

Abstract
Vascular morphogenesis is a vital process for embryonic development, normal physiologic conditions (e.g. wound healing) and pathological processes (e.g. atherosclerosis, cancer). Genetic studies of vascular anomalies have led to identification of critical genes involved in vascular morphogenesis. A susceptibility gene, VG5Q (formally named AGGF1), was cloned for Klippel-Trenaunay syndrome (KTS). AGGF1 encodes a potent angiogenic factor, and KTS-associated mutations enhance angiogenic activity of AGGF1, defining ‘increased angiogenesis’ as one molecular mechanism for the pathogenesis of KTS. Similar studies have identified other genes involved in vascular anomalies as important genes for vascular morphogenesis, including TIE2, VEGFR-3, RASA1, KRIT1, MGC4607, PDCD10, glomulin, FOXC2, NEMO, SOX18, ENG, ACVRLK1, MADH4, NDP, TIMP3, Notch3, COL3A1 and PTEN. Future studies of vascular anomaly genes will provide insights into the molecular mechanisms for vascular morphogenesis, and may lead to the development of therapeutic strategies for treating these and other angiogenesis-related diseases, including coronary artery disease and cancer.
Keywords: Blood vessels, vasculogenesis, angiogenesis, vascular anomalies and malformations, Klippel-Trenaunay syndrome (KTS), VG5Q, AGGF1
Introduction
Blood vessels are intricate networks of tubes that transport blood throughout the entire body. A closed blood vascular system efficiently carries nutrients, gases, wastes, hormones, metabolites, as well as immune cells, to and from distant actively metabolizing tissues. Blood vessel formation is a vital and dynamic physiological process for normal tissue growth, such as embryonic development, wound healing, placenta formation after fertilization and menstrual cycle. When the formation of blood vessels is unregulated or misregulated, numerous malignant, ischemic, inflammatory, infectious and immune disorders evolve. Diseases associated with pathogenic blood vessel formation can be characterized or caused by excessive or abnormal blood vessel formation, such as vascular malformations, cancer and age-related macular degeneration, or by insufficient new blood vessel formation or vessel regression such as ischemia in heart and brain, hypertension and neurodegeneration [1]. Understanding of blood vessel formation and its regulation at the cellular, molecular and genetic levels will provide information critical for the prognosis and therapy of these diseases. On the other hand, genetic and molecular studies of diseases associated with abnormal blood vessel formation will provide fundamental understanding of vascular morphogenesis, development and growth. Here, we review advances in molecular genetic studies of a congenital vascular disorder, Klippel-Trenaunay syndrome (KTS), which led to the molecular cloning of a novel angiogenic factor VG5Q. VG5Q stands for the vascular gene on chromosome 5Q, and it is the first angiogenic factor gene identified by a human genetic approach. We also update the molecular genetics of other vascular disorders. Please note that upon recent recommendation by the International Gene Nomenclature Committee, we have changed the official name for VG5Q to AGGF1 (angio genic factor with G patch and FHA domains 1).
Blood and lymphatic vessels
The vascular system consists of blood vessels and lymphatic vessels (lymphatics). The blood vascular system consists of arteries, capillaries and veins. Walls of the circulatory vessels (blood or lymphatic) are composed mainly of endothelial cells (ECs) surrounded by basement membrane and mural cells [pericytes and vascular smooth muscle cells (SMCs)] that are embedded in an extracellular matrix (ECM). They differ in the blood pressure they hold and the thickness of the vascular SMC layer.
Arteries
Arteries are strong, elastic and/or muscular vessels. Large arteries branch progressively into thinner small arteries and arterioles. The wall of a large artery consists of three layers of tunics, the intima, media and adventitia. Tunica intima is composed of ECs resting on a connective tissue membrane which is rich in elastic and collagen fibers. Tunica media has a thick layer of SMCs and elastic connective tissue. Smooth muscle fibers encircle the tube. The outer layer, tunica adventitia, consists of fibroblasts with irregularly arranged elastic and collagenous fibers. This layer attaches the artery to the surrounding tissues, which can be muscle, adipose or other types. Arterioles have also three layers but have a decreased ratio of mural cells. The wall of a pre-capillary arteriole consists of only ECs and SMCs surrounded by a small amount of elastic connective tissue. The SMCs in the walls of arteries and arterioles are innervated by the sympathetic branches of the autonomic nervous system (ANS). Vasomotor impulses cause SMCs to contract by reducing the diameter of the vessels. If these impulses are inhibited, SMCs relax and the diameter of the vessels increases, which is known as vasodilation. Changes in the diameters of arteries and arterioles greatly influence blood flow and pressure.
Capillaries
Capillaries are the most abundant blood vessel in the body. They form connections between the arterioles and the smallest venules. Capillaries consist of a single layer of ECs surrounded by basement membrane and a layer of pericytes embedded within the EC basement membrane. Because of their wall structure, they are the main site of exchange of gases, nutrients and metabolic by-products between blood and the tissue fluid surrounding the body cells. Endothelial cell-cell junctions play a role in the permeability of the capillary walls that varies from tissue to tissue depending upon the permeability requirements of perfused organs [2].
Veins
Venules continue from the capillaries and merge to form veins. The walls of most veins are similar to those of arteries in that they are composed of three distinct layers, the intima, media and adventitia. Because the middle layer is poorly developed, veins have thinner walls that contain fewer SMCs and less elastic tissue than arteries. Many veins, particularly those in the arms and legs, have flap-like valves. Valves are open as long as the blood flow is toward the heart and closed if it is in the opposite direction. Veins also function as blood reservoirs that can be drawn upon in time of need. If a hemorrhage with a drop in blood pressure occurs, the muscular walls of the veins are stimulated by the sympathetic nervous system. The venous constriction augments cardiac preload, helping to raise the blood pressure, and ensures a normal blood flow.
Lymphatics
The lymphatic system consists of lymphatic capillaries and lymphatic vessels that carry lymph and participate in the nutritional processes of organs. Interstitial or extracellular fluid is formed by leakage of blood plasma through minute pores of the capillaries. There is a continual interchange of fluids of the blood and tissue spaces with a free interchange of nutrients and other dissolved substances. Most of the tissue fluid returns to the circulatory system by means of capillaries, which feed into larger veins. However, large protein molecules, as well as white blood cells, dead cells, bacterial debris, infected substances, and larger particulate matter, pass through the porous walls of the lymphatic capillaries and, thus, enter the lymphatic circulatory system with the remainder of the tissue fluid. The interstitial fluid entering the capillaries is called lymph. The lymphatic capillary wall consists of a single layer of ECs, lymphothelium. D2-40, podoplanin, prox-1 and LYVE-1 are the new markers specific to the lymphothelium [3, 4]. Lymphatic capillaries merge to form lymphatic vessels or lymphatics. They are similar to veins in structure and also have valves. Lymphatic vessels merge to form larger lymphatic trunks and finally lymphatic ducts.
Vasculogenesis, angiogenesis, vessel maturation and lymphangiogenesis
During embryogenesis, blood vessels are formed via two processes: vasculogenesis and angiogenesis. Vasculogenesis is defined as the process in which mesoderm cells are induced to differentiate into hemangioblasts and ECs, which then assemble into a primitive tubular network called the primary capillary plexus [5, 6]. Hemangioblasts are the common progenitors of ECs and hematopoietic cells. These two cell lines carry common markers and share similar signaling pathways. Several signaling proteins, including VEGFR-1, VEGFR-2, SCL/tal-1, Cbfa2/Runx1/AML1, GATA-2, CD31 (PECAM) and CD34, are induced during vasculogenesis and hematopoiesis [7]. Then, angioblasts and other endothelial progenitors differentiate into arterial and venous ECs that form a capillary plexus. The Notch pathway was found to promote arterial fate by repressing venous differentiation [8–10]. One of the ephrin family transmembrane ligands, Eph-B2, marks future arterial but not venous endothelium cells, while one of the receptors for ephrin-B2, i.e. Eph-B4, marks venous endothelium, at the earliest stages of capillary plexus formation [11–13].
The primary capillary plexus later undergoes a remodeling and sprouting process called angiogenesis, and is transformed into a complex network. Further nonsprouting development accompanied by recruitment of smooth muscle cells and changes in size and mural structure leads to the formation of arteries, capillaries and veins, each with their own function and characteristics. Sprouting angiogenesis initiates with vasodilation, which is presumably stimulated by hypoxia, and it then involves an increase in the permeability of the ECs, allowing extravasation of plasma proteins that lay down a provisional scaffold for migrating ECs [14, 15]. To migrate, ECs need to loosen the cell-cell junctions [2] and to relieve surrounding cells and matrix support by proteolytic degradations. Proliferating ECs migrate to distant sites and a lumen is formed. Nonsprouting angiogenesis takes place by intussusceptions in which ECs proliferate within a vessel, resulting in splits in the lumen or by fusion and splitting of capillaries [16].
The maturation of nascent vessels involves stabilization of the vessels by recruiting surrounding cells (pericytes and vascular SMCs) and generating an ECM. The vascular network continues to develop and mature by growth, branching, remodeling and pruning of its different segments in response to the demands of specific tissues and organs. Another step of vessel maturation is tissue- and organ-specific specialization of wall and network structure. This procedure involves arteriovenous determination, homotypic and heterotypic junction formation, and EC differentiation to form organ-specific capillary structures [17].
Lymphangiogenesis refers to the formation of lymphatic vessels. Although angiogenesis plays a critical role in the progression of tumors, lymphangiogenesis may be even more important to metastatic spread. Embryonic lymphatic vessels originate from blood vessels. Lymphatic ECs originate from the cardinal vein during embryonic development [17].
Protein factors regulating vasculogenesis, angiogenesis, vessel maturation and lymphangiogenesis
A number of protein factors that play an important role in the regulation of vasculogenesis, angiogenesis and vascular maturation have been identified by biochemical approaches or transgenic/knockout mouse studies.
