

Abstract
The striatum with a number of dopamine containing neurons, receiving projections from the substantia nigra and ventral tegmental area; plays a critical role in neurodegenerative diseases of motor and memory function. Additionally, oxidative damage to nucleic acid may be vital in the development of age-associated neurodegeneration. The metabolism of dopamine is recognized as one of the sources of reactive oxygen species through the Fenton mechanism. The proposed interactions of oxidative insults and dopamine in the striatum during the progression of diseases are the hypotheses of most interest to our study. This study investigated the possibility of significant interactions between these molecules that are involved in the late-stage of Alzheimer’s disease (AD), Parkinson disease (PD), Parkinson disease dementia (PDD), dementia with Lewy bodies (DLB), and controls using ELISA assays, autoradiography, and mRNA in situ hybridization assay. Interestingly, lower DNA/RNA oxidative adducts levels in the caudate and putamen of diseased brains were observed with the exception of an increased DNA oxidative product in the caudate of AD brains. Similar changes were found for dopamine concentration and vesicular monoamine transporter 2 densities (VMAT2). We also found that downstream presynaptic dopamine D1 Receptor binding correlated with dopamine loss in LBD groups, and RNA damage and BACE1 in the caudate of AD. This is the first demonstration of region-specific alterations of DNA/RNA oxidative damage which cannot be viewed in isolation, but rather in connection with the interrelationship between different neuronal events; chiefly DNA oxidative adducts and density of VMAT2 in AD and PD patients.
Keywords: Oxidative damage, Dopamine, Striatum, Alzheimer’s disease, Lewy body diseases
Graphical Abstract
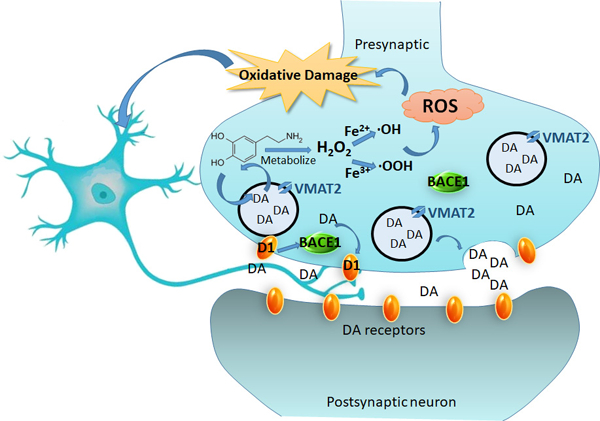
We hypothesized the following interactions between oxidative damage and dopaminergic biological molecules. One, metabolism of dopamine yields ·OH through the Fenton Reaction and leads to oxidative damage of DNA and RNA molecules. Two, VMAT2 plays a vital anti-oxidation role. Three, D1R plays a compensatory role against dopamine loss and takes on damage effects in the regulation of BACE1 during these biochemical events. We believe these interactions are involved in the late-stages of neurodegenerative diseases - resulting in striatal neuron dysfunction.
Introduction
Dementia with Lewy bodies (DLB), Parkinson disease dementia (PDD), and Parkinson disease (PD) share clinical and neuropathological similarities, and these diseases have been aggregated conceptually under the broad umbrella of Lewy body disease (LBD) (Lippa et al. 2007). Alzheimer’s disease (AD) and LBD cause progressive cognitive and motor dysfunction to varying degrees (Gan et al. 2018). AD typically begins as an amnestic syndrome though motor dysfunction may also occur (Buchman et al. 2013). LBD includes motor and cognitive manifestations that some classify as DLB if dementia occurs within or before the first year of motor Parkinsonism versus PDD if the dementia starts later in the course of the disease (Emre et al. 2007; McKeith et al. 2005). This rather arbitrary division blurs even more after realizing that 20–40% of people with PD have cognitive dysfunction at the time of initial motor symptoms (Foltynie et al. 2004; Aarsland & Kurz 2010; Goldman et al. 2015). Although the topographic distribution of pathology differs between AD and LBD both may involve dysfunction of striatal dopaminergic neurons. While degeneration of nigrostriatal dopaminergic neurons is the classic pathology of PD; striatal dopaminergic dysfunction may also contribute to the motor manifestations of AD. The striatum can be sectioned into sub-regions - caudate and putamen - based on the input it receives from different cortical areas (Draganski et al. 2008). The caudate nucleus is involved in a variety of behaviors including procedural learning (Seger & Cincotta 2005) and working memory (Hannan et al. 2010). The dorsal posterior putamen receives its primary input from the motor and sensorimotor cortices and is substantially involved in the regulation of motor circuits (Del Campo et al. 2016).
Recent studies indicate that neuroinflammation may contribute to degeneration of the nigrostriatal dopaminergic pathway in PD and possibly in AD as well. In PD, activation of glial cells causes the release of free radicals and cytokines which in turn increases the vulnerability of dopaminergic neurons to oxidative damage (Czlonkowska et al. 2002; Liu & Hong 2003). High concentrations of H2O2 - generated from the metabolism of dopamine - may lead to the formation of highly toxic hydroxyl radical (·OH) making dopaminergic neurons more susceptible to oxidative stress (Mytilineou et al. 1993). Mitochondrial dysfunction contributes to oxidative injury early in the course of these diseases. Furthermore, 8-oxo-7,8-dihydro-2’-deoxyguanosine (8-oxo-dG) and 8-oxo-7,8-dihydroguanosine (8-oxo-G) may act as biomarkers of oxidative damage to DNA and RNA, respectively. Postmortem brains from AD patients have higher levels of 8-oxo-G than healthy controls especially in the hippocampus (Nunomura et al. 1999; Hofer & Perry 2016). However, human studies into the biological consequence of oxidative damage to the dopaminergic system in the striatum are underdeveloped relative to the importance of this phenomenon (Venkateshappa et al. 2012; Horner et al. 2011; Tata & Yamamoto 2007). Regulation of reactive oxygen species (ROS) may provide a therapeutic target for these neurodegenerative diseases (Masoud et al. 2015; Martorana & Koch 2014; Hyun 2019).
In the current study, we hypothesized that the interrelationship of dopamine and oxidative insults in the striatum is involved crucially and differently in AD and LBD. This study analyzed human post-mortem brain samples from AD, LBD, and control cases. We selectively extracted DNA, RNA, and dopamine; and quantified the concentrations of 8-oxo-dG, 8-oxo-G, and dopamine in the caudate and putamen. Furthermore, it is known that dopamine compartmentalization by the vesicular monoamine transporter 2 (VMAT2) correlates with the relative vulnerability of dopaminergic neurons in Parkinsonian-like neurodegeneration (Hall et al. 2014). It is also documented that presynaptic dopamine D1 Receptor (D1R) binding is partly a complement to the alteration of dopamine levels and its pathological factor affects excitation/inhibition imbalances caused by β-Amyloid (Aβ) (Ren et al. 2018). In light of these factors, we measured densities of VMAT2 and D1R in the same regions using quantitative autoradiography and assessed the effects of D1R on β-site APP cleaving enzyme 1 (BACE1) activity in AD using mRNA in situ hybridization assay. Our findings provide valuable insights on the interactions of oxidative damage and dopamine in the striatum of AD and LBD brains, and will evoke meaningful discussion on the subject.
Materials and methods
Ethics statement.
Either the patient provided written consent prior to cognitive impairment or the next of kin provided it antemortem or postmortem in accordance with local Ethical Committee procedures (Washington University Institutional Review Board, Washington University School of Medicine). Use of this tissue for the postmortem receptor autoradiography and biochemistry studies was approved by the Charles F. and Joanne Knight Alzheimer’s Disease Research Center (Knight ADRC) and Movement Disorders Center (MDC) Leadership Committees (Ethics approval reference number: T1705).
Chemicals and radioligands.
Chemical reagents and standard compounds were purchased from Sigma (St. Louis, MO). [3H]SCH23390 (85 Ci/mmol, Cat. #NET930025UC CAS. #125941–87-9) was purchased from Perkin Elmer Life Sciences (Boston, MA) and [3H]Dihydrotetrabenazine ([3H]DTBZ, 20 Ci/mmol, Cat. #ART 0496) was purchased from American Radiolabeled Chemicals (St Louis, MO, USA).
Postmortem human brain cases.
Clinically and neuropathologically well-characterized human brain tissues were obtained from the Knight ADRC and the Movement Disorders Brain Bank at Washington University School of Medicine. The tissues obtained were as follows: 10 PD (7 male, 3 female) aged 69–87 (mean: 78 ± 2) years at death, 8 PDD (7 male, 1 females) aged 66–87 (mean: 77 ± 3) years at death, 10 DLB (5 male, 5 female) aged 69–89 (mean: 81 ± 2) years at death, 27 AD (13 male, 14 females) aged 62–94 (mean: 82 ± 2) years at death, and 10 age-matched normal control cases (6 males, 4 females) aged 72–93 (mean: 83 ± 2) years at death. The arbitrary clinical distinction between DLB and PDD was made using the McKeith et al. criteria (McKeith et al. 2005; Hughes et al. 1992). Dementia level was evaluated by CDR (Emre et al. 2007); PD participants with CDR≥1 - based on criteria for dementia in PD - were included in the study. AD pathological changes were assessed using Braak staging (Thal & Braak 2005). Stages of amyloid beta deposition refer to initial deposits in the basal neocortex (A), deposits that extended into the association areas of the neocortex (B), and heavy deposition throughout the entire cortex (C). Stages of neurofibrillary pathology correspond to transentorhinal (I–II), limbic (III–IV), and neocortical (V and VI). The average age and post mortem interval time did not significantly differ across these three groups. All the AD cases show neurofibrillary tangles (NFTs) (V: 13 cases; VI: 14 cases) and are significantly different from that of age-matched control cases (Average amyloid beta: A; NFTs: II). Detailed information on the clinical and pathological features are summarized in Table 1.
