

Abstract
Multiple sclerosis (MS) is a relatively common and etiologically unknown disease with no cure treatment. It is the leading cause of neurological disability in young adults, affecting over two million people worldwide. Traditionally, MS has been considered a chronic, inflammatory disorder of the central white matter in which ensuing demyelination results in physical disability. Recently, MS has become increasingly viewed as a neurodegenerative disorder in which axonal injury, neuronal loss, and atrophy of the central nervous system lead to permanent neurological and clinical disability. In this article, we discuss the latest developments on MS research, including etiology, pathology, genetic association, EAE animal models, mechanisms of neuronal injury and axonal transport and therapeutics. In this article, we also focus on the mechanisms of mitochondrial dysfunction that are involved in MS, including mitochondrial DNA defects, and mitochondrial structural/functional changes.
Keywords: Multiple sclerosis, Experimental autoimmune encephalomyelitis, Mitochondria, Oxidative stress, Myelin, Neuroprotection, NO, Gender difference
1. Introduction
Multiple sclerosis (MS) is a chronic, potentially highly disabling disorder with considerable social impact and economic consequences. Onset of MS typically occurs during early adulthood, making MS the most common neurological disease affecting people under the age of 30. It is the major cause of non-traumatic disability in young adults. The social costs associated with MS are high because of its early age of onset, patients with MS experience an early loss in productivity, they need assistance in performing activities of daily living, and they require immunomodulatory treatments and multidisciplinary health care. Currently, nearly 400,000 people are living with MS in the United States, and in the 1990s, there were at least 250,000 patients with MS in the United States .
The clinical presentation of MS is heterogeneous. Main symptoms include impaired vision, extreme fatigue, spasms and paralysis of a variety of muscle systems. In the majority of cases, MS develops in an episodic fashion, with phases of clinical disease followed by recovery. In this form of MS, called relapsing-remitting MS (RRMS), white matter lesions can typically deteriorate to permanent tissue injury that is associated with neural loss and clinical disability. Over time, RRMS patients may develop chronic lesions that promote irreversible axonal injury, resulting in the conversion of RRMS to secondary progressive MS (SPMS). SPMS is characterized by minimal or no intermittent recovery of function. Cognitive impairment is also common in MS, occurring at all stages of disease progression. Dysfunction in free recall from long-term memory, speed of information processing, working memory, and abstract reasoning are frequently observed in MS .
In recent years, basic research in MS has elucidated the mechanisms and processes underlying the disease, the development of imaging techniques (such as magnetic resonance imaging: MRI), and the development of immunomodulatory drugs which, for the first time, are altering disease outcome. However, basic research in MS has not help explain many disorders associated with MS, such as depression, which is the most frequent psychiatric disorder in MS patients. The cause of depression is multifactorial and is likely associated with psychosocial stress, focal demyelinating lesions, and immune dysfunction. Early intervention in depression can prevent a decline in quality of life that typically characterizes MS patients and has even prevented suicide. Despite advances in reducing clinical symptoms in patients with MS through the use of immunomodulating pharmacotherapy, not all respond well to these treatments, especially when the patient is in SPMS, probably due to disease heterogeneity and multi-local, multi-cell damage throughout not only the white matter, but also the gray matter of the central nervous system (CNS). Gray matter involvement has been detected in the earliest stages of MS, and cortical gray matter atrophy has been found to occur at a faster rate than white matter atrophy early in disease progression, suggesting that other mechanisms may be involved in MS development and progression. This hypothesis challenges current research on MS that has focused on white matter. To deny or confirm this hypothesis, additional research is needed. It argues for the development of new approaches and therapies.
The purpose of this article is 2-fold: 1) to review latest developments in MS research, particularly causal factors and therapeutic approaches, and 2) to review the mechanisms of mitochondrial function/dysfunction including mitochondrial DNA defects, mitochondrial structural and functional changes, mitochondrial DNA repair events, and mitochondrial therapeutics that are involved in MS patients and EAE mouse models.
2. Etiology and Pathology of MS
To date, the exact cause of MS is still unclear, but it is believed to result from an abnormal response of the immune system to one or more myelin antigens that develops in genetically susceptible individuals after their exposure to an as-yet undefined causal agent. It has been characterized by an accumulation of macrophages (microglia in the brain) and lymphocytes in the CNS (the white matter and the gray matter), leading to demyelination and destruction of axons. Figure 1 summarizes the possible causal factors of MS.
Figure 1.
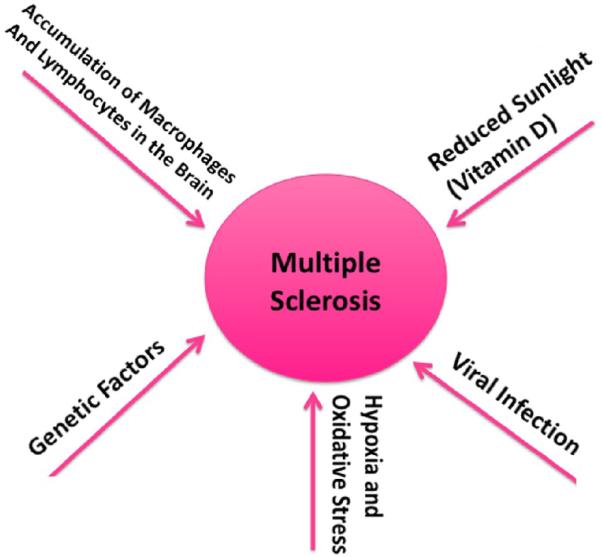
The factors that may contribute to the development and progression of multiple sclerosis. The precise causal factors of multiple sclerosis are unknown. However, it is possible that multiple factors are involved in causing multiple sclerosis, including DNA defects in nuclear and mitochondrial genomes, viral infection, hypoxia and oxidative stress, lack of sunlight or sufficient levels of vitamin D, and increased macrophages and lymphocytes in the brain.
2.1 Genetics of MS
The identification and characterization of MS susceptibility genes likely define the basic etiology of the disease, to improve risk assessment, and to influence therapies. The past 10 years have seen some progress in defining the genetic basis of MS.
The increased risk of occurrence within families indicates genetic factors may play a role in MS etiology. MS is more likely to strike siblings than the general population, and it is more likely to strike monozygotic compared to dizygotic twins. Recently, whole genome screens were conducted in different populations and identified discrete chromosomal regions potentially harboring MS susceptibility genes. However, with the exception of the major histocompatibility complex on 6p21, no single locus generated overwhelming evidence of genetic linkage. These results suggest a complex genetic etiology for MS, including multiple genes of small to moderate effect and probable genetic heterogeneity. On the other hand, the human leukocyte antigen (HLA) was found to control immune response genes in MS, with HLA associations indicating the involvement of autoimmunity. Further, MS was one of the first diseases proven to be HLA-associated, primarily linked to HLA class II factors. The HLA-DRB1*1501 molecule may explain about 50% of MS cases. Furthermore, CD45 or protein tyrosine phosphatase receptor-type C (PTPRC) has been reported as a candidate in some families with MS, 77C-->G PTPRC polymorphism is present and preferentially transmitted in a small subgroup of MS families, which may only be detected with complementary methods of analysis .
Recently, large international research collaborations have provided strong evidence for the involvement of the polymorphism of two cytokine receptor genes in MS pathogenesis: the interleukin 7 receptor alpha chain gene (IL7RA) on chromosome 5p13 and the interleukin 2 receptor alpha chain gene (IL2RA (=CD25)) on chromosome 10p15. It is estimated that the C allele of a single nucleotide polymorphism, rs6897932, within the alternative spliced exon 6 of IL7RA is involved in about 30% of MS cases. These investigations indicate that MS has a strong genetic component. Interestingly, some of these findings (such as HLA-DRB1 and IL2RA) were confirmed by recent pathway and network-based genome-wide association studies (GWAS). In GWAS, neural pathways, namely axon guidance and synaptic potentiation, were also over-represented in genes from MS patients. In addition to identifying immunological pathways previously identified, for the first time GWAS described the potential involvement of neural pathways in MS susceptibility. For example, GWAS revealed more comprehensive and extensive immune antigens, cell adhesion, and signaling molecules associated with MS, such as CD4, CD11b, CD58, CD82, ITGB2, and STAT3, as well as glutamate receptors, multimeric scaffold molecular DLG1, and DLG2. Using a pooling-based, genome-wide approach, and high-density, single-nucleotide polymorphisms arrays, GWAS also identified a novel risk locus for MS on chromosome 13, in addition of the HLA class II genes (such as HLA-DRB1) .
