

Abstract
This is the second of two invited articles reviewing the development of nucleoside analogue antiviral drugs, written for a target audience of virologists and other non-chemists, as well as chemists who may not be familiar with the field. As with the first paper, rather than providing a chronological account, we have chosen to examine particular examples of structural modifications made to nucleoside analogues that have proven fruitful as various antiviral, anticancer, and other therapeutics. The first review covered the more common, and in most cases, single modifications to the sugar and base moieties of the nucleoside scaffold. This paper focuses on more recent developments, especially nucleoside analogues that contain more than one modification to the nucleoside scaffold. We hope that these two articles will provide an informative historical perspective of some of the successfully designed analogues, as well as many candidate compounds that encountered obstacles.
Keywords: Nucleoside analogues, antiviral, prodrugs, anti-cancer, structural modifications
1. Introduction
This is the second of two invited articles reviewing the development of nucleoside analogue antiviral drugs, written for a target audience of virologists and other non-chemists, as well as chemists who may not be familiar with the field. As with the first paper, rather than providing a chronological account, we have chosen to examine particular examples of structural modifications made to nucleoside analogues that have proven fruitful as various antiviral, anticancer, and other therapeutics. The first review covered the more common, and in most cases, single modifications to the base and sugar moieties of the nucleosides scaffold. This second paper focuses on more recent developments in the field, especially nucleoside analogues that contain more than one modification to the nucleoside scaffold.
The term “nucleoside” was first used by Levene and Jacobs in 1909.1 A nucleoside is composed of a sugar moiety and nucleobase, whereas a nucleotide is composed of a sugar, nucleobase, and at least one phosphate (or phosphate-like) group (Figure 1). Both nucleosides and nucleotides play important roles in the replication and transcription of genetic information, and, as such, have been utilized for decades for chemotherapy, antiparasitic, antibacterial or antiviral therapeutics.2–6 Ideally, a nucleoside/tide analogue would mimic the structure of a natural nucleoside enough to be recognized by cellular or viral enzymes and be incorporated into the DNA or RNA replication cycle, however, these analogues would possess one or more modifications that would then lead to the disruption and/or termination of replication.7–9 Over the years, numerous modifications to the nucleos(t)ide scaffold have been made, including alterations to the sugar, nucleobase, glycosidic bond, and phosphate group (Figure 1). As described in the first paper, these modifications range from adding a substituent or group to the heterocyclic base or sugar, replacing an atom in either moiety, by moving an atom to a different position, or a combination of these approaches.3, 10 More recently, researchers have employed the latter, utilizing a combination of many different types of modifications, which has led to the development of a wide array of potent nucleoside therapeutics, with complex structures. For convenience, this review is organized based on modifications to the various positions on the sugar moiety, however many of the nucleoside analogues discussed also contain modifications to the nucleobase moiety as well.
Figure 1.
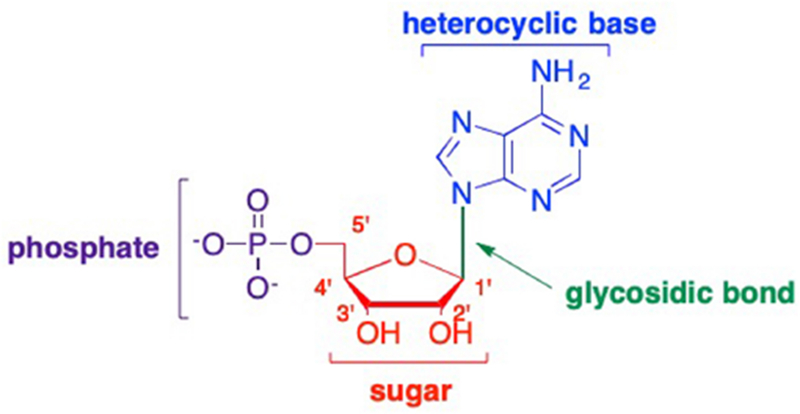
Sites for potential modifications to nucleos(t)ide analogues.
2. 1’-Sugar Modifications
The sugar modifications explored in the first review featured nucleosides with 2’-OH, 3’-OH, a combination of 2’ and 3’-OH modifications, carbocyclic nucleosides, alternative ring sizes, acyclic nucleosides, and acyclic nucleoside phosphonates. More recently, modifications to the 1’-carbon of the sugar, some in combination with prodrug strategies, have been pursued. This section details these analogues and later, their prodrugs.
2.1. Early 1’-Modifications
Structure-activity relationship (SAR) studies are common in drug design and typically involve “walking” around the nucleoside’s scaffold making a particular change and observing its subsequent effect on biological activity. One such study by Siddiqi et. al. found that moving a methyl substituent to the different positions on the sugar of an adenosine nucleoside yielded profound differences in activity.11 For example, replacement of the 4’-hydrogen with a methyl group in adenosine analogues led to a large decrease in activity against adenosine receptors, whereas replacing the 3’-hydrogen with a methyl group increased activity.11 Further analysis by Cappellacci et al. found that the replacement of the 1’-hydrogen with a methyl group had a different effect on different adenosine receptors, most notably, A1 and A2A, however, overall the 1’-methyl analogues demonstrated little or no activity (Figure 2).12
Figure 2.

Examples of early 1’-modified nucleoside analogues.
Other early 1’-modified nucleoside analogues included the 1’-fluoromethyladenosine analogues originally synthesized by Damont et al. (Figure 2).13 These analogues utilized electrophilic fluorination of an exocyclic double bond at the C-1 carbon in order to install the fluoromethyl group at the 1’ position.13 Unfortunately, like the 1’-methyl analogues, the 1’-fluoromethyl analogues also did not demonstrate any antiviral activity against bovine viral diarrhea virus (BVDV) or against hepatitis C virus (HCV) in a subgenomic replicon assay, however notably, the analogues were not toxic.13 Due to the lack of any antiviral activity, they were not pursued further.
2.2. 1’-Cyano Analogues
Further research led scientists to believe that the lack of activity demonstrated by the early 1’-analogues was due to instability of the glycosidic bond with the addition of the 1’-methyl group.12, 14 In general, the glycosidic bond is stable under physiological conditions, however, cleavage of this bond can occur and is dependent on various factors including pH, type of nucleobase, and 1’-substituents.14–18 Since the glycosidic bond cleavage occurs either by nucleophillic attack on the 1’ carbon of the sugar or by stabilization of the leaving group, changing the substituent from a hydrogen to any other group at the 1’ position could have a profound effect on glycosidic bond cleavage, either through steric or electronic effects.15, 19, 20 Scientists reasoned however, that if they replaced the hemiaminal (O-C-N) glycosidic bond with the O-C-C bond found in C-nucleosides, then they would be able to add 1’ substituents without compromising the integrity of the glycosidic bond.15, 21–23 This subsequently led to the development of a number of 1’-substituted C-nucleoside analogues.
In that regard, some of the most promising l’-substituted C-nucleosides pursued recently were the 1’-substituted 4-aza-7,9-dideazaadenosine C-nucleosides developed by Gilead (Figure 3).14, 23–28 An SAR study focused on various 1’-substituted analogues found that the 1’-cyano analogue displayed a broader spectrum of antiviral activity against viruses such as HCV, yellow fever virus (YFV), dengue-2 virus (DENV-2), influenza A, parainfluenza 3, Ebola virus (EBOV) and severe acute respiratory syndrome coronavirus (SARS-CoV), with the best antiviral activity against EBOV in HMVEC cells (EC50 = 0.78 μM).14, 23 These findings were especially interesting since both the 1’-methyl and 1’-vinyl analogues showed reduced potency, a much narrower spectrum of antiviral activity, and in some instances, higher toxicity compared to the 1’-cyano analogue.14,23 In contrast, the 1’-ethynyl analogue demonstrated no antiviral activity.14
Figure 3.

Gilead’s first generation 1’-substituted 4-aza-7,9-dideazaadeonosine C-nucleosides.
Due to the surprising broad-spectrum antiviral activity found with the 1’-cyano analogue, researchers at Gilead performed a computational docking study with the triphosphate analogue of the 1’-cyano analogue and various RNA virus polymerases. It was found that the 1’-cyano group occupies a unique pocket present in only the viral polymerase binding site, which leads to the increased selectivity of the 1’-cyano analogues for viral polymerases over human polymerases.23, 27 Moreover, in order to increase the delivery of this analogue, they also employed the McGuigan ProTide (PROdrug nucleoTIDE) approach.29–34
The ProTide approach has proven extremely valuable for delivery of nucleotide analogues, as well as to overcome the rate-limiting first phosphorylation step. During DNA/RNA replication, nucleosides (and nucleoside analogues) are phosphorylated by various host cell or viral kinases into their triphosphate form, which are then recognized by DNA polymerases, RNA polymerases, or reverse transcriptase and incorporated into the growing chain.7, 35 Since the triphosphate cannot be administered directly due to the highly charged nature of the phosphate groups, the prodrug helps deliver the nucleotide into the cell. A second limitation associated with nucleoside drugs is that the first phosphorylation step is often highly specific and rate-limiting, thus the nucleoside analogue is often not recognized and appears inactive.30–32, 36–38 To overcome this obstacle, McGuigan et al. created ProTides that would efficiently deliver the monophosphate nucleoside analogue into the target cell, bypassing the rate-limiting first phosphorylation step.23, 30–34, 39 These ProTides utilize a unique structure, with three “tunable” positions - the aryl, the amino acid, and the ester groups (Figure 4).23, 30–32 The aryl group and the amino acid ester mask the negative charges on the monophosphate, allowing the ProTide to efficiently cross the cell membrane.23, 30, 32–34 Following metabolism by various host enzymes, the monophosphate nucleotide analogue is successfully delivered and is subsequently phosphorylated into the active triphosphate.23, 32–34
Figure 4.
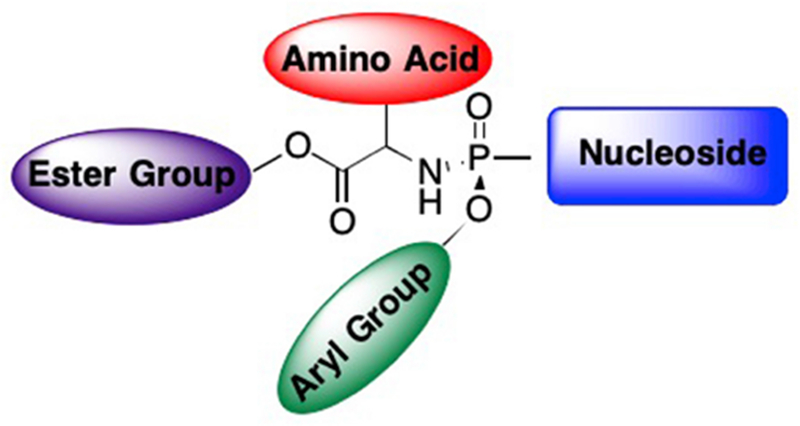
General structure of a McGuigan ProTide.
Using the McGuigan ProTide approach with the l’-cyano compound produced GS-5734 (Remdesivir), which increased the overall anti-EBOV activity (EC50 = 0.06 μM compared to 0.78 μM for the parent). In addition, this also increased the spectrum of the antiviral activity to include viruses that the parent nucleoside was not active against, including West Nile virus (WNV), Lassa fever virus, and Middle East respiratory syndrome coronavirus (MERS-CoV) (Figure 5).25–27 Further studies found that GS-5734 was an effective post-exposure therapeutic in EBOV-infected rhesus monkeys at 10 mg/kg.23, 27 Studies with healthy human volunteers are currently underway in order to evaluate pharmacokinetics and clinical safety, specifically in male Ebola survivors with EBOV persistence in semen (NCT02818582).
Figure 5.

Structure of the 1’-CN parent analogue and the McGuigan ProTide GS-5734.
3. 2’-Modifications
The first review focused on 2’-OH modifications, including the arabinose or “Ara” analogues in which the configuration of the 2’-OH is inverted, as well as the mono- and di-fluoro substituted 2’-modified nucleoside analogues. This was due to their role in the development of some of the first medically relevant nucleoside analogues, which greatly impacted the field of drug design. In this second article, we focus on more recent 2’-modifications, as well as 2’-modified analogues that contain additional modifications, especially to the nucleobase scaffold.
3.1. 2’-Methyl Modifications
Of the numerous 2’ modified nucleoside analogues that have been developed, some of the most promising have been the 2’-methyl analogues, particularly those that have exhibited activity against HCV.40, 41 The presence of this 2’-methyl group alters the configuration of the sugar which prevents the binding of subsequent nucleotides to the enzyme active site, thus these analogues are considered non-obligate chain terminators.42 Of these, 2’-methyl adenosine, 2’-methyl guanosine, and 2’-methyl cytidine (NM107) were initially pursued and all displayed micromolar levels of activity against HCV in whole-cell replicon assays (Figure 6).40, 41, 43–45 The adenosine analogue proved to be the most potent, with an EC50 of 0.26 μM compared to 3.5 μM for the guanosine analogue and 1.23 μM for the cytidine analogue.43–45 Interestingly, none of these compounds demonstrated any cytotoxicity in vitro.41, 43–45 Unfortunately, they displayed low bioavailability as well as a high rate of deamination of the nucleobase in the 2’-methyl adenosine analogues and increased glycosidic bond cleavage by purine nucleoside pyrophosphorylase.43, 44 The guanosine analogue also exhibited low bioavailability due to insufficient phosphorylation and decreased cellular uptake.43, 44, 46, 47
Figure 6.
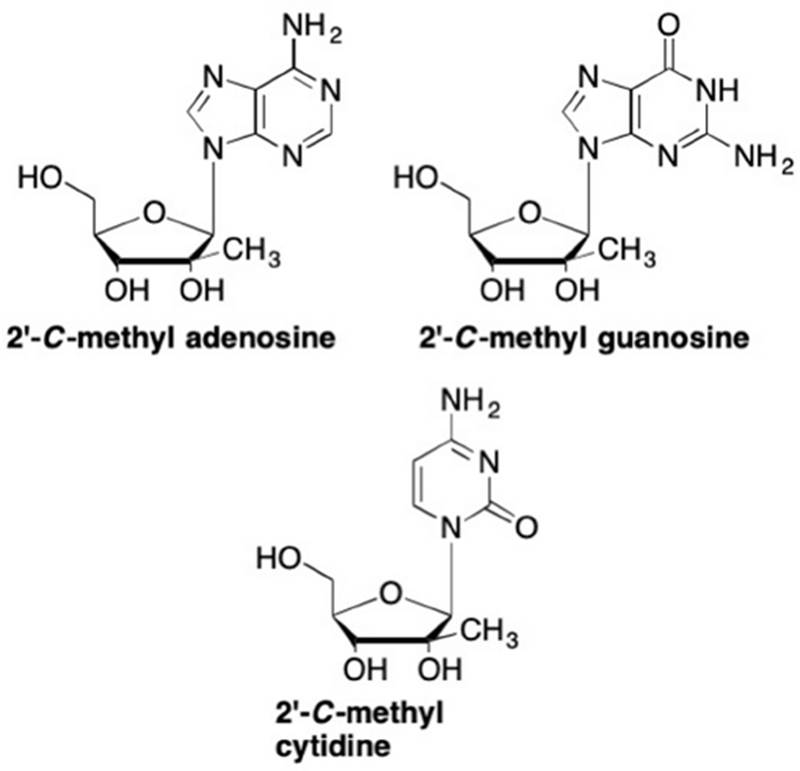
First generation 2’-methyl nucleoside analogues for HCV therapy.
While these initial studies were disappointing, they provided researchers with a starting point for developing the next generation of HCV nucleoside therapeutics. Through the initial studies with 2’-methyl adenosine and 2’-methyl guanosine, researchers determined that the bioavailability of these analogues could potentially be increased by removing the N7 of the purine ring system to yield a “deaza” analogue. Deazapurine nucleosides are naturally found in secondary metabolites produced by Streptomyces bacteria, and, due to their strong resemblance to natural purine nucleosides, the deazapurines have been shown to disrupt various biological functions, thereby leading to potent therapeutics.48 Of the initial deazamethyl purines synthesized, the most promising was 7-deaza-2’-methyl adenosine (MK-0608) (Figure 7), which demonstrated broad-spectrum activity against numerous flaviviruses, including HCV, DENV, Zika virus (ZIKV), tick-borne encephalitis (TBEV), and YFV with EC50 values ranging from 5 to 15 μM.49–55 Further analysis of MK-0608 found that this analogue was not associated with cellular toxicity at 100 μM after 24 or 72 hours in Huh7 cells, and HCV RNA replication was inhibited at 0.3 μM in a subgenomic replicon assay.50, 56, 57 In comparison to the parent analogue 2’-methyl adenosine, MK-0608 demonstrated a dramatic increase in half-life and oral bioavailability, since MK-0608 was no longer susceptible to deamination or cleavage by adenosine-metabolizing enzymes.50, 58
Figure 7.

Modified 7-deaza-2’-methyl analogues.
Unfortunately, this approach proved ineffective with the corresponding guanosine analogues once viral strain mutations were introduced, thus only the 7-deaza-2’-methyl adenosine analogues were originally pursued.50, 56 Subsequently, however, researchers found that the addition of prodrug moieties, such as the aforementioned McGuigan ProTides, greatly enhanced the antiviral activity of 2’-methyl guanosine.46, 59 Of these, one of the most successful analogues was IDX184 (Figure 8), a 2’-methylguanosine prodrug originally developed by Idenix, which utilized an S-pivaloyl-2-thioethyl (tBuSATE) moiety functionalized with an N-benzylphosphoramidate group (O-(HO)tBuSATE N-benzylphosphoramidate) to impart a significant increase in anti-HCV activity compared to the parent compound.59,60 Through an SAR study, Sizun et. al. found that the (O-(HO)tBuSATE N-benzylphosphoramidate) derivative IDX184 was the most potent analogue, with an EC50 of 0.2 ± 0.03 μM and no associated cytotoxicity in an HCV subgenomic luciferase replicon system.59, 60 Further analysis revealed that IDX184 was well tolerated in a chimpanzee model, thus researchers turned to testing IDX184 in human patients.61 In phase II clinical trials, it was determined that IDX184 was effective in patients with chronic HCV infections, thus supporting further study for clinical application.59, 62, 63
Figure 8.