The best known class of angiogenic growth factors is the vascular endothelial growth factors (VEGFs), including VEGF-A, VEGF-B, VEGF-C and VEGF-D. VEGF-A is one of the most important angiogenic factors, and is required for the earliest stages of vasculogenesis. VEGF-A knockout mice fail to develop blood islands, ECs and major vessels [18]. The concentration of VEGF-A in cells is strictly regulated as the deletion of one single copy of the gene causes embryonic lethality in mice.
VEGF-A carries out its biological functions by binding to its receptors, VEGFR-1 and VEGFR-2. VEGFR-2 is an early embryonic marker for the formation of vasculature. Mouse embryos lacking VEGFR-2 die in utero as a result of an early defect in the development of hematopoietic and endothelial cells [19]. VEGFR-1 plays a role later. Mice lacking VEGFR-1 produce angioblasts, but cannot assemble angioblasts into functional blood vessels [20]. VEGF-B was recently shown to be capable of promoting angiogenesis through its receptor VEGFR-1 and the activation of Akt and eNOS-related pathways [21]. VEGF-C is a lymphatic-specific growth factor, and VEGF-D is involved in both angiogenesis and lymphangiogenesis, discussed in detail below.
Receptor tyrosine kinases Tie-1 and Tie-2, and Tie-2 ligands angiopoietin-1 (Ang-1) and angiopoietin-2 (Ang-2) are another class of molecules important for blood vessel formation. Ang-1, its receptor Tie2 and Ang-2 (the natural antagonist for Tie2) do not seem to be essential for vasculogenesis but instead important for angiogenesis [22–27]. Disruption of the Tie2 gene, Ang-1 or transgenic overexpression of Ang-2 all lead to embryonic lethality as a result of defects in angiogenesis. Ang-1 stabilizes vessel walls and makes them less leaky by promoting interactions between ECs and the surrounding cells and ECM [27]. In the absence of VEGF, Ang2 acts as an antagonist of Ang1 and destabilizes vessels, leading to vessel regression. In the presence of VEGF, Ang2 facilitates vascular sprouting [17]. Tie-1 promotes EC survival possibly through activation of phosphatidylinositol-3-kinase (PI3K) and Akt [7]. The ligand for Tie-1 has not yet been identified.
Integrins and their ligands form another class of regulatory proteins which are critical for EC survival and vascular development. They mediate cell-cell interactions and cell-ECM adhesion [7].
Other important angiogenic factors include fibroblast growth factor-2 (basic FGF, bFGF, FGF-2), platelet-derived growth factor (PDGF) and transforming growth factor-beta.
Angiogenesis is dependent on a delicate balance between activators and inhibitors during vessel formation. In addition to angiogenic growth factors (described above) that strongly promote angiogenesis, there are antiangiogenic factors that block angiogenesis. Examples include thrombospondin-1 (TSP-1), metalloproteinase inhibitors (TIMPs), angiostatin and endostatin [17, 28–33].
The VEGF-C/VEGF-D/VEGFR-3 signaling pathway plays a central role in control of lymphangiogenesis [34]. LYVE-1 (lymphatic vessel endothelial hyaluronan receptor) and VEGFR-3 mark lymphatic vessels in the embryo and adult [35]. The homeobox gene Prox-1 regulates lymphatic sprouting and budding [36]. The Syk-SLP76 pathway triggers separation of embryonic lymphatic and blood vessels [37]. Ang2 is thought to be involved in the maturation and patterning of lymph vessels. It was also shown that neuropilin 2 (NRP2) is required for formation of small lymphatic vessels and capillaries [38]. For an overview of the molecular mechanisms involved in lymphangiogenesis, see [39] and [40].
Vascular anomalies
The processes of vessel formation and vascular morphogenesis are precisely regulated, and disruption of these processes or developmental errors affecting them leads to a heterogeneous group of vascular anomalies, including KTS (discussed later).
Vascular anomalies are broadly classified into vascular tumors and vascular malformations [41–43]. Vascular tumors include hemangiomas of infancy, tufted angiomas, Kaposiform hemangioendotheliomas, infantile hemangio-endotheliomas and spindle cell hemangioendotheliomas. Vascular malformations include telangiectasia, capillary, venous, arterial and lymphatic malformations, and combined or mixed vascular malformations, such as Klippel-Trenaunay, Parkes Weber and Servelle Martorell syndromes.
Hemangiomas are benign vascular tumors of infants, and are in fact the most common tumor of infancy, occurring in 1.1–2.6% of newborns. For infants at 1 year of age, the prevalence rate is as high as 10–12%. Hemangiomas of infancy usually are not present at birth, become visible within 1–4 weeks of neonatal life, and grow rapidly (proliferative) up to 18 months of age and then begin to regress (involute). Hemangiomas have endothelial hyperplasia. They can be superficial, deep or both.
Vascular malformations are usually, but not always, obvious at birth, grow proportionally with the patient and rarely, if ever, regress. They have a single layer of endothelium. Except for some lymphatic lesions, they do not respond to steroids. Vascular malformations can be subcategorized according to the channel type and rheology as either slow-flow or fast-flow [44]. They can be single-channel (arterial, venous, capillary or lymphatic) or combined-channel (arteriovenous, capillary-lymphatic, capillary-venous, capillary-lymphatic-venous or lymphatic-venous) malformations. The combined vascular malformations are often associated with bony and/or soft tissue overgrowth (hypertrophy). KTS is an extensive combined malformation comprising capillary, lymphatic and venous malformations associated with overgrowth of the affected extremity.
Clinical features of KTS
KTS (MIM 149000) [45–48] is defined as a congenital vascular disorder comprised of (i) capillary malformations, (ii) venous malformations or extensive distribution and early onset of the varicose veins, and (iii) hypertrophy of the affected tissues. Lymphatic malformations also occur in some KTS patients (11% of cases). The presence of two of the three cardinal features is sufficient to make a KTS diagnosis [47]. Figure 1 shows some features with which the KTS patients typically present. Vascular malformations in KTS are the slow-flow type. Significant arteriovenous fistulae do not occur in KTS [49].
Figure 1.
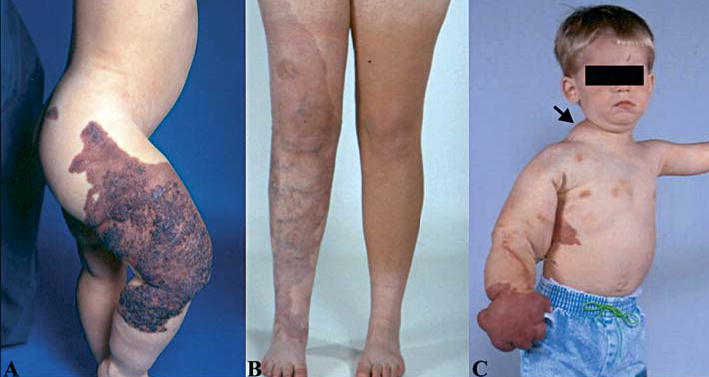
Klippel-Trenaunay syndrome (KTS). (A) Extensive combined capillary (portwine satin)-lymphatico-venousomal formation of the right lower extremity. These may be flat, or elevated as in this case. (B) Capillary malformation (portwine stain) and varicose veins. (C) Hypertrophic right arm and trunk together with capillary and venous malformations. Subcutaneous hypertrophy also is present (arrow).
Capillary malformations (CMs, also known as portwine stains) are the most common cutaneous vascular malformation in KTS, and occur in 98% of KTS patients [47]. CMs are red to purplish in color and can be present on any part of the body. They consist of an increased number of abnormal ectatic capillaries in the papillary dermis. The walls of the capillaries are thin, and the ECs are flat [44]. Capillary malformations in KTS can be accompanied by lymphatic and venous malformations. Lymphatic malformations occur in 11% of KTS patients, and 10% may degenerate into lymphedema [47]. Lymphatic malformations are a defect of cutaneous and subcutaneous lymphatic vessels. A lymphatic malformation is composed of dilated lymphatic vessels filled with clear proteinaceous fluid, and not connected to normal lymphatic vessels [44].
Varicose veins occur in 72% of patients with KTS [47]. Persistence of the embryonic lateral vein is very common in KTS (56%) [47]. Deep vein anomalies include aneuryismal dilation, duplication, hypoplasia, aplasia, and external compression of veins by fibrous bands or anomalous vessels [50–54]. Visceral vascular malformations can involve the liver, kidney, bladder, rectum and lower gastrointestinal (GI) tract, retroperitoneum, pericardium, spine and lung. They can cause severe bleeding.
Limb hypertrophy occurs in 67% of the KTS patients, of whom 88% involve the lower extremities [47]. The hypertrophy can involve girth and/or length. Hypertrophy also can occur in the thorax, pelvis, abdomen, head and neck. Macrodactyly can occur.
Genetics of KTS
Most KTS cases are sporadic. However, capillary malformations and varicose veins have been reported among family members of the KTS patients [49, 55–57]. In addition, there have been three reported chromosomal abnormalities in three different KTS patients, two balanced translocations t(5;11)(q13.3;p15.1) and t(8;14)(q22.3;q13), and an extra supernumerary ring chromosome 18 [58–60]. Translocation t(8;14)(q22.3;q13) and the ring chromosome 18 were shown to arise de novo. These findings suggest that genetic factors contribute to the pathogenesis of KTS. The finding of three different cytogenetic defects associated with KTS may suggest that KTS is genetically heterogeneous, and several different genes may be involved in different cases of KTS.
Due to the sporadic nature of KTS, it is technically challenging to identify or clone a gene for KTS using the large family-based positional cloning approach. Thus, we focused on translocation t(5;11)(q13.3;p15.1) (fig. 2A) for identifying a gene for KTS [58]. Our hypothesis was that translocation t(5;11) alters the structure or expression of a gene at one of the two chromosomal breakpoints during the formation of the translocation, which then leads to the development of KTS. No genes were identified in a 100-kb area of the chromosome 11p15.1 breakpoint; however, the chromosome 5q13.3 breakpoint was found to be located in the promoter region of a novel gene, which we named as VG5Q (vascular or vasculogenesis gene on 5q; now renamed AGFF1, angiogenic factor with G-patch and FHA domains 1) [61]. The 5q13.3 breakpoint is 1644 bp upstream from the start codon (fig. 2B) and was shown to increase the expression of AGGF1 by threefold [61].