Table 1.
Baseline information and clinical features of the study subjects. PMI: Post Mortem Interval; Braak NFT stage: Braak neurofibrillary tangle stage; Braak Aβ stage: Braak amyloid beta plaque stage.
| Control | PD | PDD | DLB | AD | P | |
|---|---|---|---|---|---|---|
| Participants | 10 | 10 | 8 | 10 | 27 | |
| Male/Female | 6/4 | 7/3 | 7/1 | 5/5 | 13/14 | NA |
| Age | 83±2 | 78±2 | 77±3 | 81±2 | 82±2 | NA |
| PMI (h) | 18.8±5 | 16.0±3.2 | 10.8±1.6 | 18.0±3.7 | 10.6±1 | NA |
| Brain weight (g) | 1326±60 | 1299±40 | 1346±41 | 1273±38 | 1127±47 | NA |
| Onset | 65±3 | 60±3 | 66±4 | 71±2 | NA | |
| Progression | 14±1 | 16±3 | 14±3 | 10±1 | NA | |
| Braak NFT stage | Stage 0 :1 | Stage I :5 | Stage I :3 | Stage I :7 | Stage V :13 | * |
| Stage I :2 | Stage II :2 | Stage II :2 | Stage II :2 | stage VI :14 | ||
| Stage II :4 | Stage III :3 | Stage III :3 | Stage V :1 | |||
| Stage III :3 | ||||||
| Braak Aβ stage | All normal | Normal :3 | Normal :1 | Normal :1 | All stage C | NA |
| Stage A :1 | Stage A :2 | Stage A :1 | ||||
| Stage B :2 | Stage B :1 | Stage B :2 | ||||
| Stage C :5 | Stage C :4 | Stage C :6 | ||||
| L-Dopa response | Yes: 9 | Yes: 6 | Yes: 9 | |||
| Modest:1 | Modest:2 | Modest:1 |
indicates p < 0.05 vs. the controls.
Tissue collection.
Brains were collected at the time of autopsy and the right hemisphere was coronally sectioned and snap-frozen by contact with Teflon-coated aluminum plates cooled in liquid nitrogen vapor. Tissue blocks were subsequently placed in airtight zip-lock plastic bags and stored at −80 °C until used. Microscopic examination to establish neuropathology was performed using established rating scales. For autoradiography and mRNA in situ hybridization studies: frozen coronal sections (20 µm) were cut with a Microm cryotome and mounted on Superfrost Plus glass slides (Fisher Scientific, Pittsburgh, PA, USA. Cat. #1255015). Striatal sub-areas - caudate and putamen - were tested separately.
8-oxo-7,8-dihydro-2’-deoxyguanosine (8-oxo-dG) Assay.
Total DNA in the caudate and putamen from study brains was extracted using the Qiagen QIAamp DNA Mini Kit according to the manufacturer’s instruction (Qiagen, Valencia, CA, USA. Cat. #51304). A NanoDrop 1000 spectrophotometer (Thermo Fisher, Pittsburgh, PA, USA) was utilized to measure DNA integrity and purity. Extracted gDNA was converted to single-strand DNA by incubating the sample at 95 °C for 5 min and rapidly chilling it on ice. DNA single-strand digestion used the nuclease P1 (NP1) and alkaline phosphatase (AP) enzymes (Huang et al. 2001). Preparation of enzymes solution was as follows: NP1 from Penicillium citrinum (1mg 1000 units of 3’-phosphomonoesterase activity, AdipoGen Life Sciences, San Diego, CA, USA. Cat. #501015753) was dissolved in 100 µL 20 mmol/L sodium acetate buffer (pH 5.2) it was further diluted 10 times to a final concentration of 1 U/µL in the acetate buffer and AP from calf intestine (1 U/µL, Thermo Scientific™, Grand Island, NY, USA. Cat. #FEREF0651) was stored in 25 mmol/L Tris HCl (pH 7.6), 1 mmol/L MgCl2, and 50% glycerol (w/v). Digestion of the DNA was carried out in the following manner: after acidification with 1 µL 3 mol/L acetate buffer (pH 5.2) the DNA reaction mixture was subjected to 1 µL of NP1 (1 U/µL) digestion for 2 h at 37 °C. After 2 h of incubation, 10 µL of 1M Tris-HCl (pH 8.0) was used to bring pH back to 7.4 followed by treatment with 1 µL of AP (1 U/µL) for 1 h at 37 °C. The reaction mixture was centrifuged for 1 min at 8,000 g and the supernatant was collected for the 8-oxo-dG assay using the OxiSelect oxidative DNA damage ELISA kit (Cell Biolabs, Inc., San Diego, CA, USA. Cat. #STA-320) according to the manufacturer’s instructions. Each prepared tissue sample was added to the assay in duplicate. Known standards were also included in the assay in triplicate to allow for accurate quantification.
8-oxo-7,8-dihydroguanosine (8-oxo-G) Assay.
Whole RNA in the caudate and putamen from study brains was extracted using the Qiagen RNeasy Plus Micro Kit according to the manufacturer’s instruction (Qiagen, Valencia, CA, USA. Cat. #74034). RNA integrity and purity was measured using NanoDrop 1000 spectrophotometer. RNA samples were digested to nucleosides by incubating the samples with 1 µL of NP1 (1 U/µL) and 1 µL 3 mol/L acetate buffer (pH 5.2) for 2 h at 37 °C. Following incubation they were treated with 10 µL of 1M Tris-HCl (pH 8.0) and 1 µL of AP (1 U/µL) for 1 h at 37 °C. The reaction mixture was centrifuged for 1 min at 8,000 g, and the supernatant was collected for the 8-oxo-G assay using the OxiSelect oxidative RNA damage ELISA kit (Cell Biolabs, Inc., San Diego, CA, USA. Cat. #STA-325–5) according to the protocol provided by the manufacturer. Each prepared tissue sample was added to the assay in duplicate. Known standards were also included in the assay in triplicate to allow for accurate quantification.
Dopamine Assay.
Dopamine concentrations in the caudate and putamen from snap-frozen study brains were measured by the commercially available Dopamine (DA) ELISA Kit (BioVision, Inc., Milpitas, CA, USA. Cat. #K4219) according to the user’s manual provided by the manufacturer. Samples (100 mg) were rinsed with 1×PBS (Fisher Scientific, Pittsburgh, PA, USA, Cat. #50–983-207) and homogenized in 0.9 mL of 1×PBS. The homogenates were centrifuged for 5 min at 5000g at 2–8°C. The supernatant was collected for use with the dopamine assay kit. Known standards were added to the assay in triplicate for accurate quantification and each tissue extract was determined in duplicate by a four-parameter logistic (4-PL) regression model. The detection range for dopamine was 1.56 −100 ng/mL and the sensitivity was 0.938 ng/mL. All data were obtained from a standard curve with r>0.99.
Quantitative autoradiography protocol.
To ensure the removal of endogenous dopamine, sections for dopamine D1R binding tissue were pretreated in buffer (50 mmol/L Tris buffer, pH 7.4, containing 120 mmol/L NaCl, 5 mmol/L KCl) for 20 min at room temperature (RT). After a 30 min incubation in an open staining jar with their respective radiotracer, slides were then rinsed five times at 1 min intervals with ice-cold buffer. The free radio ligand concentration loss was determined to be < 5% as previously described (Xu et al. 2010).
Quantification of total radioactivity.
Dried slides were made conductive by covering the free side with copper foil tape. Slides were then placed into a gas chamber containing a mixture of argon and triethylamine (Sigma-Aldrich, St. Louis, MO, USA. Cat. #BP616–500) as part of a gaseous detector system - the Beta Imager 2000Z Digital Beta Imaging System (Biospace, France) – for which there is a 0.07dpm/mm2 sensitivity limit. After the gas was well mixed and a homogenous state achieved, further exposure for 20 h yielded high-quality images. A [3H]microscale with a known amount of radioactivity (ranging from 0 to 36.3 nCi/mg) was counted with each section and used to create a standard curve; in each case the standard curve had a correlation coefficient (R) > 0.99. Quantitative analysis was performed with the program Beta-Vision Plus (BioSpace, France) for each anatomical region of interest.
Vesicular monoamine transporter 2 (VMAT2) binding.
VMAT2 binding sites were labeled with [3H]DTBZ. Brain sections were incubated at RT for 30 min in buffer solution containing 4 nmol/L [3H]DTBZ. Nonspecific binding was determined in the presence of 1 µM S-(–) tetrabenazine (Sigma-Aldrich, St. Louis, MO, USA. Cat. #T2952–10MG, CAS. #58–46-8) as previously described (Sun et al. 2013b; Sun et al. 2013a).
Dopamine D1R binding.
D1R binding sites were labeled with [3H]SCH23390 using the procedure described by Savasta with minor modifications (Savasta et al. 1986). After removing endogenous dopamine, sections were incubated for 30 min at RT in buffer solution containing 1.5 nmol/L [3H]SCH23390 and 30 nM ketanserin tartrate (Tocris Bioscience, Ellisville, Missouri, USA. Cat. #0908, CAS. #83846–83-7) to block 5-HT2 receptors. Nonspecific binding was determined in the presence of 1 µM (+)-butaclamol (Sigma-Aldrich, St. Louis, MO, USA. Cat. #D033–5MG, CAS. #55528–07-9) as previously described (Novick et al. 2008; Lim et al. 2011).