Second generation 2’-methylguanosine analogues.
In related studies by McGuigan et al, it was found that the addition of a ProTide moiety to 2’-methylguanosine resulted in a 10-fold increase in activity against HCV in Huh 5-2 cells, as well as a lack of cytotoxicity at 50 μM.46 Further investigation by McGuigan led to the development of INX-189 (BMS-986094), which utilized a phosphoramidate ProTide moiety with a naphthyl group as the aromatic component, an L-alanine as the amino acid component, and a t-BuCH2 group as the aliphatic component (Figure 8).47, 64 This analogue was considered a double prodrug, since the carbonyl group at the C6 position of the the guanine base was replaced with a methoxy group, which, following hydrolysis, reverts to the carbonyl.64 This modification proved to be essential, in that it consistently improved the activity against the HCV replicon across all ProTide derivatives, compared to the parent 2’-methylguanosine.64 Further analysis revealed INX-189 to be the most potent analogue with an EC50 value of 0.01 μM and a CC50 value of 7 μM.34, 64 Most importantly, this approach delivered substantially higher levels of the 5’-triphosphate of 2’-methylguanosine compared to the parent analogue, and increased the half-life to over 24 hours.64, 65 Similarly, studies found that combination therapy with INX-189 and ribavirin resulted in significant synergistic anti-DENV activity in vitro, with a synergy score of 2.2 ± 0.048.66 These promising results prompted further clinical analysis of INX-189, including Phase III trials of INX-189 and another anti-HCV experimental drug daclastasvir.65 Unfortunately, severe cardiotoxicity complications were soon discovered, thus INX-189 was withdrawn from further clinical study.67–69 For similar toxicity reasons, and due to the fact that both IDX184 and INX-189 share the same parent analogue, IDX184 was also pulled from clinical trials.69, 70
3.2. 2’-Methyl-Fluoro Modification
As mentioned in the first review, researchers have capitalized on the unique properties that fluorine imparts to nucleoside analogues for decades.71–73 Fluorine is often used as an isosteric replacement since it is similar in size to hydrogen, but is also similar in electronegativity to the hydroxyl groups found in nucleosides.71 The presence of fluorine on the sugar ring greatly influences the conformation of the “sugar pucker” in the ring system, which in turn has an effect on the overall conformation of the entire nucleoside, as well as recognition by different enzymes.74–76 Furthermore, studies have demonstrated that the presence of a fluorine at the 2’-position of the sugar greatly decreases the nucleoside’s susceptibility to enzymatic cleavage of the glycosidic bond.71, 76–80 One early successful 2’-fluorine analogue was 2’-deoxy-2’-fluorocytidine, which demonstrated potent HCV inhibition with an EC90 of 5 μM and no cytotoxicity up to 100 μM.81 Unfortunately, while 2’-deoxy-2’-fluorocytidine targeted the HCV non-structural NS5B polymerase, it was also shown to target cellular polymerases, thus was not pursued further.81–83
Due to the success of the 2’-methyl analogues, as well as the known impact of using fluorine in nucleoside drug design, Clark et. al. sought to combine these two modifications to yield 2’-deoxy-2’-fluoro-2’-methyl analogues (Figure 9).84 The hope was that by combining both 2’ substituents these novel analogues would demonstrate potent antiviral activity, and that the addition of the 2’-methyl group would target these analogues to the viral polymerases rather than the human polymerases, thus resulting in lower cytotoxicity compared to the 2’-deoxy-2’-fluoro analogues. In an HCV replicon assay, the cytidine analogue demonstrated an EC90 of 5.40 ± 2.6 μM with no associated cytotoxicity up to 100 μM while the uridine analogue was inactive, although also not cytotoxic.84
Figure 9.

First generation 2’-deoxy-2’-fluoro-2’-methyl analogues.
As mentioned previously, one of the limitations of nucleoside analogues is the first phosphorylation step by viral or cellular kinases. Interestingly, while the 2’-deoxy-2’-fluoro-2’-methyluridine analogue did not demonstrate any antiviral activity in the HCV replicon assays, the triphosphate of this analogue demonstrated potent inhibitory activity against the HCV NS5B, with an IC50 of 1.19 μM.85, 86 This suggested that the 2’-deoxy-2’-fluoro-2’-methyluridine analogue was not efficiently phosphorylated to the monophosphate, thus the use of the phosphoramidate ProTide method could potentially increase antiviral activity. Through an extensive SAR study, Sofia et. al. found that a phosphoramidate structure with an isopropyl alkyl chain, L-alanine, and phenyl aromatic substituent (Figure 10) greatly increased antiviral activity of the uridine analogue, PS-7851, with an EC90 of 0.52 μM.87, 88 Further analysis to determine safety found that this analogue demonstrated no cytotoxicity up to 100 μM against numerous cell lines including the human hepatocyte cell lines Huh7 and HepG2, the human pancreatic cell line BxBC3, and the human T-lymphoblast cell line CEM.87 As PS-7851 is a mixture of diastereomers at the phosphorus center, it was important to determine which isomer demonstrated a greater antiviral activity, was potentially more toxic, or, whether the two isomers worked synergistically. Sofia et. al. found that the Sp isomer (PS-7977, sofosbuvir) was indeed more active, with an EC90 of 0.42 μM while the Rp isomer (PS-7976) had an EC90 of 7.5 μM.87 Neither analogue demonstrated cytotoxicity at concentrations up to 100 μM.87 Most importantly, sofosbuvir consistently produced high levels of triphosphate in liver cells across all species tested.87–89 These promising results led to the development of sofosbuvir in clinical trials, and ultimately sofosbuvir was approved by the FDA (under the name ®Sovaldi). Moreover, sofosbuvir was also approved combination with other drugs such as ribavirin and ledipasvir for use in prophylaxis after liver transplantation, as a treatment for recurring HCV infection, as well as against numerous HCV genotypes.90–97
Figure 10.
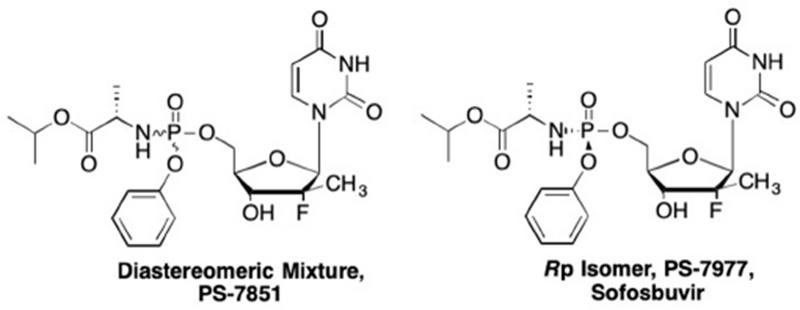
Phosphoramidate prodrug of 2’-deoxy-2’-fluoro-2’-methyluridine.
3.3. Other 2’-Combination Approaches
The field of nucleoside analogue drug design has greatly benefitted from combination approaches, whereby researchers merged different structural modifications that proved essential for antiviral activity into one analogue. This was highlighted with GS-5734, which combines a 1’-sugar modification, a deaza purine nucleobase, substitution of the N9 with a carbon to create a C-nucleoside, and the McGuigan ProTide technology.23, 25, 26 Another analogue to use this strategy is GS-6620 (Figure 11), a C-nucleoside adenosine analogue originally developed by researchers at Gilead Sciences for HCV treatment.24 As mentioned previously, the 1’-cyano group occupies a pocket in the viral polymerase binding site, which leads to the increased selectivity of the 1’-CN analogues for viral over human polymerases.23, 27 Like GS-5734, employing a C-nucleoside replaces the hemiaminal (O-C-N) glycosidic bond with an O-C-C bond, thus decreasing the nucleoside analogue’s susceptibility to enzymatic glycosidic bond cleavage.15, 21–23 This was also observed in studies with 7-deaza-2’-methyladenosine analogues, which exhibited an increase in antiviral activity, half-life, and oral bioavailability, since the 7-deaza analogues were no longer susceptible to deamination or cleavage by adenosine-metabolizing enzymes.49, 50, 56, 58
Figure 11.

Structure of the parent analogue, first generation phosphoramidate prodrug, and the double prodrug GS-6620.
As previously mentioned, addition of a 2’-methyl group has also proved fruitful, since once incorporated into the growing strand, analogues with this modification prevent incoming nucleoside triphosphates from binding to the active site of HCV NS5B, thus acting as non-obligate chain terminators.24, 42 With these modifications in mind, Cho et. al. synthesized a novel 1’-cyano-2’-methyl-7,9-deaza adenosine analogue (Figure 11) and screened it for activity against HCV.24 Unfortunately, this analogue failed to display antiviral activity up to 89 μM in whole-cell replicon assays. This was subsequently shown to be due to the presence of the 1’-cyano group, which limited the first phosphorylation step, likely due to the changes in the sugar pucker, which can affect recognition by kinases (and other enzymes).14, 24 Surprisingly however, the corresponding triphosphate displayed an IC50 value of 0.29 μM, thus the ProTide approach was employed to overcome the rate-limiting phosphorylation step, as well as to increase the amount of triphosphate delivered to the target cell.24 This proved successful, and it was found that the phosphoramidate prodrug (Figure 11) demonstrated moderate HCV activity in the replicon assay (EC50 = 1.05 μM) and was efficiently converted to the active triphosphate.24 Subsequent pharmacokinetic studies to determine the triphosphate levels in hamster liver revealed that the phosphoramidate analogue did not yield adequate amounts of triphosphate following oral absorption.24 In order to overcome this limitation, Cho et. al. then utilized a double prodrug approach, in which the 3’-OH was protected using an isobutyrate group (Figure 11).24 This modification not only increased lipophilicity and oral absorption, it also resulted in much higher levels of triphosphate in primary human hepatocytes.24 Similar to sofosbuvir, the double-prodrug analogue was a diastereomeric mixture, thus further analysis found that the Sp isomer was 6-fold more potent in HCV replicon assays and delivered 3-fold higher levels of triphosphate to primary human hepatocytes as compared to the Rp isomer.24 Due to the promising antiviral profile of the Sp isomer (later known as GS-6620), this analogue was chosen for further clinical evaluation.24, 98, 99
3.4. 2’-Cyano Modification
Not just confined to the 1’-position, the cyano group has also found use at other positions on the nucleoside sugar moiety. As mentioned previously, some of the first nucleosides discovered to have medicinal properties were the arabinose or “Ara” analogues, such as Ara-C (Figure 12), which demonstrated potent activity against numerous cancers including non-Hodgkin’s lymphoma, myeloid leukemia, acute myeloblastic leukemia, and many others.100–104 Unfortunately, there are numerous drawbacks to using Ara-C in anticancer treatment including a short half-life in plasma, inactivation by deamination to the inactive uracil metabolite by cytidine deaminase, development of resistance, and ineffectiveness against solid tumors.105–108 It was later shown that adding various groups to the 2’-β position of 2’-deoxycytidine could have profound effects on the stability of this analogue by decreasing its susceptibility to cytidine deaminase. Studies found that the introduction of an electron-withdrawing substituent at the 2’-β position increased the acidity of the 2’-α proton, thus β-elimination could occur, resulting in DNA strand breaks.109–113 This was particularly interesting, due to the hypothesis that radiation therapy causes DNA strand breaks, leading to tumor cell death.114 Thus, the addition of a 2’-β-cyano group was employed to yield 2’-C-cyano-2’-deoxy-1-β-D-arabinofuranosylcytosine (CNDAC, Figure 12) which demonstrated potent in vitro activity against numerous human tumor cells such as sarcomas, osteosarcomas, fibroblastomas, and carcinomas.109–111, 113, 115–117 In comparison to the parent analogue Ara-C, CNDAC demonstrated potent cytotoxicity in 14 tumor cell lines, with IC50 values from 0.04 to 6.8 μg/mL, whereas Ara-C was only active against 6 of these cell lines, with IC50 values of 0.09 to 4.5 μg/mL.111 Furthermore, CNDAC is very effective against solid tumors, whereas Ara-C and the widely used 5-fluorouracil and 5’-deoxy-5-fluorouridine were not active against these solid tumors.110, 111, 117 This led to a series of clinical trials, including one in which CNDAC is currently in Phase I/II trials against acute myeloid leukemia and acute lymphatic leukemia (NCT01702155),118–120 and another in which a prodrug of CNDAC, sapacitabine (Figure 12), is in Phase III trials for acute myeloid leukemia and myelodysplastic syndrome (NCT01303796).121, 122
Figure 12.
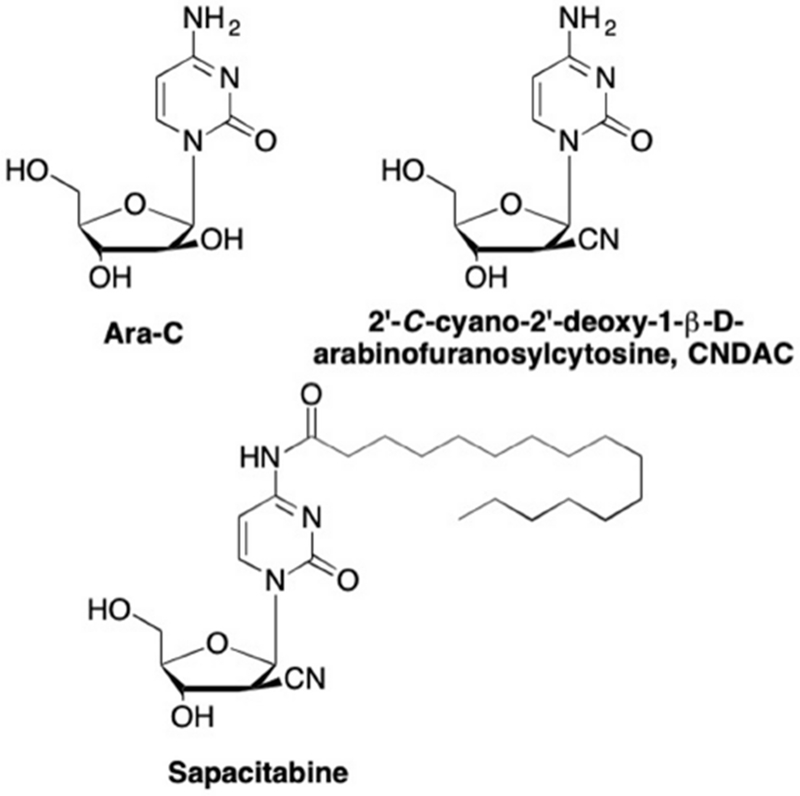
Structure of 2’-modified 2’-deoxycytosine analogues Ara-C, CNDAC, and the prodrug Sapacitabine.
3.5. 2’-Ethynyl Modification
From the studies with 2’-cyano groups, researchers found that acetylene-derived analogues were an important sugar modification associated with potent antiviral effects. Furthermore, modification at the N7 position of 2’-acetylene adenosine analogues, such as removal of the nitrogen to yield a 7-deaza analogue or addition of a carbamoyl moiety, led to profound antiviral activity against DENV.123–125 While the analogue, NITD449, demonstrated moderate anti-DENV activity (EC50 = 2.0 μM), it was also associated with cytotoxicity.124 Interestingly, the 7-deaza analogue NITD008 (Figure 13) demonstrated similar potency without the associated cytotoxicity.126–128 Studies found that this analogue was not cytotoxic up to 50 μM across numerous cell lines and inhibited DENV-2 with an EC50 of 0.64 μM.126
Figure 13.

Structure of the adenosine based analogues NITD008 and NITD449.
More importantly, NITD008 was also effective against the other three serotypes.126 This is critical, because a patient is more likely to develop severe dengue hemorrhagic fever or dengue shock syndrome when infected a second time, by a different serotype, thus an analogue with activity against all serotypes is highly sought after.129, 130 Furthermore, studies found that NITD008 was also effective against other flaviviruses including WNV, YFV, HCV, ZIKV, and TBEV, as well as against enterovirus 71.125–128, 131, 132 Elucidation of the mechanism of action determined that the triphosphate of NITD008 interacted with the flavivirus RdRp with an IC50 of 0.31 μM and served as a chain terminator in a similar fashion as the 2’-methyl analogues, thereby halting viral RNA elongation.125, 126 Other in vivo studies determined NITD008 could suppress peak viremia in infected mice and completely protected them from death.126 Further studies with NITD008 and other flaviviruses are currently under way to determine if NITD008 could prove to be a broad-spectrum therapeutic against a variety of flaviviruses.125, 128, 131
4. 3’ Modifications
Early examples of modifications at the 3’ position of nucleoside sugars led to the development of novel analogues such as the 3’-methyl nucleosides,133–135 however, the 2’, 3’-dideoxy nucleosides136–138 and 2’-deoxy-3’-modified nucleosides139, 140 ultimately proved to be more promising. The development of these analogues was covered extensively in the previous review and is not discussed here.141
5. 4’ Modifications
Until the discovery of naturally occurring 4’-modified nucleoside analogues,142 modifications to the 4’ position of the furanose ring was rather uncommon in drug design, mainly due to the synthetic challenges. As more facile synthetic routes were developed, more researchers began to pursue these interesting analogues. Researchers soon noted that modifications to the 4’ position of the furanose ring changed the sugar pucker from a C2’-exo/C3’-endo “north” conformation, as is common in natural RNA nucleosides,143 to a C2’-endo/C3’-exo “south” conformation.144 As mentioned previously, this affects recognition by different enzymes, thus can have a significant impact on their biological activity.74, 75
5.1. 4’-Fluoro Modification
As mentioned previously, fluorine substitution has been utilized at a number of positions on both the nucleobase and the sugar moiety, however, one of the lesser explored positions has been substitution at the 4’-carbon of nucleoside sugars. One of the first 4’-modified nucleoside analogues was isolated from Streptomyces calvus in 1957,142 but it wasn’t until 1969 that the correct structure of this analogue, later named nucleocidin, was elucidated (Figure 14).145–148 While this analogue was one of the first examples of a 4’-modified furanose sugar, it also possessed a novel 5’-sulfamoyl group.145–148 This unique structure endowed nucleocidin with a broad antiparasitic spectrum, particularly against trypanosomes, however, the practical use of this analogue as a therapeutic was severely limited due to its toxicity.142, 149
Figure 14.

Structure of the first 4’-modified furanose nucleoside Nucleocidin.
The initial discovery of nucleocidin prompted researchers to pursue other 4’-fluoro modified nucleoside analogues in an effort to decrease the overall toxicity. One such example was developed by Guillerm et al. as a potential S-adenosyl-L-homocysteine hydrolase (SAHase) inhibitor (Figure 15).150 Based on the mechanism of action of SAHase, it was hypothesized that the lack of a 4’-proton on 4’-fluoroadenosine would completely inhibit further catalysis, however, when tested against SAHase it was determined that 4’-fluoroadenosine had a 100-fold lower affinity for SAHase compared to natural adenosine, thus this modification proved unsuccessful.150
Figure 15.

