

Abstract
In traumatic brain injury (TBI), cellular loss from initial impact as well as secondary neurodegeneration leads to increased cholesterol and lipid debris at the site of injury. Cholesterol accumulation in the periphery can trigger inflammatory mechanisms while cholesterol clearance may be anti-inflammatory. Here we investigated whether TBI altered the regulation of cholesterol 24S-hydroxylase (Cyp46), an enzyme that converts cholesterol to the more hydrophilic 24S-hydroxycholesterol. We examined by Western blot and immunohistochemistry changes in Cyp46 expression following fluid percussion injury. Under normal conditions, most Cyp46 was present in neurons, with very little measurable in glia. Cyp46 levels were significantly increased at 7 days post-injury, and cell type specific analysis at 3 days post-injury showed a significant increase in levels of Cyp46 (84%) in microglia. Since 24-hydroxycholesterol induces activation of genes through the liver X receptor (LXR), we examined protein levels of ATP-binding cassette transporter A1 and apolipoprotein E, two LXR regulated cholesterol homeostasis proteins. Apolipoprotein E and ATP-binding cassette transporter A1 were increased at 7 days post-injury, indicating that increased LXR activity coincided with increased Cyp46 levels. We found that activation of primary rat microglia by LPS in vitro caused increased Cyp46 levels. These data suggest that increased microglial Cyp46 activity is part of a system for removal of damaged cell membranes post-injury, by conversion of cholesterol to 24-hydroxycholesterol and by activation of LXR-regulated gene transcription.
Keywords: glia cell response to injury, neurodegenerative disorders, traumatic brain injury
Introduction
Traumatic brain injury (TBI) is the cause of more than 50,000 deaths annually in the United States (Langlois et al., 2004). Although many patients survive TBI, it is estimated that 5 million people in the United States live with long-term disabilities resulting from TBI (NINDS, 2002). Long-term effects may include epilepsy, as well as disabilities in cognition, sensory processing, verbal and non-verbal communication, and mental health (NINDS, 2002). In addition, TBI increases the risk, later in life, of neurodegenerative diseases such as Alzheimer’s disease (AD) (Plassman et al., 2000) and Parkinson’s disease (PD) (Goldman et al., 2006). Neurodegeneration beyond the initial impact may be due to the inflammatory processes following TBI (Hoane et al., 2007; Laskowitz et al., 2007; Pan et al., 2007; Zhang et al., 2008), since inflammation contributes to the chronic neurodegeneration seen in AD (Akiyama et al., 2000; Bogdanovic et al., 2001; Fassbender et al., 2001; McGeer and McGeer, 2001; Bonotis et al., 2008; Rojo et al., 2008) and PD (McGeer and McGeer, 2008; Reynolds et al., 2008; Bar-On et al., 2008; Kim et al., 2008).
In the periphery, high cholesterol levels are known to trigger inflammatory mechanisms (Hansson et al., 2006), and cholesterol clearance mechanisms are thought to be anti-inflammatory (Ansell et al., 2007; Navab et al., 2007; Sanossian et al., 2007). In the central nervous system (CNS), cholesterol homeostasis is poorly understood. Although the brain is very cholesterol-rich, cholesterol is not imported from the periphery, but rather synthesized in situ (Jurevics and Morell, 1995; Turley et al., 1996, 1998; Lund et al., 2003; Xie et al., 2003). Cholesterol removal from the brain can occur through several mechanisms. Cholesterol 24S-hydroxylase (Cyp46) is a endoplasmic reticulum associated enzyme responsible for converting cholesterol into 24S-hydroxycholesterol (Lund et al., 1999; Ramirez et al., 2008), which is removed from the CNS to the periphery. 24S-Hydroxycholesterol is also an activator of the nuclear transcription factor liver X receptor (LXR) (Lehmann et al., 1997). LXR activation induces the upregulation of genes involved in cholesterol efflux, including the ATP-binding cassette transporter A1 (ABCA1) (Fukumoto et al., 2002) and apolipoprotein E (ApoE) (Whitney et al., 2002; Liang et al., 2004). These multiple roles of 24S-hydroxycholesterol emphasize the importance of Cyp46 in maintaining cholesterol homeostasis at both the cellular level and in the brain as a whole.
In both humans and rodents, Cyp46 mRNA is primarily found in the brain with trace amounts found in testis (Lund et al., 1999; Nishimura et al., 2003). Immunostaining of cortex and cerebellum has indicated that Cyp46 is mainly expressed in neurons (Lund et al., 1999; Ramirez et al., 2008). Expression of Cyp46 is first seen at birth and increases rapidly; then it remains stable throughout adulthood in both human and rodent (Lund et al., 1999; Ohyama et al., 2006). The expression of Cyp46 is not affected by a variety of hormones or oxysterols, including 24S-hydroxycholesterol, or by the inhibition of cholesterol synthesis via statins or knockout of the cholesterol synthesis enzyme delta 24-reductase (Ohyama et al., 2006). In contrast, Cyp46 promoter activity in vitro was induced by oxidative stress and a dexamethasone/interleukin-6 treatment (Ohyama et al., 2006).
TBI is an acute form of cellular injury that may necessitate a redistribution of cholesterol due to damage to neuronal membranes and the proliferation of glial cells (Cernak et al., 2004; Di Giovanni et al., 2005). Following TBI, patients show an increase in cholesterol in the CSF (Kay et al., 2003). This excess cholesterol is likely due to injury-induced neuronal damage and the resulting membrane debris (Faden, 1996; Gasparovic et al., 2001; Kamada et al., 2003). Cyp46 conversion of cholesterol to 24S-hydroxycholesterol is a possible mechanism by which this excess cholesterol could be cleared from the extracellular space. Both ABCA1 and ApoE, cholesterol homeostasis factors downstream of Cyp46 activity, have been shown to increase at the site of acute brain injury (Poirier et al., 1991; Page et al., 1998; Fukumoto et al., 2002). Here we examined Cyp46 expression in response to a moderate TBI model that has been well characterized and contains many of the characteristics of moderate TBI found in clinical settings (Natale et al., 2003; Cernak et al., 2004; Di Giovanni et al., 2005). We found that Cyp46 expression was upregulated at the site of injury, primarily in activated microglia.
Methods
Animals
Twenty-four male Sprague-Dawley rats were used in these experiments. All protocols involving animals were approved by the Georgetown University Institutional Animal Use and Care Committee, and were in compliance with the standards stated in the Committee on Care and the Use of Laboratory Animals of the Institution of Laboratory Resources DHEW Publication (no. [NIH] 85-23 1985). The CYP46 knock-out mouse was generated by Dr. David Russell and has been previously described (Lund et al., 2003). A targeting vector was used to delete exon 1 of the Cyp46 gene. CYP46 knockout and wild-type background (C57Bl/6J;129S6/SvEv) control whole brain tissue homogenized in RIPA was generously provided by Dr. Russell.
Rat Lateral Fluid Percussion Trauma Model
This model has been previously described in detail (Natale et al., 2003). Briefly, rats were anesthetized with sodium pentobarbital and intubated. A 5-mm craniotomy was created between the lambda and bregma sutures over the left parietal cortex where a female leur-loc was cemented in place. An isotonic saline-filled fluid percussion device with a 5-mm tube was attached by means of a male leur-loc fitting. A brief 2.5-atmosphere pressure pulse was given when a pendulum struck a piston at the opposite end of the device. This procedure led to moderate brain injury of parietal cortex as previously defined (Faden et al., 2003). Sham animals received a similarly located craniotomy but no percussion injury. Either 3 or 7 days after injury or sham surgery, animals were anesthetized with sodium pentobarbital. Animals used for immunohistochemistry were sacrificed following perfusion with isotonic saline and then 4% paraformaldehyde (PFA). These brains were frozen and sectioned coronally at 16 μm. There were three animals in each group. Separate animals (3 or 7 days post-surgery [sham] or post-injury) were sacrificed for Western blot. These animals were perfused with saline. Fresh brains were removed, and the cortex was dissected out. Parietal cortex on the side of injury (or the sham surgery) was isolated and homogenized in RIPA buffer. There were three animals per group dedicated to this Western blot method. In total, there were six animals in each group, with three being dedicated for immunohistochemisty and three for Western blot.