β-site APP cleaving enzyme 1 (BACE1) mRNA in situ hybridization assay.
In situ hybridization (ISH) was performed using the RNAscope 2.5 HD Chromogenic Assay kit (Advanced Cell Diagnostics, Inc. Newark, CA, USA. Cat. #322350) with a slightly modified protocol. Slides were removed from the −80 °C freezer and immediately placed in jar containing ice cold 10% Neutral Buffered Formalin (NBF, Fisher Scientific, Pittsburgh, PA, USA, Cat. #22–050-104) and fixed for 15 min at 4 °C. Then slides were placed in 50% EtOH (Ethanol 100%, Fisher Scientific, Pittsburgh, PA, USA, Cat. #04–355-720) for 5 min followed by 70% EtOH for 5 min. Finally, slides were placed in 100% EtOH for 5 min followed by fresh 100% EtOH for 5 min. These procedures were carried out at RT. Slides were left to dry for 5 min at RT then a hydrophobic barrier was drawn around each section (ImmEdge Hydrophobic Barrier Pen, ACD, Newark, CA, USA. Cat. #310018). Sections were then pretreated with Protease IV (Universal Pretreatment Reagents, ACD, Newark, CA, USA. Cat. #322380) for 15 min at RT and rinsed in PBS. Briefly, the tissue sections were incubated in a custom human gene-specific RNAscope Hs-BACE1 probe (Gene Alias: ASP2; Target Region: 1393–2418, ACD, Newark, CA, USA. Cat. #422541), a positive control probe (human Cyclophilin B (PPIB), ACD, Newark, CA, USA. Cat. #476701) and a negative control probe (bacterial dapB, ACD, Newark, CA, USA. Cat. #310043); for 2 h at 40 °C in the RNAscope oven (ACD HybEZ™ II Hybridization System, ACD, Newark, CA, USA. Cat. #321711) then washed with buffer to remove probes. Sections were sequentially hybridized to a cascade of amplification molecules, culminating in binding to HRP-labeled probes; only with modification of incubation with Amp5 for 45 min at RT using the HybEZ humidity control tray and slide rack to maintain humidity. ISH signal was detected by diluting Fast RED-B in Fast RED-A solution (1:60 ratio) and incubating sections in this solution for 15 min. Slides were washed in water 2 times to stop the reaction. Then the slides were counterstained with 50% Hematoxylin (Gills Hematoxylin 1, Fisher Scientific, Pittsburgh, PA, USA, Cat. #NC1000827) and washed in water 3–5 times. Then the slides were placed into 0.01% Ammonia water (28 to 30% Ammonium hydroxide solution, Fisher Scientific, Pittsburgh, PA, USA, Cat. #MAX13036) for 20 seconds and washed with water 3–5 times. After drying at 60 °C for at least 20 min, the slides were dipped into xylene (Sigma-Aldrich, St. Louis, MO, USA. Cat. #XX0055–6) and immediately placed mount media (EcoMount, Fisher Scientific, Pittsburgh, PA, USA, Cat. #EM897L) and coverslips. The high-resolution images of single colorimetric ISH tissue sections were acquired using a digital whole slide scanner (Nanozoomer 2-HT, Hamamatsu. Bridgewater, NJ) using a 20×/0.75 lens (Olympus, Center Valley, PA). We used the NDP.view2 (Hamamatsu Photonics, Japan) software and viewed the digital slides. The hybridization signals were then quantified using the image analysis module of the digital pathology software Visiomorph (VisioPharm, Broomfield, CO). The caudate and putamen under examination were delineated by a region of interest (ROI) in the software and the signals were quantified as the average red dots count per mm2.
Statistical analysis.
Continuous variables were expressed as the means ± SEM. One outlying sample in the PDD group and one outlying sample in the AD group were excluded from the analysis. Their results deviated more than 150% from the means in the 8-oxo-dG and dopamine assays and produced negative value in the D1 receptor density assay. More detail on these examples can be found in the supplementary materials (Figure S1–Figure S4). The statistical analyses were carried out using all data except these two outliers without any further normalization. One-way ANOVA and two-way ANOVA were used to estimate the overall significance followed by Bonferroni multiple comparison test. Student’s unpaired t-test was used to assess the difference between groups. Spearman’s correlation coefficient (rs) was calculated to verify the strength of correlation between continuous variables. Analyses of the correlations between dopamine concentration and L-Dopa response were performed using Kendall’s tau_b test. Statistical analyses were performed using GraphPad Prism 6.0 (RRID: SCR_002798) for Windows and IBM SPSS Statistics version 23 (RRID: SCR_002865). p < 0.05 was considered statistically significant. No blinding, randomization, or sample size calculations were performed during experimentation and statistical analyses.
Results
Baseline information and clinical features of study subjects
In the present study, human brains were collected at autopsy between 3 and 47 h postmortem. Table 1 recapitulates the baseline information and clinical features of the study subjects. No significant difference were found in age at death, PMI, brain weight, onset, and progression of the disease suggesting that our results were not affected by these factors. For the Braak NFT stage factor there was a significant difference between the AD and Control groups. The demographic information on the Table 1 also shows the levodopa response of the PD, PDD, and DLB patients; however, the brains were collected during too large time span to textually research for the dose and duration of levodopa treatment.
8-oxo-dG levels in the caudate and putamen of the different groups
To ascertain 8-oxo-dG levels in the caudate and putamen of the disease groups and age-matched controls we utilized the ELISA assay following the protocol mentioned above. As shown in Figure 1, we found that the levels of 8-oxo-dG in the caudate of the PD cases (11.22 ± 1.46) decreased remarkably by 45.7% compared to controls (20.67 ± 2.58) but without statistical significance. Additionally, a marked increase of 8-oxo-dG concentration in the caudate of AD patients (24.19 ± 2.94) was observed compared to PD patients (119.3%, p = 0.0039). The results in the putamen vastly differed from the caudate. In the putamen, the concentrations of 8-oxo-dG were similar to the controls except for a nonsignificant slight reduction in the AD group (Control: 16.59 ± 1.93; PD: 16.60 ± 1.78; PDD: 18.10 ± 1.23; DLB: 14.84 ± 1.83; AD: 13.87 ± 0.73). Two-way ANOVA analysis shows there is a dramatic difference with statistical significance (p = 0.0008) between the elevated 8-oxo-dG levels in the caudate with the decreased levels in the putamen of AD brains (Figure S5). This suggested that the caudate is most likely more susceptible to DNA damage than the putamen in the late-stage of the AD patients.
Figure 1.

8-oxo-dG levels in the caudate and putamen from patients with diseases (PD: n=10, PDD: n=7, DLB: n=10, AD: n=26) and age-matched controls (n=10). Value shown are means ± SEM as the concentration of 8-oxo-dG (pg) per total DNA (µg). A p value of <0.05 was considered significant. The only statistical significance is between the PD vs AD (p = 0.0039) as demonstrated with the bracket.
8-oxo-G levels in the caudate and putamen of the different groups
Figure 2 shows the RNA damage status in the caudate and putamen of the disease groups and age-matched controls using ELISA assay; the levels of 8-oxo-G in the caudate did not significantly differ between controls and the disease groups (Control: 33.08 ± 4.31; PD: 21.05 ± 3.34; PDD: 31.91 ± 5.68; DLB: 23.93 ± 2.92; AD: 27.88 ± 2.76). By contrast, all of the LBD groups had significantly lower concentrations of 8-oxo-G in the putamen compared to controls. The DLB subjects had the lowest concentrations with 67.4% reduction (DLB: 15.83 ± 2.04, 95% CI: 14.04 to 51.12, p < 0.0001; Controls: 48.41 ± 6.11). The 56.4% and 60.3% reductions were found for PD (21.12 ± 3.64, 95% CI: 8.24 to 46.34, p < 0.01) and PDD (19.17 ± 2.82, 95% CI: 7.83 to 50.65, p < 0.01) cases, respectively. The concentration of oxidative modified RNA in the putamen and caudate of AD (40.45 ± 3.12) brains decreased. There were two other interesting findings here to note. One, the level of 8-oxo-G in the putamen (48.41 ± 6.11) of health aging brain was higher than in caudate (33.08 ± 4.31), indicating that putamen was likely more susceptible to RNA oxidative damage than caudate. Additionally, greater oxidation to RNA than to DNA was found in caudate and putamen samples from all study cases, suggesting that RNA may be more vulnerable to oxidative insults than DNA.
Figure 2.

8-oxo-G levels in the caudate and putamen from patients with diseases (PD: n=10, PDD: n=7, DLB: n=10, AD: n=26) and age-matched controls (n=10). Value shown are means ± SEM as the concentration of 8-oxo-G (pg) per total RNA (µg). A p value of <0.05 was considered significant: ** indicates p < 0.01, *** indicates p < 0.001, **** indicates p < 0.0001, vs. the controls. Significant differences between two non-control groups are indicated with brackets and corresponding p-values [Putamen: PD vs AD (p = 0.0086), PDD vs AD (p = 0.0162), DLB vs AD (p = 0.002)].
Dopamine levels in the caudate and putamen of the different groups
ELISA assay was used to determine the DA concentrations in the caudate and putamen samples that were taken from the disease groups and age-matched controls, as shown in Figure 3A. DA levels in the caudate and putamen for control and diseases patients did not significantly differ. However, we observed trends of decreasing and increasing DA concentrations in the LBD and AD cohorts, respectively. These observations warrant further investigation in a larger population of patients. Looking at the correlation between dopamine and oxidative damage more closely, we see that Figure 3B shows a significant negative association between dopamine levels and the concentration of 8-oxo-dG in the caudate from patients with AD (rs = −0.454, p = 0.026). These data suggest the ability of dopamine in the caudate of AD to enhance the oxidative degradation through Fenton and Fenton-like reactions (Melin et al. 2015). Additionally, the concentration of dopamine showed a significant positive correlation with VMAT2 expression in the putamen of DLB brains (Figure 3C, rs = 0.667, p = 0.050), further proving in all likelihood that dopamine accumulates in the cytosol by means of dopamine transporter (DAT) followed by sequestration into the synaptic storage vesicles by VMAT2.
