

Abstract
Purpose:
The aim of this study was to determine the genetic cause of autosomal dominant non-syndromic hearing loss segregating in a multi-generational family.
Methods:
Clinical examination, genome-wide linkage analysis, and exome sequencing were carried out on the family.
Results:
Affected individuals presented with early-onset progressive mild hearing impairment with a fairly flat, gently downsloping or U-shaped audiogram configuration. Detailed clinical examination excluded any additional symptoms. Linkage analysis detected an interval on chromosome 1p21 with a LOD score of 8.29: designated locus DFNA37. Exome sequencing identified a novel canonical acceptor splice-site variant c.652–2A>C in the COL11A1 gene within the DFNA37 locus. Genotyping of all 48 family members confirmed segregation of this variant with the deafness phenotype in the extended family. The c.652–2A>C variant is novel, highly conserved, and confirmed in vitro to alter RNA-splicing.
Conclusion:
We have identified COL11A1 as the gene responsible for deafness at the DFNA37 locus. Previously, COL11A1 was solely associated with Marshall and Stickler syndromes. This study expands its phenotypic spectrum to include non-syndromic deafness. The implications of this discovery are valuable in the clinical diagnosis, prognosis, and treatment of patients with COL11A1 pathogenic variants.
Keywords: COL11A1, DFNA37, non-syndromic hearing loss, splice-site variant, exome sequencing
INTRODUCTION
Hereditary hearing loss is a genetically heterogeneous disorder with over 150 genes implicated. Mirroring the genetic complexity is the breadth of phenotypic manifestations associated with pathogenic variants in these genes as more than 20% exhibit an extraordinary pleiotropy: they can give rise to either autosomal dominant non-syndromic hearing loss (ADNSHL) or autosomal recessive non-syndromic hearing loss (ARNSHL) (eg. TECTA and TMC1) and they can cause syndromic hearing loss or non-syndromic hearing loss (NSHL) (eg. Usher type 1-causing genes, WFS1, TBC1D24, and COLL11A2)1–6.
The collagen family is diverse and consists of more than 20 genetically distinct genes. Collagens are fibrous structural proteins involved in the construction of skin, cartilage, bone, eye, and other tissues7,8. All collagen molecules are comprised of three α-chain subunits tightly wrapped into a triple helix. The composition of each triple helix either contains one, two, or three different types of α-chains. For instance, the α-chains encoded by COL11A1, COL11A2, and COL2A1 comprise a unique collagen fibril that is essential for proper skeletal and cartilage formation as well as ocular and auditory function6,9,10.
The COL11A1 gene, located on chromosome 1p21.1, consists of 67 exons spanning 232 Kb11. It encodes a peptide consisting of N- and C-terminal propeptides surrounding a collagen α-chain following the typical collagen Gly-X-Y repeat configuration. In the inner ear type XI collagens localize to the tectorial membrane, a gelatinous sheet-like structure anchored to the apex of the interdental cells. The tectorial membrane lays on top of sensory hair cells and is comprised of four distinct types of collagen (types II, V, IX, and XI) and three primary non-collagenous glycoproteins (α-tectorin; Tecta, β-tectorin; Tectb, and Otogelin)12.
Pathogenic variants in COL11A1 have been linked to specific genetic disorders of the connective tissue, namely Marshall syndrome (MRSHS),13 Stickler syndrome type II (STL2),14,15 and fibrochondrogenesis (FBCG1)16. Fibrochondrogenesis is an ultra-rare disorder inherited in an autosomal recessive fashion. Affected individuals have severe skeletal defects characterized by pear-shaped vertebral bodies and broad long-bone metaphyses. Both Marshall and Stickler type 2 syndromes are rare autosomal dominant disorders. Clinically, there is much overlap between the two disorders as both include less severe vertebral and long-bone abnormalities, midface hypoplasia which may include cleft palate, myopia with beaded vitreous, and mild to moderate hearing loss.
Until now, pathogenic variants in COL11A1 have been exclusively linked to syndromic deafness. In this study, we present a novel splice-site altering variant in COL11A1 which segregates in a large family with post-lingual progressive ADNSHL and thus expands the phenotypic spectrum of pathogenic variants in COL11A1.
MATERIALS AND METHODS
Patients and clinical data
A four-generation family of European descent was ascertained as part of a genetic study of dominant progressive hearing loss at Boys Town National Research Hospital (BTNRH) between the years 1990 and 2000. After obtaining written informed consent from all participants with approval by the Institutional Review Board of BTNRH, pure tone audiograms and medical information were collected from participating family members. Clinical examination of the subjects excluded any additional syndromic findings. Blood samples from 48 family members were obtained and initial linkage studies were performed. The current studies were approved by the Institutional Review Boards at the University of Nebraska Medical Center (UNMC) and the University of Iowa.
