

Abstract
Cardiac arrhythmias cause 400 000 sudden deaths annually in the United States alone. Mutations in the cardiac sodium channel gene SCN5A on chromosome 3p21 cause cardiac arrhythmias and sudden death. In this study, we define an SCN5A mutation, S1103Y, in a white family associated with syncope, ventricular fibrillation, and sudden death. A very recent study reported the same mutation in 13.2% of African Americans, but not in the white population. Our study shows that mutation S1103Y does exist in the white population, and it is associated with a considerable risk of syncope, ventricular arrhythmia, ventricular fibrillation, and sudden death in this population.
SCN5A encodes the cardiac sodium channel, a member of the voltage gated sodium channel family. 1 Although SCN5A is primarily expressed in heart,1 its expression has been detected in brain regions including neurones.2 The SCN5A protein contains 2016 amino acids and has a calculated molecular weight of 227 kDa. It consists of four homologous domains (DI–DIV) joined by short linking intracellular domains (ID I–II, ID II–III, and ID III–IV).3 Each transmembrane domain contains six segments (S1–S6).3 Mutations in SCN5A cause syncope, seizures, and sudden death triggered by lethal cardiac arrhythmias associated with long QT syndrome (LQTS), idiopathic ventricular fibrillation including Brugada syndrome, and cardiac conduction disease.4–7
A single nucleotide polymorphism (SNP) in SCN5A, S1103Y, was identified in 13.2% of African Americans and was found to increase the risk of acquired arrhythmia in this population (note that this mutation occurred at codon 1103, but was designated S1102Y in their report).8 However, Splawski et al8 did not find S1103Y SNP in 511 white people. In this study, we report the cosegregation of SCN5A mutation S1103Y with arrhythmia and sudden death in a white family.
METHODS
Identification of LQTS patients and isolation of genomic DNA
Patients with LQTS and arrhythmias were identified at the Department of Cardiovascular Medicine at the Cleveland Clinic Foundation. Diagnosis of LQTS was as described previously.9 Informed consent was obtained from all participants or their guardians, in accordance with standards established by the Cleveland Clinic Foundation Institutional Review Board on Human Subjects. Genomic DNA was prepared from the patients’ whole blood with the DNA Isolation Kit for Mammalian Blood (Roche Diagnostic Co).
Mutation analysis
Mutation analysis was carried out using single strand conformational polymorphism (SSCP) analysis as described previously.9 The aberrant SSCP conformer was cut directly from dried gels, rehydrated in water, reamplified by PCR, and sequenced by an ABI-3100 Genetic Analyzer.
Ancestry analysis
Ancestry analysis (ANCESTRY 1.0) was performed for three family members in kindred QW1446 (II.1, II.3, and II.5 in fig 1A) by DNAPrint Genomics (Sarasota, FL). ANCESTRY 1.0 facilitates the estimation of biogeographical ancestry on three axes, Native American, West African, and European. In this analysis each subject was genotyped for 31 Ancestry Informative Markers. Using maximum likelihood, the individual ancestral proportions of these three populations in the resulting multilocus genotype was computed.10
Figure 1.
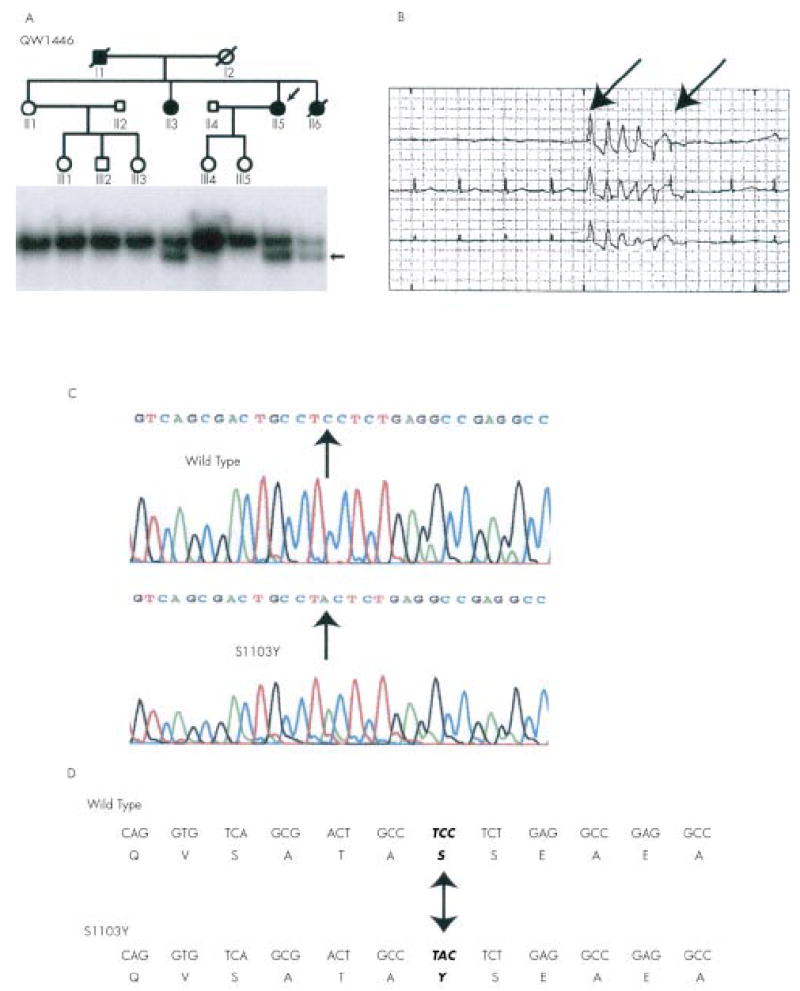
