

Abstract
The potential modulation of TRPV1 nociceptive activity by the CB1 receptor was investigated here using CB1 wildtype (WT) and knock-out (KO) mice as well as selective CB1 inverse agonists. No significant differences were detected in baseline thermal thresholds of ICR, CB1WT or CB1KO mice. Intraplantar capsaicin produced dose- and time-related paw flinch responses in ICR and CB1WT mice and induced plasma extravasation yet minimal responses were seen in CB1KO animals with no apparent differences in TRPV1 channel expression. Capsaicin-evoked CGRP release from spinal cord tissue and capsaicin-evoked action potentials on isolated skin-nerve preparation were significantly decreased in CB1KO mice. Pretreatment with intraplantar galanin and bradykinin, compounds known to sensitize TRPV1 receptors, restored capsaicin-induced flinching in CB1KO mice. The possibility that constitutive activity at the CB1 receptor is required to maintain the TRPV1 receptor in a “sensitized” state was tested using CB1 inverse agonists. The CB1 inverse agonists elicited concentration-related inhibition of capsaicin-induced calcium influx in F-11 cells and produced dose-related inhibition of capsaicin-induced flinching in ICR mice. These data suggest that constitutive activity at the CB1 receptor maintains the TRPV1 channel in a sensitized state responsive to noxious chemical stimuli. Treatment with CB1 inverse agonists may promote desensitization of the channel resulting in antinociceptive actions against chemical stimulus modalities. These studies propose possible therapeutic exploitation of a novel mechanism providing pain relief by CB1 inverse agonists.
Keywords: TRPV1, CB1, Capsaicin, PIP2, phospholipase C, knockout mouse
Introduction
The transient receptor potential vanilloid 1 (TRPV1) channel has been established as a molecular sensor of noxious heat and chemicals including capsaicin (Caterina et al., 1997) and can be directly activated by protons (Tominaga et al., 1998), lipoxygenase products (Hwang et al., 2000) and endocannabinoids (Zygmunt et al., 1999). This channel is also an important integrator of nociceptive stimuli resulting from activation of multiple receptors on nociceptors (Caterina et al., 2000; Davis et al., 2000) leading to intense efforts in developing TRPV1 antagonists as pain relieving drugs.
In keeping with a role as a molecular integrator of multiple signaling pathways, the responsiveness of the TRPV1 channel to capsaicin can also be modulated by lipids (Zygmunt et al., 1999; Hwang et al., 2000; Chuang et al., 2001), heat >43°C or elevated concentration of protons (Caterina et al., 1997; Tominaga et al., 1998) and endogenous endovanilloids (Hermann et al., 2003; Price et al., 2004; Zygmunt et al., 1999). Additionally, activity at numerous G-protein coupled receptors (GPCRs) including the galanin R1 (Jimenez-Andrade et al., 2004), somatostatin S2 (Carlton et al., 2004), bradykinin B2 (Cesare and McNaughton, 1996; Chuang et al., 2001), and cannabinoid CB1 (Jeske et al., 2006; Patwardhan et al., 2006; Hermann et al., 2003) has been demonstrated to modulate TRPV1 function. Chimeric and site-directed mutation studies of the TRPV1 channel have identified unique amino acid sites that are phosphorylated (Bhave et al., 2002, 2003; Jung et al., 2004) resulting in increased responsiveness of the channel to stimuli including capsaicin. Likewise, the dephosphorylation of TRPV1 channels can lead to pharmacological desensitization of the channel (Koplas et al., 1997; Mohapatra and Nau, 2005).
Endogenous and exogenous cannabinoids have been shown to inhibit pain behaviors (Pertwee, 1997; Richardson et al., 1998a,b) as well as capsaicin-induced release of the pain neurotransmitter CGRP (Ahluwalia et al., 2003; Richardson et al., 1998a,b). However more recent studies have demonstrated how some cannabinoids may enhance TRPV1 channel activity (Zygmunt et al., 1999; Hermann et al., 2003). The pretreatment using NGF results in an increased anandamide-evoked CGRP release in cultured trigeminal (Price et al., 2005) and dorsal root ganglia neurons (Evans et al., 2007) demonstrating an excitatory event induced by anandamide. Recent reports strongly support the likelihood of expression of CB1 and TRPV1 proteins on the same cells (Ahluwalia et al., 2000, 2002; Binzen et al., 2006; Mitrirattanakul et al., 2006) providing an anatomical basis for modulation of TRPV1 function by compounds interacting at the CB1 receptor. While cannabinoid modulation of the TRPV1 channel has been demonstrated with WIN 55,212-2 which is thought to inhibit capsaicin activity via the activation of calcineurin (Jeske et al., 2006; Patwardhan et al., 2006), the precise pathway(s) by which CB1 receptors modulate TRPV1 function remain to be established. Studies using CB1 receptor inverse agonists including SR141716A and AM251 have suggested constitutive activity at the CB1 receptor in animals (Gifford and Ashby, Jr., 1996; Zhou and Shearman, 2004; Costa et al., 2005) and humans (Despres et al., 2005; Huestis et al., 2007). Recently it has been shown that repeated administration of SR141716A is effective in alleviating thermal and mechanical hyperalgesia in a rat model of nerve injury (Costa et al., 2005). Here, we further investigate modulation of TRPV1 responsiveness through compounds acting at CB1 receptors, or through intracellular pathways associated with the TRPV1 receptor. Additionally, we explored the possibility that CB1 inverse agonists may negatively modulate TRPV1 responses to produce antinociception relevant to clinical pain states.
Materials and Methods
Animals
Male ICR mice (25–35g) were purchased from Harlan (Indianapolis, IN). Male CB1 receptor wildtype (WT) and knockout (KO) mice were obtained as a generous gift from Dr. Debra Cockayne, Roche Biosciences and bred in house. All animals were housed on a regular 12 h light/dark cycle (lights on at 07:00 am) in a climate-controlled room with food and water ad libitum. All studies were performed while animals were on their light cycle between the times of 7:00 am and 7:00 pm. Mice and rats were housed three to a cage and each animal was used only once per experiment. All procedures were approved by the University of Arizona Animal Care and Use Committee and were in accordance with the guidelines of the International Association for the Study of Pain.
Materials
Morphine sulfate and SR141716A were provided by the National Institute on Drug Abuse (Bethesda, MD). AM251 was purchased from Tocris. U73122 was obtained from Biomol International and m-3M3FBS was purchased from EMD Biosciences. Capsaicin, capsazepine, galanin, formalin, mustard oil, ethanol, DMSO, Tween 80 and PEG 400 were obtained from Sigma. Bradykinin was purchased from Bachem. Evans blue dye was purchased from Eastman Kodak Co. All solutions were prepared on the day of each experiment.
Capsaicin-induced paw flinching in ICR mice
Mice were placed separately in plexiglass boxes for a 20 to 30 min habituation period. Post habituation, capsaicin (1, 5 or 10 µg) in ethanol (100%) was injected into the plantar side of the mouse left hind paw, using a 30 gauge needle attached to a 25 µl Hamilton syringe in a volume of 5 µl; this volume was used for all intraplantar injections unless otherwise specified. In all studies, flinches of the capsaicin-injected hindpaw were recorded in 1 min bins for 5 min. In all studies of modulation of capsaicin-induced flinching, the dose of intraplantar capsaicin was 10 µg. A flinch was defined as a rapid jerk of the injected paw. Control experiments were performed in all studies using the capsaicin vehicle (100% ethanol); the vehicle did not produce any significant nociceptive behaviors when injected into the hindpaw at this volume.
Modulation of capsaicin-induced flinching in ICR mice
To investigate the effects of an opioid agonist on capsaicin-induced flinching, morphine (0.3, 1 and 3 mg/kg) was dissolved in saline and administered intraperitoneally (i.p.) using a 27 gauge needle, 15 min prior to intraplantar capsaicin. Separate animals were used for the different doses of morphine. Control experiments included animals that received saline by the i.p. route and capsaicin by the intraplantar route 15 min post saline. Intraperitoneal injections were performed by holding the animals in a supine position and inserting the extremity of a 27 gauge disposable needle attached to a 1cc disposable syringe into the peritoneal cavity in the lower left quadrant of the abdomen. SR141716A (0.03, 0.3, 1.0 and 3 mg/kg) or AM251 (0.03, 0.1, 0.3 mg/kg) were dissolved in PEG 400 and always administered i.p. 15 min prior to intraplantar capsaicin. Control animals received the PEG 400 vehicle by the i.p. route 15 min prior to capsaicin. U73122 (10 and 30 mg/kg), a PLC inhibitor (Hou et al., 2004), was administered i.p. in ICR mice 1 hr prior to intraplantar injection of capsaicin. In separate groups of animals, m-3M3FBS a PLC activator (Bae et al., 2003), was administered by the i.p. route at 5 mg/kg 30 min prior to treatment with i.p. SR141716A (0.3 mg/kg) and 45 min prior to challenge with intraplantar capsaicin. Both U73122 and m-3M3FBS were dissolved in 1:1:8 DMSO/Tween 80/saline.
Capsaicin-induced flinching in CB1WT and CB1KO mice
CB1WT or KO mice respectively received 1, 5 and 10 µg or 1, 5, 10, 30 and 100 µg of capsaicin in the plantar side of the left hindpaw. Separate animals received vehicle (100% ethanol) concurrently. The number of flinches was recorded in 1 min bins for 5 min.
Modulation of capsaicin-induced flinching in CB1WT and KO mice
To test whether galanin would restore reduced capsaicin-induced flinching in CB1KO mice, galanin (0.1 ng/20 µl) was administered as in the dose and volume described by Jimenez-Andrade et al. (2004) using ICR, CB1WT and KO mice. Intraplantar capsaicin was administered in the same paw immediately following galanin injection. As a control, galanin administration was followed by intraplantar vehicle (100% ethanol) and flinching behaviors recorded in all strains of mice. Bradykinin was administered by the intraplantar route at 3 µg in a volume of 5 µl in ICR, CB1WT and KO mice. As this dose bradykinin itself induced a small number of flinches, which were recorded but no longer present within 5 min. Intraplantar capsaicin was then injected into the same hindpaw, 10 min after the initial bradykinin injection. As a control, intraplantar bradykinin was followed by a challenge with intraplantar vehicle (100% ethanol) as described for capsaicin.
Cell culture of F-11 cells
F-11 cells (gift from Dr. Fishman, see (Platika et al., 1985) (mouse neuroblastoma × rat dorsal root ganglion neuron hybrid cell line) were cultured and maintained in 75cm2 flasks at 37°C and 5%CO2 in Ham’s F-12 supplemented with 15% fetal bovine serum, 5% newborn calf serum, 1% penicillin/streptomycin and 1% HAT. For calcium imaging experiments, 5 × 104 cells were plated in the area of cloning ring attached on the Delta-T dishes (Bioptechs). Cell differentiation was initiated 24 h after plating by replacing culture medium with the following medium: Ham’s F-12 supplemented with 1% FBS, 50 ng/ml NGF, 2 µM retinoic acid, 0.5 mM cAMP, 10 µM IBMX, 125 µg/ml insulin, 10 µg/ml transferrin, and 50 IU/ml penicillin/streptomycin. Cultures were maintained for 72 h in this medium prior to experiments.