2.2 Virus infections
A long-standing hypothesis about MS etiology is that MS is an infectious disease by a micro-organism. However, after decades of research, no specific infectious agents have been identified in MS, yet many neurologists and researchers still remain open to an infectious origin for MS. In particular, much interest has focused on a potential role for the Epstein-Barr virus (EBV). Recent findings from a population-based investigation support the implication of the EBV in MS susceptibility. It has been reported that a clinical history of infectious mononucleosis conspicuously associated with increased MS susceptibility (). Other studies have of progressive MS cases found the EBV present within B cells that infiltrate the meninges (membranes that envelop the CNS) and white matter - strong evidence for the involvement of EBV in MS through B cells as triggers.
Another type of virus, corona viruses, has also been found in the brains of MS patients. Corona viruses, important human and animal pathogens of the order Nidovirales, usually cause respiratory and gastrointestinal illnesses, including SARS (severe acute respiratory syndrome). However, their localization in the brains of MS patients indicate they may be a possible MS pathogen their neurotropism and immune system attack. Viruses have been found to induce demyelinating diseases in animals. That viruses can induce demyelinating diseases in animals strongly supports the hypothesis that MS may have a viral origin.
2.3 Gender Differences and Other Factors in MS Susceptibility
Females, Caucasians, and people of northern European ancestry are at an increased risk for MS. The incidence of MS in persons of any of these 3 ancestries has considerably increased over the last century, with the increase greatest in women. A large multicenter clinical trial of glatiramer acetate in primary progressive multiple sclerosis indicates that there exist differences in the rates of clinical diseases between men and women with MS. Sex dimorphism in MS may be explained by the effects of sex chromosomes and of sex steroid hormones on the immune system, blood brain barrier, and parenchymal CNS cells. Both clinical and experimental studies have found that sex steroid supplementation may be beneficial in MS patients in order to reduce symptoms. Interestingly, beneficial neuroprotective effects of MS were noted in clinical studies for elevated levels of hormones in both female and male hormones (estrogens, progesterone, and androgen), an elevation that could be related to anti-inflammatory actions on the immune system or the CNS or related to direct neuroprotective properties. It should be mentioned that these actions can also be seen in estrogen receptor regulators in animal model. These observations may further stimulate current clinical studies to determine the efficacy of and tolerance to sex steroid therapeutic approaches for MS as well as other related diseases.
Interestingly, a gender-based method that uses sex-specific and genotype-specific primary cultures was recently established. Astrocytes, a main type of glial cells, showed sex differences against oxygen-glucose deprivation (OGD). Wild-type female astrocytes were more resistant to OGD than were wild-type male cells, but this sex difference disappeared in aromatase knockout cells. In combination, these data suggest a critical role of the androgen-aromatase-estrogen network in protecting cells under stress conditions. However, sex differences in oligodendrocyte, another glial cell and the original target of MS, has not been reported. Therefore, a sex-specific oligodendrodyte study may help further our understanding of the role of gender difference in MS etiology and in MS therapeutics.
The prevalence of MS was higher in Scandinavia, Iceland, the British Isles, and North America (∼1-2 per 1,000) than in southern Europe (with the notable exception of Sardinia). According to some observers, this geographical distribution implicates an environmental disease pathogen that may not be ubiquitously distributed. However, the geographic distribution of MS might also be explained, at least in part, by regional variations in genetic risk factors. Interestingly, residential or occupational exposure of MS patients to sunlight may be associated with a lower mortality rate from MS slower progression with vitamin D mediating this effect. Since ultraviolet radiation is the principal catalyst for endogenous vitamin D3 synthesis in humans, and low levels of vitamin D3 are more common at northern latitudes than at southern latitudes, this may be another reason for persons in southern European countries having lower rates of MS.
2.4 MS pathophysiology
2.4.1 Autoimmune Attacks, Pre-active Lesions, and MS Lesions
Pro-inflammatory cytokines, such as interferon and tumor necrosis factor beta released by activated Th1 cells may upregulate the expression of cell-surface molecules on neighboring lymphocytes and antigen-presenting cells. The binding of putative MS antigens may trigger an enhanced immune response against the bound antigens. Such putative MS antigens include components of myelin, such as myelin basic protein, myelin-associated basic glycoprotein, myelin oligodendrocyte glycoprotein (MOG), proteolipid protein (PLP), and others in the trimolecular complex, the T cell receptor, and major histocompatibility complex class II molecules on APCs may trigger either an enhanced immune response against the bound antigens .
In addition to the autoimmune response, oligodendrocyte death, axon damage, and even neuronal loss have also been associated with MS inflammatory attacks on the CNS. However, the reason for these attacks is largely unknown, although genetic factors may influence immune-mediated inflammation as well as neuronal and glial survival by modulating the MS phenotype (). Therefore, autoreactive T cells are thought to be generated in response to the interplay of (environmental) triggers and genetic susceptibility factors. Differentiation of such CD4+ T cells results in pro-inflammatory Th1, Th17 cells and/or regulatory Th2 cells, all of which produce cytokines such as interferon-gamma, IL-17, IL-4 and IL-10. After activation, myelin-specific T cells are able to cross the blood-brain- barrier via interaction of adhesion molecules, such as vascular cell adhesion molecule-1. In the CNS, including the cerebral cortex, reactivation of these T cells involves local APCs. These APCs initiate a detrimental cascade that typically involves the attraction of microglia, macrophages, CD8+ T cells, and plasma cells, which produce myelin-specific antibodies. It may be that, in MS, these tiered mechanisms in combination may lead to mitochondrial dysfunction, neuronal demyelination, and irreversible tissue damage characterized by axonal loss and gliosis. Recent evidence showed that myelin-specific T cells also recognize neuronal autoantigen in a mouse model of MS, further indicating that multiple autoantigens may be involved in spontaneously developing human MS disease.
These features of tissue damage were found in brain and spinal cord tissue from classic MS lesions, termed reactive lesions. However, recently a new concept, termed preactive lesions, has been used to refer to early pathological changes that occur before the actual development of the reactive (active, demyelinating) lesion .
Indeed, focal disorder has been documented in normal-appearing white matter of MS patients months to years before the appearance of gadolinium-enhancing lesions. Clusters of activated microglia cells have been identified in these lesions through MRI and immunohistochemistry, notably in the absence of demyelination and clear leukocyte infiltration; distinguishing them from the traditional demyelinating active lesions and chronic active lesions [50-52]. Preactive lesions can also be seen in the grey matter, particularly in this part there may be variable degrees of demyelination, along with regions that will eventually become overtly lesion containing and areas of remyelination .
The activated state of microglia cells was also reflected by increased expression of human leukocyte antigen-DR (HLA-DR) and CD68. In addition, foamy macrophages were occasionally found in some of the clusters. Together, these features strongly suggested that the progression of MS may include a stage that actually precedes what has been termed the traditional reactive MS lesion. Although events that give rise to preactive lesions are still to be identified, oligodendrocyte abnormalities appear to be crucially involved (. Importantly, preactive lesions do not always develop into demyelinating lesions. Therefore, preactive lesions in MS may represent early stages in the development of MS lesions. As many of them spontaneously resolve, they are expected to hold important clues to halt the inflammatory demyelinating process in MS. While the activation of pro-inflammatory mechanisms in microglia may favour disease progression, the upregulation of genes involved in anti-inflammatory and antioxidative mechanisms driven by oligodendrocytes and astrocytes may protect the CNS environment and thus limit lesion formation. Interestingly, a dysfunction of mitochondria in lesions as well as in the normal-appearing white and grey matter is increasingly recognized in MS and could be an important determinant of axonal dysfunction and degeneration. Together, these observations indicate that mitochondria and mitochondrial-targeted antioxidant agents may have the potential for the disease, in addition to anti-inflammatory.