Mutation S1103Y in SCN5A associated with arrhythmia and sudden death in kindred QW1446. (A) The pedigree of QW1446 is shown at the top. Normal subjects are represented by open circles (females) or squares (males), and the affected subjects are represented by solid symbols. Results of SSCP analysis are shown below the pedigree structure. The arrow indicates the abnormal SSCP band. The affected subjects carried the abnormal SSCP band but the normal family members did not. (B) Polymorphic ventricular tachycardia (arrows) recorded in II.6 by Holter monitoring. (C) Sequence analysis of normal (wild type) and abnormal (S1103Y) SSCP conformers. The mutation spot is indicated. (D) The substitution of C by A at the second position of codon 1103 of SCN5A led to substitution of a serine residue by a tyrosine residue (S1103Y). Mutation S1103Y is located in the intracellular loop between domains II and III of the cardiac sodium channel SCN5A.
RESULTS AND DISCUSSION
Using SSCP analysis, we identified an anomalous SSCP conformer in a white family (kindred QW1446) (fig 1A). Kindred QW1446 is associated with a history of ventricular tachycardia (fig 1B), ventricular fibrillation, syncope, sudden death, and LQTS. To confirm that kindred QW1446 has a European descent, the biogeographical ancestry proportions of three of the sibs (II.1, II.3, II.5 in fig 1A) from the second generation were estimated.10 The maximum likelihood point estimates for each of these sibs were 100% European, 0% African, and 0% Native American. These results strongly suggest that kindred QW1446 is a white family.
DNA sequence analysis of the normal and the aberrant SSCP conformers showed that the abnormal SSCP conformer contained a single base substitution (C to A) at the second nucleotide of codon S1103 of SCN5A (GeneBank accession No M77235) (fig 1C). This substitution resulted in a nonconservative replacement of serine with tyrosine (S1103Y) in the intracellular linker between transmembrane domains II and III of the cardiac sodium channel (fig 1D).
The S1103Y mutation was found in only the three affected subjects in kindred QW1446, but not in the normal family members. These genetic data suggest that S1103Y is a pathogenic mutation. Mutation S1103Y was not detected in 200 white controls. These data suggest that although mutation S1103Y exists in the white population, its prevalence rate is low in the normal white population.
The proband in the family (II.5) had a baseline QTc of 0.520 seconds, and she developed two episodes of syncope at the age of 49 years. The first episode was triggered by emotion and excitement. The second episode occurred in the setting of amiodarone and low serum potassium, and it degenerated into ventricular fibrillation and cardiac arrest. She was resuscitated by cardioversion. II.6 in the family had a QTc of 0.431 seconds, and died suddenly when awakening from sleep at the age of 44 years. II.3 had a QTc of 0.452 seconds, developed one episode of syncope at the age of 33 years, and had complained of palpitations all her life. The father of the three mutation carriers died suddenly at the age of 50 years in his sleep. Subjects without mutation S1103Y had a normal QTc. Echocardiography, MRI, and catheterisation did not identify significant structural abnormalities in the heart, brain, or coronary arteries in II.1, II.5, or II.6. In II.3, catheterisation showed normal coronary arteries, but echocardiography showed left ventricular dysfunction with an ejection fraction of 30% and a dilated left atrium and ventricle (dilated cardiomyopathy), which may have been a contributing factor to the development of syncope in this person.
