

Abstract
In the past decades, the cardiovascular community has laid out the fundamental signaling cascades that become awry in the cardiomyocyte during the process of pathologic cardiac remodeling. These pathways are initiated at the cell membrane and work their way to the nucleus to mediate gene expression. Complexity is multiplied as the cardiomyocyte is subjected to cross-talk with other cells as well as a barrage of extracellular stimuli and mechanical stresses. In this review, we summarize the signaling cascades that play key roles in cardiac function and then we proceed to describe emerging concepts of how the cardiomyocyte senses the mechanical and environmental stimuli to transition to the deleterious genetic program that defines pathologic cardiac remodeling. As a highlighting example of these processes, we illustrate the transition from a compensated hypertrophied myocardium to a decompensated failing myocardium, which is clinically manifest as decompensated heart failure.
INTRODUCTION
The cardiomyocyte constitutes approximately one-third of the cellular makeup of the heart, but accounts for three quarters of the myocardial volume [1]. The heart, and in the particular the cardiomyocyte, is subjected to a barrage of stimuli; the response of the cardiomyocyte to these stimuli dictates the function of the organ, and ultimately the host. The cardiomyocyte is a terminally differentiated cell. Upon insult such as infarction, the remaining cardiomyocytes cannot undergo hyperplasia. Instead, the remaining cardiomyocytes can increase protein synthesis, cell size, the number of sarcomeres, the number of contractile proteins, and the number of mitochondria [2]. The hypertrophy and elongation of the cardiomyocyte is reflected in the increased mass of the ventricle.
The adult heart undergoes “physiological hypertrophy” in response to pregnancy and exercise. Physiologic and pathophysiologic stimuli act upon membrane-bound receptors that in turn activate intracellular signal transduction pathways such as the mitogen-activated protein kinase (MAPK) cascade, calcineurin-NFAT (nuclear factor of activated T cells), insulin-like growth factor-I–phosphatidylinositol 3-kinase–Akt/protein kinase B, amongst others. The cascades elicited by these stimuli are ultimately reflected at the gene transcription level. [3–8]
The pathologic responses that the heart can be exposed to include, but are not limited to, inflammation (myocarditis), ischemia, ischemia-reperfusion injury, biomechanical stress, excess neurohormonal activation, excess afterload (hypertension, aortic stenosis), and cytokine storm amongst others [2]. Cardiac remodeling is a complex process that summarizes the alterations in cardiac structure, shape, and function that result from pathologic stimuli. Cardiac remodeling encompasses the changes at the cellular and gene expression that occur upon insult. Grossly, the size, shape, and thickness changes that constitute cardiac remodeling influence the ventricular function and prognosis following insult [9–11]. The extracellular matrix, cellular architecture (comprising the sarcomeres), and genetic programs all contribute to this process. Remodeling encompasses both the acute events following insult (within ninety minutes of an ST-Elevation Myocardial Infarction) as well as the long-standing events (lasting up to months or even years following the insult) [1]. Cardiac remodeling is mediated by cardiomyocytes, fibroblasts, myofibroblasts, immune cells, and the endothelial cells [12].
There are two manifestations of hemodynamic overload mediated cardiac hypertrophy: [9]
Hypertension and aortic stenosis impart Pressure Overload. At the cellular level, this leads to addition of sarcomeres in parallel, increased myocyte cross-sectional area, and pronounced ventricular wall thickening. Grossly, clinicians refer to this as concentric hypertrophy.
Mitral regurgitation and aortic regurgitation impart Volume Overload. This is grossly manifest as eccentric hypertrophy, and reflects ventricular dilation mediated by an increase in myocyte length and the addition of sarcomeres in series.
The failing left ventricle, at clinical presentation, is dilated with variable wall thickness. Histologically, the cardiomyocyte at this point is elongated [13].
The prime example of cardiac remodeling occurs after myocardial infarction. The rearrangement of cardiomyocytes in surviving myocardium results in ventricular dilatation with thinning of the wall [infarct expansion]. The non-infarcted (or viable) region, in response to the newly acquired workload, undergoes hyperplasia [12]. The end-result of these pathologic stimuli include the reactivation of the fetal gene program and ultimately altered protein synthesis, and alteration in cardiomyocyte shape and structure [14]. Initially, these changes can be beneficial [repair of the necrotic area in the case of infarct followed by scar formation], and the cardiac output is maintained at the expense of larger volumes. However, the diastolic and systolic stresses eventually overwhelm the system, with decline of ejection fraction. The culmination of these remodeling events is impaired cardiac contractility, manifesting clinically as heart failure [15, 16]. The remodeling process may itself induce further stress upon the ventricular wall, inciting a vicious cycle of stress and remodeling. Clinically, guidelines are in place to curtail the pathologic remodeling process (specifically, the institution of beta-blocker prior to discharge for those patients who have suffered myocardial infarction) [17].
The cardiomyocyte undergoes crosstalk with the surrounding endothelial cells, fibroblasts, and inflammatory cells (neutrophils, macrophages, lymphocytes) depending on the context [12, 18]. The translation of biomechanical forces to gene expression presumably integrates signals from extracellular matrix, the cell membrane, membrane-bound ion channels, the cytoskeleton, sarcomere, mitochondria, and endoplasmic reticulum [19, 20]. Complexity is multiplied at the cytoplasmic/nucleus interface and then the nucleus itself, upon which the signals are converged upon gene expression; even at this point, there is fine-tuning of the message by microRNA, enzymatic modification, post-translational modification, amongst others. The reemergence of the fetal gene program is defined by induction of mRNA for atrial natriuretic factor, b-type natriuretic peptide, skeletal α-actin, and β-myosin heavy chain[3, 5].
In addition to the elaboration of paracrine and autocrine signaling elicited by mechanical stretch, the stimulus leads to the activation of cardiomyocyte angiotensin II type I receptor directly (without the involvement of angiotensin II) [16, 21]. Signaling pathways converge on a limited number of intracellular signaling cascades including mitogen-activated protein kinases, the PI3K-Akt- GSK-3 pathway, calcium/calmodulin-dependent calcineurin phosphorylation, or small GTPases such as Ras, Rac, or RhoA [22–24]. In this review, we will focus on how pathophysiological stimuli influence the adult cardiomyocyte for remodeling.
KEY CARDIOMYOCYTE MOLECULAR INTEGRATORS OF PATHOLOGIC REMODELING
G-Protein Coupled Receptors
Integral to the process of signal induced cardiac remodeling is altered intracellular calcium homeostasis and the effected pathways. Cardiomyocyte G-protein coupled receptors [β1-adrenoreceptor, α1-adrenoreceptor, and Angiotensin II type 1 receptor] act via Gα heterotrimeric G proteins to effect signaling cascades which mediate intracellular calcium handling and ultimately pathologic hypertrophy [25]. Specifically, β1-adrenoreceptor is coupled to Gαs; stimulation of this pathway is mediated by Adenylyl Cyclase, cyclic AMP, and Protein Kinase A. Protein Kinase A (PKA) exerts its effects on the sarcoplasmic reticulum (and subsequent release of calcium); PKA also dictates pathologic hypertrophy via its effects on gene transcription in the nucleus [26]. α1-adrenoreceptor is coupled to Gαq/11. Stimulation of this pathway leads to activation of Diacylglycerol (DAG) and Inositol-1,4,5-triphosphate (IP3) [27]. DAG activates Protein Kinase Cα, which acts upon the sarcoplasmic reticulum (SR) to release calcium [28]. IP3 directly bids to its receptor on the SR to mediate the release of calcium [29]. Angiotensin II binds to its receptor (Angiotensin II type 1 Receptor) and the associated Gαq/11. This activates Phospholipase-Cβ which activates IP3, resulting in calcium release. Angiotensin II, via Gαq/11 also stimulates a signaling cascade resulting in the activation of c-Jun N terminal kinase (JNK) and p38, which have direct effects on gene expression leading to pathologic hypertrophy (Figure 1) [30].
Figure 1. G-protein coupled receptors and the IGF-1 signaling in cardiac remodeling.
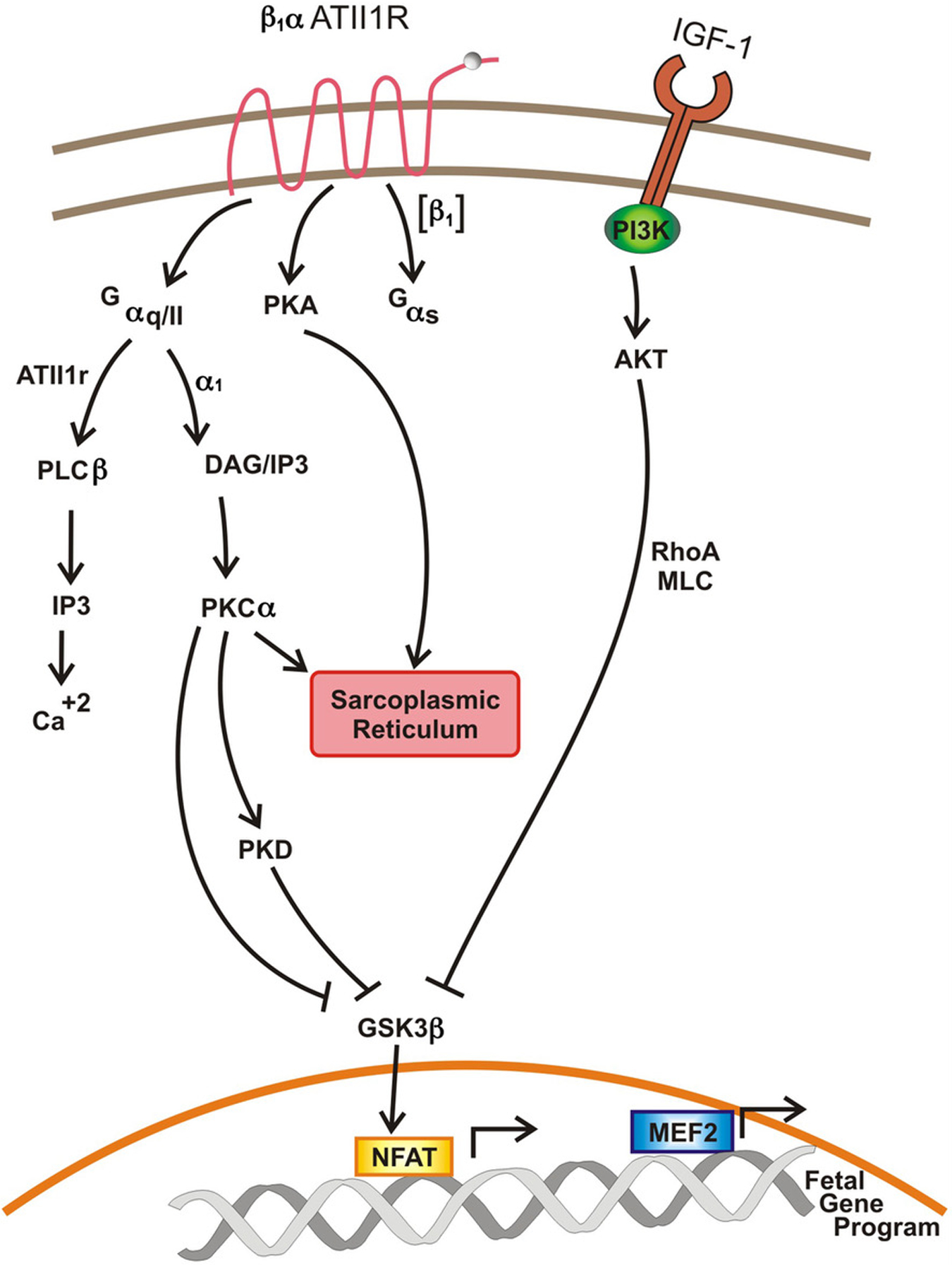
Aberrant calcium handling and exuberant activation of second messenger mediated signaling cascades culminate in increased transcriptional activity and upregulation of the fetal gene program, with resultant cardiac remodeling.
RhoA
RhoA is a key integrator of signal transduction, relaying pleiotropic signals from the plasma membrane ranging from intracellular calcium handling to gene expression. The role of RhoA in cardiac fibroblasts is well established [31], yet this key regulator of the actin cytoskeleton is less defined in the cardiomyocyte. The cardiomyocyte-specific null RhoA mice that Lauriol et. al.[14] generated had no overt phenotype at basal conditions. However, upon subjection to transverse aortic constriction (TAC), which results in chronic pressure overload, the mice developed an accentuated dilated cardiomyopathy and accelerated decline in cardiac contractility. RhoA in cardiomyocytes is required for modulation of calcium handling and phosphorylation of MLC (Myosin Light Chain). Furthermore, RhoA deletion obliterated the phosphorylation of ERK1/2 and AKT [both of which are pro-hypertrophic and pro-survival signals]. Finally, the authors demonstrated the integral role of RhoA in the cardiomyocyte in the fibrotic response, via the nuclear transcription factors MRTFa and SRF, to chronic pressure overload. In sum, RhoA in cardiomyocytes is essential for protection against dilated cardiomyopathy, whereas it promotes the myocardial fibrotic response in response to chronic pressure overload.