2.4.2 Cellular Ionic Imbalance
Intracellular environments, especially ionic balance, are critical for maintaining neuronal functions. Ionic imbalance has been hypothesized to be a key mechanism of MS pathophysiology. In the progression of MS, inflammatory mediators, including cytokines, oxidants, and nitric oxide, are released by microglia or are generated by hypoxia, which is secondary to tissue damage and which is believed to result in a malfunction of oxidative metabolism in the demyelinated axon. These mediators deplete ATP and perturb mitochondrial function, causing failure of the Na+-K+ ATPase, the enzyme that is responsible for rapidly correcting Na+ and K+ levels and for extruding Na+ from the axon and preventing a pathological influx of Na+ in both resting and active axons .
Hypoxia is considered to be a physiological stress that induces a replication-associated DNA damage response. It has been shown that hypoxia can inhibit Na+-K+ ATPase activity and ROS increases Na+-K+ ATPase degredation. Even in normal appearing white matter in MS, microarray analysis revealed that transcription factor HIF-1alpha, a key regulator of hypoxia-induced gene regulation, and its downstream genes were significantly and consistently upregulated, indicating a hypoxia condition in this area. As shown in studies of anoxia, the high intra-axonal Na+ concentration that results from this failure will cause increased activity of the Na+-Ca2+ exchange channel, with the efflux of Na+ requiring a higher degree of Ca2+ influx. This, in turn, activates intra-axonal proteases, resulting in neurofilament fragmentation and perturbation of axon transport and integrity, ultimately leading to neuronal degeneration. In fact, Na+-K+ ATPase enzymatic activity and distribution were reduced or undetectable in chronic MS patients (. It appears that chronically demyelinated axons that lack Na+-K+ ATPase cannot exchange axoplasmic Na+ for K+ and are incapable of nerve transmission. Therefore loss of axonal Na+-K+ ATPase is likely to be a major contributor to continuous neurological decline in chronic stages of MS.
2.4.3 Dysfunction of cellular clearance systems
In experimental models of demyelinating disease in aged animals, as well as in multiple sclerosis, oligodendrocyte precursor cells (OPCs) differentiation appears to be impaired. This is due, at least in part, to changes in environmental signals governing remyelination. In particular, myelin debris within lesions appears to contain powerful inhibitors of precursor cell differentiation. It has been shown that the glycosaminoglycan hyaluronan (HA) accumulates in demyelinated lesions from patients with MS and in mice with EAE, and that HA can prevent remyelination by inhibiting OPC maturation. Efficient removal of such molecules and myelin debris by macrophages (microglia) and other functional systems may thus facilitate OPCs differentiation and permit successful remyelination of damaged axons. Interestingly, the elimination of myelin debris is extremely efficient in young animals, whereas old animals show very poor clearance of myelin debris. Systemic progesterone administration could reverse partially this age-associated decline in CNS remyelination in male rats. These observations indicate that the inhibitors of remyelination are increased and/or clearance systems are not efficient in aged animals and steroid hormones and tissue/neurotrophic factors may be involved in this process. Further identifying signaling molecules in this network (the myelin sheath and myelin debris) probably represents very promising therapeutic targets for pharmacological strategies aimed at enhancing remyelination.
Autophagy is a newly recognized cellular functional system that delivers cytoplasmic materials to lysosomes for degradation. The formation of autophagosomes is controlled by a specific set of autophagy-related genes, called atg genes. Autophagy is thought to be a major, evolutionarily conserved response to nutrient and bioenergetic stresses. Autophagy has been hypothesized to remove aggregated proteins and damaged organelles, such as mitochondria. Recent studies have provided evidence that autophagy is another mechanism of programmed cell death, termed autophagic programmed cell death or secondary programmed cell death to distinguish from apoptosis, thereby possessing important housekeeping and quality-control functions that contribute to health and longevity.
Autophagy also plays a role in innate and adaptive immunity, apoptosis, neurodegeneration, and aging, as well as the prevention of cancer. However, excessive or imbalanced induction of autophagic recycling can actively contribute to neuronal atrophy, neurite degeneration, and cell death .
The role of autophagy in T cells was recently examined in mouse CD4+ T cells. Interestingly, resting naive CD4+ T cells do not contain detectable autophagosomes. Autophagy can be observed in activated CD4+ T cells upon TCR stimulation, cytokine culturing, and prolonged serum starvation. Induction of autophagy in T cells requires JNK and the class III PI3K. Autophagy is inhibited by caspases and mammalian target of rapamycin in T cells and more Th2 cells than Th1 cells undergo autophagy. Th2 cells become more resistant to growth factor-withdrawal cell death when autophagy is blocked using either chemical inhibitors 3-methyladenine, or by RNA interference knockdown of Atg7 and beclin. Therefore, autophagy is an important mechanism that controls homeostasis of CD4+ T cells .
Very recently, Alirezaei and colleagues examined the expression of Atg5 genes in T cells using both a mouse model of autoimmune demyelination as well as blood and brain tissues from MS patients. Quantitative real-time PCR analysis of RNA isolated from blood samples of the experimental autoimmune encephalitis (EAE) mice revealed a strong correlation between Atg5 expression and clinical disability. Analysis of protein extracted from the T cells confirmed both the upregulation and post-translational modification of Atg5 genes, the latter of which was positively correlated with EAE severity. Analysis of RNA extracted from T cells isolated by negative selection indicated that Atg5 expression was significantly elevated in patients with active RRMS compared to non-diseased controls. Brain tissue sections from RRMS patients, examined by immunofluorescent histochemistry, suggested that encephalitogenic T cells may be a source of Atg5 expression in MS brains. Together, these data suggest that increased T cell expression of Atg5 may contribute to inflammatory demyelination in MS .
Another clearance machinery is the ubiquitin-proteasome system (UPS). The destruction of proteins is as important as their synthesis for the maintenance of protein homeostasis in cells. In eukaryotes, the ubiquitin-proteasome system is responsible for most protein degradation: the small protein ubiquitin acts as a death warrant, tagging and targeting other proteins to the large proteolytic chamber of the proteasome. It is now known that ubiquitin-mediated destruction plays a crucial part in many basic cell functions. Given the central role of UPS in diverse cellular processes, it is not surprising that its dysfunction contributes to neurodegenerative and immunological disorders, either as a primary cause or secondary consequence (. Importantly the proteolysis system is ATP-dependent. Recently it has been shown that assembly of the proteasome base is a rapid yet highly orchestrated process, and proteasome regulatory particle is chaperone-mediated .
The autoimmune process of PLP139-151-induced relapsing experimental autoimmune encephalomyelitis is regulated, in part, by the transcription factor nuclear factor (NF)-kappaB, which is activated via the UPS. Administration of PS-519, a selective inhibitor of the ubiquitin-proteasome pathway, during the remission phase of MS following an acute attack was effective in significantly reducing the incidence of clinical relapses, CNS histopathology, and T cell responses to both the initiating and the relapse-associated PLP epitopes. The inhibition of clinical disease was dependent on a continuous administration of PS-519 in that recovery of T cell function and onset of disease relapses developed within 10-14 days of drug withdrawal (. The data indicated that UPS is involved in relapsing EAE, and they suggested that targeting the UPS, in particular the NF-kappaB, may offer a novel and efficacious approach for decreasing progressive autoimmune diseases, including MS.