Figure 2.
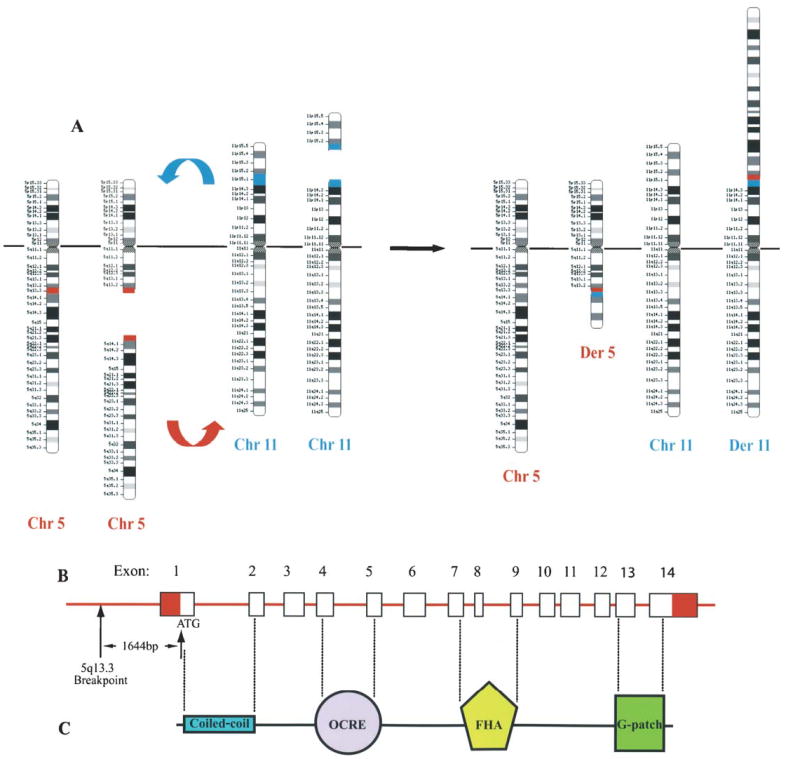
(A) Translocation, t(5;11)(q13.3;p15.1), associated with KTS. Breakages occur on one of the chromosomes 5 and 11 at the bands q13.3 (red) and p15.1 (blue), respectively. Broken pieces are exchanged between chromosomes involved, resulting in the formation of two abnormal chromosomes, derivative chromosomes 5 and 11 (Chr, chromosome; Der, derivative). (B) AGGF1 at the 5q13.3 breakpoint. The 5q13.3 breakpoint is 1644 bp upstream from the start codon (ATG) of AGGF1. AGGF1 contains 14 exons. (C) AGGF1 protein structure. There are four putative functional domains; coiled-coil, OCRE (octamer repeat), FHA (Forkhead-associated) and G-patch domains.
A case-control association study was then carried out with 130 KTS patients and 200 matched controls. A mutation in AGGF1, E133K, was identified in 5 of 130 KTS patients, but not in 200 normal controls [61]. The clinical features of the five gene carriers are shown in table 1. A statistically significant association between mutation E133K and the risk to the development of KTS was established (P = 0.009). Mutation E133K was later shown to increase the angiogenic activity of AGGF1 [61]. These results demonstrate that mutation E133K of AGGF1 is a functional mutation that acts by a gain-of-function mechanism (increased angiogenesis). These results establish AGGF1 as the first susceptibility gene that confers a risk for development of KTS.
Table 1.
Clinical findings of KTS patients with AGGF1 mutation E133K.
| Patient ID | CM | VM | LM | Hypertrophy | Others |
|---|---|---|---|---|---|
| QW576 | + | + | N/A | left leg; girth | hemorrhage in right knee |
| QW731 | + | + | + | right foot; soft tissue hypertrophy | – |
| QW1251 | + | + | + | right leg; girth and length | café au lait spots |
| QW1441 | + | + | N/A | right leg; length | – |
| QW1592 | + | + | N/A | right leg; girth and length | – |
CM, capillary malformations; VM, vascular malformations; LM, lymphatic malformations; N/A, data not available.
Translocation t(8;14)(q22.3;q13) arose de novo [59], which suggests that another pathogenic gene for KTS may be located on chromosome 8q22.3 or 14q13. The breakpoint on chromosome 8q22.3 has been defined to a <5-cM interval flanked by markers AFMA082TG9 and GATA25E10, and the 14q13 breakpoint within a 1-cM region between sequence-tagged sites (STSs) WI-6583 and D14S989 [59]. The specific vascular gene at either 8q22.3 or 14q13 is expected to be identified in the near future. The KTS-associated de novo mosaic supernumerary ring chromosome 18 implicates a potential vascular and/or overgrowth gene located on the chromosome 18 [60]. The ring chromosome 18 r(18) was mostly derived from the short arm of chromosome 18, and its size is estimated to be approximately 10 cM. Further analyses of the genes on the r(18) may lead to the identification of a new KTS gene.
AGGF1 is a novel angiogenic factor
The full-length AGGF1 complementary DNA (cDNA) encodes a novel protein with 714 amino acids with a high level of expression in ECs, vascular smooth muscle cells (VSMCs) and osteoblasts (MG-63) [61]. Strong AGGF1 protein expression was detected in blood vessels embedded in various tissues, including the heart, kidney, tail and limb. AGGF1 protein contains a coiled-coil motif (amino acids 19–85), a forkhead-associated domain (FHA, amino acids 435–508) and a G-patch domain (amino acids 619–663) (fig. 2C). The coiled-coil motif may be involved in protein-protein interactions. The roles of the FHA domain and G-patch domain in AGGF1 are not clear. The FHA domain may be involved in phospho-dependent protein-protein interactions [62], whereas G-patch domains have been implicated as RNA-interacting modules [63]. Recently, an OCRE (OCtamer REpeat) motif (amino acids 197–256) (fig. 2C) was identified in AGGF1, and the authors suggested that this motif may be involved in RNA metabolism and/or in signaling pathways activated by the tumor necrosis factor (TNF) superfamily of cytokines [64].
At the cellular level, AGGF1 is mainly localized in the cytoplasm and around the nucleus, though signal was also detected inside the nucleus [61]. Multiple assays showed that AGGF1 secreted outside the endothelial cell when angiogenesis starts (when endothelial cells are grown on matrigel-coated plates), although weak secretion signal was also detected with endothelial cells cultured on plastic dishes [61]. Our recent results suggest that AGGF1 can secrete outside bacterial cells containing overexpressed recombinant AGGF1 [X. Tian and Q. Wang, unpublished data]. The molecular mechanisms for trafficking and secretion of AGGF1 remain to be established.
With the chick chorioallantoic membrane (CAM) angiogenesis assay, we found that the purified AGGF1 protein (75 ng/μl) promoted strong angiogenesis (fig. 3A) [61]. Angiogenesis was also observed around the discs which were spotted with VEGF-A (100 ng/μl) as a positive control (fig. 3A) [61]. These results suggest that, similar to VEGF-A, AGGF1 strongly promotes angiogenesis, indicating that AGGF1 is a potent angiogenic factor [61]. This conclusion is supported by our recent finding that AGGF1 delivered as a transgene by an adenovirus vector promoted strong angiogenesis in a matrigel angiogenesis assay and in a mouse skeletal muscle angiogenesis assay in vivo [S. You and Q. Wang, unpublished data]. The molecular mechanism for AGGF1-mediated angiogenesis is not clear. On the cellular level, AGGF1 can promote weak endothelial cell proliferation and can bind to endothelial cells (cell adhesion); on the molecular level, AGGF1 can bind to another angiogenic factor, TWEAK, and there may be a cell surface receptor for AGGF1 [61]. These hypotheses warrant future studies.
Figure 3.
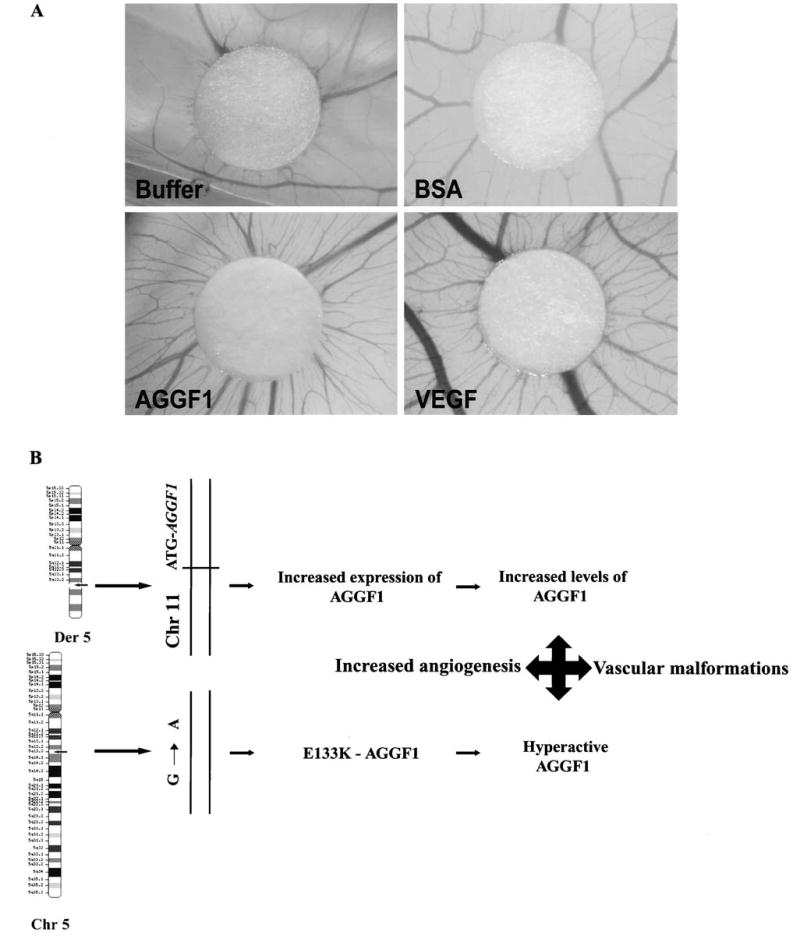
(A) AGGF1 is an angiogenic factor in chick chorioallantoic membrane (CAM) angiogenesis assays. Buffer (the same elution fraction as purified AGGF1, but from bacteria with the empty expression vector) and BSA were negative controls, and VEGF-A (100 ng/μl) was used as a positive control. AGGF1 protein (75 ng/μl) promoted strong angiogenesis. (Adapted from Tian et al. [61] with the permission of Nature Publishing Group). (B) A molecular mechanism for the pathogenesis of KTS. Small arrows on the chromosomes show the location of AGGF1. Translocation of the chromosome 11 sequences upstream of AGGF1 causes a threefold increase in expression of AGGF1, resulting in increased levels of the protein. Mutation E133K increases the angiogenic activity of AGGF1. Both effects are expected to result in increased angiogenesis, which leads to the development of vascular malformations in KTS patients (Chr, chromosome; Der, derivative).
