

Abstract
Painful temporomandibular disorders (TMD) is the leading cause of chronic orofacial pain, but its underlying molecular mechanisms remain obscure. While many environmental factors have been associated with higher risk of developing painful TMD, family and twin studies support a heritable genetic component as well. We performed a GWAS assuming an additive genetic model of TMD in a discovery cohort of 999 cases and 2031 TMD-free controls from the Orofacial Pain: Prospective Evaluation and Risk Assessment (OPPERA) study. Using logistic models adjusted for sex, age, enrollment site, and race, we identified three distinct loci that were significant in combined or sex-segregated analyses. A single nucleotide polymorphism (SNP) on chromosome 3 (rs13078961) was significantly associated with TMD in males only (odds ratio [OR]=2.9, 95% CI: 2.02–4.27, P=2.2×10−8). This association was nominally replicated in a meta-analysis of seven independent orofacial pain cohorts including 160,194 participants (OR=1.16, 95% CI: 1.0–1.35, P = 2.3×10−2). Functional analysis in human dorsal root ganglia (DRG) and blood indicated this variant is an expression quantitative trait locus (eQTL), with the minor allele associated with decreased expression of the nearby muscle RAS oncogene homolog (MRAS) gene (beta = −0.51, P = 2.43×10−5). Male mice, but not female mice, with a null mutation of Mras displayed persistent mechanical allodynia in a model of inflammatory pain. Genetic and behavioral evidence support a novel mechanism by which genetically-determined MRAS expression moderates the resiliency to chronic pain. This effect is male-specific and may contribute to the lower rates of painful TMD in men.
Summary
A polymorphic locus associated with a protective effect against painful temporomandibular disorder in males regulates the expression of the muscle RAS oncogene homolog (MRAS) gene.
INTRODUCTION
Painful temporomandibular disorder (TMD) is the most common cause of chronic orofacial pain[8] and is characterized by pain (including allodynia and hyperalgesia), jaw dysfunction, fatigue, and psychological disturbance. [7] TMDs affect about 5% of adults in the U.S.,[13; 29] with an annual incidence of 3.9% for pain-related TMD.[38] A broad range of factors predict higher risk of developing painful TMD, including: greater age, presence of comorbid pain conditions,[1] greater frequency of somatic symptoms,[10] and poor sleep quality.[34]
Evidence supports a role for genetic contributions to TMD, with twin studies demonstrating heritability of 27%.[30] In a case-control study using a candidate panel of 350 genes relevant to nociceptive, inflammatory, immunological, and affective processes, we found that TMD was associated with single nucleotide polymorphisms (SNPs) in seven genes: COMT, an enzyme catabolizing catecholamines; HTR2A, one of the receptors for serotonin; NR3C1, the glucocorticoid receptor; CAMK4, the calcium/calmodulin-dependent protein kinase; CHRM2, the muscarinic acetylcholine receptor; IFRD1, the interferon-related developmental regulator; and GRK5, the G protein-coupled receptor kinase.[41] To date, only one genome-wide association study (GWAS) of orofacial pain has been published.[35] Using self-reported orofacial pain as the phenotype, it found two female-specific genome-wide significant associations, which were subsequently replicated in independent cohorts. One SNP was near the gene encoding relaxin/insulin-like family peptide receptor 2, RXP2; the second SNP was in the BAH domain and coiled-coil containing gene, BAHCC1.
The aims of the present study were to undertake the first discovery GWAS of examiner-verified chronic TMD, evaluate generalizability of the findings through independent replication, and investigate the functional biology of genetic markers that emerged from discovery and replication phases. The discovery sample included 3,104 racially diverse participants from the Orofacial Pain: Prospective Evaluation and Risk Assessment (OPPERA) study, where painful TMD was determined by clinical examination using modified Research Diagnostic Criteria for TMD (RDC/TMD) and genotyping was conducted using the Illumina HumanOmni 2.5M bead chip. SNPs that achieved genome-wide significance (P < 5×10−8) in the discovery cohort were carried forward for replication in seven additional cohorts. Replication studies included TMD case-control studies as well as population-based, cross-sectional studies, and were combined by meta-analysis to report size and direction of genetic effects. We then investigated the functional consequences of associated SNPs on etiological pathways using bioinformatic and transgenic approaches.
METHODS
An overview of the study design, including discovery, replication, and validation phases, is provided in Fig.1.
Fig. 1. Study design overview diagram.
Discovery GWAS cohort
The discovery case-control cohort included 3104 subjects selected for genome-wide genotyping, drawn from the OPPERA study (n=2,868) and a separate case-control study of chronic TMD (n=236). The OPPERA subjects were males and females (61.9% females) between the ages of 18–44 years, recruited at four sites in the eastern United States (Buffalo, NY; Baltimore, MD; Chapel Hill, NC, and Gainesville, FL); enrollment of this cohort has been described previously.[37] The OPPERA subjects were racially and ethnically diverse: 57.5% white, 25.7% black, and 16.8% other/not reported (Supplementary Table 1). From the full OPPERA cohort, subjects selected for GWAS analysis were determined randomly, leaving approximately 1000 subjects for later replication studies. The additional 236 subjects were from a separate case-control study of chronic TMD conducted by the same OPPERA study team.[39] Subjects were all non-Hispanic white females, aged 18–44 years, recruited at the Chapel Hill, NC (UNC) site. The combined group reported in this paper consisted of 1,011 TMD cases and 2,093 TMD-free controls. All participants provided informed, signed consent for all study procedures. The OPPERA study was reviewed and approved by institutional review boards at each of the 4 study sites and at the data coordinating center, Battelle Memorial Institute. The TMD case-control study was approved by the Biomedical Institutional Review Board of the University of North Carolina at Chapel Hill.
Case classification
The classification of TMD[27] was based on the RDC/TMD.[7] Cases met all 3 of the following criteria: a) pain in the cheeks, jaw muscles, temples, or jaw joints that occurred for at least five days per month during the preceding 6 months, including at least 15 days in the month prior to enrollment; b) pain reported in the examiner-defined orofacial region for at least 5 days out of the prior 30 days; and 3) pain evoked by palpation of the orofacial muscles or maneuver of the jaw that occurred in at least 3 masticatory muscles or at least 1 temporomandibular joint or both. Examiners were trained in standardized procedures used during the examination and annual evaluation of inter-examiner reliability revealed consistently excellent levels of examiner agreement (kappa > 0.75).
Genotyping and imputation
Study participants provided a sample of whole blood which was stored in 5 ml EDTA-containing polyethylene vacutainers. Genomic DNA purification was performed using the Qiagen™ Extraction Kit following the manufacturer’s recommended protocol. Genotyping was performed by the Center for Inherited Disease Research using the Illumina HumanOmni 2.5 Exome Bead Chip platform (Illumina, Inc., San Diego, CA). Genetic data cleaning was accomplished by the Genetic Analysis Center at the University of Washington following their established pipeline.[19] Batch effects were assessed by comparing missing call rate and chi-square test for allelic frequency between genotyping batches. Sample identity and sample quality analyses included checks for missing call rate, chromosomal anomalies, cryptic relatedness, autosomal heterozygosity outliers, gender mismatch, and genetic ancestry. Consistency of genotyping calls was assessed using 69 duplicates of study samples and 66 stock samples from HapMap reference subjects. SNP level quality checks included assessments of missing call rate, duplicate discordance, and Mendelian errors. Because of the mixed population structure of the OPPERA cohort, Hardy-Weinberg equilibrium (HWE) was tested in homogenous European- and African-ancestry groups separately.
Cleaned genotypes were then imputed to the 1000 Genomes Project phase 3 reference panel using the software packages SHAPEIT[6] for pre-phasing and IMPUTE2[11] for imputation. Instead of using the quality metric provided by IMPUTE2, imputation quality was instead assessed by calculating the OEvar statistic, which is the ratio of the observed variance of imputed dosages to the expected binomial variance[21], and which ranges from 0 to 1. Imputed SNPs were excluded from analysis based on a filter for effectively small sample size, taking into account the number of minor alleles as well as imputation quality. This filter was calculated using the formula: 2 * MAF * (1-MAF) * N * OEvar ≥ 50, where N is the number of TMD cases in the cohort (i.e., 999), MAF is the minor allele frequency, and the threshold for inclusion was set at 50 for binary traits.
Data Availability
Study data have been deposited and made publicly available at the Database of Genotypes and Phenotypes (dbGaP) public repository (accession number phs000796.v1.p1).
