

Abstract
Alcohol abuse is a major cause of liver disease and mortality worldwide and is a significant public health issue. Patients with alcoholic liver disease (ALD) have severe hepatic lipid accumulation, inflammation, and fibrosis. Therapies for ALD are very limited and even abstinence from alcohol consumption does not necessarily protect patients from progression of the disease. We sought to evaluate the efficacy of a liver X receptor (LXR) inverse agonist, SR9238, in an animal model of ALD. SR9238 suppresses hepatic lipogenesis, a pathological hallmark of ALD, and we hypothesized that targeting suppression of hepatic metabolic pathways that are activated in ALD may be an effective treatment for the disease. A chronic ethanol diet with or without a final ethanol binge treatment was used to induce ALD in mice. Mice were administered the liver specific LXR inverse agonist SR9238 for 4 weeks after the mice had been maintained on the ethanol diet for 14 days. Mice developed all the hallmarks of advanced ALD demonstrating significant pathophysiology and hepatotoxicity. SR9238 significantly attenuated liver injury and hepatic steatosis and fibrosis was nearly eliminated in SR9238 treated mice. SR9238 treatment reversed the damage associated with chronic ethanol use returning the liver to near normal morphology. These results indicate that inhibiting LXR activity using the inverse agonist has a hepatoprotective effect in rodent models of ALD; thus, this pharmacological approach may be efficacious for treatment of ALD in humans.
Keywords: liver disease, fatty liver, NASH, alcohol, LXR, nuclear receptor
Graphical abstract
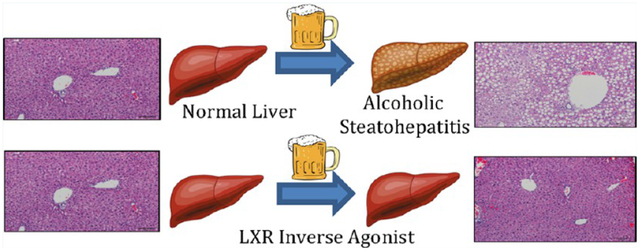
INTRODUCTION
Alcoholic beverages are some of the most commonly abused substances, and thus, alcoholic liver disease (ALD) is a significant health concern. Alcoholism or alcohol dependence is a common disease in the United States and, according to the Mayo Clinic, more than 3 million cases of alcohol dependence are identified every year. Excessive alcohol consumption is responsible for nearly 88,000 deaths annually at the cost of $223.5 billion each year.1 The liver is the main organ responsible for metabolizing ethanol, and ethanol itself is a strong hepatotoxin and can severely injure hepatocytes via its toxic metabolites such as acetaldehyde and free radicals. One of the major byproducts of ethanol metabolism is acetaldehyde, which is highly reactive and toxic, producing tissue damage, and also has been implicated in addiction.2 Currently, abstinence from alcohol and various anti-inflammatory, anticytokine therapies are available for the treatment of ALD. However, these treatments are not very effective, as abstinence does not carry complete resistance to continued progression of ALD, and almost 15% of the individuals will continue to have deterioration of liver function in spite of abstinence. For people with end-stage or advanced alcoholic liver disease (alcoholic cirrhosis), the only treatment option is liver transplant. Yet most candidates with advanced stage ALD do not qualify as suitable organ recipients especially when they are battling alcoholism, which is a significant limitation to transplant intervention.3 Clinical manifestations of ALD include uncontrolled lipid accumulation in hepatocytes thus reducing their functionality, severe oxidative damage leading to cell death and inflammation that finally culminates into alcohol-induced fatty liver disease. With continued abuse, the liver starts to lose its regenerative capacity leading to the formation of scar tissue which is permanent. There is an urgent need for the development of novel therapeutics that would effectively reverse or stop the progression of ALD and prevent any subsequent relapse of the disease.
Ligand regulated nuclear receptors have emerged as effective targets for treating metabolic diseases. The liver x receptors, LXRα and LXRβ, were discovered between 1994 and 1995 initially as orphan members of the nuclear receptor super-family,4–8 but they have subsequently been demonstrated to act as ligand-dependent transcriptional regulators. LXRα is highly expressed in liver, kidney, intestines, fat tissues, macrophages, lung, and spleen, whereas LXRβ is ubiquitously expressed. LXRs are important regulators of cholesterol homeostasis and also play a pivotal role in fatty acid and glucose metabolism. Oxysterols, which are oxygenated derivatives of cholesterol, (22-(R)-hydroxycholesterol, 27-hydroxycholesterol, and cholestenoic acid) have been identified as the endogenous ligands of LXR.9 LXR forms obligate heterodimers with the retinoid X receptor (RXR) and the LXR-RXR heterodimer binds to specific DNA response elements (LXR response elements: LXREs) in the regulatory regions of target genes. LXR’s target genes play a major role in lipid metabolism by regulating uptake, transport, absorption, and excretion of cholesterol and lipids in a tissue specific manner. LXR mediated activation of target genes such as sterol-regulatory element-binding protein 1C (SREBP1c) and fatty acid synthase (FASN). LXRα null mice display defective expression of SREPB1c and FASN, which indicates that LXR plays an important role in the lipogenic programs.10 Many investigators have developed synthetic agonists that effectively target the LXRs,11–13 and these compounds are well-characterized for their ability to induce hepatic steatosis due to increase lipogenesis.10,14–16 More recently, we developed synthetic LXR inverse agonists that specifically target LXR in vivo and suppress the expression of lipogenic enzymes and thus lipogenesis.17–19 We previously demonstrated that the liver selective LXR inverse agonist SR9238 significantly reduced hepatic steatosis and development of nonalcoholic steatohepatitis (NASH) in obese, high fat diet fed mice by suppressing hepatic lipogenesis and inflammation.17,18 Given the similarities in the pathophysiology and progression of nonalcoholic fatty liver disease (NAFLD) and ALD, we hypothesized that this LXR inverse agonist, SR9238 may be able to alleviate hepatic steatosis and inflammation caused by chronic alcohol consumption and subsequently reverse alcohol induced liver injury.