Molecular mechanism for the pathogenesis of KTS
Histological studies showed an increase in both the number and diameter of the venules in the dermis and subdermal fat and widespread hypertrophy of the smooth muscle in the walls of subcutaneous veins [65]. The blood flow in the affected limb is greater than the unaffected one, and the increased blood flow is related to the presence of a nevus on the affected limb [65]. MRI angiography data also revealed the distorted architecture of the vascular system, indicating a defect in the process of vascular growth and remodeling [55, 66]. Our results showing that translocation t(5;11) increases expression of AGGF1, and that KTS mutation E133K in AGGF1 promotes stronger angiogenesis than wild-type AGGF1, suggest that the molecular mechanism for the pathogenesis of KTS is ‘increased’ angiogenesis (fig. 3B). The increased angiogenesis theory is supported by the histological features of KTS and magnetic resonance imaging (MRI) findings described above.
The sporadic occurrence of KTS and the mosaic pattern of KTS features may be explained genetically by the concept of paradominant inheritance proposed by Happle [67–69]. Based on this hypothesis, KTS would be caused by a defect in a lethal gene. Homozygotes for the mutation cannot survive, and they die during early embryogenesis. Heterozygous individuals are phenotypically normal. Therefore, the gene can be transmitted unperceived through many generations until a somatic mutation or ‘second hit’ occurs in the developing embryo, causing loss of heterozygosity and leading to the formation of a cell population being homozygous or heterozygous for the mutation.
Genetics of other vascular and vascular/overgrowth anomalies
Molecular genetics studies have identified several disease-causing genes for other vascular anomalies. Table 2 lists some important genes identified for several vascular and vascular/overgrowth anomalies. Two of these genes have been linked to vascular morphogenesis before the human genetics studies, and they are the TIE2 gene on chromosome 9p21 and the VEGFR3 gene on 5q35.3. Gain-of-function mutations in TIE2 cause multiple cutaneous and mucosal venous malformation (VMCM) (MIM 600195) [70]. Inactivating mutations in VEGFR3 cause lymphedema type I [71]. The link of genes in table 2 to vascular morphogenesis was all initially identified by the human genetics approach.
Table 2.
Genetics of several vascular and vascular/overgrowth anomalies.
| Disease | Chromosome | Gene |
|---|---|---|
| Venous malformations, multiple cutaneous and mucosal (VMCM) | 9p21 | TIE2 [70] |
| Lymphedema type I (Nonne-Milroy lymphedema) | 5q35.3 | VEGFR3 [71] |
| Capillary malformation-arteriovenous malformation (CMAVM) | 5q13.3 | RASA 1 [73] |
| Cerebral capillary malformation (CCM) | ||
| CCM1 | 7q11.2-q21 | KRIT1 [82, 83] |
| CCM2 | 7p15-p13 | MGC4607 [88, 89] |
| CCM3 | 3q25.2-27 | PDCD10 [90] |
| Glomuvenous malformation (GVM) | 1p22-p21 | glomulin [91] |
| Lymphedema-distichiasis (LD) syndrome | 16q24.3 | FOXC2 [92] |
| OL-EDA-ID | Xq28 | NEMO [93] |
| Hypotrichosis-lymphedema-telangiectasia syndrome (HLTS) | 20q13.33 | SOX18 [94] |
| Hereditary hemorrhagic telangiectasia (HHT) (Osler-Rendu-Weber disease) | ||
| type 1 (HHT1) | 9q34.1 | endoglin (ENG) [95] |
| type 2 (HHT2) | 12q11-q14 | ACVRLK1 [96] |
| Juvenile polyposis/hereditary hemorrhagic telangiectasia | 18q21.1 | MADH4 [97] |
| Coats’ disease (retinal telangiectasis) | Xp11.4 | NDP [98] |
| Age-related macular degeneration: Sorsby’s fundus dystrophy (SFD) | 22q12.1-q13.2 | TIMP3 [102] |
| Cerebral arteriopathy (CADASIL) | 19p13.2-p13.1 | Notch3 [104] |
| Ehlers-Danlos syndrome, vascular type (type IV) | 2q31 | COL3A1 [105] |
| Proteus and Proteus-like syndromes | 10q23.31 | PTEN [107–110] |
| Macrocephaly, multiple lipomas, hemangiomata | 10q23.31 | PTEN [112] |
| Bannayan-Zonana syndrome (BZS) | ||
| Bannayan-Riley-Ruvalcaba syndrome (BRSS) | ||
| Riley-Smith syndrome (RSS) | ||
| Ruvalcaba-Myhre-Smith syndrome (RMSS) | ||
| Klippel-Trenaunay syndrome (KTS) | 5q13.3 | AGGF1 [61] |
CMs (MIM 163000) are the most common cutaneous vascular malformation, present in 0.3% of the newborns [72]. CMs also occur in several combined vascular anomalies associated with hypertrophy, as in the case of KTS. Heterozygous inactivating RASA1 mutations were detected in families with CMs with either arteriovenous malformation, arteriovenous fistula or Parkes Weber syndrome (CM-arteriovenous malformation, CMAVM) (MIM 608354) [73]. RASA1 codes for p120-RasGTPase-activating protein (p-120-RasGAP) which catalyzes intrinsic GTPase activity of Ras. It is a downregulator of the Ras/mitogen-activated protein kinase (MAPK)-signaling pathway, which mediates cellular growth, differentiation and proliferation from various receptor tyrosine kinases on cell surfaces [74, 75]. The p120-RasGTPase also binds to p190-RhoGAP which directs signaling to the cytoskeleton [76], and to Rap1a, which is involved in integrin signaling-mediated cellular adhesion [77, 78].
As a single entity, Parkes Weber syndrome (PKWS, MIM 608355) [79] is very similar to KTS. It is characterized by cutaneous CMAVMs in association with hypertrophy of the affected limbs [80]. Several differences distinguish these two syndromes [81]. In contrast to KTS, the vascular malformations in PKWS are fast flow and involve arterial malformations. Lymphatic malformations are rare in PKWS. The hypertrophy involved in PKWS occurs mostly in the length of the extremities.
Type 1 cerebral CMs (CCM1) (MIM 116860) are caused by loss-of-function mutations in the KRIT1 gene [82, 83]. KRIT1 or CCM1 gene codes for Krev interaction trapped-1 protein, which was identified as a binding partner of Rap1a [84], an antagonist of Ras transformation [85]. KRIT1 also binds to ICAP (integrin cytoplasmic domain-associated proten-1), implying a process of integrin signaling-mediated cellular adhesion in the pathogenesis of CCM [86]. It is worth noting that some CCM family members with KRIT1 mutations also have hyperkeratotic cutaneous capillary-venous malformations [73, 87]. The second CCM gene was identified as a novel gene, MGC4607 on 7p15-p13, which encodes a protein, malcavernin, with a phosphotyrosine binding domain, a domain found in ICAP1 α (a binding partner of KRIT1). Malcavernin and KRIT1 may be members of a protein complex involved in integrin signaling [88, 89]. The third CCM gene, CCM3, has been identified as PDCD10 (programmed cell death 10, TFAR15) on 3q26-27 [90]. The role of the PDCD10 gene in vascular morphogenesis remains to be investigated.
Glomuvenous malformations or glomangiomas (venous malformations with smooth muscle-like glomus cells) (MIM 138000) are caused by loss-of function mutations in the glomulin (FAP48) gene coding for an FK506-binding protein (FKBP)-associated protein of 48 kD [91].
One form of hereditary lympedema is the lymphedema-distichiasis syndrome (LD) (MIM 153400). It involves lymphedema together with the presence of double rows of eyelashes. Other complications may include cardiac and skeletal abnormalities. LD is caused by inactivating mutations in the FOXC2 (MFH-1) gene, which codes for a forkhead transcription factor [92]. On the other hand, lymphedema with osteoporosis, ectodermal dysplasia (anhidrotic) and immunodeficiency, called OL-EDA-ID (MIM 300301), was found to be caused by a mutation in the stop codon of the nuclear factor-kappaB (NF-κB) essential modulator gene (NEMO) [93]. The mutation results in expression of a protein that is 27 amino acids longer than the wild-type protein. It was shown that reduced NF-κB signaling is a possible mechanism for OL-EDA-ID [93]. Another interesting combination of lymphedema with hypotrichosis (sparse hair) and telangiectasia (abnormal dilation of capillaries and arterioles) (MIM 607823) was found to be caused by inactivating mutations in the SOX18 gene, which belongs to the SOX (Sry-type high-mobility group box) gene family. This gene family codes for transcription factors required for diverse developmental processes such as cardiac development, sex and neural determination [94].