Figure 3.

A: Concentration of dopamine in the caudate and putamen from patients with diseases (PD: n=10, PDD: n=7, DLB: n=10, AD: n=26) and age-matched controls (n=10). Value shown are means ± SEM. B: Concentration of dopamine vs level of 8-oxo-dG in the caudate from diseases brains, significantly association was observed only in AD group (p = 0.026). C: Concentration of dopamine vs VMAT2 expression in the putamen from diseases brains, significantly association was observed only in DLB group (p = 0.050). rs, the Spearman’s rank correlation coefficient.
Vesicular monoamine transporter 2 (VMAT2) density in the caudate and putamen of the different groups
We determined the striatal VMAT2 density of the disease groups and age-matched controls using quantitative autoradiography. Compared to the controls, lower levels of striatal VMAT2 binding were obtained in the caudate of LBD cases with a similar reduction (PD: −40.8%; PDD: −43.7%; DLB: −42.4%) of that shown in Figure 4A, statistical significance was reached vs. AD. The lowest VMAT2 striatal binding was found in the putamen of PD patients; it was found to have a 58.1% reduction – compared to the controls (p < 0.0001 vs. AD). On the other hand, a marked increase in VMAT2 density both in the caudate (33.3% increase) and putamen (47.8% increase) of AD brains were found in the present study. Both dopamine levels and VMAT2 density - compared to controls - showed similar changes in the caudate and putamen of the patients with diseases in the current study. Regarding the role of VMAT2 in packing cytosolic dopamine into synaptic vesicles to prevent its autoxidation and the subsequent degeneration of dopamine neurons; Spearman analyses reveal significant negative correlations between 8-oxo-dG levels and VMAT2 density in the caudate (rs = −0.451, p = 0.027) and putamen (rs = −0.516, p = 0.024) of AD brains, as well as in the caudate (rs = −0.683, p = 0.042) of PD brains (Figure 4C). This might explain the reduction of vesicular storage and increase of dopamine release, and the yielding of hydrogen peroxide from MAO-catalyzed dopamine metabolism (Golembiowska & Dziubina 2012).
Figure 4.

Quantitative autoradiographic analysis of VMAT2 density in the caudate and putamen from patients with diseases (PD: n=10, PDD: n=7, DLB: n=10, AD: n=26) and age-matched controls (n=10). A: Quantitative analysis of the VMAT2 density (fmol/mg) in the caudate and putamen from subjects. Value shown are means ± SEM. Statistical significances between two disease groups are indicated with brackets and corresponding p-values (p < 0.0001 were found for PD vs AD, PDD vs AD, and DLB vs AD in the caudate, as well as PD vs AD in the putamen). B: Autoradiograms show total binding of 4 nmol/L [3H]DTBZ (Panel B top row) and nonspecific binding in the presence of 1 µM S(−)-tetrabenazine (Panel B bottom row) in the striatal regions of 5 representative subjects. The numbers 1 and 2 designate the following regions: caudate (1) and putamen (2). C: [3H]Microscale standards (ranging from 0 to 36.3 nCi/mg) were also counted. D: Density of VMAT2 as concentration of 8-oxo-dG in the caudate and putamen from AD brains (p = 0.027 and p = 0.024, respectively), as well as that in the caudate from PD brains (p = 0.042). rs, the Spearman’s rank correlation coefficient.
Dopamine D1 Receptor (D1R) density in the caudate and putamen of the different groups
Although there is no report on the biological consequence of oxidative damage on D1R, D1R density is partly regulated by dopamine signal and should be investigated to establish the downstream consequence of damage to nucleic acids. We quantified D1R density of the disease groups and age-matched controls using quantitative autoradiography. As shown in Figure 5A - in the striatal regions - the distribution of D1R was abundant and no regional differences of receptor binding were found in the caudate and putamen of the controls (caudate: 25.28 ± 3.41; putamen: 24.63 ± 3.32). Substantial D1R density increases with statistically significance were found for the caudate and putamen from the patients with LBD when compared to controls. The greatest increase was found in the caudate for PD cases (66.09 ± 3.32, 161.4%, p < 0.001). The greatest increase was found in the putamen for PDD cases (70.23 ± 8.57, 185.1%, p < 0.001). The least increases of D1R bonding were found for the caudate (36.83 ± 1.54, 45.7% increase) and putamen (31.98 ± 2.14, 29.8% increase) of AD cases - compared to that of LBD cases.
Figure 5.

Quantitative autoradiographic analysis of D1R density in the caudate and putamen of patients with diseases (PD: n=10, PDD: n=7, DLB: n=10, AD: n=26) and age-matched controls (n=10). A: Quantitative analysis of the D1R density (fmol/mg) in the caudate and putamen of subjects. Value shown are means ± SEM. Statistical significances between two disease groups are indicated with brackets and corresponding p-values [Caudate: PD vs AD (p = 0.0021); Putamen: PD vs AD (p = 0.0005), PDD vs AD (p = 0.0004), DLB vs AD (p = 0.0041)]. A p value of < 0.05 was considered significant: ** indicates p < 0.01, *** indicates p < 0.001, vs. the controls. B: Autoradiograms show total binding of 1.5 nM [3H]SCH23390 (Panel B top row) and nonspecific binding in the presence of 1 µM (+) butaclamol (Panel B bottom row) in the striatal regions of the same 5 representative subjects. The numbers 1 and 2 designate the following regions: caudate (1) and putamen (2). C: [3H]Microscale standards (ranging from 0 to 36.3 nCi/mg) were also counted.
mRNA level of β-site APP cleaving enzyme 1 (BACE1) in the caudate and putamen of the ten AD cases and age-matched controls
To examine the effects of D1R on regulation of BACE1 activity, we designed RNAscope ISH probe to detect BACE1 gene mRNA expression from the 10 cases from AD brains with the highest D1R density ≥ 40 fmol/mg. We only analyzed striatal tissue sections in these 10 cases from AD brains along with 10 age-matched control brains by labeling them with the probe targeting BACE1 mRNA. Meanwhile, a positive control probe and negative control probe were performed under the same conditions as the RNA quality control. In our analysis we observed prominent neuropathological features of AD: Aβ plaques and neurofibrillary tauopathy consisting of threads and tangles (NFT) (Figure 6A left). Targeting BACE1 mRNA was visualized as cytoplasmic red dots of variable size which were sometimes fused to form larger foci of staining, and were located mostly on the Aβ plaque accumulation more often found in the putamen (Figure 6A middle). Semi-quantitative regional analysis results of ISH signal in ROI (Figure 6C) showed that both increased transcriptional expression in the caudate and putamen were observed in patients with AD compared to controls. The results concerning the putamen were of special note as they were statistically significant and deserved further investigation. (3.025 ± 0.10, 13.5% increase, p = 0.0197). This result can be observed visually in Figure 6B showing positive cells in the caudate and putamen of the exact AD case with the same D1R density in two brain areas and control subjects. Spearman analyses reveal positive correlations between BACE1 mRNA expression and the density of D1R in the caudate of the AD brains with statistical significance (rs = 0.750, p = 0.020) and the controls (rs = 0.119, p = 0.779) without statistical significance. Conversely, negative correlations in the putamen (rs = −0.546, p = 0.160) of the AD brains and the controls (rs = −0.690, p = 0.058) without statistical significance were observed (Figure 6D). Our results were consistent with previous reports that Aβ1–42 oligomers damage synapse function and interact with D1/D5 dopamine receptor inducing pro-epileptogenic effects, and in these reports D1R antagonist were studied as potential therapeutics (Costa et al. 2016).
Figure 6.

RNAscope in situ hybridization (ISH) analysis of BACE1 transcriptional expression in the caudate and putamen from patients with AD (n=10) and age-matched controls (n=10). A: RNAscope in situ hybridization (ISH) labeling for BACE1 mRNA in the caudate and putamen from the AD subject included in the previous group of 5 representative subjects. Scale bar in the whole slide section: 10 mm. Scale bar in the high magnification mode Aβ plaque and NFT sections: 500 µm. Rectangle drawn in the whole slide section is magnified to highlight AB plaques and labeled as AD putamen image directly to the right. The numbers 1 and 2 designate the following regions: caudate (1) and putamen (2). B: Positive cells (red with arrow pointing to them) in the caudate and putamen of AD and control subjects with scale bar: 100 µm. C: semi-quantitative regional analysis of ISH signal in ROI, the signals were quantified as the average red dots count per mm2. Value shown are means ± SEM. T-test was used. A p value of < 0.05 was considered significant, *indicates p < 0.05 vs. the controls. D: BACE1 mRNA expression as density of D1R in the caudate and putamen from patients with AD and age-matched controls. rs, the Spearman’s rank correlation coefficient. The only significant correlation was in the caudate of the AD group (p = 0.02).