Cell culture
Primary rat neurons
Hippocampal neurons were cultured from embryonic day 18–19 Sprague-Dawley rats at 150 cells/mm2 as described previously (Pak et al., 2001). Cells were plated on poly-D-lysine (30 μg/mL) and laminin (2 μg/mL) coated coverslips. Cells were maintained in neurobasal media (Gibco) with B27 (Gibco), 0.5 mM glutamine, and 12.5 μM glutamate until 9 days in vitro (DIV 9) or DIV 16.
Primary rat microglia
Cortical and midbrain tissue was taken from P2 rats and dissociated by pipetting in Leibowitz media (Gibco) containing 0.1% bovine serum albumin (BSA), 1000 unit/mL penicillin, and 1000 μg/mL streptomycin (1× pen/strep; Gibco). Cells were poured through a 100-μm mesh, spun at 2000 rpm for 5 min, and resuspended in growth media (DMEM high-glucose media [Gibco] containing 2mM L-glutamine [Gibco], 1mM sodium pyruvate [Gibco], 1× pen/strep [Gibco], and 10% fetal calf serum [FCS; Bio Whittaker]). Cells were plated on uncoated flasks and incubated for 1 week. Media was changed at day 8, 10, and 12 post-plating. On day 15 post-plating, flasks were paraffin sealed and shaken at 100 rpm for 1 h at 37°C. Unattached cells were collected and spun at 2000 rpm for 5 min. Microglia in the cell pellet were resuspended in growth media (DMEM high-glucose media [Gibco] with 2mM L-glutamine, 1mM sodium pyruvate, 1× pen/strep, and 10% fetal horse serum [FHS; Sigma]). Microglia were plated at 200,000 cells per milliliter onto uncoated coverslips. Purity of cultures was confirmed to be greater than 98% by Ox-42 immunostaining.
Lipopolysaccharide activation of microglia
Lipopolysaccharide (LPS; Calbiochem) stock solution was dissolved in distilled water at a concentration of 5 μg/mL. At 24 h after plating cells, LPS was added to the media at a final concentration of 50 ng/mL. At 24 h after treatment, cells were fixed in 4% PFA. These LPS treatment experiments were done in triplicate and were repeated on three separate microglial cell isolations.
Cyp46 analysis
The human Cyp46 cDNA as well as wild-type and Cyp46 knockout mouse brain lysate were the generous gifts of Dr. David W. Russell (University of Texas Southwestern Medical Center). Cyp46 cDNA was transferred into the pExchange-6a vector (Stratagene), which contains a myc-tag; identity to the published sequence (AF094480) was confirmed by sequencing. COS-7 cells were either plated onto coverslips for immunohistochemistry or onto six-well plates for Western blot and maintained in Opti-Mem media (Gibco) containing 10% fetal bovine serum (FBS; Gibco). When cells were 80% confluent, they were transfected with Fugene 6 (Roche) according to manufacturer’s directions, with either the myc-tagged human Cyp46 sequence or the pExchange-6a vector as control. The polyclonal anti-Cyp46 antibody (Nkk2) was produced in the rabbit against the sequence GKDWVQRRREALKRGED, which are amino acids 254–270 of the mouse Cyp46 sequence. This antibody was the generous gift of Dr. Suzana Petanceska (Nathan Kline Institute).
Western blot
Cells in culture were washed in phosphate-buffered saline (PBS) and then lysed in RIPA buffer (Pierce Biotechnology) with protease inhibitor (Roche) for protein extraction for 10 min on ice. Lysates were spun at 14,000 rpm for 10 min at room temperature (RT). For rat cortical injury or sham tissue, cortex at the location of the procedure was dissected out and homogenized in RIPA containing protease inhibitor cocktail. Lysates were measured for protein concentration using the Bradford method according to manufacturer’s directions (Biorad). Lysates were mixed 1:1 with loading buffer (Biorad), heated for 5 min at 95°C, spun at 14,000 rpm for 1 min and loaded at 20 μg/well onto polyacrylamide gels. Protein was then transferred onto PVDF membrane and blocked for 1 h at RT in 5% milk in PBS. Membranes were probed with anti-Cyp46 antibody (1:1000), ApoE antibody (Abcam, 1:1000), or ABCA1 antibody (Novus, 1:1000) overnight at 4°C. Membranes were washed three times for 10 min in PBS and then incubated with goat anti-rabbit HRP tagged antibody at 1:10,000 dilution. Membranes were washed three times for 10 min at RT and incubated with DURA (Pierce) for 5 min and exposed to Kodak MR film. Membranes were reprobed with anti-beta-actin antibody (1:5000) to control for protein loading. Bands were quantified using Scion Image. Local background levels were subtracted and levels were adjusted for minor variations in loading controls.
Analysis of variance (ANOVA) statistical analysis was performed using Graphpad Prism 4. All p values were calculated using the Newman-Keuls Multiple Comparison Test.
Immunohistochemistry
Coronal sections were rinsed in PBS twice and incubated in 0.03% hydrogen peroxide for 30 min. Sections were washed 10 times in PBS and blocked 1 h in 10% FBS (Gibco) in PBS with 0.01% Triton X-100 (Fisher; PBST-100). Slides were incubated overnight with anti-Cyp46 antibody (1:1000). DAB staining was performed using the rabbit ABC kit (Vector Lab). All washes were done 10 times with PBST-100 unless specified otherwise. Slides were washed and incubated for 3 h at RT with anti-rabbit antibody (1:1000). Slides were washed, incubated with AB solution for 30 min, washed again, and incubated with DAB solution for 5 min. Finally, they were washed with PBST-100 and then 10 times in PBS. Slides were allowed to dry overnight and then mounted with Permount (Fisher Scientific). Images were taken with a Zeiss Axiophot Microscope.
Fluorescence immunostaining
Cells on coverslips were fixed in 4% PFA in PBS for 10 min at RT, rinsed twice in PBS, and stored at 4°C until further use. Rat brain sections (16 μm) or fixed cells were incubated in PBST-100 for 10 min at RT, washed 10 times with PBS, and blocked for 1 h at RT in 10% FBS plus 0.01% sodium azide. Tissue was incubated overnight at 4°C with anti-Cyp46 antibody (1:1000), washed 10 times in PBS and counterstained with mouse monoclonal antibodies against neuronal marker MAP-2 (1:100, Chemicon), microglial marker Ox42 (1:100, Serotec), astrocytic marker GFAP (1:100, Sigma), or the nuclear marker ToPro-3 (1:1000, Molecular Probes). Tissue was washed 10 times in PBS and incubated 1 h with Alexa 488 anti-rabbit antibody (1:100, Molecular Probes) and Alexa 546 anti-mouse antibody (1:100, Molecular Probes). Tissue was washed 10 times in PBS and mounted with Fluoramount G (Electron Microscopy Sciences). Staining was visualized with a Zeiss 510 Met confocal laser scanning microscope using an argon ion laser emitting at 488 nm and helium-neon lasers emitting at 543 and 633 nm.
Relative Cyp46 levels
For analysis of Cyp46 fluorescent immunostains, a single 1-micron slice at the greatest intensity was obtained for a field of cells using Zeiss LSM software. Signal from Alexa 488 (Cyp46) was converted to grayscale and inverted using Adobe Photoshop. Relative intensity levels at the cell body were measured using a constant sampling area for all cells within an experimental set. An equal number of fields were obtained for each condition. For primary microglial experiments, all cells in the field were measured regardless of activation level. For in vivo measurements following TBI, an equal number of fields were obtained from each animal (n = 3). Cell type specific grayscale measurements were guided using a grid and identical fields in color, including both Cyp46 signal (Alexa 488, green) and cellular marker (Alexa 546, red). For each cell type measurement, all cells of that type were measured.