Hereditary hemorrhagic telangiectasia (HHT) or Osler-Rendu-Weber disease is a vascular dysplasia leading to telangiectases and AVMs of skin, mucosa and viscera (lung, liver and brain). The mucosal complications involve epistaxis and GI bleeding. There are two types of the disease, HHT1 (MIM 187300) and HHT2 (MIM 600376). HHT1 patients have an earlier onset of epix-taxis and telengiectasis than those with HHT2, and only HHT1 patients have a high frequency of pulmonary AVMs. The inactivating mutations in the endoglin (ENG) gene, which encodes an accessory TGFβ receptor, were found to cause HTT1 [95]. HHT2 was caused by loss-of function mutations in the ACVRLK1 (ALK1) gene, which codes for activin receptor-like kinase type 1 [96]. Both proteins are involved in the TGFβ signaling pathway. A combined syndrome of juvenile polyposis and HHT (MIM 175050) was found to be associated with mutations in the MADH4 gene, which encodes SMAD4 [97]. This protein is an integral downstream effector of TGFβ signal transduction.
Another telangiectasia-related disease, Coats’ disease or retinal telangiectasis, was shown to be caused by a somatic mutation in the NDP gene, which encodes Norrie disease protein (Norrin) [98]. The disease is characterized by a defect of retinal vascular development, leading to vessel leakage, subretinal exudation and retinal detachment. The consequent retinal detachment often results in progressive visual loss. Norrie disease (MIM 310600), on the other hand, is an X-linked recessive disorder in which affected males are blind at birth or in early infancy. The ocular findings involve bilateral retinal folds, retinal detachment, vitreous hemorrhage and bilateral retrolental masses consisting of hemorrhagic vascular and glial tissue (vitreoretinal dysplasia). Patients also develop progressive sensorineural deafness and varying degrees of developmental delay. More than 100 different mutations of the NDP gene have been identified in Norrie disease, suggesting an angiogenic role for NDP [99]. It has been proposed that retinal telangiectasis is secondary to somatic mutation in the NDP gene [98]. Molecular modeling of NDP revealed a protein structure similar to that of TGFβ and other cysteine-knot growth factors [100].
The hereditary macular dystrophies are progressive degenerations of the central retina and a cause of irreversible visual loss. Among these disorders, Sorsby’s fundus dystrophy (SFD) (MIM 136900) is very similar to age-related macular degeneration (AMD), a major cause of blindness in the elderly population of Western countries. The bilateral central visual loss occurs during the fourth or fifth decade of life. Sorsby et al. described five families with a fundus dystrophy [101]. The dystrophy became manifest at about the age of 40 years; the earliest manifestations were color vision deficits and abnormal yellow-white deposits, followed by a central macular lesion with edema, hemorrhage and exudates. In subsequent years, atrophy with pigmentation of the central area and extension occurred peripherally. The choroidal vessels became exposed and appeared somewhat sclerotic. Within about 35 years after onset, the entire fundus was involved. The choroidal vessels disappeared by this stage, and the terminal picture was one of extensive choroidal atrophy. The disease is caused by mutations, likely inactivating, in the TIMP3 gene [102]. The gene codes for a protein called metalloproteinase-3, which is a matrix-bound inhibitor of matrix metalloproteinases (MMPs). MMPs play a major role in angiogenesis by degrading the ECM and activating growth factors through their degradative activity, thus facilitating EC migration. TIMP3 inhibits angiogenesis by blocking the binding of VEGF to its receptor VEGFR-2 [103].
Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) (MIM 125310) is thought to be the most common form of hereditary stroke disorder. It is characterized by recurrent transient ischemic attacks, strokes and vascular dementia. Other symptoms involve migraine, mostly with aura, psychiatric disturbance, cognitive decline and epilepsy. Arteriopathy is characterized by progressive degeneration of vascular SMCs of small arteries and the accumulation of granular osmiophilic material (GOM) and Notch3 protein within the cell surface of these cells. Notch3 is a member of Notch transmembrane receptors. Its expression is highly restricted to vascular SMCs. Mutations in the Notch3 gene were found to cause CADASIL [104]. However, the molecular mechanisms by which the mutant Notch3 proteins lead to the disease are still not clear.
The vascular form of Ehlers-Danlos syndrome (type IV) (MIM 130050) is characterized by joint and dermal manifestations and proneness to spontaneous rupture of bowel, intestine and large arteries (e.g. splenic, pulmonary, renal). Patients have also a striking facial appearance, easy bruising and translucent skin with visible veins. Some patients have cerebral vascular complications, including intracranial aneurysms, spontaneous carotid cavernous sinus fistulas and dissection of cervical arteries. The disease is caused by mutations in the COL3A1 gene, which encodes type III procollagen [105]. Collagen III is an important component of the arterial walls that provides tensile strength to the tissues.
Proteus syndrome (PS) is a complex hamartomatous disorder characterized by asymmetric and disproportionate overgrowth of body parts (macrocephaly, gigantism of hands and feet), connective tissue nevi, epidermal nevi, dysregulated adipose tissue and vascular malformations. Vascular malformations include vascular hamartomas and capillary-venous malformations [106]. Interestingly, germline mutations in the tumor suppressor gene PTEN (phosphatase, tensin homologue, deleted on chromosome TEN) were identified in up to 20% of PS cases (MIM 176920) and around 50% of Proteus-like syndrome (PSL) patients [107–110]. PTEN encodes a lipid phosphatase that mediates cell cycle arrest and apoptosis [107]. It was fist described in Cowden syndrome (CS) (MIM 158350), which is characterized by multiple hamartomas and a risk of breast, thyroid and endometrial carcinomas [111]. PTEN mutations are also associated with other syndromes (MIM 153480) that clinically overlap and are characterized by macrocephaly, lipomatosis, hemangiomata, arteriovenous malformations and developmental delay [112] (table 2).
Summary
Significant progress has been made in the molecular genetics studies of a number of vascular anomalies. Disease-causing or susceptibility genes have been identified for multiple cutaneous and mucosal venous malformations, lymphedema type I, capillary malformations associated with arteriovenous malformations, cerebral capillary malformations, glomuvenous malformations, hereditary hemorrhagic telangiectasia, some forms of lymphedema and telangiectasia, SFD, cerebral arteriopathy, vascular-type Ehlers-Danlos syndrome, Proteus and Proteus-like syndromes and some clinically overlapping syndromes associated with macrocephaly, lipomas and hemangiomata, and, finally, KTS (table 2). These findings make genetic testing possible for some patients and/or families with these diseases. Identification of the disease genes for vascular anomalies also offers interesting targets for investigating the molecular mechanisms involved in vascular morphogenesis, growth and development. The functional roles of TIE2, VEGFR-3 and AGGF1 in vasculogenesis and angiogenesis have been identified, but the clear functions of other vascular anomaly genes listed in table 2 remain to be identified. The functional studies of vascular anomaly genes may lead to the development of therapeutic options for treating these vascular malformations as well as more common diseases, including cancer, diabetic blindness, age-related macular degeneration, rheumatoid arthritis, psoriasis and other conditions with excessive angiogenesis, or coronary artery disease, stroke and delayed wound healing with insufficient angiogenesis or abnormal vessel regression. For example, our finding that AGGF1 is an angiogenic factor may facilitate the development of therapeutic angiogenesis, in which AGGF1 is delivered as a recombinant protein or as a transgene by adenovirus vectors to promote collateral growth of blood vessels for patients with ischemic heart disease or peripheral vascular diseases. Our finding that knockdown of AGGF1 expression blocks vessel tube formation may lead to the development of antiangiogenic therapies designed to block angiogenesis and to treat cancer and macular degeneration with excessive angiogenesis.
Acknowledgments
This work was supported by NIH grants R01 HL65630, R01 HL66251 and R01 HL73817 (to Q.W.) and an American Heart Association established Investigator award (to Q.W.).