Protein Kinase C
There are 10 different isoforms of the calcium- or lipid- activated PKC, leading to complexity and variation in physiologic function [32]. Ytrehus et. al. demonstrated that PKC–dependent phosphorylation was required for protection against ischemia/reperfusion injury [33]. Subsequent studies demonstrated that PKCε activation provided cardioprotection, whereas PKCδ activation mediates acute injury in the setting of myocardial ischemia. Angiotensin II, Endothelin, and catecholamines bind to α-adrenergic receptors on cardiomyocytes, which are coupled to the heterotrimeric G proteins [34]. Initiation of this pathway stimulates the activation and membrane translocation of PKCα by the bound DAG and calcium. PKCα, the isoform predominantly expressed in the human and mouse heart, is activated during hypertrophy in the setting of heart failure as well as following myocardial infarction. PKCα regulates cardiac contractility. PKCα−/− mice demonstrate increased contractility following exposure to stimuli such as β-adrenergic receptor stimulation. Mechanistically, this occurs through increased Ca2+ loading in the sarcoplasmic reticulum. Deletion of PKCα in mice provided protection against pressure-overload and myocardial infarction. In contradistinction, PKCα transgenic mice demonstrated reduced cardiac performance at the age of 4 months, and hypertrophy at 6 months. Although several convincing mechanisms have been proposed to clarify PKC’s role in cardiac contractility, the mechanism regarding PKC’s role in cardiac remodeling has yet to be concretely laid out [35, 36].
Protein Kinase D
The protein kinase (PKD) family consists of 3 isoforms, and were first identified in myocardium. PKC phosphorylates and activates PKD. Stimuli leading to the activation of PKD include phenylephrine, ET-1, angiotensin II, and aldosterone [37]. Pathophysiologically, myocardial PKD is activated by hypertension, pressure overload, and chronic neurohormonal signaling [38]. The activities of PKD include myofilament Troponin I phosphorylation (resulting in reduced calcium sensitivity) and mediation of transcriptional activity [39]. PKD is activated by G protein-coupled receptors; in parallel with the Ca2+/calmodulin-dependent kinase mediated cascade, these stimuli converge upon histone deacetylases (HDACs). The resultant phosphorylation of class II HDACs leads to their nuclear export, exposing and freeing the MEF2 family of transcription factors for transcriptional activation of the pathologic cardiac remodeling gene program. Fielitz et. al. generated cardiac-specific knockout of PKD1, and demonstrated attenuation of hypertrophy following transverse aortic constriction (TAC), Angiotensin II infusion, and isoproterenol infusion [38]. Furthermore, the mice had curtailed fetal gene expression upon exposure to pathologic stimuli.
Insulin and the Insulin-Like Growth Factor 1
Peptide growth factors [e.g. Insulin and Insulin-Like Growth Factor 1 (IGF1)] activate the phosphatidylinositol 3’-kinase (PI3K)- signaling cascade to mediate physiologic cardiac hypertrophy; however, perturbations of these pathways can lead to pathologic maladaptation [16]. Insulin, IGF-1 and angiotensin II can activate the mitogen-activated protein kinase (MAPK) and/or the PI3K-Akt signaling cascades. Insulin controls the entry of glucose into the cardiomyocyte; the often seen insulin resistance in heart failure limits the glucose availability and hence ATP synthesis [40]. Insulin bids to the Insulin Receptor (tyrosine kinase), leading to the activation of the PI3-AKT signaling cascade. Cardiac-specific insulin receptor null mice exhibit exaggerated systolic dysfunction and dilation 4 weeks post aortic banding; this is accompanied by increased interstitial collagen deposition in comparison to wild-type mice [41]. In conditions of health, insulin and IGF-1 stimulate the PI3K/PKB/Akt to potentiate cardiac growth and physiologic hypertrophy in response to exercise. In pathophysiologic situations such as the metabolic syndrome, it is suggested that hyperinsulinemia activates angiotensin II and overstimulates MAPK signaling, with resultant ventricular hypertrophy, dysfunction, and fibrosis [42–44]. IGF-1 is the hepatic metabolic product of growth hormone. High-fat diets reduce IGF-1 levels; in animal models, exogenous IGF-1 prevents impaired myocardial contractility in the setting of high-fat diet mediated cardiomyopathy [45]. IGF-1 acts on its transmembrane tyrosine kinase receptor with subsequent activation of the PI3K-AKT cascade [46]. In this instance, the AKT activates phosphoinositide-dependent protein kinase 1 (PDK1) and subsequently glycogen synthase kinase 3β (GSK-3β), which modulates the hypertrophic gene program via NFAT transcription factors.
Glycogen Synthase Kinase-3 (GSK-3)
Glycogen synthase kinase-3 (GSK-3) is a serine/threonine kinase that was originally named for its regulation of glycogen synthase [47]. The role of GSK-3β is most well-known in the Wnt signaling pathway. Specifically, the binding of Wnt ligand to the Frizzled receptor culminates in the stabilization of the potent nuclear transactivator, β-catenin. GSK-3β is actually active in unstimulated cells, and its role is to degrade β-catenin and prevent aberrant transcription. The activation of the Wnt signaling cascade leads to the degradation of GSK-3β, thereby resulting in the nuclear accumulation and activation of β-catenin and lymphoid enhancer factor-1 (LEF)/T-cell factor (TCF) transcription factor mediated transcriptional program. This entire program becomes awry in colorectal cancer, amongst other tumors [48]. With regard to the heart, this pathway is required for cardiac hypertrophy and fibrosis. Although GSK-3β has received the majority of the scientific community’s attention, it is now apparent that GSK-3α is critical to cardiomyocyte function. Upon activation of hypertrophic signaling cascades initiated by the actors stated above (GPCRs, PKA, RhoA, PKC, and PKD), GSK-3 is phosphorylated (and hence its inhibitory activity is shut down). The inactivation of GSK-3 results in the activation of potent nuclear transcription factors invoked in the hypertrophic response [NFAT, β-catenin, and translational elongation factors [eIF2B] [49, 50].
In their seminal paper, the Force group demonstrated that GSK-3β was inhibited by hypertrophic stimuli in vitro (Endothelin-1 and IGF-1) as well as in vivo (post-aortic banding). They further demonstrated that the fetal gene expression program was mediated by this inhibition, via activation of NFAT [51]. Studies over the succeeding 15 years have narrowed down GSK-3β as the dominant isoform regulating hypertrophic response to ischemic insult; cardiac-specific deletion of GSK-3β protected the mouse against left ventricular dilatation following experimentally induced infarction. However, the same group demonstrated that GSK-3β was not implicated in the hypertrophic response to pressure overload (in a TAC model) [49, 50].
The GSK-3β null mouse was determined to be embryonically lethal, whereas deletion of GSK-3α did not affect development. Using mice with cardiomyocyte specific deletion of GSK-3α, it was determined that this molecule promotes cardiac remodeling and infarct expansion following experimental infarction. Cardiac-specific deletion of GSK-3α protected against ventricular chamber dilation and depressed ejection fraction in the infarct model [50, 52].
Calcium
The failing heart is characterized abnormal contraction and relaxation during periods of tachycardia. This is thought to be a direct result of decreased intracellular calcium in the diseased cardiomyocyte. Cardiomyocyte contraction is due to a transient rise in cytosolic calcium, as it causes movement of contractile myofibrils leading to cell contraction. Calcium enters the cardiomyocyte through L-type calcium channels. This entry triggers a larger wave of calcium release by the sarcoplasmic reticulum (SR) into the cytosol through the ryanodine receptor (RyR). The aforementioned process is termed calcium-induced-calcium release (CICR) from the SR. RyRs open during early systole and are closed during diastole. Ca2+ activated Sarco-Endoplasmic Reticulum ATPase (SERCA) translocates Ca2+ across the SR membrane against a concentration gradient; the Ca2+ ATPase of SR sequesters calcium from the cytoplasm. The removal of calcium from the cytoplasm into the SR and the forward mode sarcolemmal Na+/Ca2+ exchanger (NCX), which is located along T-tubules, mediates physiologic relaxation [9, 53–56].
Abnormalities in calcium handling during heart failure are attributed to:
Impaired calcium re‐uptake defective or reduced SERCA [57].
Calcium leak through dysfunctional of RyRs that remain open in diastole [58]
Redistribution of NCX from predominantly the T-tubules to a more even distribution amongst the T-tubules and sarcolemma [59].
Abnormal intracellular calcium handling leads to activation of the calmodulin–dependent phosphatase calcineurin, a necessity in the progression of maladaptive hypertrophy [16]. The components of calcineurin include a catalytic subunit (CnA) and a regulatory subunit (CnB) [3]. The serine-threonine protein phosphatase calcineurin dephosphorylates nuclear factor of activated T cells (NFAT) transcription factors, leading to their nuclear translocation. Transgenic overexpression of CnA in mice results in aberrant dephosphorylation of NFAT, with subsequent translocation of the transcription factor to the nucleus. Once in the nucleus, NFAT associates with the GATA4 and myocyte enhancer factor-2 (MEF2) transcription factors to drive the maladaptive hypertrophic gene program [60, 61].
Calmodulin Kinase II (CaMKII) is a serine/threonine protein kinase, activated by Ca2+/calmodulin, and an integrator of divergent signaling pathways. Specific to cardiac hypertrophy, CAMKII activity is stimulated by neurohormonal, β-adrenergic, and Angiotensin II signals. CaMKII exerts its effects through its locations in cytoplasm as well as the nucleus; of the four isoforms, CaMKIIδ and CAMKIIγ are expressed in the heart. CaMKII phosphorylates the ryanodine receptor RyR2, causing sarcoplasmic reticulum leak of calcium with resultant heart failure [62]. Weinreuter et al. demonstrated that CAMKII mediates infiltration of CD45+ cells and pro-inflammatory chemoattractant signals (CCL2 and CCL3). Attenuation of this signaling pathway, as demonstrated in cardiac-specific knockout of both CaMKIIδ and CAMKIIγ, lead to decreased scar formation and cardiac function 5 weeks post ischemia-reperfusion injury. Overall, these data demonstrated CaMKIIδ and CAMKIIγ regulation of deleterious remodeling at the latter stages of cardiac remodeling via recruitment of inflammatory mediators [63]. Kreusser et. al. further cemented the role of CAMKII in cardiac remodeling by demonstrating that CaMKII inactivates histone deacetylase 4 (HDAC4), freeing up and activating MEF2 [with resultant maladaptive hypertrophy] in vivo. Unexpectedly, however, the same group demonstrated that CaMKII phosphorylation of calcineurin A represses the calcineurin-NFAT pathway, in effect executing an anti-hypertrophic mechanism [61].
MAP Kinases
Mitogen-Activated Protein Kinases (MAPKs) stimulate cell proliferation, differentiation, and ultimately can activate the fetal gene program. The three terminal effectors of this signaling cascade [p38 kinase, c-Jun N-terminal kinase (JNK), and extracellular regulated protein kinase 1/2 (ERK1/2)] are acted upon by upstream MAPK kinases (MEKs). This pathway is hyperactive in human heart failure. Progressive studies demonstrated numerous instigators of this signaling cascade: GPCRs [Angiotensin II, endothelin-1, adrenergic receptors], receptor tyrosine kinases (IGF-1), reactive oxygen species, ultraviolet irradiation, osmotic stress, and mechanical stretch stimuli. Traditionally, ERKs have been thought to mediate mitogenic and growth stimuli, whereas the p38 and JNK are referred to as the Stress Activated Protein Kinases [64, 65]. Constitutive activation of ERK1/2 results in the development of concentric hypertrophy; ERK1/2 act upon NFAT to mediate cardiac hypertrophy [3]. p38 appears to have both functions that are both detrimental to the heart as well as cardioprotective [66]. It has become evident that the MAPK pathway cross-talks with many other signaling cascades to regulate the function of cardiomyocytes.
THE NUCLEAR INTEGRATORS OF PATHOPHYSIOLOGICAL REMODELING
Transcriptional control and epigenetic mediated regulation of gene expression play a central role in a variety of biological and pathological processes. It is not surprised that functions of cardiomyocyte expressed transcription and epigenetic factors are linked to cardiac remodeling and disease. Histone deacetylases (HDACs) are key transducers of extracellular stimuli that impact the hypertrophic gene response. HDACs prevent a favorable chromatin structure for transcription by opposing the activity of histone acetyl transferases (HAT) [67]. Amongst the type II HDACs, HDAC5 and HDAC9 are signal-responsive repressors of MEF2. In the unstimulated state, HDAC5 is bound to MEF2, preventing a MEF2-directed gene program (Figure 2) [68, 69]. Neurohormonal stimuli upon cardiomyocytes via G-protein receptors trigger CAMKII or protein kinase D-mediated phosphorylation and subsequent nuclear export of HDAC5. This series of events allows a MEF2-directed gene program. Deletion of either HDAC5 or HDAC9 in mice results in spontaneous cardiac hypertrophy at 8 months of age, and further demonstrated exaggerated hypertrophic response to pressure overload, increased MEF2 activity and activation of the fetal gene program [68, 70]
Figure 2. Regulation of fetal gene program by the calcineurin-NFAT-mediated transcription in hypertrophic hearts.