Statistical analysis was performed using Graphpad Prism 4 and Student’s t-test.
Results
To test whether our anti-Cyp46 antibody recognized Cyp46 in immunostains and Western blots, we examined COS-7 cells transfected with either control vector or human Cyp46 cDNA. We used Western blot to examine whole rat brain lysate as well as COS-7 cells transfected with control vector or myc-tagged Cyp46. Cyp46 is 57 kDa, while the myc tagged version of human Cyp46 is expected to be an additional 2 kDa. A Cyp46-reactive band was found at the expected size in rat brain lysate with additional smaller bands (Fig. 1A, first lane). No bands were seen in untransfected COS-7 cells (Fig. 1A, second lane), but Cyp46 immunoreactivity was observed at the expected size in Cyp46-myc transfected cells (Fig. 1A, third lane). We also examined antibody specificity in Western blot looking at wildtype versus Cyp46 knockout mouse brain lysates (Fig. 1B). The expected 57 kDa band was found in wildtype but not Cyp46 knockout brain indicating that this band is authentic Cyp46. Smaller bands similar to those found in rat brain lysate were observed in both wildtype and knockout mouse brains, indicating these bands are unrelated to Cyp46. To test specificity of anti-Cyp46 antibody in immunohistochemistry, we transfected COS-7 cells with either control vector or Cyp46 cDNA. Control cells showed little Cyp46 immunostaining (Fig. 2A), while Cyp46-transfected cells showed strong immunostaining in over 20% of cells (Fig. 2B). In order to test whether the anti-Cyp46 antibody recognized endogenous Cyp46, we cultured primary neurons in vitro since Cyp46 is primarily expressed in neurons (Lund et al., 1999; Ramirez et al., 2008). In DIV16 primary neurons, we found strong expression of Cyp46 especially in the soma but also continuing throughout the processes (Fig. 2C). Similar results were seen at DIV9 (data not shown). MAP-2 neuronal staining indicated that there were also non-neuronal cells in the cultures that expressed Cyp46 at much lower levels (Fig. 2C, arrow). Thus, the anti-Cyp46 antibody displayed specificity in both immunoblots and immunohistochemistry.
FIG. 1.

Cyp46 antibody recognizes Cyp46 in immunoblots. (A) Cyp46 expression in whole rat brain lysate was compared to COS-7 cells following transfection for 48 h with control vector or myc-tagged human Cyp46 using immunoblot (arrow, endogenous Cyp46 57-kD band; asterix, myc-tagged huCyp46 59-kDa band). Molecular weight markers are shown on the right. (B) Cyp46 expression was compared in wildtype mouse versus Cyp46 knock-out mouse whole brain lysate (arrow, endogenous Cyp46 57-kD band). Molecular weight markers are shown on the right. Both mouse and rat brain lysate show non-specific bands below the 57-kD Cyp46 band, while these bands were not present in COS-7 cell lysate.
FIG. 2.
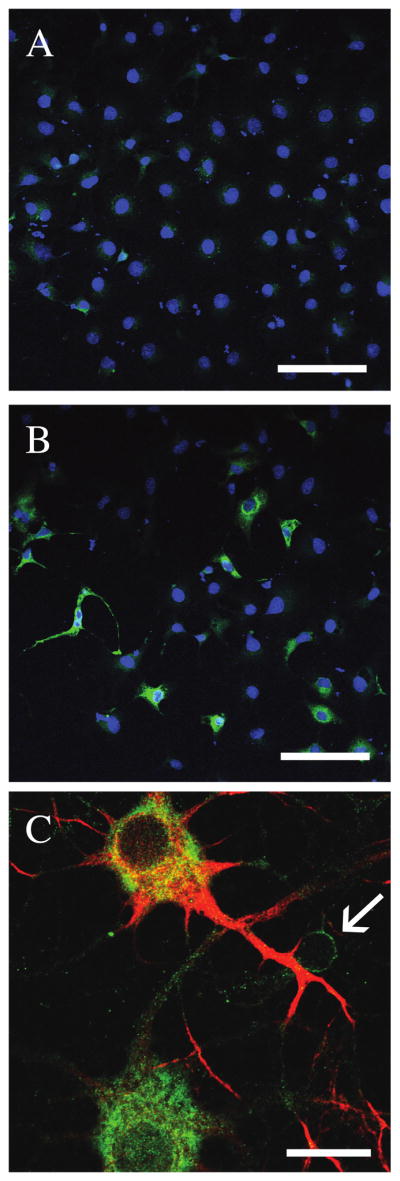
Cyp46 antibody recognizes Cyp46 by immunohistochemistry. COS7 cells were transfected with either control vector (A) or human Cyp46 (B) for 48 h. Immunostaining was performed using anti-Cyp46 antibody and Alexa 488 secondary antibody (green). Cell nuclei were counterstained with Topro-3 (blue), 20×. Scale bar = 100 μm. (C) In rat DIV16 hippocampal neurons, Cyp46 levels (green) were most pronounced in large, well-connected neurons with very high expression in the cytoplasm. Neuronal cells were identified using anti-MAP-2 antibody and Alexa 546 secondary (red). Some MAP-2-negative cells show low levels of Cyp46 expression (arrow), 63×. Scale bar = 20 μm.
Using this anti-Cyp46 antibody, we examined whether Cyp46 expression was altered following an acute brain trauma, using moderate fluid percussion injury of the cortex. Previous studies of microglial phagocytosis of lipids have shown that lipid debris begins to co-localize with microglia 2 days post-injury and reaches a maximum at 7 days post-injury (Gasparovic et al., 2001; Kamada et al., 2003). We therefore investigated possible changes in Cyp46 levels at 3 and 7 days after TBI. Sham animals received a craniotomy but no percussion injury. Either 3 or 7 days after injury animals were sacrificed and perfused with 4% PFA for immunohistochemistry. Brains were frozen and sectioned coronally at 16 μm. There were three animals in each group.
At 3 days post-injury, Cyp46 immunostaining was increased in the cortex at the site of injury in comparison to the contralateral cortex in all three rats at this time point (Fig. 3A,B). At 7 days post-injury, the site of injury included more cortical area than at 3 days post-injury (data not shown), and Cyp46 levels were again increased in comparison to contralateral cortex, again in all three animals (Fig. 3C,D). Sham tissue at 7 days after craniotomy showed cortical Cyp46 expression consistent with neuronal staining and comparable to that seen in sham tissue at 3 days after craniotomy and in contralateral cortex of injured animals (data not shown). Control staining lacking primary antibody showed no signal in sham tissue (data not shown) or at the injury site 7 days post-injury (Fig. 3F).
FIG. 3.

Cyp46 levels are increased at site of injury by immunohistochemistry. DAB immunostaining against Cyp46: (A) Contralateral cortex 3 days post-injury, 20×. Scale bar = 100 μm. (B) Site of injury 3 days post-injury, 20×. Scale bar = 100 μm. (C) Contralateral cortex 7 days post-injury, 20×. Scale bar = 100 μm. (D) Site of injury 7 days post-injury, 20×. Scale bar = 100 μm. Cyp46 levels appeared to be upregulated at both 3 and 7 days post-injury in comparison to contralateral cortex. (E) Sham cortex 7 days after craniotomy, 20×. Scale bar = 100 μm. Expression patterns and levels appeared similar to that seen in contralateral cortex of injured animals. (F) Secondary control staining, site of injury 7 days post-injury, 20×. Scale bar = 100 μm. No staining was visible with secondary antibody alone.