References
- 1.Carmeliet P. Angiogenesis in health and disease. Nat Med. 2003;9:653–660. doi: 10.1038/nm0603-653. [DOI] [PubMed] [Google Scholar]
- 2.Bazzoni G, Dejana E. Endothelial cell-to-cell junctions: molecular organization and role in vascular home-ostasis. Physiol Rev. 2004;84:869–901. doi: 10.1152/physrev.00035.2003. [DOI] [PubMed] [Google Scholar]
- 3.Kahn HJ, Bailey D, Marks A. Monoclonal antibody D2-40, a new marker of lymphatic endothelium, reacts with Kaposi’s sarcoma and a subset of angiocarcinomas. Mod Pathol. 2002;15:434–440. doi: 10.1038/modpathol.3880543. [DOI] [PubMed] [Google Scholar]
- 4.Al-Rawi MA, Mansel RE, Jiang WG. Molecular and cellular mechanisms of lymphangiogenesis. Eur J Surg Oncol. 2005;31:117–121. doi: 10.1016/j.ejso.2004.08.015. [DOI] [PubMed] [Google Scholar]
- 5.Baron M. Induction of embryonic hematopoietic and endothelial stem/progenitor cells by hedgehog-mediated signals. Differentiation. 2001;68:175–185. doi: 10.1046/j.1432-0436.2001.680405.x. [DOI] [PubMed] [Google Scholar]
- 6.Choi K. The hemangioblast: a common progenitor of hematopoietic and endothelial cells. J Hematother Stem Cell Res. 2002;11:91–101. doi: 10.1089/152581602753448568. [DOI] [PubMed] [Google Scholar]
- 7.Tang DG, Conti CJ. Endothelial cell development, vasculogenesis, angiogenesis and tumor neovascularization: an update. Semin Thromb Hemost. 2004;30:109–117. doi: 10.1055/s-2004-822975. [DOI] [PubMed] [Google Scholar]
- 8.Lawson ND, Scheer N, Pham VN, Kim CH, Chitnis AB, Campos-Ortega JA, et al. Notch signaling is required for arterial-venous differentiation during embryonic vascular development. Development. 2001;128:3675–3683. doi: 10.1242/dev.128.19.3675. [DOI] [PubMed] [Google Scholar]
- 9.Shawber CJ, Kitajewski J. Notch function in the vasculature: insights from zebrafish, mouse and man. Bioessays. 2004;26:225–234. doi: 10.1002/bies.20004. [DOI] [PubMed] [Google Scholar]
- 10.Zhong TP, Childs S, Leu JP, Fishman MC. Gridlock signalling pathway fashions the first embryonic artery. Nature. 2001;414:216–220. doi: 10.1038/35102599. [DOI] [PubMed] [Google Scholar]
- 11.Wang HU, Chen ZF, Anderson DJ. Molecular distinction and angiogenic interaction between embryonic arteries and veins revealed by ephrin-B2 and its receptor Eph-B4. Cell. 1998;93:741–753. doi: 10.1016/s0092-8674(00)81436-1. [DOI] [PubMed] [Google Scholar]
- 12.Adams RH, Wilkinson GA, Weiss C, Diella F, Gale NW, Deutsch U, et al. Roles of ephrinB ligands and EphB receptors in cardiovascular development: demarcation of arterial/venous domains, vascular morphogenesis and sprouting angiogenesis. Genes Dev. 1999;13:295–306. doi: 10.1101/gad.13.3.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang XQ, Takakura N, Oike Y, Inada T, Gale NW, Yancopoulos GD, et al. Stromal cells expressing ephrin-B2 promote the growth and sprouting of ephrin-B2(+) endothelial cells. Blood. 2001;98:1028–1037. doi: 10.1182/blood.v98.4.1028. [DOI] [PubMed] [Google Scholar]
- 14.Carmeliet P. Mechanisms of angiogenesis and arteriogenesis. Nat Med. 2000;6:389–395. doi: 10.1038/74651. [DOI] [PubMed] [Google Scholar]
- 15.Pugh CW, Ratcliffe PJ. Regulation of angiogenesis by hypoxia: role of the HIF system. Nat Med. 2003;9:677–684. doi: 10.1038/nm0603-677. [DOI] [PubMed] [Google Scholar]
- 16.Scappaticci FA. Mechanisms and future directions for angiogenesis-based cancer therapies. J Clin Oncol. 2002;20:3906–3927. doi: 10.1200/JCO.2002.01.033. [DOI] [PubMed] [Google Scholar]
- 17.Jain RK. Molecular regulation of vessel maturation. Nat Med. 2003;9:685–693. doi: 10.1038/nm0603-685. [DOI] [PubMed] [Google Scholar]
- 18.Ferrara N, Carver-Moore K, Chen H, Dowd M, Lu L, O’Shea KS, et al. Heterozygous embryonic lethality induced by targeted inactivation of the VEGF gene. Nature. 1996;380:439–442. doi: 10.1038/380439a0. [DOI] [PubMed] [Google Scholar]
- 19.Shalaby F, Rossant J, Yamaguchi TP, Gertsenstein M, Wu XF, Breitman ML, et al. Failure of blood-island formation and vasculogenesis in Flk-1-deficient mice. Nature. 1995;376:62–66. doi: 10.1038/376062a0. [DOI] [PubMed] [Google Scholar]
- 20.Fong GH, Rossant J, Gertsenstein M, Breitman ML. Role of the Flt-1 receptor tyrosine kinase in regulating the assembly of vascular endothelium. Nature. 1995;376:66–70. doi: 10.1038/376066a0. [DOI] [PubMed] [Google Scholar]
- 21.Silvestre JS, Tamarat R, Ebrahimian TG, Le-Roux A, Clergue M, Emmanuel F, et al. Vascular endothelial growth factor-B promotes in vivo angiogenesis. Circ Res. 2003;93:114–123. doi: 10.1161/01.RES.0000081594.21764.44. [DOI] [PubMed] [Google Scholar]
- 22.Dumont DJ, Gradwohl G, Fong GH, Puri MC, Gertsenstein M, Auerbach A, et al. Dominant-negative and targeted null mutations in the endothelial receptor tyrosine kinase, tek, reveal a critical role in vasculogenesis of the embryo. Genes Dev. 1994;8:1897–1909. doi: 10.1101/gad.8.16.1897. [DOI] [PubMed] [Google Scholar]
- 23.Jones N, Dumont DJ. Tek/Tie2 signaling: new and old partners. Cancer Metastasis Rev. 2000;19:13–17. doi: 10.1023/a:1026555121511. [DOI] [PubMed] [Google Scholar]
- 24.Maisonpierre PC, Suri C, Jones PF, Bartunkova S, Wiegand SJ, Radziejewski, et al. Angiopoietin-2, a natural antagonist for Tie2 that disrupts in vivo angiogenesis. Science. 1997;277:55–60. doi: 10.1126/science.277.5322.55. [DOI] [PubMed] [Google Scholar]
- 25.Sato TN, Tozawa Y, Deutsch U, Wolburg-Buchholz K, Fujiwara Y, Gendron-Maguire M, et al. Distinct roles of the receptor tyrosine kinases Tie-1 and Tie-2 in blood vessel formation. Nature. 1995;376:70–74. doi: 10.1038/376070a0. [DOI] [PubMed] [Google Scholar]
- 26.Suri C, Jones PF, Patan S, Bartunkova S, Maisonpierre PC, Davis S, et al. Requisite role of angiopoietin-1, a ligand for the TIE2 receptor, during embryonic angiogenesis. Cell. 1996;87:1171–1180. doi: 10.1016/s0092-8674(00)81813-9. [DOI] [PubMed] [Google Scholar]
- 27.Suri C, McClain J, Thurston G, McDonald DM, Zhou H, Oldmixon EH, et al. Increased vascularization in mice overexpressing angiopoietin-1. Science. 1998;282:468–471. doi: 10.1126/science.282.5388.468. [DOI] [PubMed] [Google Scholar]
- 28.Jimenez B, Volpert OV, Crawford SE, Febbraio M, Silverstein RL, Bouck N. Signals leading to apoptosis-dependent inhibition of neovascularization by thrombospondin-1. Nat Med. 2000;6:41–48. doi: 10.1038/71517. [DOI] [PubMed] [Google Scholar]
- 29.Nor JE, Mitra RS, Sutorik MM, Mooney DJ, Castle VP, Polverini PJ. Thrombospondin-1 induces endothelial cell apoptosis and inhibits angiogenesis by activating the caspase death pathway. J Vasc Res. 2000;37:209–218. doi: 10.1159/000025733. [DOI] [PubMed] [Google Scholar]
- 30.Chavakis E, Dimmeler S. Regulation of endothelial cell survival and apoptosis during angiogenesis. Arterioscler Thromb Vasc Biol. 2002;22:887–893. doi: 10.1161/01.atv.0000017728.55907.a9. [DOI] [PubMed] [Google Scholar]
- 31.Volpert OV, Zaichuk T, Zhou W, Reiher F, Ferguson TA, Stuart PM, et al. Inducer-stimulated Fas targets activated endothelium for destruction by anti-angiogenic thrombospondin-1 and pigment epithelium-derived factor. Nat Med. 2002;8:349–357. doi: 10.1038/nm0402-349. [DOI] [PubMed] [Google Scholar]
- 32.Tonini T, Rossi F, Claudio PP. Molecular basis of angiogenesis and cancer. Oncogene. 2003;22:6549–6556. doi: 10.1038/sj.onc.1206816. [DOI] [PubMed] [Google Scholar]
- 33.Qi JH, Ebrahem Q, Moore N, Murphy G, Claesson-Welsh L, et al. A novel function for tissue inhibitor of metalloproteinases-3 (TIMP3): inhibition of angiogenesis by blockage of VEGF binding to VEGF receptor-2. Nat Med. 2003;9:407–415. doi: 10.1038/nm846. [DOI] [PubMed] [Google Scholar]
- 34.Scavelli C, Weber E, Agliano M, Cirulli T, Nico B, Vacca A, et al. Lymphatics at the crossroads of angiogenesis and lymphangiogenesis. J Anat. 2004;204:433–449. doi: 10.1111/j.0021-8782.2004.00293.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Taipale J, Makinen T, Arighi E, Kukk E, Karkkainen M, Alitalo K. Vascular endothelial growth factor receptor-3. Curr Top Microbiol Immunol. 1999;237:85–96. doi: 10.1007/978-3-642-59953-8_5. [DOI] [PubMed] [Google Scholar]
- 36.Wigle JT, Oliver G. Prox1 function is required for the development of the murine lymphatic system. Cell. 1999;98:769–778. doi: 10.1016/s0092-8674(00)81511-1. [DOI] [PubMed] [Google Scholar]
- 37.Jain RK, Padera TP. Development. Lymphatics make the break Science. 2003;299:209–210. doi: 10.1126/science.1081345. [DOI] [PubMed] [Google Scholar]
- 38.Yuan L, Moyon D, Pardanaud L, Breant C, Karkkainen MJ, Alitalo K, et al. Abnormal lymphatic vessel development in neuropilin 2 mutant mice. Development. 2002;129:4797–4806. doi: 10.1242/dev.129.20.4797. [DOI] [PubMed] [Google Scholar]