Audiograms and data analysis
Pure tone audiometry was performed according to current standards to determine air conduction thresholds at 0.25, 0.5, 1, 2, 4, 6 and 8 kHz. Bone conduction thresholds were determined at some frequencies in some patients to exclude conductive hearing impairment. After validating binaural symmetry, the binaural mean air conduction threshold (dB Hearing Level, HL) at each frequency was used for further analyses. Conduction loss was recorded for some individuals. For this reason the threshold data from individual IV:12 were excluded, as well as those obtained at age 6 years from individual III:17, and those obtained from the left ear in individual III:4.
Linear regression analyses of threshold on age were used to evaluate progression of hearing impairment at the separate audio frequencies. These analyses comprised both individual longitudinal data derived from serial audiograms, and overall, cross-sectional last-visit data17. Progression was called significant if the 95% confidence interval (95%CI) for slope did not include zero at two or more frequencies (out of 6 or 7, which is significant at P < 0.05 according to binomial distribution statistics, with p = 0.025 and q = 0.975 for positive correlations). The same applies to the threshold intercept. Progression was expressed in dB per year, and designated Annual Threshold Deterioration (ATD). It was checked that the cross-sectional regression data conformed to the individual longitudinal regression data. Following that check, the regression data bearing on the last-visit thresholds were used to derive Age-Related Typical Audiograms (ARTA), which show the expected thresholds for a number of decade steps in age17.
Cross-sectional linear regression analyses were repeated for plots of threshold –P50presby against age, where P50presby is the median presbyacusis predicted by the ISO 7029 norm18 according to each patient’s gender and the age at which the audiogram was obtained.
Linkage analysis
Genome wide linkage analysis was first conducted with microsatellite markers using an ABI Prism linkage mapping set and results were confirmed using the Affymetrix Xba chip (Affymetrix, Santa Clara, CA) with 50,000 SNP markers19. Genotype calls were made with the BRLMM algorithm. Parametric multipoint linkage analysis was carried out using the Merlin program.
Exome Sequencing and Bioinformatic analysis pipeline
Four affected individuals (II.6, II.9, II.11 and III.9) underwent Exome Sequencing (ES) using the Agilent SureSelect Human All Exonv5 Kit (Agilent Technologies, Santa Clara, CA) as described2,20 (Figure 1A). Prepared libraries were pooled and sequenced using an Illumina Hiseq 2000 (Illumina, Inc., San Diego,CA). We analyzed ES data using a custom bioinformatic pipeline. Reads were aligned to human reference genome (Human GRCh37/hg19) using BWA. Variant calling was performed two ways: initially with the conservative Genome Analysis Toolkit (Broad Institute, Cambridge, MA), and later with the more inclusive SAMtool’s mpileup. Variants were annotated for conservation and deleteriousness using dbNSFP v2.021, and for minor allele frequency (MAF) using the 1000 Genomes Project database, the Exome Aggregation Consortium database (ExAC) (http://exac.broadinstitute.org/) and the Genome Aggregation Database (gnomAD) (http://gnomad.broadinstitute.org/). Variant predicted effects on splicing were assessed using Human Splicing Finder (http://www.umd.be/HSF3/).
Figure 1.
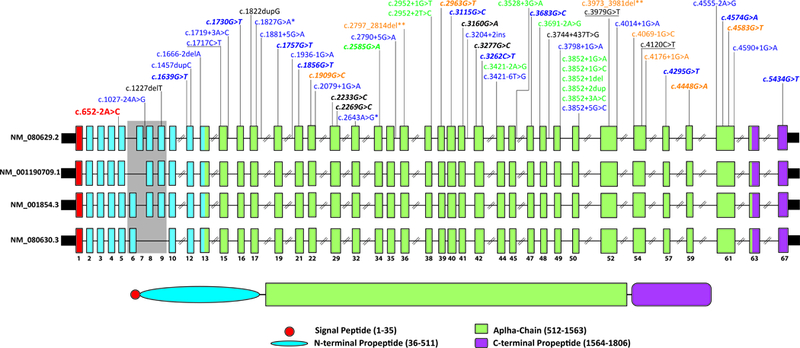
A) Family pedigree showing the segregation of the c.652–2A>C variant in COL11A1. Red and bold indicates the mutant allele. Circles and squares represent females and males, respectively. Filled symbols denote individuals with non-syndromic hearing loss (NSHL) and non-filled symbols show individuals with normal hearing. B) Age-related typical audiograms (ARTA). Binaural mean air conduction thresholds (dB HL) are presented for the ages 10–60 years. C) Parametric linkage analysis plot of chromosome 1. D) Representative chromatograms from wild-type and mutant sequences. E) Gel electrophoresis of Wild-type COL11A1 exon 5, c.652–2A>C mutation and the empty pET01 vector. The inclusion of exon 5 results in a 372 bp product and its exclusion results in a 234 bp band. Sequence chromatograms show read through at each exon junction. Results shown from COS7 experiments.