Data analysis of all assay results of the caudate and putamen of the ten AD cases chosen for BACE1 mRNA analysis and age-matched controls
Re-analysis of all assays data involving the caudate and putamen of the only ten AD cases chosen for BACE1 mRNA assay and age-matched controls is consistent with the results previously discussed. However, compared to the control cases, there were some more apparent findings than in the previously reported data: an exceptional increase of 8-oxo-dG in the caudate, and decrease in the putamen; decreased 8-oxo-G levels, increased dopamine concentration, VMAT2, and D1R density were found both in the caudate and putamen of AD cases (Figure 7A, Figure 7B and Table 2). Spearman analyses reveal significant negative correlations between density of VMAT2 and DNA oxidative adducts levels (rs = −0.678, p = 0.015), density of VMAT2 and RNA oxidative adducts levels (rs = −0.717, p = 0.030), as well as density of D1R and RNA oxidative adducts levels (rs = −0.750, p = 0.020) in the caudate of AD cases (Figure 7C). These results indicate that dopamine storage ability plays a critical role in enhancing the oxidative degradation through Fenton and Fenton-like reactions and that interactions between D1R and RNA oxidative damage is still unclear and needs to be explored in greater detail. Greater amounts of DNA oxidative adducts in the caudate and opposite changes in the putamen were of note, indicating that the caudate of AD patients are likely more susceptible to DNA damage.
Figure 7.

Data analysis of all assay results of the caudate and putamen of the AD cases chosen for BACE1 mRNA analysis (n=10) and age-matched controls (n=10). A: Oxidative damage of nucleic acid in the caudate and putamen from 10 AD cases and age-matched controls. A p value of < 0.05 was considered significant: *indicates p < 0.05 and ** indicates p < 0.01, vs. the controls. B: Biological consequence of oxidative damage to dopamine system in the caudate and putamen from 10 AD cases and age-matched controls. A p value of < 0.05 was considered significant, *indicates p < 0.05 vs. the controls. C: Density of VMAT2 and DNA oxidative adducts levels (rs = −0.678, p = 0.015), density of VMAT2 and RNA oxidative adducts levels (rs = −0.717, p = 0.030), as well as density of D1R and RNA oxidative adducts levels (rs = −0.750, p = 0.020) in the caudate from 10 AD cases. rs, the Spearman’s rank correlation coefficient.
Table 2.
Data analysis of all assay results of the caudate and putamen of the ten AD cases chosen for BACE1 mRNA analysis and age-matched controls
| Assays | Caudate | Putamen | |||
|---|---|---|---|---|---|
| Mean ± SME | p value | Mean ± SME | p value | ||
| 8-oxo-dG (pg/μg total DNA) | Control | 20.67 ± 2.581 | 16.59 ± 1.928 | ||
| AD | 26.49 ± 7.545 | ˃ 0.05 | 14.17 ± 0.823 | ˃ 0.05 | |
| 8-oxo-G (pg/μg total DNA) | Control | 33.08 ± 4.313 | 48.41 ± 6.115 | ||
| AD | 22.07 ± 1.838* | 0.0376 | 37.46 ± 4.851 | ˃ 0.05 | |
| Dopamine (ng/g wet wt) | Control | 19.29 ± 6.986 | 17.21 ± 5.083 | ||
| AD | 29.66 ± 9.108 | ˃ 0.05 | 25.89 ± 4.060 | ˃ 0.05 | |
| VMAT2 Density (fmol/mg) | Control | 133.5 ± 10.28 | 123.4 ± 10.62 | ||
| AD | 176.6 ± 17.08 | 0.051 | 152.5 ± 18.34 | ˃ 0.05 | |
| D1 Receptor Density (fmol/mg) | Control | 25.28 ± 3.410 | 24.63 ± 3.318 | ||
| AD | 43.98 ± 1.292**** | < 0.0001 | 40.38 ± 2.904** | 0.0030 | |
| BACE1 mRNA Levels | Control | 2.804 ± 0.1252 | 2.664 ± 0.092 | ||
| AD | 2.948 ± 0.1615 | ˃ 0.05 | 3.025 ± 0.101* | 0.0197 | |
Discussion
Oxidative DNA insults have received much attention - as the brain has a high oxygen demand, a relatively high metabolic rate, and is thought to have a decreased ratio of antioxidant enzymes, all of which result in elevated oxidative stress and ROS generation (Madabhushi et al. 2014). DNA damage could continuously alter chromatin conformation and gene expression patterns with age. Lesions to mitochondrial DNA have also been found in patients with PD (Bender et al. 2006). On the other hand, RNA molecules are likely oxidized by hydroxyl radical generated from the reaction of H2O2 with transition metals through the Fenton reaction. Furthermore, RNA bases are not protected by hydrogen bonding and specific proteins. These problems underlie the reasons that make RNA more vulnerable to oxidative damage than DNA (Fimognari 2015). While elevated oxidative stress in the cerebral cortex in patients with early-stage PD has been described (Ferrer 2009); RNA oxidation has been prominently observed in postmortem AD brains with lesser volumes of Aβ plaque deposition or shorter disease duration (Nunomura et al. 2001).
Oxidative damage of nucleic acid in the caudate and putamen from patients with neurodegenerative diseases and age-matched controls
Transition metal ions - iron or copper - and hydrogen peroxide are able to oxidize a wide range of substrates resulting in biological damage. The reaction referred to as the Fenton reaction:
is capable of yielding both hydroxyl radical and higher oxidation states of the iron (Winterbourn 1995). Excessive hydroxyl radical attacks adjacent to mitochondrial DNA strands and cytoplasmic RNA single-strands eventually generate a wide variety of oxidative adducts. The major products of the oxidation of DNA/RNA are 8-oxo-dG/8-oxo-G - as guanine in DNA/RNA is more sensitive to ROS attacks than other bases - can be stable and relatively easily formed as biomarkers (Che et al. 2010). Enzymatically, metabolism of dopamine yields plenty of H2O2, leading conversely to dopaminergic neurons being more exposed to oxidative damage. Given the above, we can rethink how the metabolism and concentration of dopamine further plays a critical role in nucleic oxidation as a major source of ROS. Also, the progressive dysfunction or eventual death of dopaminergic neurons may be in part a result of an imbalance between clearance and generation of ROS. This theory can be supported further by a significant negative correlation between dopamine levels and DNA oxidative adducts levels in the caudate of AD subjects studied currently; dopamine is a good metal chelator and electron donor that can be capable of reacting with iron and manganese (Kong & Lin 2010).
Several studies have shown increased 8-OHdG level in patients with PD (Kikuchi et al. 2002; Chen et al. 2009), and a higher level of 8-oxo-dG in cerebrospinal fluid (CSF) of patients with PD compared to controls (Abe et al. 2003). However, in the present study focusing on the late-stage of the disease, noticeable reductions of 8-oxo-dG level in the caudate of LBD cases were observed. The urinary 8-OHdG levels in MFB 6-hydroxydopamine (6-OHDA) lesion model started to increase as early as day 3 with significant increases to day 7 and gradually reverting back to near baseline levels at day 42 (Kikuchi et al. 2011). Also, the increased 8-oxo-dG levels in caudate of AD patients correlated with the increase of dopamine levels of the same cases, which are consistent with elevated 8-oxo-dG levels in the hippocampus of patients with AD (Hofer & Perry 2016). We observed a significant negative association between dopamine levels and the concentration of 8-oxo-dG in the caudate of patients with AD (rs = −0.454, p = 0.026). A significant negative correlation between 8-oxo-dG levels and VMAT2 density in the caudate (rs = −0.451, p = 0.027) of AD brains was also observed. These results are most likely owing to the Fenton oxidation reaction taking place in the caudate of the AD - as a response to dopamine concentration (Youdim 2018) and dopamine compartmentalization by VMAT2 association with DNA oxidative damage. Furthermore, decreased 8-oxo-dG level and increased Aβ plaque deposition in the putamen of AD brains were unexpectedly observed in the present study. In cell culture experiments, deposition of Aβ42 is associated with a decrease in the level of neuronal oxidative stress (Nunomura et al. 2000).
As oxidized RNAs turns-over rapidly, the pattern of RNA oxidation - a “steady-state” marker of oxidative damage rather than history - is prominent in neurons without pathology and is present in lesser amounts in neurons containing pathology (Nunomura et al. 2001). The prominent 8-oxo-G immunoreactivity were found in the hippocampus, subiculum, entorhinal cortex, and temporal neocortex of DLB cases (Nunomura et al. 2002). Previous reports describe that 8-oxo-dG might be used as an “early-stage” marker, whereas the decrease of 8-oxo-G in CSF might be an indicator of the degree of neurodegeneration during the PD disease progression (Gmitterova et al. 2018). As reported, a 58% decrease in striatal dopamine concentration was observed in the mouse model of Parkinson Disease (Xu et al. 2017). In the present LBD patients, notable deficiencies in dopamine levels in the putamen - resulting in decreased oxidative damage to RNA - could be explained by the decreased metabolism of dopamine through Fenton Reaction thus leading to a lower yield of ·OH. The exception being for the metabolism of dopamine itself, other sources for the generation of ROS: mitochondrial dysfunction, iron, neuroinflammatory cells, calcium, and aging - need to be explored more in future research. We found the changes of dopamine levels in the caudate and putamen did not correlated with that of 8-oxo-G levels in the same brain areas of AD cases, indicating that alteration of dopamine was insufficient to fully explain the RNA oxidative damage. For patients with AD, the Apolipoprotein E (ApoE) ϵ4 was mainly responsible for increased amounts of Aβ deposits, as well as the strong negative correlation between RNA oxidative damage and Aβ deposits (Nunomura et al. 2001). Therefore, the same degrees of reduction of 8-oxo-G levels in the caudate (−15.7%) and putamen (−16.4%) were observed in AD brains despite the elevated dopamine levels in these brain areas. This is consistent with the previously reported decreased 8-oxo-G levels in disease-affected areas: the hippocampus, the inferior parietal lobule, and the superior and middle temporal gyri (Weidner et al. 2011).