Ca2+ imaging assay
F-11 cells were washed three times with bath solution and loaded with 5 µM of fura-2/AM (Molecular Probes) at 37°C for 40 min. The bath solution (Hanks’ balanced salt solution) contained 136 mM NaCl, 5.4 mM KCl, 2 mM CaCl2, 1 mM MgSO4, 5.5 mM glucose, 10 mM HEPES (pH 7.4). Cells were washed twice and incubated at 37°C for another 10 min. The cells were maintained at 37°C throughout the experiment with the Delta TC3 open culture dish system (Bioptechs). Image acquisition was carried out using a Nikon TE200 outfitted with a plan fluor 40X oil N.A. 1.3 objective lens, Xenon burner, an ORCA Hi binning 12 bit digital camera, filter wheel with fura-2 filter set controlled by Mutech image master digital workstation and Metafluor imaging software (Universal Imaging). The ratiometric fluorescence images were captured at 6 s intervals. The changes in the fluorescence level of individual cells over time are obtained by digitizing the integrated optical density of fura-2 emission at 510 nm as a ratio of 340 nm/380 nm excitation associated with a designated cell area of the serial images. Individual experiments were performed in triplicate (Lai et al., 2006). SR141716A (10 and 100 nM) or AM251 (10 or 100 nM) were dissolved in PEG 400 and administered 10 min prior to capsaicin (10 nM).
Western blot analysis
The L4, L5 and L6 dorsal root ganglia (DRG)s and dorsal horns (lumbar enlargement) were removed from mice and homogenized separately in an ice-cold 10 mM sodium phosphate buffer (pH 7.4) containing 100 µM PMSF, bestatin (30 µM) and captopril (10 µM). The membrane fractions of sequentially centrifugal extractions were performed as follows. Briefly, the homogenate was centrifuged at 2000 × g for 10 min at 4°C. The supernatant was removed and centrifuged at 16,500 rpm for 1 h at 4°C. The pellet was solubilized in the 10 mM sodium phosphate buffer containing 2% Trition X-100, 4% SDS and the cocktail of protease inhibitors. The resultant membrane protein was refined by passage through 23 gauge and 27.5 gauge needle several times. The protein concentration was determined with a standard Lowry assay as described previously (Bilsky et al., 1996). Protein samples were separated on a SDS-PAGE gradient gel (4%–15%; Bio-Rad) and transferred to the nitrocellulose membrane. The blots were blocked with 3% non-fat dry milk, 2% goat serum and 2% mouse serum for 30 min and incubated with rabbit-anti-rat vanilloid receptor subtype 1 (TRPV1) antibodies (Alpha Diagnostic, TX) (3 µg/ml) for 1.5 h at room temperature. The blots were then incubated in goat-anti-rabbit HRP-conjugated secondary antibody 1:5,000 (Jackson ImmunoResearch, PA) for 1 h at RT, developed in ECL solution (Amersham Pharmacia) for 1 min, and exposed onto X-films (Hyperfilm; Amersham) for 30 min. The cross-reactivity of the antibody to mouse TRPV1 is proven by the detection of TRPV1 protein in the DRGs from ICR mice. Gaq antibodies (Santa Cruz, CA) 1:500 were applied on the same membrane as an internal loading control although Western blots were not used for quantitative purposes.
Reverse transcription – polymerase chain reaction (RT-PCR)
Total RNA was isolated from F-11 cells and rat DRGs using RNAqueous-total RNA isolation kit (Ambion). First strand cDNA was reverse transcribed using RETROscript™ kit (Ambion) following manufacturer's instructions. For multiplex PCR amplifications, 3 µl of the first-strand cDNA template were used in 20 µl of reactions containing 1X PCR buffer, 0.25 µM of each sense and antisense primer, 125 µM of each dNTP, and 1 unit of Taq DNA polymerase (Invitrogen). 1X PCR buffer contained 10 mM Tris-HCl, 50 mM KCl, 0.1% Triton X-100, and 1.5 mM MgCl2. PCR was performed in PCR Express Thermal Cycler (Thermo Hybrid) as follows: 1) hold at 94°C for 2 min; 2) 35 cycles at 94°C for 40 sec, 55°C for 40 sec, and 72°C for 1 min; 3) hold at 4°C. Primer sequences for CB1 were 5’-AAGAGGATCGTCACCAGG -3’ (sense) and 5’-CCAGCCTAATGTCCATGC -3’ (antisense); and that for TRPV1 were 5’-CAACGCAAGGAGTATGTGG -3’ (sense) and 5’- GAGTTACCTGGCTTGCAG -3’. GAPDH (sense primer: 5’-CACGGCAAGTTCAATGGC -3’; antisense primer: 5’-GCCTGCTTCACCACCTTC -3’) was co-amplified with CB1 or TRPV1 cDNA as an internal control. The PCR products were run on a 2% agarose gel containing ethidium bromide, visualized by UV illumination, and then photographed. Primers used for PCR amplification were synthesized from Midland (Midland, TX).
Skin nerve electrophysiological recordings
Male ICR mice, CB1WT or CB1KO mice were recorded from in random order, and the investigator was blinded as to their genotype. The saphenous nerve and innervated skin were removed from mice and placed in an organ bath and bathed with synthetic interstitial fluid (SIF) buffer as previously described (Stucky et al., 2004). The nerve was placed in a separate chamber filled with mineral oil, desheathed, and teased into fine filaments. Single units were identified by mechanical stimulation with a glass probe. Conduction velocity was determined by applying electrical pulses to the receptive field via a tungsten needle electrode. Only unmyelinated C fibers were recorded for this study and C fibers were identified as units conducting slower than 1.2 m/s. Mechanical threshold was determined by using calibrated von Frey filaments (range 0.04–147 mN). Data were recorded using Chart (AD Instruments). For chemical stimulation, a stock solution of capsaicin (10 mM) was made in 1-methyl-2-pyrrolidinone (Sigma), a solvent that has no effect on fibers in the skin nerve preparation (Stucky et al., 2004) and stored at −20°C. On the day of recording, the capsaicin stock was diluted in 32°C SIF buffer just prior to application. The receptive field of the C fiber was isolated with a stainless steel ring (6 mm ID) that was sealed to the skin with a thin layer of vacuum grease. First, a control was performed by applying 1:100 1-methyl-2-pyrrolidinone:SIF buffer (32°C) for 1 min. Next, 100 µM capsaicin was applied for 1 min and the number of evoked action potentials were counted. Capsaicin was applied at 100 µM because this concentration maximally activates C fibers (approximately 58% of all C fibers) in the skin nerve preparation. The criterion for a positive response to chemical or mechanical stimuli was at least three evoked action potentials. If any action potentials occurred during the application of control solution, these action potentials were subtracted from the total capsaicin-evoked action potentials. To confirm that non-capsaicin responsive fibers were still active, each fiber was stimulated mechanically with a glass rod at the end of recording. Fibers that were unresponsive to the terminal mechanical test were excluded from the data. Only fibers with non-overlapping receptive fields were used from the same skin preparation.
Plasma Extravasation
Extravasation of plasma albumin in mice was assessed by the Evans blue leakage method. Evan’s blue dye (100 mg/kg) in saline was injected using a 30 gauge needle attached to a 1 ml syringe in the tail 15 min after capsaicin (10 µg) administration in the left hind paw. Vehicle (100% ethanol) was injected in the contralateral paw (right), so that each mouse served as its own control. Two hours after Evan’s blue injection animals were sacrificed under ether and hind paw tissue was removed with an 8 mm hole-puncher. The tissue was incubated in 100% formamide, 50oC water bath for 2 h and samples were centrifuged at 14000 rpm for 20 min. The supernatant was collected and measured by spectrophotometry at 620 nm (Beckman DU-62 Spectrophotometer). The absorbance values were calculated from a calibration curve. The amount of extracted dye was expressed as fmol of dye per gram of wet tissue, as described (Trevisani et al., 2002).
Calcitonin Gene Related Peptide (CGRP) Release Assay
Tissue extraction and preparation
Mice were deeply anesthetized using CO2 and decapitated. The spinal cord was severed at the pelvic girdle. Hydraulic extrusion was performed by inserting a 16 gauge needle into the sacral vertebral canal and expelled with ice-cold saline. The spinal cord was immediately placed on ice in a glass Petri dish, and the dorsal half of the lumbar cord was dissected, weighed and chopped into 0.2 mm cubes with a McIwain tissue chopper (Mickle Laboratory Engineering, Gomshall, UK).
CGRP assays
Chopped lumbar spinal cord tissue was placed in a 1 cc superfusion chamber and continuously superfused with oxygenated modified Krebs’ buffer (135 mM NaCl, 3.5 mM KCl, 20 mM NaHCO3, 1 mM NaHPO4, 2.5 mM CaCl2, 3.3 mM dextrose, 0.1 mM ascorbic acid, 10 mM thiorphan and 0.1%bovine serum albumin) maintained at 37°C, pH 7.4, at a rate of 0.5 ml/min with a Brandel Superfusion Pump (Brandel Gaithersburg, MD). The tissues were equilibrated for 30 min. Superfusate samples were collected into test tubes using a fraction collector (Gilson, Middleton, WI) every 3 min and measured for CGRP release. A total of 5 fractions (15 min) were collected to establish baseline levels of CGRP release before capsaicin (1 µM) was applied for 2 fractions (6 min). Superfusate was then collected for an additional 8 fractions (24 min). The superfusate obtained from the release assay was preincubated with 100 µl of a C-terminus directed anti-CGRP antibody (Peninsula Lab.) for 24 hr at 4°C. The samples were each mixed with 50 µl of goat anti-rabbit antiserum coupled to ferric beads and 100 µl of [125I-Tyr0]CGRP28–37 ( at ∼25,000 cpm per assay tube) and incubated for an additional 24 hr. The [125I]CGRP bound to the CGRP antibody was separated from the free tracer through immunomagnetic separation (PerSeptive Diagnostics, Cambridge, MA). The immunoprecipitates were determined by gamma counting. Standard curves were generated and CGRP content was determined through logit-log analysis. This assay has a minimal detection limit of 1–3 fmol/tube. The CGRP antiserum used in these experiments binds near the C-terminal end of CGRP and does not cross react with cholecystokinin, neuropeptide Y, or other peptides with similar C-terminal residues. The CGRP concentrations were plotted against time in 3 min intervals. Evoked release was calculated as the total amount of CGRP released (i.e., CGRP-IR) during the capsaicin infusion above the basal release of CGRP.
Statistical Analysis
All data were expressed as mean ± SEM. For behavioral experiments, differences among several means relative to a single baseline group were determined with one-way ANOVA, followed by Fisher’s least significant difference test. Fisher’s least significant difference test is based on using ANOVA to determine a given value where, if the difference between two means is equal to or greater than that value, the difference is significant. Differences among several treatment groups were evaluated with ANOVA followed by Student-Neuman-Keuls post-hoc test. Differences between two individual means were analyzed with Student’s t-test. A p<0.05 was considered significant and is indicated by a star (*).