The following criteria were used for variant filtering: quality (Depth≥10X, QD>5, Quality>30), MAF<0.0001, coding effect (non-synonymous, indels, and splice-site variants), heterozygosity, and allele sharing amongst sequenced affected individuals.
Segregation Analysis
Sanger sequencing was completed in available family members to confirm segregation of all candidate variants (Table S1); c.652–2A>C in COL11A1 gene (MIM 120280; NM_080629.2); c.70C>T:p.R24W in ARHGEF16 (NM_014448.3); and c.847G>A:p.E283K in TRABD (NM_001320484.1).
Mini-gene Splicing Assay
In vitro mini-gene assays were carried out as described22. Briefly, wild-type exon 5 and ~120 base pairs of each flanking intron of COL11A1 was PCR amplified and ligated into the pET01 vector (MoBiTec, Goettingen, Germany). The c.652–2A>C variant was introduced to the wild-type vector using the QuikChange Lightning Site-Directed Mutagenesis kit (Agilent, Santa Clara, CA, USA) according to the manufacture’s protocol. Wild-type or mutant vectors were transfected into COS7 and HEK293 cells in triplicate. Total RNA was harvested 48 hours post-transfection and cDNA was transcribed according to the manufacture protocol. PCR using primers specific to the 5′ and 3′ native exons of the pET01 vector was performed and products were visualized on an agarose gel. Gel products were extracted and Sanger sequenced.
RESULTS
Clinical presentation
The family ascertained in this study is a four-generation kindred of European descent segregating hearing loss as an autosomal dominant trait (Figure 1A). Pure tone audiometric evaluation of affected members showed bilateral, post-lingual, progressive sensorineural hearing loss (Figure 1B). The hearing loss was mild-to-moderate and progressed slowly. The finding of a significant threshold intercept at age 0 years (Figure S2) at all frequencies except 8 kHz (Figure S2) suggests the presence of a substantial congenital component (of 12 to 23 dB) to the SNHL. As shown by the ARTA depicted in Figure 1B, the mean audiogram configuration developed from U-shaped (mid-frequency type) to flat with advancing age up to ~40 years. At more advanced ages it remained flat or became very gently downsloping (Figure 1B, S1).
To evaluate progression of hearing impairment at each frequency, we performed linear regression analyses of threshold on age. The resulting ATD (progression) was significant at 5 out of 7 frequencies (significant): 0.25–1 kHz, 4 and 8 kHz. It varied between 0.2 to 0.8 dB per year (Figure S2). This variation is also reflected by the ARTA (Figure 1B). One-way analysis of variance (ANOVA) followed by Tukey’s multiple comparison test showed that the ATD at 2 kHz was significantly smaller than the ATD at all other frequencies, whereas the ATD at 8 kHz was significantly greater than the ATD at all other frequencies, except for 0.25 and 4 kHz. The ATD for the thresholds corrected for median presbyacusis was significantly positive at 0.25–1kHz (Figure S3). This suggests SNHL progression beyond presbyacusis at these lower frequencies. The ATD at the higher frequencies did not differ significantly from zero, even though the values at 6–8 kHz were negative. This implies that progression at 2–8 kHz conformed with expected presbyacusis. Clinical examination of affected members excluded any additional syndromic features usually associated with Marshall Syndrome and Stickler Syndrome as the craniofacial features of affected family members were normal. X-ray images of the long bones of the proband (III-19) were also normal. There was no history of ocular abnormalities or cleft palate in the family.
Linkage analysis
The initial linkage analysis identified a single region on chromosome 1p21 spanning ~12Mb between markers D1S497 and D1S2651 with a maximum LOD score of 8.29 for marker D1S19519. This linked interval was designated DFNA37. A second genome-wide linkage using the Affymetrix Xba chip narrowed down the linked interval to a ~8.4Mb region between markers rs724480 and rs6667402 (Figure 1C, S4).
