

Abstract
Ovarian cancer (OvCa) exhibits a specific predilection for metastasis to the omentum. Our earlier studies highlighted the tumor suppressor effect of SPARC (secreted protein acidic and rich in cysteine) in OvCa through multifaceted roles inhibiting cancer cell interactions within the peritoneal milieu. The goal of this study is to investigate the role of SPARC in OvCa interactions with omental adipocytes and its role in OvCa colonization in the omentum. We employed multipronged approach using primary omental adipocytes from Sparc knockout mice, genetically-engineered human omental adipocytes in 3D co-cultures with OvCa cells as well as treatment with recombinant SPARC protein. We show that SPARC suppresses multistep cascade in OvCa omental metastasis. SPARC inhibited in vivo and adipocyte-induced homing, proliferation and invasion of OvCa cells. SPARC suppressed metabolic programming of both adipocytes and OvCa cells and exerted an inhibitory effect of adipocyte differentiation and their phenotypic switch to cancer associated phenotype. Mechanistic studies revealed that this effect is mediated through inhibition of cEBPβ-NFkB-AP-1 transcription machinery. These findings define a novel and functionally important role of SPARC in OvCa and not only bridge the knowledge gap but highlight the need to consider SPARC protein expression in therapeutic development.
Keywords: SPARC, ovarian cancer, adipocytes, omentum, metastases
INTRODUCTION
Epithelial Ovarian cancer (OvCa) is the leading cause of death from gynecologic malignancies in the United States1. More than 75% of patients are diagnosed at late stages with high mortality rates2. The main site of OvCa metastasis within the abdomen is the omentum, a vascular and adipocyte-rich tissue that drapes abdominal organs2. The role of omental adipocytes in promoting OvCa metastasis to the omentum has been recently established as they represent a significant source of factors that substantially promote OvCa-omental tropism, and colonization3–8. Adipocytes adjacent to cancer cells have been named cancer-associated adipocytes (CAA) as they not only promote tumour growth, survival and chemo-resistance9, but they revert from mature, differentiated adipocytes to pre-adipocytes stage releasing their lipids to provide energy to cancer cells3. This phenotypic plasticity is controlled by factors that orchestrate differentiation, inflammation, and metabolic reprogramming in metabolic diseases, obesity and cancer10–15. Strong evidence exists for a link between inflammation and adipocyte plasticity and their phenotypic switch to CAA; however, little is known about the signaling networks involved. Co-regulation and trans-activation of CCAAT/enhancer-binding protein beta (C/EBPβ), NFκB and AP-1 have been reported in the context of obesity, insulin-resistance, and inflammation16–19 as they are involved in the up-regulation of inflammatory mediators20, 21. In addition, the three transcription factors have been reported as crucial factors with transactivation circuitry in, inflammation and cancer22. However, their pivotal role in OvCa-peritoneal dissemination is relatively unexplored.
Secreted Protein Acidic and Rich in Cysteine (SPARC), also termed osteonectin and BM-40, is an extracellular matrix (ECM) protein that exhibits contextual highly regulated expression in remodeling tissues to maintain tissue homeostasis (summarized in23). In this respect, SPARC has been shown to regulate the differentiation of mesenchymal, stem cells including adipocytes24–26. SPARC has been reported to inhibit adipogenesis as evidenced by the phenotype of Sparc null mice exhibiting osteoporosis and fatty bone marrow24–26. We have earlier reported that SPARC is an OvCa suppressor5–8. We reported that SPARC inhibited OvCa cell adhesion to various ECM proteins enriched in the peritoneal tumor microenvironment (TME) and peritoneal mesothelial cells5–7. SPARC exhibited an anti-proliferative effect that was attributed to inhibition of integrin- and growth factor-mediated survival signaling pathways6–8. We also reported that SPARC normalizes the TME through anti-inflammatory properties through suppression of the bi-directional cross-talk between cancer cells and macrophages and mesothelial cells5–8, 27. In addition, we reported that in the immunocompetent Sparc knockout mice (will be referred to as SP−/−), the enhanced peritoneal carcinomatosis was characterized by high levels and biological activity of pro-inflammatory mediators in tumors and ascitic fluid6–8, 27. These pro-inflammatory factors are reciprocated by cancer cells and stromal cells7, 27, 28 and are correlated with advanced human disease, chemo-resistance and poor prognosis28. Given the specific predilection of OvCa cells to the omentum and the reported inhibitory effects of SPARC on adipocyte differentiation25, 29, we sought to investigate the role of SPARC in the bidirectional cross-talk between OvCa cells and omental adipocytes. We present evidence, for the first time, that the tumor suppressor role of SPARC in OvCa is mediated through inhibition of OvCa cells-adipocytes interactions, the phenotypic plasticity of omental adipocytes and metabolic programing.
Results:
SPARC inhibits OvCa cell homing to the omentum in vivo and in vitro.