To quantify cortical changes in Cyp46 levels following TBI, we analyzed lysate of parietal cortex on the side of injury compared to parietal cortex on the side of craniotomy from sham controls. Separate sets of animals (n = 3 for each condition) were treated and sacrificed for these Western blots. Shams at 3 and 7 days showed no signs of cortical injury by immunohistochemical staining of coronal sections and were comparable (data not shown). Therefore, Western blot analysis included only those sham animals sacrificed 3 days after surgery. Cyp46 levels were clearly increased 7 days after injury compared to sham controls (Fig. 4A). Lower molecular weight bands were observed but showed no changes with brain injury (data not shown). Quantification of band intensity following correction for small variability in loading demonstrated that Cyp46 levels were significantly increased 28% by 7 days post-injury (Fig. 4B, p < 0.05) but that there was no significant change in Cyp46 levels 3 days post-injury.
FIG. 4.
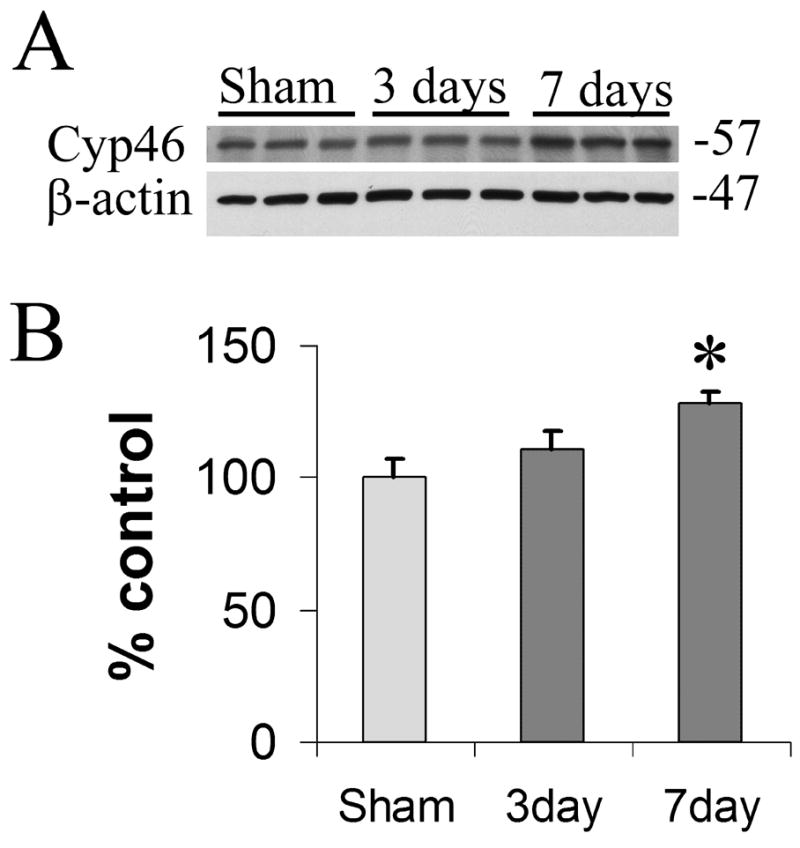
Cyp46 levels increase with injury by immunoblot. (A) Three and 7 days after TBI, Cyp46 protein levels increased in comparison to sham controls (upper panel). Beta-actin levels were unchanged (lower panel). (B) Quantification of levels seen in (A) following correction for each beta-actin loading control indicated a significant increase in Cyp46 levels at 7 days (28%) post-injury compared to sham levels according to Newman-Keuls Post-hoc Multiple Comparison Test (*p < 0.05). Changes between sham and 3 days post injury, or 3 versus 7 days post-injury did not reach significance (overall ANOVA: p = 0.0505, F = 5.116, df [treatment] = 2, n = 3). Error bars (standard error of the mean).
Cyp46 levels were increased locally at 3 and 7 days post-injury (Fig. 3A,B) and showed significant regional increases at 7 days post-injury (Fig. 4B). To determine whether these increases translated into increased LXR activity downstream, we examined levels of ABCA1 and ApoE, two LXR regulated cholesterol homeostasis proteins. Levels of ApoE were increased at 3 and 7 days post-injury, while ABCA1 levels were increased 7 days post-injury (Fig. 5), indicating that increased Cyp46 enzymatic expression correlates with increased LXR activity following TBI.
FIG. 5.

ABCA1 and ApoE levels increase with cortical injury. (A) ABCA1 protein levels (upper panel) with corresponding β-actin levels (lower panel). ABCA1 levels increased 7 days (7d) after TBI in comparison to sham controls (S). Each lane represents a separate animal (n = 2). (B) ApoE protein levels (upper panel) with corresponding β-actin levels (lower panel). ApoE protein levels increased at 3 days (3d) and 7 days (7d) after TBI in comparison to sham controls (S).
Cyp46 expression has been shown to be unaffected by most regulatory axes known to influence cholesterol homeostasis but has been shown to be regulated by oxidative stress and the inflammatory signals in vitro (Ohyama et al., 2006). These same signals would also lead to the microglial and astrocytic activation seen following TBI (Morganti-Kossmann et al., 2007). Although Cyp46 expression in normal brain is primarily neuronal, we have found non-neuronal expression of Cyp46 in primary cell culture (Fig. 2C) and other studies have indicated that astrocytes may express Cyp46 in AD brains (Bogdanovic et al., 2001; Brown III et al., 2004) or following kainate injury (He et al., 2006). In a model of multiple schlerosis (MS), Cyp46 expression was seen in macrophages (Teunissen et al., 2007). To determine whether changes seen with immunohistochemistry may be limited to a subset of cells, we performed co-staining of Cyp46 and cellular markers for neurons, microglia, and astrocytes on coronal sections from rat brain 3 days post-injury. As expected, Cyp46 expression colocalized with Map2-positive neurons in contralateral cortex and at the site of injury (Fig. 6, row A). In the contralateral cortex, Cyp46-positive cells were neuronal in morphology and were generally evenly spaced, and orientated in alignment with the cortical layers (Fig. 6, row A, left panel). However, this alignment was disrupted at both the edge and even more at the center of the injury site, while Map2 expression decreased (Fig. 6, row A, right panel). Very few Ox42-positive micoglia showed Cyp46 expression in contralateral cortex (Fig. 6, row B, left panel). However, there was clear colocalization of Cyp46 with microglia at the site of injury (Fig. 6, row B, right panel). In addition, the microglia at the site of injury appeared larger and more numerous, indicating the activation and proliferation of this cell type at the site of injury. Faint Ox42 expression was seen in contralateral cortex with the star-like staining pattern characteristic of resting microglia (Fig. 6, row C, left panel). Increased levels of Ox42 staining were seen at the center of the injury site, indicating an increase in microglial activation (Fig. 6, row C, right panel). There was minimal colocalization of Cyp46 to GFAP-positive astrocytes in contralateral cortex (Fig. 6, row C, left panel). At the site of injury the number of GFAP-positive astrocytes was increased indicating activation and proliferation. However, only occasional astrocytes showed Cyp46-positive staining at the site of injury (Fig. 6, row C, right panel), indicating that astrocytes are only a minor source of Cyp46 expression following injury.
FIG. 6.

Cyp46 expression in neurons, microglia, and astrocytes 3 days following cortical injury. Cyp46 levels in MAP-2-positive neurons (row A), Ox42-positive microglia (row B), or GFAP-positive astrocytes (row C) in contralateral cortex (left column) and at the injury site (right column); Cyp46 (Alexa 488, green), MAP-2 (row A), Ox42 (row B), and GFAP (row C) (Alexa 546, red), 63×. Scale bar = 50 μm. While Cyp46 expression occurred in neurons in both contralateral cortex and at the site of injury, there was minimal microglial expression of Cyp46 in contralateral cortex, but clear colocalization of expression at the site of injury (arrow). Cyp46 expression was not present in contralateral cortex astrocytes. At the site of injury, some astrocytes showed Cyp46-positive staining, but most did not.