Exome Sequencing and variant assessment
Capitalizing on the advances made in sequencing technologies, we used ES to screen four affected individuals (II.6, II.9, II.11 and III.9). An average depth of coverage of 114 reads was obtained with 92% of targeted regions covered at ≥30X (Table S2). After filtering for quality, MAF, coding effect (non-synonymous, indels and splice-site variants), heterozygosity, and allele sharing amongst sequenced affected individuals in the DFNA37 locus, only one variant in COL11A1 was identified. The variant NM_080629.2; c.652–2A>C (chr1:103496802T>G) affects the canonical splice-site in intron 4 resulting in the alteration of the acceptor site (AG to CG) confirmed by analysis with Human Splicing Finder 3.0 and NNSPLICE 0.9. Sanger sequencing performed on all family members showed the segregation of c.652–2A>C variant at a heterozygous state with the deafness phenotype in the extended family (Figure 1A and 1D). This splice-site variant is predicted to cause skipping of exon 5 and a production of a protein lacking residues 218–260 in the N-propeptide domain.
Splicing Analysis
To characterize the impact of the c.652–2A>C variant on RNA-splicing, we cloned the wild-type and mutant sequences of COL11A1 exon 5 and flanking introns into the pET01 exon trap vector and transfected them into two different cell lines. Visualization of the splicing products revealed that cells transfected with the wild-type vector yielded the expected 372 bp band containing exon 5 (Figure 1E). In contrast, cells transfected with the mutant vector yielded 2 bands; one at 372 bp corresponding to the wild-type allele and the second at 234 bp lacking exon 5. These results were identical across both cell lines. Sequencing all bands confirmed break points and splicing events.
DISCUSSION
Coupling linkage analysis and ES, we identified a novel splice-site altering variant (c.652–2A>C) in the COL11A1 gene segregating in a large European-American pedigree with post-lingual progressive ADNSHL. The DFNA37 locus was mapped almost two decades ago but screening methodologies at the time failed to detect causative alterations in COL11A119. Since mapping of DFNA37, knowledge of genomic sequences has improved substantially. In addition, development of next generation sequencing has proven useful in elucidating the genetic causes of Mendelian disorders and notably, hereditary deafness23. We capitalized on these new technologies to reevaluate the DFNA37 locus for pathogenic variants and identified the genetic cause underlying hearing loss in this family.
The heterozygous c.652–2A>C variant in COL11A1 we identified segregating with the NSHL in this family is novel (absent from all population databases), highly conserved, and predicted to abolish the acceptor splice-site of exon 5 by in silico analysis. We confirmed aberrant splicing of exon 5 due to c.652–2A>C in vitro using a mini-gene splicing assay (Figure 1E). Interestingly, the c.652–2A>C variant seems to affect splicing efficiency rather than completely abolishing it. These findings suggest the c.652–2A>C variant creates a leaky acceptor splice-site that allows for some expression of a normal spliced transcript. This might explain the audiometric variability seen among some affected individuals, such as the manifestation of conductive hearing loss seen in III.4 and IV.12 or the diversity of audiometric configurations between individuals which could be flat, gently downsloping or U-shaped (Figure S1). Exon 5 is the last exon before the start of the variable region (exons 6, 7, 8 and 9) thus the c.652–2A>C variant would affect splicing in all five transcripts of COL11A1 (Figure 2). Aberrant splicing leading to exon 5 skipping would result in an inframe deletion and COL11A1 peptides lacking residues 218–260 in the N-terminal propeptide (Figure 2). It is also possible that other splicing events occur that could not be detected with the current mini-gene design. Analysis of RNA from individuals harboring the c.652–2A>C variant would better define the splicing defects resulting from this variant.
Figure 2.