Results
Thermal and mechanical testing in CB1WT and KO mice
No significant differences in response latencies to three different temperatures were observed in CB1WT and CB1KO mice. Tail withdrawal latencies from water at 48°C for WT was 25.8 ± 3.1 sec and KO 23.6 ± 2.6 sec, from water set at 52°C for WT was 8.9 ± 1.2 sec and KO 9.3 ± 1.5 sec and from water set at 55°C for WT was 4.2 ± 0.2 sec and KO 4.3 ± 0.5 sec. Non-noxious mechanical thresholds using von Frey filaments also showed no significant differences between WT mice 1.8 ± 0.3 g and KO mice 2.0 ± 0.4 g (n=6 to 8 in all experiments).
Capsaicin-induced paw flinching
Intraplantar administration of capsaicin produced a time- and dose-related increase in flinching behaviors in ICR mice. The dose producing a 50% response (and 95% C.I.) based on the highest total flinching responses observed throughout all experiments was 2.5 (1.3 – 3.4) µg (Fig. 1A, B). At the 10 µg dose, the total (cumulative) number of flinches after 1, 5 and 10 min were 19.0 ± 1.7, 37.7 ± 2.8 and 44.5 ± 1.9. The time course for all subsequent capsaicin-induced paw flinch experiments was shortened to 5 min, since the majority of paw flinches occurred during this period.
Figure 1.
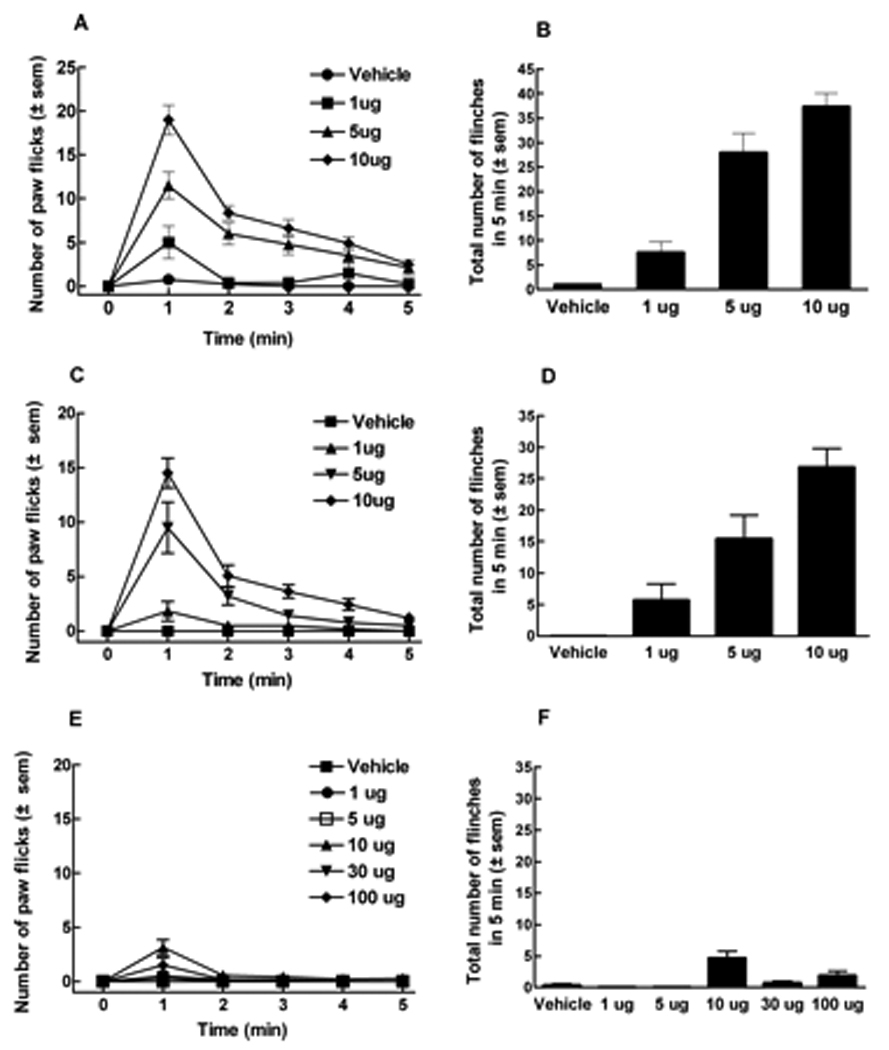
ICR, CB1 wildtype (CB1WT) or knockout (CB1KO) mice received capsaicin or vehicle (100% ethanol) into the plantar surface of the left hind paw. Volume of i.paw administration was 5 µl. (A) Number of flinches induced by capsaicin (1, 5 or 10 µg) or vehicle in ICR mice every minute for 5 min. (B) Total number of flinches evoked by capsaicin or vehicle in 5 min in ICR mice. Capsaicin induced paw flinches in a dose-dependent manner, resulting in A50 of 2.5 µg. (C) Number of flinches induced by capsaicin (1, 5 or 10 µg) or vehicle in CB1WT mice every minute for 5 min. (D) Total number of flinches evoked by capsaicin or vehicle in 5 min in CB1WT mice. Capsaicin induced paw flinches in a dose-dependent manner, resulting in A50 of 3.2 µg. (E) Number of flinches induced by capsaicin (1, 5, 10, 30, 100 µg) or vehicle in CB1KO mice every minute for 5 min. (F) Total number of flinches evoked by the different doses of capsaicin in 5 min was significantly abolished in CB1KO mice (n = 6 to 12 in all studies).
Systemic morphine significantly and dose-dependently inhibited capsaicin-induced paw flinching in ICR mice. Morphine pre-treatment at 1 and 3 mg/kg reduced the total number of flinches to 8.7 ± 1.2 (n=6) and 2.3 ± 0.3 (n=6) respectively, when compared to control (vehicle administered) animals receiving capsaicin (24.9 ± 1.5, n=12) (p < 0.001).
Capsaicin also elicited time and dose-related flinching behaviors in CB1WT mice with responses similar to those observed in ICR mice. The dose producing a 50% response (and 95% C.I.) was 3.2 (2.1 – 4.4) µg (Fig. 1 C, D). After 5 min, the number of flinches induced by capsaicin (10 µg) in CB1WT mice was 26.9 ± 3.0. However, in CB1KO mice, capsaicin elicited significantly fewer flinching responses compared to either CB1WT or ICR control mice. Even when substantially higher doses were administered to CB1KO mice, very few flinches were observed, such that a 50% response could not be calculated (Fig. 1E, F). The total number of flinches following capsaicin at 10 or 100 µg in CB1KO mice were 4.6 ± 1.1 and 1.8 ± 0.8, respectively; these responses were significantly lower than those seen in either ICR or WT mice (p < 0.001, n=6 to 12 in all experiments). No responses were seen at the lowest doses of capsaicin (e.g., 1 and 5 µg) suggesting that the lack of response seen at higher doses was not due to desensitization.
Reduced plasma extravasation is observed in CB1KO mice
Capsaicin resulted in extravasation of Evan’s Blue dye following injection into the hindpaw of CB1WT (60.6 ± 3.1 fmol/g of tissue) or ICR mice (50.6 ± 2.3 fmol/g of tissue) (Fig. 2). In contrast, tissues from CB1KO mice showed significantly decreased extravasation in the capsaicin-injected paw (1.9 ± 0.3 fmol/g of tissue). In all animals, Evan’s blue dye extravasation was minimal in the contralateral (non-capsaicin injected) paw (p< 0.005, n=5 to 10 in all studies) (Fig. 2).
Figure 2.
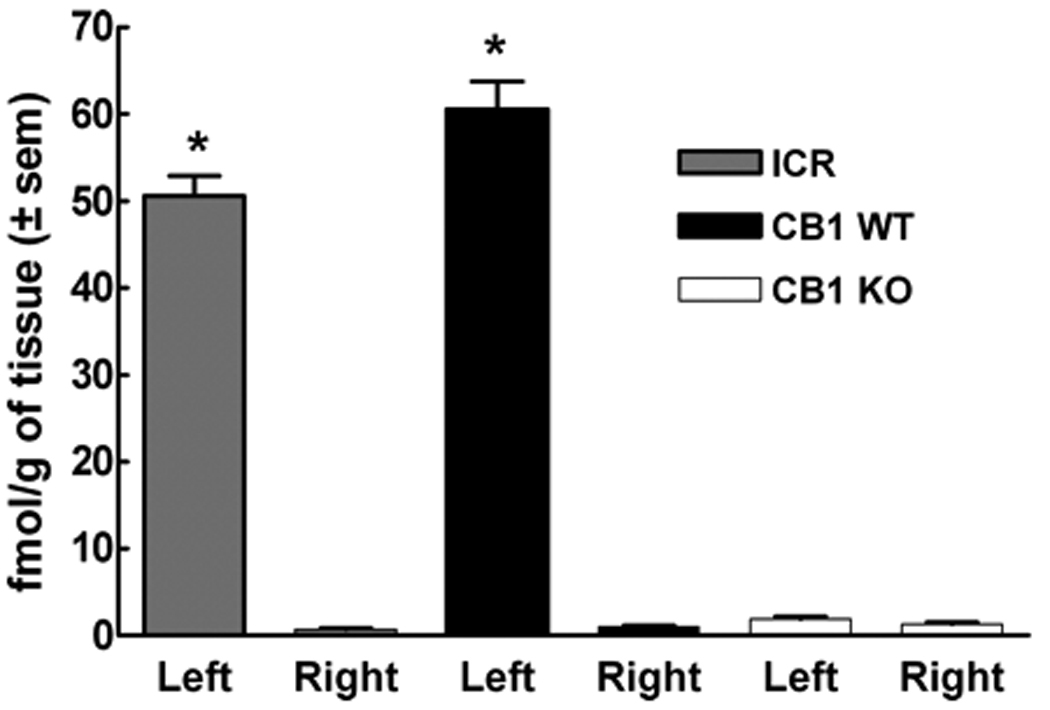
ICR, CB1WT and CB1KO mice received an intraplantar (left paw) injection of capsaicin (10 µg). Intravenous (tail vein) injection of Evan’s blue dye (100 mg/kg) was performed 15 min after capsaicin administration. Vehicle for capsaicin (100% ethanol) was injected into the contralateral paw (right paw). Pronounced plasma extravasation expressed as fmol/g of tissue was seen in the capsaicin-injected left paw of ICR and CB1WT mice, as opposed to CB1KO, in which no capsaicin evoked plasma extravasation was detected (*p< 0.005, n= 5 to 10).
Reduced capsaicin-evoked release of i-CGRP in dorsal spinal cord of CB1KO mice
Baseline levels of CGRP in ICR and CB1WT just prior to capsaicin administration were 10.3 ± 1.7 and 9.1 ± 1.1 fmol/mg tissue, respectively. Similarly, baseline levels of CGRP in spinal cord tissue from CB1KO mice prior to capsaicin administration were 6.0 ± 0.2 fmol/mg tissue; these values were not significantly different from those of the ICR or WT tissues. Application of capsaicin (1 µM) for 6 min evoked CGRP release in spinal cord tissue from ICR and CB1WT mice. Capsaicin-evoked CGRP release was significantly reduced in spinal cord tissue from CB1KO mice. Evoked CGRP release was calculated as 17.6 ± 1.9, 16.6 ± 3.1 and 5.0 ± 2.5, fmol/mg in ICR, WT and KO tissues respectively (p< 0.05, n= 6 in all groups) (Fig. 3).