Genotype Quality Assessment
The discovery dataset included the maximal set of unrelated individuals passing all quality checks, and consisted of 999 TMD cases and 2031 controls. Initial genotyping results were provided for 2.57 million SNPs; a total of 2.53 million SNPs passed quality filters including call rate < 2%; > 1 discordant calls in 69 study duplicates; >1 Mendelian error in 9 HapMap trios; HWE P value < 10−4 in either white or black subjects; and sex differences in rates of allele frequency ≥ 0.2 or of heterozygosity > 0.3 (see Supplementary Table 2 for SNP filters). The SNP filters removed 1.4% of SNPs for quality purposes. An additional 5.9% of SNPs that were monomorphic were likewise removed. The final MAF filtering was based on MAF and sample size considerations. The final cleaned dataset used for analysis and imputation included 1.83 million SNPs, or 71.3% of the total. Imputation quality was assessed using the masked/imputed concordance feature of IMPUTE2; all chromosomes exceeded the recommended 95% overall concordance rate. After applying the OEvar filter for MAF and imputation quality, 9.70 million SNPs were retained for analysis.
Genetic Association Analysis
Genome-wide association was tested using PLINK 1.07 software,[32] on discrete genotype and (for imputed SNPs) allele dosage datasets. Logistic regression models were constructed using additive SNP effects and included sex, age, race, and enrollment site. To account for population stratification, genetic ancestry was modeled using the first three principal components of the genotypes. The threshold for genome-wide significance was p < 5.0×10−8. In addition to analysis in the full cohort, we also performed tests of association stratified by sex, as TMD has much greater prevalence in females than in males and is thought to have some sex-specific pathophysiologic mechanisms.
To assess the possible effects of population stratification on association results, we examined all associated SNPs using two additional approaches. We first subdivided the overall cohort and male and female groups separately into homogeneous racial groups representing European- and African-American ancestry. We assessed association effect size and direction in these sex- and race-specific strata using logistic regression, as described previously. We combined the race-specific results by fixed-effects meta-analysis using PLINK. Statistical significance was also assessed within strata and in the meta-analysis, but was considered of secondary importance given the study was not powered to fully explore stratum-specific associations. In a separate approach, we used the software package GEMMA[52] to apply a univariate logistic mixed model analysis to the overall cohort or to sex-stratified subgroups, as appropriate for the observed associations. This model accounted for population stratification by introducing a term describing an n-vector of random effects, using an n × n matrix where each row and column represent each study participant, and each value describes the relatedness between the two corresponding individuals. This relatedness matrix was calculated in GEMMA using the “-gk 2” function on a standardized genotype matrix, as standardization is considered to improve performance when SNPs with lower MAF tend to have larger effects. The model also included covariates as described for the logistic regression analyses; significance was assessed by Wald test.
Power calculations were carried out according to the method of Kuo and Zaykin,[17] which estimates the number of true positives expected among the top ranked p-values (see Supplementary Table 3 and its legend for further details). Assuming a truly etiological SNP to have an average OR = 1.2 with MAF = 0.15, and the number of such true SNPs to be 25, there is 80% chance that one or more of these true positives will score among the top 41 SNPs ranked by order of association p-value. For 50 such true loci, there is 80% power that at least one will be found in the top 10 ranked p-values. Assumptions about the number of loci and the effect size were guided by distributions for other complex diseases.
Estimation of Heritability
Heritability of susceptibility to TMD was estimated using genome-wide SNP coverage using the GREML method implemented in the software package Genome-Wide Complex Trait Analysis (GCTA).[48] This method estimates the contribution of additive SNP effects to observed variance to estimate narrow-sense heritability, or the proportion of phenotypic variance attributable to additive allelic factors. This analysis was performed on a homogenous group of subjects with European ancestry (n = 1,627), as indicated by the principal components analysis of the genotypes.
Meta-analysis
Studies
Seven independent cohorts were used for replication analysis (Table 1). Further details on study design, recruitment, subject characteristics, and phenotyping for each study are provided in Supplementary Materials.
Table 1.
Demographic data for the discovery and replication studies.
| DISCOVERY COHORT | REPLICATION COHORTS | |||||||
|---|---|---|---|---|---|---|---|---|
| STUDY NAME | Orofacial Pain: Prospective Evaluation and Risk Assessment | Study of Health in Pomerania | Northern Finland Birth Cohort | São Paulo Brazil TMD Case-Control | OPPERA-II Chronic TMD Replication | Complex Persistent Pain Conditions | Hispanic Community Health Study/Study of Latinos | UK Biobank |
| ACRONYM | OPPERA | SHIP | NFBC | SPB | OP2 | CPPC | HCHS/SOL | UKB |
| COUNTRY | USA | Germany | Finland | Brazil | United States | United States | United States | United Kingdom |
| N (% FEMALE) | 3,030 (64.6%) | 4,051 (50.7) | 1,497 (55.2) | 636 (100.0) | 1,342 (66.0) | 490 (85.7) | 11,975 (58.7) | 137,173 (51.3) |
| CASES / CONTROLS | 999/2,031 | 607/3,044 | 161/1,336 | 144/492 | 444/898 | 266/224 | 2,597/9,378 | 1,320/135,853 |
| ANCESTRY (% WHITE) | 61 | 100 | 100 | 100 | 79 | 68 | NA1 | 100 |
| AGE RANGE | 18–44 | 20–81 | 462 | 18–44 | 18–74 | 18–64 | 18–74 | 40–69 |
| RDC EXAM | Yes | Yes | Yes | Yes | Yes | Yes | No | No |
All percentages are a function of the total number of individuals in the respective cohort. Country corresponds to the country where study enrollment was conducted. Ancestry indicates the percentage of participants who described themselves as white or Caucasian.
All subjects were Hispanic/Latino; see {Sanders, 2017 #2581} for full background information
Data collected at 46-year time point
Analyses on all replication cohorts were performed with approval from the respective institutional review boards at each site. DNA collected in each replication study was genotyped separately before imputation to the 1000 Genomes Project phase 3 reference panel. Due to differences in genotyping platforms and racial/ethnic composition, quality control procedures were performed separately for each cohort. Tests for association between orofacial pain phenotypes and SNPs were performed using logistic regression, with study-specific covariates. Filtering for MAF and imputation quality was performed using the same OEvar method used for OPPERA.
Meta-analysis statistical methods
Fixed-effects meta-analysis was performed using the metafor package in R version 3.2.2.[45] Allelic log odds ratios (OR), standard errors (SE), and P values were calculated separately for each SNP-phenotype association from each study included in the meta-analysis. The threshold of statistical significance for replication was P < 0.05, divided by the number of independent loci carried forward (i.e., 3) using the Bonferroni correction for multiple testing, for the one-tailed test of association. Study-specific SNP filters for imputation quality and MAF were imposed, as described for each study. The minor allele on the forward strand was used as the effect allele, or reference to standardize direction of ORs. The Cochrane’s Q statistic P value was used as a measure of heterogeneity of associations among studies.
Functional Analysis: eQTL
As ganglia neurons relay sensory information to the brain, they represent a highly relevant tissue in which to study cellular mechanisms of chronic pain. Given the strong correlation in gene expression patterns between trigeminal and dorsal root ganglia (DRG),[3] we examined the correlation of TMD-associated SNPs with gene expression in primary afferent cells from DRG tissue. DRG specimens were collected post-mortem from donors who died following asystole by the CORE/CORID repository at the University of Pittsburgh. DRG tissues were harvested from bilateral lumbar L4 and L5 and snap-frozen for storage. Thawed samples were genotyped using Illumina’s Human Omni Express chip (~1M probes), while RNA levels were measured with Affymetrix’s Human Transcriptome Array 2.0 (~70K gene-level probes, ~900K isoform-level probes), which provides for both gene- and exon-level assessment of expression quantitative trait loci (eQTLs) (see diatchenko.lab.mcgill.ca:/DRG-eQTLs/).[28] Both analyses were performed at the Genomics Research Core facility of the University of Pittsburgh. Associations were designated as cis-acting if the distance between the SNP and the gene was less than 1 Mb for gene-level probes, and if the distance was less than 5 kb for exon-level probes. A total of 214 samples with matched genotype/RNA levels was used in this study. We performed eQTL analyses using all samples, and then stratified analyses by sex. Donors’ median age was 52; 199 were white, 105 female and 109 male.
To relate eQTLs observed in DRG specimens (for which TMD status is not available) to RNA regulation in TMD cases and controls, we performed eQTL analysis using whole blood collected in the OPPERA study. Peripheral blood samples were stored in PAXgene tubes and frozen for subsequent processing. Genotyping was done on the Illumina Human Omni Express chip (~1M probes), while RNA levels were measured with Affymetrix’s Human Transcriptome Array 2.0 (~70K gene-level probes, ~900K isoform-level probes) at the McGill University and Génome Québec Innovation Centre. A total of 167 samples with matched genotype / RNA levels was used. Subjects’ median age was 27 years. 117 were white, 29 African-American, and 9 Hispanic; 115 subjects were female and 52 male. Analyses were performed on the full set of samples as well as in strata subdivided by sex. Genotype differences in probe means were assessed using linear regression to generate p-values, and a false discovery rate (FDR) of 1% was set to control for multiple testing.