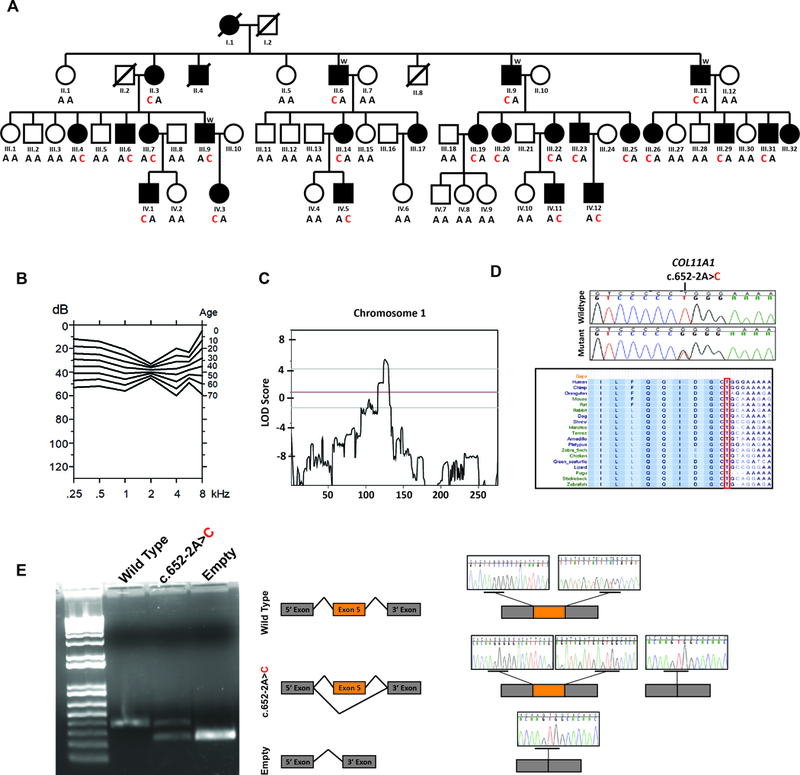
COL11A1 gene and protein schematic denoting reported pathogenic variants and their associated phenotypes. All variants were collected from the Deafness Variation Database (DVD; http://deafnessvariationdatabase.org/). The grey box shows the alternatively spliced exons. Text colored blue, black, green and orange indicates the phenotypes associated with pathogenic variants in COL11A1: STL2, FBCG1, MRSHS and STL2/MRSHS, respectively. Missense variants are in italics and nonsense variants are underlined. An “*” denotes a synonymous change, while “**” represents in-frame indels. The position of the c.652–2A>C mutation is shown in red and bold. Nucleotide numbering: the A of the ATG translation initiation site is noted as +1 using transcript NM_001854.3.
The exact function of N-terminal propeptide remains unclear. However, it is known to have a role in regulating the shape and size of the collagen fiber24. Lacking the amino acids encoded by exon 5 could result in a collagen fibril with a different diameter or shape, due to loss of key N-terminal propeptide regulatory sequences, such as the heparan sulfate binding motif that is located between residues 147–15225,26. Alternatively, it may result in protein mis-folding as this domain also houses two structurally important cysteine residues at amino acids 236 and 243 which are important to disulfide bond formation with other cysteines at positions 182 and 61. In the inner ear, COL11A1 is expressed in the tectorial membrane. Within the tectorial membrane, collagens form two networks of fibers: one unbranched, parallel and coordinated mainly by non-collagenous proteoglycans. The second, a striated sheet structure, orientated via the cross bridges and glycoproteins. Loss of this organization at either the collagenous or non-collagenous level causes hearing loss in mice and humans5,27. Since the N-propeptide domain plays a role in the establishment of molecular interactions with several extracellular matrix molecules and cellular proteins such as heparan sulfate proteoglycans and calcium, its alteration might impair its binding affinity for these molecules25,28. This could hamper interactions between cells and the surrounding extracellular matrix, as well as interactions between the diverse constituents of the extracellular matrix.
Regardless of the variant effect, it is clear the residues encoded by exon 5 are essential for proper auditory function. The absence of any other phenotypic manifestations in the described family is remarkable, given all previously reported autosomal dominant pathogenic variants (>50) have been only associated with either STL2 or MRSHS (Figure 2). However, the splice-site variant identified in this study is the first pathogenic alteration reported in the non-variable region of the N-propeptide domain. The majority of pathogenic variants in COL11A1 are splice-altering located in the triple-helical domain and thought to exert their effect via a dominant–negative mechanism. This is further supported by studies in mouse showing that homozygous for a spontaneous frameshift pathogenic variant in Col11a1 have severe chondrodysplasia and die at birth. Heterozygous mice escape lethality, develop osteoarthritis and have normal auditory responses up to 10 months postnatally29. The lack of an auditory phenotype in the heterozygous mouse, suggests haploinsufficiency is not the pathogenic mechanism underlying COL11A1-related auditory defects in humans. Pleiotropy associated with deafness-causing genes, where pathogenic variants in the same gene could cause either syndromic or non-syndromic hearing loss, has been demonstrated for several other genes such as the genes involved in Usher syndrome (PCDH15; DFNB23/USH1F, USH1C; DFNB18A/USH1C, WHRN; DFNB31/USH2D, MYO7A; DFNB2/DFNA11/USH1B, CDH23; DFNB12/USH1D), WFS1 (DFNA6/14/38/Wolfram Syndrome), TBC1D24 (DFNA65/DFNB86/DOORS syndrome) and COLL11A2 (DFNB53/DFNA13/STL3)1–3,5.