Figure 3.
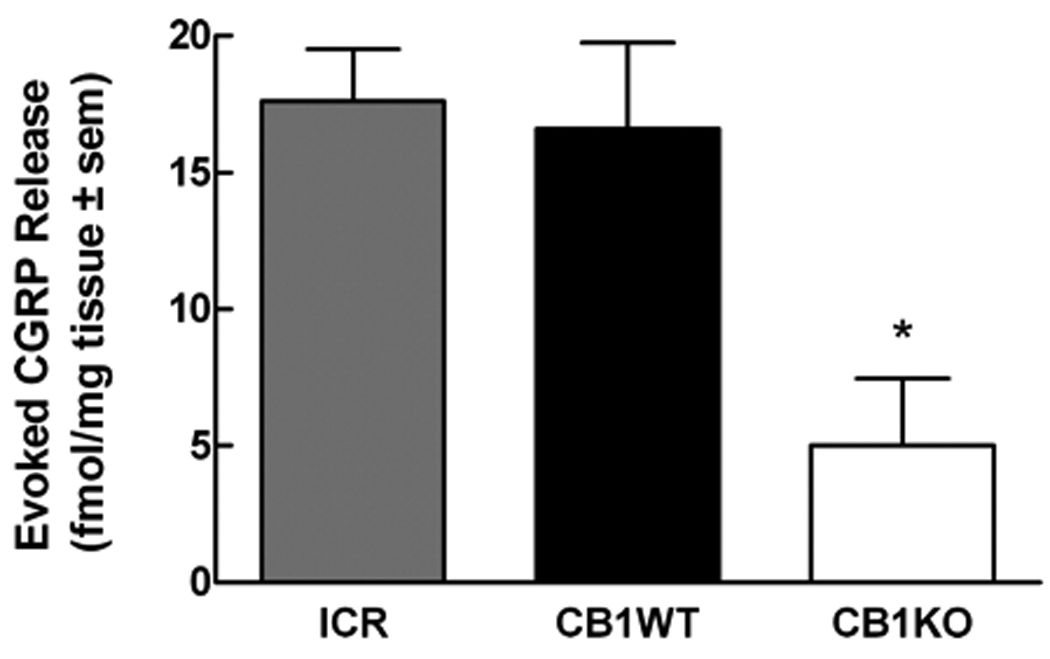
Calcitonin gene related peptide (CGRP) was measured from the lumbar spinal cord tissue of ICR, CB1WT and CB1KO mice. After a 45 min equilibration period in the perfusion chambers, followed by a 12 min collection of perfusate for determination of baseline values, capsaicin (1 µM) was added to the perfusion medium. CGRP content was quantified by radioimmunoassay. Evoked release was defined as the amount of CGRP above the basal values. Capsaicin-evoked CGRP release from spinal cord tissue of ICR and CB1WT mice was significantly higher when compared to spinal cord tissue from CB1KO mice (*p< 0.05, n= 6 in all groups).
Capsaicin-evoked responses are reduced in C-fibers from CB1KO mice
Capsaicin-sensitivity of single cutaneous C-fibers was evaluated in skin-nerve preparations from CB1KO and CB1WT mice. Whereas nearly 60% (16/27) of C fibers from CB1WT mice responded to 100 µM capsaicin, only 37% (12/32) (1-tailed t-test p=0.07) of C-fibers from CB1KO mice responded to capsaicin with 3 or more action potentials during the capsaicin application. More importantly, the number of capsaicin-evoked action potentials in C-fibers from CB1KO mice (14.6 ± 4.3 spikes/min) was approximately 50% less than that in WT mice (25.2 ± 5.6 spikes/min; 1-tailed t-test p = 0.05). There were no differences between the genotypes in C-fiber conduction velocity (WT: 0.57 ± 0.04 m/s; KO: 0.57 ± 0.03 m/s) or von Frey thresholds (WT: median 6.8 mN, lower and upper quartiles 4.0, 11.7 mN; KO: median 9.25 mN, lower and upper quartiles 5.4 mN, 12.8 mN).
TRPV1 receptors are expressed in DRG of CB1WT and CB1KO mice
The presence of TRPV1 protein in tissues from CB1WT and KO mice was evaluated via Western blot analysis. TRPV1 protein was present in DRG of both CB1WT and CB1KO mice with a molecular weight of approximately 95 KDa (Fig 4). Gqα was used as an internal loading control for membrane proteins. Although Western blots were not performed for quantitative purposes, Gqα bands with a molecular weight of 42 KDa, were identified in both genotypes. Figure 4 is representative of three separate Western blots using DRGs from CB1WT and CB1KO mice.
Figure 4.
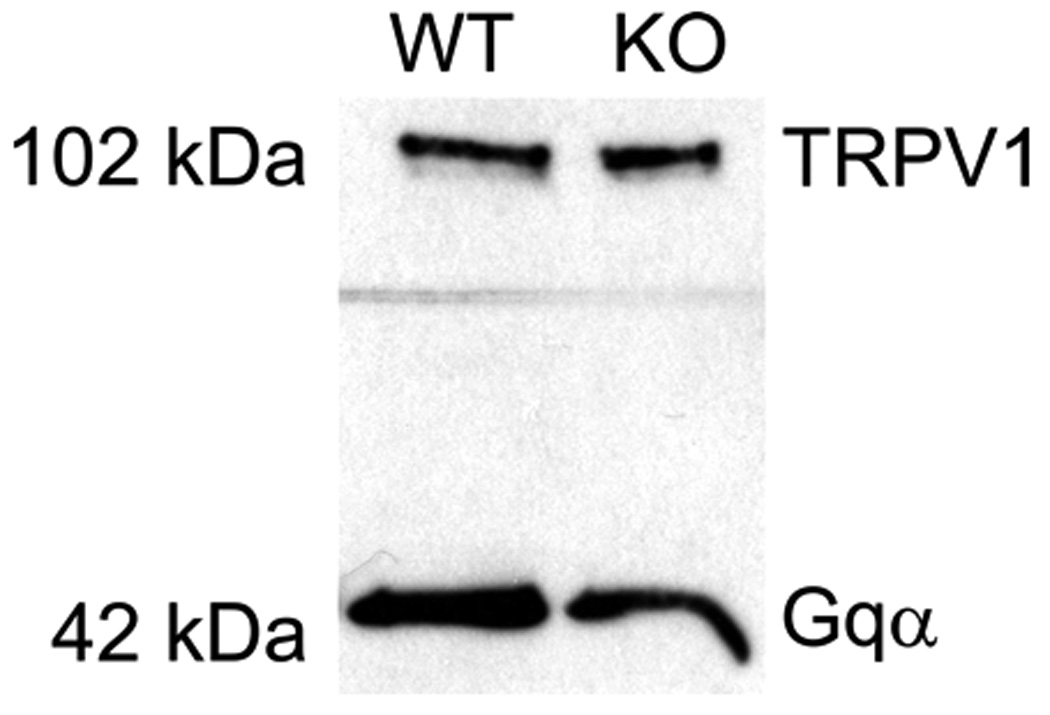
Western blot for TRPV1 protein (95KDa – the figure shows 102KDa) was performed using lumbar spinal cord tissue or dorsal root ganglia (DRG) from either CB1WT or CB1KO mice. Gqα bands with the molecular weight of 42 KDa were identified and used as an internal loading control for membrane proteins. Picture represents a total of three separate Western blots using DRGs from CB1WT and CB1KO mice.
Pretreatment with galanin or bradykinin restores capsaicin-induced flinching in CB1KO mice
Intraplantar galanin (0.1 ng), a peptide known to sensitize TRPV1 channels, followed by intraplantar vehicle did not result in significant flinching responses in ICR, WT or KO mice. Intraplantar galanin followed by capsaicin challenge in ICR and WT mice resulted in an increase in the total number of flinches. In ICR and CB1WT mice, capsaicin elicited 31.6 ± 1.6 and 21.8 ± 2.2 flinches, whereas in CB1KO mice, capsaicin only evoked 3.9 ± 1.4 flinches. Pretreatment with galanin resulted in 40.8 ± 3.2 and 28.9 ± 1.6 flinches in ICR and CB1WT mice, respectively, values which were significantly higher than in the vehicle pretreated group (p<0.05). More strikingly, in CB1KO mice, galanin pretreatment markedly enhanced capsaicin-evoked flinching from 3.9 ± 1.4 to 33.7 ± 3.1 (p<0.001). The capsaicin response of galanin pretreated CB1KO mice was not different from that observed in galanin-pretreated ICR or WT mice (Fig. 5A).
Figure 5.
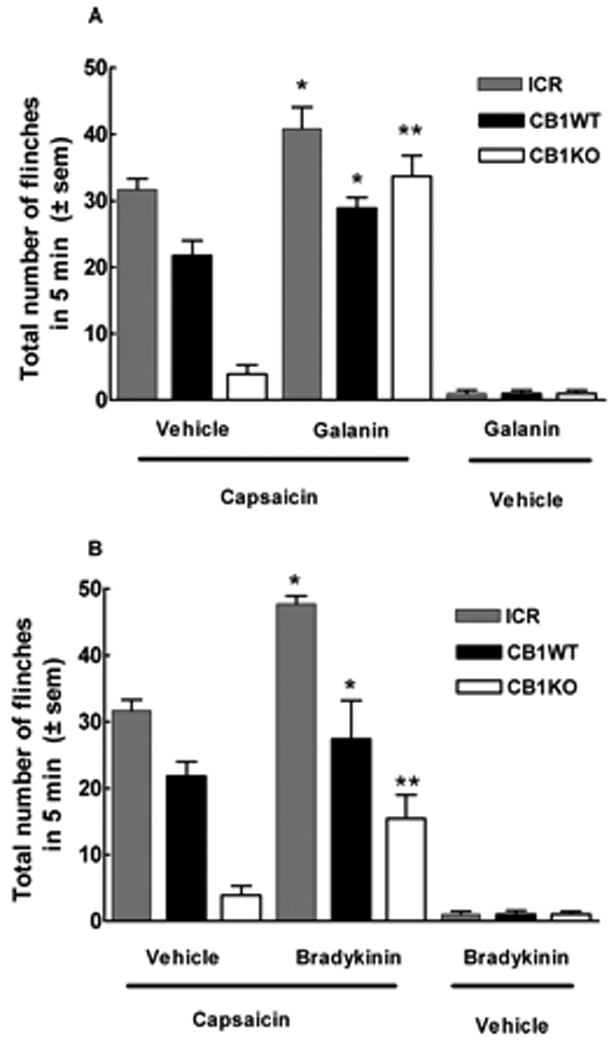
Compounds known to sensitize the TRPV1 channel restore flinching in CB1KO mice. (A) ICR, CB1WT and CB1KO mice were administered galanin (0.1 ng) in the ventral side of the left hind paw followed by capsaicin (10 µg) administered in the same paw. In ICR and CB1WT mice, the total number of capsaicin-induced flinches in 5 min was increased in the presence of galanin, when compared to vehicle pre-treated animals. Surprisingly, in CB1KO mice, galanin completely restored the capsaicin-evoked response, (**p<0.001, n=6 to 10). (B) ICR, CB1WT and CB1KO mice were administered bradykinin (3 µg) in the ventral side of the left hind paw 10 min prior to intraplantar capsaicin (10 µg) administration in the same paw. In ICR and CB1WT mice, the total number of capsaicin-induced flinches in 5 min was increased in the presence of bradykinin, when compared to vehicle pre-treated animals. CB1KO mice pre-treated with bradykinin resulted in a significant increase in the capsaicin-evoked flinching behavior (** p<0.05, n=8 to 10).