Second generation 4’-fluoro analogues.
Another 4’-fluoro analogue related to nucleocidin was 5’-deoxy-4’,5-difluorouridine (Figure 15), a 5-fluorouracil analogue that demonstrated similar inhibition of growth of L1210 mouse leukemia cells as the parent 5-fluorouracil, but 10-fold greater activity than previously reported prodrugs.151 Since 5’-deoxy-4’,5-difluorouridine does not have a 5’-hydroxyl group, this analogue cannot be converted into the nucleotide and be incorporated by polymerases. Instead, the presence of the 4’-fluoro group makes the glycosidic bond unusually more acid-labile and increases glycosidic bond cleavage by uridine phosphorylase, thus delivering increased amounts of 5-fluorouracil to tumor sites.151, 152 In comparison to another 5-fluorouracil prodrug, 5’-deoxy-5-fluorouridine (discussed below), 5’-deoxy-4’,5-difluorouridine demonstrated an IC50 of 0.3 μM and a 500-fold increase in glycosidic bond hydrolysis, whereas the IC50 of 5’-deoxy-5-fluorouridine was 3.0 μM, and it failed to undergo significant hydrolysis.152 While this analogue appeared promising as a potential anticancer agent, further studies have yet to be reported.
5.2. 4’-Methyl Modification
Another common isosteric modification in nucleoside drug design is the substitution of a methyl group for a hydrogen. While this modification has been explored at the 1’,12 2’,40–45 and 3’133–135, 153 positions on the furanose ring, little research had been reported on the presence of a methyl group at the 4’ position. As mentioned previously, it was found that the addition of a 4’-modification altered the reactivities of the 3’ and 5’-hydroxyl groups compared to natural nucleosides due to steric effects,144 thus 4’-methyl modifications were considered an interesting avenue to pursue.
Synthesis of 4’-methyl analogues was pioneered by Waga et al. in the early 90s for use as potential anticancer and/or antiviral therapeutics.144, 154 While the change in sugar pucker due to the presence of the 4’-methyl substituent imparted these analogues with interesting characteristics, Waga et al. also hypothesized that the 4’-methyl modification could act synergistically with other sugar modifications (Figure 16).154 Combining the 4’-methyl modification with dideoxy sugars, 2’-deoxy sugars, and saturated dideoxy sugars with various nucleobases, the resulting analogues were screened against HIV-1 in MT-4 cells.154 Most of the analogues failed to display any notable antiviral activity, with IC50 values ranging from 21 μM to over 500 μM, however, the 4’-methyl-thymidine analogue and the 4’-methyl-2’-deoxycytidine analogue displayed potent activity with IC50 values of 7.2 μM and 0.072 μM respectively.154, 155 While the thymidine analogue did not display cytotoxicity up to 100 μM, the cytidine analogue was quite toxic with a CC50 value of 0.13 μM, thus these analogues were not pursued further.154, 155
Figure 16.

Examples of 4’-methyl nucleoside analogues.
Years later, the 4’-methyl modification was revisited when Gosselin et al. synthesized a ribose series that focused on nucleobase modifications instead of the sugar modifications seen in the study by Waga et al. (Figure 16).156 Like the analogues synthesized by Waga, the 4’-methyl ribose analogues were evaluated for their inhibitory effects against HIV-1 replication in MT-4 cells, however, none demonstrated any meaningful antiviral activity.156 These analogues were also tested for activity against other viruses including HBV in HBV DNA-transfected Hep-G2 cells (2.2.15 cells) and against YFV in BHK cells. Again, no antiviral activity or cytotoxicity was observed with any of the analogues against either virus,156 so pursuit of the 4’-methyl modification approach was abandoned.
5.3. 4’-Azido Modifications
With the early success of 2’-deoxy-3’-azidothymidine (AZT, zidovudine),139, 140, 157, 158 which bears a 3’-azido modification, researchers sought to utilize this modification at different positions on the sugar ring to either increase antiviral activity, or, perhaps most importantly, decrease cellular toxicity. One example that garnered early attention was the addition of the azide group at the 4’-position of the sugar, due to the result the electron-withdrawing effects of the azide group on the sugar pucker.159 Thus, a series of 2’-deoxy-4’-azido nucleosides was synthesized and demonstrated potent anti-HIV activity. These were the first nucleoside analogues with potent anti-HIV activity that had an azido group in any position other than the 3’ position (Figure 17).159, 160 Interestingly, the presence of the 4’-azido group in these analogues affected the 3’ and 5’-hydroxyl groups in a way that brought these two groups into closer proximity.159, 160 Due to its electronegativity, the 4’-azido group prefers a pseudoaxial orientation, which then forces both the 3’ and 5’-hydroxyl groups into pseudo-equatorial positions, causing the sugar to adopt a C3’-endo/C2’-exo RNA-type conformation.159 Thus, while these analogues retain the 3’-hydroxyl moiety in contrast to AZT and other HIV chain terminators, the change in sugar pucker allows the 4’-azido analogues to still act as DNA chain terminators, however designated as pseudo-obligate chain terminators.159–161
Figure 17.
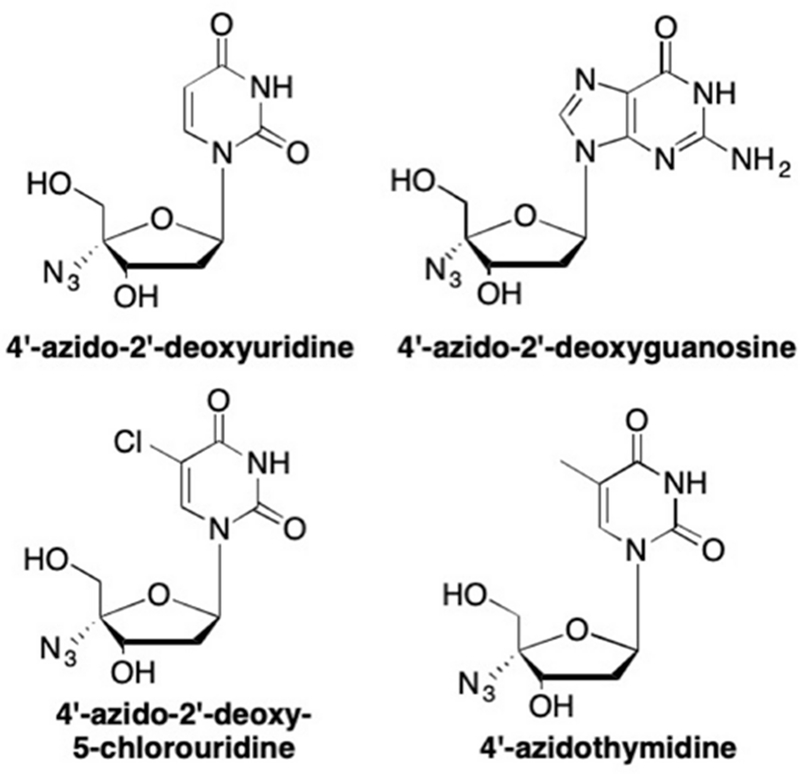
Novel 4’-azido nucleoside analogues with potent anti-HIV activity.
While none of the analogues was more active than AZT against HIV-1 isolates, they did demonstrate potent activity.159, 160 The IC50 for the 4’-azido-2’-deoxyuridine analogue was 0.8 μM with no associated cytotoxicity up to 200 μM, whereas the 4’-azido-2’-deoxy-5-chlorouridine analogue possessed an IC50 value of 0.056 μM, also with no cytotoxicity.159 Furthermore, the 4’-azidothymidine analogue demonstrated equipotent activity against HIV-1 with an IC50 value of 0.01 μM compared to AZT in A3.01 cells.160 Even more significant was the observation that 4’-azidothymidine was effective against strains of HIV-1 that were AZT-resistant.159, 160 Unfortunately, this analogue proved to have increased cytotoxicity compared to AZT, and never progressed to the clinic.
In contrast, one of the most successful 4’-azido analogues was 4’-azidocytidine, later termed R1479, which demonstrated potent anti-HCV activity (Figure 18).162, 163 This analogue was originally discovered during a 4’-modification SAR study by Roche, where it was found that 4’-azidocytidine showed an IC50 of 1.28 μM in an HCV replicon proliferation assay, and the triphosphate of 4’-azidocytidine inhibited the HCV NS5B with an IC50 of 0.30 μM.162 Further analysis found that R1479 was also active against mutant strains of HCV.163 These promising results, along with low cytotoxicity, led researchers to further investigate 4’-azidocytidine against other viruses. It was found that R1479 was effective against HCV and against DENV serotypes 1, 2, and 4 (EC50 range 1.3–3.2 μM) in primary human macrophages,164 as well as against respiratory syncytial virus (RSV), for which R1479-TP had an IC50 of 0.24 μM.165, 166 More recent studies found micromolar activity against the henipaviruses, Nipah and Hendra.165 Unfortunately, like many nucleoside analogues, R1479 suffered from low bioavailability, thus a tri-isobutyl ester prodrug moiety was introduced to give balapiravir (R1626, Figure 18).167–170 Although balapiravir demonstrated increased antiviral activity compared to the parent analogue R1479 and was efficacious in clinical trials against HCV,167, 168, 171–173 the development of more potent nucleoside analogues, such as sofosbuvir, as well as adverse toxicity,174 halted further advancement of this analogue as an HCV therapeutic. Later, balapiravir was analyzed in a clinical study against DENV,164, 175 however, treatment was not well tolerated due to adverse effects, nor did it decrease viral load or fever clearance time, thus clinical trials were terminated.
Figure 18.
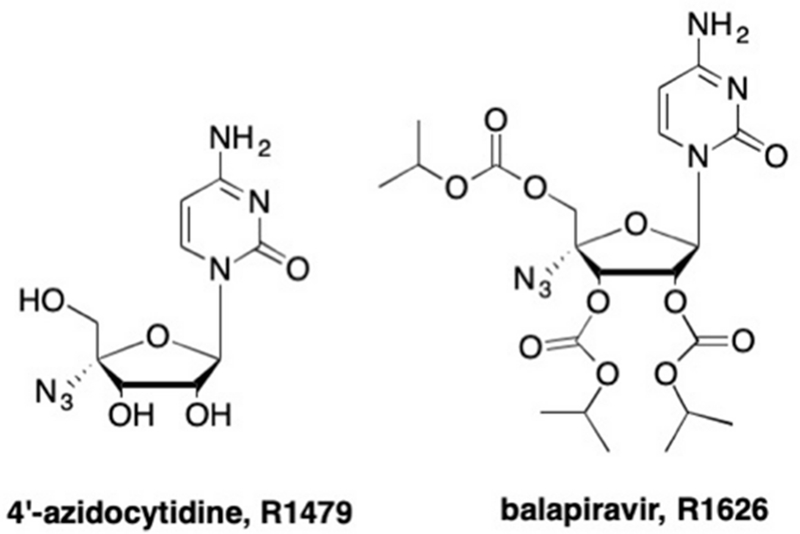
Structure of 4’-azidocytidine and its tri-isobutyl ester prodrug balapiravir.
Other 4’-azido analogues of early interest include 4’-azido-aracytidine (RO-9187) and 4’-azido-2’-methyl nucleosides (Figure 19) for use as anti-HCV analogues.55, 176–179 RO-9187 features a 4’-azide group and the 2’ ara, or “up” hydroxyl group and was discovered through an SAR study in which the authors were attempting to develop more potent 4’-azido ribonucleosides against HCV.162, 176 Interestingly, RO-9187 proved to be the most potent analogue tested, with an IC50 of 0.171 μM, and no associated cytotoxicity up to 1000 μM.162, 176 Furthermore, studies found that not only was it a potent anti-HCV analogue, but it also was effective against TBEV with an EC50 of 0.3 ± 0.01 μM and no associated cytotoxicity up to 50 μM.55 Similarly, researchers analyzed the change in antiviral activity when the ara hydroxyl group was substituted with a methyl group, yielding 2’-methyl analogues such as 4’-azido-2’-methylcytidine (Figure 19).178, 179 Unfortunately, it was determined that this analogue did not demonstrate potent anti-HCV activity, however, the addition of a phosphoramidate prodrug moiety greatly increased antiviral activity, with EC50 values ranging from 3.0 to 4.9 μM.178, 179 This increase in activity has led researchers to pursue other phosphoramidate modifications in order to increase the anti-HCV activity even further, and these studies are currently under way.
Figure 19.

Novel structure of 4’-azido-aracytidine and 4’-azido-2’-methylcytidine.
5.4. 4’-Cyano Modifications
Due to the initial successes of the various 4’-azido analogues, scientists sought more potent candidates, utilizing other moieties with terminal nitrogen atoms such as a 4’-cyano group. This led to the early development of 4’-cyanothymidine (Figure 20), which demonstrated activity against HIV, with an EC50 of 0.002 μM.180 Unfortunately, when studied in a mouse model, 4’-cyanothymidine demonstrated toxicity at 0.3 mg/kg dose per day,180 thus studies were discontinued.
Figure 20.

Potent HIV inhibitor 4’-cyanothymidine.
After these initial findings of toxicity, researchers sought additional 4’-cyano analogues in an effort to retain their biological activity and decrease their cytotoxicity. In an SAR study focusing on different 4’-modifications, Nomura et al. determined that the 4’-cyano-2’-deoxycytidine analogue (Figure 21) demonstrated activity against L1210 tumor cells, herpes simplex virus (HSV) 1, HSV-2, and very potent activity against HIV-1 (EC50 = 0.0012 μM) with no accompanying cytotoxicity.155 Interestingly, the corresponding ribose derivative, 4’-cyanocytidine, demonstrated similar antiviral activity but exhibited much greater levels of cytotoxicity,162 thus the deoxyribose analogues were selected for further studies.
Figure 21.

Second generation 4’-cyano nucleoside analogues.
Synthesis of these analogues initially was challenging, since 4’-modified sugars exhibit low reactivity in glycosylation reactions, thus making it difficult to add the necessary heterocyclic bases, but researchers subsequently developed novel synthetic approaches by modifying naturally occurring deoxyribonucleosides as starting materials, which allowed for more readily available 4’-C-modifed analogues.155, 181 Subsequent studies showed that 4’-cyano-2’-deoxyguanosine (CdG) and 4’-cyano-2’-deoxy-2,6-diaminopurine-ribonucleoside (CAdA) analogues (Figure 21) exhibited sub-nanomolar levels of activity against HIV-1 (Ec50 = 0.19 nM and EC50 = 0.79 nM respectively), but were associated with significant toxicity.181 By comparison, the 4’-cyano-2’-deoxyadenosine (CdA) and 4’-cyano-2’-deoxyinosine analogues (CdI) (Figure 21) exhibited sub-micromolar activity (Ec50 = 0.05 μM for both) with little to no cytotoxicity.181 Both CAdA and CdG were analyzed further by Takamatsu et al., who found that both analogues demonstrated potent activity against HIV-1, but also had potent activity against hepatitis B virus (HBV) (EC50 = 0.4 nM for both analogues) with less cytotoxicity than previously reported.181, 182
5.5. 4’-Combination Approach
Before the discovery of the anti-HIV activity of 4’-modified nucleoside analogues, many scientists believed that only HIV therapeutics lacking a 3’-hydroxyl group could act as chain terminators. While the absence of a 3’-hydroxyl group did impart potent anti-HIV effects, there were also several disadvantages including poor phosphorylation and reduced recognition by the polymerases.183–186 Researchers therefore attempted to design novel analogues that would retain antiviral potency, as well as the recognition required for activation and incorporation.
One such analogue was 4’-ethylnyl-2-fluoro-2’-deoxyadenosine (EFdA, Figure 22). Notably, EFdA demonstrated potent anti-HIV activity with an EC50 of 0.05 nM, but also significant activity against nucleoside reverse transcriptase inhibitor (NRTI)-resistant strains.183, 185, 187, 188 The design of EFdA’s scaffold was a result of combining several strategic structural modifications. For example, the fluorine moiety at the 2-position of the adenosine nucleobase was chosen due to the observation that a fluorine or another halogen at the 2-position of 4’-modified nucleoside analogues significantly enhances antiviral activity.183, 189 This increase in activity was due to the decreased susceptibility of EFdA to adenosine deaminase, as a result of the highly electronegative fluorine at the 2-position.183, 189 The addition of the 4’-ethynyl moiety also plays a role in decreasing deamination of EFdA by adenosine deaminase, thus the two modifications work synergistically.183
Figure 22.

Unique structure of 4’-ethynyl-2-fluoro-2’-deoxyadenosine.
The second modification was the retention of the 3’-OH to ensure recognition by the kinases, as this was also known to be important. Related, to this, addition of the 4’-ethynyl group leads EFdA to strongly favor the “north” (C2’-exo/C3’-endo) conformation. A number of studies have determined that HIV-1 reverse transcriptase (RT) prefers NRTIs and/or incoming nucleotides with a north confirmation, thus EFdA binding with HIV-1 RT is optimized.183, 190–193 Furthermore, while EFdA is readily recognized and incorporated by HIV-1 RT, it does not inhibit human DNA polymerases α or β, or mitochondrial DNA polymerase γ, thus EFdA displays a superior toxicity profile compared to other HIV-1 NRTIs.187, 188, 193, 194
While the addition of the 4’-ethynyl group had a profound effect on the sugar pucker, and thus the increased binding affinity with HIV-1 RT, the 4’-ethynyl moiety also endowed EFdA with two unique mechanisms of action. Instead of obligate, non-obligate, delayed or pseudo-obligate chain termination typical of other nucleoside analogues, EFdA acts as a translocation-defective reverse transcriptase inhibitor (TDRTI).193, 195, 196 Once EFdA-TP is incorporated by RT at the 3’-primer terminus, the unique structure of EFdA blocks translocation of the primer strand on the viral polymerase, thus further nucleotides cannot be incorporated.193, 195, 196 Interestingly, EFdA can also act as a traditional non-obligate chain terminator since it retains the 3’-OH.193, 195, 196 Due to its low toxicity profile and highly potent anti-HIV activity, EFdA has progressed to clinical trials under the name MK-8591, sponsored by Merck (NCT03272347, NCT02217904).
6. 5’ Modifications
While modifications to various positions of the furanose ring are very common, the importance of the 5’-hydroxyl group in nucleotide incorporation for both DNA and RNA synthesis initially caused researchers to avoid modifications at the 5’-position. In some instances however, as seen with the 5’-deoxy, 5’-nor, and truncated carbocyclic nucleosides related to aristeromycin and neplanocin developed by Schneller, Seley, Borchardt, and others, removal or replacement of the 5’-methylene group and/or 5’-hydroxyl group proved beneficial since these analogues could no longer be phosphorylated, thus the toxicity observed with the parent analogues aristeromycin and neplanocin did not occur with the truncated analogues.141, 197–207 Other researchers have also utilized these approaches to their advantage in order to decrease overall toxicity of various nucleoside analogues.
6.1. 5’-Deoxy-5-fluorouridine
One early nucleoside analogue that incorporated the 5’ substitution was 5’-deoxy-5-fluorouridine (5’-dFUrd, doxifluridine, Figure 23), in which instead of a 5’-hydroxymethylene group, 5’-dFUrd has a methyl at that position.208–211 Like other 5’-modified analogues, 5’-dFUrd cannot be phosphorylated and converted into the corresponding triphosphate. Instead, 5’-dFUrd exerts its effect by acting as a prodrug and releases 5-fluorouracil as a result of glycosidic bond cleavage by human phosphorylases.212,213 This analogue has demonstrated a broad range activity against numerous cancers including colorectal, leukemia, and melanomas.208, 209, 211, 213–216 Furthermore, compared to other fluorinated pyrimidines, 5’-dFUrd exhibits a higher therapeutic index and is less immunosuppressive.208, 209, 215, 217
Figure 23.
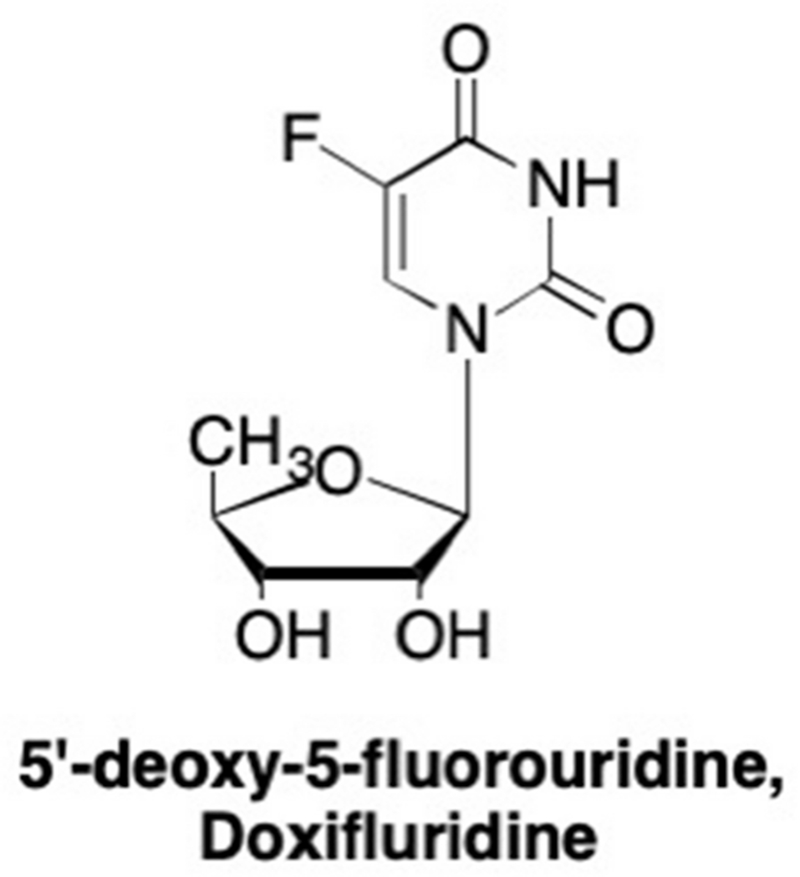
Structure of one of the first 5’-truncated nucleoside analogues, 5’-deoxy-5-fluorouridine.
Since its initial discovery in the mid 1970’s, 5’-FdUrd has progressed from Phase I to numerous Phase II clinical trials for treatment of squamous cell carcinomas, advanced breast cancer, advanced colorectal adenocarcinoma and others.218–221 Despite these successes, the FDA has yet to approve 5’-dFUrd as an anticancer treatment in the United States, thus more research is needed in order to better determine potential side effects that could be associated with this analogue.
7. Additional Modifications
This final section is dedicated to describing several types of nucleoside analogues that utilize novel and unique structural modifications, as well as explaining how some are used in unconventional ways.
7.1. Tricyclic Analogues
In the first article, various expanded purine nucleobases were described, including Nelson Leonard’s benzyl-expanded nucleosides,235–238 and Seley-Radtke’s thieno-expanded nucleosides.239–242 Other examples of interesting tricyclic analogues are the dual-faced bases or the Janus nucleosides, named after Janus, the Roman god of gates and doors (Figure 24).243, 244 These unique nucleosides can present Watson-Crick donor/acceptor base pairing from two different faces of the nucleobase through rotation about the glycosidic bond, thus forming stable base pairs with more than one complementary nucleoside.243, 244 Synthesis of both ribose and 2’-methyl Janus-type nucleosides met with challenges, however, the ribose J-AU and J-AG analogues demonstrated moderate activity against HCV, with EC50 values of 5.7 μM and 3 μM respectively.243 Unfortunately, they all demonstrated significant toxicity in numerous cell lines,243 thus were not extensively pursued.
Figure 24.