Some proteinases may be involved in the proteolysis of immune antigens and may be involved in the progression of MS. It is generally accepted that the processing of MHC 1 antigens is mediated by UPS pathway. In addition, matrix metalloproteinase proteolysis plays a significant role in the fragmentation of MBP. The classic MBP isoforms are predominantly expressed in the oligodendrocytes of the CNS. A recent in vitro cleavage study determined that MBP, and its splice variants, are highly sensitive to redundant matrix metalloproteinases proteolysis. MT6-MMP (initially called leukolysin), however, was superior over all of the other MMPs in cleaving the MBP isoforms. This study demonstrated that matrix metalloproteinase proteolysis of the MBP and its isoforms is a source of immunogenic peptides in autoimmune MS. If this is the case in vivo, in some cases matrix metalloproteinase proteolysis may directly destroy MBP and initiate demyelination in MS or EAE.
Protease-activated receptors are G protein-coupled receptors that regulate the cellular response to extracellular serine proteases. The PAR family consists of four members: PAR-1, -3, and -4 as thrombin receptors, and PAR-2 as the trypsin/tryptase receptor. These four members are abundantly expressed in the brain throughout development of MS. The expression of PARs in the brain is differentially upregulated or downregulated under pathological conditions in neurodegenerative disorders, including MS (. Noorbakhsh et al. found that PAR2 expression was significantly increased on astrocytes and infiltrating macrophages in human MS and murine EAE CNS white matter. Indeed, PAR2 wild-type mice showed markedly greater microglial activation and T lymphocyte infiltration accompanied by worsened demyelination and axonal injury in the CNS compared to their PAR2 knockout littermates. Enhanced neuropathological changes were associated with a more severe, progressive relapsing disease phenotype in wild-type mice. These studies revealed pathogenic interactions between CNS PAR2 expression and neuroinflammation with ensuing demyelination and axonal injury. Therefore, PARs are capable of mediating either neurodegeneration or neuroprotection in MS as well as other neurodegenerative disorders, and they represent attractive therapeutic targets for treatment of these diseases.
3. Experimental autoimmune encephalitis model of MS
Currently there are no genetically engineered mouse models available to study MS progression in mice. However, several induced mouse models have been generated, particularly EAE in mouse. The EAE can be induced by immunization of susceptible animals with a number of myelin antigens including myelin basic protein, PLP, and MOG. The origins of EAE date back to the 1920s, when Koritschoner and Schweinburg induced spinal cord inflammation in rabbits by inoculating with tissue from a human spinal cord. Since then, EAE has been developed in many different species, including rodents and primates. EAE is an animal model of MS that exhibits the functional characteristics of human immune molecules in vivo. The ‘humanized MS animal models allow the functional characterization of human immune molecules in vivo. We emphasized that MOG although a minor component of the myelin sheath, is a potent encephalitogenic protein that induces EAE in many strains and species of experimental animals, particularly monkey model of MS that may closely mimic human disease, may provide a unique experimental platform to understand the mechanisms of disease process, and also to develop therapeutic strategies for MS .
It is clear that EAE can mimic many of the clinical, neuropathological, and immunological aspects of MS. In particular, EAE appears to mimic most closely the disability-related axonal loss seen in MS and may provide a convenient opportunity to study axon-damaging mechanisms of relevance to MS. More importantly, EAE has led directly to the development of three therapies approved for use in MS: glatiramer acetate (copaxone), mitoxantrone, and natalizumab. Several new approaches to studying MS in clinical trials have also been based on preclinical work relying on EAE.
There are a few limitations in using EAE as a research model for MS. First, MS is a spontaneous disease, while EAE is induced by active sensitization with brain tissue antigens and strong immune adjuvants. Second, genetic heterogeneity of MS in the human populations. To understand the disease progression and pathology of MS in mice, it is important to study multiple mouse models of EAE that may provide more human MS features. Therefore, when used appropriately, the EAE model provides a crucial tool for improving our understanding of and treatment of MS.
In contrast, the compelling MS in vitro model has not been developed thus far. However, a few related systems were reported for the MS/EAE mechanism study in some degree using oligodendrocyte or neuron co-culture with microglial cells .
4. Multiple sclerosis/experimental autoimmune encephalitis is a neurodegenerative disorder
Axonal loss occurs in MS and is responsible for the permanent disability characterizing the later chronic progressive stages of the disease. Immunohistochemistry of brain tissues showed that the expression of amyloid precursor protein, a sensitive marker of axonal damage, occurs in axons within acute MS lesions and in the active borders of less acute lesions that had not been identified as MS. Recently, evidence for widespread axonal damage even at the earliest clinical stages of MS has been reported, leading to the hypothesis that MS is a neurodegenerative disorder in which axonal injury, neuronal loss, and atrophy of the CNS begin in the earliest stages of the disease and then intensify over time, even axonal loss could be found in normal-appearing white matter in a patient with acute MS. Such evidence has called into question the previously long-held hypothesis that axonal pathology is the end-stage result of repeated inflammatory events in MS and argues strongly in favor of early neuroprotective intervention .
Axon loss has also been found in animal models of MS, especially in the EAE model, and has been found to correlate with permanent neurological disability in the animal models. This chronic-relapsing EAE model provides an excellent platform for two critical research objectives: determining mechanisms of axon loss in MS and evaluating the efficacy of neuroprotective therapies.
5. Mitochondria dysfunction and ROS as causes of neuronal degeneration in MS
5.1 Mitochondria, neurodegenerative diseases and MS
Figure 2 summarizes the involvement of mitochondrial abnormalities in patients with MS and EAE mouse models. As shown, current research revealed that the following mitochondrial abnormalities are involved in the development and progression of multiple sclerosis: 1) mitochondrial DNA defects, 2) abnormal mitochondrial gene expression, 3) defective mitochondrial enzyme activities, 4) deficient mitochondrial DNA repair activity 5) and mitochondrial dysfunction. We propose that abnormal mitochondrial dynamics (increased fission and decreased fusion in neurons affected by MS). Further, we also propose that mitochondrial abnormalities and mitochondrial energy failure may impact other cellular pathways, including increased demyelination and inflammation in neurons and tissues that are affected by multiple sclerosis. The details are given below.
Figure 2.
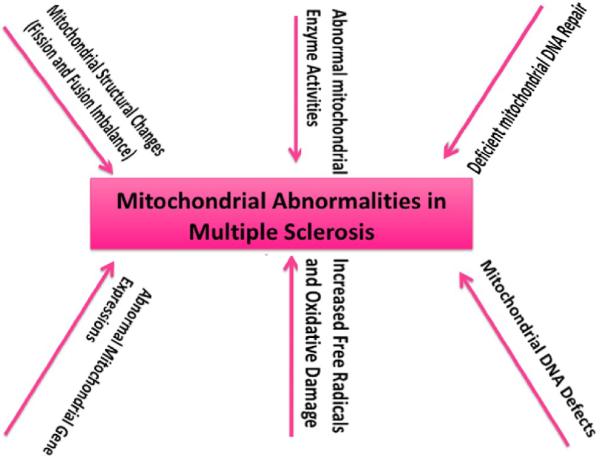
Mitochondrial abnormalities in patients with multiple sclerosis and EAE mouse models. Based on current research, we propose that mitochondrial abnormalities are involved in the development and progression of multiple sclerosis, including mitochondrial DNA defects, abnormal mitochondrial gene expression, defective mitochondrial enzyme activities, abnormal or deficient mitochondrial DNA repair mechanisms, and mitochondrial dysfunction. We propose that abnormal mitochondrial dynamics (imbalance in mitochondrial fission and fusion) plays a key role in tissues affected by multiple sclerosis. We also propose that mitochondrial abnormalities and mitochondrial energy failure may impact other cellular pathways including increased demyelination and inflammation in neurons and tissues that are affected by multiple sclerosis.