Although cis-regulation is presumed to result from the direct action of regulatory elements on nearby genes, trans-regulation likely involves downstream effects within biological pathways. We selected expressed genes (eGenes) at FDR 1% level (n=1,231) from p-values of association between genes and cis- (nearby genes) and trans- (distal genes) acting eQTLs of interest in human DRG. This dataset was submitted to pathway analysis approaches, using QIAGEN’s Ingenuity® Pathway Analysis (IPA®, QIAGEN, Redwood City) and Pathway Studio.[26] Core Analysis was applied as a primary IPA® analytic tool for identifying cellular pathways, bio-toxicology functions, diseases, and known biomarkers associated with the trans-acting gene set. Pathway Studio pathway analyses were performed using the online web server. Analyses were restricted to Gene Ontology (GO) Consortium’s biological processes. Statistical significance was assessed based on a FDR of 1% to account for multiple testing and redundancy in gene composition between pathways.
Mras Null Mutant Mouse Behavioral Assays
A rodent gene-knockout experiment was designed to test functional consequences of genetic variants implicated with TMD in the preceding GWAS and eQTL studies. Mras (muscle RAS oncogene homolog) null mutant mice (B6; 129S5-MrasGt(OST108223)Lex/Ieg) were generated by the Wellcome Trust Knockout Mouse Resource and frozen embryos were obtained from the European Mouse Mutant Archive (EMMA) repository (EM:02322). Cryorecovery was performed by the Goodman Cancer Research Center Transgenic Core Facility (McGill University) and heterozygous mice were mated in house to generate homozygous, heterozygous, and wildtype mice. All animals used in this experiment were bred on site, and littermates representing the three Mras genotypes were introduced into the study as soon as they matured to 8 weeks of age, accounting for the unbalanced numbers between males and females. All mice were housed in standard polycarbonate cages in groups of three or four same-sex, same-genotype mice in a temperature-controlled (20 ± 1°C) environment (14:10-hour light/dark cycle; lights on at 07:00 h); tap water and food (Harlan Teklad 8604) were available ad libitum. Experimenters were blinded to genotype throughout the experiment. All procedures were approved by the Downtown Animal Care Committee at McGill University, and conformed to ethical guidelines of the Canadian Council on Animal Care.
All experiments took place during the light cycle, no earlier than 09:00 h and no later than 16:00 h. Naïve mice were placed in custom-made Plexiglas cubicles (5 × 8.5 × 6 cm) on a perforated metal floor, and were habituated for at least 1 h before testing. The up-down method of Dixon was used to estimate 50% withdrawal thresholds using calibrated von Frey nylon monofilaments[4] (Stoelting Touch Test). Filaments were applied to the plantar surface of the hind paw for 3 s and responses were recorded. At least two consecutive measures were taken on each hind paw at each time point, and averaged. Although data were collected on both hind paws, only data from the hind paw injected with complete Freund’s adjuvant (CFA, see below) are presented, as no significant genotype effects on the contralateral paw were observed.
All mice received 20 μl unilateral injections of 50% CFA (Sigma) into the plantar surface of the left hind paw on day 0. Mice were tested for mechanical sensitivity using von Frey fibers before at baseline (one day before CFA injection) and 3, 7, 10, 14 and 21 days post-injection to quantify mechanical allodynia.
Statistical analyses were conducted using an α level of 0.05. Data were analyzed in Systat v. 13 by repeated-measures two-way ANOVA followed by a posthoc Sidak’s test with reported P values adjusted for multiple comparisons, and graphed with GraphPad Prism v.7.
RESULTS
Discovery Genome-wide Association Scan
After quality control exclusions and genome-wide imputation (see Supplementary Tables 1 and 2), 999 painful TMD cases and 2031 pain-free controls were analyzed for association with ~9.7M single nucleotide polymorphisms (Supplementary Fig. 1), using logistic regression under an additive model in both the full cohort (Fig. 2a) and in females (Fig. 2b) and males (Fig. 2c) separately. The GREML method implemented in the software package GCTA was used to estimate the variance explained by autosomal SNPs in order to estimate the narrow-sense heritability of chronic TMD. This analysis indicated that about 17% of the phenotypic variance was accounted for by additive genetic factors.
Fig. 2. Manhattan plots for the test of association with TMD.

All imputed SNPs passing the effective sample size filter are included. Threshold lines indicate suggestive (yellow, p=1×10−5) and genome-wide significant (red, p=5×10−8) values; statistically significant SNPs are labeled. a. All OPPERA subjects, n = 3030. b. Females only, n = 1956. c. Males only, n = 1074.
All SNPs that exceeded the whole genome-corrected threshold for significance are noted in Table 2, including the closest gene features. An imputed insertion/deletion (indel) polymorphism on chromosome 4, rs5862730, achieved genome-wide statistical significance (OR = 1.4, 95% CI: 1.26–1.61, P = 2.82×10−8, Fig. 3a). In a females-only analysis (Fig. 2b), rs5862730 on chromosome 4 was likewise associated with TMD (OR = 1.54, 95% CI: 1.33–1.79, P = 1.7×10−8). Another SNP, rs10092633 on chromosome 8, was also significant in females (OR = 4.12, 95% CI: 2.50–6.79, P = 2.9×10−8, MAF = 0.04); this SNP appears to be specific to African-Americans (minor allele A frequency in whites = 0.0, MAF in African-Americans = 0.15; see Supplementary Table 4 for an investigation of potential population stratification effects).
Table 2.
OPPERA TMD genome-wide significant associations.
| Group | rsID | Chr | Position | nearest genes | EA | EAF | N | OR | SE | P |
|---|---|---|---|---|---|---|---|---|---|---|
| all | rs5862730 | 4 | 146211844 | OTUD4, SMAD1 | D | 0.33 | 3030 | 1.42 | 0.06 | 2.82×10−8 |
| fem | rs5862730 | 4 | 146211844 | OTUD4, SMAD1 | D | 0.33 | 1956 | 1.54 | 0.08 | 1.70×10−8 |
| fem | rs10092633 | 8 | 41123732 | SFRP1 (intronic) | A | 0.03 | 1956 | 4.12 | 0.25 | 2.91×10−8 |
| male | rs34612513 | 3 | 137541085 | SOX14, CLDN18* | A | 0.08 | 1074 | 3.00 | 0.19 | 1.49×10−8 |
| male | rs28865059 | 3 | 137687399 | SOX14, CLDN18* | C | 0.09 | 1074 | 2.94 | 0.19 | 2.21×10−8 |
| male | rs13078961 | 3 | 137687685 | SOX14, CLDN18* | C | 0.09 | 1074 | 2.94 | 0.19 | 2.22×10−8 |
rsID: SNP name; Chr: chromosome; EA: effect allele; EAF: effect allele frequency; N: number of subjects in analysis; OR: odds ratio; SE: standard error; P: P-value; D: deletion allele of an insertion/deletion polymorphism. Chromosome and position are from GRCh37/hg19 (build 37).
While we report the two genes flanking this intergenic locus, eQTL analysis indicated functional effects of the locus at the MRAS gene, located at 138,067,508–138,124,377 bp.
Fig. 3. Regional plots for genome-wide associated SNPs.
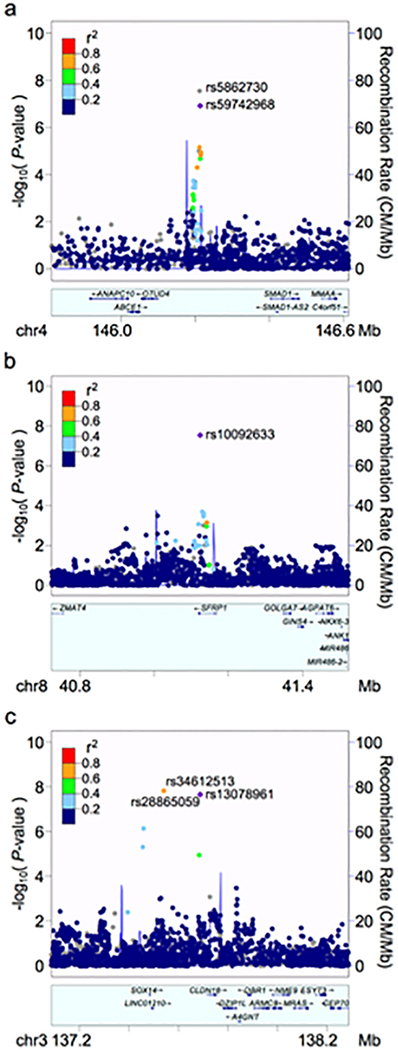
Plots were generated using LocusZoom. The index SNP for each plot is shown as a purple diamond. Linkage disequilibrium (using values obtained from hg19/1000 Genomes EUR data) with other SNPs is indicated by dot color, according to r2 values as indicated in the legend; grey values indicate no pairwise LD information is available for the index SNP. a. Chromosome 4. The most strongly associated SNP, rs5862730, was not included in the reference panel used for pairwise LD information with neighboring SNPs, so rs59742968 (r2 = 0.95 with rs5862730) was used as the index SNP for this plot. b. Chromosome 8, females only. c. Chromosome 3, males only.