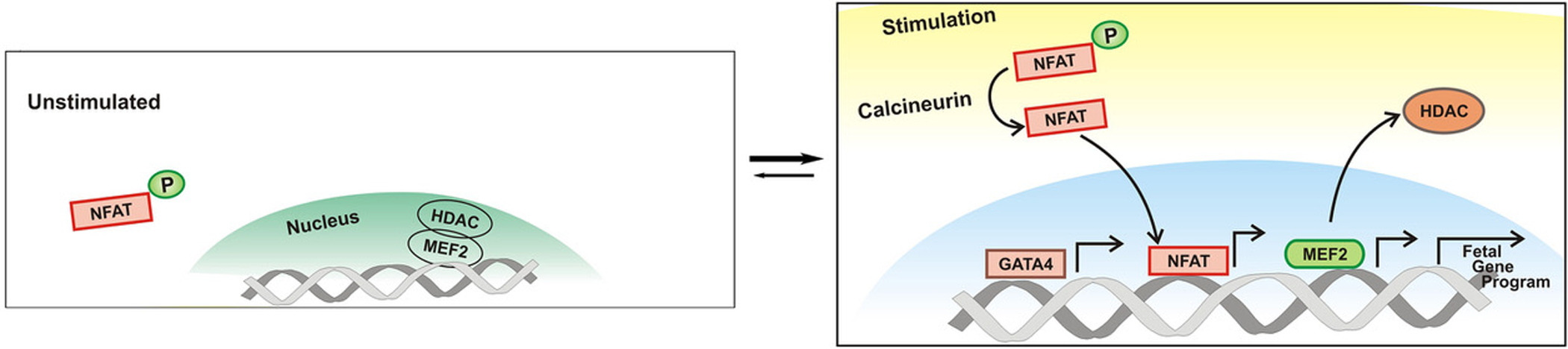
In the activated state, calcineurin mediates the dephosphorylation of NFAT and its subsequent nuclear translocation. The displacement of HDAC from the nucleus allows MEF2 mediated transcriptional activation. The culmination of these events results in activation of the fetal gene program, and ultimately cardiac remodeling.
The MEF2 family of MADS (MCM1, agamous, deficiens, serum response factor) box transcription factors are essential to heart development as well as cardiac remodeling in the pathological state. They integrate stress signals and dictate gene expression. MEF2a and MEF2c genes are essential for cardiac contractility and development respectively, as determined by studies of null mice. In the adult heart, MEF2A and MEF2D heterodimerize and serve as the major MEF2 isoforms. As above, CAMKII or PKD alleviation of HDAC repression allows expression of MEF2 transcriptional activity. Additionally, MEF2 transcription factors are directly activated by phosphorylation induced by MAPK signaling. Mice lacking MEF2d display an attenuation of cardiac remodeling in response to pressure overload or chronic β-adrenergic stimulation. In contradistinction, forced cardiac overexpression of MEF2d provoked severe cardiomyopathy [71].
The nuclear factors of activated T-cells (NFATs) belong to the Rel family transcription factors and are populated by 5 family members (NFATc1, NFATc2, NFATc3, NFATc4, and NFAT5) [72]. Wilkins et. al. demonstrated that calcineurin–NFAT signaling was integral to pathologic cardiac remodeling [73]. Interestingly, this group observed suppression of NFAT activity upon physiologic cardiac remodeling (i.e. exercise-induced hypertrophy). Upon activation, calcineurin binds and dephosphorylates the hyperphosphorylated and sequestered cytoplasmic NFAT transcription factors, permitting nuclear translocation and transcription of genes responsible for the onset of pathologic cardiac remodeling (Figure 2). NFAT factors only weakly bind to DNA, and are seen to interact with other transcription factors (e.g. MEF2, GATA 4/6, and NF-κB) in the execution of cardiomyocyte hypertrophic gene expression. NFATc1 null mice are embryonically lethal as a consequence of valvular defects. Deletion of NFATc2 or NFATc3 attenuates remodeling upon exposure to pathologic stimuli [3, 4].
The GATA transcription factors, of which there are 6 family members, contain DNA binding domains consisting of zinc fingers [74]. GATA-4 is expressed in embryonic hearts and GATA-4 null mice are embryonic lethal due to a failure of cardiac development. In the adult heart, GATA-4 mediates genes essential for pathologic cardiac hypertrophy. GATA-4 is directly phosphorylated by Erk1/2 and p38 mitogen-activated protein kinase. Moreover, mice bearing transgenically overexpressed GATA-4 or GATA-6 suffered heart failure and reduced cardiac contractility. GATA-4 and GATA-6 physically and functionally interact to control the expression of genes essential for cardiac remodeling. In addition, chronic pressure overload drives up the expression of GATA-6 in the mouse heart, concomitant with increased DNA-binding activity. Using GATA-6fl/fl::βMHC-cre mice to overcome the embryonic lethality of GATA-6 null mice, Molkentin’s group observed attenuation of the hypertrophic response to TAC and Angiotensin II/Phenylephrine infusion [75, 76].
CONNECTING EXTERNAL STIMULI TO THE CARDIOMYOCYTE SIGNALING CASCADES AND PATHOLOGIC REMODELING
Mechanotransduction
Mechanotransduction is the process by which the physical contacts of cardiomyocytes are converted into intracellular signals by transmembrane proteins [77, 78]. Extracellular stimuli increase intracellular Ca2+ levels by either promoting its release from intracellular organelles or its entry across the plasma membrane [60]. The plasma membranes of cardiomyocytes bear calcium-permeable cation channels known as the TRPC family of channels. Calcium influx through cardiac TRPC channels mediate calcineurin signaling and subsequently hypertrophy. Studies have been particularly focused on TRPC6, which is increased in hypertrophic hearts. The regulation of TRPC6 is controlled at the transcriptional level by NFAT. Genetically TRPC6 null mice have no detectable cardiac dysfunction at baseline. At baseline, TRPC6 is undetectable in the heart. However, stressors such as neuroendocrine stimulation, isoproterenol stimulation, and TAC-induced overload, levels of TRPC6 mRNA and protein are increased. Clinically, levels of TRPC6 are increased in failing human hearts. The anti-ageing protein, Klotho, is produced by the kidney upon stressful stimuli. Xie et. al. demonstrated that Klotho null mice exhibit exaggerated cardiac hypertrophy upon isoproterenol stimulation; overexpression of this soluble protein attenuated the cardiac hypertrophy induced by isoproterenol [60]. Mechanistically, they demonstrated soluble Klotho inhibits TRPC6 currents in isolated cardiomyocytes. Furthermore, they demonstrated that Klotho inhibits TRPC6 action by: 1. Inhibition of PI3K-mediated exocytosis of TRPC6 channels (thereby counteracting the effects of agents such as IGF-1). 2. Decreasing the cell-surface concentration of TRPC6.
Analogous to the Frank-Starling mechanism (increased cardiac contractility upon encounter with an increased preload), the Anrep effect describes enhancement of cardiac contractility upon encounter with an increased afterload [79]. In any case, the cardiomyocyte stretch is mediated by calcium sparks. In their seminal study, Petroff et. al. demonstrated that cardiomyocyte stretch induces an increase in Ca2+-spark frequency via PI(3)K-dependent phosphorylation of both Akt and the endothelial isoform of nitric oxide synthase (NOS). The group proposed that eNOS activation enabled Nitric Oxide (NO) to function as a second messenger during the cardiomyocyte stretch [80]. It was later demonstrated that cardiomyocyte stretch activates nicotinamide adenine dinucleotide phosphate oxidase 2 (NOX2) to cause Ca2+ sparks. More recently, Jian et. al. elaborated on the mechanism. Using an innovative “cell-in-gel” system, they identified nitric oxide synthase 1 (nNOS), CaMKII, and NOX2 as key mediators of mechanosensing pathways that mediate and dictate Ca2+ signaling. With specific reference to the Anrep effect, the net effect of these mediators was increased systolic Ca2+ transient in response to an increased afterload. In this study, the group singled out nNOS as the principal molecule responsible for the afterload-induced spontaneous Ca2+ sparks, as opposed to eNOS which was not responsible for this phenomenon. Current studies are aimed at delineating the roles of eNOS and nNOS in the generation of Ca2+ sparks and mediation of cardiac contractility [81].
Integrins and Mechanotransduction
Guanosine triphosphate (GTP)-binding proteins (also known as GTPases) regulate cell shape, migration, and adhesion. The Ras and Rho GTPase subfamilies mediate cardiac hypertrophy. RhoA transmits signals from the extracellular matrix and certain G-protein coupled receptors to effect adhesion, migration, and gene expression. RhoA is a key integrator of cardiomyocyte signaling, and its exact role appears to be signal and concentration dependent. Using transgenic mice, Xiang et. al. demonstrated that moderate RhoA activation in adult mouse cardiomyocytes mediated cardioprotection against ischemia/reperfusion (I/R) injury; conversely cardiac-selective deletion of RhoA led to increased pathology upon I/R injury [82]. In another study, Miyamoto et. al. demonstrated that RhoA activation in cardiomyocytes led to the induction of apoptosis and cardiomyopathy [83].
Integrins are heterodimeric cell-surface receptors that lack intrinsic enzymatic activity; integrins link the extra cellular matrix (ECM) to the cytoskeleton at focal adhesion sites, and are integral for cardiomyocyte mechanotransduction. The α-subunit of (α1, α3, α4, α6, α7, α10 and α11) confer ECM specificity, whereas β-subunit (β1, β3 and β5) interact with the cytoplasm in cardiomyocytes. As an example, angiotensin II stimulates cardiomyocyte contraction and adhesion via β1 and αvβ3 integrins. Stretch activates integrin-mediated signaling via intermediaries such as focal adhesion kinases and integrin-linked kinases. Integrins stimulate GTP binding to RhoA via Rho guanine nucleotide exchange factors (RhoGEFs). Takefuji et. al. demonstrated that RhoGEF12 integrates signals from integrins and GPCRs to mediate hypertrophic gene expression and the transition to heart failure [24]. Honing in on the mechanisms of gene expression in hypertrophy, they demonstrated that stretch-induced translocation of MRTF-A, a member of the myocardin family of transcription factors, from the cytoplasm to the nucleus was dependent on RhoGEF12; this corresponded with the expression of the hypertrophic gene program. In vivo, inactivation of RhoGEF12-dependent RhoA activation, protected against the development of hypertrophy development, cardiomyocyte apoptosis, fibrosis, and the development of chronic heart failure.
Integrin Linked Kinase (ILK) is a ubiquitous serine/threonine kinase that serves as a molecular scaffold and transducer of signaling cascades. ILK binds β1-integrin, linking the extracellular matrix to cardiomyocyte remodeling processes [84]. White et. al. demonstrated that cardiac-specific deletion of ILK resulted in dilated cardiomyopathy and sudden death at 6 weeks of age; there was decreased AKT phosphorylation and disaggregation of cardiomyocytes [85]. Conversely, overexpression of ILK in a transgenic model led to cardiac hypertrophy [86]. ILK induces nuclear translocation of β-catenin, with subsequent activation of TCF/LEF mediated transcription. In a particularly prolific manuscript, Dedhar’s group demonstrated that insulin, via its receptor, activated PI(3)K with subsequent activation of ILK. They additionally demonstrated that ILK phosphorylates GSK-3β and AKT/PKB [87]. More recently, it has been demonstrated that ILK mediates cardiomyocyte force transduction through regulation of SERCA-2a in the human heart [88].
The PKD1 gene, known for mutations in autosomal dominant polycystic kidney disease, encodes Polycystin-1 (PC1), a large membrane glycoprotein [composed of an extracellular domain, 11 transmembrane domains, and an intracellular C-terminus] integral to mechanical sensation. Pedrozo et. al. recently demonstrated that the C-terminus of PC-1 physically interacts with, and stabilizes, the L-type calcium channel [89]. Deletion of PC-1 in mice resulted in downregulation of the α1C L-type calcium channel and attenuation of pressure-overload induced hypertrophy. Extrapolation of the established role of PC-1 in other cell types as well as in vitro studies implicate PC-1 as a cardiomyocyte mechanosensor that relays extracellular stress into gene expression changes via alteration of intracellular calcium handling. As of yet, there have been no reports on the function of PC-1 in the cardiomyocyte following infarction. The role of PC1 in human cardiomyocyte disorders still remains to be established.