In order to compare cell type specific changes in Cyp46, we measured Cyp46 immunoflorescence intensity levels at the soma of cells in contralateral cortex and central to the site of injury at 3 days post-injury. Because the soma size varied, a constant size sample measurement at the soma was taken rather than a measurement of total Cyp46 expression for the cell. Cells from an equal number of fields (n = 4) were measured from each animal (n = 3). Although levels tended to decrease, relative levels of Cyp46 did not change significantly in Map2-positive neurons (Fig. 7A). An 84% increase in relative Cyp46 levels was observed in Ox42-positive microglia in comparison to microglia in contralateral cortex (Fig. 7B). In GFAP-positive astrocytes, there was no significant change in relative levels of Cyp46 with injury, although levels tended to increase (Fig. 7C).
FIG. 7.

Cyp46 levels increase in microglia and astrocytes but not neurons 3 days following cortical injury. Relative levels of Cyp46 at the cell body of cells were compared to those in the contralateral cortex. (A) MAP-2-positive neurons at the site of injury did not show a significant change in Cyp46 levels, although levels tended to decrease (contralateral cortex, n = 139; injury site, n = 88). (B) Ox42-positive microglia at the site of injury showed an increase of 84% in Cyp46 levels (*Student’s t-test, p < 0.0001; contralateral cortex, n = 62; injury site, n = 130). (C) GFAP-positive astrocytes at the site of injury did not show a significant change in Cyp46 levels, although levels tended to increase (contralateral cortex, n = 55; injury site, n = 57). Error bars (standard error of the mean).
We have shown that microglia are a significant source of Cyp46 levels following TBI. However, because one of the main roles of activated microglia is the phagocytosis of cellular debris, we wanted to determine whether activated microglia increase expression of Cyp46 or whether results seen in vivo were due to phagocytosis of the neuronally derived protein. To determine this, we activated primary microglial cells with LPS in vitro. In untreated resting microglia, we found little Cyp46 immunostaining (Fig. 8A). After a 24-h treatment of 50 ng/mL LPS, many microglia showed enlarged soma indicating microglial activation, although not all cells in a field appeared activated with this relatively mild LPS treatment. Based on Topro-3 nuclear staining, cells appeared to cluster and there were more cells in a given field, indicating LPS induced proliferation. An increase in Cyp46 expression was seen following 24-h LPS treatment, appearing not only in soma of microglia but also continuing into processes (Fig. 8B). Quantification of relative intensity levels at the soma indicated a 28% increase in relative Cyp46 levels. Because all cells in a field were measured regardless of apparent activation levels, this measurement may underestimate increases in Cyp46 with microglial activation.
FIG. 8.

Cyp46 levels increase in activated microglia. Primary rat microglia were either left untreated (A) or treated with 50 ng/mL LPS for 24 h (B). LPS resulted in a strong increase in Cyp46 expression (Alexa 488, green), as well as a proliferation of microglial cells. Cell nuclei were counter-stained with Topro-3 (blue), 20×. Scale bar = 100 μm. (C) Relative levels of Cyp46 at the cell body showed a 28% increase according to Student’s t-test (*Student’s t-test, p < 0.0001; control, n = 197; LPS treatment, n = 269). Error bars (standard error of the mean).
Discussion
In the normal brain, Cyp46 expression is a mechanism for maintaining cholesterol homeostasis (Jurevics and Morell, 1995; Turley et al., 1996, 1998; Lund et al., 2003; Xie et al., 2003). Previous research identified Cyp46 as a neuron-specific enzyme under normal conditions (Lund et al., 1999; Bretillon et al., 2007; Ramirez et al., 2008). Neuronal Cyp46 has stable expression, perhaps performing a role in maintaining stable cholesterol levels within the neuronal membrane (Lund et al., 1999; Joseph et al., 2003; Ohyama et al., 2006). However, following brain injury, there is significant neuronal loss (Cernak et al., 2004; Di Giovanni et al., 2005; Zhang et al., 2005). We expected that, with neuronal death, lipid debris would increase while neuronally expressed Cyp46 would be decreased. Instead, we observed that Cyp46 levels increased after TBI at the site of damage (Fig. 4). This finding is in line with other models of CNS injury showing increases in Cyp46 within lesion sites (He et al., 2006; Teunissen et al., 2007) and led us to examine Cyp46 in non-neuronal cells.
Previous studies looking at Cyp46 following CNS damage have seen expression in glia, especially in astrocytes. Postmortem AD brains do not exhibit gross increases in Cyp46 levels but have been shown to have Cyp46 expression in GFAP-positive astrocytes not seen in control tissue (Bogdanovic et al., 2001; Brown III et al., 2004). Following hippocampal kainate injury, Cyp46 expression was seen in astrocytes at the lesion site but not elsewhere (He et al., 2006). In TBI, we did not observe a strong up-regulation of Cyp46 in neurons or astrocytes, but we did observe Cyp46 levels increased 84% in activated microglia (Figs. 6 and 7). Microglia are the immune cells of the CNS (Streit, 2002). At a time when neurons are dying, microglia are activated and proliferating (Kamada et al., 2003; Cernak et al., 2004; Di Giovanni et al., 2005). In addition, a function of microglia following injury is the removal of lipid debris (Gasparovic et al., 2001; Kamada et al., 2003). Thus, increased expression of Cyp46 in microglia may be a necessary step in the processing of phagocytosed lipids. In a rat model of MS, Cyp46 staining has been shown to colocalize to ED1-positive macrophages within brainstem lesions (Teunissen et al., 2007). This expression was attributed to infiltrating peripheral macrophages; however the activation of peripheral peritonium derived macrophages with LPS in vitro suppressed Cyp46 expression (Teunissen et al., 2007). In contrast, we have shown that brain-derived microglia increase Cyp46 expression following LPS activation (Fig. 8). The consensus based on previous studies and our findings is that both astrocytes and microglia are capable of upregulating Cyp46 expression following neuronal injury. However, after TBI, this upregulation appears to be primarily in microglia and not in astrocytes.
There are some indications that very high levels of 24S-hydroxycholesterol might be detrimental. Human control subjects show levels of CSF levels of 24S-hydroxycholesterol levels averaging at 6.7 μM while AD subjects showed levels averaging 8.7 μM (Papassotiropoulous et al., 2002). While in vitro treatments of 50 μM 24S-hydroxycholesterol were found to be toxic to neurons in hippocampal slices, 15 μM treatment, still significantly higher than endogenous levels, did not cause damage (He et al., 2006). In co-cultures of primary human neurons and glia 10 μM 24S-hydroxycholesterol treatments increase inflammatory signals (Alexandrov et al., 2005), possibly due to the induction of changes in cholesterol homeostasis. However, after TBI neuronal membranes have already been disrupted by damage leading to neuronal death and lipid debris (Faden, 1996; Gasparovic et al., 2001; Kamada et al., 2003). Although we did not directly measure Cyp46 activity, we did see an increase in Cyp46 expression in microglia (Fig. 6) which would be expected to promote the removal of cholesterol debris following TBI.