In a males-only analysis (Fig. 2c), three SNPs on chromosome 3 were significantly associated with TMD (Fig. 3c). These SNPs, which were in high linkage disequilibrium (LD; r2 ≥ 0.8), were rs34612513 (OR = 3.00, 95% CI: 2.06–4.38, P = 1.5×10−8); rs28865059 (MAF = 0.09, OR = 2.94, 95% CI: 2.02–4.27, P = 2.2×10−8); and rs13078961 (OR = 2.94, 95% CI: 2.02–4.27, P = 2.2×10−8). Further statistical analysis revealed a significant interaction between rs13078961 (selected to be illustrative of this locus) and sex (P = 0.0001), and no association signal in the corresponding females-only analysis (OR = 1.02, P = 0.88). Altogether, the data suggest this locus is associated with TMD only in males.
Replication and Meta-analysis
Five SNPs from three distinct loci, significantly associated in either the combined or sex-stratified analyses (Table 2), were carried forward for examination in seven replication cohorts (Fig. 4). SNP rs5862730 (chromosome 4) significant in the analysis of all OPPERA subjects was not in the cleaned datasets for SHIP nor NFBC, although it was not significantly associated with TMD at P < 0.017 (one-tailed test, corrected for replication testing at 3 independent loci) in combined or female-only analyses in any of the available replication cohorts (Fig. 4a). A fixed-effects meta-analysis summarizing effects from available replication cohorts (but not the discovery cohort) also did not indicate an overall association effect of this SNP (OR = 1.0, 95% CI: 0.93–1.07).
Fig. 4. Forest plots for associated SNPs in OPPERA and the replication cohorts.

The OR and 95% C.I. for the OPPERA discovery cohort is given in the first row. OR and 95% C.I. values for all available replication cohorts are provided in subsequent rows, and the final row indicates the meta-analysis effect in replication cohorts only, not including the discovery cohort. The P value at the top of each plot pertains to the meta-analysis effect in replication cohorts only. a. chr4: rs59742968, male and female combined; for the replication meta-analysis, Cochrane’s Q P = 0.37, I2 = 5.4. b. chr8: rs10092633, females only, Cochrane’s Q P = 0.16, I2 = 38.3. c. chr3: rs13078961, males only, Cochrane’s Q P = 0.79, I2 = 0.0. d. chr3: rs34612513, males only, Cochrane’s Q P = 0.95, I2 = 0.0. e. chr3: rs28865059, males only, Cochrane’s Q P = 0.65, I2 = 0.0.
The SNP on chromosome 8, rs10092633, associated with TMD only in females in the discovery phase, was genotyped in four replication datasets (excluding SHIP and NFBC). No significant association with TMD was observed (Fig. 4b), although a suggestive effect in a direction consistent with OPPERA was observed in UKB females (OR = 1.87, 95% CI: 0.83–4.24, one-tailed P = 7.8×10−2), albeit with a low MAF (0.003).
The three SNPs on chromosome 3 with male-specific associations were not examined in the SPB cohort, which included only females. Genotypes for two SNPs were available in all six replication datasets. One SNP, rs13078961, showed nominal replication of the OPPERA association in a meta-analysis of all six replication cohorts’ males (OR=1.16, 95% CI: 1.0–1.35, one-sided test P = 2.3×10−2; Fig. 4c), although this result was not significant after Bonferroni correction for the number of independent loci. Among the individual replication cohorts, it was also nominally significantly associated (in males only) in HCHS/SOL (OR=1.23, 95% CI: 0.98–1.5, one-sided test P = 3.7×10−2; Fig. 4c). The other SNP, rs34612513, had a weaker association in the replication cohorts and did not replicate in the meta-analysis (OR=1.09, 95% CI: 0.94–1.27, one-sided P = 1.4×10−1; Fig. 4d). Genotypes for one SNP, rs28865059, were present in only four of the six replication datasets. Similar to rs13078961, the OPPERA finding replicated in the males-only meta-analysis (OR=1.17, 95% CI: 1.0–1.37, one-sided test P = 2.4×10−2; Fig. 4e) as well as HCHS/SOL males (OR = 1.23, 95% CI: 0.98–1.55, one-tailed P = 3.7×10−2; Fig. 4e), but not in the other three cohorts’ males. The FDR for replication testing of the three discoveries is 0.069, which indicates an expectation of 0.2 false discoveries among the three loci.
Thus, only the chromosome 3 SNPs were able to be independently replicated. As replication was nominal and was not significant after Bonferroni correction for the number of tested loci, we further examined this locus for potential functional relevance to pain in bioinformatic analysis.
Bioinformatic Analyses
To provide evidence for the functional significance of genetic variants that were associated with painful TMD in discovery or replication association studies, we performed further bioinformatics analysis. As disease-associated loci may alter gene function via effects on genes distal from the polymorphic variants identified in the association analysis, we first examined correlations between SNPs and gene expression. Separate analyses considered regulatory effects on expression in cis (i.e., with neighboring genes) and globally in trans (with genes located more than 1M nucleotides away); these analyses also considered probes that characterize exons constituting distinct isoforms of genes. The same five SNPs that emerged from the discovery cohort were investigated for eQTLs in both human post-mortem nerve tissue extracted from DRG, and in blood from OPPERA participants.
In the DRG experiment, none of the five SNPs were associated in cis with mRNA expression at the gene level, using a FDR threshold of 1%. However, at the exon level, the three SNPs associated with TMD in males only (rs34612513, rs13078961, and rs28865059) exhibited cis-acting eQTL effects (Supplementary Table 5). These SNPs showed significant association with expression of the gene muscle RAS oncogene homolog (MRAS, representative SNP rs13078961: beta = −0.51, P = 2.43×10−5, FDR = 5.83×10−3; Fig. 5), which has a transcription start site about 380 kb from the eQTL region (Fig. 3c). In sex-stratified analyses, significant associations between rs13078961 and several MRAS exons were observed in males (for exon 8: beta = −0.55, P = 2.25×10−4, FDR = 4.73×10−3) but not in females (beta = −0.47, P = 8.2×10−3, FDR = 2.0×10−1).
Fig. 5. Replicated male-only TMD risk SNP rs13078961 is an eQTL for the 3’UTR of MRAS gene.

(a) MRAS has 6 different splice isoforms in the RefSeq database (top), comprising 9 exons (numbered 1–9 inclusively). mRNA levels measured by 9 non-overlapping microarray probes in exons 5–9 were significantly associated with dosage of minor alleles of SNP rs13078961 in human DRGs as discovery (green), blood as replication (blue), and TMD caseness (red). (b) The relative position of microarray probes at 3’ UTR of MRAS (exon 9) associated with SNP rs13078961 in human DRGs (green), blood (blue), and TMD caseness (red). Coordinates are UCSC hg19. For brevity, only the last two digits of probe identifiers are shown; whole probe identifiers are: PSR030135xx.
For trans-acting associations, a large number of genes showed statistically relevant associations at FDR 1% with the same male-specific locus. This association was observed at both gene- and exon-levels, and in both cases was driven exclusively by males (Supplementary Table 5c). To assess if identified trans-acting genes are enriched in any biological pathway or are related to known human diseases, we applied a pathway analysis approach. Both Ingenuity ® Pathway Analysis and Pathway Studio methods identified immune response and particularly B Cell and T Cell Receptor Signaling as of highest significance (Supplementary Tables 6 and 7).