In this study, we demonstrate that by repressing the normal function of the LXR using synthetic inverse agonist, SR9238, we can effectively suppress or reverse the progression of ALD, by blocking lipogenesis, accelerating ethanol metabolism, and preserving healthy liver morphology.
MATERIALS AND METHODS
Animals and Treatments.
Eight-week-old male C57BL/6J mice were purchased from Jackson Laboratories, single housed in standard cages to ensure accurate monitoring of food intake daily. Mice were allowed a week to acclimate before placing them on the Lieber DeCarli diet. Groups were weight matched and body composition was analyzed by conducting NMR, using the Bruker BioSpin LF50 Body Composition Analyzer prior to beginning the liquid ethanol diet, to ensure homogeneity of fat, lean, and fluid mass in between groups. Mice were gradually weaned on to the Lieber DeCarli diet (BioServ, Frenchtown, NJ) with 4.5% (v/v) ethanol (32.4% ethanol-derived calories) over 10 days. Mice were maintained on diet for 2 weeks prior to beginning treatment. After 2 weeks, mice continued on the Lieber DeCarli diet and were simultaneously treated with 30 mg/kg SR9238 q.d i.p. in 10% DMSO/10% Tween-80/80% water or vehicle for 4 weeks. This dosing paradigm was based on pharmacokinetic studies previously published by our lab.17,18 Body weight and food intake were monitored daily. Non-ethanol fed controls were pair-fed receiving an equal number of calories as their alcohol-fed counterparts with the alcohol-derived calories substituted with dextran-maltose. The feeding protocol was followed according to the guidelines established by NIAAA.20 At the end of 4 weeks of dosing, a single dose of ethanol (2.5 g/kg body weight) was administered to a subgroup (ASH model) receiving the chronic ethanol diet by oral gavage at 7.00 am and sacrificed 9 h later. Adding the alcohol binge to the chronic EtOH diet helped us create a group that reliability mirrors ASH pathology with development of hepatic fibrosis. Respective controls received isocaloric dextrin-maltose as their sham binge. At the end of the study, blood was collected by cardiac puncture and analyzed using clinical chemistry (Roche COBAS c311). Whole liver tissues were rapidly excised, weighed, and a portion was immediately flash frozen in liquid nitrogen whereas the rest was placed in 10% neutral buffered formalin and 4% paraformaldehyde for histopathological analysis. Mice were monitored daily for signs of illness, pain, or severe weight loss. All mice were humanely euthanized using CO2 followed by cervical dislocation. The Institutional Animal Care and Use Committee at Saint Louis University, St. Louis, MO, approved all mouse studies.
Real-Time Quantitative PCR.
Total RNA was isolated from liver tissue using Trizol extraction method and analyzed by QPCR as previously described.18 RNA was reverse transcribed using the iScript cDNA synthesis kit (Quanta). The synthesized cDNA was then assessed for gene expression using cognate primers.
Immunoblotting.
Total protein was isolated from flash frozen liver tissue as previously described.21 Specific antibodies against FASN (Cell Signaling C20G5), SREBP1 (Novus NB600-582), ADH (Abcam ab108203), CYP2E1 (Abcam Ab19140), ALDH1/2 (SantaCruz sc-166362) were used to quantify protein expression.
Histology and Immunofluorescence.
Liver sections were excised from identical lobes and fixed in 10% neutral buffered formalin overnight and then embedded in paraffin for sectioning. Another set of livers were placed in 4% PFA overnight and then transferred to 10% sucrose in PBS for 2 days followed by 20% sucrose in PBS for another 2 days and finally 30% sucrose in PBS for cryoprotection. These tissues were then embedded in OCT and frozen in 2-methylbutane dry ice bath. Livers were sectioned at 10 μm and placed on slides for H&E and PicoSirius Red Staining (Direct Red-80; Sigma) or floated in 6-well dishes in 1X PBS for Bodipy 493/503 (Molecular Probes) staining of neutral lipids.17 Sections were stained and mounted on slides prior to counterstaining with DAPI.
Terminal Deoxynucleotidyl Transferase-Mediated dUTP Nick-End Labeling (TUNEL).
Apoptotic cells in liver tissue were detected by terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) using ApopTag In Situ Apoptosis Detection Kits (Milipore Sigma, cat. no. S7165). Liver tissues were collected and fixed as described above. Tissues were incubated for 1 h at 37 °C with TUNEL working strength TdT enzyme reaction mixture. Post three washes with PBS, tissue sections were incubated with antidigoxigenin conjugate (rhodamine) for 30 min at room temperature. Sections were counterstained with DAPI. TUNEL-positive nuclei were visualized with an Olympus fluorescence microscope using rhodamine and DAPI filters. TUNEL positive nuclei were counted manually and quantified using ImageJ.
Immunohistochemistry (IHC).