Cardiomyocytes Sensing Surrounding Necrosis
RAGE (Receptor for Advanced Glycation End Products) is a pattern recognition receptor, activated by advanced glycation end-products (AGEs), and expressed in cardiomyocytes. AGEs are released from necrotic or injured cardiomyocytes; they are the product of non-enzymatic reactions between lipids, sugars, nucleic acids, and the free amino acids of proteins [90]. Interaction of AGEs with their receptor (RAGE) incites the activation of a pro-inflammatory cascade. RAGE binds to the High-Mobility Group Box 1 (HMGB1) transcription factor in the incitement of this inflammatory cascade [91]. Ablation of RAGE resulted in decreased myocardial necrosis 48 hours post- ischemia-reperfusion injury. Zhu et. al. elegantly delineated the co-dependence of the RAGE and β-adrenergic receptor mediated signaling pathways [92]. They demonstrated a physical interaction between RAGE and β1AR [effectively uniting the Pattern Recognition Receptor and the GPCR pathways], and they demonstrated the detrimental effect on cardiomyocytes is mediated by CAMKII. Ablation of RAGE in mice blocked β1AR-mediated cardiac remodeling in response to isoproterenol infusion. The cardiomyocyte post-infarct, exposed to excess catecholamines and potentially hyperglycemia, likely marched along a deleterious pathway via the synergy of β1AR- and RAGE. The production of reactive oxygen species in metabolic disorders such as chronic hyperglycemia contribute to the formation of AGEs. The end-product of the gene transcription in such metabolic disorders include increased collagen deposition and fibrosis [90].
EMERGING CONCEPTS – SIGNALS THAT ELICIT THE TRANSITION FROM HYPERTROPHIC CARDIOMYOPATHY TO DILATED CARDIOMYOPATHY
Prolonged hypertrophic growth often leads to dilated cardiomyopathy (DCM) – a condition in which the heart is enlarged, with a thinner ventricular wall and decreased function. The molecular cues that herald the transition hypertrophic cardiomyopathy (HCM) to dilated cardiomyopathy (DCM) and heart failure (HF) are not fully understood.
The lamin A/C (LMNA) gene on chromosome 1q21.2 encodes for lamin A and lamin C; these proteins are located in the nuclear envelope, and are instrumental in nuclear stability, chromatin structure and gene expression [93]. Clinically, the hallmark of LMNA mutation induced cardiomyopathy is the coexistence of dilated cardiomyopathy (DCM) and a premature conduction system with an aggressive course that progresses to either death or necessitates cardiac transplant [94]. A-type lamins are thought to be integral to stress-induced transcriptional activity. Embryonic fibroblasts isolated from Lmna−/− mice, under conditions of mechanical strain, demonstrated deranged mechanotransduction in response to strain; furthermore, NF-kappaB response to mechanical and cytokine mediated stimulation was attenuated in these cells [95]. LMNA heterozygous mice develop adult-onset DCM at 18 months of age [96]. LMNA-null mice develop DCM at 4 to 6 weeks of age without cardiac hypertrophy and succumb to death at 8 weeks of age [97]. Although the nuclear lamina in LMNA-null cardiomyocytes is intact, the nuclei are elongated and lobulated with irregular, central chromatin clumps. In addition, increased cardiomyocyte apoptosis is observed in 4 to 6-week-old LMNA-null heart [98]. Mounkes et. al. recapitulated the asparagine-to-lysine substitution at amino acid 195 of LMNA in human dilated cardiomyopathy in a mouse model [Lmna-N195K]. These mice succumbed to dilated cardiomyopathy and arrhythmia, with histologic demonstration of a loss of organization at sarcomeres and intercalated disks [99]. Arimura et. al. recapitulated the LMNA H222P missense mutation identified in a case of familial Emery-Dreifuss Muscular Dystrophy (EDMD) in a mouse model. The LmnaH222P/H222P mice developed dilated hypokinetic ventricular chambers with conduction defects; the mice succumb to death by the age of 26 weeks [100]. In addition to the discovery of the upregulated fibrosis and fetal gene program in the hearts of the LmnaH222P/H222P mice, Worman’s group noted upregulation of the MAPK signaling branches (ERK1/2 and JNK). When subjected the LmnaH222P/H222P mice to inhibitors of the MAPK signaling cascade, the investigators were able to attenuate the cardiac defect and the fetal gene program [101, 102]. In sum, there is abundant data emanating from the clinical and basic science worlds to fuel the notion that LMNA and its interacting proteins are at the nexus of cardiomyocyte sensation of the transition to pathologic remodeling.
Studies from our laboratory have demonstrated LMNA interacts genetically with a cardiac-specific protein named CIP (Cardiac Isl1-interacting Protein) [103, 104]. The CIP transcript has multiple alternative slicing-produced isoforms, including a recently reported protein called MLIP (Muscle-enriched A-type lamin interacting protein [105]. It is demonstrated that the expression of CIP is decreased in adverse remodeling in the heart. Loss of CIP accelerated the development of DCM; in contradistinction, overexpression of CIP attenuated cardiac hypertrophy in a pressure overload model. Intriguingly, the expression of CIP was markedly reduced in dilated hearts but not in hypertrophic hearts. CIP-null mice are phenotypically normal at baseline. However, the CIP-null mice develop a dramatic cardiac dilation with an enlarged lumen and reduced wall thickness of the left ventricle after 6 weeks of transverse aortic constriction (TAC), while the control hearts are still in the hypertrophic growth phase. Following cardiac injury/stress, the CIP null mice developed DCM at time points earlier than control mice. Together, these results suggest that CIP negatively regulates cardiac hypertrophy. Given the accelerated progression from cardiac hypertrophy to dilation cardiomyopathy under stress, we propose that CIP may function as a cellular sensor of mechanical and/or pathophysiological stimuli. Together, these studies demonstrated a genetic and physical interaction between CIP and LMNA; neither one of the proteins affected the expression of the other. Given the location of CIP and LMNA on the nuclear envelope, we propose that they respond to mechanic and/or pathological stresses to modulate cardiac function. Alternatively, CIP is also located in the cytoplasm, leaving it in a key location to integrate extracellular and cytoplasmic signals into the nucleus, with the end result of gene expression modulation.
microRNAs and Cardiac Remodeling
microRNAs (miRNAs) are small RNAs that inhibit the expression of downstream genes through translational repression or mRNA degradation by base-pairing with mRNA transcripts. miRNAs serve to “fine-tune” gene expression. The reader is referred to comprehensive reviews for further information [106–109]. As an example of the pivotal role microRNAs in cardiac remodeling, our lab demonstrated that the cardiac- and skeletal- muscle enriched microRNA-22 (miR-22) is upregulated during myocyte differentiation and cardiomyocyte hypertrophy. Overexpression of miR-22 was sufficient to induce cardiomyocyte hypertrophy. We demonstrated that cardiac miR-22 was essential for hypertrophic cardiac growth in response to stress [110, 111]. Deletion of miR-22 attenuated cardiac hypertrophy and cardiac remodeling in response to isoproterenol infusion and an activated calcineurin transgene. We identified Sirt1 and Hdac4 as miR-22 targets in the heart. Most recently, we uncovered a linear genetic pathway involving miR-208a and its target in the regulation of cardiac contractile gene expression and cardiac function, highlighting the previously underestimated significance of miRNAs in cardiac function and remodeling [112]. In a particularly provocative study, Liu et. al. demonstrated that miR-222 is up-regulated by exercise and effectively represses pathologic cardiac remodeling [113–115]. Experimentally, transgenic mice bearing miR-222 were protected against adverse ventricular remodeling and cardiac dysfunction after ischemic injury. Mice genetically null for miR-222 developed dilated cardiomyopathy under conditions of stress [116]. The putative miR-222 targets have been identified as the cell-cycle inhibitor p27, HIPK-1, HIPK-2, and Hmbox1. It has become more and more evident that miRNAs are a class of novel molecules with great potential to be biomarkers and therapeutic targets for cardiac diseases.
THE FACTORS THAT DICTATE VENTRICULAR REMODELING - FOCUSING ON THE IMPLICATIONS ON THE TRANSITION FROM COMPENSATED HYPERTROPHY TO DECOMPENSATED HEART FAILURE
Physiology of the Failing Heart
Under conditions of health, increased cardiac preload leads to increased myocardial contractility and subsequent cardiac output [the Frank-Starling Law]. In heart failure, an excessive preload overwhelms the contractility of the heart, resulting in decreased cardiac output. Decompensated heart failure is defined the failure of the heart to balance venous return with cardiac output. The end result is a failure of the heart to meet the perfusion and metabolic requirements of the peripheral organs and tissues. This process is depicted as a vicious circle, in which the reduced cardiac output results in lowered renal oxygen delivery, fluid congestion in the lungs, and activation of neurohormonal signaling. Specifically, the renin-angiotensin-aldosterone system and vasopressin secretion increase tubular sodium reabsorption and fluid reabsorption (Figure 3); hence, these pathways are pharmacologically targeted in the management of decompensated heart failure [117].
Figure 3. Physiology of the Failing Heart.
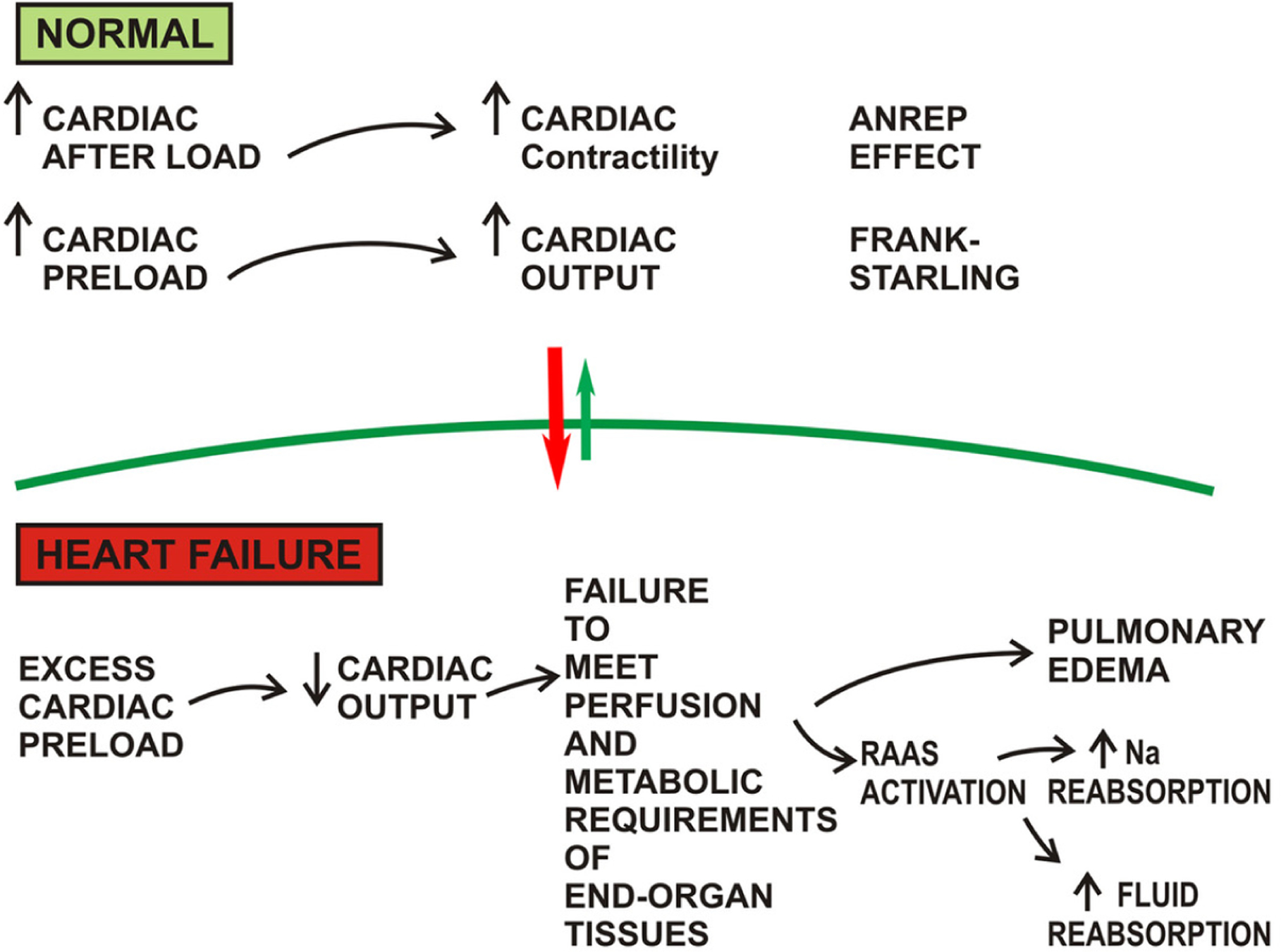
An excess load chronically imposed the heart results in failure to satisfy the metabolic requirements of end organ tissues, afflicting the patient with pulmonary edema and excess fluid and sodium retention. *RAAS = Renin-Angiotensin-Aldosterone System
Alterations in Excitation Contraction Coupling
Excitation contraction coupling refers to the process by which calcium ions lead to cardiomyocyte action potential (excitation) and contraction followed by relaxation. Heart failure is characterized by pathologic handling of calcium. Transverse tubules (T-tubules) are located along the Z-line of myofibrils, and are critical for synchronous contraction of cardiomyocytes via mediation of the release of calcium ions from the sarcoplasmic reticulum [9]. This loss and reorganization of the T-tubule system is an early event in compensated hypertrophy. Global spread of this process, with resultant massive T-tubule disruption, from the left ventricle to the right ventricle characterizes the transition from compensated hypertrophy to early and late heart failure [118]. Ca2+ influx through L-type Ca2+ channels in T-tubules triggers Ca2+ release from ryanodine receptors in the sarcoplasmic reticulum (SR). Ordered excitation-contraction relies on the integrity of this cascade. Junctional membrane complexes are the intersection of plasma membrane and the sarcoplasmic reticulum. Junctophilin 2 (JP2) bridges the plasma membrane and SR, and is essential for T-tubule maturation [119]. At a histologic level, there is documented downregulation of JP2 in the hearts of humans who have succumbed to hypertrophic cardiomyopathy, dilated cardiomyopathy, and ischemic cardiomyopathy [120]. Experimentally, knockdown or silencing of JP2 results is disorganization and loss of T-tubules, culminating in heart failure and death. Conversely, overexpression of JP2 protects the mouse against pressure-overload induced heart failure while preserving the integrity of the T-tubule. MicroRNA-24 (miR-24), is upregulated during cardiac hypertrophy and cardiac failure, as determined histologically from patient samples as well as experimentally. miR-24 has been demonstrated to downregulate JP-2. Using an antagomir directly against miR-24 in vivo in a TAC model, Li et. al. demonstrated that silencing of miR-24 prevented the transition from compensated hypertrophy to decompensated hypertrophy [121]. The suppression of miR-24 did not prevent the development of cardiac hypertrophy; in contradistinction, suppression of miR-24 protected the integrity of the T-tubule-SR junction and the progression to decompensated failure. Importantly, the suppression of miR-24 allowed maintenance (normal) expression of JP2, fueling the speculation that this axis is critical to the dysregulation of excitation-contraction coupling in the transition from compensated hypertrophy to decompensated failure.