Mitochondria contain the respiratory chain where energy in the form of ATP is most efficiently produced. The mitochondrial respiratory chain is located in the inner mitochondrial membrane and consists of five complexes (complexes I-V); the fifth complex is directly involved in ATP synthesis. The complexes of the mitochondrial respiratory chain are made up of multiple subunits, and all contain proteins encoded by nuclear DNA and mtDNA, except for complex II, which is entirely encoded by nuclear DNA. Neurons are highly dependent on oxidative energy metabolism. Axons, in particular, consume significant amounts of ATP, which it uses primarily to fuel the sodium/potassium ATPase, or sodium pump that functions to remove the sodium ions that enter the axon during impulse activity. Mitochondria are not only the energy factory for cells but also the seat of a number of important cellular functions, including essential pathways of intermediate metabolism, amino acid biosynthesis, fatty acid oxidation, steroid metabolism, calcium handling and apoptosis. Of key importance is the role of mitochondria in oxidative energy metabolism. Oxidative phosphorylation generates most of the cell’s ATP, and any impairment of the organelle’s ability to produce energy can have catastrophic consequences, not only due to the primary loss of ATP, but also due to indirect impairment of downstream events. Moreover, the production of superoxide occurs mostly within the mitochondria, mainly in complexes I and III, TCA cycle and conditionally in complex II .
Deficient mitochondrial metabolism may generate more reactive oxygen species (ROS) that can wreak havoc in the cell. Therefore, mitochondrial dysfunction is an attractive candidate for neuronal degeneration. Impairment of mitochondrial energy metabolism is the key pathogenic factor in a number of neurodegenerative disorders, such as Alzheimer’s disease and Parkinson’s disease. Hence, therapeutic approaches targeting mitochondrial dysfunction and oxidative damage in neurodegenerative diseases, including MS have great promise .
Recently, several lines of evidence suggests that mitochondrial dysfunction is present in patients with MS. Mitochondrial DNA alterations, mitochondrial structural changes, defective mitochondrial DNA repair events, abnormal mitochondrial enzyme activities, mitochondrial gene expressions, increased free radical production and oxidative damage have been reported in patients with MS and EAE mouse models (Figure 2).
5.2 Mitochondrial DNA alternations in MS
Age-related decline of mtDNA copy number is associated with late-onset MS. mtDNA mutations may increase the risk of MS. SNP analysis has shown that genetic variants of complex I genes may influence the response of tissues to inflammation in the CNS. Further, genetic alterations in uncoupling proteins are reported to be implicated in patients with MS. Uncoupling protein 2 (UCP2) is a member of the mitochondrial proton transport family that uncouples proton entry to the mitochondria from ATP synthesis. Vogler and colleagues reported that the UCP2 common -866G/A promoter polymorphism is associated with susceptibility to MS in a German population. In a study of 1,097 MS patients and 462 control subjects, they found the common G allele associated with disease susceptibility (p = 0.0015). The UCP2 -866G allele was correlated with lower levels of UCP2 expression in vitro and in vivo. Thus, UCP2 may contribute to MS susceptibility by regulating the level of UCP2 protein in the CNS and/or in the immune system .
Defects in mtDNA have been associated with late onset MS. Ban and colleagues (2008) sequenced the mtDNA from 159 patients with MS and completed a haplogroup analysis of 835 MS patients and 1,506 controls. They found a trend towards over-representation of super-haplogroup U as the only evidence for association with MS. In a parallel analysis of nuclear-encoded mitochondrial protein genes in the same subjects, they also found a trend towards association with the complex I gene, NDUFS2. Taken together, these studies have contributed to evidence suggesting that variations in mtDNA and nuclear-encoded mitochondrial protein genes may contribute to disease susceptibility in MS.
A study of MS patients in Europe showed that a potentially functional mtDNA SNP, nt13708 G/A, was significantly associated with an increased risk of MS (P = 0.0002). The study identified the nt13708A variant as a allele susceptible to MS, which may suggest a role in MS pathogenesis. Recently, Vyshkina et al. discovered an association among common variants of the mitochondrial ND2 and ATP6 genes with both MS and systemic lupus erythematosus. This finding raises the possibility of a shared mitochondrial genetic background between these two autoimmune diseases. On the other hand, an increasing number of case reports on Leber’s hereditary optic neuropathy (LHON) associated mtDNA point mutations, and some patients with MS and LHON share the same mtDNA mutation, suggesting that mitochondrial determinants may contribute to genetic susceptibility in MS and LHON .
In fact, only a very small subgroup of MS patients, usually with prominent optic neuritis, may carry pathogenic LHON mutations. This overlap between the two diseases may be related to the association of MS with an mtDNA haplotype (a set of mtDNA polymorphisms) within which pathogenic LHON mutations preferentially occur. In a recent study, 58 unrelated Bulgarian patients with RRMS and 104 randomly selected healthy individuals were analyzed for the presence of 14 mtDNA polymorphisms determining major European haplogroups as well as three (4216, 14 798, 13 708) secondary LHON mutations. Restriction enzyme analysis, used to screen patients and controls for common haplogroup-associated polymorphisms, showed that each of these changes which occurred in MS patients at a similar rate to control subjects. However, 21 of the 58 patients (36.2%) were positive for the T4 216C mutation, while only 11.3% of the controls carried this mutation (P < 0.01; OR = 4.38), suggesting that the 4216C base substitution may be a predisposing marker for MS. These findings also supported the hypothesis that particular mtDNA variants may contribute to the genetic susceptibility of some people with MS. To further study the relationship between LHON and MS, Hwang et al. tested 20 Korean MS patients for the presence of mtDNA mutations at nucleotide (nt) 11778, and nt 14484, 3460, and 15257. However, none of the MS patients exhibited any pathogenic LHON mtDNA mutations. This result is in agreement with that of Japanese MS patients. It may be the case that racial characteristics may influence the association.
5.3 Mitochondrial dysfunction in MS
Increasing evidence suggests that mitochondrial dysfunction is involved in MS. Ultrastructural analysis of demyelinated spinal cord lesions showed dramatically reduced numbers of mitochondria and microtubules, and demonstrated Ca2+-mediated destruction of chronically demyelinated axons and axonal swelling. Further, the gene expression study showed an unbalanced gene expression in MS patients. As reduced energy production is a major contributor to Ca2+-mediated axonal degeneration, authors focused on changes in oxidative phosphorylation and inhibitory neurotransmission. Compared with controls, 488 transcripts were decreased and 67 were increased in the MS cortex. Twenty-six nuclear-encoded mitochondrial genes and the functional activities of mitochondrial respiratory chain complexes I and III were decreased in the MS motor cortex. Reduced mitochondrial gene expression was specific for neurons. In addition, synaptic components of GABAergic neurotransmission and the density of inhibitory interneuron processes also were decreased in the MS cortex.
In addition, recently a number of mitochondrial respiratory chain proteins in active lesions from acute MS was analyzed using immunohistochemistry. Functionally important defects of mitochondrial respiratory chain complex IV [cytochrome c oxidase (COX)] including its catalytic component (COX-I) are present in some active MS lesions (Pattern III). The lack of immunohistochemically detected COX-I is apparent in oligodendrocytes, hypertrophied astrocytes and axons, but not in microglia. These findings suggest that hypoxia-like tissue injury in Pattern III MS lesions may be initiated from mitochondrial impairment. On the other hand, in inactive areas of chronic MS lesions the complex IV activity and mitochondrial mass, judged by porin immunoreactivity, are increased within approximately half of large chronically demyelinated axons compared with large myelinated axons in the brain and spinal cord. The axon-specific mitochondrial docking protein (syntaphilin) and phosphorylated neurofilament-H were increased in chronic lesions. These results clearly indicate an adaptive change of mitochondrial function and morphology in chronic MS.
Recently, Regenold and colleagues investigated the relationship between disturbed CNS mitochondrial energy metabolism and MS disease progression by measuring cerebrospinal fluid (CSF) concentrations of sorbitol, fructose, and lactate, all metabolites of extra-mitochondrial glucose metabolism. They found that concentrations of all three metabolites, but not concentrations of glucose or myoinositol, were significantly increased in CSF from secondary progressive and, to a lesser degree, relapsing-remitting patients, compared to healthy controls. Furthermore, CSF concentrations of sorbitol and fructose (polyol pathway metabolites), but not lactate (anaerobic glycolysis metabolite), correlated positively and significantly with Expanded Disability Status Scale (EDSS) score, an index of neurologic disability in MS patients. These findings suggest that abnormal mitochondrial glucose metabolism is increased in MS patients and is associated with disease progression .