There are plenty of reasons to believe that increases in Cyp46 expression may be beneficial following brain damage. The effects of Cyp46 are threefold, including clearance of cholesterol by hydroxylation (Lund et al., 2003), clearance of cholesterol via ABCA1 export to lipoproteins (Rebeck, 2004), and anti-inflammatory and antioxidant effects attributed to LXR regulated genes (Joseph et al., 2003; Walcher et al., 2006), especially ApoE (Hoane et al., 2007; Laskowitz et al., 2007). LXR activation has been shown to suppress inflammatory cytokine production including IL-6 (Joseph et al., 2003; Walcher et al., 2006) as well as iNOS and Cox-2 production (Joseph et al., 2003). ApoE has been shown to have anti-inflammatory effects (Stannard et al., 2001; Guo et al., 2004), possibly due to cholesterol clearance. Peptides containing the receptor binding domain of ApoE have been shown to be anti-inflammatory (Hoane et al., 2007; Laskowitz et al., 2007), indicating that ApoE may have additional anti-inflammatory properties unrelated to cholesterol clearance. Since ApoE secretion is necessary for cholesterol efflux through ABCA1 (Fagan et al., 2002), efflux of cholesterol to lipoproteins would be most effective when both ABCA1 and ApoE are upregulated. We have found that both ApoE and ABCA1 levels increase after TBI indicating that effective lipid efflux is possible (Fig. 5).
From our studies, we hypothesize that microglial expression of Cyp46 following injury may increase microglial cholesterol efflux following phagocytosis of cellular debris and also act to reduce inflammatory and oxidative signals regionally. Our results demonstrate that change in Cyp46 expression is not a marker of any specific neurodegenerative disease; rather, Cyp46 increases are a marker of CNS injury. The signals controlling Cyp46 expression have yet to be fully discerned, but increasing Cyp46 expression would be potentially beneficial following TBI. The effects of these endogenous mechanisms, although positive, may come only after significant neuronal loss has occurred. In addition, oxidative stress, inflammatory events, and glial activation all contribute to neuronal death (Fitch et al., 1999; Langley and Ratan, 2004). By treating TBI early on with moderate levels of 24S-hydroxycholesterol or a synthetic LXR agonist, such as TO901317 or GW-3965, it may be possible to confer the benefits of cholesterol efflux as well as anti-inflammatory and anti-oxidative properties in advance of microglial activation.
Acknowledgments
We thank Dr. Suzana Petanceska for providing the anti-Cyp46 antibody and Dr. David Russell for providing the Cyp46 cDNA and CYP46 knock-out brain tissue. This research was supported by the National Institutes of Health (R01-AG14473, F31-AG025676).
Footnotes
Author Disclosure Statement
No conflicting financial interests exist.
References
- Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE, Frautschy S, Griffin WS, Hampel H, Hull M, Landreth G, Lue L, Mrak R, Mackenzie IR, McGeer PL, O’Banion MK, Pachter J, Pasinetti G, Plata-Salaman C, Rogers J, Rydel R, Shen Y, Streit W, Strohmeyer R, Tooyoma I, Van Muiswinkel FL, Veerhuis R, Walker D, Webster S, Wegrzyniak B, Wenk G, Wyss-Coray T. Inflammation and Alzheimer’s disease. Neurobiol Aging. 2000;21:383–421. doi: 10.1016/s0197-4580(00)00124-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov P, Cui J, Zhao Y, Lukiw WJ. 24S-Hydroxycholesterol induces inflammatory gene expression in primary human neural cells. Neuroreport. 2005;16:909–913. doi: 10.1097/00001756-200506210-00007. [DOI] [PubMed] [Google Scholar]
- Ansell BJ, Fonarow GC, Navab M, Fogelman AM. Modifying the anti-inflammatory effects of high-density lipoprotein. Curr Atheroscler Rep. 2007;9:57–63. doi: 10.1007/BF02693941. [DOI] [PubMed] [Google Scholar]
- Bar-On P, Crews L, Koob AO, Mizuno H, Adame A, Spencer B, Masliah E. Statins reduce neuronal alpha-synuclein aggregation in in vitro models of Parkinson’s disease. J Neurochem. 2008;105:1656–1667. doi: 10.1111/j.1471-4159.2008.05254.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogdanovic N, Bretillon L, Lund EG, Diczfalusy U, Lannfelt L, Winblad B, Russell DW, Bjorkhem I. On the turnover of brain cholesterol in patients with Alzheimer’s disease. Abnormal induction of the cholesterol-catabolic enzyme CYP46 in glial cells. Neurosci Lett. 2001;314:45–48. doi: 10.1016/s0304-3940(01)02277-7. [DOI] [PubMed] [Google Scholar]
- Bonotis K, Krikki E, Holeva V, Aggouridaki C, Costa V, Baloyannis S. Systemic immune aberrations in Alzheimer’s disease patients. J Neuroimmunol. 2008;193:183–187. doi: 10.1016/j.jneuroim.2007.10.020. [DOI] [PubMed] [Google Scholar]
- Bretillon L, Diczfalusy U, Bjorkhem I, Maire MA, Martine L, Joffre C, Acar N, Bron A, Creuzot-Garcher C. Cholesterol-24S-hydroxylase (CYP46A1) is specifically expressed in neurons of the neural retina. Curr Eye Res. 2007;32:361–366. doi: 10.1080/02713680701231857. [DOI] [PubMed] [Google Scholar]
- Brown J, III, Theisler C, Silberman S, Magnuson D, Gootardi-Littell N, Lee JM, Yager D, Crowley J, Sambamuri K, Rahman MM, Reiss AB, Eckman CB, Wolozin B. Differential expression of cholesterol hydroxylases in Alzheimer’s disease. J Biol Chem. 2004;279:34674–34681. doi: 10.1074/jbc.M402324200. [DOI] [PubMed] [Google Scholar]
- Cernak I, Vink R, Zapple DN, Cruz MI, Ahmed F, Chang T, Fricke ST, Faden AI. The pathobiology of moderate diffuse traumatic brain injury as identified using a new experimental model of injury in rats. Neurobiol Dis. 2004;17:29–43. doi: 10.1016/j.nbd.2004.05.011. [DOI] [PubMed] [Google Scholar]
- Di Giovanni S, Movsesyan V, Ahmed F, Cernak I, Schinelli S, Stoica B, Faden AI. Cell cycle inhibition provides neuroprotection and reduces glial proliferation and scar formation after traumatic brain injury. Proc Natl Acad Sci USA. 2005;102:8333–8338. doi: 10.1073/pnas.0500989102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faden AI. Pharmacologic treatment of acute traumatic brain injury. JAMA. 1996;276:569–570. [PubMed] [Google Scholar]
- Faden AI, Knoblach SM, Cernak I, Fan L, Vink R, Araldi GL, Fricke ST, Roth BL, Kozikowski AP. Novel diketopiperazine enhances motor and cognitive recovery after traumatic brain injury in rats and shows neuroprotection in vitro and in vivo. J Cereb Blood Flow Metab. 2003;23:342–354. doi: 10.1097/01.WCB.0000046143.31247.FD. [DOI] [PubMed] [Google Scholar]
- Fagan AM, Watson M, Parsadanian M, Bales KR, Paul SM, Holtzman DM. Human and murine ApoE markedly alters A beta metabolism in a mouse model of Alzheimer’s disease. Neurobiol Dis. 2002;9:305–318. doi: 10.1006/nbdi.2002.0483. [DOI] [PubMed] [Google Scholar]