Janus-type nucleosides that feature two pyrimidine faces.
7.2. Fleximer Analogues
A serendipitous outcome of some of the early studies with Seley-Radtke’s thiophene-expanded tricyclic analogues led to a new class of novel nucleoside analogues. By treating the tricyclic nucleosides with Raney nickel, the sulfur in the middle ring was removed leaving the two outer rings intact, thereby resulting in a flexible nucleoside. These unique analogues were termed “fleximers” (Figure 25).253–256 They feature a purine ring that is “split” into the imidazole and pyrimidine moieties, which remain connected by a single carbon-carbon bond from the C4 of the imidazole to the C5 of the pyrimidine in proximal fleximers, or from the C5 of the imidazole to the C6 of the pyrimidine in distal fleximers (Figure 26).253–257 This design retains the hydrogen bonding pattern necessary for nucleoside-recognizing enzymes while creating an increase in flexibility that allows for alternative interactions in the enzyme binding site, resulting in a number of highly beneficial properties.253–256, 258
Figure 25.
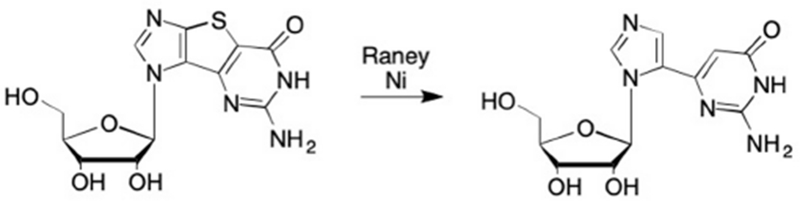
Origins of fleximer analogues from treatment of thienophene expanded nucleosides with Raney Nickel.
Figure 26.

Structures of proximal and distal fleximers.
The inherent flexibility of these analogues allows for free rotation about the carbon-carbon bond, increasing the rotational degrees of freedom and allowing the fleximer to interact with alternative amino acids in the binding pocket, that were previously unattainable by the parent nucleoside.255, 256, 258–261 Further studies found that the introduction of the flexible nucleoside scaffold corresponds to an increase in binding affinity compared to corresponding rigid inhibitors, as well as the ability to circumvent point mutations in the binding site, thus overcoming the development of drug resistance.255, 256, 258–261
To date a number of different types of fleximer and thienophene analogues of FDA-approved nucleoside analogues have been synthesized by Seley-Radtke et al.,241, 242, 255, 256, 262, 263 however, the most promising analogues are the more recent acyclic fleximers based on the FDA-approved drug acyclovir. These doubly flexible nucleosides have demonstrated potent micromolar activity against numerous RNA viruses (Figure 27).264, 265 The most potent analogue, a methoxy-prodrug of Flex-Acyclovir (HP083), has an EC50 of 8.8 ± 1.5 μM against MERS-CoV, and also has potent activity against EBOV at 2.2 μM. 264, 265 More recent studies with this analogue have found sub-micromolar activity against DENV (EC50 = 0.057 μM) and YFV (EC50 = 0.37 μM), as well as potent inhibitory activity of the corresponding triphosphate against both DENV and ZIKV (IC50 = 8.4 μM and 1.7 μM respectively).266–268 Most importantly, these analogues have demonstrated little to no cytotoxicity.264–266 Due to their broad-spectrum antiviral activity, they are currently undergoing further analysis to determine their mechanism of action.
Figure 27.