Interaction of oxidative damage and dopamine in the caudate and putamen from patients with neurodegenerative diseases and age-matched controls
The dopamine levels in the caudate and putamen of LBD groups decreased. Obviously, dopamine treatments - such as levodopa – and Aβ are known to influence dopamine levels and could account for the reason the data gathered from our assay did not reach statistical significance. As shown in Table 1, majority of LBD patients had positive levodopa response. Kendall’s tau_b analyses of the correlations between dopamine concentration and L-Dopa response in the caudate (p = 0.075) and putamen (p = 0.064) of LBD patients reveal that L-Dopa treatment was likely one of the factors that resulted in changes to dopamine levels (Figure S7). Dramatic fluctuation of dopamine levels occurs in the synaptic clefts of striatal neurons after each levodopa dose (Tomiyama 2017); these transient elevations would increase with duration of Parkinson disease resulting in changes to the downstream dopamine receptors (de la Fuente-Fernandez et al. 2004). Even though, the total synaptic dopamine levels - including endogenous and derived from exogenous levodopa - are below normal values in health subjects. What’s more, other natural variabilities - postmortem intervals and dissection of postmortem tissue – assuredly contributed to significant dispersion both in PD subjects and the controls (Buddhala et al. 2015). Beyond our expectation, the increased concentrations of dopamine in the caudate and putamen of the AD brains were observed, conflicting with the hypothesis that a portion of AD patients could be more prone to develop dopamine-deficit symptoms (Martorana & Koch 2014). Aβ stimulates dopamine release from dopaminergic axons in the anterior cingulate cortex and excessive dopamine over activates D1R of fast-spiking interneurons, thus contributing to gamma-aminobutyric acid (GABA) inhibitory and excitation/inhibition imbalance caused by Aβ (Ren et al. 2018). Furthermore, soluble Aβ in the AD mice model at 100 nM evoked the release of dopamine to ~170% of base line, which was sensitive to antagonists of α7 nicotinic receptors (Wu et al. 2007).
Although we cannot exclude the artificial oxidation of samples during analysis, we have confirmed that oxidative damage systemically changed in study groups. In neurodegenerative disease patients, metabolism of dopamine - one source of ROS - initiates the formation of free radicals through the Fenton reaction pathway; that in turn promotes oxidative damage to proteins, lipids, and nucleic acids. Paradoxically, this contributes to selective loss of dopaminergic neurons or even neuron death. ROS is thought to be the major source of oxidative damage contributing to the death of dopamine neurons in PD (Ortiz et al. 2017; Koutsilieri et al. 2002).
To better understand the interactions among oxidative damage and dopamine storage abilities the spatial control of dopamine by VMAT2 and anti-oxidation role of VMAT2 should be noted. With regard to the role of oxidative damage in the pathogenesis of PD, packing of cytosolic dopamine into synaptic vesicles by VMAT2 inhibits its autoxidation and subsequent degeneration of dopaminergic neurons (Carlsson et al. 1957; Golembiowska & Dziubina 2012). This theory is most likely proved further by the negative correlations of DNA/RNA oxidative damage and VMAT2 density in striatum of AD cases, as well as the negative correlation of DNA oxidative damage and VMAT2 density in putamen of PD brains. Decreased tissue concentrations of dopamine attenuated its uptake and transport functions altering dopamine turnover, thus VMAT2 levels correlate with the severity of Parkinsonism and (Hall et al. 2014) and cognitive impairment in DLB patients (Roselli et al. 2009). Striatal VMAT2 binding is also interpreted as reflecting the integrity of the nigro-striatal dopamine system in PD (Gao et al. 2016). Among the caudate and putamen, VMAT2 is a mark of clinical diagnosis differentiation between DLB and AD. The greatest density difference between groups was observed for the lowest posterior putamen: PD < DLB < AD ≈ Controls (Siderowf et al. 2014). This is consistent with our results in the caudate: LBD < control < AD and in the putamen: PD < AD (p < 0.0001 were found for all LBD groups vs AD in the caudate and PD vs AD in the putamen). Additionally and unexpectedly, conflicting with no striatal reductions in AD patients (Villemagne et al. 2012), elevated VMAT2 density in the caudate and putamen of patients with AD was observed. These changes are probably due to the unexpected increase of dopamine levels in these two brain areas.
Amongst dopamine receptors, the D1R variety are crucially implicated in maintaining higher cognitive functions - in particular working memory, attention, and executive functions (Bruns et al. 2018). The increases of D1R density in striatum of patients with LBD can in all likelihood be explained by the fact that dopamine receptor function is strongly associated with the compensatory mechanism to make up for the loss of excitatory D1R stimulation (Perez et al. 2017). What’s more, it is widely believed that L-Dopa treatment stimulates D1R signaling leading to a persistent D1R hypersensitivity and contributing to the genesis of long-term complications involving L-Dopa including the development of L-Dopa-induced dyskinesia (LID) (Solis et al. 2017; Corvol et al. 2004). Increased D1R binding has been previously observed in the caudate when associated with the presence of Lewy body in AD subjects (Sweet et al. 2001).
Correlation of BACE1 mRNA transcriptional expression and D1R density in the caudate and putamen of the ten AD cases and age-matched controls
In a previous report, D1R seems play a more notable role in specific aspects of cognitive function; preclinical findings indicate that D1R is involved in mediating the epileptic effect of Aβ1–42 (Costa et al. 2016). Therefore, the ameliorative effects of dopamine D1-like receptor agonist SKF38393-D1R improved cognitive dysfunction. This result was likely mediated by increased phosphorylation of cAMP response element binding protein (CREB) and expression of Bcl-2 and brain-derived neurotrophic factor (BDNF) along with reduction of BACE1 and Aβ1–42 levels in hippocampus and cortex of animal model (Zang et al. 2018). In the current study, a statistically significant positive correlation of D1R density and BACE1 mRNA transcriptional expression in the caudate of AD was observed. This suggests that D1R hypersensitivity is mediated by a complex interaction between N-methyl-D-aspartate (NMDA) receptors and dopamine D1-histamine H3 receptor heteromer (Rodriguez-Ruiz et al. 2017). Subsequently, our understanding of D1R activation mechanism in BACE1 activity needs to be further clarified.
Conclusion
In the current study, we show how region-specific alteration levels of DNA/RNA oxidative adducts and relevant dopamine levels along with dopamine storage abilities changes in the striatum of late-stage neurodegenerative diseases patients. There is a chicken and egg problem inherent to our findings when trying to correlate dopaminergic neuron dysfunction or even loss and oxidative damage. The downstream presynaptic D1R binding is associated with an alteration of dopamine levels, as well as the presence of Aβ plaque and RNA damage. When talking about the oxidative damage in the neurodegenerative diseases there are several protagonists that are involved in this story: metabolism of dopamine, mitochondrial dysfunction, and neuroinflammation are focused on as main resources of ROS (Dias et al. 2013). To our knowledge, this study is the first to investigate the interrelationship of dopamine and oxidative insults in the striatum of neurodegenerative brains. The omission of other ROS sources, limited samples, and significant dispersion are limitations of this present study. The current results do not robustly support our hypothesis. Our findings may be the tips of the iceberg in the path to understanding the interactions of oxidative damage in the striatal dopaminergic system and open new questions for research in that field.
Supplementary Material
Acknowledgments:
The authors thank Dr. Nigel Cairns, Ms. Erin E. Franklin and Mr. Michael Baxter of the Knight Alzheimer Disease Research Center Neuropathology Core at Washington University School of Medicine, for coordination of the tissue preparation and expert technical assistance. Research funded by NIH R01 NS092865, R01 AG052550, R01 NS075321, P01 AG026276, P01 AG03991 and P50 AG05681 as well as the American Parkinson Disease Association (APDA), the Greater St. Louis Chapter of the APDA, the Barnes-Jewish Hospital Foundation (Elliot Stein Family Fund and Parkinson disease fund), the Oertli Fund and the Riney Foundation. The digital whole slide scanner (Nanozoomer 2-HT) was supported by a NIH Shared Instrumentation Grant (S10 RR0227552) to Washington University for the Alafi Neuroimaging Laboratory of the Hope Center for Neurological Disorders.
This work was funded by the Greater St. Louis Chapter of the APDA, (Grant / Award Number: ) NIH, (Grant / Award Number: ‘P01 AG026276’,’P01 AG03991 ‘,’P50 AG05681’,’R01 AG052550’,’R01 NS075321’,’R01 NS092865’,’S10 RR0227552’) the American Parkinson Disease Association (APDA), (Grant / Award Number: ) the Oertli Fund and the Riney Foundation, (Grant / Award Number: ) the Barnes-Jewish Hospital Foundation (Elliot Stein Family Fund and Parkinson disease fund), (Grant / Award Number: ) (grant number ): This information is usually included already, but please add to the Acknowledgments if not.
Abbreviation:
- DLB
dementia with Lewy bodies
- PDD
Parkinson disease dementia
- PD
Parkinson disease
- LBD
Lewy body disease
- AD
Alzheimer’s disease
- 8-oxo-dG
8-oxo-7,8-dihydro-2’-deoxyguanosine
- 8-oxo-G
8-oxo-7,8-dihydroguanosine
- ROS
reactive oxygen species
- VMAT2
vesicular monoamine transporter 2
- D1R
dopamine D1 Receptor
- Aβ
β-Amyloid
- BACE1
β-site APP cleaving enzyme 1
- NFTs
neurofibrillary tangles
- DA
Dopamine
- 4-PL
four-parameter logistic
- RT
room temperature
- ISH
in situ hybridization
- ROI
region of interest
- CSF
cerebrospinal fluid
- DAT
dopamine transporter
- 6-OHDA
6-hydroxydopamine
- ApoE
Apolipoprotein E
- GABA
gamma-aminobutyric acid
- LID
L-Dopa-induced dyskinesia
- CREB
cAMP response element binding protein
- BDNF
brain-derived neurotrophic factor
- NMDA
N-methyl-D-aspartate
Footnotes
Conflict of interest
The authors have no conflicts of interest and this study was not pre-registered.