Next, we examined eQTL association using blood RNA samples from the OPPERA cohort. Similarly to the DRG eQTL analysis, the representative SNP rs13078961 was associated with MRAS mRNA expression at the exon level with a similar pattern in males and females combined (beta = −0.21, P = 3.76×10−2), and nominally significant only in males in a sex-stratified analysis (beta = −0.39, P = 2.17×10−2) (Supplementary Table 8). Importantly, in all cases, the minor allele of rs13078961 was associated with a lower level of MRAS expression. We found no statistically significant associations at FDR 1% levels for trans-acting eQTLs for either gene- or exon-level probes. Thus, our data indicate MRAS mRNA exhibits a similar cis-eQTL-dependent expression pattern in sensory DRG and whole blood in males.
Finally, we tested if whole blood mRNA levels of MRAS varied according to TMD case status. We found a statistically significant association at the gene level (beta = −0.04, P = 4.8×10−2), in males and females combined. One exon of MRAS in males also was nominally significantly associated with a much larger effect size (beta = −0.27, P = 1.7×10−2) (Supplementary Table 9). The direction of association was consistently negative, indicating levels of mRNA are lower in TMD cases, in agreement with the eQTL results.
Overall, the male-specific eQTL and differential M-RAS expression was observed mostly with different exons of the MRAS transcript, rather than with the entire gene (Fig. 5). These probes, though distinct, are situated adjacent to each other at the very end of the 3’ untranslated region (UTR), which is the most common location of eQTL regulation of mRNAs.[28; 33] Importantly, we note that we did not apply Bonferroni correction for the number of exons we tested, but such correction would be overly conservative, as expression of the vast majority of exons is not independent, and notably, our association pattern was consistently located at the last 3’ UTR exon of the gene.
Mras Null Mutant Mouse Nociceptive Assays
Genetic association and eQTL analyses both suggested MRAS expression level influenced chronic pain in males, so we tested a null mutant (knockout) mouse in which functional Mras protein was reduced or eliminated. The injection of CFA was selected as an assay relevant to a wide variety of chronic pain conditions, especially those with an inflammatory neuroimmune component, to confirm the involvement of this gene in pain. We compared the mechanical hypersensitivity (allodynia) induced by hind paw injection of CFA between Mras null mutant, heterozygote, and wild-type mice (Fig. 6). All groups exhibited a decrease in withdrawal threshold to stimulation with von Frey fibers by day 3, which resolved by day 21 (Fig. 5a). There were no genotype differences in baseline mechanical thresholds prior to CFA injection or peak mechanical allodynia observed 3 days after CFA injection. A two-between (genotype × sex) repeated-measures (RM) ANOVA (baseline to Day 21) showed a significant genotype × sex × time interaction (F10,120 = 2.1, P = 0.03). In sex-segregated one-between RM ANOVAs, there was a significant genotype × RM interaction in males (F10,80 = 2.9, P = 0.004; Fig. 5b), but not in females (F10,40 = 2.1, P = 0.06; Fig. 5c). In male but not female null mutant mice, the duration of CFA-induced mechanical allodynia was significantly prolonged, lasting for 14–21 days instead of the 3–7 day duration exhibited by wildtypes.
Fig. 6. Mechanical allodynia following CFA injury in Mras knockout mice and wild type controls.
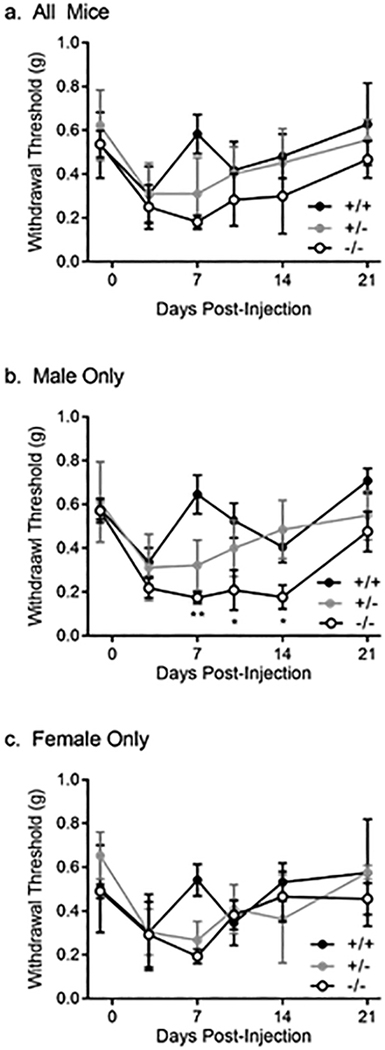
Mean ± SD reported. a. all mice (n=30); b. male mice only (n=19); c. female mice only (n=11). * p < 0.05; ** p < 0.01.
DISCUSSION
Extending beyond previous candidate gene investigations, we now report a discovery GWAS and validation study of clinically diagnosed painful TMD. In the discovery phase analysis of 3030 individuals from the OPPERA study, genome-wide significant associations with chronic TMD were found for three loci, including an association in males only at three linked SNPs on chromosome 3 (rs28865059, rs13078961, rs34612513), with OR around 3.0. In meta-analysis of up to seven independent case-control and population-based cohorts we observed a nominal replication of two SNPs in this locus. Functional studies of the chromosome 3 locus implicated allele-specific expression differences of the nearby gene MRAS for its effects, and we observed that male Mras knockout mice exhibited slower recovery from inflammatory pain hypersensitivity induced by CFA.
As in many chronic idiopathic pain conditions,[23] a higher prevalence of TMD among females has been observed, suggesting that sex-specific mechanisms may be responsible. We examined genotype effects within sex in order to discover variants and pathways potentially underlying these mechanisms. Relevant associations at the chromosome 3 locus were observed in males only, and the sex-specificity of this effect was supported by a significant sex × genotype interaction (P = 0.0001). Consistent with the primary association of these SNPs with TMD in males only, all eQTL associations were nominally significant in males in both DRG and blood RNA samples. Sex-dependent effects observed in the mouse inflammatory pain model provide insight into the nature of the susceptibility to persistent pain conferred by a decrease in Mras expression. Neither sex showed a genotype difference in baseline mechanical pain sensitivity, and all mice displayed similar levels of acute pain hypersensitivity three days after CFA treatment. In males, null mutant mice showed substantially delayed recovery from CFA at day 10 and day 14 compared to wildtypes, suggesting that low expression of Mras interferes with recuperative processes. Although the mechanism by which Mras facilitates this recovery is unknown, our results indicate that it is an important component to healing in males, whereas females appear to rely on separate pathways.
Our results demonstrate the importance of conducting sex-stratified analyses in association and behavioral studies of pain.[24; 25; 44] We identified more genome-wide significant associations when we separated our cohorts into males and females, compared to the combined cohort, despite reduced sample size.[2; 46] Our findings suggest that males and females may develop TMD through different pathophysiological pathways. An interesting question remains as to why the strongest association results, which we were able to reproduce and show functional relevance, are in male-specific loci, even though chronic TMD is much more prevalent in females. In contrast to the typical approach of explaining increased risk in females, perhaps these findings highlight the value of understanding lowered risk or resiliency in males for pain chronicity. In this regard, our discovery may have identified a pathway that protects males from developing chronic TMD.
M-RAS is a member of the Ras family of small GTPases, and is broadly expressed in many tissue types including brain, nerves, and heart. It is generally involved in cell growth and differentiation processes, and is a component of tumor necrosis factor-alpha (TNF-α)[49] and ERK-MAPK[50] signaling pathways. Because the candidate SNPs were associated with the last exon’s expression level rather than the entire MRAS mRNA, we suggest that these SNPs or haplotypes affect exon expression in a relatively less abundant alternative isoform, by creating or removing alternative exons, the polyadenylation signal, or splice sites,[18; 22; 51] or via selective miRNA-dependent degradation.[20]
The direction of the eQTL effect was identical for all three associated SNPs at this locus, such that the minor allele associated with higher risk of TMD was also associated with lower MRAS expression in all exons in both DRG and blood (Supplementary Tables 5 and 8). Consistent with this observation, we found mRNA levels of MRAS negatively correlated with TMD case status with the strongest effect again in the probe SR03013530 at the 3’ UTR (Supplementary Table 9, Fig. 5), reinforcing the direct contribution of MRAS to the TMD resistant phenotype.