Contractile Proteins in Cardiac Remodeling
β-myosin heavy chain (βMyHC) mutations are one of the finite identified anomalies in dilated cardiomyopathy (DCM); similarly, the gene is mutated in Hypertrophic Cardiomyopathy (HCM). Two major cardiac MyHC isoforms are encoded by genes located on chromosome 14: MYH6 encodes αMyHC and MYH7 encodes βMyHC. Human adult hearts express predominantly βMyHC whereas αMyHC is expressed during heart development. In non-failing human hearts, αMyHC mRNA constitutes about a third of MHC mRNA in the myocardium; in the failing heart, αMyHC mRNA levels drop to ~2% of the MHC mRNA within the myocardium. In contradistinction, βMyHC is upregulated in the failing heart [122]. This is an essential core of re-induction of fetal gene expression that is characteristic of the failing adult heart. Transcriptional and epigenetic regulation govern the expression of Myh6 and Myh7 genes [16]. Of note, and potentially confusing matters, αMyHC is the predominant isoform in the rodent heart. In the failing rodent heart, there is increased expression of βMyHC, with decreased expression of αMyHC. Our group and others have demonstrated that miR-208a and miR-208b, encoded by introns of Myh6 and Myh7, respectively, have also been involved in the regulation of α/β-MHC expression in pathological cardiac hypertrophy [123, 124]. Van Rooj et. al. demonstrated that the inhibition of miR-208a by systemic delivery of locked nucleic acid–modified antisense oligonucleotide prevented cardiac remodeling in a rat model of LV failure. This technology also prevented pathological myosin switching and cardiac remodeling, while improving cardiac function [123].
In contradistinction to the left ventricle, which can be maintained in a compensated hypertrophied state for years/decades before decompensation or transition to dilated cardiomyopathy, the right ventricle has a far narrower adaptive capacity. While investigating the transition from compensated right ventricular hypertrophy (cRVH) to decompensated right ventricular hypertrophy (dRVH), Michelakis’s group demonstrated a progressively decreasing level of miR-208a in RVH (in contrast to that described for LVH). The decreased miR-208a correlated with increased expression of βMyHC. The progressively declining miR-208a ultimately inhibits expression of the transcription factor Mef-2 (critical for the development of the RV and maintenance of the compensated fetal hypertrophy state), thus precipitating the exit from compensated RVH to decompensated RVH [125, 126]. A theme proposed by these authors, in concurrence with abundant literature, is that inflammatory cytokines [TNFα in particular] is the driving precipitant of this decompensation.
Cytoskeletal Proteins in Cardiac Remodeling
In addition to myosin, cardiomyocyte cytoskeleton consists of actin, desmin, titin, and microtubules (formed by the polymerization of α- and β-tubulin dimers). During heart failure, there is a disfiguration of the cytoskeletal arrangement, and this may have broad impact on mechanosensation, The structural remodeling leads to altered localization of the mitochondria, with resultant aberrant excitation-contraction coupling and intracellular calcium handling [127]. The microtubules, in particular, are subjected to chemotherapy (taxol) mediated cardiotoxicity [128]. At human autopsy as well as experimentally, the failed heart is characterized by an increase in microtubule density (microtubule densification) [129]. In Cooper’s original observations [130, 131], the group demonstrated marked pronouncement of microtubule densification during the transition from compensated hypertrophy (either RVH or LVH) to decompensated heart failure. The group later demonstrated that microtubule densification during the transition to decompensated heart failure was mediated by the stabilizing effects of dephosphorylated microtubule-associated protein (MAP4) [132].
Metabolic Remodeling in the Failing Heart
A hallmark of the failing heart is a reversion to fetal metabolism (the use of glucose as the primary source of energy). The use of carbohydrate as the major fuel source allows the fetal heart to withstand a low oxygen environment, a rapidly increasing hemodynamic burden, as well as the stimuli for growth. There is a rapid transition to fatty acid oxidation shortly after birth. Indeed, the adult heart is capable of utilizing all energy substrates (carbohydrate, lipid, amino acid, and ketone bodies), but the fatty acid oxidation is the key means of energy provision. Specifically, fatty acid oxidation yields ~2/3 of the ATP requirements, with ~1/3 of the requirements being yielded by glucose oxidation. In the failing heart, however, there is a metabolic re-programming imparted perhaps by the fetal gene program, that promotes a reversion back to the use of glucose as the major energy provision. This reversion not only impacts myocardial energetics, but also impacts contractility [40, 133].
The cardiomyocyte mitochondrion is the “powerhouse” responsible for regulating calcium homeostasis, effecting cellular death pathways, and generating 95% of the ATP via oxidative phosphorylation. The metabolites of fatty acid oxidation (fatty acyl-CoA) and carbohydrates (pyruvate) fuel the mitochondria [134]. The failing heart is characterized by decreased fatty acid and glucose oxidation, culminating in energy deficit. The decrease in fatty acid oxidation has been linked to decreases in the fatty acid oxidative enzymes in the diseased state.
The peroxisome proliferator-activated receptor family (PPARα, β/δ, γ) of nuclear receptor transcription factors regulate cardiac metabolism, pertinently, fatty acid utilization. PPARs and their coactivator protein PGC-1α are integral to the metabolic state of the healthy and diseased cardiomyocyte. PPAR isoforms heterodimerize with the retinoid X receptor (RXR) and bind PPAR response element (PPRE) to modulate transcription [135]. PPARα, highly expressed in the myocardium, regulates fatty acid transport and fatty acid oxidation pathways. The ligand binding domain of PPARα responds to changes in substrate availability [long-chain fatty acids and phospholipids are thought to serve as endogenous ligands]. In obesity and metabolic disorders, elevated levels of circulating free fatty acids modulate cardiac PPARα [136, 137]. Intriguingly, PPARα expression is decreased in human heart failure and animal models of pressure overload-induced cardiac hypertrophy; corresponding to the metabolic shift from fatty acid oxidation to glucose metabolism that occurs during the transition to cardiac hypertrophy and heart failure. Haemmerle et al. elegantly demonstrated that the activation of cardiac PPAR-α–PGC-1 complex is regulated by lipolytic catabolism of cellular triglyceride depots. The resulting signaling cascade regulates mitochondrial biogenesis, fatty acid metabolism and transport [124–126].
Mitochondria and Mitochondrial Remodeling in Decompensated Heart Failure
As discussed above, the biogenesis and function of mitochondria play a key role during metabolic remodeling of failing hearts. The mitochondrion is integral to energy transduction, ATP production, and calcium regulation. Mitochondria occupy 40% of the cardiomyocyte cytoplasm, with enclaves beneath the sarcolemma (subsarcolemmal mitochondria) or within the contractile apparatus (interfibrillar mitochondria) [138–140]. The healthy human heart generates an astounding 6–30kg of ATP per day in the quest to adapt to changes in demand. As above, myocardial contraction and relaxation are both highly regulated processes (and thus energy-requiring). This ATP generation is performed with little change in the intracellular ATP concentration [irrespective of the imposed workload], leaving the cell with little beat-to-beat reserve [135–139]. Oxidative phosphorylation (OXPHOS) generates >95% of the ATP in the adult myocardium. The failing heart is characterized by an inability to adapt and meet the energy requirements demanded. The failed heart is characterized by energy deficit. In animal models and in explanted human hearts, it is noted that cardiomyocyte mitochondria are altered in size, shape, DNA content, and number [138–140]. As an example, in the rat model of aortic constriction induced hypertrophy, there is increased mitochondrial size and mitochondrial DNA replication [141]. Changes in mitochondrial morphology and mitochondrial number are driven by transcription of both mitochondrial and nuclear DNA. Peroxisome proliferator-activated receptor gamma co-activator (PGC-1α) is critical to the synchronization of the two genomes. During compensated hypertrophy, the energy requirement of the hypertrophied cardiomyocyte is satisfied by mitochondrial biogenesis. In striking contradistinction, there is a downregulation of the master synchronizer PGC-1α, electron transport complex enzymes, and the entire mitochondrial biogenesis pathway in experimental models of decompensated heart failure [142].
During oxidative phosphorylation, oxygen serves as a low-energy electron acceptor of electrons derived from NADH and FADH2. The energy derived from oxidation/reduction reactions drives the phosphorylation of ADP to ATP. During this process, the electrons passage through the electron transport chain; the energy yielded from these series of reactions drive ATP synthesis [143]. In heart failure, there is a shift toward glucose oxidation from fatty acid oxidation. This metabolic remodeling in the failing heart is thought to underlie the energy-deficiency. As alluded to above, defects in the electron transport chain and fatty acid metabolite transport machinery, in addition to dysregulated PPAR and PGC-1 transcriptional activity, may be responsible for the reduction in fatty acid oxidation.
In cardiomyocytes derived from the failing heart, the calcium loses its orderly localized confinement. There is displacement of the sub-sarcolemmal mitochondria as a consequence of the altered myocardial stiffness and architecture. It is proposed that the altered configuration of the mitochondria and microtubules alters mitochondrial mechanosensor domains, with resultant abnormal calcium release in decompensated heart failure [127]. Endoplasmic reticulum release of calcium directly modulates mitochondria activity and function. Increased calcium concentration within the mitochondrion increases NADH and ATP production, and activates mitochondrial metabolic enzymes. Calcium- and phospholipid- mediated activation of PKCα modulates the level of oxidative phosphorylation. In an animal model, in which the investigators used cardiac-specific expression of calcineurin to exemplify compensated heart failure which subsequently progressed to heart failure, it is noted that the sub-sarcolemmal mitochondria exhibit decreased oxidative phosphorylation and increased superoxide production [144].
Defects in the electron transport chain and phosphorylation apparatus have been described in both humans and experimental animal models of heart failure, however how this bears out to the development and progression of HF remains to be established [140]. Indeed, Hamilton’s group isolated mitochondria from failing ventricles and determined that the mitochondria are capable of coupled respiration when supplied with external substrate, leading one to postulate whether there are issues with regard to substrate uptake and metabolism that contribute to the pathogenesis of heart failure [145].