Intraplantar bradykinin (3 µg) alone induced a total of 1.0 ± 0.5 and 1.0 ± 0.6 flinches in KO and WT mice, respectively, in the first 5 min; intraplantar saline did not elicit flinching behaviors (data not shown). Following 10 min, no remaining bradykinin-induced flinching behaviors were observed. At this time point, animals were challenged with intraplantar capsaicin or vehicle and flinching behaviors were recorded. Bradykinin pretreatment in ICR mice enhanced capsaicin-evoked flinching from 31.6 ± 1.6 to 47.7 ± 1.2. Likewise, bradykinin pretreatment enhanced capsaicin-evoked flinches in WT mice from 21.8 ± 2.2 to 27.4 ± 5.8. Pretreatment with intraplantar vehicle or bradykinin in KO mice significantly increased the flinching response from 3.9 ± 1.4 to 15.4 ± 3.6 flinches (p<0.05) (Fig. 5B). The enhancement of capsaicin-evoked flinching by bradykinin pretreatment in KO mice remained significantly lower than the response seen in WT or ICR mice.
CB1 inverse agonists inhibit capsaicin-evoked Ca2+ influx in F-11 cells
F11 cells were used in order to characterize the effects of CB1 inverse agonists on TRPV1 channels. The presence of CB1 and TRPV1 receptors were demonstrated by multiplex RT-PCR analysis of total RNA extracts from F-11 cells and L5 DRG of male Sprague Dawley rats. The 2% agarose gel electrophoresis displays PCR products demonstrating both CB1 (352bp product) and TRPV1 (281bp product) expression in F-11 cells, as well as in rat DRG. GAPDH (638bp product) as an internal control represented equal total RNA materials for amplification. A 100bp DNA ladder was used (Fig. 6inset).
Figure 6.
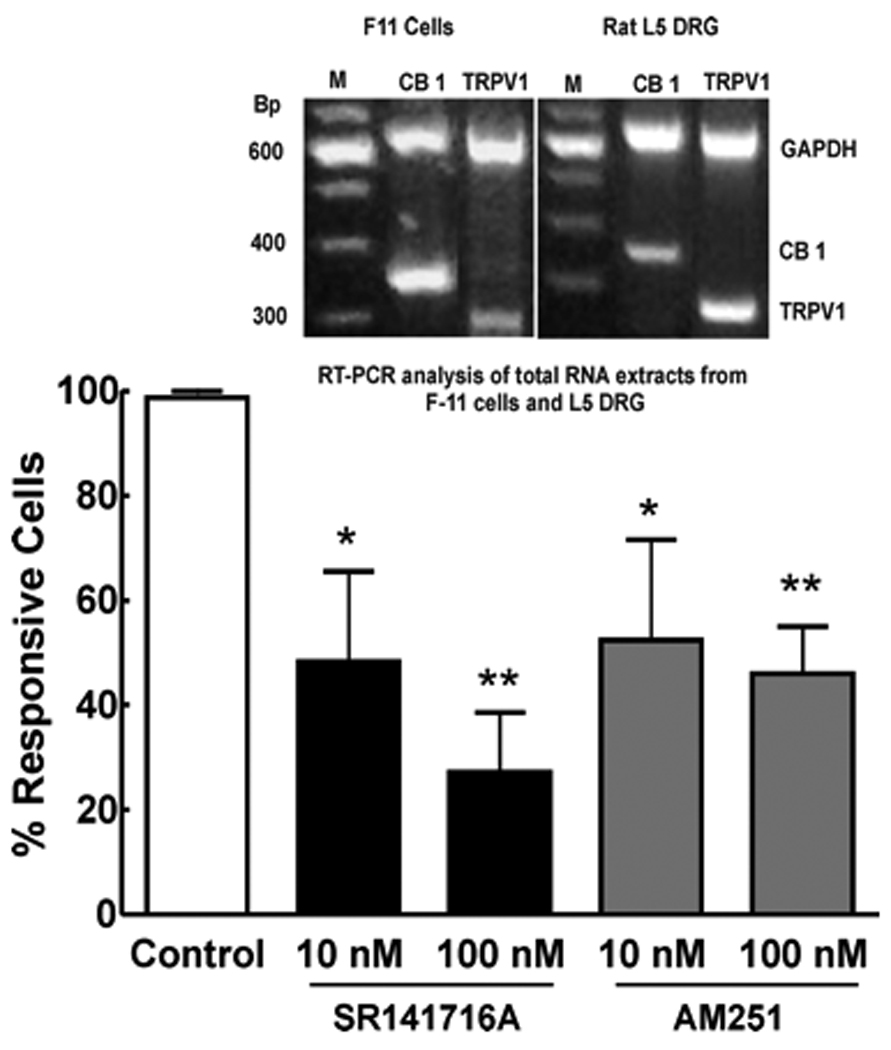
Capsaicin (10 nM)-induced transient [Ca2+] influx in F-11 cells was performed in the presence and absence of the CB1 inverse agonists SR141716A or AM251. Capsaicin-induced calcium influx in the F11 cells was significantly blocked in a concentration-dependent manner by either SR141716A (n=29 at 10 nM and n=89 at 100 nM) or AM251 (n=35 at 10 nM and n=86 at 100 nM) (*p<0.05, **p<0.001). RT-PCR was performed using Sprague Dawley rat L5 DRG or the mouse neuroblastoma rat DRG hybrid F11 cell line (inset). A 2% agarose gel electrophoresis of multiplex PCR products demonstrating both CB1 (352bp product) and TRPV1 (281bp product) expression in F-11 cells as well as in rat DRG. GAPDH (638bp product) as an internal control represented equal total RNA materials for amplification. 100bp DNA ladder (Fig 6inset).
The acute application of 10 nM of capsaicin resulted in a an increase in [Ca2+]i in 56% of the total recorded cells (322 of 577). When cells were challenged a second time with the same capsaicin test, 81% of capsaicin responsive cells were desensitized (n = 172 of 212). In contrast, allowing cells to recover for 5 min after the first capsaicin application resulted in desensitization of only 3% of capsaicin-responsive cells (n = 1 of 33) (see control group of Fig. 6). Therefore, experiments were performed using this protocol to assure capsaicin sensitive cells yet exclude the factor of capsaicin/TRPV1 desensitization. As a control to assure activity via the TRPV1 channel, effects of capsaicin on transient [Ca2+] influx were suppressed following incubation of the cells with capsazepine. Pre-incubation, 10 min, with capsazepine at 100 nM or 1 uM decreased the number of capsaicin-responsive cells by 82% (n = 42) or 93% (n = 42), respectively. SR141716A or AM251 (10 or 100 nM) significantly inhibited capsaicin-evoked transient [Ca2+] influx in F11 cells (Fig. 6). In the presence of 10 nM and 100 nM of SR141716A, number of capsaicin responsive cells was decreased by 45% and 79%, respectively. Similar results were seen with AM251 at 10 nM and 100 nM; the number of capsaicin responsive cells was decreased by 37% and 61%, respectively (Fig. 6).
CB1 inverse agonists suppress capsaicin-induced flinching in ICR mice
Intraperitoneal pretreatment with SR141617A (0.03–3 mg/kg) significantly suppressed intraplantar capsaicin-induced flinching in ICR mice. Intraplantar capsaicin elicited 24.6 ± 0.8 flinches in vehicle pretreated animals and 10.2 ± 2.9, 8.0 ± 3.6, 6.1 ± 2.0, and 0.2 ± 0.2 flinches following pretreatment with doses of 0.03, 0.3, 1.0 and 3 mg/kg of SR171416A, respectively (Fig. 7A, B). In the same manner, pre-administration of AM251 significantly decreased total capsaicin-induced flinching at the doses of 0.1 and 0.3 mg/kg resulting in 12.8 ± 0.8 and 5.5 ± 2.2 flinches, respectively (Fig. 7C, D).
Figure 7.
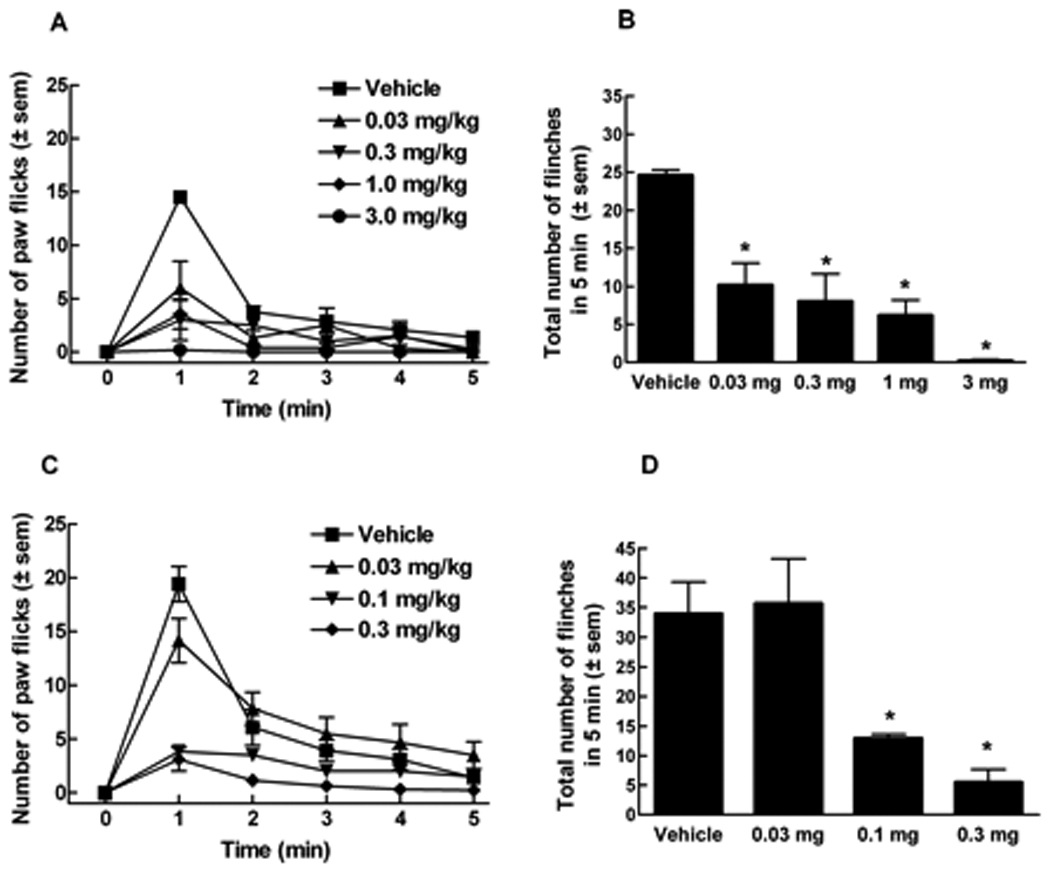
ICR mice received CB1 inverse agonist, SR141716A, or vehicle (PEG 400) i.p. 15 min prior to intraplantar capsaicin. (A) Number of flinches induced by capsaicin (10 µg) every minute for 5 min in the presence of SR141716A (0.03, 0.3, 1.0 and 3 mg/kg). (B) Total number of flinches in the presence of SR141716A. (C) Number of flinches induced by capsaicin (10 µg) every minute for 5 min in the presence of AM251 (0.03, 0.1, 0.3 mg/kg). (D) Total number of capsaicin-induced flinches in the presence of AM251. (*p<0.05, n = 6 in all groups).