Interestingly, analysis of mitochondrial enzymes on human muscle showed that in people with MS, there were fewer type I fibers, and that fibers of all types were smaller and had lower succinate dehydrogenase (SDH, component of the respiratory chain complex II) and SDH/alpha-glycerol-phosphate dehydrogenase (GPDH) but not GPDH activities, suggesting that muscle in this disease is smaller and relies more on anaerobic than aerobic-oxidative energy supply than does muscle of healthy individuals. Similar to brain, muscles are also highly dependent on mitochondrial oxidative energy metabolism, so it is reasonable that there is a weaker muscle in MS patients, indicating muscle is also one of the targets of MS. In some rare cases, MS could have a mitochondrial myopathy combination, in which MRI showed widespread white matter lesions, muscle biopsy showed ragged red fibres and COX (complex IV) deficiency, Southern blot analysis revealed a large deletion of mtDNA. Probably the severe mitochondrial genomic deletion is the key cause or initiation factor for this special case.
Another interesting key issue of mitochondria must be discussed below. The mitochondrial permeability transition leads to mitochondrial swelling, outer membrane rupture and the release of apoptotic mediators. The mitochondrial permeability transition pore (PTP) is thought to consist of the adenine nucleotide translocator, a voltage-dependent anion channel, and cyclophilin D (CyPD, the Ppif gene product), a prolyl isomerase located within the mitochondrial matrix. CyPD is a key regulator of the PTP and they are required for mediating Ca2+- and oxidative damage-induced cell death. In experimental animal MS disease model, EAE mice lacking CyPD showed that neurons missing CyPD, are resistant to oxidative agents thought to be the mediators of axonal degeneration observed in both EAE and MS and have mitochondria that are able to more effectively handle elevated Ca2+. Consistent with this neuronal resistance, animals missing CyPD are able to recover, clinically, following the induction of EAE. These results directly implicate pathological activation of the mitochondrial PTP in the axonal damage occurring during MS, in other word, PTP and mitochondria are the critical target of EAE, perhaps multiple sclerosis.
5.4 Oxidative stress in MS
Reactive oxygen species (ROS) are the by-products of cellular metabolism. Excessive ROS or an imbalance between cellular production of ROS and the ability of cells to defend against them is referred to as oxidative stress. Oxidative stress can cause cellular damage and subsequent cell death because the ROS oxidize critical cellular components, such as lipids, proteins, and DNA especially mitochondrial DNA. In neurodegenerative and neuroinflammatory disorders, there is evidence for a primary contribution of oxidative stress in neuronal death, as opposed to other diseases where oxidative stress more likely plays a secondary or by-stander role .
There is increasing evidence that oxidative stress is an important component in the pathogenesis of MS. The inflammatory environment in demyelinating lesions is conducive to the generation of reactive oxygen species. Macrophages and microglia are known to express myeloperoxidase (MPO) and generate ROS during myelin phagocytosis in the white matter. Recent research involving the cerebral cortex in MS indicates that microglial production of ROS is also likely to be involved in cortical demyelination. Protein kinase C (PKC) could induce an increased production of ROS in mononuclear cells of patients with MS compared to those of controls, and it was predominantly or exclusively generated by PKC activated NADPH oxidase. The concentrations of reactive oxygen and/or nitrogen species (e.g. superoxide, nitric oxide and peroxynitrite) can increase dramatically under conditions such as inflammation, and this can overwhelm the inherent antioxidant defences within lesions. Such oxidative and/or nitrative stress can damage the lipids, proteins and nucleic acids of cells and mitochondria, potentially causing cell death. Oligodendrocytes are more sensitive to oxidative and nitrative stress in vitro than are astrocytes and microglia, seemingly due to a diminished capacity for antioxidant defence, and the presence of raised risk factors. Oxidative and nitrative stress might therefore result in selective oligodendrocyte death, and thereby demyelination in vivo. The reactive species may also damage the myelin sheath, promoting its attack by macrophages/microglia.
Evidence for the existence of oxidative and nitrative stress within inflammatory demyelinating lesions includes the presence of both lipid peroxidation and protein peroxides (protein carbonyls), and nitrotyrosine (a marker for peroxynitrite formation). When the ROS/RNS are generated in MS and animal models of MS, products such as superoxide and peroxynitrite are formed that are highly toxic to both glia and neuronal cells. Kalman’s group has determined the level of DNA damage in MS patients using 8-hydroxy-deoxy-guanosine (8-OH-dG) as an oxidative marker, they found that a significant increase in DNA oxidation within plaques compared to NAWM specimens in MS cerebella. A tendency for increase of oxidative markers in normal appearing cortical tissues located in the proximity of MS plaques was also observed when compared to those in control cortical specimens. Also, oxidative damage to mitochondrial DNA and impaired activity of mitochondrial enzyme complexes in MS lesions suggest that inflammation can affect energy metabolism, ATP synthesis, and viability of affected cells .
A recent report showed that oxidative stress occurs in progressive as well as benign MS patients. For example, serum diene conjugate levels (a measure of lipid peroxidation) were significantly elevated in MS patients, especially patients with primary progressive phenotypes. However, serum total antioxidative activity and total antiradical activity were not different between MS patients and healthy controls. On the other hand, the chemical composition of human cerebrospinal fluid is considered to reflect brain metabolism, there is experimental evidence of a decrease in sulfhydryl groups (antioxidants) and increased content of products of lipid peroxidation, such as ultraweak chemiluminescence and liposoluble fluorescence, which was higher in the CSF and plasma of MS patients than in controls, clearly pointing out the role of oxidative stress in the pathogenesis of MS .
Recent insights into the molecular pathogenesis of progression in MS also list oxidative stress as one of main mechanisms. More recently, Van Horssen J et al reported the presence of extensive oxidative damage to proteins, lipids, and nucleotides occurring in active demyelinating MS lesions, predominantly in reactive astrocytes and myelin-laden macrophages. It is reasonable that some of structurally-damaged myelin proteins are the targets of immune system, which are recognized as foreign antigens. Hence we consider that if not all, at lease for some MS patients, oxidative stress (including nitrative stress) may be the primary mechanism in pathogenesis.
On the other hand, antioxidant enzymes, including superoxide dismutase 1 and 2, catalase, and heme oxygenase 1, are markedly upregulated in active demyelinating MS lesions compared to normal-appearing white matter and white matter tissue from nonneurological control brains. Enhanced antioxidant enzyme production in inflammatory MS lesions may reflect an adaptive defense mechanism to reduce ROS-induced cellular damage. These data and observations strongly indicate that antioxidant therapy may be a potential treatment for MS patients.
5.5 Nitric oxide and MS
As described above, human blood macrophages, astrocytes, and microglial cells make NO. NO is present at increased concentrations in acute MS lesions and is known to have a deleterious effect on mitochondria. The relationship between NO and cytochrome c oxidase (mt complex IV) has been investigated at different integration levels of the enzyme, including the in situ state, such as in mouse liver mitochondria or cultured human SY5Y neuroblastoma cells. Micromolar NO rapidly inhibits cytochrome c oxidase in turnover with physiological substrates. The respiratory chain is inhibited by NO, either supplied exogenously or produced endogenously via the NO synthase activation. Inhibition of respiration is reversible, although it remains to be clarified whether reversibility is always full and how it depends on concentration of and time of exposure to NO. At least under hypoxic condition, NO irreversibly inhibits cytochrome oxidase. NO and superoxide redical combine to form peroxynitrate (ONOO-), which breaks down to form the highly reactive radicals hydroxyl radical and nitrogen dioxide. In other words NO can enhance the cellular toxicity of ROS.