Formalin fixed paraffin embedded tissues were sectioned at 10 μm and mounted on slides. Slides were deparaffinized in xylene and rehydrated in EtOH. After performing antigen retrival with 1 × DIVA (DV2004MX, Biocare Medical), and blocking with 5% DK serum (donkey serum, 017-000-121; Jackson Immunoresearch), sections were washed and incubated with primary antibodies (anti-CD68 (Abcam Ab125212) 1:1000; anti-IL6 (Bioss BS-0782R) 1:200) overnight at 4 °C. Sections were washed and treated with secondary HRP-conjugate (DK antirabbit 1:300) for 1 h. Sections were incubated with DAB substrate until desired staining was achieved (45 s–1 min); counterstained with hematoxylin. Slides were dehydrated and mounted with Permount mounting media before imaging
Statistical Analysis.
qPCR, immunoblot, and histological quantification of fluorescent signals data were represented as mean ± SEM, and statistical significance was determined by subjecting mean values per group to Student’s t-test unless otherwise specified. A value of p 0.05 is considered statistically significant.
RESULTS
LXR Inverse Agonist Treatment Significantly Reduces Liver Injury Induced by Ethanol.
In 2013, Bertola et al. described the NIAAA mouse model of chronic and binge ethanol consumption. They showed that administration of the Lieber DeCarli diet for 4–6 weeks leads to the development of steatosis with elevated serum transaminases.20 Gao and coworkers later demonstrated that administration of a secondary insult, in the form of an ethanol binge, exacerbated the liver injury induced by chronic administration of ethanol leading to hepatic fibrosis,22 which much more reliably replicates the pathology of ALD in humans. We utilized this diet induced chronic and chronic plus binge model of alcohol abuse to test the efficacy of SR9238. Eight-week old C57BL6/J male mice were subjected to the Lieber DeCarli diet for 2 weeks and then treated with 30 mg/kg of LXR inverse agonist SR9238 for the following 4 weeks. The chronic ethanol diet or the SR9238 treatment did not affect the gross body weight of these mice (Supporting Information, Figure S-1A). The liver is an organ that is highly susceptible to xenobiotic induced injury since it is primarily involved in its metabolism. Hepatotoxicity of a xenobiotic agent such as alcohol, drugs and environmental chemicals is positively correlated to the gross liver weight to body weight ratio, thus we assessed this end point to determine if SR9238 had any therapeutic effects. There was a significant reduction in liver weight in both the chronic as well as chronic plus binge (hereafter referred to as the alcoholic steatohepatitis (ASH) model) mice that received the SR9238 treatment. However, SR9238 treatment alone did not affect the liver weights of the pair-fed controls (Figure 1A). This indicates that there was no hepatotoxicity induced in the drug treated group, and in fact, suggests that SR9238 protected against the hepatotoxic effect of ethanol. Alcohol is a well-known risk factor for obesity, and chronic alcohol abuse is positively correlated to increase in central adiposity.23,24 We observed that chronic EtOH administration increased fat mass and an EtOH-dependent decrease in lean mass (Figure 1B), which is consistent with past published studies in which loss in skeletal muscle mass was one of the effects of chronic alcohol abuse.25,26 SR9238 did not protect the mice from these effects and SR9238 did not alter body composition in the pair fed mice either (Figure 1B). Alanine aminotransferase (ALT) is an enzyme found primarily in the liver and is one of the primary diagnostic markers for liver injury and hepatocellular damage. In case of any liver disease, damage or injury, ALT is released into the bloodstream. It has been shown that mice on chronic ethanol diet for 4 weeks display elevated serum ALT and extensive hepatic steatosis which leads to an increase in mortality.27 As expected, the mice on the chronic ethanol diet had elevated plasma ALT levels (Figure 1C). Interestingly, SR9238 significantly lowered the elevated ALT levels that were induced by chronic ethanol consumption with or without the binge EtOH component (Figure 1C). SR9238 treatment also significantly reduced levels of total circulating cholesterol and specifically HDL (Figure 1D,E), which is consistent with our previous studies examining this compound in mouse models including those of hepatic steatosis and NASH.17,18,21
Figure 1.

SR9238 protects mice against ethanol induced hepatic injury. (A) Whole liver weight normalized to total body weight of mice on Lieber DeCarli ethanol diet (EtOH) and Pair Fed (PF) controls with SR9238 treatment or vehicle. Mice received a 2.5 g/kg of ethanol binge (+) vs isocaloric maltose (−) 9 h prior to sacrifice (N = 10; results presented as mean ± SEM, two way-ANOVA performed). (B) % Fat and % lean showing differences in total fat mass and total lean mass per body weight before (initial) and end of (final) study (N = 10; results presented as mean ± SEM, two way-ANOVA performed). (C) Clinical analysis of plasma showing, SR9238 protects from alcohol induced hepatocellular damage and shows therapeutic properties by reducing ALT levels (N = 10; results presented as mean ± SEM, two way-ANOVA performed). (D) Clinical analysis of plasma from mice exposed to chronic EtOH and EtOH plus binge (ASH model) with or without SR9238 treatment. Levels of circulating plasma cholesterol decreased on SR9238 treatment (N = 10; results presented as mean ± SEM, two way-ANOVA performed). (E) Clinical analysis of plasma showing effect of SR9238 treatment on HDL levels (N = 10; results presented as mean ± SEM, two way-ANOVA performed). (F) Representative images of H&E staining of mice livers showing severe hepatocellular damage and inflammation with ballooning as well as micro- and macrosteatosis in vehicle treated liver sections, was rescued by SR9238 treatment (N = 6). *p < 0.05, * *p < 0.01, * * *p < 0.001, and * * * *p < 0.0001.