Suppression of capsaicin-induced flinching by U73122, a PLC inhibitor
Pretreatment with U73122 (Hou et al., 2004) significantly attenuated capsaicin-induced flinching in ICR mice. Vehicle treated mice showed 51.6 ± 4.5 flinches. U73122 given at 10 and 30 mg/kg, resulted in 39.0 ± 4.8 and 26.3 ± 3.4 flinches, respectively indicating a 29% and 49% decrease in capsaicin-induced flinching (p <0.001) (Fig. 8A).
Figure 8.
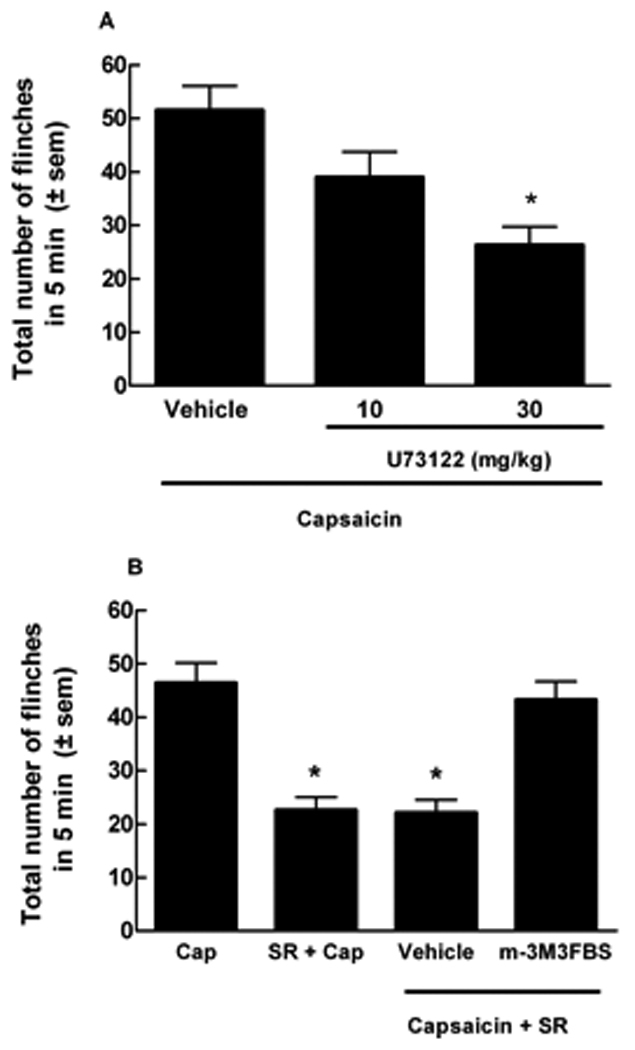
Compounds that modulate PLC, the enzyme responsible for PIP2 breakdown, affect capsaicin-induced flinching. (A) ICR mice were pre-treated with U73122 (10 or 30 mg/kg, i.p.), a PLC inhibitor, i.p. 1 hr prior to intraplantar injection of capsaicin (10 µg). U73122 produced a 29% (10 mg/kg) and 49% (30 mg/kg) blockade of total number of capsaicin-evoked flinches, when compared to vehicle pre-treated mice (*p<0.001, n = 10). (B) m-3M3FBS (5 mg/kg, i.p.), a PLC activator, was given 30 min prior to i.p. administration of SR141716A (SR, 0.3 mg/kg). SR141716A was given 15 min prior to capsaicin (Cap) injection. The total number of capsaicin-induced flinches in 5 min was significantly inhibited by pre-treatment with SR141716A (*p<0.05), yet restored by pretreatment with m-3M3FBS but not by vehicle (n = 10 to 14). m-3M3FBS alone did not enhance capsaicin-induced flinching and had no effect on vehicle (data not shown).
Blockade of SR141716A-induced antinociception by m-3M3FBS, a PLC activator
When administered alone, m-3M3FBS (5 mg/kg, (Bae et al., 2003)) did not alter responses to intraplantar vehicle (4.3 ± 0.4 flinches) or alter capsaicin-induced flinching (42.0 ± 5.1 flinches). However, pretreatment with m-3M3FBS (5 mg/kg) 30 min prior to i.p. SR141716A (0.3 mg/kg) and 45 min prior to intraplantar capsaicin injection blocked the antinociceptive effects of the CB1 inverse agonist in ICR mice. The total number of capsaicin-induced flinches (46.4 ± 3.7) was significantly inhibited by pre-treatment with SR141716A (22.6 ± 2.4), but restored by the PLC activator m-3M3FBS (43.3 ± 3.4 flinches) (Fig. 8B). Pre-treatment with vehicle did not restore capsaicin-induced flinching (22.1 ± 2.4) in animals treated with SR141716A.
Discussion
Studies presented here support the idea that tonic activity of the CB1 receptor maintains the TRPV1 channel in a “sensitized” state through a PLC-dependent process. Our studies have shown that inhibition of CB1 function either through genetic deletion of the receptor or by activity of inverse agonists, results in diminished responses to noxious chemical stimuli acting via the TRPV1 channel. In CB1KO mice, diminished capsaicin-induced responses included flinching behaviors, neurogenic inflammatory responses, generation of action potentials, and evoked release of CGRP in spinal cord tissue. Consistent with the idea of CB1 receptor modulation of TRPV1 sensitivity, our data showed that application of CB1 inverse agonists diminished responses to capsaicin. Finally, similar results could be obtained by substances known to alter PLC. Activation of PLC resulted in restoration of capsaicin sensitivity in mice pre-treated with SR141716A, an inverse agonist at the CB1 receptor, while inhibitors of PLC resulted in decreased responses to capsaicin in ICR mice. These findings suggest the possibility of modulation of TRPV1 function through means other than compounds with direct interaction at the channel.
Although our data showed diminished responses in CB1KO mice to a chemical activator of the TRPV1 channel, the responses of these mice to three different noxious temperatures was not significantly different from those in ICR and CB1WT. These results are consistent with previous reports indicating a lack of difference in tail flick responses between CB1WT and CB1KO mice (Zimmer et al., 1999) and a lack of difference in baseline responses on an increasing-temperature hotplate between TRPV1 WT and TRPV1 KO mice (Bölcskei et al., 2005), findings that may reflect the contributions of other heat-sensitive channels (Guler et al., 2002). Unlike heat, capsaicin has been shown to be selective for the TRPV1 channel (Caterina and Julius, 2001).
One reasonable explanation for such loss of capsaicin activity in the CB1KO mice is that genetic deletion of the CB1 receptor alters the expression of the TRPV1 channel. However, our findings indicate that the expression of TRPV1 was similar in tissues from WT and KO mice. The CB1 receptor has been well established as having constitutive activity both in in vitro and in vivo (Gifford and Ashby, Jr., 1996; Zygmunt et al., 1999; Zhou and Shearman, 2004). Costa and colleagues (2005) demonstrated SR141716A (Rimonabant) as having efficacy in relieving hypersensitivities in a rat model of sciatic nerve constriction. In clinical studies, Rimonabant has resulted in weight loss, reduction in low-density lipoproteins, and decrease in craving of cigarette smoking suggesting a significant constitutive activity of CB1 receptors (Despres et al., 2005; Huestis et al., 2007). Such tonic activity is maintaining an endogenous intracellular level of second messengers that can be altered by inverse agonism. Therefore, to confirm our findings in the CB1KO mice, we utilized two different CB1 inverse agonists to attenuate the constitutive activity of the CB1 receptor and measured capsaicin’s ability to provoke an effect via the TRPV1 channel.
Using F-11 cells containing functional CB1 and TRPV1 receptors we demonstrated that application of CB1 inverse agonists blocked capsaicin-induced calcium influx in a concentration-dependent fashion. Although it has been reported that SR141716A may act directly at the TRPV1 channel in concentrations above 1µM (De Petrocellis et al., 2001a; Gibson et al., 2008), our in vitro studies employed SR141716A at concentrations 10–100 fold lower, suggesting that this ligand is acting selectively at CB1 receptors. A similar blockade of capsaicin-induced calcium influx was observed with AM251, a different selective CB1 inverse agonist, which has not been shown to bind to TRPV1 channels. Taken together, our in vitro data combined with the blockade of capsaicin-induced flinching produced by SR141716A and AM251 in ICR mice confirm our findings in CB1KO, and support the idea that suppression of CB1-receptor constitutive activity leads to decreased TRPV1 sensitivity to capsaicin, perhaps through alteration of the phosphorylation state of the channel. Numerous studies have demonstrated that phosphorylation of the TRPV1 and/or removal of PIP2 results in enhanced capsaicin-induced calcium influx via the TRPV1 channel (De Petrocellis et al., 2001b; Rathee et al., 2002; Bhave et al., 2003; Jung et al., 2004).
The possibility of altering the response to capsaicin stimulation in CB1KO mice through the actions of GPCRs that are known to modulate the responsiveness of the TRPV1 channel was explored with galanin and bradykinin. Both substances have been shown to indirectly phosphorylate the TRPV1 channel, increasing its responses to capsaicin (Cesare and McNaughton, 1996; Jimenez-Andrade et al., 2004). Galanin or bradykinin, when pre-administered at low doses to CB1KO mice, restored capsaicin responses in CB1KO mice to levels similar to those observed with capsaicin alone in WT mice. These findings suggest that the TRPV1 channel is present in a desensitized state in CB1KO mice, and when known sensitizing compounds are pre-administered, re-sensitization of the TRPV1 channel to capsaicin takes place.
The CB1 receptor is known to couple with Gαi/o and inhibit adenylate cyclase activity (Bayewitch et al., 1995), yet studies have also demonstrated CB1 receptor coupling with PLC (Ho et al., 1999, 2002) and beta-gamma portions of G-proteins that activate PLC isoforms (Liu and Simon, 1996; Huang et al., 1998) suggesting that activation of CB1 receptors can result in a decrease in PIP2 and an increase in PKC and Ca+2-induced kinases, intracellular activities that sensitize TRPV1 channels to capsaicin. Capsaicin-induced calcium influx was enhanced by the administration of either bradykinin or NGF, both acting to stimulate PLC activity, decreasing PIP2 levels and increasing kinase levels (Chuang et al., 2001). Additionally, CB1 receptors can directly couple to Gαq and activate PLC (Lauckner et al., 2005), and may also stimulate cAMP/PKA pathways (Calandra et al., 1999). Although the CB1 receptor has traditionally been classified as a Gαi/o-protein coupled receptor, several studies have demonstrated CB1 coupling to multiple G protein subunits influencing several intracellular signal pathways including PLC and PKA, pathways known to sensitize the TRPV1 to capsaicin.