As MRAS has not previously been connected with a pain phenotype or disorder, we sought to generate further hypotheses regarding the downstream effects of the MRAS eQTL using pathway analysis methods. Our analysis of trans-acting associations with the three associated SNPs further confirmed the primary link of this locus with TMD was exclusive to males, as multiple trans-associated genes were found in our male-specific DRG analysis. We used two separate pathway analysis programs to understand if these genes are related to particular biological processes, and identified immune response with the focus on B/T-cell receptor signaling as a primary pathway in DRG. This finding further emphasizes a role of the immune system in neurological tissues in the pathophysiology of chronic pain and is in line with our recent evidence showing contribution of the human leukocyte antigen (HLA) locus to pain-related genetic associations[28] It is also consistent with the growing understanding of the sexually divergent roles of immune cells, as animal studies have provided evidence that pain hypersensivity in males is dependent on microglia, while females rely on a separate system likely involving T lymphocytes. [42; 43] Sex differences in M-RAS signaling may arise due to this dimorphism in male and female systems, or they may be secondary to interactions between TNF-α and hormones such as estrogen[40] or testosterone.[5; 36] Given that we identified much stronger SNP-dependent MRAS regulation in DRG than in blood (Supplementary Table 5), our pathway analysis results suggest that this association reflects interaction of the immune system with sensory neurons (i.e., neuroimmune modulation), rather than immune processes per se.
Other findings from the discovery study implicated genes with the potential for intriguing pathophysiological insights, but until they can be reproduced in independent cohorts should be considered provisional. We were not able to identify any relevant functionality for rs5862730, the indel on chromosome 4, through our bioinformatics approaches, nor were we able to conclusively pinpoint the gene responsible for the effect. The most promising nearby candidate gene is SMAD1, which is a transcriptional modulator involved in cell growth and development, most prominently via mediation of signaling by bone morphogenetic proteins (BMPs).[16] Although a clear link between SMAD1 and TMD pain has not been confirmed, other SMAD pathway genes have been linked to acute inflammatory (but not neuropathic) pain[31] and excitability of DRG neurons[14] as well as osteoarthritis of the temporomandibular joint.[47] Furthermore, our group found that TGFβ1 regulation of cytokines such as MCP-1 and IL-8, which is mediated by SMAD protein activation, is consistently inhibited in the plasma of TMD cases relative to controls.[39]
As with all genome-wide surveys, this study has a number of limitations. An estimated 88% of GWAS findings are located outside of gene coding regions,[9] a proportion consistent with the findings of this study. We have used bioinformatics tools to determine functionality of our findings, leading to the implication of the putative eQTL at the “males-only” chromosome 3 locus with MRAS expression. However, the mechanisms underlying the remainder of the putative associations must be elucidated by further study. This study was also marked by the low replication rate typically observed in GWAS studies, commonly ascribed to low power, as well as phenotypic or ancestral heterogeneity.[12; 15] We have attempted to replicate statistically significant findings from a moderately powered discovery study (n=3030) in cohorts of vastly larger size; however, greater sample size often comes at a huge cost to phenotypic homogeneity. Though we accounted for genetic ancestral variation within studies via principal components analysis adjustment, regional and ethnic differences between the replication cohorts may alter genetic effects, precluding replication. Several of the SNPs observed in the discovery cohort had relatively low MAF in particular racial groups, which would limit our power to observe effects in other cohorts, especially those with different ancestral background. Although we observed a number of significant associations, the associated SNPs account for a very small proportion of the observed phenotypic variance. As we estimated the additive heritability of TMD at 17%, there are likely many TMD susceptibility variants yet to be discovered with larger cohorts and improved phenotyping. Finally, the consequences of Mras deletion in the knockout mouse model may be quite different from the allelic effect on regulation of MRAS in humans, limiting the interpretability of the animal study.
As has been observed in GWAS meta-analyses of other complex traits, we anticipate studies combining larger cohorts in diverse populations will increase the number of loci associated with TMD. Our results provide evidence to guide the design of these studies, suggesting that different molecular mechanisms underlie pathology in males and females. These results also provide targets for investigation into novel therapeutic strategies, notably the Ras subfamily of signaling molecules, and further emphasize the contribution of immune processes in the pathophysiology of chronic pain.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank the OPPERA program staff and participants at the respective host universities, and for resources specifically provided for this project by these institutions: University at Buffalo, University of Florida, University of Maryland–Baltimore, and University of North Carolina–Chapel Hill. We also thank the staff and participants of the replication cohorts for their important contributions. We gratefully acknowledge all the donor families who agreed to donate DRG tissue samples to pain research.
Funding Sources
OPPERA was supported by the National Institute of Dental and Craniofacial Research (NIDCR; https://www.nidcr.nih.gov/): grant number U01DE017018. SBS was supported by K12DE022793. The OPPERA program also acknowledges resources specifically provided for this project by the respective host universities: University at Buffalo, University of Florida, University of Maryland–Baltimore, and University of North Carolina–Chapel Hill. Funding for genotyping was provided by NIDCR through a contract to the Center for Inherited Disease Research at Johns Hopkins University (HHSN268201200008I). Data from the OPPERA study are available through the NIH dbGaP: phs000796.v1.p1, phs000761.v1.p1.
Dr. Diatchenko and the analytical team at McGill University were supported by the Canadian Excellence Research Chairs (CERC) Program grant (http://www.cerc.gc.ca/home-accueil-eng.aspx, CERC09). CERC09 also funded genotyping and cDNA array assays for DRG and blood expression studies.
Funding for DRG cohort collection was kindly provided by the US Cancer Pain Relief Committee (Career Development Award “Neurochemistry and Physiology of Human Pain-Processing Nuclei” to I.B.). We gratefully acknowledge all the donor families who agreed to donate tissue samples to pain research, without whose participation and cooperation this work would not have been possible.
SHIP is part of the Community Medicine Research net of the University of Greifswald, Germany, by the Federal Ministry of Education and Research (https://www.bmbf.de; grant numbers 01ZZ9603, 01ZZ0103, and 01ZZ0403), the Ministry of Cultural Affairs, and the Social Ministry of the Federal State of Mecklenburg–West Pomerania and the network “Greifswald Approach to Individualized Medicine (GANI_MED),” funded by the Federal Ministry of Education and Research (grant number 03IS2061A). Genome-wide data have been supported by the Federal Ministry of Education and Research (grant number 03ZIK012) and a joint grant from Siemens Healthcare (Erlangen, Germany) and the Federal State of Mecklenburg–West Pomerania. The University of Greifswald is a member of the Caché Campus program of the InterSystems GmbH.
The Northern Finland Birth Cohort 1966 has received financial support from the Academy of Finland (http://www.aka.fi; project grants 104781, 120315, 129269, 1114194, 24300796, Center of Excellence in Complex Disease Genetics and SALVE), University Hospital Oulu, Biocenter, University of Oulu (75617), NHLBI grant 5R01HL087679-02 through the STAMPEED program (1RL1MH083268-01), NIH/National Institute of Mental Health (NIMH) (5R01MH63706:02), ENGAGE project and grant agreement HEALTH-F4-2007-201413, EU FP7 EurHEALTH Ageing-277849, the Medical Research Council (G0500539, G0600705, G1002319, PrevMetSyn/SALVE), and the MRC, Centenary Early Career Award. The program is currently being funded by the H2020-633595 DynaHEALTH action and Academy of Finland EGEAproject (285547). The DNA extractions, sample quality controls, biobank upkeeping, and aliquoting were performed in the National Public Health Institute, Biomedicum Helsinki and supported financially by the Academy of Finland and Biocentrum Helsinki. We thank the late Professor Paula Rantakallio (launch of NFBCs) and Ms Outi Tornwall and Ms Minttu Jussila (DNA biobanking). The authors would like to acknowledge the contribution of the late Academian of Science Leena Peltonen. The study has been financially supported by the Academy of Finland, the European Commission (EURO-BLCS, https://ec.europa.eu/commission; Framework 5 award QLG1-CT-2000-01643), the Sigrid Jusélius Foundation (http://sigridjuselius.fi), and US National Institute of Mental Health (https://www.nimh.nih.gov; 5R01MH 63706:02).
The Brazilian cohort has been funded by the São Paulo Research Foundation (http://www.fapesp.br; grant numbers 2006/56019-8R and 2009/02520-6), and genotyping was funded by the Canadian Excellence Research Chairs (CERC) Program (grant CERC09).
The Complex Persistent Pain Conditions: Unique and Shared Pathways of Vulnerability Program Project was supported by NIH/National Institute of Neurological Disorders and Stroke (NINDS; https://www.ninds.nih.gov) grant NS045685 to the University of North Carolina at Chapel Hill, and genotyping was funded by the Canadian Excellence Research Chairs (CERC) Program (grant CERC09).
The OPPERA II study was supported by the NIDCR under Award Number U01DE017018, and genotyping was funded by the Canadian Excellence Research Chairs (CERC) Program (grant CERC09).
The authors thank the staff and participants of HCHS/SOL for their important contributions.