- 39.Kim H, Dumont DJ. Molecular mechanisms in lymphangiogenesis: model systems and implications in human disease. Clin Genet. 2003;64:282–292. doi: 10.1034/j.1399-0004.2003.00152.x. [DOI] [PubMed] [Google Scholar]
- 40.Lohela M, Saaristo A, Veikkola T, Alitalo K. Lymphangiogenic growth factors, receptors and therapies. Thromb Haemost. 2003;90:167–184. doi: 10.1160/TH03-04-0200. [DOI] [PubMed] [Google Scholar]
- 41.Mulliken JB, Glowacki J. Hemangiomas and vascular malformations in infants and children: a classification based on endothelial characteristics. Plast Reconstr Surg. 1982;69:412–422. doi: 10.1097/00006534-198203000-00002. [DOI] [PubMed] [Google Scholar]
- 42.Mulliken JB. Cutaneous vascular anomalies. Semin Vasc Surg. 1993;6:204–218. [PubMed] [Google Scholar]
- 43.Enjolras O, Mulliken JB. Vascular tumors and vascular malformations (new issues) Adv Dermatol. 1997;13:375–423. [PubMed] [Google Scholar]
- 44.Vikkula M, Boon LM, Mulliken JB, Olsen BR. Molecular basis of vascular anomalies. Trends Cardiovasc Med. 1998;8:281–292. doi: 10.1016/s1050-1738(98)00024-3. [DOI] [PubMed] [Google Scholar]
- 45.Klippel M, Trenaunay P. Du naevus variqueux osteo-hypertrophique. Arch Gen Med. 1900;185:641–672. [Google Scholar]
- 46.Berry SA, Peterson C, Mize W, Bloom K, Zachary C, Blasco P, et al. Klippel-Trenaunay syndrome. Am J Med Genet. 1998;79:319–326. [PubMed] [Google Scholar]
- 47.Jacob AG, Driscoll DJ, Shaughnessy WJ, Stanson AW, Clay RP, Gloviczki P. Klippel-Trenaunay syndrome: spectrum and management. Mayo Clin Proc. 1998;73:28–36. doi: 10.1016/S0025-6196(11)63615-X. [DOI] [PubMed] [Google Scholar]
- 48.Cohen MM. Klippel-Trenaunay syndrome. Am J Med Genet. 2000;93:171–175. doi: 10.1002/1096-8628(20000731)93:3<171::aid-ajmg1>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 49.Lindenauer SM. The Klippel-Trenaunay syndrome: varicosity, hypertrophy and hemangioma with no arteriovenous fistula. Ann Surg. 1965;162:303–314. doi: 10.1097/00000658-196508000-00023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gloviczki P, Stanson AW, Stickler GB, Johnson CM, Toomey BJ, Meland NB, et al. Klippel-Trenaunay syndrome: the risks and benefits of vascular interventions. Surgery. 1991;110:469–479. [PubMed] [Google Scholar]
- 51.Servelle M. Klippel and Trenaunay’s syndrome. 768 operated cases. Ann Surg. 1985;201:365–373. doi: 10.1097/00000658-198503000-00020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gorenstein A, Katz S, Schiller M. Congenital angiodysplasia of the superficial venous system of the lower extremities in children. Ann Surg. 1988;207:213–218. doi: 10.1097/00000658-198802000-00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gorenstein A, Shifrin E, Gordon RL, Katz S, Schiller M. Congenital aplasia of the deep veins of lower extremities in children: the role of ascending functional phlebography. Surgery. 1986;99:414–420. [PubMed] [Google Scholar]
- 54.Paes EH, Vollmar JF. Aneurysma transformation in congenital venous angiodysplasias in lower extremities. Int Angiol. 1990;9:90–96. [PubMed] [Google Scholar]
- 55.Aelvoet GE, Jorens PG, Roelen LM. Genetic aspects of the Klippel-Trenaunay syndrome. Br J Dermatol. 1992;126:603–607. doi: 10.1111/j.1365-2133.1992.tb00107.x. [DOI] [PubMed] [Google Scholar]
- 56.Ceballos-Quintal JM, Pinto-Escalante D, Castillo-Zapata I. A new case of Klippel-Trenaunay-Weber (KTW) syndrome: evidence of autosomal dominant inheritance. Am J Med Genet. 1996;63:426–427. doi: 10.1002/(SICI)1096-8628(19960614)63:3<426::AID-AJMG2>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 57.Lorda-Sanchez I, Prieto L, Rodriguez-Pinilla E, Martinez-Frias ML. Increased parental age and number of pregnancies in Klippel-Trenaunay-Weber syndrome. Ann Hum Genet. 1998;62:235–239. doi: 10.1046/j.1469-1809.1998.6230235.x. [DOI] [PubMed] [Google Scholar]
- 58.Whelan AJ, Watson MS, Porter FD, Steiner RD. Klippel-Trenaunay-Weber syndrome associated with a 5:11 balanced translocation. Am J Med Genet. 1995;59:492–494. doi: 10.1002/ajmg.1320590416. [DOI] [PubMed] [Google Scholar]
- 59.Wang Q, Timur AA, Szafranski P, Sadgephour A, Jurecic V, Cowell J, et al. Identification and molecular characterization of de novo translocation t(8;14)(q22.3;q13) associated with a vascular and tissue overgrowth syndrome. Cytogenet Cell Genet. 2001;95:183–188. doi: 10.1159/000059343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Timur AA, Sadgephour A, Graf M, Schwartz S, Libby ED, Driscoll DJ, et al. Identification and molecular characterization of a de novo supernumerary ring chromosome 18 in a patient with Klippel-Trenaunay syndrome. Ann Hum Genet. 2004;68:353–361. doi: 10.1046/j.1529-8817.2004.00095.x. [DOI] [PubMed] [Google Scholar]
- 61.Tian XL, Kadaba R, You SA, Liu M, Timur AA, Yang L, et al. Identification of an angiogenic factor that when mutated causes susceptibility to Klippel-Trenaunay syndrome. Nature. 2004;427:640–645. doi: 10.1038/nature02320.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Durocher D, Jackson SP. The FHA domain. FEBS Lett. 2002;513:58–66. doi: 10.1016/s0014-5793(01)03294-x. [DOI] [PubMed] [Google Scholar]
- 63.Guglielmi B, Werner M. The yeast homolog of human PinX1 is involved in rRNA and small nucleolar RNA maturation, not in telemore elongation inhibition. J Biol Chem. 2002;277:35712–35719. doi: 10.1074/jbc.M205526200. [DOI] [PubMed] [Google Scholar]
- 64.Callebaut I, Mornon JP. OCRE: a novel domain made of imperfect, aromatic-rich octamer repeats. Bioinformatics. 2004 doi: 10.1093/bioinformatics/bti065. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 65.Baskerville PA, Ackroyd JS, Browse NL. The etiology of the Klippel-Trenaunay syndrome. Ann Surg. 1985;202:624–627. doi: 10.1097/00000658-198511000-00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fontana A, Olivetti L. Peripheral MR angiography of Klippel-Trenaunay syndrome. Cardiovasc Intervent Radiol. 2004;27:297–299. doi: 10.1007/s00270-003-0196-5. [DOI] [PubMed] [Google Scholar]
- 67.Happle R. Cutaneous manifestation of lethal genes. Hum Genet. 1986;72:280. doi: 10.1007/BF00291899. [DOI] [PubMed] [Google Scholar]
- 68.Happle R. Lethal genes surviving by mosaicism: a possible explanation for sporadic birth defects involving the skin. J Am Acad Dermatol. 1987;16:899–906. doi: 10.1016/s0190-9622(87)80249-9. [DOI] [PubMed] [Google Scholar]
- 69.Happle R. Klippel-Trenaunay syndrome: is it a para-dominant trait? Br J Dermatol. 1993;128:465–466. doi: 10.1111/j.1365-2133.1993.tb00214.x. [DOI] [PubMed] [Google Scholar]
- 70.Vikkula M, Boon LM, Carraway KL, III, Calvert JT, Diamonti AJ, Goumnerov B, et al. Vascular dysmorphogenesis caused by an activating mutation in the receptor tyrosine kinase TIE2. Cell. 1996;87:1181–1190. doi: 10.1016/s0092-8674(00)81814-0. [DOI] [PubMed] [Google Scholar]
- 71.Karkkainen MJ, Ferrell RE, Lawrence EC, Kimak MA, Levinson KL, McTigue MA, et al. Missense mutations interfere with VEGFR-3 signalling in primary lymphoedema. Nat Genet. 2000;25:153–159. doi: 10.1038/75997. [DOI] [PubMed] [Google Scholar]
- 72.Jacobs AH, Walton RG. The incidence of birthmarks in the neonate. Pediatrics. 1976;58:218–222. [PubMed] [Google Scholar]
- 73.Eerola I, Boon LM, Mulliken JB, Burrows PE, Dompmartin A, Watanabe S, et al. Capillary malformation arteriovenous malformation, a new clinical and genetic disorder caused by RASA1 mutations. Am J Hum Genet. 2003;73:1240–1249. doi: 10.1086/379793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gibbs JB, Marshall MS, Scolnick EM, Dixon RA, Vogel US. Modulation of guanine nucleotides bound to Ras in NIH3T3 cells by oncogenes, growth factors and the GTPase activating protein (GAP) J Biol Chem. 1990;265:20437–20442. [PubMed] [Google Scholar]
- 75.Clark GJ, Quilliam LA, Hisaka MM, Der CJ. Differential antagonism of Ras biological activity by catalytic and Src homology domains of Ras GTPase activation protein. Proc Natl Acad Sci USA. 1993;90:4887–4891. doi: 10.1073/pnas.90.11.4887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Settleman J, Narasimhan V, Foster LC, Weinberg RA. Molecular cloning of cDNAs encoding the GAP-associated protein p190: implications for a signaling pathway from ras to the nucleus. Cell. 1992;69:539–549. doi: 10.1016/0092-8674(92)90454-k. [DOI] [PubMed] [Google Scholar]
- 77.Caron E, Self AJ, Hall A. The GTPase Rap1 controls functional activation of macrophage integrin alpha-Mbeta2 by LPS and other inflammatory mediators. Curr Biol. 2000;10:974–978. doi: 10.1016/s0960-9822(00)00641-2. [DOI] [PubMed] [Google Scholar]
- 78.Reedquist KA, Ross E, Koop EA, Wolthuis RM, Zwartkruis FJ, van KY, et al. The small GTPase, Rap1, mediates CD31-induced integrin adhesion. J Cell Biol. 2000;148:1151–1158. doi: 10.1083/jcb.148.6.1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Parkes Weber F. Angioma formation in connection with hypertrophy of limbs and hemihypertrophy. Br J Dermatol. 1907;19:231. [Google Scholar]