To determine whether SPARC inhibits ID8 cells preferential homing to omental adipocytes, we injected ID8-GFP cells intraperitoneally (ip) in SP−/− and SP+/+ mice5 and determined adherent ID8 cells harvested omenta (Figure 1A) by measuring A488 fluorescence of GFP-labeled cells. We found that homing of ID8-GFP cells to SP−/− omenta was significantly higher than to the SP+/+ at 60–120 minutes. To determine whether this increased homing was SPARC-dependent, we injected recombinant murine SPARC (rSPARC 5μg/100μl PBS) ip 30 min prior to ID8 injection. We found that SPARC inhibited ID8 homing to the omentum starting at 60 min post ID8-injection and mitigated the increased ID8-GFP adhesion to SP−/− omenta (Figure 1A). To clearly distinguish the role of omental adipocyte-SPARC, independent of other sources of SPARC in the complex peritoneal milieu, we constructed 3D omental adipocyte culture composed of freshly isolated primary SP−/− and SP+/+ omental adipocytes (Supplement Figure 1) embedded in reduced-growth factor matrigel and co-cultured them with ID8-GFP cells as illustrated in Figure 1B. We first determined the effect adipocyte-SPARC on ID8-GFP cell chemotaxis/migration or homing towards SP−/− and SP+/+ omental adipocytes, and found that ID8 homing to SP−/− omental adipocytes was significantly higher than to SP+/+ adipocytes (Figure 1B). We next determined whether difference of homing of ID8 cells to adipocytes was mediated by differences in secreted factors and found that SP−/− omental adipocytes exhibited significant increase in the levels of IL-6, CCL2/MCP1, CCL3/MIP1, VEGF, TNFα, IL-2 and leptin with modest though insignificant increase in levels of CTACK/CCL27, and TIMP1 (Supplement Figure 2A). Neutralizing antibodies of the factors that exhibited significant differences between the 2 genotypes, significantly inhibited migration/homing of ID8 cells towards SP−/− and SP+/+ omental adipocytes (Supplement Figure 2B). Of note that homing of ID8 cells to adipocytes isolated from mice bearing ID8 peritoneal tumors (will be referred to as CAA) was significantly higher than to normal adipocytes (normal Adi) isolated from non-tumor-bearing mice. Homing of ID8 to SP−/− CAA was significantly higher than to SP+/+ CAA (Supplement Figure 2C). Furthermore, CAA exhibited significantly higher levels of the aforementioned inflammatory factors than normal adipocytes with SP−/− CAA exhibiting significantly higher levels than SP+/+ CAA (Supplement Figure 2D). Adhesion of GFP-fluorescent human and murine OvCa cell lines SKOV3, OVCAR3, CAOV3 and ID8 (GFP-SKOV3, GFP-OVCAR3, GFP-CAOV3 and ID8-GFP) to omental adipocytes was inhibited by exogenous recombinant human and murine SPARC (rSPARC, Figure 1C). Furthermore, rSPARC inhibited adipocyte-induced invasiveness human and murine OvCa cells (Figure 1D). In addition, overexpression and depletion of SPARC in human adipocytes (Figure 1E) significantly inhibited/increased invasiveness of OvCa cells compared to their corresponding vector control adipocytes, respectively (Figure 1F). Together these data highlight the paracrine effect of adipocyte-SPARC in inhibiting homing and invasiveness of OvCa cells through secreted inflammatory factors.
Figure 1: Effect of SPARC on homing of ovarian cancer cells to omental adipocytes.