Structure of Acyclovir compared to the potent antiviral acyclic fleximer analogue HP083.
8. Concluding remarks
As detailed in this and the previous review, nucleoside analogues remain the cornerstone of antiviral and anticancer therapeutics, particularly in combination therapies. As additional structural and biological information becomes available, new and more complex modifications will continue to be pursued. We hope that these two articles have increased our readers’ understanding of the historical modifications to the nucleoside scaffold, the justification for these modifications, and their significance in modern-day therapeutics.
AVR_2018_525 Highlights.
This is the second of two invited articles reviewing the development of nucleoside analogue antiviral drugs.
It is written for a target audience of virologists and other non-chemists, and for chemists unfamiliar with the field.
Numerous modifications have been made to the nucleoside scaffold in order to impart therapeutic benefits.
Current nucleoside analogues employ a combination approach, using multiple modifications to the scaffold.
We examine thought processes, progress in synthetic chemistry and results of antiviral testing that led to approved drugs.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
10. References
- 1.Levene PA; Jacobs WA Über Inosinsäure. Berichte der deutschen chemischen Gesellschaft 1909, 42, 1198–1203. [Google Scholar]
- 2.De Clercq E Antivirals and antiviral strategies. Nat Rev Microbiol 2004, 2, 704–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Perigaud C; Gosselin G; Imbach JL Nucleoside analogues as chemotherapeutic agents: a review. Nucleoside and Nucleotides 1992, 11, 903–945. [Google Scholar]
- 4.De Clercq E; Li G Approved Antiviral Drugs over the Past 50 Years. Clin Microbiol Rev 2016, 29, 695–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.De Clercq E Milestones in the discovery of antiviral agents: nucleosides and nucleotides. Acta Pharmaceutica Sinica B 2012, 2, 535–548. [Google Scholar]
- 6.Field HJ; De Clercq E Antiviral drugs - a short history of their discovery and development. Microbiology Today 2004, 31, 58–61. [Google Scholar]
- 7.De Clercq E; Neyts J Antiviral agents acting as DNA or RNA chain terminators. Handb Exp Pharmacol 2009, 53–84. [DOI] [PubMed] [Google Scholar]
- 8.De Clercq E Antiviral drug discovery and development: where chemistry meets with biomedicine. Antiviral Res 2005, 67, 56–75. [DOI] [PubMed] [Google Scholar]
- 9.De Clercq E Recent highlights in the development of new antiviral drugs. Curr Opin Microbiol 2005, 8, 552–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jordheim LP; Durantel D; Zoulim F; Dumontet C Advances in the development of nucleoside and nucleotide analogues for cancer and viral diseases. Nat Rev Drug Discov 2013, 12, 447–64. [DOI] [PubMed] [Google Scholar]
- 11.Siddiqi SM; Jacobson KA; Esker JL; Olah ME; Ji XD; Melman N; Tiwari KN; Secrist JA; Schneller SW; Cristalli G Search for new purine- and ribose-modified adenosine analogues as selective agonists and antagonists at adenosine receptors. J Med Chem 1995, 38, 1174–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cappellacci L; Barboni G; Palmieri M; Pasqualini M; Grifantini M; Costa B; Martini C; Franchetti P Ribose-modified nucleosides as ligands for adenosine receptors: synthesis, conformational analysis, and biological evaluation of 1’-C-methyl adenosine analogues. J Med Chem 2002, 45, 1196–202. [DOI] [PubMed] [Google Scholar]
- 13.Damont A; Dukhan D; Gosselin G; Peyronnet J; Storer R Synthesis of 1’-C-fluoromethyladenosine. Nucleosides Nucleotides Nucleic Acids 2007, 26, 1431–4. [DOI] [PubMed] [Google Scholar]
- 14.Cho A; Saunders OL; Butler T; Zhang L; Xu J; Vela JE; Feng JY; Ray AS; Kim CU Synthesis and antiviral activity of a series of 1’-substituted 4-aza-7,9-dideazaadenosine C-nucleosides. Bioorg Med Chem Lett 2012, 22, 2705–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Temburnikar K; Seley-Radtke KL Recent Advances in Synthetic Approaches to C-Nucleosides. Beilstein J Org Chem 2018, 14, 772–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rios AC; Yu HT; Tor Y Hydrolytic Fitness of N-glycosyl Bonds: Comparing the Deglycosylation Kinetics of Modified, Alternative and Native Nucleosides. J Phys Org Chem 2015, 28, 173–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lindahl T; Karlstrom O Heat-induced depyrimidination of deoxyribonucleic acid in neutral solution. Biochemistry 1973, 12, 5151–4. [DOI] [PubMed] [Google Scholar]
- 18.Levy M; Miller SL The stability of the RNA bases: implications for the origin of life. Proc Natl Acad Sci U S A 1998, 95, 7933–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Berti PJ; McCann JA Toward a detailed understanding of base excision repair enzymes: transition state and mechanistic analyses of N-glycoside hydrolysis and N-glycoside transfer. Chem Rev 2006, 106, 506–55. [DOI] [PubMed] [Google Scholar]
- 20.Lenz SA; Kohout JD; Wetmore SD Hydrolytic Glycosidic Bond Cleavage in RNA Nucleosides: Effects of the 2’-Hydroxy Group and Acid-Base Catalysis. J Phys Chem B 2016, 120, 12795–12806. [DOI] [PubMed] [Google Scholar]
- 21.De Clercq E C-Nucleosides To Be Revisited. Journal of Medicinal Chemistry 2016, 59, 2301–2311. [DOI] [PubMed] [Google Scholar]
- 22.Stambasky J; Hocek M; Kocovsky P C-nucleosides: synthetic strategies and biological applications. Chem Rev 2009, 109, 6729–64. [DOI] [PubMed] [Google Scholar]
- 23.Siegel D; Hui HC; Doerffler E; Clarke MO; Chun K; Zhang L; Neville S; Carra E; Lew W; Ross B; Wang Q; Wolfe L; Jordan R; Soloveva V; Knox J; Perry J; Perron M; Stray KM; Barauskas O; Feng JY; Xu Y; Lee G; Rheingold AL; Ray AS; Bannister R; Strickley R; Swaminathan S; Lee WA; Bavari S; Cihlar T; Lo MK; Warren TK; Mackman RL Discovery and Synthesis of a Phosphoramidate Prodrug of a Pyrrolo[2,1f][triazin-4-amino] Adenine C-Nucleoside (GS-5734) for the Treatment of Ebola and Emerging Viruses. J Med Chem 2017, 60, 1648–1661. [DOI] [PubMed] [Google Scholar]
- 24.Cho A; Zhang L; Xu J; Lee R; Butler T; Metobo S; Aktoudianakis V; Lew W; Ye H; Clarke M; Doerffler E; Byun D; Wang T; Babusis D; Carey AC; German P; Sauer D; Zhong W; Rossi S; Fenaux M; McHutchison JG; Perry J; Feng J; Ray AS; Kim CU Discovery of the first C-nucleoside HCV polymerase inhibitor (GS-6620) with demonstrated antiviral response in HCV infected patients. J Med Chem 2014, 57, 1812–25. [DOI] [PubMed] [Google Scholar]
- 25.Lo MK; Jordan R; Arvey A; Sudhamsu J; Shrivastava-Ranjan P; Hotard AL; Flint M; McMullan LK; Siegel D; Clarke MO; Mackman RL; Hui HC; Perron M; Ray AS; Cihlar T; Nichol ST; Spiropoulou CF GS-5734 and its parent nucleoside analog inhibit Filo-Pneumo-, and Paramyxoviruses. Sci Rep 2017, 7, 43395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sheahan TP; Sims AC; Graham RL; Menachery VD; Gralinski LE; Case JB; Leist SR; Pyrc K; Feng JY; Trantcheva I; Bannister R; Park Y; Babusis D; Clarke MO; Mackman RL; Spahn JE; Palmiotti CA; Siegel D; Ray AS; Cihlar T; Jordan R; Denison MR; Baric RS Broad-spectrum antiviral GS-5734 inhibits both epidemic and zoonotic coronaviruses. Sci Transl Med 2017, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Warren TK; Jordan R; Lo MK; Ray AS; Mackman RL; Soloveva V; Siegel D; Perron M; Bannister R; Hui HC; Larson N; Strickley R; Wells J; Stuthman KS; Van Tongeren SA; Garza NL; Donnelly G; Shurtleff AC; Retterer CJ; Gharaibeh D; Zamani R; Kenny T; Eaton BP; Grimes E; Welch LS; Gomba L; Wilhelmsen CL; Nichols DK; Nuss JE; Nagle ER; Kugelman JR; Palacios G; Doerffler E; Neville S; Carra E; Clarke MO; Zhang L; Lew W; Ross B; Wang Q; Chun K; Wolfe L; Babusis D; Park Y; Stray KM; Trancheva I; Feng JY; Barauskas O; Xu Y; Wong P; Braun MR; Flint M; McMullan LK; Chen SS; Fearns R; Swaminathan S; Mayers DL; Spiropoulou CF; Lee WA; Nichol ST; Cihlar T; Bavari S Therapeutic efficacy of the small molecule GS-5734 against Ebola virus in rhesus monkeys. Nature 2016, 531, 381–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Metobo S; Xu J; Saunders OL; Butler T; Aktoudianakis E; Cho A; Kim CU Practical synthesis of 1′-substituted Tubercidin C-nucleoside analogs. 2012, 53, 484–486. [Google Scholar]
- 29.Pertusati F; Serpi M; McGuigan C Medicinal chemistry of nucleoside phosphonate prodrugs for antiviral therapy. Antivir Chem Chemother 2012, 22, 181–203. [DOI] [PubMed] [Google Scholar]
- 30.McGuigan C; Hassan-Abdallah A; Srinivasan S; Wang Y; Siddiqui A; Daluge SM; Gudmundsson KS; Zhou H; McLean EW; Peckham JP Application of phosphoramidate ProTide technology significantly improves antiviral potency of carbocyclic adenosine derivatives. Journal of medicinal chemistry 2006, 49, 7215–7226. [DOI] [PubMed] [Google Scholar]
- 31.McGuigan C; Cahard D; Sheeka HM; De Clercq E; Balzarini J Aryl phosphoramidate derivatives of d4T have improved anti-HIV efficacy in tissue culture and may act by the generation of a novel intracellular metabolite. J Med Chem 1996, 39, 1748–53. [DOI] [PubMed] [Google Scholar]
- 32.Mehellou Y; Rattan HS; Balzarini J The ProTide Prodrug Technology: From the Concept to the Clinic. J Med Chem 2018, 61, 2211–2226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mehellou Y; Balzarini J; McGuigan C Aryloxy phosphoramidate triesters: a technology for delivering monophosphorylated nucleosides and sugars into cells. ChemMedChem 2009, 4, 1779–91. [DOI] [PubMed] [Google Scholar]
- 34.Mehellou Y The ProTides Boom. ChemMedChem 2016, 11, 1114–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Deval J Antimicrobial strategies: inhibition of viral polymerases by 3’-hydroxyl nucleosides. Drugs 2009, 69, 151–66. [DOI] [PubMed] [Google Scholar]
- 36.Herdewijn P; Balzarini J; De Clercq E; Pauwels R; Baba M; Broder S; Vanderhaeghe H 3’-substituted 2’,3’-dideoxynucleoside analogues as potential anti-HIV (HTLV-III/LAV) agents. J Med Chem 1987, 30, 1270–8. [DOI] [PubMed] [Google Scholar]
- 37.Balzarini J; Kang GJ; Dalal M; Herdewijn P; De Clercq E; Broder S; Johns DG The anti-HTLV-III (anti-HIV) and cytotoxic activity of 2’,3’-didehydro-2’,3’-dideoxyribonucleosides: a comparison with their parental 2’,3’-dideoxyribonucleosides. Mol Pharmacol 1987, 32, 162–7. [PubMed] [Google Scholar]
- 38.Balzarini J; Herdewijn P; De Clercq E Differential patterns of intracellular metabolism of 2’,3’-didehydro-2’,3’-dideoxythymidine and 3’-azido-2’,3’-dideoxythymidine, two potent anti-human immunodeficiency virus compounds. J Biol Chem 1989, 264, 6127–33. [PubMed] [Google Scholar]
- 39.McGuigan C; Yarnold CJ; Jones G; Velazquez S; Barucki H; Brancale A; Andrei G; Snoeck R; De Clercq E; Balzarini J Potent and selective inhibition of varicella-zoster virus (VZV) by nucleoside analogues with an unusual bicyclic base. J Med Chem 1999, 42, 4479–84. [DOI] [PubMed] [Google Scholar]
- 40.Zhang HW; Zhou L; Coats SJ; McBrayer TR; Tharnish PM; Bondada L; Detorio M; Amichai SA; Johns MD; Whitaker T; Schinazi RF Synthesis of purine modified 2’-C-methyl nucleosides as potential anti-HCV agents. Bioorg Med Chem Lett 2011, 21, 6788–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Carroll SS; Tomassini JE; Bosserman M; Getty K; Stahlhut MW; Eldrup AB; Bhat B; Hall D; Simcoe AL; LaFemina R; Rutkowski CA; Wolanski B; Yang Z; Migliaccio G; De Francesco R; Kuo LC; MacCoss M; Olsen DB Inhibition of hepatitis C virus RNA replication by 2’-modified nucleoside analogs. J Biol Chem 2003, 278, 11979–84. [DOI] [PubMed] [Google Scholar]
- 42.Migliaccio G; Tomassini JE; Carroll SS; Tomei L; Altamura S; Bhat B; Bartholomew L; Bosserman MR; Ceccacci A; Colwell LF; Cortese R; De Francesco R; Eldrup AB; Getty KL; Hou XS; LaFemina RL; Ludmerer SW; MacCoss M; McMasters DR; Stahlhut MW; Olsen DB; Hazuda DJ; Flores OA Characterization of resistance to non-obligate chain-terminating ribonucleoside analogs that inhibit hepatitis C virus replication in vitro. J Biol Chem 2003, 278, 49164–70. [DOI] [PubMed] [Google Scholar]
- 43.Eldrup AB; Prhavc M; Brooks J; Bhat B; Prakash TP; Song Q; Bera S; Bhat N; Dande P; Cook PD; Bennett CF; Carroll SS; Ball RG; Bosserman M; Burlein C; Colwell LF; Fay JF; Flores OA; Getty K; LaFemina RL; Leone J; MacCoss M; McMasters DR; Tomassini JE; Von Langen D; Wolanski B; Olsen DB Structure-activity relationship of heterobase-modified 2’-C-methyl ribonucleosides as inhibitors of hepatitis C virus RNA replication. J Med Chem 2004, 47, 5284–97. [DOI] [PubMed] [Google Scholar]
- 44.Eldrup AB; Allerson CR; Bennett CF; Bera S; Bhat B; Bhat N; Bosserman MR; Brooks J; Burlein C; Carroll SS; Cook PD; Getty KL; MacCoss M; McMasters DR; Olsen DB; Prakash TP; Prhavc M; Song Q; Tomassini JE; Xia J Structure-activity relationship of purine ribonucleosides for inhibition of hepatitis C virus RNA-dependent RNA polymerase. J Med Chem 2004, 47, 2283–95. [DOI] [PubMed] [Google Scholar]
- 45.Pierra C; Amador A; Benzaria S; Cretton-Scott E; D’Amours M; Mao J; Mathieu S; Moussa A; Bridges EG; Standring DN; Sommadossi JP; Storer R; Gosselin G Synthesis and pharmacokinetics of valopicitabine (NM283), an efficient prodrug of the potent anti-HCV agent 2’-C-methylcytidine. J Med Chem 2006, 49, 6614–20. [DOI] [PubMed] [Google Scholar]
- 46.McGuigan C; Perrone P; Madela K; Neyts J The phosphoramidate ProTide approach greatly enhances the activity of beta-2’-C-methylguanosine against hepatitis C virus. Bioorg Med Chem Lett 2009, 19, 4316–20. [DOI] [PubMed] [Google Scholar]
- 47.McGuigan C; Gilles A; Madela K; Aljarah M; Holl S; Jones S; Vernachio J; Hutchins J; Ames B; Bryant KD; Gorovits E; Ganguly B; Hunley D; Hall A; Kolykhalov A; Liu Y; Muhammad J; Raja N; Walters R; Wang J; Chamberlain S; Henson G Phosphoramidate ProTides of 2’-C-methylguanosine as highly potent inhibitors of hepatitis C virus. Study of their in vitro and in vivo properties. J Med Chem 2010, 53, 4949–57. [DOI] [PubMed] [Google Scholar]
- 48.McCarty RM; Bandarian V Deciphering deazapurine biosynthesis: pathway for pyrrolopyrimidine nucleosides toyocamycin and sangivamycin. Chem Biol 2008, 15, 790–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zmurko J; Marques RE; Schols D; Verbeken E; Kaptein SJ; Neyts J The Viral Polymerase Inhibitor 7-Deaza-2’-C-Methyladenosine Is a Potent Inhibitor of In Vitro Zika Virus Replication and Delays Disease Progression in a Robust Mouse Infection Model. PLoS Negl Trop Dis 2016, 10, e0004695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Olsen DB; Eldrup AB; Bartholomew L; Bhat B; Bosserman MR; Ceccacci A; Colwell LF; Fay JF; Flores OA; Getty KL; Grobler JA; LaFemina RL; Markel EJ; Migliaccio G; Prhavc M; Stahlhut MW; Tomassini JE; MacCoss M; Hazuda DJ; Carroll SSA 7-deaza-adenosine analog is a potent and selective inhibitor of hepatitis C virus replication with excellent pharmacokinetic properties. Antimicrob Agents Chemother 2004, 48, 3944–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schul W; Liu W; Xu HY; Flamand M; Vasudevan SG A dengue fever viremia model in mice shows reduction in viral replication and suppression of the inflammatory response after treatment with antiviral drugs. J Infect Dis 2007, 195, 665–74. [DOI] [PubMed] [Google Scholar]
- 52.Eyer L; Valdés JJ; Gil VA; Nencka R; Hřebabecký H; Šála M; Salát J; Černý J; Palus M; De Clercq E; Růžek D Nucleoside inhibitors of tick-borne encephalitis virus. Antimicrob Agents Chemother 2015, 59, 5483–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Di Francesco ME; Avolio S; Dessole G; Koch U; Pompei M; Pucci V; Rowley M; Summa V Synthesis and antiviral properties of novel tetracyclic nucleoside inhibitors of hepatitis C NS5B polymerase. Nucleosides Nucleotides Nucleic Acids 2012, 31, 592–607. [DOI] [PubMed] [Google Scholar]
- 54.Eyer L; Kondo H; Zouharova D; Hirano M; Valdes JJ; Muto M; Kastl T; Kobayashi S; Haviernik J; Igarashi M; Kariwa H; Vaculovicova M; Cerny J; Kizek R; Kroger A; Lienenklaus S; Dejmek M; Nencka R; Palus M; Salat J; De Clercq E; Yoshii K; Ruzek D Escape of tick-borne flavivirus from 2’ -. J Virol 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Eyer L; Šmídkový M; Nencka R; Neča J; Kastl T; Palus M; De Clercq E; Růžek D Structure-activity relationships of nucleoside analogues for inhibition of tick-borne encephalitis virus. Antiviral Res 2016, 133, 119–29. [DOI] [PubMed] [Google Scholar]
- 56.Di Francesco ME; Avolio S; Pompei M; Pesci S; Monteagudo E; Pucci V; Giuliano C; Fiore F; Rowley M; Summa V Synthesis and antiviral properties of novel 7-heterocyclic substituted 7-deaza-adenine nucleoside inhibitors of Hepatitis C NS5B polymerase. Bioorg Med Chem 2012, 20, 4801–11. [DOI] [PubMed] [Google Scholar]
- 57.Carroll SS; Davies ME; Handt L; Koeplinger K; Zhang R; Ludmerer SW; MacCoss M; Hazuda DJ; Olsen DB Robust suppression of viral replication by a nucleoside polymerase inhibitor in chimpanzees infected with hepatitis C virus. Hepatology 2006, 44.16799985 [Google Scholar]
- 58.Bloch A; Leonard RJ; Nichol CA On the mode of action of 7-deaza-adenosine (tubercidin). Biochim Biophys Acta 1967, 138, 10–25. [DOI] [PubMed] [Google Scholar]
- 59.Sizun G; Pierra C; Peyronnet J; Badaroux E; Rabeson C; Benzaria-Prad S; Surleraux D; Loi AG; Musiu C; Liuzzi M; Seifer M; Standring D; Sommadossi JP; Gosselin G Design, synthesis and antiviral evaluation of 2’-C-methyl branched guanosine pronucleotides: the discovery of IDX184, a potent liver-targeted HCV polymerase inhibitor. Future Med. Chem 2015, 7, 1675–1700. [DOI] [PubMed] [Google Scholar]
- 60.Cretton-Scott E; Perigaud C; Peyrottes S; Licklider L; Camire M; Larsson M; La Colla M; Hildebrand E; Lallos L; Bilello J; McCarville J; Seifer M; Liuzzi M; Pierra C; Badaroux B; Gosselin G; Surleraux D; Standring DN In vitro antiviral activity and pharmacology of IDX184, a novel and potent inhibitor of HCV replication. J Hepatol 2008, 48. [Google Scholar]
- 61.Standring DN; Lanford R; Cretton-Scott E; Licklider L; Larsson M; Pierra C; Gosselin G; Perigaud C; Surleraux D; Mayes B; Moussa A; Selden J Potent antiviral activity of second generation nucleoside inhibitors, IDX102 and IDX184 in HCV-infected chimpanzees. J Hepatol 2008, 48. [Google Scholar]