Hematoxylin and eosin staining of fixed liver tissue demonstrated that SR9238 treatment resulted in substantially improved liver morphology in the mice maintained on the chronic EtOH and ASH diet (Figure 1F). The vehicle controls show classic pathophysiological signs of alcoholic fatty liver disease with significant macro- and microvesicular steatosis, hepatocellular ballooning, and degenerating hepatocytes along with inflammatory infiltrates consisting of neutrophilic and mononuclear cells (Figure 1F). These pathophysiological signs were significantly ameliorated by SR9238 treatment (Figure 1F). These data clearly indicate that SR9238 is quite effective at protecting the liver against alcohol induced hepatotoxicity and hepatocellular damage and significantly improves hepatic morphology in both chronic EtOH and ASH models.
SR9238 Prevents Alcohol Induced Fibrosis and Lipid Accumulation in Rodent Livers.
Progression of ASH is characterized by not only severe lipid accumulation and inflammation but also collagen deposits in the liver leading to formation of scar tissue, also known as liver fibrosis. We assessed the ability of SR9238 to prevent liver fibrosis assessing Sirius red staining. Incorporation of the final binge with the chronic EtOH recapitulates all the hallmarks of an ASH model which makes the chronic plus binge an effective model of ASH. The ASH model treatment paradigm has been previously demonstrated to induce very significant liver fibrosis28 and indeed we observed significant collagen staining in liver sections with this ASH model vs the chronic EtOH only group (Figure 2A) Most importantly, SR9238 treatment significantly suppressed fibrosis in both groups as indicated by reduced collagen staining (Figure 2A). Total collagen staining was reduced nearly by 90% with SR9238 treatment. This suggests that SR9238 has a protective role in preventing and reducing fibrosis in animals with ASH. One of the main hallmarks of ALD is severe lipid accumulation in the liver which leads to liver hypoxia and impaired hepatocellular function and ultimately death of the hepatocytes.29 To visualize the extent of hepatic lipid accumulation resulting from the ASH model as well as the chronic EtOH diet we utilized Bodipy 493/503 neutral lipid staining technique. Consistent with our results, Bodipy staining 493/503 neutral lipid staining demonstrated a remarkable reduction in lipid accumulation in the liver of the mice treated with SR9238 in both of our chronic and ASH group compared to the vehicle treated (Figure 2B). In summary, these data clearly indicate that SR9238 is not only effective at protecting the liver against alcohol induced hepatocellular damage and preventing alcoholic hepatitis and hepatic fibrosis but is also efficacious in suppressing alcohol-induced lipogenesis and hepatic steatosis, which are hallmarks of severe ALD.
Figure 2.

SR9238 prevents alcohol induced fibrosis and lipid accumulation in rodent livers. (A) Representative images of PicoSirius Red stain showing marked difference in collagen staining with SR9238 treatment demonstrating SR9238’s ability to reduce and prevent alcohol induced hepatic fibrosis. %Collagen stained quantified using ImageJ (N = 6). (B) Representative images demonstrating how SR9238 treatment significantly reduces alcohol induced accumulation of neutral lipids in the liver, demonstrated by Bodipy 493/503 (N = 6). Images were quantified using ImageJ. *p < 0.05 and * * * *p < 0.0001.
SR9238 Significantly Reduces Expression of Ethanol-Induced Key Hepatic Lipogenic Markers.
Chronic alcohol abuse is known to impair lipid metabolism leading to the development of fatty liver disease. It has been shown that with just 2 weeks of chronic ethanol administration that there is an increase level of hepatic free fatty acid and a decrease in mitochondrial fatty acid oxidation.30 Given that we observed the marked reduction in lipid accumulation with SR9238 treatment, we wanted to assess the effect of SR9238 on key hepatic lipogenic markers. SR9238 was developed to suppress the endogenous lipogenic activity of LXR in the liver on a transcriptional level.17 Given that certain lipogenic LXR target genes such as Srebp-1c and Fasn are involved in the de novo lipogenesis, which is directly upregulated by the chronic administration of alcohol,31 we investigated the effect of SR9238 on these key hepatic lipogenic genes. SR9238 treatment significantly suppressed the hepatic expression of Fasn, Srebf as well as Srebp1c (Figure 3A–C). We also examined the level of protein expression of a subset of these LXR targets that are known to be major drivers of lipogenesis in ALD.31 Consistent with our gene expression results, SR9238 significantly suppressed FASN in the ASH model and SREBP1 protein expression in both chronic EtOH as well as the ASH model groups (Figure 3D). These findings suggest that SR9238 protects against alcohol induced de novo lipogenesis and the resulting hepatic steatosis by suppressing expression of key hepatic lipogenic enzymes. In summary, these results suggest that SR9238 is efficacious in suppressing alcohol-induced lipogenesis and hepatic steatosis.
Figure 3.

SR9238 treatment significantly reduces lipogenesis and hepatic steatosis. RT-QPCR showing that SR9238 significantly suppresses genes encoding enzymes of the lipogenic pathway in the liver: (A) Fasn; (B) Srebp1c; (C) Srebf; (N = 10) as well as protein levels of (D) FASN and SREBP1 (N = 4). (Right panel) quantification of the Western blots shown in panels D: *p < 0.05 and * *p < 0.01.
Inhibiting LXR Activity Leads to Altered Expression of Alcohol Metabolism Enzymes.