Here we have demonstrated that the pre-administration of the PLC inhibitor U73122 significantly reduces behavioral responses to capsaicin in ICR mice similar to levels seen in CB1KO mice or in animals pretreated with SR141716A. Consistent with a role for PLC in the mediation of TRPV1-sensitivity via the CB1 receptor, the significant reduction of capsaicin-induced flinching by the CB1 inverse agonist SR141716A was attenuated by the pre-administration of the PLC activator m-3M3FBS. Furthermore, increases in cAMP/PKA, which occur in the presence of the inverse agonist SR141716A, result in the inhibition of PLC activity (Wen et al., 1992; Liu and Simon, 1996). These results suggest that CB1 receptors are constitutively active and maintain PLC activity, which results in the breakdown of products such as PIP2 that inhibits TRPV1 and in the modulation of levels of kinases that “sensitize” TRPV1 function. This hypothesis is further supported by the administration of agonists that activate the PLC pathway such as bradykinin or galanin resulting in TRPV1 sensitization (Chuang et al., 2001; Jimenez-Andrade et al., 2004). In addition, it has been shown that PLC is responsible for the synthesis of endogenous cannabinoids and/or endovanilloids (Starowicz et al., 2007) resulting in a constitutive pathway for the production of endogenous agonists for CB1 and TRPV1 receptors, respectively. The inhibition of PLC by U73122 in our studies could potentially decrease the generation of such endogenous ligands that directly activate the CB1 and/or TRPV1 receptors. In the case of endocannabinoids, such interpretations would support our hypothesis that the CB1 receptors are being constitutively activated, maintaining a capsaicin-sensitive TRPV1. In the case of PLC generating endovanilloids, we observed that blockade of PLC suppressed TRPV1 activation by its exogenous agonist capsaicin whose agonistic effect’s are independent from endovanilloid availability. Since the possible decrease in endovanilloids by blocking PLC has not been shown to alter capsaicin’s ability to activate the TRPV1 channel it seems likely that PLC maintains TRPV1 sensitization through intracellular pathways. Nevertheless, such studies strengthen the possibility of a crosstalk between CB1 and TRPV1 receptors via PLC, whose activation by CB1 receptors lead to changes in the intracellular milieu restoring the TRPV1 from its desensitized state in CB1KO mice.
The recent development of TRPV1 antagonists for acute and inflammatory pain have resulted in some unwanted side effects including increasing body temperature (Gavva et al., 2007) and some uncertainty in such antagonists for drug development. Although the TRPV1 channel has demonstrated to be a very promiscuous channel with multiple intracellular messengers and surface receptors modulating its function, such complimentary pathways may lead to novel pharmaceutical targets. The endogenous cannabinoid system may be one such pathway that when attenuated may result in the inhibition of some types of inflammatory pain. Thus far the CB1 receptor has demonstrated a propensity to be constitutively activated and is plausible as a receptor that is maintaining an intracellular environment that results in a capsaicin-sensitized TRPV1 channel. These studies support an antihyperalgesic role for CB1 inverse agonists in conditions of inflammatory pain in which TRPV1 receptors have been shown to promote hypersensitivity and suggest that strategies to indirectly modulate TRPV1-function warrant increased exploration.
Contributor Information
Beatriz Fioravanti, Department of Pharmacology and Anesthesiology, College of Medicine, University of Arizona, Tucson, Arizona 85724..
Milena De Felice, Department of Pharmacology and Anesthesiology, College of Medicine, University of Arizona, Tucson, Arizona 85724..
Cheryl L. Stucky, Department of Cell Biology, Neurobiology and Anatomy, Medical College of Wisconsin, Milwaukee, WI 53226-0509
Karen A. Medler, Department of Cell Biology, Neurobiology and Anatomy, Medical College of Wisconsin, Milwaukee, WI 53226-0509
Miaw-Chyi Luo, Department of Pharmacology and Anesthesiology, College of Medicine, University of Arizona, Tucson, Arizona 85724..
Luis R. Gardell, Department of Pharmacology and Anesthesiology, College of Medicine, University of Arizona, Tucson, Arizona 85724.
Mohab Ibrahim, Department of Pharmacology and Anesthesiology, College of Medicine, University of Arizona, Tucson, Arizona 85724..
T. Phil Malan, Jr, Department of Pharmacology and Anesthesiology, College of Medicine, University of Arizona, Tucson, Arizona 85724..
Henry I. Yamamura, Department of Pharmacology and Anesthesiology, College of Medicine, University of Arizona, Tucson, Arizona 85724.
Michael H. Ossipov, Department of Pharmacology and Anesthesiology, College of Medicine, University of Arizona, Tucson, Arizona 85724.
Tamara King, Department of Pharmacology and Anesthesiology, College of Medicine, University of Arizona, Tucson, Arizona 85724..
Josephine Lai, Department of Pharmacology and Anesthesiology, College of Medicine, University of Arizona, Tucson, Arizona 85724..
Frank Porreca, Department of Pharmacology and Anesthesiology, College of Medicine, University of Arizona, Tucson, Arizona 85724..
Todd W. Vanderah, Department of Pharmacology and Anesthesiology, College of Medicine, University of Arizona, Tucson, Arizona 85724.
Reference List
- Ahluwalia J, Urban L, Bevan S, Capogna M, Nagy I. Cannabinoid 1 receptors are expressed by nerve growth factor-and glial cell-derived neurotrophic factor-responsive primary sensory neurones. Neuroscience. 2002;110:747–753. doi: 10.1016/s0306-4522(01)00601-7. [DOI] [PubMed] [Google Scholar]
- Ahluwalia J, Urban L, Capogna M, Bevan S, Nagy I. Cannabinoid 1 receptors are expressed in nociceptive primary sensory neurons. Neuroscience. 2000;100:685–688. doi: 10.1016/s0306-4522(00)00389-4. [DOI] [PubMed] [Google Scholar]
- Bae YS, Lee TG, Park JC, Hur JH, Kim Y, Heo K, Kwak JY, Suh PG, Ryu SH. Identification of a compound that directly stimulates phospholipase C activity. Mol Pharmacol. 2003;63:1043–1050. doi: 10.1124/mol.63.5.1043. [DOI] [PubMed] [Google Scholar]
- Bayewitch M, vidor-Reiss T, Levy R, Barg J, Mechoulam R, Vogel Z. The peripheral cannabinoid receptor: adenylate cyclase inhibition and G protein coupling. FEBS Lett. 1995;375:143–147. doi: 10.1016/0014-5793(95)01207-u. [DOI] [PubMed] [Google Scholar]
- Bhave G, Hu HJ, Glauner KS, Zhu W, Wang H, Brasier DJ, Oxford GS, Gereau RW. Protein kinase C phosphorylation sensitizes but does not activate the capsaicin receptor transient receptor potential vanilloid 1 (TRPV1) Proc Natl Acad Sci U S A. 2003;100:12480–12485. doi: 10.1073/pnas.2032100100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhave G, Zhu W, Wang H, Brasier DJ, Oxford GS, Gereau RW. cAMP-dependent protein kinase regulates desensitization of the capsaicin receptor (VR1) by direct phosphorylation. Neuron. 2002;35:721–731. doi: 10.1016/s0896-6273(02)00802-4. [DOI] [PubMed] [Google Scholar]
- Bilsky EJ, Bernstein RN, Hruby VJ, Rothman RB, Lai J, Porreca F. Characterization of antinociception to opioid receptor selective agonists after antisense oligodeoxynucleotide-mediated “knock-down” of opioid receptor in vivo. J Pharmacol Exp Ther. 1996;277:491–501. [PubMed] [Google Scholar]
- Binzen U, Greffrath W, Hennessy S, Bausen M, Saaler-Reinhardt S, Treede RD. Co-expression of the voltage-gated potassium channel Kv1.4 with transient receptor potential channels (TRPV1 and TRPV2) and the cannabinoid receptor CB1 in rat dorsal root ganglion neurons. Neuroscience. 2006;142:527–539. doi: 10.1016/j.neuroscience.2006.06.020. [DOI] [PubMed] [Google Scholar]
- Bölcskei K, Helyes Z, Szabo A, Sandor K, Elekes K, Nemeth J, Almasi R, Pinter E, Petho G, Szolcsanyi J. Investigation of the role of TRPV1 receptors in acute and chronic nociceptive processes using gene-deficient mice. Pain. 2005;117:368–376. doi: 10.1016/j.pain.2005.06.024. [DOI] [PubMed] [Google Scholar]
- Calandra B, Portier M, Kerneis A, Delpech M, Carillon C, Le FG, Ferrara P, Shire D. Dual intracellular signaling pathways mediated by the human cannabinoid CB1 receptor. Eur J Pharmacol. 1999;374:445–455. doi: 10.1016/s0014-2999(99)00349-0. [DOI] [PubMed] [Google Scholar]
- Carlton SM, Zhou S, Du J, Hargett GL, Ji G, Coggeshall RE. Somatostatin modulates the transient receptor potential vanilloid 1 (TRPV1) ion channel. Pain. 2004;110:616–627. doi: 10.1016/j.pain.2004.04.042. [DOI] [PubMed] [Google Scholar]
- Caterina MJ, Julius D. The vanilloid receptor: a molecular gateway to the pain pathway. Annu Rev Neurosci. 2001;24:487–517. doi: 10.1146/annurev.neuro.24.1.487. [DOI] [PubMed] [Google Scholar]
- Caterina MJ, Leffler A, Malmberg AB, Martin WJ, Trafton J, Petersen-Zeitz KR, Koltzenburg M, Basbaum AI, Julius D. Impaired nociception and pain sensation in mice lacking the capsaicin receptor. Science. 2000;288:306–313. doi: 10.1126/science.288.5464.306. [DOI] [PubMed] [Google Scholar]
- Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D. The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature. 1997;389:816–824. doi: 10.1038/39807. [DOI] [PubMed] [Google Scholar]
- Cesare P, McNaughton P. A novel heat-activated current in nociceptive neurons and its sensitization by bradykinin. Proc Natl Acad Sci U S A. 1996;93:15435–15439. doi: 10.1073/pnas.93.26.15435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang HH, Prescott ED, Kong H, Shields S, Jordt SE, Basbaum AI, Chao MV, Julius D. Bradykinin and nerve growth factor release the capsaicin receptor from PtdIns(4,5)P2-mediated inhibition. Nature. 2001;411:957–962. doi: 10.1038/35082088. [DOI] [PubMed] [Google Scholar]