- Fassbender K, Simon M, Bergmann C, Stroick M, Lutjohann D, Keller P, Runz H, Bertsch T, von Bergmann K, Hennerici M, Beyreuther K, Hartmann T. Simvastatin strongly reduces levels of Alzheimer’s disease beta-amyloid peptides A beta 42 and A beta 40 in vitro and in vivo. Proc Natl Acad Sci USA. 2001;98:5856–5861. doi: 10.1073/pnas.081620098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitch MT, Doller C, Combs CK, Landreth GE, Silver J. Cellular and molecular mechanisms of glial scarring and progressive cavitation: in vivo and in vitro analysis of inflammation-induced secondary injury after CNS trauma. J Neurosci. 1999;19:8182–8198. doi: 10.1523/JNEUROSCI.19-19-08182.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukumoto H, Deng A, Irizarry MC, Fitzgerald ML, Rebeck GW. Induction of the cholesterol transporter ABCA1 in central nervous system cells by liver X receptor agonists increases secreted A beta levels. J Biol Chem. 2002;277:48508–48513. doi: 10.1074/jbc.M209085200. [DOI] [PubMed] [Google Scholar]
- Gasparovic C, Rosenberg GA, Wallace JA, Estrada EY, Roberts K, Pastuszyn A, Ahmed W, Graham GD. Magnetic resonance lipid signals in rat brain after experimental stroke correlate with neutral lipid accumulation. Neurosci Lett. 2001;301:87–90. doi: 10.1016/s0304-3940(01)01616-0. [DOI] [PubMed] [Google Scholar]
- Goldman SM, Tanner CM, Oakes D, Bhudhikanok GS, Gupta A, Langston JW. Head injury and Parkinson’s disease risk in twins. Ann Neurol. 2006;60:65–72. doi: 10.1002/ana.20882. [DOI] [PubMed] [Google Scholar]
- Guo L, LaDu MJ, Van Eldik LJ. A dual role for apolipoprotein E in neuroinflammation: anti- and pro-inflammatory activity. J Mol Neurosci. 2004;23:205–212. doi: 10.1385/JMN:23:3:205. [DOI] [PubMed] [Google Scholar]
- Hansson GK, Robertson AK, Soderberg-Naucler C. Inflammation and atherosclerosis. Annu Rev Pathol. 2006;1:297–329. doi: 10.1146/annurev.pathol.1.110304.100100. [DOI] [PubMed] [Google Scholar]
- He X, Jenner AM, Ong WY, Farooqui AA, Patel SC. Lovastatin modulates increased cholesterol and oxysterol levels and has a neuroprotective effect on rat hippocampal neurons after kainate injury. J Neuropathol Exp Neurol. 2006;65:652–663. doi: 10.1097/01.jnen.0000225906.82428.69. [DOI] [PubMed] [Google Scholar]
- Hoane MR, Pierce JL, Holland MA, Birky ND, Dang T, Vitek MP, McKenna SE. The novel apolipoprotein E-based peptide COG1410 improves sensorimotor performance and reduces injury magnitude following cortical contusion injury. J Neurotrauma. 2007;24:1108–1118. doi: 10.1089/neu.2006.0254. [DOI] [PubMed] [Google Scholar]
- Joseph SB, Castrillo A, Laffitte BA, Mangelsdorf DJ, Tontonoz P. Reciprocal regulation of inflammation and lipid metabolism by liver X receptors. Nat Med. 2003;9:213–219. doi: 10.1038/nm820. [DOI] [PubMed] [Google Scholar]
- Jurevics H, Morell P. Cholesterol for synthesis of myelin is made locally, not imported into brain. J Neurochem. 1995;64:895–901. doi: 10.1046/j.1471-4159.1995.64020895.x. [DOI] [PubMed] [Google Scholar]
- Kamada H, Sato K, Iwai M, Zhang WR, Nagano I, Manabe Y, Shoji M, Abe K. Temporal and spatial changes of free cholesterol and neutral lipids in rat brain after transient middle cerebral artery occlusion. Neurosci Res. 2003;45:91–100. doi: 10.1016/s0168-0102(02)00203-1. [DOI] [PubMed] [Google Scholar]
- Kay AD, Day SP, Kerr M, Nicoll JA, Packard CJ, Caslake MJ. Remodeling of cerebrospinal fluid lipoprotein particles after human traumatic brain injury. J Neurotrauma. 2003;20:717–723. doi: 10.1089/089771503767869953. [DOI] [PubMed] [Google Scholar]
- Kim HJ, Fan X, Gabbi C, Yakimchuk K, Parini P, Warner M, Gustafsson JA. Liver X receptor beta (LXR-beta): a link between beta-sitosterol and amyotrophic lateral sclerosis-Parkinson’s dementia. Proc Natl Acad Sci USA. 2008;105:2094–2099. doi: 10.1073/pnas.0711599105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langley B, Ratan RR. Oxidative stress-induced death in the nervous system: cell cycle dependent or independent? J Neurosci Res. 2004;77:621–629. doi: 10.1002/jnr.20210. [DOI] [PubMed] [Google Scholar]
- Langlois JA, Rutland-Brown W, Thomas KE. Traumatic Brain Injury in the United States: Emergency Department Visits, Hospitalizations, and Deaths. Centers for Disease Control and Prevention, National Center for Injury Prevention and Control; Atlanta: 2004. [Google Scholar]
- Laskowitz DT, McKenna SE, Song P, Wang H, Durham L, Yeung N, Christensen D, Vitek MP. COG1410, a novel apolipoprotein E-based peptide, improves functional recovery in a murine model of traumatic brain injury. J Neurotrauma. 2007;24:1093–1107. doi: 10.1089/neu.2006.0192. [DOI] [PubMed] [Google Scholar]
- Lehmann JM, Kliewer SA, Moore LB, Smith-Oliver TA, Oliver BB, Su JL, Sundseth SS, Winegar DA, Blanchard DE, Spencer TA, Willson TM. Activation of the nuclear receptor LXR by oxysterols defines a new hormone response pathway. J Biol Chem. 1997;272:3137–3140. doi: 10.1074/jbc.272.6.3137. [DOI] [PubMed] [Google Scholar]
- Liang Y, Lin S, Beyer TP, Zhang Y, Wu X, Bales KR, De-Mattos RB, May PC, Li SD, Jiang XC, Eacho PI, Cao G, Paul SM. A liver X receptor and retinoid X receptor heterodimer mediates apolipoprotein E expression, secretion and cholesterol homeostasis in astrocytes. J Neurochem. 2004;88:623–634. doi: 10.1111/j.1471-4159.2004.02183.x. [DOI] [PubMed] [Google Scholar]
- Lund EG, Guileyardo JM, Russell DW. cDNA cloning of cholesterol 24–hydroxylase, a mediator of cholesterol homeostasis in the brain. Proc Natl Acad Sci USA. 1999;96:7238–7243. doi: 10.1073/pnas.96.13.7238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund EG, Xie C, Kotti T, Turley SD, Dietschy JM, Russell DW. Knockout of the cholesterol 24–hydroxylase gene in mice reveals a brain-specific mechanism of cholesterol turnover. J Biol Chem. 2003;278:22980–22988. doi: 10.1074/jbc.M303415200. [DOI] [PubMed] [Google Scholar]
- McGeer PL, McGeer EG. Inflammation, autotoxicity and Alzheimer disease. Neurobiol Aging. 2001;22:799–809. doi: 10.1016/s0197-4580(01)00289-5. [DOI] [PubMed] [Google Scholar]
- McGeer PL, McGeer EG. Glial reactions in Parkinson’s disease. Mov Disord. 2008;23:474–483. doi: 10.1002/mds.21751. [DOI] [PubMed] [Google Scholar]
- Morganti-Kossmann MC, Satgunaseelan L, Bye N, Kossmann T. Modulation of immune response by head injury. Injury. 2007;38:1392–1400. doi: 10.1016/j.injury.2007.10.005. [DOI] [PubMed] [Google Scholar]
- Natale JE, Ahmed F, Cernak I, Stoica B, Faden AI. Gene expression profile changes are commonly modulated across models and species after traumatic brain injury. J Neurotrauma. 2003;20:907–927. doi: 10.1089/089771503770195777. [DOI] [PubMed] [Google Scholar]