Fatty acids, the preferred metabolic fuel for the healthy adult heart, are metabolized to acetyl-CoA which enters the Krebs cycle, generating further amounts of NADH and FADH2. The less utilized substrate in times of health, glucose, is metabolized via the glycolytic pathway, producing pyruvate. The pyruvate enters the Krebs cycle, similarly producing NADH and FADH2. The NADH and FADH2 enter into the electron transport chain, yielding ATP by oxidative phosphorylation in the mitochondria. Oxygen is the acceptor of low-energy electrons, and is critical to the function of the electron transport chain [146]. Under physiologic conditions, the aforementioned processes are synchronized (coupled). However, oxidative phosphorylation yields Reactive Oxygen Species (ROS) byproducts such as •O2− (superoxide anion radical), H2O2 (hydrogen peroxide), and •OH (hydroxyl radical). Electron leakage from the ETC generates •O2−. The generation of these ROS is exaggerated under conditions of imbalanced oxidant/antioxidant ratios. Detailed discussion of the mechanism and biochemistry of ROS is beyond the scope of this review, and the reader is referred to comprehensive reviews [146, 147].
H2O2 is a membrane-permeable ROS that functions as a second messenger. Manganese superoxide dismutase (MnSOD), an anti-oxidant enzyme encoded by the SOD2 gene, catalyzes the dismutation of O2·− to H2O2 [148]. The H2O2 scavenger, glutathione peroxidase (GPx), converts H2O2 to H2O [149]. In addition to the mitochondria, other sources of ROS production in the heart include NAD(P)H oxidases, cytochrome P450, and xanthine oxidase. Indeed, ROS at a regulated level mediate critical processes such as excitation-contraction coupling, calcium release, and cardiomyocyte growth. However, upon excess ROS production with an overwhelming of the antioxidant system, the cardiomyocyte is subjected to oxidative stress. This pathologic process is exacerbated under conditions of inflammation (TNFα and IL-1 stimulation), pathologic neurohormonal stimulation, and exuberant angiotensin (Ang) II stimulation [9, 150, 151]. Angiotensin II is thought to mediated cardiac hypertrophy via production of ROS and a subsequent deleterious gene transcription program mediated by NF-κB and AP-1 [146]. In a particularly provocative study, Rabinovitch and colleagues [152] demonstrated that mitochondrial ROS was critical to the Ang II mediated cardiac hypertrophy. They further demonstrated that targeting catalase specifically to the mitochondria, ameliorated LV hypertrophy, fibrosis and diastolic dysfunction induced by 4 weeks of Ang II. These effects were not seen when the catalase was overexpressed peroxisomes, reinforcing the essential role of mitochondrial ROS in the pathologic process. A vicious cycle of events is proposed, in which the ROS cause perturbation of mitochondrial DNA, further propagating ROS. When compounded by persistent Ang II and its mediation of ROS production, the hypertrophied heart may transition to decompensated heart failure.
CONCLUSION AND FUTURE DIRECTIONS
In the past two decades, the key signaling pathways and molecular integrators of adverse cardiac remodeling have been identified. An acquisition of detailed knowledge regarding the mechanisms by which adult cardiomyocytes sense and are directed to activate these deleterious signaling cascades will enable timely and targeted therapy. The arsenal for preventing adverse remodeling consists of beta-blockers, ACE-inhibitors, and angiotensin-receptor blockers, mineralocorticoid receptor blockers, hydralazine, nitrates, and cardiac resynchronization therapy. This leaves the vast majority of signaling cascades highlighted in this article untouched. Specifically, the LMNA/RhoA/MRTFA/SRF, MAPK, calcineurin-NFAT, and IGF1-PI3K–Akt cascades remain untouched pharmacologically. The data thus far suggest that the switch between physiological hypertrophy and pathologic hypertrophy is made at the nuclear level. Further data will have to either prove or disprove this notion. As of yet, there have been no reports directly implicating miRNA as mechanosensors in the cardiomyocyte. We have yet to exploit the potential of recently described proteins such as CIP and PC1 in cardiac remodeling and disease.
The factors that facilitate the cardiomyocyte to a remodeled state are likely at work in the clinically devastating and costly decompensated states. Therefore, there is much room for expansion in the therapeutic strategies against cardiomyocyte remodeling and decompensated heart failure. For example, abnormal calcium handling has a footprint in the diseased myocardium; the clinically relevant arrhythmogenic and decompensated heart failure states are mediated by aberrant calcium handling. Pharmacologically, in the instance of decompensated heart failure, there has been a paradigm switch; the focus has transitioned from “calcium mobilizers” to “calcium sensitizers.,” in essence forgoing the iatrogenic risks associated with changing intracellular calcium levels [153]. In Europe, the calcium sensitizing agent, Levosimendan, is recommended as first-line therapy for the patient in decompensated heart failure who receives beta-blockade and whose urinary output is low in the face of diuretic therapy. This agent stabilizes troponin C, prolonging its binding to calcium, and thus prolonging systolic actin-myosin interaction with the outcome of increased myocardial contractility. This agent has yet to be used in the USA [154]. Omecamtiv mecarbil belongs to a novel class of agents known as cardiac myosin activators. The drug binds directly to β-myosin heavy chain and increases the transition rate of myosin into the actin-bound force-generating state, thus enhancing cardiac contractility. This action is accomplished without raising levels of intracellular calcium [155, 156]. Thiazolidinediones (TZDs) activate peroxisome proliferator-activated receptor gamma (PPARγ) and are widely used in the therapy of Diabetes Mellitus II. However, they have been demonstrated to promote edema and exacerbated congestive heart failure likely as a result of renal tubular reabsorption of sodium. Fueled by the observations that 25% of heart failure patients with reduced ejection fraction exhibit hyperuricemia, the investigators of the EXACT-HF trial sought to quell Xanthine oxidase, a potential source of oxidative stress, using Allopurinol. Unfortunately, this therapy failed to demonstrate any clinical benefit yielded by the drug. Nonetheless, this study serves as only one of the initial steps in targeting aberrant ROS production and deleterious effects [157]. Although an agent acting upon the GSK-3 molecule is currently used in psychiatric disorders such as bipolar disorder (Lithium), one can imagine complications such as nephrogenic diabetes insipidus and mood alteration being limitations to the use of this drug in cardiac pathologies. Provided we are armed with the specific pharmacologic agents targeting the intended isoform, we have yet to tap into the potential of GSK-3 inhibitors and activators. As of now, it can be stated that the surgical and mechanical technology have excelled in the therapy of both preventing remodeling and the same deleterious forces from the progression to decompensated heart failure. Although there have been vigorous attempts to intervene on some of the pathways detailed above, we have yet to make pharmaceutical advances with regard to factors at the cytoplasm/nucleus interface, nuclear integrators, and microRNA.
Acknowledgements
We thank Zhanpeng Huang for critical input into this manuscript. We thank members of the Wang lab for their insight. We are grateful to Mohammad Aslam (Chroma enterprises) for helping with figures. We apologize to our colleagues whose work could not be cited here due to space limitations. Work in the Wang lab is supported by the American Heart Association, Muscular Dystrophy Association and the NIH (HL085635, HL116919, HL125925). Z.K. Haque is supported by NIH T32HL007572.
References:
- 1.Jugdutt BI, Dhalla NS, and Springer Link, Cardiac Remodeling Molecular Mechanisms, in Advances in Biochemistry in Health and Disease 5. 2013, Springer New York : Imprint: Springer,: New York, NY: p. XI, 569 p. 80 illus., 48 illus. in color. [Google Scholar]
- 2.Kumar V, Abbas AK, and Aster JC, Robbins and Cotran pathologic basis of disease. Ninth edition ed. 2015, Philadelphia, PA: Elsevier/Saunders; xvi, 1391 pages. [Google Scholar]
- 3.Molkentin JD, Parsing good versus bad signaling pathways in the heart: role of calcineurin-nuclear factor of activated T-cells. Circ Res, 2013. 113(1): p. 16–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu Q, et al. , Interaction between NFkappaB and NFAT coordinates cardiac hypertrophy and pathological remodeling. Circ Res, 2012. 110(8): p. 1077–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van Berlo JH, Maillet M, and Molkentin JD, Signaling effectors underlying pathologic growth and remodeling of the heart. J Clin Invest, 2013. 123(1): p. 37–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burchfield JS, Xie M, and Hill JA, Pathological ventricular remodeling: mechanisms: part 1 of 2. Circulation, 2013. 128(4): p. 388–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shiojima I and Walsh K, Regulation of cardiac growth and coronary angiogenesis by the Akt/PKB signaling pathway. Genes Dev, 2006. 20(24): p. 3347–65. [DOI] [PubMed] [Google Scholar]
- 8.Xie M, Burchfield JS, and Hill JA, Pathological ventricular remodeling: therapies: part 2 of 2. Circulation, 2013. 128(9): p. 1021–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mann DL, et al. , Braunwald’s heart disease : a textbook of cardiovascular medicine. 2015, Elsevier/Saunders,: Philadelphia, PA: p. 1 online resource (xxvii, 1943, 3 pages). [Google Scholar]
- 10.Grossman W and Paulus WJ, Myocardial stress and hypertrophy: a complex interface between biophysics and cardiac remodeling. J Clin Invest, 2013. 123(9): p. 3701–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gerdes AM, Cardiac myocyte remodeling in hypertrophy and progression to failure. J Card Fail, 2002. 8(6 Suppl): p. S264–8. [DOI] [PubMed] [Google Scholar]
- 12.Frangogiannis NG, Pathophysiology of Myocardial Infarction. Compr Physiol, 2015. 5(4): p. 1841–75. [DOI] [PubMed] [Google Scholar]
- 13.Francis GS, Changing the remodeling process in heart failure: basic mechanisms and laboratory results. Curr Opin Cardiol, 1998. 13(3): p. 156–61. [PubMed] [Google Scholar]
- 14.Lauriol J, et al. , RhoA signaling in cardiomyocytes protects against stress-induced heart failure but facilitates cardiac fibrosis. Sci Signal, 2014. 7(348): p. ra100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cohn JN, Ferrari R, and Sharpe N, Cardiac remodeling--concepts and clinical implications: a consensus paper from an international forum on cardiac remodeling. Behalf of an International Forum on Cardiac Remodeling. J Am Coll Cardiol, 2000. 35(3): p. 569–82. [DOI] [PubMed] [Google Scholar]
- 16.Hill JA and Olson EN, Cardiac plasticity. N Engl J Med, 2008. 358(13): p. 1370–80. [DOI] [PubMed] [Google Scholar]
- 17.Yang JH, et al. , Association of beta-blocker therapy at discharge with clinical outcomes in patients with ST-segment elevation myocardial infarction undergoing primary percutaneous coronary intervention. JACC Cardiovasc Interv, 2014. 7(6): p. 592–601. [DOI] [PubMed] [Google Scholar]
- 18.Frangogiannis NG, Inflammation in cardiac injury, repair and regeneration. Curr Opin Cardiol, 2015. 30(3): p. 240–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Doroudgar S and Glembotski CC, New concepts of endoplasmic reticulum function in the heart: programmed to conserve. J Mol Cell Cardiol, 2013. 55: p. 85–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Glembotski CC, Roles for ATF6 and the sarco/endoplasmic reticulum protein quality control system in the heart. J Mol Cell Cardiol, 2014. 71: p. 11–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zou Y, et al. , Mechanical stress activates angiotensin II type 1 receptor without the involvement of angiotensin II. Nat Cell Biol, 2004. 6(6): p. 499–506. [DOI] [PubMed] [Google Scholar]
- 22.Frey N and Olson EN, Cardiac hypertrophy: the good, the bad, and the ugly. Annu Rev Physiol, 2003. 65: p. 45–79. [DOI] [PubMed] [Google Scholar]
- 23.Heineke J and Molkentin JD, Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat Rev Mol Cell Biol, 2006. 7(8): p. 589–600. [DOI] [PubMed] [Google Scholar]
- 24.Takefuji M, et al. , RhoGEF12 controls cardiac remodeling by integrating G protein- and integrin-dependent signaling cascades. J Exp Med, 2013. 210(4): p. 665–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Capote LA, Mendez Perez R, and Lymperopoulos A, GPCR signaling and cardiac function. Eur J Pharmacol, 2015. 763(Pt B): p. 143–8. [DOI] [PubMed] [Google Scholar]
- 26.Xiao RP, Beta-adrenergic signaling in the heart: dual coupling of the beta2-adrenergic receptor to G(s) and G(i) proteins. Sci STKE, 2001. 2001(104): p. re15. [DOI] [PubMed] [Google Scholar]