- Costa B, Trovato AE, Colleoni M, Giagnoni G, Zarini E, Croci T. Effect of the cannabinoid CB1 receptor antagonist, SR141716, on nociceptive response and nerve demyelination in rodents with chronic constriction injury of the sciatic nerve. Pain. 2005;116:52–61. doi: 10.1016/j.pain.2005.03.043. [DOI] [PubMed] [Google Scholar]
- Davis JB, Gray J, Gunthorpe MJ, Hatcher JP, Davey PT, Overend P, Harries MH, Latcham J, Clapham C, Atkinson K, Hughes SA, Rance K, Grau E, Harper AJ, Pugh PL, Rogers DC, Bingham S, Randall A, Sheardown SA. Vanilloid receptor-1 is essential for inflammatory thermal hyperalgesia. Nature. 2000;405:183–187. doi: 10.1038/35012076. [DOI] [PubMed] [Google Scholar]
- De Petrocellis L, Bisogno T, Maccarrone M, Davis JB, Finazzi-Agrò A, Di Marzo V. The activity of anandamide at vanilloid VR1 receptors requires facilitated transport across the cell membrane and is limited by intracellular metabolism. J Biol Chem. 2001a;276:12856–12863. doi: 10.1074/jbc.M008555200. [DOI] [PubMed] [Google Scholar]
- De Petrocellis L, Harrison S, Bisogno T, Tognetto M, Brandi I, Smith GD, Creminon C, Davis JB, Geppetti P, Di Marzo V. The vanilloid receptor (VR1)-mediated effects of anandamide are potently enhanced by the cAMP-dependent protein kinase. J Neurochem. 2001b;77:1660–1663. doi: 10.1046/j.1471-4159.2001.00406.x. [DOI] [PubMed] [Google Scholar]
- Despres JP, Golay A, Sjostrom L. Effects of rimonabant on metabolic risk factors in overweight patients with dyslipidemia. N Engl J Med. 2005;353:2121–2134. doi: 10.1056/NEJMoa044537. [DOI] [PubMed] [Google Scholar]
- Gavva NR, Bannon AW, Surapaneni S, Hovland DN, Jr, Lehto SG, Gore A, Juan T, Deng H, Han B, Klionsky L, Kuang R, Le A, Tamir R, Wang J, Youngblood B, Zhu D, Norman MH, Magal E, Treanor JJ, Louis JC. The vanilloid receptor TRPV1 is tonically activated in vivo and involved in body temperature regulation. J Neurosci. 2007;27:3366–3374. doi: 10.1523/JNEUROSCI.4833-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson HE, Edwards JG, Page RS, Van Hook MJ, Kauer JA. TRPV1 channels mediate long-term depression at synapses on hippocampal interneurons. Neuron. 2008;57:746–759. doi: 10.1016/j.neuron.2007.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gifford AN, Ashby CR., Jr Electrically evoked acetylcholine release from hippocampal slices is inhibited by the cannabinoid receptor agonist, WIN 55212-2, and is potentiated by the cannabinoid antagonist, SR 141716A. J Pharmacol Exp Ther. 1996;277:1431–1436. [PubMed] [Google Scholar]
- Guler AD, Lee H, Iida T, Shimizu I, Tominaga M, Caterina M. Heat-evoked activation of the ion channel, TRPV4. J Neurosci. 2002;22:6408–6414. doi: 10.1523/JNEUROSCI.22-15-06408.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho BY, Current L, Drewett JG. Role of intracellular loops of cannabinoid CB(1) receptor in functional interaction with G(alpha16) FEBS Lett. 2002;522:130–134. doi: 10.1016/s0014-5793(02)02917-4. [DOI] [PubMed] [Google Scholar]
- Ho BY, Uezono Y, Takada S, Takase I, Izumi F. Coupling of the expressed cannabinoid CB1 and CB2 receptors to phospholipase C and G protein-coupled inwardly rectifying K+ channels. Receptors Channels. 1999;6:363–374. [PubMed] [Google Scholar]
- Hou C, Kirchner T, Singer M, Matheis M, Argentieri D, Cavender D. In vivo activity of a phospholipase C inhibitor, 1-(6-((17beta-3-methoxyestra-1,3,5(10)-trien-17-yl)amino)hexyl)-1H-pyrrole -2,5-dione (U73122), in acute and chronic inflammatory reactions. J Pharmacol Exp Ther. 2004;309:697–704. doi: 10.1124/jpet.103.060574. [DOI] [PubMed] [Google Scholar]
- Huang CL, Feng S, Hilgemann DW. Direct activation of inward rectifier potassium channels by PIP2 and its stabilization by Gbetagamma. Nature. 1998;391:803–806. doi: 10.1038/35882. [DOI] [PubMed] [Google Scholar]
- Huestis MA, Boyd SJ, Heishman SJ, Preston KL, Bonnet D, Le FG, Gorelick DA. Single and multiple doses of rimonabant antagonize acute effects of smoked cannabis in male cannabis users. Psychopharmacology (Berl) 2007 doi: 10.1007/s00213-007-0861-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang SW, Cho H, Kwak J, Lee SY, Kang CJ, Jung J, Cho S, Min KH, Suh YG, Kim D, Oh U. Direct activation of capsaicin receptors by products of lipoxygenases: endogenous capsaicin-like substances. Proc Natl Acad Sci U S A. 2000;97:6155–6160. doi: 10.1073/pnas.97.11.6155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeske NA, Patwardhan AM, Gamper N, Price TJ, Akopian AN, Hargreaves KM. Cannabinoid WIN 55,212-2 regulates TRPV1 phosphorylation in sensory neurons. J Biol Chem. 2006;281:32879–32890. doi: 10.1074/jbc.M603220200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez-Andrade JM, Zhou S, Du J, Yamani A, Grady JJ, Castaneda-Hernandez G, Carlton SM. Pro-nociceptive role of peripheral galanin in inflammatory pain. Pain. 2004;110:10–21. doi: 10.1016/j.pain.2004.02.032. [DOI] [PubMed] [Google Scholar]
- Jung J, Shin JS, Lee SY, Hwang SW, Koo J, Cho H, Oh U. Phosphorylation of vanilloid receptor 1 by Ca2+/calmodulin-dependent kinase II regulates its vanilloid binding. J Biol Chem. 2004;279:7048–7054. doi: 10.1074/jbc.M311448200. [DOI] [PubMed] [Google Scholar]
- Koplas PA, Rosenberg RL, Oxford GS. The role of calcium in the desensitization of capsaicin responses in rat dorsal root ganglion neurons. J Neurosci. 1997;17:3525–3537. doi: 10.1523/JNEUROSCI.17-10-03525.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai J, Luo MC, Chen Q, Ma S, Gardell LR, Ossipov MH, Porreca F. Dynorphin A activates bradykinin receptors to maintain neuropathic pain. Nat Neurosci. 2006;9:1534–1540. doi: 10.1038/nn1804. [DOI] [PubMed] [Google Scholar]
- Lauckner JE, Hille B, Mackie K. The cannabinoid agonist WIN 55,212-2 increases intracellular calcium via CB1 receptor coupling to Gq/11 G proteins. Proc Natl Acad Sci U S A. 2005;102:19144–19149. doi: 10.1073/pnas.0509588102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Simon MI. Regulation by cAMP-dependent protein kinease of a G-protein-mediated phospholipase C. Nature. 1996;382:83–87. doi: 10.1038/382083a0. [DOI] [PubMed] [Google Scholar]
- Mitrirattanakul S, Ramakul N, Guerrero AV, Matsuka Y, Ono T, Iwase H, Mackie K, Faull KF, Spigelman I. Site-specific increases in peripheral cannabinoid receptors and their endogenous ligands in a model of neuropathic pain. Pain. 2006;126:102–114. doi: 10.1016/j.pain.2006.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohapatra DP, Nau C. Regulation of Ca2+-dependent desensitization in the vanilloid receptor TRPV1 by calcineurin and cAMP-dependent protein kinase. J Biol Chem. 2005;280:13424–13432. doi: 10.1074/jbc.M410917200. [DOI] [PubMed] [Google Scholar]
- Patwardhan AM, Jeske NA, Price TJ, Gamper N, Akopian AN, Hargreaves KM. The cannabinoid WIN 55,212-2 inhibits transient receptor potential vanilloid 1 (TRPV1) and evokes peripheral antihyperalgesia via calcineurin. Proc Natl Acad Sci U S A. 2006;103:11393–11398. doi: 10.1073/pnas.0603861103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platika D, Boulos MH, Baizer L, Fishman MC. Neuronal traits of clonal cell lines derived by fusion of dorsal root ganglia neurons with neuroblastoma cells. Proc Natl Acad Sci U S A. 1985;82:3499–3503. doi: 10.1073/pnas.82.10.3499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathee PK, Distler C, Obreja O, Neuhuber W, Wang GK, Wang SY, Nau C, Kress M. PKA/AKAP/VR-1 module: A common link of Gs-mediated signaling to thermal hyperalgesia. J Neurosci. 2002;22:4740–4745. doi: 10.1523/JNEUROSCI.22-11-04740.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starowicz K, Santosh N, Di Marzo V. Biochemistry and pharmacology of endovanilloids. Pharmacol Ther. 2007;114:13–33. doi: 10.1016/j.pharmthera.2007.01.005. [DOI] [PubMed] [Google Scholar]
- Stucky CL, Medler KA, Molliver DC. The P2Y agonist UTP activates cutaneous afferent fibers. Pain. 2004;109:36–44. doi: 10.1016/j.pain.2004.01.007. [DOI] [PubMed] [Google Scholar]
- Tominaga M, Caterina MJ, Malmberg AB, Rosen TA, Gilbert H, Skinner K, Raumann BE, Basbaum AI, Julius D. The cloned capsaicin receptor integrates multiple pain-producing stimuli. Neuron. 1998;21:531–543. doi: 10.1016/s0896-6273(00)80564-4. [DOI] [PubMed] [Google Scholar]
- Trevisani M, Smart D, Gunthorpe MJ, Tognetto M, Barbieri M, Campi B, Amadesi S, Gray J, Jerman JC, Brough SJ, Owen D, Smith GD, Randall AD, Harrison S, Bianchi A, Davis JB, Geppetti P. Ethanol elicits and potentiates nociceptor responses via the vanilloid receptor-1. Nat Neurosci. 2002;5:546–551. doi: 10.1038/nn0602-852. [DOI] [PubMed] [Google Scholar]
- Wen Y, Anwer K, Singh SP, Sanborn BM. Protein kinase-A inhibits phospholipase-C activity and alters protein phosphorylation in rat myometrial plasma membranes. Endocrinology. 1992;131:1377–1382. doi: 10.1210/endo.131.3.1324160. [DOI] [PubMed] [Google Scholar]
- Zhou D, Shearman LP. Voluntary exercise augments acute effects of CB1-receptor inverse agonist on body weight loss in obese and lean mice. Pharmacol Biochem Behav. 2004;77:117–125. doi: 10.1016/j.pbb.2003.10.015. [DOI] [PubMed] [Google Scholar]
- Zimmer A, Zimmer AM, Hohmann AG, Herkenham M, Bonner TI. Increased mortality, hypoactivity, and hypoalgesia in cannabinoid CB1 receptor knockout mice. Proc Natl Acad Sci U S A. 1999;96:5780–5785. doi: 10.1073/pnas.96.10.5780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zygmunt PM, Petersson J, Andersson DA, Chuang H, Sorgard M, Di Marzo V, Julius D, Hogestatt ED. Vanilloid receptors on sensory nerves mediate the vasodilator action of anandamide. Nature. 1999;400:452–457. doi: 10.1038/22761. [DOI] [PubMed] [Google Scholar]