- Navab M, Yu R, Gharavi N, Huang W, Ezra N, Lotfizadeh A, Anantharamaiah GM, Alipour N, Van Lenten BJ, Reddy ST, Marelli D. High-density lipoprotein: antioxidant and anti-inflammatory properties. Curr Atheroscler Rep. 2007;9:244–248. doi: 10.1007/s11883-007-0026-3. [DOI] [PubMed] [Google Scholar]
- NINDS. National Institute of Neurological Disorders and Stroke (NINDS). Traumatic Brain Injury: Hope Through Research. NIH . National Institutes of Health; Bethesda, MD: 2002. Publication No. 02–158. [Google Scholar]
- Nishimura M, Yaguti H, Yoshitsugo H, Naito S, Satoh T. Tissue distribution of mRNA expression of human cytochrome P450 isoforms assessed by high-sensitivity real-time reverse transcription PCR. Yakugaku Zasshi. 2003;123:369–375. doi: 10.1248/yakushi.123.369. [DOI] [PubMed] [Google Scholar]
- Ohyama Y, Meaney S, Heverin M, Ekstrom L, Brafman A, Shafir M, Andersson U, Olin M, Eggertsen G, Diczfalusy U, Feinstein E, Bjorkhem I. Studies on the transcriptional regulation of cholesterol 24–hydroxylase (CYP46A1): marked insensitivity toward different regulatory axes. J Biol Chem. 2006;281:3810–3820. doi: 10.1074/jbc.M505179200. [DOI] [PubMed] [Google Scholar]
- Page KJ, Hollister RD, Hyman BT. Dissociation of apolipoprotein and apolipoprotein receptor response to lesion in the rat brain: an in situ hybridization study. Neuroscience. 1998;85:1161–1171. doi: 10.1016/s0306-4522(97)00661-1. [DOI] [PubMed] [Google Scholar]
- Pak DT, Yang S, Rudolph-Correia S, Kim E, Sheng M. Regulation of dendritic spine morphology by SPAR, a PSD-95–associated RapGAP. Neuron. 2001;31:289–303. doi: 10.1016/s0896-6273(01)00355-5. [DOI] [PubMed] [Google Scholar]
- Pan DS, Liu WG, Yang XF, Cao F. Inhibitory effect of progesterone on inflammatory factors after experimental traumatic brain injury. Biomed Environ Sci. 2007;20:432–438. [PubMed] [Google Scholar]
- Papassotiropoulous A, Lutjohann D, Bagli M, Locatelli S, Jessen F, Buschfort R, Ptok U, Bjorkhem I, von Bergmann K, Heun R. 24S-Hydroxycholesterol in cerebrospinal fluid is elevated in early stages of dementia. J Psychiatr Res. 2002;36:27–32. doi: 10.1016/s0022-3956(01)00050-4. [DOI] [PubMed] [Google Scholar]
- Plassman BL, Havlik RJ, Steffens DC, Helms MJ, Newman TN, Drosdick D, Phillips C, Gau BA, Welsh-Bohmer KA, Burke JR, Guralnik JM, Breitner JCS. Documented head injury in early adulthood and risk of Alzheimer’s disease and other dementias. Neurology. 2000;55:1158–1166. doi: 10.1212/wnl.55.8.1158. [DOI] [PubMed] [Google Scholar]
- Poirier J, Hess M, May PC, Finch CE. Astrocytic apolipoprotein E mRNA and GFAP mRNA in hippocampus after entorhinal cortex lesioning. Brain Res Mol Brain Res. 1991;11:97–106. doi: 10.1016/0169-328x(91)90111-a. [DOI] [PubMed] [Google Scholar]
- Ramirez DM, Andersson S, Russell DW. Neuronal expression and subcellular localization of cholesterol 24–hydroxylase in the mouse brain. J Comp Neurol. 2008;507:1676–1693. doi: 10.1002/cne.21605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebeck GW. Induction of cholesterol efflux in the CNS. Neurobiol Lipids. 2004;3:1–3. [Google Scholar]
- Reynolds AD, Glanzer JG, Kadiu I, Ricardo-Dukelow M, Chaudhuri A, Ciborowski P, Cerny R, Gelman B, Thomas MP, Mosley RL, Gendelman HE. Nitrated alpha-synuclein-activated microglial profiling for Parkinson’s disease. J Neurochem. 2008;104:1504–1525. doi: 10.1111/j.1471-4159.2007.05087.x. [DOI] [PubMed] [Google Scholar]
- Rojo LE, Fernandez JA, Maccioni AA, Jimenez JM, Maccioni RB. Neuroinflammation: implications for the pathogenesis and molecular diagnosis of Alzheimer’s disease. Arch Med Res. 2008;39:1–16. doi: 10.1016/j.arcmed.2007.10.001. [DOI] [PubMed] [Google Scholar]
- Sanossian N, Saver JL, Navab M, Ovbiagele B. High-density lipoprotein cholesterol: an emerging target for stroke treatment. Stroke. 2007;38:1104–1109. doi: 10.1161/01.STR.0000258347.19449.0f. [DOI] [PubMed] [Google Scholar]
- Stannard AK, Riddell DR, Sacre SM, Tagalakis AD, Langer C, von Eckardstein A, Cullen P, Athanasopoulos T, Dickson G, Owen JS. Cell-derived apolipoprotein E (ApoE) particles inhibit vascular cell adhesion molecule—1 (VCAM-1) expression in human endothelial cells. J Biol Chem. 2001;276:46011–46016. doi: 10.1074/jbc.M104812200. [DOI] [PubMed] [Google Scholar]
- Streit WJ. Microglia as neuroprotective, immunocompetent cells of the CNS. Glia. 2002;40:133–139. doi: 10.1002/glia.10154. [DOI] [PubMed] [Google Scholar]
- Teunissen CE, Floris S, Sonke M, Dijkstra CD, De Vries HE, Lutjohann D. 24S-Hydroxycholesterol in relation to disease manifestations of acute experimental autoimmune encephalomyelitis. J Neurosci Res. 2007;85:1499–1505. doi: 10.1002/jnr.21266. [DOI] [PubMed] [Google Scholar]
- Turley SD, Burns DK, Dietschy JM. Preferential utilization of newly synthesized cholesterol for brain growth in neonatal lambs. Am J Physiol. 1998;274:E1099–E1105. doi: 10.1152/ajpendo.1998.274.6.E1099. [DOI] [PubMed] [Google Scholar]
- Turley SD, Burns DK, Rosenfeld CR, Dietschy JM. Brain does not utilize low density lipoprotein-cholesterol during fetal and neonatal development in the sheep. J Lipid Res. 1996;37:1953–1961. [PubMed] [Google Scholar]
- Walcher D, Kummel A, Kehrle B, Bach H, Grub M, Durst R, Hombach V, Marx N. LXR activation reduces proinflammatory cytokine expression in human CD4–positive lymphocytes. Arterioscler Thromb Vasc Biol. 2006;26:1022–1028. doi: 10.1161/01.ATV.0000210278.67076.8f. [DOI] [PubMed] [Google Scholar]
- Whitney KD, Watson MA, Collins JL, Benson WG, Stone TM, Numerick MJ, Tippin TK, Wilson JG, Winegar DA, Kliewer SA. Regulation of cholesterol homeostasis by the liver X receptors in the central nervous system. Mol Endocrinol. 2002;16:1378–1385. doi: 10.1210/mend.16.6.0835. [DOI] [PubMed] [Google Scholar]
- Xie C, Lund EG, Turley SD, Russell DW, Dietschy JM. Quantitation of two pathways for cholesterol excretion from the brain in normal mice and mice with neurodegeneration. J Lipid Res. 2003;44:1780–1789. doi: 10.1194/jlr.M300164-JLR200. [DOI] [PubMed] [Google Scholar]
- Zhang X, Chen Y, Jenkins LW, Kochanek PM, Clark RS. Bench-to-bedside review: apoptosis/programmed cell death triggered by traumatic brain injury. Crit Care. 2005;9:66–75. doi: 10.1186/cc2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Fauser U, Schluesener HJ. Early attenuation of lesional interleukin-16 up-regulation by dexamethasone and FTY720 in experimental traumatic brain injury. Neuropathol Appl Neurobiol. 2008;34:330–339. doi: 10.1111/j.1365-2990.2007.00893.x. [DOI] [PubMed] [Google Scholar]