- 80.Mulliken JB, Young AE, editors. W. B. Saunders; Philadelphia: 1988. Vascular Birthmarks: Hemangiomas and Vascular Malformations. [Google Scholar]
- 81.Cohen MM., Jr Vasculogenesis, angiogenesis, hemangiomas and vascular malformations. Am J Med Genet. 2002;108:265–274. doi: 10.1002/ajmg.10260. [DOI] [PubMed] [Google Scholar]
- 82.Laberge-Le CS, Jung HH, Labauge P, Houtteville JP, Lescoat C, Cecillon M, et al. Truncating mutations in CCM1, encoding KRIT1, cause hereditary cavernous angiomas. Nat Genet. 1999;23:189–193. doi: 10.1038/13815. [DOI] [PubMed] [Google Scholar]
- 83.Sahoo T, Johnson EW, Thomas JW, Kuehl PM, Jones TL, Dokken CG, et al. Mutations in the gene encoding KRIT1, a Krev-1/rap1a binding protein, cause cerebral cavernous malformations (CCM1) Hum Mol Genet. 1999;8:2325–2333. doi: 10.1093/hmg/8.12.2325. [DOI] [PubMed] [Google Scholar]
- 84.Serebriiskii I, Estojak J, Sonoda G, Testa JR, Golemis EA. Association of Krev-1/rap1a with Krit1, a novel ankyrin repeat-containing protein encoded by a gene mapping to 7q21-22. Oncogene. 1997;15:1043–1049. doi: 10.1038/sj.onc.1201268. [DOI] [PubMed] [Google Scholar]
- 85.Kitayama H, Sugimoto Y, Matsuzaki T, Ikawa Y, Noda M. A ras-related gene with transformation suppressor activity. Cell. 1989;56:77–84. doi: 10.1016/0092-8674(89)90985-9. [DOI] [PubMed] [Google Scholar]
- 86.Zawistowski JS, Serebriiskii IG, Lee MF, Golemis EA, Marchuk DA. KRIT1 association with the integrin-binding protein ICAP-1: a new direction in the elucidation of cerebral cavernous malformations (CCM1) pathogenesis. Hum Mol Genet. 2002;11:389–396. doi: 10.1093/hmg/11.4.389. [DOI] [PubMed] [Google Scholar]
- 87.Eerola I, Plate KH, Spiegel R, Boon LM, Mulliken JB, Vikkula M. KRIT1 is mutated in hyperkeratotic cutaneous capillary-venous malformation associated with cerebral capillary malformation. Hum Mol Genet. 2000;9:1351–1355. doi: 10.1093/hmg/9.9.1351. [DOI] [PubMed] [Google Scholar]
- 88.Denier C, Goutagny S, Labauge P, Krivosic V, Arnoult M, Cousin A, et al. Mutations within the MGC4607 gene cause cerebral cavernous malformations. Am J Hum Genet. 2004;74:326–337. doi: 10.1086/381718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Liquori CL, Berg MJ, Siegel AM, Huang E, Zawistowski JS, Stoffer T, et al. Mutations in a gene encoding a novel protein containing a phosphotyrosine-binding domain cause type 2 cerebral cavernous malformations. Am J Hum Genet. 2003;73:1459–1464. doi: 10.1086/380314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bergametti F, Denier C, Labauge P, Arnoult M, Boetto S, Clanet M, et al. Mutations within the programmed cell death 10 gene cause cerebral cavernous malformations. Am J Hum Genet. 2004;76:42–51. doi: 10.1086/426952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Brouillard P, Boon LM, Mulliken JB, Enjolras O, Ghassibe M, Warman ML, et al. Mutations in a novel factor, glomulin, are responsible for glomuvenous malformations (‘glomangiomas’) Am J Hum Genet. 2002;70:866–874. doi: 10.1086/339492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Fang J, Dagenais SL, Erickson RP, Arlt MF, Glynn MW, Gorski JL, et al. Mutations in FOXC2 (MFH-1), a forkhead family transcription factor, are responsible for the hereditary lymphedema-distichiasis syndrome. Am J HumGenet. 2000;67:1382–1388. doi: 10.1086/316915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Doffinger R, Smahi A, Bessia C, Geissmann F, Feinberg J, Durandy A, et al. X-linked anhidrotic ectodermal dysplasia with immunodeficiency caused by impaired NF-kappaB signaling. Nat Genet. 2001;27:277–285. doi: 10.1038/85837. [DOI] [PubMed] [Google Scholar]
- 94.Irrthum A, Devriendt K, Chitayat D, Matthijs G, Glade C, Steijlen PM, et al. Mutations in the transcription factor gene SOX18 underlie recessive and dominant forms of hypotrichosis-lymphedema-telangiectasia. Am J Hum Genet. 2003;72:1470–1478. doi: 10.1086/375614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.McAllister KA, Grogg KM, Johnson DW, Gallione CJ, Baldwin MA, Jackson CE, et al. Endoglin, a TGF-beta binding protein of endothelial cells, is the gene for hereditary haemorrhagic telangiectasia type 1. Nat Genet. 1994;8:345–351. doi: 10.1038/ng1294-345. [DOI] [PubMed] [Google Scholar]
- 96.Johnson DW, Berg JN, Baldwin MA, Gallione CJ, Marondel I, Yoon SJ, et al. Nat Genet. Vol. 13. 1996. Mutations in the activin receptor-like kinase 1 gene in hereditary haemorrhagic telangiectasia type 2; pp. 189–195. [DOI] [PubMed] [Google Scholar]
- 97.Gallione CJ, Repetto GM, Legius E, Rustgi AK, Schelley SL, Tejpar S, et al. A combined syndrome of juvenile polyposis and hereditary haemorrhagic telangiectasia associated with mutations in MADH4 (SMAD4) Lancet. 2004;363:852–859. doi: 10.1016/S0140-6736(04)15732-2. [DOI] [PubMed] [Google Scholar]
- 98.Black GC, Perveen R, Bonshek R, Cahill M, Clayton-Smith J, Lloyd IC, et al. Coats’ disease of the retina (unilateral retinal telangiectasis) caused by somatic mutation in the NDP gene: a role for norrin in retinal angiogenesis. Hum Mol Genet. 1999;8:2031–2035. doi: 10.1093/hmg/8.11.2031. [DOI] [PubMed] [Google Scholar]
- 99.Black G, Redmond RM. The molecular biology of Norrie’s disease. Eye. 1994;8:491–496. doi: 10.1038/eye.1994.124. [DOI] [PubMed] [Google Scholar]
- 100.Meitinger T, Meindl A, Bork P, Rost B, Sander C, Haasemann M, et al. Molecular modelling of the Norrie disease protein predicts a cystine knot growth factor tertiary structure. Nat Genet. 1993;5:376–380. doi: 10.1038/ng1293-376. [DOI] [PubMed] [Google Scholar]
- 101.Sorsby A. A fundus dystrophy with unusual features (late onset and dominant inheritance of a central retinal lesion showing oedema, haemorrhage and exudates developing into generalized choroidal atrophy with massive pigment proliferation) Brit J Opthal. 1949;33:67–97. doi: 10.1136/bjo.33.2.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Weber BH. Mutations in the tissue inhibitor of metalloproteinases-3 (TIMP3) in patients with Sorsby’s fundus dystrophy. Nat Genet. 1994;8:352–356. doi: 10.1038/ng1294-352. [DOI] [PubMed] [Google Scholar]
- 103.Qi JH, Ebrahem Q, Moore N, Murphy G, Claesson-Welsh L, Bond M, et al. A novel function for tissue inhibitor of metalloproteninases-3 (TIMP3): inhibition of angiogenesis by blockage of VEGF binding to VEGF receptor-2. Nat Med. 2003;9:407–415. doi: 10.1038/nm846. [DOI] [PubMed] [Google Scholar]
- 104.Joutel A, Corpechot C, Ducros A, Vahedi K, Chabriat H, Mouton P, et al. Notch3 mutations in CADASIL, a hereditary adult-onset condition causing stroke and dementia. Nature. 1996;383:707–710. doi: 10.1038/383707a0. [DOI] [PubMed] [Google Scholar]
- 105.Superti-Furga A, Gugler E, Gitzelmann R, Steinmann B. Ehlers-Danlos syndrome type IV: a multi-exon deletion in one of the two COL3A1 alleles affecting structure, stability and processing of type III procollagen. J Biol Chem. 1988;263:6226–6232. [PubMed] [Google Scholar]
- 106.Hoeger PH, Martinez A, Maerker J, Harper JI. Vascular anomalies in Proteus syndrome. Clin Exp Dermatol. 2004;29:222–230. doi: 10.1111/j.1365-2230.2004.01513.x. [DOI] [PubMed] [Google Scholar]
- 107.Eng C. PTEN: one gene, many syndromes. Hum Mutat. 2003;22:183–198. doi: 10.1002/humu.10257. [DOI] [PubMed] [Google Scholar]
- 108.Zhou XP, Marsh DJ, Hampel H, Mulliken JB, Gimm O, Eng C. Germline and germline mosaic PTEN mutations associated with a Proteus-like syndrome of hemi-hypertrophy, lower limb asymmetry, arteriovenous malformations and lipomatosis. Hum Mol Genet. 2000;9:765–768. doi: 10.1093/hmg/9.5.765. [DOI] [PubMed] [Google Scholar]
- 109.Zhou X, Hampel H, Thiele H, Gorlin RJ, Hennekam RC, Parisi M, et al. Association of germline mutation in the PTEN tumour suppressor gene and Proteus and Proteus-like syndromes. Lancet. 2001;358:210–211. doi: 10.1016/s0140-6736(01)05412-5. [DOI] [PubMed] [Google Scholar]
- 110.Smith JM, Kirk EP, Theodosopoulos G, Marshall GM, Walker J, Rogers M, et al. Germline mutation of the tumour suppressor PTEN in Proteus syndrome. J Med Genet. 2002;39:937–940. doi: 10.1136/jmg.39.12.937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Nelen MR, van Staveren WC, Peeters EA, Hassel MB, Gorlin RJ, Hamm H, et al. Germline mutations in the PTEN/MMAC1 gene in patients with Cowden disease. Hum Mol Genet. 1997;6:1383–1387. doi: 10.1093/hmg/6.8.1383. [DOI] [PubMed] [Google Scholar]
- 112.Marsh DJ, Dahia PL, Zheng Z, Liaw D, Parsons R, Gorlin RJ, et al. Germline mutations in PTEN are present in Bannayan-Zonana syndrome. Nat Genet. 1997;16:333–334. doi: 10.1038/ng0897-333. [DOI] [PubMed] [Google Scholar]