It is interesting to note that all three known mutation carriers in kindred QW1446 developed symptoms, even though the cardiac events developed at advanced ages (33, 44, 49, and 50 years for II.3, II.6, II.5, and I.1, respectively). We speculate that other factors in the family may have contributed to their high rate of cardiac events. The left ventricular dysfunction may have been a modulating factor for the development of syncope in II.3, whereas amiodarone and hypokalaemia may have been contributing factors to sudden cardiac arrest in II.5. The cardiac events may also have been precipitated by obesity (all three mutation carriers were obese), modifier genes, or environmental or other unknown factors.
Electrophysiological characterisation of SCN5A channel with S1103Y suggested that the mutant channels had accelerated channel activation.8 Mutation S1103Y was found subtly to increase the risk of arrhythmia in African Americans and was not identified in a group of 511 white subjects in a previously study.8 Our study reported here shows that mutation S1103Y does exist in the white population and that it is associated with a considerable risk for syncope, ventricular arrhythmia, ventricular fibrillation, and sudden death.
Acknowledgments
We thank Amy Moore for help with the manuscript. This study was supported by a grant in aid from the American Heart Association Ohio-Affiliate and NIH grant R01 HL66251 (to QW).
References
- 1.Gellens ME, George AL, Jr, Chen LQ, Chahine M, Horn R, Barchi RL, Kallen RG. Primary structure and functional expression of the human cardiac tetrodotoxin-insensitive voltage-dependent sodium channel. Proc Natl Acad Sci USA. 1992;89:554–8. doi: 10.1073/pnas.89.2.554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wu L, Nishiyama K, Hollyfield JG, Wang Q. Localization of Nav1.5 sodium channel protein in the mouse brain. NeuroReport. doi: 10.1097/01.wnr.0000052322.62862.a5. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Balser JR. Structure and function of the cardiac sodium channels. Cardiovasc Res. 1999;42:327–38. doi: 10.1016/s0008-6363(99)00031-0. [DOI] [PubMed] [Google Scholar]
- 4.Wang Q, Shen J, Li Z, Timothy K, Vincent GM, Priori SG, Schwartz PJ, Keating MT. Cardiac sodium channel mutations in patients with long QT syndrome, an inherited cardiac arrhythmia. Hum Mol Genet. 1995;4:1603–7. doi: 10.1093/hmg/4.9.1603. [DOI] [PubMed] [Google Scholar]
- 5.Wang Q, Shen J, Splawski I, Atkinson D, Li Z, Robinson JL, Moss AJ, Towbin JA, Keating MT. SCN5A mutations associated with an inherited cardiac arrhythmia, long QT syndrome. Cell. 1995;80:805–11. doi: 10.1016/0092-8674(95)90359-3. [DOI] [PubMed] [Google Scholar]
- 6.Tan HL, Bink-Boelkens MTE, Bezzina CR, Viswanathan PC, Beaufort-Krol GCM, van Tintelen PJ, van den Berg MP, Wilde AAM, Balser JR. A sodium-channel mutation causes isolated cardiac conduction disease. Nature. 2001;409:1043–7. doi: 10.1038/35059090. [DOI] [PubMed] [Google Scholar]
- 7.Chen Q, Kirsch GE, Zhang D, Brugada R, Brugada J, Brugada P, Potenza D, Moya A, Borggrefe M, Breithardt G, Ortiz-Lopez R, Wang Z, Antzelevitch C, O’Brien RE, Schulze-Bahr E, Keating MT, Towbin JA, Wang Q. Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature. 1998;392:293–6. doi: 10.1038/32675. [DOI] [PubMed] [Google Scholar]
- 8.Splawski I, Timothy K, Tateyama M, Clancy CE, Malhotra A, Beggs AH, Cappuccio FP, Sagnella GA, Kass R, Keating M. Variant of SCN5A sodium channel implicated in risk of cardiac arrhythmia. Science. 2002;297:1333–6. doi: 10.1126/science.1073569. [DOI] [PubMed] [Google Scholar]
- 9.Wang Q, Curran ME, Splawski I, Burn TC, Millholland JM, VanRaay TJ, Shen J, Timothy KW, Vincent GM, de Jager T, Schwartz PJ, Toubin JA, Moss AJ, Atkinson DL, Landes GM, Connors TD, Keating MT. Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nat Genet. 1996;12:17–23. doi: 10.1038/ng0196-17. [DOI] [PubMed] [Google Scholar]
- 10.Shriver MD, Smith MW, Jin L, Marcini A, Akey JM, Deka R, Ferrell RE. Ethnic-affiliation estimation by use of population-specific DNA markers. Am J Hum Genet. 1997;60:957–64. [PMC free article] [PubMed] [Google Scholar]