The Hispanic Community Health Study/Study of Latinos is a collaborative study supported by contracts from the National Heart, Lung, and Blood Institute (NHLBI; https://www.nhlbi.nih.gov) to the University of North Carolina (HHSN268201300001I / N01-HC-65233), University of Miami (HHSN268201300004I / N01-HC-65234), Albert Einstein College of Medicine (HHSN268201300002I / N01-HC-65235), University of Illinois at Chicago – HHSN268201300003I / N01-HC-65236 Northwestern Univ), and San Diego State University (HHSN268201300005I / N01-HC-65237). The following Institutes/Centers/Offices have contributed to the HCHS/SOL through a transfer of funds to the NHLBI: National Institute on Minority Health and Health Disparities, National Institute on Deafness and Other Communication Disorders, National Institute of Dental and Craniofacial Research, National Institute of Diabetes and Digestive and Kidney Diseases, National Institute of Neurological Disorders and Stroke, NIH Institution-Office of Dietary Supplements. Data from HCHS/SOL are available through the NIH database of Genotypes and Phenotypes (dbGaP): phs000810. v1.p1.
The current study was conducted under UK Biobank (http://www.ukbiobank.ac.uk)application number 20802.
Dr. Belfer contributed to this article in her personal capacity. The views expressed are her own and do not necessarily represent the views of the National Institutes of Health or the United States Government.
Dr. Zaykin was supported in part by the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences.
Footnotes
Conflict of Interest
Luda Diatchenko and William Maixner are cofounders and equity stock holders in Algynomics, Inc., a company providing research services in personalized pain medication and diagnostics. Shad B. Smith, Gary D. Slade, and Roger B. Fillingim are equity stock holders in Algynomics, Inc. The other authors declare no conflicts of interest.
REFERENCES
- [1].Bair E, Ohrbach R, Fillingim RB, Greenspan JD, Dubner R, Diatchenko L, Helgeson E, Knott C, Maixner W, Slade GD. Multivariable modeling of phenotypic risk factors for first-onset TMD: the OPPERA prospective cohort study. J Pain 2013;14(12 Suppl):T102–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Belfer I, Segall SK, Lariviere WR, Smith SB, Dai F, Slade GD, Rashid NU, Mogil JS, Campbell CM, Edwards RR, Liu Q, Bair E, Maixner W, Diatchenko L. Pain modality- and sex-specific effects of COMT genetic functional variants. Pain 2013;154(8):1368–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Benita Y, Cao Z, Giallourakis C, Li C, Gardet A, Xavier RJ. Gene enrichment profiles reveal T-cell development, differentiation, and lineage-specific transcription factors including ZBTB25 as a novel NF-AT repressor. Blood 2010;115(26):5376–5384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Chaplan S, Bach F, Pogrel J, Chung J, Yaksh T. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Meth 1994;53:55–63. [DOI] [PubMed] [Google Scholar]
- [5].D’Agostino P, Milano S, Barbera C, Di Bella G, La Rosa M, Ferlazzo V, Farruggio R, Miceli DM, Miele M, Castagnetta L, Cillari E. Sex hormones modulate inflammatory mediators produced by macrophages. Annals of the New York Academy of Sciences 1999;876:426–429. [DOI] [PubMed] [Google Scholar]
- [6].Delaneau O, Marchini J, Zagury JF. A linear complexity phasing method for thousands of genomes. Nat Meth 2012;9(2):179–181. [DOI] [PubMed] [Google Scholar]
- [7].Dworkin S, LeResche L. Research diagnostic criteria for temporomandibular disorders: review, criteria, examinations and specifications, critique. J Craniomandib Disord 1992;6(4):301–355. [PubMed] [Google Scholar]
- [8].Dworkin SF, Huggins KH, Le Resche L, Von Korff M, Howard J, Truelove E, Sommers E. Epidemiology of signs and symptoms in temporomandibular disorders: clinical signs in cases and controls. J Am Dent Assoc 1990;120(3):273–281. [DOI] [PubMed] [Google Scholar]
- [9].Edwards SL, Beesley J, French JD, Dunning AM. Beyond GWASs: illuminating the dark road from association to function. Am J Hum Genet 2013;93(5):779–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Fillingim RB, Ohrbach R, Greenspan JD, Knott C, Diatchenko L, Dubner R, Bair E, Baraian C, Mack N, Slade GD, Maixner W. Psychological factors associated with development of TMD: the OPPERA prospective cohort study. J Pain 2013;14(12 Suppl):T75–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Howie BN, Donnelly P, Marchini J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet 2009;5(6):e1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Ioannidis JPA. Non-replication and inconsistency in the genome-wide association setting. Hum Hered 2007;64(4):203–213. [DOI] [PubMed] [Google Scholar]
- [13].Isong U, Gansky SA, Plesh O. Temporomandibular joint and muscle disorder-type pain in U.S. adults: the National Health Interview Survey. J Orofac Pain 2008;22(4):317–322. [PMC free article] [PubMed] [Google Scholar]
- [14].Jeub M, Emrich M, Pradier B, Taha O, Gailus-Durner V, Fuchs H, de Angelis MH, Huylebroeck D, Zimmer A, Beck H, Racz I. The transcription factor Smad-interacting protein 1 controls pain sensitivity via modulation of DRG neuron excitability. Pain 2011;152(10):2384–2398. [DOI] [PubMed] [Google Scholar]
- [15].Kraft P, Zeggini E, Ioannidis JPA. Replication in genome-wide association studies. Statistical Science 2009;24(4):561–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Kretzschmar M, Liu F, Hata A, Doody J, Massague J. The TGF-beta family mediator Smad1 is phosphorylated directly and activated functionally by the BMP receptor kinase. Genes Dev 1997;11(8):984–995. [DOI] [PubMed] [Google Scholar]
- [17].Kuo C-L, Zaykin DV. Novel rank-based approaches for discovery and replication in genome wide association studies. Genetics 2011;189(1):329–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Lalonde E, Ha KC, Wang Z, Bemmo A, Kleinman CL, Kwan T, Pastinen T, Majewski J. RNA sequencing reveals the role of splicing polymorphisms in regulating human gene expression. Genome Res 2011;21(4):545–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Laurie CC, Doheny KF, Mirel DB, Pugh EW, Bierut LJ, Bhangale T, Boehm F, Caporaso NE, Cornelis MC, Edenberg HJ, Gabriel SB, Harris EL, Hu FB, Jacobs KB, Kraft P, Landi MT, Lumley T, Manolio TA, McHugh C, Painter I, Paschall J, Rice JP, Rice KM, Zheng X, Weir BS, Investigators ftG. Quality control and quality assurance in genotypic data for genome-wide association studies. Genet Epidemiol 2010;34(6):591–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 2005;120(1):15–20. [DOI] [PubMed] [Google Scholar]
- [21].Li Y, Willer CJ, Ding J, Scheet P, Abecasis GaR. MaCH: using sequence and genotype data to estimate haplotypes and unobserved genotypes. Genet Epidemiol 2010;34(8):816–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Li YI, van de Geijn B, Raj A, Knowles DA, Petti AA, Golan D, Gilad Y, Pritchard JK. RNA splicing is a primary link between genetic variation and disease. Science 2016;352(6285):600–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Maixner W, Fillingim RB, Williams DA, Smith SB, Slade GD. Overlapping chronic pain conditions: Implications for diagnosis and classification. J Pain 2016;17(9 Suppl):T93–t107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Melchior M, Poisbeau P, Gaumond I, Marchand S. Insights into the mechanisms and the emergence of sex-differences in pain. Neuroscience 2016;338:63–80. [DOI] [PubMed] [Google Scholar]
- [25].Mogil JS, Bailey AL. Sex and gender differences in pain and analgesia. Progress in brain research 2010;186:141–157. [DOI] [PubMed] [Google Scholar]
- [26].Nikitin A, Egorov S, Daraselia N, Mazo I. Pathway studio--the analysis and navigation of molecular networks. Bioinformatics 2003;19(16):2155–2157. [DOI] [PubMed] [Google Scholar]