- 62.Zhou XJ; Pietropaolo K; Chen J; Khan S; Sullivan-Bólyai J; Mayers D Safety and pharmacokinetics of IDX184, a liver-targeted nucleotide polymerase inhibitor of hepatitis C virus, in healthy subjects. Antimicrob Agents Chemother 2011, 55, 76–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lalezari J; Asmuth D; Casiró A; Vargas H; Lawrence S; Dubuc-Patrick G; Chen J; McCarville J; Pietropaolo K; Zhou XJ; Sullivan-Bólyai J; Mayers D Short-term monotherapy with IDX184, a liver-targeted nucleotide polymerase inhibitor, in patients with chronic hepatitis C virus infection. Antimicrob Agents Chemother 2012, 56, 6372–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.McGuigan C; Madela K; Aljarah M; Gilles A; Brancale A; Zonta N; Chamberlain S; Vernachio J; Hutchins J; Hall A; Ames B; Gorovits E; Ganguly B; Kolykhalov A; Wang J; Muhammad J; Patti JM; Henson G Design, synthesis and evaluation of a novel double prodrug: INX-08189. A new clinical candidate for hepatitis C virus. Bioorg Med Chem Lett 2010, 20, 4850–4. [DOI] [PubMed] [Google Scholar]
- 65.Vernachio JH; Bleiman B; Bryant KD; Chamberlain S; Hunley D; Hutchins J; Ames B; Gorovits E; Ganguly B; Hall A; Kolykhalov A; Liu Y; Muhammad J; Raja N; Walters CR; Wang J; Williams K; Patti JM; Henson G; Madela K; Aljarah M; Gilles A; McGuigan C INX-08189, a phosphoramidate prodrug of 6-O-methyl-2’-C-methyl guanosine, is a potent inhibitor of hepatitis C virus replication with excellent pharmacokinetic and pharmacodynamic properties. Antimicrob Agents Chemother 2011, 55, 1843–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yeo KL; Chen YL; Xu HY; Dong H; Wang QY; Yokokawa F; Shi PY Synergistic suppression of dengue virus replication using a combination of nucleoside analogs and nucleoside synthesis inhibitors. Antimicrob Agents Chemother 2015, 59, 2086–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Baumgart BR; Wang F; Kwagh J; Storck C; Euler C; Fuller M; Simic D; Sharma S; Arnold JJ; Cameron CE; Van Vleet TR; Flint O; Bunch RT; Davies MH; Graziano MJ; Sanderson TP Effects of BMS-986094, a Guanosine Nucleotide Analogue, on Mitochondrial DNA Synthesis and Function. Toxicol Sci 2016, 153, 396–408. [DOI] [PubMed] [Google Scholar]
- 68.Feng JY; Tay CH; Ray AS Role of Mitochondrial Toxicity in BMS-986094-Induced Toxicity. Toxicol Sci 2017, 155, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gentile I; Buonomo AR; Zappulo E; Borgia G Discontinued drugs in 2012 – 2013: hepatitis C virus infection. Expert Opin Investig Drugs 2015, 24, 239–51. [DOI] [PubMed] [Google Scholar]
- 70.Luo S; Rush R; Standring D Single- and repeat-dose toxicity of IDX14184, a nucleotide prodrug with antiviral activity for hepatitis C viral infection, in mice, rats, and monkeys. Hum Exp Toxicol 2016, 35, 472–90. [DOI] [PubMed] [Google Scholar]
- 71.Liu P; Sharon A; Chu CK Fluorinated Nucleosides: Synthesis and Biological Implication. J Fluor Chem 2008, 129, 743–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pankiewicz KW; Watanabe KA Synthesis of 2’-β-fluoro-substituted nucleosides by a direct approach. Elsevier: Amsterdam, PAYS-BAS, 1993; Vol. 64. [Google Scholar]
- 73.Pankiewicz KW Fluorinated nucleosides. Carbohydr Res 2000, 327, 87–105. [DOI] [PubMed] [Google Scholar]
- 74.Saenger W Defining Terms for the Nucleic Acids In Principles of Nucleic Acid Structure, Cantor CR, Ed. Springer-Verlag New York Inc: 1984; pp 9–28. [Google Scholar]
- 75.Ikeda H; Fernandez R; Wilk A; Barchi JJ; Huang X; Marquez VE The effect of two antipodal fluorine-induced sugar puckers on the conformation and stability of the Dickerson-Drew dodecamer duplex [d(CGCGAATTCGCG)]2. Nucleic Acids Res 1998, 26, 2237–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wojtowicz-Rajchel H Synthesis and applications of fluorinated nucleoside analogues. J Fluorine Chem 2012, 143, 11–48. [Google Scholar]
- 77.Bohm H; Banner D; Bendels S; Kansy M; Kuhn B; Muller K; Obst-Sander U; Stahl M Fluorine in medicinal chemistry. Chembiochem 2004, 5, 637–643. [DOI] [PubMed] [Google Scholar]
- 78.Kirk K Fluorine in medicinal chemistry: Recent therapeutic applications of fluorinated small molecules. J Fluorine Chem 2006, 127, 1013–1029. [Google Scholar]
- 79.Park B; Kitteringham N; O’Neill P Metabolism of fluorine-containing drugs. Annu Rev Pharmacol Toxicol 2001, 41, 443–470. [DOI] [PubMed] [Google Scholar]
- 80.Gudmundsson KS; Freeman GA; Drach JC; Townsend LB Synthesis of fluorosugar analogues of 2,5,6-trichloro-1-(beta-D-ribofuranosyl)benzimidazole as antivirals with potentially increased glycosidic bond stability. J Med Chem 2000, 43, 2473–8. [DOI] [PubMed] [Google Scholar]
- 81.Stuyver LJ; McBrayer TR; Whitaker T; Tharnish PM; Ramesh M; Lostia S; Cartee L; Shi J; Hobbs A; Schinazi RF; Watanabe KA; Otto MJ Inhibition of the subgenomic hepatitis C virus replicon in huh-7 cells by 2’-deoxy-2’-fluorocytidine. Antimicrob Agents Chemother 2004, 48, 651–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Brox LW; LePage GA; Hendler SS; Shannahoff DH Studies on the growth inhibition and metabolism of 2’-deoxy-2’-fluorocytidine in cultured human lymphoblasts. Cancer Res 1974, 34, 1838–42. [PubMed] [Google Scholar]
- 83.Richardson FC; Kuchta RD; Mazurkiewicz A; Richardson KA Polymerization of 2’-fluoro- and 2’-O-methyl-dNTPs by human DNA polymerase alpha, polymerase gamma, and primase. Biochem Pharmacol 2000, 59, 1045–52. [DOI] [PubMed] [Google Scholar]
- 84.Clark JL; Hollecker L; Mason JC; Stuyver LJ; Tharnish PM; Lostia S; McBrayer TR; Schinazi RF; Watanabe KA; Otto MJ; Furman PA; Stec WJ; Patterson SE; Pankiewicz KW Design, synthesis, and antiviral activity of 2’-deoxy-2’-fluoro-2’-C-methylcytidine, a potent inhibitor of hepatitis C virus replication. J Med Chem 2005, 48, 5504–8. [DOI] [PubMed] [Google Scholar]
- 85.Ma H; Jiang WR; Robledo N; Leveque V; Ali S; Lara-Jaime T; Masjedizadeh M; Smith DB; Cammack N; Klumpp K; Symons J Characterization of the metabolic activation of hepatitis C virus nucleoside inhibitor beta-D-2’-Deoxy-2’-fluoro-2’-C-methylcytidine (PSI-6130) and identification of a novel active 5’-triphosphate species. J Biol Chem 2007, 282, 29812–20. [DOI] [PubMed] [Google Scholar]
- 86.Murakami E; Niu C; Bao H; Micolochick Steuer HM; Whitaker T; Nachman T; Sofia MA; Wang P; Otto MJ; Furman PA The mechanism of action of beta-D-2’-deoxy-2’-fluoro-2’-C-methylcytidine involves a second metabolic pathway leading to beta-D-2’-deoxy-2’-fluoro-2’-C-methyluridine 5’-triphosphate, a potent inhibitor of the hepatitis C virus RNA-dependent RNA polymerase. Antimicrob Agents Chemother 2008, 52, 458–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sofia MJ; Bao D; Chang W; Du J; Nagarathnam D; Rachakonda S; Reddy PG; Ross BS; Wang P; Zhang HR; Bansal S; Espiritu C; Keilman M; Lam AM; Steuer HM; Niu C; Otto MJ; Furman PA Discovery of a beta-d-2’-deoxy-2’-alpha-fluoro-2’-beta-C-methyluridine nucleotide prodrug (PSI-7977) for the treatment of hepatitis C virus. J Med Chem 2010, 53, 7202–18. [DOI] [PubMed] [Google Scholar]
- 88.Lam AM; Murakami E; Espiritu C; Steuer HM; Niu C; Keilman M; Bao H; Zennou V; Bourne N; Julander JG; Morrey JD; Smee DF; Frick DN; Heck JA; Wang P; Nagarathnam D; Ross BS; Sofia MJ; Otto MJ; Furman PA PSI-7851, a pronucleotide of beta-D-2’-deoxy-2’-fluoro-2’-C-methyluridine monophosphate, is a potent and pan-enotype inhibitor of hepatitis C virus replication. Antimicrob Agents Chemother 2010, 54, 3187–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Murakami E; Tolstykh T; Bao H; Niu C; Steuer HM; Bao D; Chang W; Espiritu C; Bansal S; Lam AM; Otto MJ; Sofia MJ; Furman PA Mechanism of activation of PSI-7851 and its diastereoisomer PSI-7977. J Biol Chem 2010, 285, 34337–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Charlton M; Gane E; Manns MP; Brown RS; Curry MP; Kwo PY; Fontana RJ; Gilroy R; Teperman L; Muir AJ; McHutchison JG; Symonds WT; Brainard D; Kirby B; Dvory-Sobol H; Denning J; Arterburn S; Samuel D; Forns X; Terrault NA Sofosbuvir and ribavirin for treatment of compensated recurrent hepatitis C virus infection after liver transplantation. Gastroenterology 2015, 148, 108–17. [DOI] [PubMed] [Google Scholar]
- 91.Curry MP; Forns X; Chung RT; Terrault NA; Brown R; Fenkel JM; Gordon F; O’Leary J; Kuo A; Schiano T; Everson G; Schiff E; Befeler A; Gane E; Saab S; McHutchison JG; Subramanian GM; Symonds WT; Denning J; McNair L; Arterburn S; Svarovskaia E; Moonka D; Afdhal N Sofosbuvir and ribavirin prevent recurrence of HCV infection after liver transplantation: an open-label study. Gastroenterology 2015, 148, 100–107.e1. [DOI] [PubMed] [Google Scholar]
- 92.Desnoyer A; Pospai D; Lê MP; Gervais A; Heurgué-Berlot A; Laradi A; Harent S; Pinto A; Salmon D; Hillaire S; Fontaine H; Zucman D; Simonpoli AM; Muret P; Larrouy L; Bernard Chabert B; Descamps D; Yazdanpanah Y; Peytavin G Pharmacokinetics, safety and efficacy of a full dose sofosbuvir-based regimen given daily in hemodialysis patients with chronic hepatitis C. J Hepatol 2016, 65, 40–47. [DOI] [PubMed] [Google Scholar]
- 93.Foster GR; Pianko S; Brown A; Forton D; Nahass RG; George J; Barnes E; Brainard DM; Massetto B; Lin M; Han B; McHutchison JG; Subramanian GM; Cooper C; Agarwal K; Group BS Efficacy of sofosbuvir plus ribavirin with or without peginterferon-alfa in patients with hepatitis C virus genotype 3 infection and treatment-experienced patients with cirrhosis and hepatitis C virus genotype 2 infection. Gastroenterology 2015, 149, 1462–70. [DOI] [PubMed] [Google Scholar]
- 94.Gane EJ; Stedman CA; Hyland RH; Ding X; Svarovskaia E; Symonds WT; Hindes RG; Berrey MM Nucleotide polymerase inhibitor sofosbuvir plus ribavirin for hepatitis C. N Engl J Med 2013, 368, 34–44. [DOI] [PubMed] [Google Scholar]
- 95.Gane EJ; Stedman CA; Hyland RH; Ding X; Svarovskaia E; Subramanian GM; Symonds WT; McHutchison JG; Pang PS Efficacy of nucleotide polymerase inhibitor sofosbuvir plus the NS5A inhibitor ledipasvir or the NS5B non-nucleoside inhibitor GS-9669 against HCV genotype 1 infection. Gastroenterology 2014, 146, 736–743.e1. [DOI] [PubMed] [Google Scholar]
- 96.Jacobson IM; Gordon SC; Kowdley KV; Yoshida EM; Rodriguez-Torres M; Sulkowski MS; Shiffman ML; Lawitz E; Everson G; Bennett M; Schiff E; Al-Assi MT; Subramanian GM; An D; Lin M; McNally J; Brainard D; Symonds WT; McHutchison JG; Patel K; Feld J; Pianko S; Nelson DR; Study P; Study F Sofosbuvir for hepatitis C genotype 2 or 3 in patients without treatment options. N Engl J Med 2013, 368, 1867–77. [DOI] [PubMed] [Google Scholar]
- 97.Lawitz E; Mangia A; Wyles D; Rodriguez-Torres M; Hassanein T; Gordon SC; Schultz M; Davis MN; Kayali Z; Reddy KR; Jacobson IM; Kowdley KV; Nyberg L; Subramanian GM; Hyland RH; Arterburn S; Jiang D; McNally J; Brainard D; Symonds WT; McHutchison JG; Sheikh AM; Younossi Z; Gane EJ Sofosbuvir for previously untreated chronic hepatitis C infection. N Engl J Med 2013, 368, 1878–87. [DOI] [PubMed] [Google Scholar]
- 98.Feng JY; Cheng G; Perry J; Barauskas O; Xu Y; Fenaux M; Eng S; Tirunagari N; Peng B; Yu M; Tian Y; Lee YJ; Stepan G; Lagpacan LL; Jin D; Hung M; Ku KS; Han B; Kitrinos K; Perron M; Birkus G; Wong KA; Zhong W; Kim CU; Carey A; Cho A; Ray AS Inhibition of hepatitis C virus replication by GS-6620, a potent C-nucleoside monophosphate prodrug. Antimicrob Agents Chemother 2014, 58, 1930–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Murakami E; Wang T; Babusis D; Lepist EI; Sauer D; Park Y; Vela JE; Shih R; Birkus G; Stefanidis D; Kim CU; Cho A; Ray AS Metabolism and pharmacokinetics of the anti-hepatitis C virus nucleotide prodrug GS-6620. Antimicrob Agents Chemother 2014, 58, 1943–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Reese ND; Schiller GJ High-dose cytarabine (HD araC) in the treatment of leukemias: a review. Curr Hematol Malig Rep 2013, 8, 141–8. [DOI] [PubMed] [Google Scholar]
- 101.Schilsky RL; Williams SF; Ultmann JE; Watson S Sequential hydroxyurea cytarabine chemotherapy for refractory non-Hodgkin’s lymphoma. J Clin Oncol 1987, 5, 419–425. [DOI] [PubMed] [Google Scholar]
- 102.Bodey GP; Freireich EJ; Monto RW; Hewlett JS Cytosine arabinoside (NSC-63878) therapy for acute leukemia in adults. Cancer Chemother Rep 1969, 53, 59–66. [PubMed] [Google Scholar]
- 103.Hiddemann W Cytosine arabinoside in the treatment of acute myeloid leukemia: the role and place of high-dose regimens. Ann Hematol 1991, 62, 119–28. [DOI] [PubMed] [Google Scholar]
- 104.Livingston RB; Carter SK Cytosine arabinoside (NSC-63878)--clinical brochure. Cancer Chemother Rep 3 1968, 1, 179–205. [PubMed] [Google Scholar]
- 105.Chabner BA; Hande KR; Drake JC Ara-C metabolism: implications for drug resistance and drug interactions. Bull Cancer 1979, 66, 89–92. [PubMed] [Google Scholar]
- 106.Prince HN; Grunberg E; Buck M; Cleeland R A Comparative Study of the Antitumor and Antiviral Activity of 1-β-D-Arabinofuranosyl-5-fluorocytosine (FCA) and 1-β-D-Arabinofuranosylcytosine (CA). Proceedings of the Society for Experimental Biology and Medicine 1969, 130, 1080–1086. [DOI] [PubMed] [Google Scholar]
- 107.Ho DH Distribution of kinase and deaminase of 1-beta-D-arabinofuranosylcytosine in tissues of man and mouse. Cancer Res 1973, 33, 2816–20. [PubMed] [Google Scholar]
- 108.Plunkett W; Gandhi V Cellular pharmacodynamics of anticancer drugs. Semin Oncol 1993, 20, 50–63. [PubMed] [Google Scholar]
- 109.Azuma A; Nakajima Y; Nishizono N; Minakawa N; Suzuki M; Hanaoka K; Kobayashi T; Tanaka M; Sasaki T; Matsuda A Nucleosides and nucleotides. 122. 2’-C-cyano-2’-deoxy-1-beta-D-arabinofuranosylcytosine and its derivatives. A new class of nucleoside with a broad antitumor spectrum. J Med Chem 1993, 36, 4183–9. [DOI] [PubMed] [Google Scholar]
- 110.Azuma A; Hanaoka K; Kurihara A; Kobayashi T; Miyauchi S; Kamo N; Tanaka M; Sasaki T; Matsuda A Nucleosides and nucleotides. 141. Chemical stability of a new antitumor nucleoside, 2’-C-cyano-2’-deoxy-1-beta-D-arabino-pentofuranosylcytosine in alkaline medium: formation of 2’-C-cyano-2’-deoxy-1-beta-D-ribo-pentofuranosylcytosine and its antitumor activity. J Med Chem 1995, 38, 3391–7. [DOI] [PubMed] [Google Scholar]
- 111.Matsuda A; Nakajima Y; Azuma A; Tanaka M; Sasaki T Nucleosides and nucleotides. 100. 2’-C-cyano-2’-deoxy-1-beta-D-arabinofuranosyl-cytosine (CNDAC): design of a potential mechanism-based DNA-strand-breaking antineoplastic nucleoside. J Med Chem 1991, 34, 2917–9. [DOI] [PubMed] [Google Scholar]
- 112.Matsuda A; Takenuki K; Tanaka M; Sasaki T; Ueda T Nucleosides and nucleotides. 97. Synthesis of new broad spectrum antineoplastic nucleosides, 2’-deoxy-2’-methylidenecytidine (DMDC) and its derivatives. J Med Chem 1991, 34, 812–9. [DOI] [PubMed] [Google Scholar]
- 113.Matsuda A; Azuma A 2′-C-Cyano-2′-deoxy-1-β-D-arabinofuranosylcytosine (CNDAC): A Mechanism-Based DNA-Strand-Breaking Antitumor Nucleoside1. Nucleosides and Nucleotides 1995, 14, 461–471. [Google Scholar]
- 114.Baskar R; Lee KA; Yeo R; Yeoh KW Cancer and radiation therapy: current advances and future directions. Int J Med Sci 2012, 9, 193–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Azuma A; Huang P; Matsuda A; Plunkett W Intracellular Metabolism and Action of an Antitumor Nucleoside, 2′-C-Cyano-2′-Deoxy-1-β-d-arabinofuranosylcytosine (CNDAC). Nucleosides and Nucleotides 1997, 16, 1037–1039. [Google Scholar]
- 116.Azuma A; Huang P; Matsuda A; Plunkett W Cellular pharmacokinetics and pharmacodynamics of the deoxycytidine analog 2’-C-cyano-2’-deoxy-1-beta-D-arabinopentofuranosylcytosine (CNDAC). Biochem Pharmacol 2001, 61, 1497–507. [DOI] [PubMed] [Google Scholar]
- 117.Tanaka M; Matsuda A; Terao T; Sasaki T Antitumor activity of a novel nucleoside, 2’-C-cyano-2’-deoxy-1-beta-D-arabinofuranosylcytosine (CNDAC) against murine and human tumors. Cancer Lett 1992, 64, 67–74. [DOI] [PubMed] [Google Scholar]
- 118.Kantarjian H; Jabbour E; Garcia-Manero G; Kadia T; DiNardo C; Daver N; Borthakur G; Jain N; Waukau J; Kwari M; Anderson BD; Iizuka K; Jin C; Plunkett W Results of a Phase I/II Study of DFP-10917, a Nucleoside Analog, Given By Continuous Infusion (CI) in Patients (pts) with Relapsed or Refractory Acute Leukemia. Blood 2015, 126, 3804–804. [Google Scholar]
- 119.Kantarjian HM; Jabbour EJ; Garcia-Manero G; Kadia TM; DiNardo CD; Daver NG; Borthakur G; Jain N; Waukau J; Kwari M; Anderson BD; Iizuka K; Jin C; Zhang C; Ravandi F; Plunkett W Phase I/II Study of DFP-10917 in Relapsed/Refractory AML Demonstrates Efficacy and Safety Profile Suitable for Phase III Study. Blood 2016, 128.27827828 [Google Scholar]
- 120.Kantarjian HM; Jabbour E; Garcia-Manero G; Kadia TM; Dinardo CD; Daver NG; Borthakur G; Jain N; Waukau J; Kwari M; Anderson BD; Iizuka K; Jin C; Plunkett W First report of a phase I/II study of DFP-10917, a nucleoside analog, given by continuous infusion (CI) in patients (pts) with relapsed or refractory acute leukemia. Journal of Clinical Oncology 2015, 33, 7077–7077. [Google Scholar]
- 121.Kantarjian H; Garcia-Manero G; O’Brien S; Faderl S; Ravandi F; Westwood R; Green SR; Chiao JH; Boone PA; Cortes J; Plunkett W Phase I clinical and pharmacokinetic study of oral sapacitabine in patients with acute leukemia and myelodysplastic syndrome. J Clin Oncol 2010, 28, 285–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kantarjian H; Faderl S; Garcia-Manero G; Luger S; Venugopal P; Maness L; Wetzler M; Coutre S; Stock W; Claxton D; Goldberg SL; Arellano M; Strickland SA; Seiter K; Schiller G; Jabbour E; Chiao J; Plunkett W Oral sapacitabine for the treatment of acute myeloid leukaemia in elderly patients: a randomised phase 2 study. Lancet Oncol 2012, 13, 1096–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Chen YL; Yin Z; Lakshminarayana SB; Qing M; Schul W; Duraiswamy J; Kondreddi RR; Goh A; Xu HY; Yip A; Liu B; Weaver M; Dartois V; Keller TH; Shi PY Inhibition of dengue virus by an ester prodrug of an adenosine analog. Antimicrob Agents Chemother 2010, 54, 3255–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Chen YL; Yin Z; Duraiswamy J; Schul W; Lim CC; Liu B; Xu HY; Qing M; Yip A; Wang G; Chan WL; Tan HP; Lo M; Liung S; Kondreddi RR; Rao R; Gu H; He H; Keller TH; Shi PY Inhibition of dengue virus RNA synthesis by an adenosine nucleoside. Antimicrob Agents Chemother 2010, 54, 2932–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Deng YQ; Zhang NN; Li CF; Tian M; Hao JN; Xie XP; Shi PY; Qin CF Adenosine Analog NITD008 Is a Potent Inhibitor of Zika Virus. Open Forum Infect Dis 2016, 3, ofw175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Yin Z; Chen YL; Schul W; Wang QY; Gu F; Duraiswamy J; Kondreddi RR; Niyomrattanakit P; Lakshminarayana SB; Goh A; Xu HY; Liu W; Liu B; Lim JY; Ng CY; Qing M; Lim CC; Yip A; Wang G; Chan WL; Tan HP; Lin K; Zhang B; Zou G; Bernard KA; Garrett C; Beltz K; Dong M; Weaver M; He H; Pichota A; Dartois V; Keller TH; Shi PY An adenosine nucleoside inhibitor of dengue virus. Proc Natl Acad Sci U S A 2009, 106, 20435–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Shang L; Wang Y; Qing J; Shu B; Cao L; Lou Z; Gong P; Sun Y; Yin Z An adenosine nucleoside analogue NITD008 inhibits EV71 proliferation. Antiviral Res 2014, 112, 47–58. [DOI] [PubMed] [Google Scholar]