Chronic and or binge ethanol abuse leads to increased oxidative damage due to production of free radicals in the liver through the process of ethanol metabolism. Production of acetaldehyde from the metabolism of alcohol results in production of reactive oxygen species (ROS) that leads to severe oxidative stress in the liver. Oxidation of ethanol also leads to the production of NADH that alters the NADH/NAD+ ratio, which results in suppression of gluconeogenesis and subsequently acetyl-CoA being diverted to ketogenesis and fatty acid synthesis. The disrupted NADH/NAD+ ratio also suppresses mitochondrial fatty acid β-oxidation, thereby resulting in fat accumulation in the liver.32 At high concentrations, when alcohol needs to be eliminated rapidly, an alternate pathway, known as the microsomal ethanol oxidizing system, comes into play that depends on the activity of the enzyme CYP2E1.2 Interestingly, in many cases SR9238-treated mice that were on the chronic as well as the ASH diet showed elevated levels of ethanol metabolizing enzymes CYP2E1 and alcohol dehydrogenase (ADH) at both the mRNA and protein levels (Figure 4A–D). We did not observe an increase in the expression of these gene in mice receiving the control diet thus the induction does not appear to be through direct regulation by the LXR inverse agonist (Figure S-2a,b,c). Given that the SR9238-treated mice were subjected to considerably less hepatocellular damage, we postulated that the elevated levels of these enzymes were likely due to preservation of hepatocellular function. Intriguingly, in the chronic EtOH model, SR9238 treatment increased Cyp2e1 gene expression while the enzyme expression was suppressed (Figure 2a,c). It is unclear what the cause of this discordance may be, but it appears to be specific for CYP2E1 since a similar trend was not observed with ADH. Additionally, the SR9238-dependent increase in ADH was lost following the binge and it is unclear why this would occur for ADH, but not CYP2E1 expression. Aldehyde dehydrogenase 1 and 2 (ALDH1/2) are the two most important enzymes involved in the conversion of acetaldehyde, the primary toxic metabolite of ethanol metabolism, to nontoxic acetate. ALDH1/2 expression was significantly upregulated in the SR9238-treated mice compared to its vehicle controls (Figure 4D), likely due to improved liver function as indicated above. There were no changes in the expression of Aldh genes, however, suggesting that the alterations in expression of ALDH1/2 may have been due to post-transcriptional events (data not shown).
Figure 4.

SR9238 enhances expression of alcohol metabolizing enzymes. RT-QPCR showing that SR9238 treatment significantly increases the expression of markers of ethanol metabolism on a transcriptional level: (A) Cyp2e1; (B) Adh2; (C) Adh3; (N = 10) as well as on a post translational level; (D) CYP2E1, ADH, ALDH1/2 (N = 4). (Bottom panels) quantification of the Western blot shown in panel D: *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001
SR9238 Prevents Apoptosis of Hepatocytes Induced by Chronic and Binge Alcohol Exposure.
Apoptosis is a well identified pathological feature contributing to ethanol induced tissue injury. In the liver, alcohol-induced apoptosis is a hallmark of alcoholic steatohepatitis. To assess the level of apoptosis in the livers of mice receiving various treatments we utilized TUNEL staining. It is well established that EtOH treatment increases the number of apoptotic nuclei in the liver as detected by TUNEL staining.28,33–35 We observed a significant increase in the number of TUNEL positive cells in the ASH model-vehicle treated group versus the corresponding SR9238-treated group (Figure 5A,B). These data are consistent with the concept that SR9238 exhibits hepatoprotective effects in this model of ALD. In the chronic EtOH group without the binge, the level of TUNEL positive cells under these conditions was markedly less than ASH model animals. Interestingly, SR9238 treatment did not alter the rate of apoptosis in the livers of mice in the chronic EtOH group suggesting that SR9238 may selectively protect against binge induced acute alcohol toxicity.
Figure 5.

SR9238 prevents hepatocyte apoptosis caused by ethanol diet. Representative images of apoptosis staining showing TUNEL positive cells (green arrows) in mouse liver. TUNEL positive cells were manually counted and quantified using ImageJ software. Representative images were taken at 20× magnification. (N = 5) **p < 0.01.
SR9238 Treatment Prevents Inflammation Induced by Chronic As Well As Chronic Plus Binge Alcohol Exposure.
Along with hepatic lipid accumulation and apoptosis, inflammation is another key hallmark of alcoholic steatohepatitis. Progression of ALD is not due to one pathological factor such as lipid accumulation but a synergistic effect of excessive lipid accumulation in the hepatocytes leading to inflammation followed by apoptosis. Multiple studies conducted in both rodents as well as human subjects have shown significant upregulation of pro-inflammatory markers in the liver such as IL-6 and CD68.36 CD68 is a key marker for hepatic macrophages (Kupffer cells) that propagate pro-inflammatory signals in response to chronic and acute alcohol induced liver injury. It has been shown that depletion or inactivation of Kupffer cells in the liver prevents alcohol induced liver injury.37–39 We performed immunohistochemical staining on liver sections to assess the effect of SR9238 treatment on pro-inflammatory IL6 expression and Kupffer cell activity. SR9238 treatment significantly suppressed Kupffer cells activation (Figure 6A,C,D) as indicated by reduced hepatic CD68+ cell numbers and diminished CD68 marker expression (Figure 6A,C,D). Expression of the pro-inflammatory cytokine IL-6, known to be involved in the pathogenesis of ALD was also significantly reduced in the livers of mice treated with SR9238 (Figure 6B,E). These findings suggest that LXR inverse agonist, SR9238 is able to suppress alcohol induced inflammation and lipogenesis which has a combined hepatoprotective effect through the reduction of hepatocyte cell-death.