- 27.Snabaitis AK, et al. , Regulation of the extracellular signal-regulated kinase pathway in adult myocardium: differential roles of G(q/11), Gi and G(12/13) proteins in signalling by alpha1-adrenergic, endothelin-1 and thrombin-sensitive protease-activated receptors. Cell Signal, 2005. 17(5): p. 655–64. [DOI] [PubMed] [Google Scholar]
- 28.Takeishi Y, Goto K, and Kubota I, Role of diacylglycerol kinase in cellular regulatory processes: a new regulator for cardiomyocyte hypertrophy. Pharmacol Ther, 2007. 115(3): p. 352–9. [DOI] [PubMed] [Google Scholar]
- 29.Domeier TL, et al. , IP3 receptor-dependent Ca2+ release modulates excitation-contraction coupling in rabbit ventricular myocytes. Am J Physiol Heart Circ Physiol, 2008. 294(2): p. H596–604. [DOI] [PubMed] [Google Scholar]
- 30.Branco AF and Allen BG, G protein-coupled receptor signaling in cardiac nuclear membranes. J Cardiovasc Pharmacol, 2015. 65(2): p. 101–9. [DOI] [PubMed] [Google Scholar]
- 31.Small EM, The actin-MRTF-SRF gene regulatory axis and myofibroblast differentiation. J Cardiovasc Transl Res, 2012. 5(6): p. 794–804. [DOI] [PubMed] [Google Scholar]
- 32.Ferreira JC, Mochly-Rosen D, and Boutjdir M, Regulation of cardiac excitability by protein kinase C isozymes. Front Biosci (Schol Ed), 2012. 4: p. 532–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ytrehus K, Liu Y, and Downey JM, Preconditioning protects ischemic rabbit heart by protein kinase C activation. Am J Physiol, 1994. 266(3 Pt 2): p. H1145–52. [DOI] [PubMed] [Google Scholar]
- 34.O’Connell TD, et al. , Cardiac alpha1-adrenergic receptors: novel aspects of expression, signaling mechanisms, physiologic function, and clinical importance. Pharmacol Rev, 2014. 66(1): p. 308–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Braz JC, et al. , PKC-alpha regulates cardiac contractility and propensity toward heart failure. Nat Med, 2004. 10(3): p. 248–54. [DOI] [PubMed] [Google Scholar]
- 36.Liu Q and Molkentin JD, Protein kinase Calpha as a heart failure therapeutic target. J Mol Cell Cardiol, 2011. 51(4): p. 474–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guo J, et al. , Protein kinase D isoforms are activated in an agonist-specific manner in cardiomyocytes. J Biol Chem, 2011. 286(8): p. 6500–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fielitz J, et al. , Requirement of protein kinase D1 for pathological cardiac remodeling. Proc Natl Acad Sci U S A, 2008. 105(8): p. 3059–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Avkiran M, et al. , Protein kinase d in the cardiovascular system: emerging roles in health and disease. Circ Res, 2008. 102(2): p. 157–63. [DOI] [PubMed] [Google Scholar]
- 40.Kolwicz SC Jr., Purohit S, and Tian R, Cardiac metabolism and its interactions with contraction, growth, and survival of cardiomyocytes. Circ Res, 2013. 113(5): p. 603–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hu P, et al. , Minimally invasive aortic banding in mice: effects of altered cardiomyocyte insulin signaling during pressure overload. Am J Physiol Heart Circ Physiol, 2003. 285(3): p. H1261–9. [DOI] [PubMed] [Google Scholar]
- 42.Asrih M, et al. , Role of mitogen-activated protein kinase pathways in multifactorial adverse cardiac remodeling associated with metabolic syndrome. Mediators Inflamm, 2013. 2013: p. 367245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yu W, et al. , Insulin signaling: a possible pathogenesis of cardiac hypertrophy. Cardiovasc Ther, 2010. 28(2): p. 101–5. [DOI] [PubMed] [Google Scholar]
- 44.Samuelsson AM, et al. , Hyperinsulinemia: effect on cardiac mass/function, angiotensin II receptor expression, and insulin signaling pathways. Am J Physiol Heart Circ Physiol, 2006. 291(2): p. H787–96. [DOI] [PubMed] [Google Scholar]
- 45.Zhang Y, et al. , Insulin-like growth factor 1 alleviates high-fat diet-induced myocardial contractile dysfunction: role of insulin signaling and mitochondrial function. Hypertension, 2012. 59(3): p. 680–93. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 46.Damilano F, Perino A, and Hirsch E, PI3K kinase and scaffold functions in heart. Ann N Y Acad Sci, 2010. 1188: p. 39–45. [DOI] [PubMed] [Google Scholar]
- 47.Embi N, Rylatt DB, and Cohen P, Glycogen synthase kinase-3 from rabbit skeletal muscle. Separation from cyclic-AMP-dependent protein kinase and phosphorylase kinase. Eur J Biochem, 1980. 107(2): p. 519–27. [PubMed] [Google Scholar]
- 48.Sampson EM, et al. , Negative regulation of the Wnt-beta-catenin pathway by the transcriptional repressor HBP1. EMBO J, 2001. 20(16): p. 4500–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Woulfe KC, et al. , Glycogen synthase kinase-3beta regulates post-myocardial infarction remodeling and stress-induced cardiomyocyte proliferation in vivo. Circ Res, 2010. 106(10): p. 1635–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lal H, et al. , The GSK-3 family as therapeutic target for myocardial diseases. Circ Res, 2015. 116(1): p. 138–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Haq S, et al. , Glycogen synthase kinase-3beta is a negative regulator of cardiomyocyte hypertrophy. J Cell Biol, 2000. 151(1): p. 117–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ahmad F, et al. , Cardiomyocyte-specific deletion of Gsk3alpha mitigates post-myocardial infarction remodeling, contractile dysfunction, and heart failure. J Am Coll Cardiol, 2014. 64(7): p. 696–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sankaranarayanan R, et al. , Biphasic decay of the Ca transient results from increased sarcoplasmic reticulum Ca leak. J Physiol, 2016. 594(3): p. 611–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Woodier J, et al. , Intracellular Zinc Modulates Cardiac Ryanodine Receptor-mediated Calcium Release. J Biol Chem, 2015. 290(28): p. 17599–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Inesi G, Prasad AM, and Pilankatta R, The Ca2+ ATPase of cardiac sarcoplasmic reticulum: Physiological role and relevance to diseases. Biochem Biophys Res Commun, 2008. 369(1): p. 182–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liu T and O’Rourke B, Regulation of the Na+/Ca2+ exchanger by pyridine nucleotide redox potential in ventricular myocytes. J Biol Chem, 2013. 288(44): p. 31984–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pieske B, et al. , Alterations in intracellular calcium handling associated with the inverse force-frequency relation in human dilated cardiomyopathy. Circulation, 1995. 92(5): p. 1169–78. [DOI] [PubMed] [Google Scholar]
- 58.Marx SO, et al. , Coupled gating between cardiac calcium release channels (ryanodine receptors). Circ Res, 2001. 88(11): p. 1151–8. [DOI] [PubMed] [Google Scholar]
- 59.Gadeberg HC, et al. , Altered Na/Ca exchange distribution in ventricular myocytes from failing hearts. Am J Physiol Heart Circ Physiol, 2016. 310(2): p. H262–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Xie J, et al. , Cardioprotection by Klotho through downregulation of TRPC6 channels in the mouse heart. Nat Commun, 2012. 3: p. 1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kreusser MM, et al. , Cardiac CaM Kinase II genes delta and gamma contribute to adverse remodeling but redundantly inhibit calcineurin-induced myocardial hypertrophy. Circulation, 2014. 130(15): p. 1262–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ling H, et al. , Requirement for Ca2+/calmodulin-dependent kinase II in the transition from pressure overload-induced cardiac hypertrophy to heart failure in mice. J Clin Invest, 2009. 119(5): p. 1230–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Weinreuter M, et al. , CaM Kinase II mediates maladaptive post-infarct remodeling and pro-inflammatory chemoattractant signaling but not acute myocardial ischemia/reperfusion injury. EMBO Mol Med, 2014. 6(10): p. 1231–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Aikawa R, et al. , Integrins play a critical role in mechanical stress-induced p38 MAPK activation. Hypertension, 2002. 39(2): p. 233–8. [DOI] [PubMed] [Google Scholar]
- 65.Purcell NH, et al. , Genetic inhibition of cardiac ERK1/2 promotes stress-induced apoptosis and heart failure but has no effect on hypertrophy in vivo. Proc Natl Acad Sci U S A, 2007. 104(35): p. 14074–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Martin ED, Bassi R, and Marber MS, p38 MAPK in cardioprotection - are we there yet? Br J Pharmacol, 2015. 172(8): p. 2101–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mathiyalagan P, et al. , Chromatin modifications remodel cardiac gene expression. Cardiovasc Res, 2014. 103(1): p. 7–16. [DOI] [PubMed] [Google Scholar]
- 68.Weeks KL and Avkiran M, Roles and post-translational regulation of cardiac class IIa histone deacetylase isoforms. J Physiol, 2015. 593(8): p. 1785–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Xie M and Hill JA, HDAC-dependent ventricular remodeling. Trends Cardiovasc Med, 2013. 23(6): p. 229–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.McKinsey TA, et al. , Signal-dependent nuclear export of a histone deacetylase regulates muscle differentiation. Nature, 2000. 408(6808): p. 106–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kim Y, et al. , The MEF2D transcription factor mediates stress-dependent cardiac remodeling in mice. J Clin Invest, 2008. 118(1): p. 124–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hogan PG, et al. , Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev, 2003. 17(18): p. 2205–32. [DOI] [PubMed] [Google Scholar]
- 73.Wilkins BJ, et al. , Calcineurin/NFAT coupling participates in pathological, but not physiological, cardiac hypertrophy. Circ Res, 2004. 94(1): p. 110–8. [DOI] [PubMed] [Google Scholar]
- 74.Pikkarainen S, et al. , GATA transcription factors in the developing and adult heart. Cardiovasc Res, 2004. 63(2): p. 196–207. [DOI] [PubMed] [Google Scholar]
- 75.van Berlo JH, et al. , The transcription factor GATA-6 regulates pathological cardiac hypertrophy. Circ Res, 2010. 107(8): p. 1032–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Prendiville TW, et al. , Novel Roles of GATA4/6 in the Postnatal Heart Identified through Temporally Controlled, Cardiomyocyte-Specific Gene Inactivation by Adeno-Associated Virus Delivery of Cre Recombinase. PLoS One, 2015. 10(5): p. e0128105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lyon RC, et al. , Mechanotransduction in cardiac hypertrophy and failure. Circ Res, 2015. 116(8): p. 1462–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.McCain ML and Parker KK, Mechanotransduction: the role of mechanical stress, myocyte shape, and cytoskeletal architecture on cardiac function. Pflugers Arch, 2011. 462(1): p. 89–104. [DOI] [PubMed] [Google Scholar]
- 79.Cingolani HE, et al. , The Anrep effect: 100 years later. Am J Physiol Heart Circ Physiol, 2013. 304(2): p. H175–82. [DOI] [PubMed] [Google Scholar]
- 80.Petroff MG, et al. , Endogenous nitric oxide mechanisms mediate the stretch dependence of Ca2+ release in cardiomyocytes. Nat Cell Biol, 2001. 3(10): p. 867–73. [DOI] [PubMed] [Google Scholar]
- 81.Jian Z, et al. , Mechanochemotransduction during cardiomyocyte contraction is mediated by localized nitric oxide signaling. Sci Signal, 2014. 7(317): p. ra27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Xiang SY, et al. , RhoA protects the mouse heart against ischemia/reperfusion injury. J Clin Invest, 2011. 121(8): p. 3269–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Miyamoto S, et al. , Revisited and revised: is RhoA always a villain in cardiac pathophysiology? J Cardiovasc Transl Res, 2010. 3(4): p. 330–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Dostal DE, et al. , Mechanosensing and Regulation of Cardiac Function. J Clin Exp Cardiolog, 2014. 5(6): p. 314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.White DE, et al. , Targeted ablation of ILK from the murine heart results in dilated cardiomyopathy and spontaneous heart failure. Genes Dev, 2006. 20(17): p. 2355–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lu H, et al. , Integrin-linked kinase expression is elevated in human cardiac hypertrophy and induces hypertrophy in transgenic mice. Circulation, 2006. 114(21): p. 2271–9. [DOI] [PubMed] [Google Scholar]
- 87.Delcommenne M, et al. , Phosphoinositide-3-OH kinase-dependent regulation of glycogen synthase kinase 3 and protein kinase B/AKT by the integrin-linked kinase. Proc Natl Acad Sci U S A, 1998. 95(19): p. 11211–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Traister A, et al. , Integrin-linked kinase mediates force transduction in cardiomyocytes by modulating SERCA2a/PLN function. Nat Commun, 2014. 5: p. 4533. [DOI] [PubMed] [Google Scholar]
- 89.Pedrozo Z, et al. , Polycystin-1 Is a Cardiomyocyte Mechanosensor That Governs L-Type Ca2+ Channel Protein Stability. Circulation, 2015. 131(24): p. 2131–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Jia G, DeMarco VG, and Sowers JR, Insulin resistance and hyperinsulinaemia in diabetic cardiomyopathy. Nat Rev Endocrinol, 2016. 12(3): p. 144–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Liu W, et al. , High-mobility group box 1 (HMGB1) downregulates cardiac transient outward potassium current (Ito) through downregulation of Kv4.2 and Kv4.3 channel transcripts and proteins. J Mol Cell Cardiol, 2010. 49(3): p. 438–48. [DOI] [PubMed] [Google Scholar]