- 128.Lo MK; Shi PY; Chen YL; Flint M; Spiropoulou CF In vitro antiviral activity of adenosine analog NITD008 against tick-borne flaviviruses. Antiviral Res 2016, 130, 46–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Katzelnick LC; Coloma J; Harris E Dengue: knowledge gaps, unmet needs, and research priorities. Lancet Infect Dis 2017, 17, e88–e100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Low JG; Ooi EE; Vasudevan SG Current Status of Dengue Therapeutics Research and Development. J Infect Dis 2017, 215, S96–S102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Qing J; Luo R; Wang Y; Nong J; Wu M; Shao Y; Tang R; Yu X; Yin Z; Sun Y Resistance analysis and characterization of NITD008 as an adenosine analog inhibitor against hepatitis C virus. Antiviral Res 2016, 126, 43–54. [DOI] [PubMed] [Google Scholar]
- 132.Deng CL; Yeo H; Ye HQ; Liu SQ; Shang BD; Gong P; Alonso S; Shi PY; Zhang B Inhibition of enterovirus 71 by adenosine analog NITD008. J Virol 2014, 88, 11915–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Mikhailov SN; Beigelman LN; Gurskaya GV; Padyukova NS; Yakovlev GI; Karpeisky MY Synthesis and properties of 3′-C-methylnucleosides and their phosphoric esters. Carb Res 1983, 124, 75–96. [Google Scholar]
- 134.Franchetti P; Cappellacci L; Pasqualini M; Petrelli R; Vita P; Jayaram HN; Horvath Z; Szekeres T; Grifantini M Antitumor activity of C-methyl-beta-D-ribofuranosyladenine nucleoside ribonucleotide reductase inhibitors. J Med Chem 2005, 48, 4983–9. [DOI] [PubMed] [Google Scholar]
- 135.Cappellacci L; Franchetti P; Petrelli R; Riccioni S; Vita PN; Jayaram H; Grifantini M Purine and Pyrimidine Nucleoside Analogs of 3’-C-Methyladenosine as Antitumor Agents. Collect of Czechoslov Chem Comm 2006, 71. [Google Scholar]
- 136.Veal GJ; Barry MG; Back DJ Zalcitabine (DDC) Phosphorylation and Drug-Interactions. Antiviral Chem Chemother 1995, 6, 379–384. [Google Scholar]
- 137.Horwitz JP; Chua J; Noel M; Donatti JT Nucleosides. XI. 2’,3’-dideoxycytidine. J Org Chem 1967, 32, 817–8. [DOI] [PubMed] [Google Scholar]
- 138.Mitsuya H; Broder S Inhibition of the in vitro infectivity and cytopathic effect of human T-lymphotrophic virus type III/lymphadenopathy-associated virus (HTLV-III/LAV) by 2’,3’-dideoxynucleosides. Proc Natl Acad Sci U S A 1986, 83, 1911–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Smith RA; Gottlieb GS; Anderson DJ; Pyrak CL; Preston BD Human immunodeficiency virus types 1 and 2 exhibit comparable sensitivities to Zidovudine and other nucleoside analog inhibitors in vitro. Antimicrob Agents Chemother 2008, 52, 329–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Hostetler KY; Richman DD; Sridhar CN; Felgner PL; Felgner J; Ricci J; Gardner MF; Selleseth DW; Ellis MN Phosphatidylazidothymidine and phosphatidyl-ddC: assessment of uptake in mouse lymphoid tissues and antiviral activities in human immunodeficiency virus-infected cells and in Rauscher leukemia virus-infected mice. Antimicrob Agents Chemother 1994, 38, 2792–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Seley-Radtke KL; Yates MK The evolution of nucleoside analogue antivirals: A review for chemists and non-chemists. Part 1: Early structural modifications to the nucleoside scaffold. Antiviral Res 2018, 154, 66–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Thomas SO; Singleton VL; Lowery JA; Sharpe RW; Pruess LM; Porter JN; Mowat JH; Bohonos N Nucleocidin, a new antibiotic with activity against Trypanosomes. Antibiot Annu 1956, 716–21. [PubMed] [Google Scholar]
- 143.Koole LH; Plavec J; Liu H; Vincent BR; Dyson MR; Coe PL; Walker RT; Hardy GW; Rahim SG; Chattopadhyaya J Conformation of two 4’-thio-2’-deoxynucleoside analogs studied by 5000-MHz proton NMR spectroscopy and x-ray crystallography. Journal of the American Chemical Society 1992, 114, 9936–9943. [Google Scholar]
- 144.Waga T; Nishizaki T; Miyakawa I; Ohrui H; Meguro H Synthesis of 4’-C-methylnucleosides. Biosci Biotechnol Biochem 1993, 57, 1433–8. [DOI] [PubMed] [Google Scholar]
- 145.Jenkins ID; Verheyden JP; Moffatt JG 4’-Substituted nucleosides. 2. Synthesis of the nucleoside antibiotic nucleocidin. J Am Chem Soc 1976, 98, 3346–57. [DOI] [PubMed] [Google Scholar]
- 146.Morton GO; Lancaster JE; Van Lear GE; Fulmor W; Meyer WE The structure of nucleocidin. 3. (A new structure). J Am Chem Soc 1969, 91, 1535–7. [DOI] [PubMed] [Google Scholar]
- 147.Jenkins ID; Verheyden JP; Moffatt JG Synthesis of the nucleoside antibiotic nucleocidin. J Am Chem Soc 1971, 93, 4323–4. [DOI] [PubMed] [Google Scholar]
- 148.Owen GR a. V. J. P. H. a. M. J. G. 4’-Substituted nucleosides. 3. Synthesis of some 4’-fluorouridine derivatives. The Journal of Organic Chemistry 1976, 41, 3010–3017. [DOI] [PubMed] [Google Scholar]
- 149.Hewitt RI; Gumble AR; Taylor LH; Wallace WS The activity of a new antibiotic, nucleocidin, in experimental infections with Trypanosoma equiperdum. Antibiot Annu 1956, 722–9. [PubMed] [Google Scholar]
- 150.Guillerm D; Muzard M; Allart B; Guillerm G Synthesis of 4′-fluoroadenosine as an inhibitor of S-adenosyl-L-homocysteine hydrolase. Bioorganic & Medicinal Chemistry Letters 1995, 5, 1455–1460. [Google Scholar]
- 151.Ajmera S; Bapat AR; Danenberg PV; Stephanian E Synthesis and interaction with uridine phosphorylase of 5’-deoxy-4’,5-difluorouridine, a new prodrug of 5-fluorouracil. Journal of Medicinal Chemistry 1988, 31, 1094–1098. [DOI] [PubMed] [Google Scholar]
- 152.Ajmera S a. B. A. R. a. D. P. V. a. S. E. Synthesis and interaction with uridine phosphorylase of 5’-deoxy-4’,5-difluorouridine, a new prodrug of 5-fluorouracil. Journal of Medicinal Chemistry 1988, 31, 1094–1098. [DOI] [PubMed] [Google Scholar]
- 153.Orlov AA; Drenichev MS; Oslovsky VE; Kurochkin NN; Solyev PN; Kozlovskaya LI; Palyulin VA; Karganova GG; Mikhailov SN; Osolodkin DI New tools in nucleoside toolbox of tick-borne encephalitis virus reproduction inhibitors. Bioorg Med Chem Lett 2017, 27, 1267–1273. [DOI] [PubMed] [Google Scholar]
- 154.Waga T; Ohrui H; Meguro H Synthesis and Biological Evaluation of 4′-C-Methyl Nucleosides. Nucleosides and Nucleotides 1996, 15, 287–304. [Google Scholar]
- 155.Nomura M; Shuto S; Tanaka M; Sasaki T; Mori S; Shigeta S; Matsuda A Nucleosides and Nucleotides. 185. Synthesis and Biological Activities of 4’α-C-Branched-Chain Sugar Pyrimidine Nucleosides. Journal of Medicinal Chemistry 1999, 42, 2901–2908. [DOI] [PubMed] [Google Scholar]
- 156.Griffon JF; Dukhan D; Pierra C; Benzaria S; Loi AG; La Colla P; Sommadossi JP; Gosselin G 4’-C-methyl-beta-D-ribofuranosyl purine and pyrimidine nucleosides revisited. Nucleosides Nucleotides Nucleic Acids 2003, 22, 707–9. [DOI] [PubMed] [Google Scholar]
- 157.Shirasaka T; Chokekijchai S; Yamada A; Gosselin G; Imbach JL; Mitsuya H Comparative analysis of anti-human immunodeficiency virus type 1 activities of dideoxynucleoside analogs in resting and activated peripheral blood mononuclear cells. Antimicrob Agents Chemother 1995, 39, 2555–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Horwitz JP; Chua J; Noel M Nucleosides. V. The Monomesylates of 1-(2’-Deoxy-β-D-lyxofuranosyl) thymine. J. Org. Chem 1964, 29, 2076–2078. [Google Scholar]
- 159.Maag H; Rydzewski RM; McRoberts MJ; Crawford-Ruth D; Verheyden JP; Prisbe EJ Synthesis and anti-HIV activity of 4’-azido- and 4’-methoxynucleosides. J Med Chem 1992, 35, 1440–51. [DOI] [PubMed] [Google Scholar]
- 160.Chen MS; Suttmann RT; Wu JC; Prisbe EJ Metabolism of 4’-azidothymidine. A compound with potent and selective activity against the human immunodeficiency virus. J Biol Chem 1992, 267, 257–60. [PubMed] [Google Scholar]
- 161.Yamada K; Wahba AS; Bernatchez JA; Ilina T; Martinez-Montero S; Habibian M; Deleavey GF; Götte M; Parniak MA; Damha MJ Nucleotide Sugar Pucker Preference Mitigates Excision by HIV-1 RT. ACS Chem Biol 2015, 10, 2024–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Smith DB; Martin JA; Klumpp K; Baker SJ; Blomgren PA; Devos R; Granycome C; Hang J; Hobbs CJ; Jiang WR; Laxton C; Le Pogam S; Leveque V; Ma H; Maile G; Merrett JH; Pichota A; Sarma K; Smith M; Swallow S; Symons J; Vesey D; Najera I; Cammack N Design, synthesis, and antiviral properties of 4’-substituted ribonucleosides as inhibitors of hepatitis C virus replication: the discovery of R1479. Bioorg Med Chem Lett 2007, 17, 2570–6. [DOI] [PubMed] [Google Scholar]
- 163.Klumpp K; Leveque V; Le Pogam S; Ma H; Jiang WR; Kang H; Granycome C; Singer M; Laxton C; Hang JQ; Sarma K; Smith DB; Heindl D; Hobbs CJ; Merrett JH; Symons J; Cammack N; Martin JA; Devos R; Najera I The novel nucleoside analog R1479 (4’-azidocytidine) is a potent inhibitor of NS5B-dependent RNA synthesis and hepatitis C virus replication in cell culture. J Biol Chem 2006, 281, 3793–9. [DOI] [PubMed] [Google Scholar]
- 164.Nguyen NM; Tran CN; Phung LK; Duong KT; Huynh H. e. A.; Farrar J; Nguyen QT; Tran HT; Nguyen CV; Merson L; Hoang LT; Hibberd ML; Aw PP; Wilm A; Nagarajan N; Nguyen DT; Pham MP; Nguyen TT; Javanbakht H; Klumpp K; Hammond J; Petric R; Wolbers M; Nguyen CT; Simmons CP A randomized, double-blind placebo controlled trial of balapiravir, a polymerase inhibitor, in adult dengue patients. J Infect Dis 2013, 207, 1442–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Hotard AL; He B; Nichol ST; Spiropoulou CF; Lo MK 4’-Azidocytidine (R1479) inhibits henipaviruses and other paramyxoviruses with high potency. Antiviral Res 2017, 144, 147–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Wang G; Deval J; Hong J; Dyatkina N; Prhavc M; Taylor J; Fung A; Jin Z; Stevens SK; Serebryany V; Liu J; Zhang Q; Tam Y; Chanda SM; Smith DB; Symons JA; Blatt LM; Beigelman L Discovery of 4’-chloromethyl-2’-deoxy-3’,5’-di-O-isobutyryl-2’-fluorocytidine (ALS-8176), a first-in-class RSV polymerase inhibitor for treatment of human respiratory syncytial virus infection. J Med Chem 2015, 58, 1862–78. [DOI] [PubMed] [Google Scholar]
- 167.Klumpp K; Smith DB Discovery and Clinical Evaluation of the Nucleoside Analog Balapiravir (R1626) for the Treatment of HCV Infection. 2011, 287–304. [Google Scholar]
- 168.Roberts SK; Cooksley G; Dore GJ; Robson R; Shaw D; Berns H; Hill G; Klumpp K; Najera I; Washington C Robust antiviral activity of R1626, a novel nucleoside analog: a randomized, placebo-controlled study in patients with chronic hepatitis C. Hepatology 2008, 48, 398–406. [DOI] [PubMed] [Google Scholar]
- 169.Toniutto P; Fabris C; Bitetto D; Fumolo E; Fornasiere E; Pirisi M R-1626, a specific oral NS5B polymerase inhibitor of hepatitis C virus. IDrugs 2008, 11, 738–49. [PubMed] [Google Scholar]
- 170.Brandl M; Wu X; Holper M; Hong L; Jia Z; Birudaraj R; Reddy M; Alfredson T; Tran T; Larrabee S; Hadig X; Sarma K; Washington C; Hill G; Smith DB Physicochemical properties of the nucleoside prodrug R1626 leading to high oral bioavailability. Drug Dev Ind Pharm 2008, 34, 683–91. [DOI] [PubMed] [Google Scholar]
- 171.Roberts R; Cooksley W; Shaw D; Berns KH; Brandl TM; Fettner HS; Hill G; Ipe D; Klumpp K; Mannino M; O’mara E; Tuo Y; Washington BC Interim results of a multiple ascending dose study of R1626, a novel nucleoside analog targeting HCV polymerase in chronic hcv patients. 2006, 44. [Google Scholar]
- 172.Robson R; Berns H; Brandl M; Fettner S; Hill G; Ipe D; Love J; Mannino M; O’mara E; Tu Y Safety, Tolerability And Pharmacokinetics Of R1626, A Novel Nucleoside Analog Targeting Hcv Polymerase: Results From A Phase 1 Single Dose Escalation Trial In Healthy Subjects.: piii-41. Clinical Pharmacology & Therapeutics 2007, 81, S98. [Google Scholar]
- 173.Pockros PJ; Nelson D; Godofsky E; Rodriguez-Torres M; Everson GT; Fried MW; Ghalib R; Harrison S; Nyberg L; Shiffman ML; Najera I; Chan A; Hill G R1626 plus peginterferon Alfa-2a provides potent suppression of hepatitis C virus RNA and significant antiviral synergy in combination with ribavirin. Hepatology 2008, 48, 385–97. [DOI] [PubMed] [Google Scholar]
- 174.Pockros P; Nelson D; Godofsky E; Rodriguez-Torres M; Everson GT; Fried MW; Ghalib R; Harrison S; Nyberg L; Shiffman ML; Chan A; Hill G High relapse rate seen at week 72 for patients treated with R1626 combination therapy. Hepatology 2008, 48, 1349–50. [DOI] [PubMed] [Google Scholar]
- 175.Chen YL; Abdul Ghafar N; Karuna R; Fu Y; Lim SP; Schul W; Gu F; Herve M; Yokohama F; Wang G; Cerny D; Fink K; Blasco F; Shi PY Activation of peripheral blood mononuclear cells by dengue virus infection depotentiates balapiravir. J Virol 2014, 88, 1740–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Klumpp K; Kalayanov G; Ma H; Le Pogam S; Leveque V; Jiang WR; Inocencio N; De Witte A; Rajyaguru S; Tai E; Chanda S; Irwin MR; Sund C; Winqist A; Maltseva T; Eriksson S; Usova E; Smith M; Alker A; Najera I; Cammack N; Martin JA; Johansson NG; Smith DB 2’-deoxy-4’-azido nucleoside analogs are highly potent inhibitors of hepatitis C virus replication despite the lack of 2’-alpha-hydroxyl groups. J Biol Chem 2008, 283, 2167–75. [DOI] [PubMed] [Google Scholar]
- 177.Smith DB; Kalayanov G; Sund C; Winqvist A; Pinho P; Maltseva T; Morisson V; Leveque V; Rajyaguru S; Le Pogam S; Najera I; Benkestock K; Zhou XX; Maag H; Cammack N; Martin JA; Swallow S; Johansson NG; Klumpp K; Smith M The design, synthesis, and antiviral activity of 4’-azidocytidine analogues against hepatitis C virus replication: the discovery of 4’-azidoarabinocytidine. J Med Chem 2009, 52, 219–23. [DOI] [PubMed] [Google Scholar]
- 178.Rondla R; Coats SJ; McBrayer TR; Grier J; Johns M; Tharnish PM; Whitaker T; Zhou L; Schinazi RF Anti-hepatitis C virus activity of novel beta-d-2’-C-methyl-4’-azido pyrimidine nucleoside phosphoramidate prodrugs. Antivir Chem Chemother 2009, 20, 99–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Nilsson M; Kalayanov G; Winqvist A; Pinho P; Sund C; Zhou XX; Wähling H; Belfrage AK; Pelcman M; Agback T; Benckestock K; Wikström K; Boothee M; Lindqvist A; Rydegård C; Jonckers TH; Vandyck K; Raboisson P; Lin TI; Lachau-Durand S; de Kock H; Smith DB; Martin JA; Klumpp K; Simmen K; Vrang L; Terelius Y; Samuelsson B; Rosenquist S; Johansson NG Discovery of 4’-azido-2’-deoxy-2’-C-methyl cytidine and prodrugs thereof: a potent inhibitor of Hepatitis C virus replication. Bioorg Med Chem Lett 2012, 22, 3265–8. [DOI] [PubMed] [Google Scholar]
- 180.O-Yang C; Wu HY; Fraser-Smith WB; Walker KAM Synthesis of 4′-cyanothymidine and analogs as potent inhibitors of HIV. Tetrahedron Letters 1992, 33, 37–40. [Google Scholar]
- 181.Kohgo S; Yamada K; Kitano K; Iwai Y; Sakata S; Ashida N; Hayakawa H; Nameki D; Kodama E; Matsuoka M; Mitsuya H; Ohrui H Design, efficient synthesis, and anti-HIV activity of 4’-C-cyano- and 4’-C-ethynyl-2’-deoxy purine nucleosides. Nucleosides Nucleotides Nucleic Acids 2004, 23, 671–90. [DOI] [PubMed] [Google Scholar]
- 182.Takamatsu Y; Tanaka Y; Kohgo S; Murakami S; Singh K; Das D; Venzon DJ; Amano M; Higashi-Kuwata N; Aoki M; Delino NS; Hayashi S; Takahashi S; Sukenaga Y; Haraguchi K; Sarafianos SG; Maeda K; Mitsuya H 4’-modified nucleoside analogs: potent inhibitors active against entecavir-resistant hepatitis B virus. Hepatology 2015, 62, 1024–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Kirby KA; Michailidis E; Fetterly TL; Steinbach MA; Singh K; Marchand B; Leslie MD; Hagedorn AN; Kodama EN; Marquez VE; Hughes SH; Mitsuya H; Parniak MA; Sarafianos SG Effects of substitutions at the 4’ and 2 positions on the bioactivity of 4’-ethynyl-2-fluoro-2’-deoxyadenosine. Antimicrob Agents Chemother 2013, 57, 6254–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Gallois-Montbrun S; Schneider B; Chen Y; Giacomoni-Fernandes V; Mulard L; Morera S; Janin J; Deville-Bonne D; Veron M Improving nucleoside diphosphate kinase for antiviral nucleotide analogs activation. J Biol Chem 2002, 277, 39953–9. [DOI] [PubMed] [Google Scholar]
- 185.Hattori S; Ide K; Nakata H; Harada H; Suzu S; Ashida N; Kohgo S; Hayakawa H; Mitsuya H; Okada S Potent activity of a nucleoside reverse transcriptase inhibitor, 4’-ethynyl-2-fluoro-2’-deoxyadenosine, against human immunodeficiency virus type 1 infection in a model using human peripheral blood mononuclear cell-transplanted NOD/SCID Janus kinase 3 knockout mice. Antimicrob Agents Chemother 2009, 53, 3887–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Nakata H; Amano M; Koh Y; Kodama E; Yang G; Bailey CM; Kohgo S; Hayakawa H; Matsuoka M; Anderson KS; Cheng YC; Mitsuya H Activity against human immunodeficiency virus type 1, intracellular metabolism, and effects on human DNA polymerases of 4’-ethynyl-2-fluoro-2’-deoxyadenosine. Antimicrob Agents Chemother 2007, 51, 2701–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Ohrui H 2’-deoxy-4’-C-ethynyl-2-fluoroadenosine, a nucleoside reverse transcriptase inhibitor, is highly potent against all human immunodeficiency viruses type 1 and has low toxicity. Chem Rec 2006, 6, 133–43. [DOI] [PubMed] [Google Scholar]
- 188.Ohrui H; Kohgo S; Hayakawa H; Kodama E; Matsuoka M; Nakata T; Mitsuya H 2’-deoxy-4’-C-ethynyl-2-fluoroadenosine: a nucleoside reverse transcriptase inhibitor with highly potent activity against wide spectrum of HIV-1 strains, favorable toxic profiles, and stability in plasma. Nucleosides Nucleotides Nucleic Acids 2007, 26, 1543–6. [DOI] [PubMed] [Google Scholar]
- 189.Kawamoto A; Kodama E; Sarafianos SG; Sakagami Y; Kohgo S; Kitano K; Ashida N; Iwai Y; Hayakawa H; Nakata H; Mitsuya H; Arnold E; Matsuoka M 2’-deoxy-4’-C-ethynyl-2-halo-adenosines active against drug-resistant human immunodeficiency virus type 1 variants. Int J Biochem Cell Biol 2008, 40, 2410–20. [DOI] [PubMed] [Google Scholar]