Figure 6.

SR9238 treatment prevents inflammation caused by ethanol diet. Representative images showing SR9238 treatment causes reduction in inflammation. (A) Immunohistochemical expression of CD68 positive cells (black arrows) in mouse liver. (B) Immunohistochemical expression of IL-6 (brown) shown in mouse livers. (Inset: Higher magnification showing clustering of IL-6 around the vessels). (C) Quantification of the number of CD68+ cells and (D) mean color intensity (MCI) of the positively stained punctae shown in panel A. CD68 positive cells/MCI were counted and quantified using Olympus CellSens Dimensions 1.16 software. Representative images were taken at 20× magnification (N = 4). (E) Quantification of the intensity of IL-6 staining represented as Integrated Optical Density) IL-6 staining was quantified using ImageJ software. Representative images were taken at 20× magnification (N = 4).
DISCUSSION
Alcohol abuse including binge drinking is a significant public health issue that is leading to an increased incidence of ALD. ALD has a wide range of manifestations consisting of steatosis, steatohepatitis, cirrhosis and carcinoma. Current therapeutics for ALD include corticosteroid and anticytokine therapy that only target the inflammatory aspect of the disease. Corticosteroids have been used over 35 years for the treatment of ALD and they have the potential to inhibit pro-inflammatory transcription factors such as activator protein-1 (AP-1) and nuclear factor-κB, which may be beneficial in the treatment of ALD. There are several limitations associated with corticosteroids and anticytokine therapy including general immunosuppression and metabolic and bone side effects. In this study, we demonstrated that by targeting inhibition of de novo lipogenesis with an LXR inverse agonist we can effectively treat ALD. SR9238 treatment eliminated hepatosteatosis associated with chronic EtOH diet with or without a final binge EtOH treatment (ASH model). Furthermore, liver fibrosis, which was particularly evident in the chronic EtOH plus binge treatment group, was also substantially reduced by SR9238 treatment. Consistent with these observations liver weights were also reduced by SR9238 treatment as well as plasma ALT levels.
Unexpectedly, in our study, we discovered that mice treated with SR9238 displayed higher hepatic CYP2E1, ADH, and ALDH1/2 expression than in vehicle-treated mice when maintained on EtOH diets. These enzymes play a critical role in metabolism of EtOH and are induced by EtOH exposure. ADHs and CYP2E1 are involved in its conversion to acetaldehyde and ALDHs convert acetaldehyde to acetate and increased expression of these enzymes is associated with a protective effect against the EtOH induced hepatotoxicity.40 Interestingly, SR9238 treatment did not increase the expression of these genes in mice receiving a non-EtOH diet (pair-fed) suggesting that CYP2E1 induction was due to direct regulation by LXR or a xenobiotic response to SR9238; rather we believe that the SR9238 was effective enough in protecting the hepatocytes from the toxic effects of EtOH that the elevated enzyme levels were due to the greater number of functional hepatocytes present in the SR9238 treated mice. This idea is also consistent with our findings that SR9238 was protective against EtOH-induced hepatocyte apoptosis.
It has been shown that in models of alcoholic as well nonalcoholic fatty liver disease, macrophage activity leads to rapid apoptotic hepatic cell death.41 LXRs have a well-documented role in regulating macrophage activity and inflammation. We show here that SR9238 stymied stellate cell activity and hepatic IL6 production. Whether the hepatoprotective effect of LXR suppression is predominantly dependent on the antilipogenic effects or vice versa is unclear and should be the subject of further investigations. All these findings suggest that suppression of LXR activity promoted a strong hepatoprotective effect by both also reducing hepatic inflammation.
In summary, targeting LXR with an inverse agonist displays efficacy in a model of ALD and, most importantly, targets the key pathological hallmarks of the disease. SR9238 treatment considerably improves histopathology associated with alcohol induced liver injury. SR9238, therapeutically targets ALD by suppressing de novo lipogenesis and reducing lipid accumulation in the hepatocytes by >90%, which is sufficient to not only prevent excessive lipid accumulation in the liver, but also restores liver function by preventing excessive inflammation, rapid apoptosis, and protecting against chronic and ethanol-binge induced liver injury without any noted toxic side effects. Alcoholism and alcohol abuse is a growing global health concern and it has deleterious effects on the liver. We propose that by repressing the normal function of the liver X receptor using synthetic inverse agonist, such as SR9238, we can effectively suppress or reverse the progression of ALD. Given the paucity of therapies to treat or prevent liver damage due to ALD, the LXR inverse agonists represent a potentially important new pathway for therapeutic intervention.
Supplementary Material
Footnotes
The authors declare no competing financial interest.
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsptsci.8b00003.