Peroxynitrite and other reactive nitrogen oxide species exert a toxic effect on neurons, axons and glia cells and enhance apoptosis. In addition, they increase the blood-brain-barrier (BBB) permeability and can therefore promote invasion of inflammatory cells into the CNS. On the other hand, uric acid, a purine metabolite and peroxynitrite scavenger inhibits blood-CNS-barrier permeability changes, CNS inflammation and tissue damage in EAE and in mice with spinal cord injury. More recently the concentrations of uric acid, purine profile and creatinine in samples of cerebrospinal fluid and serum of MS patients were measured in detail by HPLC. The values of all compounds assayed were significantly higher in both biological fluids of MS patients with respect to values measured in controls. In particular, serum hypoxanthine, xanthine, uric acid and sum of oxypurines were, respectively, 3.17, 3.11, 1.23 and 1.27-fold higher in these patients than corresponding values recorded in controls. Though differently from what previously reported, these data clearly demonstrate that all purine compounds, including uric acid, are elevated in biological fluids of MS patients. Reinforced by the trend observed for creatinine, this corroborates the notion of sustained purine catabolism, possibly due to imbalance in ATP homeostasis, under these pathological conditions. As observed in other pathological states, uric acid, purine compounds and creatinine, can be considered markers of metabolic energy imbalance rather than of reactive oxygen species, even in MS.
Nitric oxide synthases (NOS) also play an important role under physiological as well as pathological conditions. Active iNOS enzyme has been demonstrated in astrocytes in acute and chronic-active MS lesions at the lesion edge where de myelination is occurring. QRT-PCR analysis detected significant upregulation of the neuronal form of NOS (nNOS), in most of the MS normal-appearing white matter tissue samples, this change together with the upregulation of HIF-1 in oligodendrocytes and neurons supports the view of oligodendrocyte and/or neuronal dysfunction in this non-lesion containing tissue as a possible primary cause .
6. Development of therapeutic approaches in MS
6.1 immunomodulatory treatments
The major development of the past decades is that MS has changed from an untreatable to a treatable disease. Four immunomodulatory treatments (glatiramer acetate and three interferon-b preparations) and two immunosuppressants (natalizumab and mitoxantrone) are now approved for the treatment of MS and allow the frequency of attacks to be diminished and the progression of disability to be slowed, at least in the shorten medium-term. In addition, a number of new strategies were developed for the treatment of the disease, such as novel immunomodulators (including antibodies/immunosuppressants), therapeutic strategies targeting leukocyte differentiation molecules, costimulatory molecules, anti-adhesion molecules, chemotaxis, autologous stem cell transplantation, anti-infectious therapies and strategies for neuroprotection neurorepair and remyelination (for detail, see previous reviews). Furthermore, there are good scientific rationales for the use of combination therapy in MS, and the clinical trials are also currently underway to establish the therapeutic efficacy and safety of various combination therapies for MS patients. Recently a small scale, open-label, 7-month trial of combination therapy with intramuscular interferon beta-1a and oral doxycycline, a potent inhibitor of matrix metalloproteinases, in patients with RRMS was successfully completed. The combination of doxycycline and interferon beta-1a treatment resulted in reductions in contrast-enhancing lesion numbers and posttreatment Expanded Disability Status Scale values (P < 0.001 for both). Totally this combination treatment was effective, safe, and well tolerated. To evaluate the efficacy and tolerability of this combination, more important work is double-blind, randomized, placebo-controlled clinical trials in larger cohorts of patients with MS. This hopeful trial may encourage related reasonable combination study in the near future. In EAE animal models, some novel combination or cocktail therapy studies already showed the therapeutic efficacy. For instance, multiple Ag peptides (MAPs) containing 8 PLP (139-151) peptides arranged around a dendrimeric branched lysine core were tested to influence the expression and development of relapsing EAE in SJL mice. The PLP (139-151) MAPs were very efficient agents in preventing the development of clinical disease when administered after immunization. The treatment effect with these MAPs was peptide specific and long lasted over a 60-day observation period. Such effective cocktail agents should be tested in different EAE models (including non-human primate model) prior to clinical patient trial. Hence evaluation of specific combination therapies in the controlled setting of preclinical studies and clinical trials should be a priority in MS research.
6.2 Complementary and alternative medicine for MS
MS is a chronic, unpredictable neurological disease that mainly affects the CNS and has no known cure. Because of this, many people with MS often seek complementary and alternative medicine (CAM) therapies to manage their disease symptoms. Results from the literature showed that around 63% of people with MS reported that they used one or more CAM therapies. The major reasons for choosing CAM were as follows: conventional treatment was not effective, anecdotal reports of CAM’s help, and doctor referral. Common CAM therapies that people use include dietary modification, nutritional and herbal supplementation, and mind-body therapies, including acupuncture and massage. There is a revival of interest among MS researchers about the therapeutic potential of low-fat diet and essential fatty acid supplementation in MS. The efficacy of specific vitamin supplementation remains unclear. Recently, cannabis and yoga have been studied in more controlled studies and have provided evidence that they may have some benefit. The research on CAM therapies in MS is still exploratory, but considering peoples’ interest and common use of these therapies, further research in this area is clearly warranted .
Traditional Chinese medicine (TCM) has an intrinsic system for treatment of patients. It has been used for several thousands of years and today it is still an important part of the Medicine in China. Interestingly, the Journal of the Integration of Chinese Medicine and Western Medicine showed that the most MS patients could be divided to two main types by syndrome typing according to their clinical manifestations, the Gan-Shen yin-deficiency (GSYD) type and the both yin-yang deficiency (YYD) type. They found that the age of first attack was later, level of MBP in cerebrospinal fluid was higher, in the YYD type than those in the GSYD type. Besides, the relapsing time in GSYD type, and the blood-brain barrier index and level of MBP in YYD type showed an ascending trend (P = 0.056, 0.074, 0.093, respectively). The distinguishing and classification of patients with MS is the first step for treatment of patients with the disease by using TCM. However, more important issue is randomized, well-controlled clinical trial on the MS patients. This is a difficult task but it should be completed as soon as possible. On the other hand, the existed immunological difference between the MS patients of GSYD type and those of YYD type indicates that traditional Chinese medicine has scientific basis even it is not fully understood. Recently, an Iranian herbal-marine medicine, MS14, was reported to ameliorates experimental allergic encephalomyelitis in mice. Hence the CAM exploration of both basic and clinical study is widely performed and it would be expected to provide more useful therapeutic agents for treatment of MS patients.
6.3 Mitochondria targeted approaches in MS
As described above, multiple sclerosis is very complex, it considered to be an autoimmune and neurodegenerative disease. Current treatments for MS include immunomodulatory agents but no neuroprotective or regenerative therapy is available. Especially not all patients with MS respond well to treatment with these agents. From various animal models, we have learned that remyelination in the CNS is a potent neuroprotective mechanism. The knowledge about oligodendrocyte biology and the process of remyelination has greatly increased in recent years; however, the precise mechanisms are far from being understood, and remyelination, neurogeneration as well as neuroprotection occur only in animal models. Although remyelination is, in principle, also possible in the diseased MS brain, it is not clear why it fails in many MS patients. The clinical trials performed so far either failed to show an effect or were insufficient in design. Thus, further knowledge about the molecular mechanisms of the repair processes and MS pathophysiology is required to achieve the ultimate goal of a neuroprotective and neuroregenerative treatment in MS.
As described above, CyPD knockout mouse studies have provided evidence of direct links between mitochondrial function, Ca2+ overload, and axonal destruction during EAE, and by extension, MS. Hence, compounds that are able to inhibit the PTP by inactivation of CyPD specifically or by other mechanisms that modulate the PTP, would seem to warrant investigation as neuroprotective therapies in MS. Intravenous mitoxantrone (novantrone) treatment improved neurological disability and delayed progression of MS in patients with worsening relapsing-remitting or secondary-progressive disease. Regarding the action mechanisms of this approved immunosuppressant mitoxantrone, a synthetic analog of anthraquinone, at least in tumor cells, the mitochondrial PTP targeted mechanism is involved .