- [27].Ohrbach R, Fillingim RB, Mulkey F, Gonzalez Y, Gordon S, Gremillion H, Lim P-F, Ribeiro-Dasilva M, Greenspan JD, Knott C, Maixner W, Slade G. Clinical findings and pain symptoms as potential risk factors for chronic TMD: descriptive data and empirically identified domains from the OPPERA Case-Control Study. J Pain 2011;12(11, Supplement):T27–T45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Parisien M, Khoury S, Chabot-Dore A-J, Sotocinal SG, Slade GD, Smith SB, Fillingim RB, Ohrbach R, Greenspan J, Maixner W, Mogil JS, Belfer I, Diatchenko L. Effect of human genetic variability on gene expression in dorsal root ganglia and association with pain phenotypes. Cell Rep 2017;19(9):1940–1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Plesh O, Adams SH, Gansky SA. Temporomandibular joint and muscle disorder-type pain and comorbid pains in a national US sample. J Orofac Pain 2011;25(3):190–198. [PMC free article] [PubMed] [Google Scholar]
- [30].Plesh O, Noonan C, Buchwald DS, Goldberg J, Afari N. Temporomandibular disorder–type pain and migraine headache in women: A preliminary twin study J Orofac Pain 2012;26(2):91–98. [PubMed] [Google Scholar]
- [31].Pradier B, Jeub M, Markert A, Mauer D, Tolksdorf K, Van de Putte T, Seuntjens E, Gailus-Durner V, Fuchs H, Hrabe de Angelis M, Huylebroeck D, Beck H, Zimmer A, Racz I. Smad-interacting protein 1 affects acute and tonic, but not chronic pain. Eur J Pain 2014;18(2):249–257. [DOI] [PubMed] [Google Scholar]
- [32].Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira M, Bender D, Maller J, Sklar P, de Bakker P, Daly M, Sham P. PLINK: a toolset for whole-genome association and population-based linkage analysis. Am J Hum Genet 2007;81(3):559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Ramasamy A, Trabzuni D, Guelfi S, Varghese V, Smith C, Walker R, De T, Coin L, de Silva R, Cookson MR, Singleton AB, Hardy J, Ryten M, Weale ME. Genetic variability in the regulation of gene expression in ten regions of the human brain. Nat Neurosci 2014;17(10):1418–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Sanders AE, Essick GK, Fillingim R, Knott C, Ohrbach R, Greenspan JD, Diatchenko L, Maixner W, Dubner R, Bair E, Miller VE, Slade GD. Sleep apnea symptoms and risk of temporomandibular disorder: OPPERA cohort. J Dent Res 2013;92(7 suppl):S70–S77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Sanders AE, Jain D, Sofer T, Kerr KF, Laurie CC, Shaffer JR, Marazita ML, Kaste LM, Slade GD, Fillingim RB, Ohrbach R, Maixner W, Kocher T, Bernhardt O, Teumer A, Schwahn C, Sipila K, Lahdesmaki R, Mannikko M, Pesonen P, Jarvelin M, Rizzatti-Barbosa CM, Meloto CB, Ribeiro-Dasilva M, Diatchenko L, Serrano P, Smith SB. GWAS identifies new loci for painful temporomandibular disorder. J Dent Res 2017;96(3):277–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Santos-Galindo M, Acaz-Fonseca E, Bellini MJ, Garcia-Segura LM. Sex differences in the inflammatory response of primary astrocytes to lipopolysaccharide. Biol Sex Differ 2011;2:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Slade GD, Bair E, By K, Mulkey F, Baraian C, Rothwell R, Reynolds M, Miller V, Gonzalez Y, Gordon S, Ribeiro-Dasilva M, Lim PF, Greenspan JD, Dubner R, Fillingim RB, Diatchenko L, Maixner W, Dampier D, Knott C, Ohrbach R. Study methods, recruitment, sociodemographic findings, and demographic representativeness in the OPPERA Study. J Pain 2011;12(11, Supplement):T12–T26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Slade GD, Bair E, Greenspan JD, Dubner R, Fillingim RB, Diatchenko L, Maixner W, Knott C, Ohrbach R. Signs and symptoms of first-onset TMD and sociodemographic predictors of its development: the OPPERA prospective cohort study. J Pain 2013;14(12 Suppl):T20–32.e21–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Slade GD, Conrad MS, Diatchenko L, Rashid NU, Zhong S, Smith SB, Rhodes J, Medvedev A, Makarov S, Maixner W, Nackley AG. Cytokine biomarkers and chronic pain: Association of genes, transcription, and circulating proteins with temporomandibular disorders and widespread palpation tenderness. Pain 2011;152(12):2802–2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Smith JA, Das A, Butler JT, Ray SK, Banik NL. Estrogen or estrogen receptor agonist inhibits lipopolysaccharide induced microglial activation and death. Neurochem Res 2011;36(9):1587–1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Smith SB, Maixner DW, Greenspan JD, Dubner R, Fillingim RB, Ohrbach R, Knott C, Slade GD, Bair E, Gibson DG, Zaykin DV, Weir BS, Maixner W, Diatchenko L. Potential genetic risk factors for chronic TMD: Genetic associations from the OPPERA Case Control Study. J Pain 2011;12(11, Supplement):T92–T101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Sorge RE, LaCroix-Fralish ML, Tuttle AH, Sotocinal SG, Austin J-S, Ritchie J, Chanda ML, Graham AC, Topham L, Beggs S, Salter MW, Mogil JS. Spinal cord toll-like receptor 4 mediates inflammatory and neuropathic hypersensitivity in male but not female mice. J Neurosci 2011;31(43):15450–15454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Sorge RE, Mapplebeck JC, Rosen S, Beggs S, Taves S, Alexander JK, Martin LJ, Austin JS, Sotocinal SG, Chen D, Yang M, Shi XQ, Huang H, Pillon NJ, Bilan PJ, Tu Y, Klip A, Ji RR, Zhang J, Salter MW, Mogil JS. Different immune cells mediate mechanical pain hypersensitivity in male and female mice. Nat Neurosci 2015;18(8):1081–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Sorge RE, Totsch SK. Sex differences in pain. Journal of neuroscience research 2017;95(6):1271–1281. [DOI] [PubMed] [Google Scholar]
- [45].Viechtbauer W Conducting meta-analyses in R with the metafor package. J Stat Softw 2010;36(3):48. [Google Scholar]
- [46].Wieskopf JS, Mathur J, Limapichat W, Post MR, Al-Qazzaz M, Sorge RE, Martin LJ, Zaykin DV, Smith SB, Freitas K, Austin JS, Dai F, Zhang J, Marcovitz J, Tuttle AH, Slepian PM, Clarke S, Drenan RM, Janes J, Al Sharari S, Segall SK, Aasvang EK, Lai W, Bittner R, Richards CI, Slade GD, Kehlet H, Walker J, Maskos U, Changeux JP, Devor M, Maixner W, Diatchenko L, Belfer I, Dougherty DA, Su AI, Lummis SC, Imad Damaj M, Lester HA, Patapoutian A, Mogil JS. The nicotinic alpha6 subunit gene determines variability in chronic pain sensitivity via cross-inhibition of P2X2/3 receptors. Sci Transl Med 2015;7(287):287ra272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Xiao JL, Meng JH, Gan YH, Zhou CY, Ma XC. Association of GDF5, SMAD3 and RUNX2 polymorphisms with temporomandibular joint osteoarthritis in female Han Chinese. J Oral Rehabil 2015;42(7):529–536. [DOI] [PubMed] [Google Scholar]
- [48].Yang J, Lee SH, Goddard ME, Visscher PM. GCTA: A tool for genome-wide complex trait analysis. Am J Hum Genet 2011;88(1):76–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Yoshikawa Y, Satoh T, Tamura T, Wei P, Bilasy SE, Edamatsu H, Aiba A, Katagiri K, Kinashi T, Nakao K, Kataoka T. The M-Ras-RA-GEF-2-Rap1 pathway mediates tumor necrosis factor-alpha dependent regulation of integrin activation in splenocytes. Mol Biol Cell 2007;18(8):2949–2959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Young LC, Hartig N, Munoz-Alegre M, Oses-Prieto JA, Durdu S, Bender S, Vijayakumar V, Vietri Rudan M, Gewinner C, Henderson S, Jathoul AP, Ghatrora R, Lythgoe MF, Burlingame AL, Rodriguez-Viciana P. An MRAS, SHOC2, and SCRIB complex coordinates ERK pathway activation with polarity and tumorigenic growth. Mol Cell 2013;52(5):679–692. [DOI] [PubMed] [Google Scholar]
- [51].Zhernakova DV, de Klerk E, Westra HJ, Mastrokolias A, Amini S, Ariyurek Y, Jansen R, Penninx BW, Hottenga JJ, Willemsen G, de Geus EJ, Boomsma DI, Veldink JH, van den Berg LH, Wijmenga C, den Dunnen JT, van Ommen GJ, t Hoen PA, Franke L. DeepSAGE reveals genetic variants associated with alternative polyadenylation and expression of coding and non-coding transcripts. PLoS Genet 2013;9(6):e1003594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Zhou X, Stephens M. Genome-wide efficient mixed-model analysis for association studies. Nat Genet 2012;44(7):821–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Study data have been deposited and made publicly available at the Database of Genotypes and Phenotypes (dbGaP) public repository (accession number phs000796.v1.p1).