- 92.Zhu W, et al. , Interaction of beta1-adrenoceptor with RAGE mediates cardiomyopathy via CaMKII signaling. JCI Insight, 2016. 1(1): p. e84969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Genschel J and Schmidt HH, Mutations in the LMNA gene encoding lamin A/C. Hum Mutat, 2000. 16(6): p. 451–9. [DOI] [PubMed] [Google Scholar]
- 94.Lu JT, et al. , LMNA cardiomyopathy: cell biology and genetics meet clinical medicine. Dis Model Mech, 2011. 4(5): p. 562–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lammerding J, et al. , Lamin A/C deficiency causes defective nuclear mechanics and mechanotransduction. J Clin Invest, 2004. 113(3): p. 370–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Chandar S, et al. , Effects of mechanical stress and carvedilol in lamin A/C-deficient dilated cardiomyopathy. Circ Res, 2010. 106(3): p. 573–82. [DOI] [PubMed] [Google Scholar]
- 97.Cupesi M, et al. , Attenuated hypertrophic response to pressure overload in a lamin A/C haploinsufficiency mouse. J Mol Cell Cardiol, 2010. 48(6): p. 1290–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sylvius N and Tesson F, Lamin A/C and cardiac diseases. Curr Opin Cardiol, 2006. 21(3): p. 159–65. [DOI] [PubMed] [Google Scholar]
- 99.Mounkes LC, et al. , Expression of an LMNA-N195K variant of A-type lamins results in cardiac conduction defects and death in mice. Hum Mol Genet, 2005. 14(15): p. 2167–80. [DOI] [PubMed] [Google Scholar]
- 100.Arimura T, et al. , Mouse model carrying H222P-Lmna mutation develops muscular dystrophy and dilated cardiomyopathy similar to human striated muscle laminopathies. Hum Mol Genet, 2005. 14(1): p. 155–69. [DOI] [PubMed] [Google Scholar]
- 101.Muchir A, Wu W, and Worman HJ, Mitogen-activated protein kinase inhibitor regulation of heart function and fibrosis in cardiomyopathy caused by lamin A/C gene mutation. Trends Cardiovasc Med, 2010. 20(7): p. 217–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wu W, et al. , Mitogen-activated protein kinase inhibitors improve heart function and prevent fibrosis in cardiomyopathy caused by mutation in lamin A/C gene. Circulation, 2011. 123(1): p. 53–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Huang ZP, et al. , Cardiomyocyte-enriched protein CIP protects against pathophysiological stresses and regulates cardiac homeostasis. J Clin Invest, 2015. 125(11): p. 4122–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Huang ZP, et al. , CIP, a cardiac Isl1-interacting protein, represses cardiomyocyte hypertrophy. Circ Res, 2012. 110(6): p. 818–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Cattin ME, et al. , Deletion of MLIP (muscle-enriched A-type lamin-interacting protein) leads to cardiac hyperactivation of Akt/mammalian target of rapamycin (mTOR) and impaired cardiac adaptation. J Biol Chem, 2015. 290(44): p. 26699–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Chen J and Wang DZ, microRNAs in cardiovascular development. J Mol Cell Cardiol, 2012. 52(5): p. 949–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wu G, Huang ZP, and Wang DZ, MicroRNAs in cardiac regeneration and cardiovascular disease. Sci China Life Sci, 2013. 56(10): p. 907–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Neppl RL and Wang DZ, The myriad essential roles of microRNAs in cardiovascular homeostasis and disease. Genes Dis, 2014. 1(1): p. 18–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kataoka M and Wang DZ, Non-Coding RNAs Including miRNAs and lncRNAs in Cardiovascular Biology and Disease. Cells, 2014. 3(3): p. 883–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Huang ZP, et al. , MicroRNA-22 regulates cardiac hypertrophy and remodeling in response to stress. Circ Res, 2013. 112(9): p. 1234–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Huang ZP and Wang DZ, miR-22 in cardiac remodeling and disease. Trends Cardiovasc Med, 2014. 24(7): p. 267–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ding J, et al. , Trbp regulates heart function through microRNA-mediated Sox6 repression. Nat Genet, 2015. 47(7): p. 776–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Liu X, et al. , miR-222 is necessary for exercise-induced cardiac growth and protects against pathological cardiac remodeling. Cell Metab, 2015. 21(4): p. 584–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Uchida S and Dimmeler S, Exercise controls non-coding RNAs. Cell Metab, 2015. 21(4): p. 511–2. [DOI] [PubMed] [Google Scholar]
- 115.Hill JA, Braking bad hypertrophy. N Engl J Med, 2015. 372(22): p. 2160–2. [DOI] [PubMed] [Google Scholar]
- 116.Chen J, et al. , mir-17–92 cluster is required for and sufficient to induce cardiomyocyte proliferation in postnatal and adult hearts. Circ Res, 2013. 112(12): p. 1557–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Jonsson S, et al. , Renal neurohormonal regulation in heart failure decompensation. Am J Physiol Regul Integr Comp Physiol, 2014. 307(5): p. R493–7. [DOI] [PubMed] [Google Scholar]
- 118.Wei S, et al. , T-tubule remodeling during transition from hypertrophy to heart failure. Circ Res, 2010. 107(4): p. 520–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Takeshima H, Hoshijima M, and Song LS, Ca(2)(+) microdomains organized by junctophilins. Cell Calcium, 2015. 58(4): p. 349–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Zhang HB, et al. , Ultrastructural uncoupling between T-tubules and sarcoplasmic reticulum in human heart failure. Cardiovasc Res, 2013. 98(2): p. 269–76. [DOI] [PubMed] [Google Scholar]
- 121.Li RC, et al. , In vivo suppression of microRNA-24 prevents the transition toward decompensated hypertrophy in aortic-constricted mice. Circ Res, 2013. 112(4): p. 601–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Carniel E, et al. , Alpha-myosin heavy chain: a sarcomeric gene associated with dilated and hypertrophic phenotypes of cardiomyopathy. Circulation, 2005. 112(1): p. 54–9. [DOI] [PubMed] [Google Scholar]
- 123.Montgomery RL, et al. , Therapeutic inhibition of miR-208a improves cardiac function and survival during heart failure. Circulation, 2011. 124(14): p. 1537–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Callis TE, et al. , MicroRNA-208a is a regulator of cardiac hypertrophy and conduction in mice. J Clin Invest, 2009. 119(9): p. 2772–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Paulin R, et al. , A miR-208-Mef2 axis drives the decompensation of right ventricular function in pulmonary hypertension. Circ Res, 2015. 116(1): p. 56–69. [DOI] [PubMed] [Google Scholar]
- 126.Tang H, Fernandez RA, and Yuan JX, miRNA208/Mef2 and TNF-alpha in right ventricular dysfunction: the transition from hypertrophy to failure. Circ Res, 2015. 116(1): p. 6–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Miragoli M, et al. , Microtubule-Dependent Mitochondria Alignment Regulates Calcium Release in Response to Nanomechanical Stimulus in Heart Myocytes. Cell Rep, 2016. 14(1): p. 140–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Zhang C, et al. , Microtubule-mediated defects in junctophilin-2 trafficking contribute to myocyte transverse-tubule remodeling and Ca2+ handling dysfunction in heart failure. Circulation, 2014. 129(17): p. 1742–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Kostin S, et al. , The cytoskeleton and related proteins in the human failing heart. Heart Fail Rev, 2000. 5(3): p. 271–80. [DOI] [PubMed] [Google Scholar]
- 130.Tsutsui H, Ishihara K, and Cooper G.t., Cytoskeletal role in the contractile dysfunction of hypertrophied myocardium. Science, 1993. 260(5108): p. 682–7. [DOI] [PubMed] [Google Scholar]
- 131.Tsutsui H, et al. , Role of microtubules in contractile dysfunction of hypertrophied cardiocytes. Circulation, 1994. 90(1): p. 533–55. [DOI] [PubMed] [Google Scholar]
- 132.Sato H, et al. , Microtubule stabilization in pressure overload cardiac hypertrophy. J Cell Biol, 1997. 139(4): p. 963–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Rees ML, et al. , A PKM2 signature in the failing heart. Biochem Biophys Res Commun, 2015. 459(3): p. 430–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Sankaralingam S and Lopaschuk GD, Cardiac energy metabolic alterations in pressure overload-induced left and right heart failure (2013 Grover Conference Series). Pulm Circ, 2015. 5(1): p. 15–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Aubert G, Vega RB, and Kelly DP, Perturbations in the gene regulatory pathways controlling mitochondrial energy production in the failing heart. Biochim Biophys Acta, 2013. 1833(4): p. 840–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Haemmerle G, et al. , ATGL-mediated fat catabolism regulates cardiac mitochondrial function via PPAR-alpha and PGC-1. Nat Med, 2011. 17(9): p. 1076–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Wu SP, et al. , Increased COUP-TFII expression in adult hearts induces mitochondrial dysfunction resulting in heart failure. Nat Commun, 2015. 6: p. 8245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Dorn GW 2nd, Vega RB, and Kelly DP, Mitochondrial biogenesis and dynamics in the developing and diseased heart. Genes Dev, 2015. 29(19): p. 1981–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Guzun R, et al. , Modular organization of cardiac energy metabolism: energy conversion, transfer and feedback regulation. Acta Physiol (Oxf), 2015. 213(1): p. 84–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Rosca MG and Hoppel CL, Mitochondria in heart failure. Cardiovasc Res, 2010. 88(1): p. 40–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Zak R, et al. , Mitochondrial proliferation in cardiac hypertrophy. Basic Res Cardiol, 1980. 75(1): p. 171–8. [DOI] [PubMed] [Google Scholar]
- 142.Rowe GC, Jiang A, and Arany Z, PGC-1 coactivators in cardiac development and disease. Circ Res, 2010. 107(7): p. 825–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Cooper GM and Hausman RE, The cell : a molecular approach. Third edition ed. xx, 713 pages. [Google Scholar]
- 144.Sayen MR, et al. , Calcineurin transgenic mice have mitochondrial dysfunction and elevated superoxide production. Am J Physiol Cell Physiol, 2003. 284(2): p. C562–70. [DOI] [PubMed] [Google Scholar]
- 145.Cordero-Reyes AM, et al. , Freshly isolated mitochondria from failing human hearts exhibit preserved respiratory function. J Mol Cell Cardiol, 2014. 68: p. 98–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Giordano FJ, Oxygen, oxidative stress, hypoxia, and heart failure. J Clin Invest, 2005. 115(3): p. 500–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Chen YR and Zweier JL, Cardiac mitochondria and reactive oxygen species generation. Circ Res, 2014. 114(3): p. 524–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Lee S, et al. , Mitochondrial H2O2 generated from electron transport chain complex I stimulates muscle differentiation. Cell Res, 2011. 21(5): p. 817–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Aon MA, et al. , Glutathione/thioredoxin systems modulate mitochondrial H2O2 emission: an experimental-computational study. J Gen Physiol, 2012. 139(6): p. 479–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Zablocki D and Sadoshima J, Angiotensin II and oxidative stress in the failing heart. Antioxid Redox Signal, 2013. 19(10): p. 1095–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Burgoyne JR, et al. , Redox signaling in cardiac physiology and pathology. Circ Res, 2012. 111(8): p. 1091–106. [DOI] [PubMed] [Google Scholar]
- 152.Dai DF, et al. , Mitochondrial oxidative stress mediates angiotensin II-induced cardiac hypertrophy and Galphaq overexpression-induced heart failure. Circ Res, 2011. 108(7): p. 837–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Pollesello P, Papp Z, and Papp JG, Calcium sensitizers: What have we learned over the last 25 years? Int J Cardiol, 2016. 203: p. 543–8. [DOI] [PubMed] [Google Scholar]
- 154.Nieminen MS, et al. , The role of levosimendan in acute heart failure complicating acute coronary syndrome: A review and expert consensus opinion. Int J Cardiol, 2016. 218: p. 150–7. [DOI] [PubMed] [Google Scholar]
- 155.Greenberg BH, et al. , Safety and tolerability of omecamtiv mecarbil during exercise in patients with ischemic cardiomyopathy and angina. JACC Heart Fail, 2015. 3(1): p. 22–9. [DOI] [PubMed] [Google Scholar]
- 156.Nanasi P Jr., Vaczi K, and Papp Z, The myosin activator omecamtiv mecarbil: a promising new inotropic agent. Can J Physiol Pharmacol, 2016: p. 1–7. [DOI] [PubMed] [Google Scholar]
- 157.Givertz MM, et al. , Effects of Xanthine Oxidase Inhibition in Hyperuricemic Heart Failure Patients: The Xanthine Oxidase Inhibition for Hyperuricemic Heart Failure Patients (EXACT-HF) Study. Circulation, 2015. 131(20): p. 1763–71. [DOI] [PMC free article] [PubMed] [Google Scholar]