- 190.Kirby KA; Singh K; Michailidis E; Marchand B; Kodama EN; Ashida N; Mitsuya H; Parniak MA; Sarafianos SG The sugar ring conformation of 4’-ethynyl-2-fluoro-2’-deoxyadenosine and its recognition by the polymerase active site of HIV reverse transcriptase. Cell Mol Biol (Noisy-le-grand) 2011, 57, 40–6. [PMC free article] [PubMed] [Google Scholar]
- 191.Boyer PL; Julias JG; Marquez VE; Hughes SH Fixed conformation nucleoside analogs effectively inhibit excision-proficient HIV-1 reverse transcriptases. J Mol Biol 2005, 345, 441–50. [DOI] [PubMed] [Google Scholar]
- 192.Marquez VE; Ezzitouni A; Russ P; Siddiqui MA; Ford H Jr.; Feldman RJ; Mitsuya H; George C; Barchi J, Lessons JJ from the Pseudorotational Cycle: Conformationally Rigid AZT Carbocyclic Nucleosides and Their Interaction with Reverse Transcriptase. Nucleosides and Nucleotides 1998, 17, 1881–1884. [Google Scholar]
- 193.Michailidis E; Marchand B; Kodama EN; Singh K; Matsuoka M; Kirby KA; Ryan EM; Sawani AM; Nagy E; Ashida N; Mitsuya H; Parniak MA; Sarafianos SG Mechanism of inhibition of HIV-1 reverse transcriptase by 4’-Ethynyl-2-fluoro-2’-deoxyadenosine triphosphate, a translocation-defective reverse transcriptase inhibitor. J Biol Chem 2009, 284, 35681–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Sohl CD; Singh K; Kasiviswanathan R; Copeland WC; Mitsuya H; Sarafianos SG; Anderson KS Mechanism of interaction of human mitochondrial DNA polymerase γ with the novel nucleoside reverse transcriptase inhibitor 4’-ethynyl-2-fluoro-2’-deoxyadenosine indicates a low potential for host toxicity. Antimicrob Agents Chemother 2012, 56, 1630–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Muftuoglu Y; Sohl CD; Mislak AC; Mitsuya H; Sarafianos SG; Anderson KS Probing the molecular mechanism of action of the HIV-1 reverse transcriptase inhibitor 4’-ethynyl-2-fluoro-2’-deoxyadenosine (EFdA) using pre-steady-state kinetics. Antiviral Res 2014, 106, 1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Salie ZL; Kirby KA; Michailidis E; Marchand B; Singh K; Rohan LC; Kodama EN; Mitsuya H; Parniak MA; Sarafianos SG Structural basis of HIV inhibition by translocation-defective RT inhibitor 4’-ethynyl-2-fluoro-2’-deoxyadenosine (EFdA). Proc Natl Acad Sci U S A 2016, 113, 9274–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Das SR; Schneller SW The 5’-nor aristeromycin analogues of 5’-deoxy-5’-methylthioadenosine and 5’-deoxy-5’-thiophenyladenosine. Nucleosides Nucleotides Nucleic Acids 2014, 33, 668–77. [DOI] [PubMed] [Google Scholar]
- 198.Barnard DL; Stowell VD; Seley KL; Hegde VR; Das SR; Rajappan VP; Schneller SW; Smee DF; Sidwell RW Inhibition of measles virus replication by 5’-nor carbocyclic adenosine analogues. Antivir Chem Chemother 2001, 12, 241–50. [DOI] [PubMed] [Google Scholar]
- 199.Hegde VR; Seley KL; Schneller SW Carbocyclic 5’-noruridine. Nucleosides Nucleotides Nucleic Acids 2000, 19, 269–73. [DOI] [PubMed] [Google Scholar]
- 200.Hegde VR; Seley KL; Chen X; Schneller SW The Synthesis of Carbocyclic 5′-Nor Thymidine and an Isomer as Oligonucleotide Monomers. Nucleosides and Nucleotides 1999, 18, 1905–1910. [Google Scholar]
- 201.Seley KL; Schneller SW; Rattendi D; Lane S; Bacchi CJ Synthesis and anti-trypanosomal activities of a series of 7-deaza-5’-noraristeromycin derivatives with variations in the cyclopentyl ring substituents. Antimicrob Agents Chemother 1997, 41, 1658–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Seley KL; Schneller SW; Rattendi D; Bacchi CJ (+)-7-Deaza-5’-noraristeromycin as an Anti-Trypanosomal Agent. J Med Chem 1997, 40, 622–624. [DOI] [PubMed] [Google Scholar]
- 203.Seley KL; Schneller SW; Rattendi D; Lane S; Bacchi CJ Synthesis and antitrypanosomal activity of various 8-aza-7-deaza-5’noraristeromycin derivatives. J Med Chem 1997, 40, 625–9. [DOI] [PubMed] [Google Scholar]
- 204.Seley KL; Schneller SW; Rattendi D; Lane S; Bacchi CJ Synthesis and Anti-Trypanosomal Activity of Various 8-Aza-7-deaza-5’-noraristeromycin Derivatives. J Med Chem 1997, 40, 625–629. [DOI] [PubMed] [Google Scholar]
- 205.Seley KL; Schneller SW; De Clercq E A Methylated Derivative of 5’-Noraristeromycin. J Org Chem 1997, 62, 5645–5646. [Google Scholar]
- 206.Seley KL; Schneller SW; Korba BA 5′-Noraristeromycin Enantiomer with Activity Towards Hepatitis B Virus. Nucleosides and Nucleotides 1997, 16, 2095–2099. [Google Scholar]
- 207.Hasobe M; McKee JG; Borcherding DR; Borchardt RT 9-(trans-2’,trans-3’-Dihydroxycyclopent-4’-enyl)-adenine and -3-deazaadenine: analogs of neplanocin A which retain potent antiviral activity but exhibit reduced cytotoxicity. Antimicrob Agents Chemother 1987, 31, 1849–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Armstrong RD; Diasio RB Selective activation of 5’-deoxy-5-fluorouridine by tumor cells as a basis for an improved therapeutic index. Cancer Res 1981, 41, 4891–4. [PubMed] [Google Scholar]
- 209.Armstrong RD; Diasio RB Metabolism and biological activity of 5’-deoxy-5-fluorouridine, a novel fluoropyrimidine. Cancer Res 1980, 40, 3333–8. [PubMed] [Google Scholar]
- 210.Bollag W; Hartmann HR Tumor inhibitory effects of a new fluorouracil derivative: 5’-deoxy-5-fluorouridine. Eur J Cancer 1980, 16, 427–32. [DOI] [PubMed] [Google Scholar]
- 211.Cook AF; Holman MJ; Kramer MJ; Trown PW Fluorinated pyrimidine nucleosides. 3. Synthesis and antitumor activity of a series of 5’-deoxy-5-fluoropyrimidine nucleosides. Journal of Medicinal Chemistry 1979, 22, 1330–1335. [DOI] [PubMed] [Google Scholar]
- 212.Kono A; Hara Y; Sugata S; Karube Y; Matsushima Y; Ishitsuka H Activation of 5’-deoxy-5-fluorouridine by thymidine phosphorylase in human tumors. Chem Pharm Bull (Tokyo) 1983, 31, 175–8. [DOI] [PubMed] [Google Scholar]
- 213.Sommadossi JP; Aubert C; Cano JP; Gouveia J; Ribaud P; Mathe G Kinetics and Metabolism of a New Fluoropyrimidine, 5′-Deoxy-5-fluorouridine, in Humans. Cancer Research 1983, 43, 930–933. [PubMed] [Google Scholar]
- 214.Ishitsuka H; Miwa M; Takemoto K; Fukuoa K; Itoga A; Maruyama HB Role of Uridine Phosphorylase for Antitumor Activity of 5’-deoxy-5-fluorouridine. Gann 1980, 71. [PubMed] [Google Scholar]
- 215.Kramer MJ; Trown PW; Cleeland R; Cook AF; Grunberg E 5’-deoxy-5-fluorouridine-a New orally active antitumor agent. Comparative activity with 5-fluorouracil, 2’-deoxy-5-fluorouridine and ftorafur against transplant-able tumors in mice and rats. Subject Strain Bibliography 1979. 1979, 1519. [Google Scholar]
- 216.Tevaearai HT; Givel JC; Laurent PL; Suardet L; Eliason JF; Odartchenko N Interactions of interferon-α2a with 5′-deoxy-5-fluorouridine in colorectal cancer cells in vitro. European Journal of Cancer 1992, 28, 368–372. [DOI] [PubMed] [Google Scholar]
- 217.Ohta Y; Sueki K; Kitta K; Takemoto K; Ishitsuka H; Yagi Y Comparative studies on the immunosuppressive effect among 5’-deoxy-5-fluorouridine, ftorafur, and 5-fluorouracil. Gann 1980, 71. [PubMed] [Google Scholar]
- 218.Alberto P; Jungi WF; Siegenthaler P; Mermillod B; Obrecht JP; Decoster G; Cavalli F A phase II study of doxifluridine in patients with advanced breast cancer. European Journal of Cancer 1988, 24, 565–566. [DOI] [PubMed] [Google Scholar]
- 219.Abele R; Kaplan E; Grossenbacher R; Schmid HJ; Cavalli F Phase II study of doxifluridine in advanced squamous cell carcinoma of the head and neck. European Journal of Cancer and Clinical Oncology 1984, 20, 333–336. [DOI] [PubMed] [Google Scholar]
- 220.Abele R; Alberto P; Kaplan S; Siegenthaler P; Hofmann V; Ryssel HJ; Hartmann D; Holdener EE; Cavalli F Phase II study of doxifluridine in advanced colorectal adenocarcinoma. J Clin Oncol 1983, 1, 750–4. [DOI] [PubMed] [Google Scholar]
- 221.van Oosterom AT; ten Bokkel Huinink WW; van der Burg ME; Vermorken JB; Willemse PH; Neijt JP Phase II clinical trial of doxifluridine in patients with advanced ovarian cancer. Eur J Cancer 1991, 27, 747–9. [DOI] [PubMed] [Google Scholar]
- 222.Debarge S; Balzarini J; Maguire AR Design and synthesis of α-carboxy phosphononucleosides. J Org Chem 2011, 76, 105–26. [DOI] [PubMed] [Google Scholar]
- 223.Balzarini J; Das K; Bernatchez JA; Martinez SE; Ngure M; Keane S; Ford A; Maguire N; Mullins N; John J; Kim Y; Dehaen W; Vande Voorde J; Liekens S; Naesens L; Götte M; Maguire AR; Arnold E Alpha-carboxy nucleoside phosphonates as universal nucleoside triphosphate mimics. Proc Natl Acad Sci U S A 2015, 112, 3475–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.John J; Kim Y; Bennett N; Das K; Liekens S; Naesens L; Arnold E; Maguire AR; Götte M; Dehaen W; Balzarini J Pronounced Inhibition Shift from HIV Reverse Transcriptase to Herpetic DNA Polymerases by Increasing the Flexibility of α-Carboxy Nucleoside Phosphonates. J Med Chem 2015, 58, 8110–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Keane SJ; Ford A; Mullins ND; Maguire NM; Legigan T; Balzarini J; Maguire AR Design and synthesis of α-carboxy nucleoside phosphonate analogues and evaluation as HIV-1 reverse transcriptase-targeting agents. J Org Chem 2015, 80, 2479–93. [DOI] [PubMed] [Google Scholar]
- 226.Maguire NM; Ford A; Balzarini J; Maguire AR Synthesis of Guanine α-Carboxy Nucleoside Phosphonate (G-α-CNP), a Direct Inhibitor of Multiple Viral DNA Polymerases. J Org Chem 2018, 83, 10510–10517. [DOI] [PubMed] [Google Scholar]
- 227.Stoeckler JD; Cambor C; Parks RE Human erythrocytic purine nucleoside phosphorylase: reaction with sugar-modified nucleoside substrates. Biochem 1980, 19, 102–7. [DOI] [PubMed] [Google Scholar]
- 228.Marquez VE; Lim MI Carbocyclic nucleosides. Med Res Rev 1986, 6, 1–40. [DOI] [PubMed] [Google Scholar]
- 229.Marquez VE a. L. M.-I. Carbocyclic nucleosides. Medicinal Research Reviews 1986, 6, 1–40. [DOI] [PubMed] [Google Scholar]
- 230.Marquez VE Carbocyclic nucleosides. Advances in Antiviral Drug Design 1996, 2, 89–146. [Google Scholar]
- 231.Schinazi RF; Chu CK; Peck A; McMillan A; Mathis R; Cannon D; Jeong LS; Beach JW; Choi WB; Yeola S Activities of the four optical isomers of 2’,3’-dideoxy-3’-thiacytidine (BCH-189) against human immunodeficiency virus type 1 in human lymphocytes. Antimicrob Agents Chemother 1992, 36, 672–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Schinazi RF; McMillan A; Cannon D; Mathis R; Lloyd RM; Peck A; Sommadossi JP; St Clair M; Wilson J; Furman PA Selective inhibition of human immunodeficiency viruses by racemates and enantiomers of cis-5-fluoro-1-[2-(hydroxymethyl)-1,3-oxathiolan-5-yl]cytosine. Antimicrob Agents Chemother 1992, 36, 2423–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Gumina G; Song GY; Chu CK L-Nucleosides as chemotherapeutic agents. FEMS Microbiol Lett 2001, 202, 9–15. [DOI] [PubMed] [Google Scholar]
- 234.Jarvis B; Faulds D Lamivudine. A review of its therapeutic potential in chronic hepatitis B. Drugs 1999, 58, 101–41. [DOI] [PubMed] [Google Scholar]
- 235.Leonard NL; Morrice AG; Sprecker MA Linear benzoadenine. A stretched-out analog of adenine. J Org Chem 1975, 40, 356–63. [DOI] [PubMed] [Google Scholar]
- 236.Leonard NJ; Sprecker MA; Morrice AG Defined dimensional changes in enzyme substrates and cofactors. Synthesis of lin-benzoadenosine and enzymatic evaluation of derivatives of the benzopurines. J Am Chem Soc 1976, 98, 3987–94. [DOI] [PubMed] [Google Scholar]
- 237.Leonard NJ; Scopes DI; VanDerLijn P; Barrio JR Dimensional probes of the enzyme binding sites of adenine nucleotides. Biological effects of widening the adenine ring by 2.4 A. Biochemistry 1978, 17, 3677–85. [DOI] [PubMed] [Google Scholar]
- 238.Leonard NJ Dimensional Probes of Enzyme-Coenzyme Binding-Sites. Accounts of Chemical Research 1982, 15, 128–135. [Google Scholar]
- 239.Seley-Radtke KL; Zhang Z; Wauchope OR; Zimmermann SC; Ivanov A; Korba B Hetero-expanded purine nucleosides. Design, synthesis and preliminary biological activity. Nucleic Acids Symp Ser (Oxf) 2008, 635–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Wauchope OR; Johnson C; Krishnamoorthy P; Andrei G; Snoeck R; Balzarini J; Seley-Radtke KL Synthesis and biological evaluation of a series of thieno-expanded tricyclic purine 2’-deoxy nucleoside analogues. Bioorg Med Chem 2012, 20, 3009–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Chen Z; Jochmans D; Ku T; Paeshuyse J; Neyts J; Seley-Radtke KL Bicyclic and Tricyclic “Expanded” Nucleobase Analogues of Sofosbuvir: New Scaffolds for Hepatitis C Therapies. ACS Infect Dis 2015, 1, 357–66. [DOI] [PubMed] [Google Scholar]
- 242.Chen Z; Ku TC; Seley-Radtke KL Thiophene-expanded guanosine analogues of Gemcitabine. Bioorg Med Chem Lett 2015, 25, 4274–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Zhou L; Amblard F; Zhang H; McBrayer TR; Detorio MA; Whitaker T; Coats SJ; Schinazi RF Synthesis and evaluation of Janus type nucleosides as potential HCV NS5B polymerase inhibitors. Bioorg Med Chem Lett 2013, 23, 3385–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Chung FL; Schram KH; Panzica RP; Earl RA; Wotring LL; Townsend LB Synthesis of certain [6:5:6] linear tricyclic nucleosides as potential antitumor agents. J Med Chem 1980, 23, 1158–66. [DOI] [PubMed] [Google Scholar]
- 245.Burns DD; Teppang KL; Lee RW; Lokensgard ME; Purse BW Fluorescence Turn-On Sensing of DNA Duplex Formation by a Tricyclic Cytidine Analogue. J Am Chem Soc 2017, 139, 1372–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Rodgers BJ; Elsharif NA; Vashisht N; Mingus MM; Mulvahill MA; Stengel G; Kuchta RD; Purse BW Functionalized tricyclic cytosine analogues provide nucleoside fluorophores with improved photophysical properties and a range of solvent sensitivities. Chemistry 2014, 20, 2010–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Stengel G; Urban M; Purse BW; Kuchta RD Incorporation of the fluorescent ribonucleotide analogue tCTP by T7 RNA polymerase. Anal Chem 2010, 82, 1082–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Stengel G; Purse BW; Kuchta RD Effect of transition metal ions on the fluorescence and Taq-catalyzed polymerase chain reaction of tricyclic cytidine analogs. Anal Biochem 2011, 416, 53–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Lin KY; Jones RJ; Matteucci M Tricyclic 2’-Deoxycytidine Analogs: Syntheses and Incorporation into Oligodeoxynucleotides Which Have Enhanced Binding to Complementary RNA. Journal of the American Chemical Society 1995, 117, 3873–3874. [Google Scholar]
- 250.Stengel G; Urban M; Purse BW; Kuchta RD High density labeling of polymerase chain reaction products with the fluorescent base analogue tCo. Anal Chem 2009, 81, 9079–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Flanagan WM; Wolf JJ; Olson P; Grant D; Lin KY; Wagner RW; Matteucci MD A cytosine analog that confers enhanced potency to antisense oligonucleotides. Proc Natl Acad Sci U S A 1999, 96, 3513–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Sandin P; Wilhelmsson LM; Lincoln P; Powers VE; Brown T; Albinsson B Fluorescent properties of DNA base analogue tC upon incorporation into DNA--negligible influence of neighbouring bases on fluorescence quantum yield. Nucleic Acids Res 2005, 33, 5019–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Seley KL; Zhang L; Hagos A “Fleximers”. Design and synthesis of two novel split nucleosides. Org Lett 2001, 3, 3209–10. [DOI] [PubMed] [Google Scholar]
- 254.Seley KL; Zhang L; Hagos A; Quirk S “Fleximers”. Design and synthesis of a new class of novel shape-modified nucleosides(1). J Org Chem 2002, 67, 3365–73. [DOI] [PubMed] [Google Scholar]
- 255.Seley KL; Salim S; Zhang L “Molecular chameleons”. Design and synthesis of C-4-substituted imidazole fleximers. Org Lett 2005, 7, 63–6. [DOI] [PubMed] [Google Scholar]
- 256.Seley KL; Salim S; Zhang L; O’Daniel PI “Molecular chameleons”. Design and synthesis of a second series of flexible nucleosides. J Org Chem 2005, 70, 1612–9. [DOI] [PubMed] [Google Scholar]
- 257.Wauchope OR; Velasquez M; Seley-Radtke K Synthetic Routes to a Series of Proximal and Distal 2’-Deoxy Fleximers. Synthesis (Stuttg) 2012, 44, 3496–3504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Seley KL; Quirk S; Salim S; Zhang L; Hagos A Unexpected inhibition of S-adenosyl-L-homocysteine hydrolase by a guanosine nucleoside. Bioorg Med Chem Lett 2003, 13, 1985–8. [DOI] [PubMed] [Google Scholar]
- 259.Polak M; Seley KL; Plavec J Conformational properties of shape modified nucleosides--fleximers. J Am Chem Soc 2004, 126, 8159–66. [DOI] [PubMed] [Google Scholar]
- 260.Quirk S; Seley KL Substrate discrimination by the human GTP fucose pyrophosphorylase. Biochemistry 2005, 44, 10854–63. [DOI] [PubMed] [Google Scholar]
- 261.Quirk S; Seley KL Identification of catalytic amino acids in the human GTP fucose pyrophosphorylase active site. Biochem 2005, 44, 13172–8. [DOI] [PubMed] [Google Scholar]
- 262.Zimmermann SC; Sadler JM; O’Daniel PI; Kim NT; Seley-Radtke KL “Reverse” carbocyclic fleximers: synthesis of a new class of adenosine deaminase inhibitors. Nucleosides Nucleotides Nucleic Acids 2013, 32, 137–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Zimmermann SC; O’Neill E; Ebiloma GU; Wallace LJ; De Koning HP; Seley-Radtke KL Design and synthesis of a series of truncated neplanocin fleximers. Molecules 2014, 19, 21200–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Yates MK; Raje MR; Chatterjee P; Spiropoulou CF; Bavari S; Flint M; Soloveva V; Seley-Radtke KL Flex-nucleoside analogues - Novel therapeutics against filoviruses. Bioorg Med Chem Lett 2017, 27, 2800–2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Peters HL; Jochmans D; de Wilde AH; Posthuma CC; Snijder EJ; Neyts J; Seley-Radtke KL Design, synthesis and evaluation of a series of acyclic fleximer nucleoside analogues with anti-coronavirus activity. Bioorg Med Chem Lett 2015, 25, 2923–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Yates MK; Vallé C; Martin B; Aoudai W; Selisko B; Decroly E; Canard B; Day C; Falat A; Bassetto M; Brancale A; Seley-Radtke KL Synthesis and biological evaluation of novel flexible nucleoside analogues that inhibit flavivirus replication in vitro. In preparation 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Seley-Radtke KL Flex-Nucleoside Analogues, Novel Therapeutics Against Filoviruses and Flaviviruses. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Seley-Radtke KL Design, Synthesis and Methods of Use of Acyclic Nucleoside Analogues Having Anti-coronavirus Activity. 2018. [Google Scholar]