Supporting information
One outlying sample in the PDD group and one outlying sample in the AD group were excluded from the analysis; two-way ANOVA analysis of 8-oxo-dG/8-oxo-G levels in the caudate and putamen from patients with diseases; Kendall’s tau_b analysis of correlation between dopamine concentration and L-Dopa response in the caudate and putamen of LBD patients, more detail on these examples can be found in the supplement materials. It may be found online in the Supporting Information section at the end of the article.
References
- Aarsland D and Kurz MW (2010) The epidemiology of dementia associated with Parkinson disease. Journal of the neurological sciences 289, 18–22. [DOI] [PubMed] [Google Scholar]
- Abe T, Isobe C, Murata T, Sato C and Tohgi H (2003) Alteration of 8-hydroxyguanosine concentrations in the cerebrospinal fluid and serum from patients with Parkinson’s disease. Neuroscience letters 336, 105–108. [DOI] [PubMed] [Google Scholar]
- Bender A, Krishnan KJ, Morris CM et al. (2006) High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nature genetics 38, 515–517. [DOI] [PubMed] [Google Scholar]
- Bruns RF, Mitchell SN, Wafford KA et al. (2018) Preclinical profile of a dopamine D1 potentiator suggests therapeutic utility in neurological and psychiatric disorders. Neuropharmacology 128, 351–365. [DOI] [PubMed] [Google Scholar]
- Buchman AS, Yu L, Wilson RS, Schneider JA and Bennett DA (2013) Association of brain pathology with the progression of frailty in older adults. Neurology 80, 2055–2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buddhala C, Loftin SK, Kuley BM, Cairns NJ, Campbell MC, Perlmutter JS and Kotzbauer PT (2015) Dopaminergic, serotonergic, and noradrenergic deficits in Parkinson disease. Ann Clin Transl Neurol 2, 949–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlsson A, Lindqvist M and Magnusson T (1957) 3,4-Dihydroxyphenylalanine and 5-hydroxytryptophan as reserpine antagonists. Nature 180, 1200. [DOI] [PubMed] [Google Scholar]
- Che Y, Wang JF, Shao L and Young T (2010) Oxidative damage to RNA but not DNA in the hippocampus of patients with major mental illness. Journal of psychiatry & neuroscience : JPN 35, 296–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CM, Liu JL, Wu YR, Chen YC, Cheng HS, Cheng ML and Chiu DT (2009) Increased oxidative damage in peripheral blood correlates with severity of Parkinson’s disease. Neurobiol Dis 33, 429–435. [DOI] [PubMed] [Google Scholar]
- Corvol JC, Muriel MP, Valjent E, Feger J, Hanoun N, Girault JA, Hirsch EC and Herve D (2004) Persistent increase in olfactory type G-protein alpha subunit levels may underlie D1 receptor functional hypersensitivity in Parkinson disease. J Neurosci 24, 7007–7014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa C, Parnetti L, D’Amelio M et al. (2016) Epilepsy, amyloid-beta, and D1 dopamine receptors: a possible pathogenetic link? Neurobiology of aging 48, 161–171. [DOI] [PubMed] [Google Scholar]
- Czlonkowska A, Kurkowska-Jastrzebska I, Czlonkowski A, Peter D and Stefano GB (2002) Immune processes in the pathogenesis of Parkinson’s disease - a potential role for microglia and nitric oxide. Medical science monitor : international medical journal of experimental and clinical research 8, Ra165–177. [PubMed] [Google Scholar]
- de la Fuente-Fernandez R, Sossi V, Huang Z, Furtado S, Lu JQ, Calne DB, Ruth TJ and Stoessl AJ (2004) Levodopa-induced changes in synaptic dopamine levels increase with progression of Parkinson’s disease: implications for dyskinesias. Brain 127, 2747–2754. [DOI] [PubMed] [Google Scholar]
- Del Campo N, Payoux P, Djilali A et al. (2016) Relationship of regional brain beta-amyloid to gait speed. Neurology 86, 36–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dias V, Junn E and Mouradian MM (2013) The role of oxidative stress in Parkinson’s disease. Journal of Parkinson’s disease 3, 461–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Draganski B, Kherif F, Kloppel S, Cook PA, Alexander DC, Parker GJ, Deichmann R, Ashburner J and Frackowiak RS (2008) Evidence for segregated and integrative connectivity patterns in the human Basal Ganglia. J Neurosci 28, 7143–7152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emre M, Aarsland D, Brown R et al. (2007) Clinical diagnostic criteria for dementia associated with Parkinson’s disease. Movement disorders : official journal of the Movement Disorder Society 22, 1689–1707; quiz 1837. [DOI] [PubMed] [Google Scholar]
- Ferrer I (2009) Early involvement of the cerebral cortex in Parkinson’s disease: convergence of multiple metabolic defects. Progress in neurobiology 88, 89–103. [DOI] [PubMed] [Google Scholar]
- Fimognari C (2015) Role of Oxidative RNA Damage in Chronic-Degenerative Diseases. Oxidative medicine and cellular longevity 2015, 358713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foltynie T, Brayne CE, Robbins TW and Barker RA (2004) The cognitive ability of an incident cohort of Parkinson’s patients in the UK. The CamPaIGN study. Brain 127, 550–560. [DOI] [PubMed] [Google Scholar]
- Gan L, Cookson MR, Petrucelli L and La Spada AR (2018) Converging pathways in neurodegeneration, from genetics to mechanisms. Nature neuroscience 21, 1300–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao R, Zhang G, Chen X, Yang A, Smith G, Wong DF and Zhou Y (2016) CSF Biomarkers and Its Associations with 18F-AV133 Cerebral VMAT2 Binding in Parkinson’s Disease-A Preliminary Report. PLoS One 11, e0164762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gmitterova K, Gawinecka J, Heinemann U, Valkovic P and Zerr I (2018) DNA versus RNA oxidation in Parkinson’s disease: Which is more important? Neuroscience letters 662, 22–28. [DOI] [PubMed] [Google Scholar]
- Goldman JG, Aggarwal NT and Schroeder CD (2015) Mild cognitive impairment: an update in Parkinson’s disease and lessons learned from Alzheimer’s disease. Neurodegenerative disease management 5, 425–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golembiowska K and Dziubina A (2012) The effect of adenosine A(2A) receptor antagonists on hydroxyl radical, dopamine, and glutamate in the striatum of rats with altered function of VMAT2. Neurotoxicity research 22, 150–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall FS, Itokawa K, Schmitt A, Moessner R, Sora I, Lesch KP and Uhl GR (2014) Decreased vesicular monoamine transporter 2 (VMAT2) and dopamine transporter (DAT) function in knockout mice affects aging of dopaminergic systems. Neuropharmacology 76 Pt A, 146–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannan KL, Wood SJ, Yung AR, Velakoulis D, Phillips LJ, Soulsby B, Berger G, McGorry PD and Pantelis C (2010) Caudate nucleus volume in individuals at ultra-high risk of psychosis: a cross-sectional magnetic resonance imaging study. Psychiatry research 182, 223–230. [DOI] [PubMed] [Google Scholar]
- Hofer T and Perry G (2016) Nucleic acid oxidative damage in Alzheimer’s disease-explained by the hepcidin-ferroportin neuronal iron overload hypothesis? Journal of trace elements in medicine and biology : organ of the Society for Minerals and Trace Elements (GMS) 38, 1–9. [DOI] [PubMed] [Google Scholar]
- Horner KA, Gilbert YE and Cline SD (2011) Widespread increases in malondialdehyde immunoreactivity in dopamine-rich and dopamine-poor regions of rat brain following multiple, high doses of methamphetamine. Front Syst Neurosci 5, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Powell J, Mooney LA, Li C and Frenkel K (2001) Importance of complete DNA digestion in minimizing variability of 8-oxo-dG analyses. Free radical biology & medicine 31, 1341–1351. [DOI] [PubMed] [Google Scholar]
- Hughes AJ, Daniel SE, Kilford L and Lees AJ (1992) Accuracy of clinical diagnosis of idiopathic Parkinson’s disease: a clinico-pathological study of 100 cases. Journal of neurology, neurosurgery, and psychiatry 55, 181–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyun DH (2019) Plasma membrane redox enzymes: new therapeutic targets for neurodegenerative diseases. Archives of pharmacal research. [DOI] [PubMed]
- Kikuchi A, Takeda A, Onodera H et al. (2002) Systemic increase of oxidative nucleic acid damage in Parkinson’s disease and multiple system atrophy. Neurobiol Dis 9, 244–248. [DOI] [PubMed] [Google Scholar]
- Kikuchi Y, Yasuhara T, Agari T et al. (2011) Urinary 8-OHdG elevations in a partial lesion rat model of Parkinson’s disease correlate with behavioral symptoms and nigrostriatal dopaminergic depletion. Journal of cellular physiology 226, 1390–1398. [DOI] [PubMed] [Google Scholar]
- Kong Q and Lin CL (2010) Oxidative damage to RNA: mechanisms, consequences, and diseases. Cell Mol Life Sci 67, 1817–1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koutsilieri E, Scheller C, Tribl F and Riederer P (2002) Degeneration of neuronal cells due to oxidative stress--microglial contribution. Parkinsonism & related disorders 8, 401–406. [DOI] [PubMed] [Google Scholar]