The present DFNA37 patients with a COL11A1 pathogenic variant showed fairly similar audiograms to those reported for DFNA traits with a mid-frequency type of hearing impairment: DFNA8/12 (TECTA), and DFNA13 (COL11A2)30,31.The DFNB phenotypes that are allelic to these traits also show mid-frequency-like types of audiograms, but usually at substantially higher thresholds: DFNB21 (TECTA), and DFNB53 (COL11A2)32,33. Patients with collagenopathies that include hearing impairment due to deleterious variants in COL11A1 (STL2 and Marshall syndrome) and COL11A2 (STL3), also show remarkably similar types of audiograms34–36. The results of psychophysical tests at suprathreshold levels in DFNA8/12, DFNA13 and STL3 (COL11A2) patients revealed that the type of hearing impairment is compatible with intra-cochlear conduction loss36–38. This is in line with the notion that disease-causing variants in COL11A2 affect tectorial membrane function31,39. Recent work on cochlear (micro)mechanics involving animal models with genetic modifications has provided compelling evidence about the relevant mechanical changes that can be involved5,12,39.
In summary, here we report the clinical and genetic characteristics associated with deafness at the DFNA37 locus and we expand the spectrum of COL11A1-associated phenotypes to include ADNSHL. This study illustrates another example of the pleiotropy exhibited by other deafness-causing genes and highlights the complexity associated with providing the correct genetic diagnosis.
Supplementary Material
Acknowledgements
We would like to thank all family members reported here for their collaboration in this study. ZT also acknowledges the helpful advice of Stefan Stamm, Ph.D. (University of Kentucky) in the interpretation of splice site variants. Funding for this study was provided by NIDCD R01s DC02942 to SD Smith; RO1s DC003544, DC002842 and DC012049 to RJH Smith and the Omaha Community Foundation (SD Smith).The UNMC DNA Sequencing Core facility is also supported by P30 GM110768: Core B Sequencing (JD Eudy, SD Smith).
Footnotes
DISCLOSURE
The authors do not have any conflicts of interest regarding the information in this manuscript.
Co-first authors
REFERENCES
- 1.Bork JM, Peters LM, Riazuddin S, Bernstein SL, Ahmed ZM, Ness SL, Polomeno R, Ramesh A, Schloss M, Srisailpathy CRS, et al. (2001). Usher Syndrome 1D and Nonsyndromic Autosomal Recessive Deafness DFNB12 Are Caused by Allelic Mutations of the Novel Cadherin-Like Gene CDH23. Am. J. Hum. Genet. 68, 26–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Azaiez H, Booth KT, Bu F, Huygen P, Shibata SB, Shearer AE, Kolbe D, Meyer N, Black-Ziegelbein EA, and Smith RJH (2014). TBC1D24 Mutation Causes Autosomal-Dominant Nonsyndromic Hearing Loss. Hum. Mutat. 35, 819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cryns K, Sivakumaran TA, Van den Ouweland JMW, Pennings RJE, Cremers CWRJ, Flothmann K, Young TL, Smith RJH, Lesperance MM, and Van Camp G (2003). Mutational spectrum of the WFS1 gene in Wolfram syndrome, nonsyndromic hearing impairment, diabetes mellitus, and psychiatric disease. Hum. Mutat. 22, 275–287. [DOI] [PubMed] [Google Scholar]
- 4.Rigoli L, Lombardo F, and Di Bella C (2011). Wolfram syndrome and WFS1 gene. Clin. Genet. 79, 103–117. [DOI] [PubMed] [Google Scholar]
- 5.Richardson GP, de Monvel JB, and Petit C (2011). How the genetics of deafness illuminates auditory physiology. Annu. Rev. Physiol. 73, 311–334. [DOI] [PubMed] [Google Scholar]
- 6.Acke FRE, Dhooge IJM, Malfait F, and De Leenheer EMR (2012). Hearing impairment in Stickler syndrome: a systematic review. Orphanet J. Rare Dis. 7, 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ricard-Blum S (2011). The Collagen Family. Cold Spring Harb. Perspect. Biol. 3, 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Spranger J (1998). The type XI collagenopathies. Pediatr. Radiol. 28, 745–750. [DOI] [PubMed] [Google Scholar]