Figure S-1: SR9238 treatment did not severely effect body weight, expression of alcohol metabolism in mice not exposed to ethanol. Figure S-2: SR9238 treatment does not affect inflammatory marker activity in mice not exposed to ethanol (PDF)
REFERENCES
- (1).Esser MB, Hedden SL, Kanny D, Brewer RD, Gfroerer JC, and Naimi TS (2014) Prevalence of alcohol dependence among US adult drinkers, 2009–2011. Preventing chronic disease 11, 140329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Zakhari S (2006) Overview: how is alcohol metabolized by the body? Alcohol research & health: the journal of the National Institute on Alcohol Abuse and Alcoholism 29 (4), 245–254. [PMC free article] [PubMed] [Google Scholar]
- (3).Alcohol Related Liver Disease Brochure 2017; American Liver Foundation, 2017. [Google Scholar]
- (4).Apfel R, Benbrook D, Lernhardt E, Ortiz MA, Salbert G, and Pfahl M (1994) A novel orphan receptor specific for a subset of thyroid hormone responsive elements and its interaction with the retinoid/thyroid hormone receptor subfamily. Mol. Cell. Biol 14 (10), 7025–7035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Willy PJ, Umesono K, Ong ES, Evans RM, Heyman RA, and Mangelsdorf DJ (1995) Lxr, a Nuclear Receptor That Defines a Distinct Retinoid Response Pathway. Genes Dev. 9 (9), 1033–1045. [DOI] [PubMed] [Google Scholar]
- (6).Teboul M, Enmark E, Li Q, Wikstrom AC, Peltohuikko M, and Gustafsson JA (1995) Or-1, a Member of the Nuclear Receptor Superfamily That Interacts with the 9-Cis-Retinoic Acid Receptor. Proc. Natl. Acad. Sci. U. S. A 92 (6), 2096–2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Shinar DM, Endo N, Rutledge SJ, Vogel R, Rodan GA, and Schmidt A (1994) Ner, a New Member of the Gene Family Encoding the Human Steroid-Hormone Nuclear Receptor. Gene 147 (2), 273–276. [DOI] [PubMed] [Google Scholar]
- (8).Song C, Kokontis JM, Hiipakka RA, and Liao SS (1994) Ubiquitous Receptor - a Receptor That Modulates Gene Activation by Retinoic Acid and Thyroid-Hormone Receptors. Proc. Natl. Acad. Sci. U. S. A 91 (23), 10809–10813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Janowski BA, Willy PJ, Devi TR, Falck JR, and Mangelsdorf DJ (1996) An oxysterol signalling pathway mediated by the nuclear receptor LXR alpha. Nature 383 (6602), 728–31. [DOI] [PubMed] [Google Scholar]
- (10).Repa JJ, Liang G, Ou J, Bashmakov Y, Lobaccaro JM, Shimomura I, Shan B, Brown MS, Goldstein JL, and Mangelsdorf DJ (2000) Regulation of mouse sterol regulatory element-binding protein-1c gene (SREBP-1c) by oxysterol receptors, LXRalpha and LXRbeta. Genes Dev 14 (22), 2819–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Michael LF, Schkeryantz JM, and Burris TP (2005) The pharmacology of LXR. Mini-Rev. Med. Chem 5 (8), 729–40. [DOI] [PubMed] [Google Scholar]
- (12).Komati R, Spadoni D, Zheng S, Sridhar J, Riley KE, and Wang G (2017) Ligands of Therapeutic Utility for the Liver X Receptors. Molecules 22 (1), 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Hong C, and Tontonoz P (2014) Liver X receptors in lipid metabolism: opportunities for drug discovery. Nat. Rev. Drug Discovery 13 (6), 433–44. [DOI] [PubMed] [Google Scholar]
- (14).Gao M, Bu L, Ma Y, and Liu D (2013) Concurrent activation of liver X receptor and peroxisome proliferator-activated receptor alpha exacerbates hepatic steatosis in high fat diet-induced obese mice. PLoS One 8 (6), e65641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Grefhorst A, Elzinga BM, Voshol PJ, Plosch T, Kok T, Bloks VW, van der Sluijs FH, Havekes LM, Romijn JA, Verkade HJ, and Kuipers F (2002) Stimulation of lipogenesis by pharmacological activation of the liver X receptor leads to production of large, triglyceride-rich very low density lipoprotein particles. J. Biol. Chem 277 (37), 34182–90. [DOI] [PubMed] [Google Scholar]
- (16).Chisholm JW, Hong J, Mills SA, and Lawn RM (2003) The LXR ligand T0901317 induces severe lipogenesis in the db/db diabetic mouse. J. Lipid Res 44 (11), 2039–2048. [DOI] [PubMed] [Google Scholar]
- (17).Griffett K, Solt LA, El-Gendy Bel D, Kamenecka TM, and Burris TP (2013) A liver-selective LXR inverse agonist that suppresses hepatic steatosis. ACS Chem. Biol 8 (3), 559–567. [DOI] [PubMed] [Google Scholar]
- (18).Griffett K, Welch RD, Flaveny CA, Kolar GR, Neuschwander-Tetri BA, and Burris TP (2015) The LXR inverse agonist SR9238 suppresses fibrosis in a model of non-alcoholic steatohepatitis. Mol. Metab 4 (4), 353–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Flaveny CA, Griffett K, El-Gendy BE, Kazantzis M, Sengupta M, Amelio AL, Chatterjee A, Walker J, Solt LA, Kamenecka TM, and Burris TP (2015) Broad Anti-tumor Activity of a Small Molecule that Selectively Targets the Warburg Effect and Lipogenesis. Cancer Cell 28, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Bertola A, Mathews S, Ki SH, Wang H, and Gao B (2013) Mouse model of chronic and binge ethanol feeding (the NIAAA model). Nat. Protoc 8 (3), 627–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Flaveny CA, Griffett K, El-Gendy BE-DM, Kazantzis M, Sengupta M, Amelio AL, Chatterjee A, Walker J, Solt LA, Kamenecka TM, and Burris TP (2015) Broad Anti-tumor Activity of a Small Molecule that Selectively Targets the Warburg Effect and Lipogenesis. Cancer Cell 28 (1), 42–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Xu MJ, Cai Y, Wang H, Altamirano J, Chang B, Bertola A, Odena G, Lu J, Tanaka N, Matsusue K, Matsubara T, Mukhopadhyay P, Kimura S, Pacher P, Gonzalez FJ, Bataller R, and Gao B (2015) Fat-specific Protein 27/CIDEC Promotes Development of Alcoholic Steatohepatitis in Mice and Humans. Gastroenterology 149 (4), 1030–1041.