- Lim MM, Xu J, Holtzman DM and Mach RH (2011) Sleep deprivation differentially affects dopamine receptor subtypes in mouse striatum. Neuroreport 22, 489–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippa CF, Duda JE, Grossman M et al. (2007) DLB and PDD boundary issues: diagnosis, treatment, molecular pathology, and biomarkers. Neurology 68, 812–819. [DOI] [PubMed] [Google Scholar]
- Liu B and Hong JS (2003) Role of microglia in inflammation-mediated neurodegenerative diseases: mechanisms and strategies for therapeutic intervention. The Journal of pharmacology and experimental therapeutics 304, 1–7. [DOI] [PubMed] [Google Scholar]
- Madabhushi R, Pan L and Tsai LH (2014) DNA damage and its links to neurodegeneration. Neuron 83, 266–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martorana A and Koch G (2014) “Is dopamine involved in Alzheimer’s disease?”. Frontiers in aging neuroscience 6, 252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masoud ST, Vecchio LM, Bergeron Y et al. (2015) Increased expression of the dopamine transporter leads to loss of dopamine neurons, oxidative stress and l-DOPA reversible motor deficits. Neurobiol Dis 74, 66–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKeith IG, Dickson DW, Lowe J et al. (2005) Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology 65, 1863–1872. [DOI] [PubMed] [Google Scholar]
- Melin V, Henriquez A, Freer J and Contreras D (2015) Reactivity of catecholamine-driven Fenton reaction and its relationships with iron(III) speciation. Redox report : communications in free radical research 20, 89–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mytilineou C, Han SK and Cohen G (1993) Toxic and protective effects of L-dopa on mesencephalic cell cultures. J Neurochem 61, 1470–1478. [DOI] [PubMed] [Google Scholar]
- Novick A, Yaroslavsky I and Tejani-Butt S (2008) Strain differences in the expression of dopamine D1 receptors in Wistar-Kyoto (WKY) and Wistar rats. Life Sci 83, 74–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunomura A, Chiba S, Kosaka K, Takeda A, Castellani RJ, Smith MA and Perry G (2002) Neuronal RNA oxidation is a prominent feature of dementia with Lewy bodies. Neuroreport 13, 2035–2039. [DOI] [PubMed] [Google Scholar]
- Nunomura A, Perry G, Aliev G et al. (2001) Oxidative damage is the earliest event in Alzheimer disease. Journal of neuropathology and experimental neurology 60, 759–767. [DOI] [PubMed] [Google Scholar]
- Nunomura A, Perry G, Pappolla MA, Friedland RP, Hirai K, Chiba S and Smith MA (2000) Neuronal oxidative stress precedes amyloid-beta deposition in Down syndrome. Journal of neuropathology and experimental neurology 59, 1011–1017. [DOI] [PubMed] [Google Scholar]
- Nunomura A, Perry G, Pappolla MA, Wade R, Hirai K, Chiba S and Smith MA (1999) RNA oxidation is a prominent feature of vulnerable neurons in Alzheimer’s disease. J Neurosci 19, 1959–1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortiz GG, Pacheco Moises FP, Mireles-Ramirez M et al. (2017) Oxidative Stress: Love and Hate History in Central Nervous System. Advances in protein chemistry and structural biology 108, 1–31. [DOI] [PubMed] [Google Scholar]
- Perez XA, Zhang D, Bordia T and Quik M (2017) Striatal D1 medium spiny neuron activation induces dyskinesias in parkinsonian mice. Movement disorders : official journal of the Movement Disorder Society 32, 538–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren SQ, Yao W, Yan JZ, Jin C, Yin JJ, Yuan J, Yu S and Cheng Z (2018) Amyloid beta causes excitation/inhibition imbalance through dopamine receptor 1-dependent disruption of fast-spiking GABAergic input in anterior cingulate cortex. Sci Rep 8, 302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Ruiz M, Moreno E, Moreno-Delgado D et al. (2017) Heteroreceptor Complexes Formed by Dopamine D1, Histamine H3, and N-Methyl-D-Aspartate Glutamate Receptors as Targets to Prevent Neuronal Death in Alzheimer’s Disease. Mol Neurobiol 54, 4537–4550. [DOI] [PubMed] [Google Scholar]
- Roselli F, Pisciotta NM, Perneczky R et al. (2009) Severity of neuropsychiatric symptoms and dopamine transporter levels in dementia with Lewy bodies: a 123I-FP-CIT SPECT study. Movement disorders : official journal of the Movement Disorder Society 24, 2097–2103. [DOI] [PubMed] [Google Scholar]
- Savasta M, Dubois A and Scatton B (1986) Autoradiographic localization of D1 dopamine receptors in the rat brain with [3H]SCH 23390. Brain Res 375, 291–301. [DOI] [PubMed] [Google Scholar]
- Seger CA and Cincotta CM (2005) The roles of the caudate nucleus in human classification learning. J Neurosci 25, 2941–2951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siderowf A, Pontecorvo MJ, Shill HA et al. (2014) PET imaging of amyloid with Florbetapir F 18 and PET imaging of dopamine degeneration with 18F-AV-133 (florbenazine) in patients with Alzheimer’s disease and Lewy body disorders. BMC neurology 14, 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solis O, Garcia-Montes JR, Gonzalez-Granillo A, Xu M and Moratalla R (2017) Dopamine D3 Receptor Modulates l-DOPA-Induced Dyskinesia by Targeting D1 Receptor-Mediated Striatal Signaling. Cerebral cortex (New York, N.Y. : 1991) 27, 435–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Cairns NJ, Perlmutter JS, Mach RH and Xu J (2013a) Regulation of dopamine D(3) receptor in the striatal regions and substantia nigra in diffuse Lewy body disease. Neuroscience 248, 112–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Kouranova E, Cui X, Mach RH and Xu J (2013b) Regulation of dopamine presynaptic markers and receptors in the striatum of DJ-1 and Pink1 knockout rats. Neuroscience letters 557 Pt B, 123–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweet RA, Hamilton RL, Healy MT, Wisniewski SR, Henteleff R, Pollock BG, Lewis DA and DeKosky ST (2001) Alterations of striatal dopamine receptor binding in Alzheimer disease are associated with Lewy body pathology and antemortem psychosis. Archives of neurology 58, 466–472. [DOI] [PubMed] [Google Scholar]
- Tata DA and Yamamoto BK (2007) Interactions between methamphetamine and environmental stress: role of oxidative stress, glutamate and mitochondrial dysfunction. Addiction 102 Suppl 1, 49–60. [DOI] [PubMed] [Google Scholar]
- Thal DR and Braak H (2005) [Post-mortem diagnosis of Alzheimer’s disease]. Der Pathologe 26, 201–213. [DOI] [PubMed] [Google Scholar]
- Tomiyama M (2017) [Symptoms and Pathophysiology of Dyskinesias]. Brain Nerve 69, 1409–1416. [DOI] [PubMed] [Google Scholar]
- Venkateshappa C, Harish G, Mythri RB, Mahadevan A, Bharath MM and Shankar SK (2012) Increased oxidative damage and decreased antioxidant function in aging human substantia nigra compared to striatum: implications for Parkinson’s disease. Neurochem Res 37, 358–369. [DOI] [PubMed] [Google Scholar]
- Villemagne VL, Okamura N, Pejoska S et al. (2012) Differential diagnosis in Alzheimer’s disease and dementia with Lewy bodies via VMAT2 and amyloid imaging. Neuro-degenerative diseases 10, 161–165. [DOI] [PubMed] [Google Scholar]
- Weidner AM, Bradley MA, Beckett TL, Niedowicz DM, Dowling AL, Matveev SV, LeVine H 3rd, Lovell MA and Murphy MP (2011) RNA oxidation adducts 8-OHG and 8-OHA change with Abeta42 levels in late-stage Alzheimer’s disease. PLoS One 6, e24930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winterbourn CC (1995) Toxicity of iron and hydrogen peroxide: the Fenton reaction. Toxicol Lett 82–83, 969–974. [DOI] [PubMed] [Google Scholar]
- Wu J, Khan GM and Nichols RA (2007) Dopamine release in prefrontal cortex in response to beta-amyloid activation of alpha7 * nicotinic receptors. Brain Res 1182, 82–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Hassanzadeh B, Chu W, Tu Z, Jones LA, Luedtke RR, Perlmutter JS, Mintun MA and Mach RH (2010) [3H]4-(dimethylamino)-N-(4-(4-(2-methoxyphenyl)piperazin-1-yl) butyl)benzamide: a selective radioligand for dopamine D(3) receptors. II. Quantitative analysis of dopamine D(3) and D(2) receptor density ratio in the caudate-putamen. Synapse (New York, N.Y.) 64, 449–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q, Langley M, Kanthasamy AG and Reddy MB (2017) Epigallocatechin Gallate Has a Neurorescue Effect in a Mouse Model of Parkinson Disease. The Journal of nutrition 147, 1926–1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youdim MBH (2018) Monoamine oxidase inhibitors, and iron chelators in depressive illness and neurodegenerative diseases. Journal of neural transmission (Vienna, Austria : 1996) 125, 1719–1733. [DOI] [PubMed] [Google Scholar]
- Zang X, Cheng ZY, Sun Y, Hua N, Zhu LH and He L (2018) The ameliorative effects and underlying mechanisms of dopamine D1-like receptor agonist SKF38393 on Abeta1–42-induced cognitive impairment. Progress in neuro-psychopharmacology & biological psychiatry 81, 250–261. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.