- 9.Myllyharju J, and Kivirikko KI (2001). Collagens and collagen-related diseases. Ann. Med. 33, 7–21. [DOI] [PubMed] [Google Scholar]
- 10.Keene DR, Oxford JT, and Morris NP (1995). Ultrastructural localization of collagen types II, IX, and XI in the growth plate of human rib and fetal bovine epiphyseal cartilage: type XI collagen is restricted to thin fibrils. J. Histochem. Cytochem. 43, 967–979. [DOI] [PubMed] [Google Scholar]
- 11.Henry I, Bernheim a, Bernard M, van der Rest M, Kimura T, Jeanpierre C, Barichard F, Berger R, Olsen BR, and Ramirez F (1988). Mapping of a human fibrillar collagen gene, pro alpha 1 (XI) (COL11A1), to the p21 region of chromosome 1. Genomics 3, 87–90. [DOI] [PubMed] [Google Scholar]
- 12.Richardson GP, Lukashkin AN, and Russell IJ (2008). The tectorial membrane: one slice of a complex cochlear sandwich. Curr. Opin. Otolaryngol. Head Neck Surg. 16, 458–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Annunen S, Körkkö J, Czarny M, Warman ML, Brunner HG, Kääriäinen H, Mulliken JB, Tranebjaerg L, Brooks DG, Cox GF, et al. (1999). Splicing mutations of 54-bp exons in the COL11A1 gene cause Marshall syndrome, but other mutations cause overlapping Marshall/Stickler phenotypes. Am. J. Hum. Genet. 65, 974–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Majava M, Hoornaert KP, Bartholdi D, Bouma MC, Bouman K, Carrera M, Devriendt K, Hurst J, Kitsos G, Niedrist D, et al. (2007). A Report on 10 New Patients With Heterozygous Mutations in the COL11A1 Gene and a Review of Genotype–Phenotype Correlations in Type XI Collagenopathies. Am. J. Med. Genet. A 143A, 258–264. [DOI] [PubMed] [Google Scholar]
- 15.Rose PS, Levy HP, Liberfarb RM, Davis J, Szymko-Bennett Y, Rubin BI, Tsilou E, Griffith AJ, and Francomano CA (2005). Stickler syndrome: Clinical characteristics and diagnostic criteria. Am. J. Med. Genet. Part A 138A, 199–207. [DOI] [PubMed] [Google Scholar]
- 16.Tompson SW, Bacino CA, Safina NP, Bober MB, Proud VK, Funari T, Wangler MF, Nevarez L, Ala-Kokko L, Wilcox WR, et al. (2010). Fibrochondrogenesis results from mutations in the COL11A1 type XI collagen gene. Am. J. Hum. Genet. 87, 708–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huygen PLM, Pennings RJE, and Cremers CWRJ (2003). Characterizing and Distinguishing Progressive Phenotypes in Nonsyndromic Autosomal Dominant Hearing Impairment. Audiol. Med. 1, 37–46. [Google Scholar]
- 18.Stenklev NC, and Laukli E (2004). Presbyacusis - Hearing thresholds and the ISO 7029. Int. J. Audiol. 43, 295–306. [DOI] [PubMed] [Google Scholar]
- 19.Talebizadeh Z, Kenyon J, Askew J, and Smith S (2000). A new locus for dominant progressive hearing loss DFNA37 mapped to chromosome 1p21. In American Society of Human Genetics, p. 314. [Google Scholar]
- 20.Azaiez H, Decker AR, Booth KT, Simpson AC, Shearer AE, Huygen PLM, Bu F, Hildebrand MS, Ranum PT, Shibata SB, et al. (2015). HOMER2, a stereociliary scaffolding protein, is essential for normal hearing in humans and mice. PLoS Genet. 11, e1005137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu X, Jian X, and Boerwinkle E (2013). dbNSFP v2.0: A database of human non-synonymous SNVs and their functional predictions and annotations. Hum. Mutat. 34, 2393–2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Booth KT, Azaiez H, Kahrizi K, Wang D, Zhang Y, Frees K, Nishimura C, Najmabadi H, and Smith RJ (2018). Exonic mutations and exon skipping: Lessons learned from DFNA5. Hum. Mutat. 39, 433–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vona B, Nanda I, Hofrichter MAH, Shehata-Dieler W, and Haaf T (2015). Non-syndromic hearing loss gene identification: A brief history and glimpse into the future. Mol. Cell. Probes 29, 260–270. [DOI] [PubMed] [Google Scholar]
- 24.Hulmes DJS (2002). Building collagen molecules, fibrils, and suprafibrillar structures. J. Struct. Biol. 137, 2–10. [DOI] [PubMed] [Google Scholar]