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Traversy G, and Chaput JP (2015) Alcohol Consumption and Obesity: An Update. Current Obesity Reports 4 (1), 122–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Wannamethee SG, Shaper AG, and Whincup PH (2005) Alcohol and adiposity: effects of quantity and type of drink and time relation with meals. Int. J. Obes 29 (12), 1436–44. [DOI] [PubMed] [Google Scholar]
- (25).Steiner JL, and Lang CH (2015) Alcohol intoxication following muscle contraction in mice decreases muscle protein synthesis but not mTOR signal transduction. Alcohol.: Clin. Exp. Res 39 (1), 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Thapaliya S, Runkana A, McMullen MR, Nagy LE, McDonald C, Naga Prasad SV, and Dasarathy S (2014) Alcohol-induced autophagy contributes to loss in skeletal muscle mass. Autophagy 10 (4), 677–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Petrasek J, Bala S, Csak T, Lippai D, Kodys K, Menashy V, Barrieau M, Min SY, Kurt-Jones EA, and Szabo G (2012) IL-1 receptor antagonist ameliorates inflammasome-dependent alcoholic steatohepatitis in mice. J. Clin. Invest 122 (10), 3476–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Wu D, and Cederbaum AI (1999) Ethanol-induced apoptosis to stable HepG2 cell lines expressing human cytochrome P-4502E1. Alcohol.: Clin. Exp. Res 23 (1), 67–76. [PubMed] [Google Scholar]
- (29).Nath B, Levin I, Csak T, Petrasek J, Mueller C, Kodys K, Catalano D, Mandrekar P, and Szabo G (2011) Hepatocyte-specific hypoxia-inducible factor-1alpha is a determinant of lipid accumulation and liver injury in alcohol-induced steatosis in mice. Hepatology 53 (5), 1526–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Clugston RD, Jiang H, Lee MX, Piantedosi R, Yuen JJ, Ramakrishnan R, Lewis MJ, Gottesman ME, Huang LS, Goldberg IJ, Berk PD, and Blaner WS (2011) Altered hepatic lipid metabolism in C57BL/6 mice fed alcohol: a targeted lipidomic and gene expression study. J. Lipid Res 52 (11), 2021–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Sozio M, and Crabb DW (2008) Alcohol and lipid metabolism. American journal of physiology. Endocrinology and metabolism 295 (1), E10–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Stewart S, Jones D, and Day CP (2001) Alcoholic liver disease: new insights into mechanisms and preventative strategies. Trends Mol. Med 7 (9), 408–13. [DOI] [PubMed] [Google Scholar]
- (33).Higuchi H, Adachi M, Miura S, Gores GJ, and Ishii H (2001) The mitochondrial permeability transition contributes to acute ethanol-induced apoptosis in rat hepatocytes. Hepatology 34 (2), 320–328. [DOI] [PubMed] [Google Scholar]
- (34).Higuchi H, Kurose I, Kato S, Miura S, and Ishii H (1996) Ethanol-induced apoptosis and oxidative stress in hepatocytes. Alcohol.: Clin. Exp. Res 20 (s9), 340a–346a. [PubMed] [Google Scholar]
- (35).Zhou Z, Sun X, and Kang YJ (2001) Ethanol-induced apoptosis in mouse liver: Fas- and cytochrome c-mediated caspase-3 activation pathway. Am. J. Pathol 159 (1), 329–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Wang HJ, Gao B, Zakhari S, and Nagy LE (2012) Inflammation in Alcoholic Liver Disease. Annu. Rev. Nutr 32, 343–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Adachi Y, Bradford BU, Gao W, Bojes HK, and Thurman RG (1994) Inactivation of Kupffer cells prevents early alcohol-induced liver injury. Hepatology 20 (2), 453–60. [PubMed] [Google Scholar]
- (38).Thurman RG (1998) Alcoholic liver injury involves activation of Kupffer cells by endotoxin. American journal of physiology 275 (4 Pt 1), G605–11. [DOI] [PubMed] [Google Scholar]
- (39).Dong D, Zhong W, Sun Q, Zhang W, Sun X, and Zhou Z (2016) Oxidative products from alcohol metabolism differentially modulate pro-inflammatory cytokine expression in Kupffer cells and hepatocytes. Cytokine+ 85, 109–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Zhong W, Zhang W, Li Q, Xie G, Sun Q, Sun X, Tan X, Sun X, Jia W, and Zhou Z (2015) Pharmacological activation of aldehyde dehydrogenase 2 by Alda-1 reverses alcohol-induced hepatic steatosis and cell death in mice. J. Hepatol 62 (6), 1375–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Wan J, Benkdane M, Teixeira-Clerc F, Bonnafous S, Louvet A, Lafdil F, Pecker F, Tran A, Gual P, Mallat A, Lotersztajn S, and Pavoine C (2014) M2 Kupffer cells promote M1 Kupffer cell apoptosis: a protective mechanism against alcoholic and nonalcoholic fatty liver disease. Hepatology 59 (1), 130–42. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.