Mitochondrial DNA repair is a new and important part for neuroprotection in aging related diseases including MS. Multiple pathways of DNA repair have been elucidated for nuclear DNA. However, it appears that only base excision repair is functioning in mitochondria. This repair pathway is responsible for the removal of most endogenous damage including alkylation damage and oxidative damage. Interestingly, Astrocytes exhibit efficient repair, whereas, other glial cell types and neuronal cells exhibit a reduced ability to remove lesions from mtDNA. A strategy of targeting DNA repair proteins to mitochondria to enhance mtDNA repair capacity was developed. Enhancement of mtDNA repair in oligodendrocytes provided protection from reactive oxygen species- and cytokine-induced apoptosis. These experiments provide a novel strategy for protecting sensitive CNS cells and thus provide new treatment options for neurodegenerative diseases, such as MS.
As described in Pathology section, ion channels and ionic imbalance are involved in the MS initiation and development, some channel blockers should protect axons from inflammatory mediators. In fact, Na+ channel blocker tetrodotoxin, which can block Na+ influx throughout the anoxic period, has been shown to preserve ATP levels, concurrent with protecting white matter axons from NO-induced injury. Another Na+ channel blockers phenytoin and flecainide have been shown to be protective in mouse and rat models of EAE, and the protective effect on axons and clinical improvement persists for as long as 180 days in mice treated regularly with phenytoin .
Neurotrophic factors such as insulin-like growth factor-I, platelet-derived growth factor (PDGF), fibroblast growth factor and ciliary neurotrophic factor (CNTF) are multifunctional growth factors which are found in the CNS. In vitro and in vivo studies have shown that neurotrophic factors influence proliferation, differentiation, survival, and regeneration of mature oligodendrocytes and oligodendroglial precursors in favor of a myelin repair. Since myelin breakdown is often severe in MS, the possibility of growth factors use in the treatment of MS has been considered and recently, some have been shown to reduce lesion severity and promote myelin regeneration in EAE. To investigate the role of endogenous CNTF in inflammatory demyelinating disease, MOG-induced EAE in CNTF-deficient and wild-type C57BL/6 mice was studied. Disease was more severe in CNTF-deficient mice and recovery was poor, with a 60% decrease in the number of proliferating oligodendrocyte precursor cells and a more than 50% increase in the rate of oligodendrocyte apoptosis. In addition, vacuolar dystrophy of myelin and axonal damage were more severe in CNTF-deficient mice. These specific pathological features could be prevented by treatment with an antiserum against tumor necrosis factor-alpha, suggesting that endogenous CNTF may counterbalance this effect of TNF-α. Hence CNTF modulates, in an inflammatory environment, glial cell survival and is an outcome determinant of EAE and it may be a hopeful agent for treatment of MS patients. More recently the neuroprotective role of CTNF was confirmed by overexpression of CNTF in EAE mice, possibly by exerting their immunoregulatory activity, inhibiting inflammation, reducing demyelination, and stimulating oligodendrogenesis .
Oligodendroglia are the cells that form and maintain myelin sheaths, the oligodendroglial lineage targeted approaches would be very interesting. Notably remyelination is regulated by stimulators and inhibitors. Leucine-rich repeats and Ig domain-containing, neurite outgrowth inhibitor receptor-interacting protein-1 (LINGO-1) is a potent negative regulator of axonal myelination. Loss of LINGO-1 function by Lingo1 gene knockout or by treatment with an antibody antagonist of LINGO-1 function leads to functional recovery from EAE. This remyelination role is associated with enhanced OPCs differentiation. Therefore antagonism of LINGO-1 or its pathway is a promising approach for the treatment of MS .
Since oxidative damage has been known to be involved in inflammatory and autoimmune-mediated tissue destruction in which, modulation of oxygen free radical production represents a new approach to the treatment of inflammatory and autoimmune diseases. Although a few antioxidants showed some efficacy in animal models, there is limited and conflicting evidence of potential therapeutic effects of antioxidants such as vitamins C and E in treating MS. Otherwise, little information is available on the effect of treatments with some antioxidants in patients with MS .
In the initial phase of MS lesion formation, ROS are known to mediate the transendothelial migration of monocytes and induce a dysfunction of the BBB. The beneficial effect of the antioxidant alpha-lipoic acid (LA) on these phenomena has been investigated. Interestingly, LA dose-dependently prevented the development of clinical signs in a rat model for MS, and clinical improvement was coupled to a decrease in leukocyte infiltration into the CNS, in particular monocytes. Live cell imaging assessed that ROS are produced within minutes upon the interaction of monocytes with brain endothelium. Monocyte adhesion to an in vitro model of the BBB subsequently induced enhanced permeability, which could be inhibited by LA. Hence LA has a protective effect on EAE not only by affecting the migratory capacity of monocytes, but also by stabilization of the BBB, making LA an attractive therapeutic agent for the treatment of MS.
As discussed above, the mitochondrial-targeted antioxidants, particularly MitoQ, SS31, CART may have potential role in EAE model or MS disease. Recently we discovered a potential direct interaction between neuropeptide CART and subunit B of the mitochondrial enzyme succinate dehydrogenase (SDHB). We found that CART significantly increased SDH function, mt complex II activity and ATP generation in purified mitochondria and primary cultured neurons. Furthermore, pretreatment with CART enhanced mitochondrial mechanisms of neuronal survival and prevented the decline in SDH and CII activities and ATP production after OGD. The findings suggest that CART’s neuroprotective mechanism of action may be linked to preservation of mitochondrial function and prevention of energy failure after ischemia-reperfusion injury. Importantly, CART has a transcription activity and stimulates brain-derived neurotrophic factor production in cultured neuronal cells. More recently we found that CART has a protective role in a mouse EAE model (unpublished data). This observation further indicates that mitochondrial targeted neuroprotection benefit EAE and probably MS patients.
A double-blind, placebo-controlled, parallel group designed clinical trial of the effect of Ginkgo biloba, a popular herbal medicine in China and Japan, on functional measures in MS has been completed. The Ginkgo group (patients received 240 mg per day of a Ginkgo special extract, EGb 761 had significantly more individuals showing improvement on four or more measures with improvements associated with significantly larger effect sizes on measures of fatigue, symptom severity, and functionality. The Ginkgo group also exhibited less fatigue at follow-up compared with the placebo group. This exploratory pilot study also showed that no adverse events or side effects were reported. Therefore Ginkgo exerted modest beneficial effects on select functional measures (eg, fatigue) among some individuals with MS. Even though the study may need verification from a multiple-center and large scale study, the exciting functional data indicate CAM and traditional medicine are worth further exploring for the treatment of MS, as well as other related diseases.
7. Conclusions and future directions
In summary, MS is a complicated autoimmune and neurodegenerative disease and causal factors are still unknown. Several cellular mechanisms have been proposed, including genetic factors, viral infections, autoimmune attack, demyelination, mitochondrial dysfunction, free radicals production, ionic imbalance and cellular clearance system dysfunction leading to final neuron loss. These factors work either as a primary cause or secondary consequence, however in many cases they work together to cause the MS disease. On-going efforts across world in searching genetic loci that may cause vast majority of patients with MS may provide some important clues to understand the disease process. Given the central role of the mitochondria in many important cellular functions including energy production, it is reasonable that its dysfunction is the key contributor to neurodegenerative process of this disease. We propose that the inflammation may be initiated by infection or by intrinsic imbalance (such as cellular energy failure and increased oxidative stress in neurons or oligodendrocytes), and tissue response to this inflammation that controlled by DNA defects (in nuclear and mitochondrial genomes) in MS patients. Therefore, mitochondrial targeted and neuroprotective treatments, or combination of neuroprotection and immunomodulatory may represent new and correct approach of MS therapy.
Acknowledgments
This work was supported by grants from NIH (AG028072 AG026051) and Vertex Pharmaceuticals to P.H.R. and by American Heart Association Award 0565527Z to P.M.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.