- 25.Warner LR, Brown RJ, Yingst SMC, and Oxford JT (2006). Isoform-specific heparan sulfate binding within the amino-terminal noncollagenous domain of collagen alpha1(XI). J. Biol. Chem. 281, 39507–39516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McDougal OM, Warner LR, Mallory C, and Oxford JT (2011). PREDICTED STRUCTURE AND BINDING MOTIFS OF COLLAGEN α1(XI). GSTF Int. J. Bioinforma. Biotechnol. 1, 43–48. [PMC free article] [PubMed] [Google Scholar]
- 27.Petit C, and Richardson GP (2009). Linking genes underlying deafness to hair-bundle development and function. Nat. Neurosci. 12, 703–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kearney HM, Thorland EC, Brown KK, Quintero-Rivera F, and South ST (2011). American College of Medical Genetics standards and guidelines for interpretation and reporting of postnatal constitutional copy number variants. Genet. Med. 13, 680–685. [DOI] [PubMed] [Google Scholar]
- 29.Rodriguez RR, Seegmiller RE, Stark MR, and Bridgewater LC (2004). A type XI collagen mutation leads to increased degradation of type II collagen in articular cartilage. Osteoarthr. Cartil. 12, 314–320. [DOI] [PubMed] [Google Scholar]
- 30.Hildebrand MS, Morín M, Meyer NC, Mayo F, Modamio-Hoybjor S, Mencía A, Olavarrieta L, Morales-Angulo C, Nishimura CJ, Workman H, et al. (2011). DFNA8/12 caused by TECTA mutations is the most identified subtype of nonsyndromic autosomal dominant hearing loss. Hum. Mutat. 32, 825–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McGuirt WT, Prasad SD, Griffith AJ, Kunst HPM, Green GE, Shpargel KB, Runge C, Huybrechts C, Mueller RF, Lynch E, et al. (1999). Mutations in COL11A2 cause non-syndromic hearing loss (DFNA13). Nat. Genet. 23, 413–419. [DOI] [PubMed] [Google Scholar]
- 32.Meyer NC, Alasti F, Nishimura CJ, Imanirad P, Kahrizi K, Riazalhosseini Y, Malekpour M, Kochakian N, Jamali P, Van Camp G, et al. (2007). Identification of three novel TECTA mutations in Iranian families with autosomal recessive nonsyndromic hearing impairment at the DFNB21 locus. Am. J. Med. Genet. Part A 143, 1623–1629. [DOI] [PubMed] [Google Scholar]
- 33.Chen W (2005). Mutation of COL11A2 causes autosomal recessive non-syndromic hearing loss at the DFNB53 locus. J. Med. Genet. 42, e61–e61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Acke FR, Swinnen FK, Malfait F, Dhooge IJ, and De Leenheer EMR (2016). Auditory phenotype in Stickler syndrome: results of audiometric analysis in 20 patients. Eur. Arch. Oto-Rhino-Laryngology 273, 3025–3034. [DOI] [PubMed] [Google Scholar]
- 35.Griffith AJ, Gebarski SS, Shepard NT, and Kileny PR (2000). Audiovestibular phenotype associated with a COL11A1 mutation in Marshall syndrome. Arch. Otolaryngol. Head. Neck Surg. 126, 891–894. [DOI] [PubMed] [Google Scholar]
- 36.van Beelen E, Leijendeckers JM, Huygen PLM, Admiraal RJC, Hoefsloot LH, Lichtenbelt KD, St??be L, Pennings RJE, Leuwer R, Snik AFM, et al. (2012). Audiometric characteristics of two Dutch families with non-ocular Stickler syndrome (COL11A2). Hear. Res. 291, 15–23. [DOI] [PubMed] [Google Scholar]
- 37.Plantinga RF, Cremers CWRJ, Huygen PLM, Kunst HPM, and Bosman AJ (2007). Audiological evaluation of affected members from a Dutch DFNA8/12 (TECTA) family. JARO - J. Assoc. Res. Otolaryngol. 8, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.De Leenheer EMR, Busman AJ, Huygen PLM, Kunst HPM, and Cremers CWRJ (2004). Audiological characteristics of some affected members of a Dutch DFNA13/COL11A2 family. Ann. Otol. Rhinol. Laryngol. 113, 922–929. [DOI] [PubMed] [Google Scholar]
- 39.Masaki K, Gu JW, Ghaffari R, Chan G, Smith RJH, Freeman DM, and Aranyosi AJ (2009). Col11a2 deletion reveals the molecular basis for tectorial membrane mechanical anisotropy. Biophys. J. 96, 4717–4724. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.