, 204.6, 112.4 | 79.36, 106.21, 151.71 | 94.81, 139.22, 180.98 |
| α, β, γ (°) | 90, 90, 90 | 90, 90, 90 | 76.44, 80.06, 71.37 | 90, 103.34, 90 |
| Mosaicity (°) | 0.08 | 0.15 | 0.17 | 0.08 |
| Resolution range (Å) | 2.15–39.5 | 102.31–2.26 | 39.06–2.32 | 39.49–2.42 |
| Total No. of reflections | 628809 | 555901 | 684187 | 1210143 |
| No. of unique reflections | 150477 | 84688 | 175876 | 172562 |
| Completeness (%) | 99.0 (95.5) | 90.4 (98.8) # | 89.4 (76.9) * | 98.9 (92.1) |
| Redundancy | 4.2 (4.2) | 6.6 (6.1) | 3.9 (3.4) | 7.0 (5.8) |
| 〈 I/σ(I)〉 | 10.3 (2.0) | 14.9 (2.8) | 12.2 (2.0) | 16.4 (2.0) |
| Rr.i.m. | 7.7 (64.6) | 9.3 (69.3) | 8.2 (65.6) | 8.9 (94.7) |
| Overall B factor from Wilson plot (Å2) | 34.47 | 29.92 | 38.45 | 45.68 |
Processed using ellipsoidal resolution cut-off to account for observed anisotropy. Ellipsoidal completeness in the outer shell exceeds 98% at 2.26 Å. Spherical completeness in the inner shell exceeds 93% and outer shell exceeds 98% at 2.55 Å.
Completeness fell below 90% for this P1 crystal despite collecting 360˚ of data. We have been unable to collect additional data.
Synopsis.
Structural characterization of hLPYK variants demonstrating allosteric loop dynamics.
Acknowledgements
This work was supported by NIH Grant GM115340 to AWF, NIH Grant GM127655 to ALL and NSF Grant CHE1403293 to ALL. JSM and TAR were supported by the NIH Graduate Training Program in the Dynamic Aspects of Chemical Biology Grant T32 GM008545 and JSM was supported by an American Heart Association Predoctoral Fellowship PRE33960374. Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract DE-AC02–76SF00515. The Stanford Synchrotron Radiation Lightsource Structural Molecular Biology Program is supported by the U.S. Department of Energy Office of Biological and Environmental Research and by NIGMS, NIH Grant P41GM103393. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. Thank you to Lauren Arney and Azel King for their work on PYK variant purification trials.
Funding information National Institutes of Health, National Institute of General Medical Sciences (grant No. GM115340 to Aron W. Fenton; grant No. GM127655 to Audrey L. Lamb; studentship No. GM008545 to Jeffrey S. McFarlane, Trey A. Ronnebaum); National Science Foundation, Division of Chemistry (grant No. CHE1403293 to Audrey L. Lamb); American Heart Association (studentship No. PRE33960374 to Jeffrey S. McFarlane).
References
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC & Zwart PH (2010). Acta Crystallogr D Biol Crystallogr 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson GM & Fenton AW (2016). Biophys J 110, 1912–1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen VB, Arendall WB 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS & Richardson DC (2010). Acta Crystallogr D Biol Crystallogr 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dombrauckas JD, Santarsiero BD & Mesecar AD (2005). Biochemistry 44, 9417–9429. [DOI] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG & Cowtan K (2010). Acta Crystallogr D Biol Crystallogr 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans PR & Murshudov GN (2013). Acta Crystallogr D Biol Crystallogr 69, 1204–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenton AW & Alontaga AY (2009). Biothermodynamics, Part B, pp. 83–107. [Google Scholar]
- Girdlestone C & Hayward S (2016). J Comput Biol 23, 21–26. [DOI] [PubMed] [Google Scholar]
- Holyoak T, Zhang B, Deng J, Tang Q, Prasannan CB & Fenton AW (2013). Biochemistry 52, 466–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishwar A, Tang Q & Fenton AW (2015). Biochemistry 54, 1516–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurica MS, Mesecar A, Heath PJ, Shi W, Nowak T & Stoddard BL (1998). Structure 6, 16. [DOI] [PubMed] [Google Scholar]
- Kabsch W (2010). Acta Crystallogr D Biol Crystallogr 66, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krissinel E & Henrick K (2004). Acta Crystallogr D Biol Crystallogr 60, 2256–2268. [DOI] [PubMed] [Google Scholar]
- Krissinel E & Henrick K (2007). Journal of Molecular Biology 372, 774–797. [DOI] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC & Read RJ (2007). J Appl Crystallogr 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPhillips TM, McPhillips SE, Chiu H-J, Cohen AE, Deacon AM, Ellis PJ, Garman E, Gonzalez A, Sauter NK, Phizackerley RP, Soltis SM & Kuhn P (2002). Journal of Synchrotron Radiation 9, 401–406. [DOI] [PubMed] [Google Scholar]
- Morgan HP, McNae IW, Nowicki MW, Hannaert V, Michels PA, Fothergill-Gilmore LA & Walkinshaw MD (2010). J Biol Chem 285, 12892–12898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan HP, O’Reilly FJ, Wear MA, O’Neill JR, Fothergill-Gilmore LA, Hupp T & Walkinshaw MD (2013). Proc Natl Acad Sci U S A 110, 5881–5886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valentini G, Chiarelli LR, Fortin R, Dolzan M, Galizzi A, Abraham DJ, Wang C, Bianchi P, Zanella A & Mattevi A (2002). J Biol Chem 277, 23807–23814. [DOI] [PubMed] [Google Scholar]
- Vonrhein C, Flensburg C, Keller P, Sharff A, Smart O, Paciorek W, Womack T & Bricogne G (2011). Acta Crystallogr D Biol Crystallogr 67, 293–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.