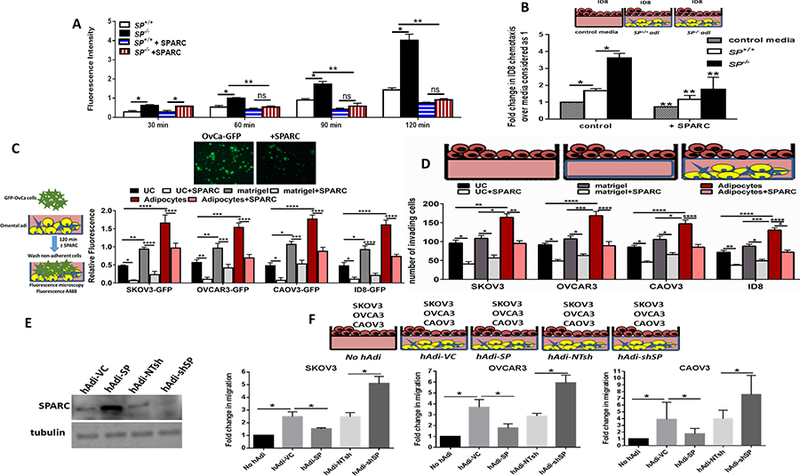
A. In vivo homing of ID8-GFP cells to SP+/+ and SP−/− omenta in the presence or absence of prior injection of 5μg/ml SPARC. Bars represent means ± SEM of fluorescence intensity of adherent cells to omenta harvested at the indicated time points. B. Schema of the in vitro homing/chemotaxis of ID8 cells towards SP+/+ and SP−/− omental adipocytes. Bars represent means ± SEM of fluorescence intensity of ID8 cells that migrated through trans-wells towards adipocytes. Complete growth media were used as controls for migration (n=4). C. Schema of the adhesion assay of GFP-OvCa cells overlayed on top of adipocytes for 120 min (left). Bars represent means ± SEM of fluorescence intensity of adherent cells (n=6/experimental condition). *p<0.05. Photomicrographs of fluorescent adherent cells (top, 5x). D. Schema of adipocyte-induced OvCa invasiveness through trans-well inserts towards omental adipocytes in the bottom chamber. Bars represent the means ± SEM of migrated cells counted in 5 random fields/insert, (n=3). *p<0.05 Student’s t-test. E. Western blots showing the expression of SPARC after overexpression and knockdown in primary human omental adipocytes (hAdi). F. Schema of the in vitro homing assay with GFP-labeled human OvCa cell lines. Bars represent means ± SEM of fold change of OvCa cells that migrated through trans-wells towards genetically engineered hAdi compared to cells migrated to control media (without adipocytes) considered as 1. (n=4/experimental condition. Experiments were repeated 3 times). *p<0.05 Student’s t-test.
SPARC inhibits adipocyte-induced OvCa cell proliferation in vitro and in vivo.
To further investigate the adipocyte-SPARC on OvCa cell proliferation, we incubated ID8-GFP cells in direct contact with SP−/− and SP+/+ omental adipocytes and found that ID8 proliferation was significantly higher (~3 folds) compared to those incubated with the SP+/+ as determined by measuring the GFP fluorescence over 72h. This effect was partially mitigated by treating co-cultures by rSPARC (Figure 2A–B). Similar results were obtained by parallel experiments in which OvCa cells were separated from adipocytes by 0.4μm transwell inserts placed in direct contact with the differentiated adipocytes for 72h. Adipocytes and OvCa cells were collected, trypsinized and counted by trypan blue exclusion at the same time points. Consistent results were observed with OvCa counting and measuring fluorescent intensity of GFP, whereas the number of viable adipocyte did not exhibit significant change during the experiment (data not shown). To further confirm the role of adipocyte-SPARC on ovarian tumour growth in vivo, we injected ID8 cells with SP+/+ and SP−/− omental adipocytes (1:2, cancer cell: adipocyte ratio) subcutaneously in 6-weeks-old female athymic nude mice. We found that ID8 cells injected with SP−/− adipocytes produced ~ 3.5 times larger tumors than those injected with SP+/+ adipocytes (Figure 2C). Similar effect of adipocyte-SPARC was observed with early passage differentiated primary human omental (pre)adipocytes genetically manipulated SPARC expression (by overexpression and depletion, respectively) incubated with SKOV3-GFP, OVCAR3-GFP and CAOV3-GFP (Figure 2D). Furthermore, exogenous rSPARC inhibited adipocyte-induced OvCa cell proliferation (Figure 2E). It is noteworthy that OvCa cell proliferation was significantly higher when plated on adipocyte/matrigel plugs compared to plating on matrigel alone or on uncoated wells; an effect that was inhibited by rSPARC (Figure 2E). These results further support the paracrine effect of adipocyte-SPARC and rSPARC inhibiting adipocyte-induced OvCa cell proliferation, homing, adhesion and invasiveness.
Figure 2. Effect of SPARC on omental adipocyte induced OvCa cell proliferation.

A. Schema of the effect of human and murine omental adipocytes on GFP-OvCa cells proliferation. B. Line graphs representing means ± SEM of changes in the proliferation (GFP fluorescence) of ID8-GFP cells over-layed on top of SP+/+ and SP−/− adipocytes in the presence or absence of 5μg/ml SPARC, over 72h. C. Means ± SEM of changes of tumor volumes after ID8 SC injection either alone or with SP+/+ and SP−/− adipocytes (1:2) in athymic nude mice. *p<0.05, between experimental conditions starting at week 3 post-injection (n=8/group; two-way ANOVA with Tuckey’s multiple comparison test). D. Means ± SEM of changes in the proliferation (GFP fluorescence) of GFP-labeled human OvCa cells on top of genetically engineered human adipocytes over 72h. (n=6/experimental condition). E. Changes in the proliferation (GFP fluorescence) of OvCa cells over-layed on top of adipocytes in presence or absence of 5μg/ml SPARC over 72h. (n=6/experimental condition). *p<0.05 one way ANOVA.
Effect of SPARC on co-culture induced inflammatory chemokines in OvCa cells and adipocytes.
To further determine the effect of adipocyte-SPARC on the expression of cytokines in both tumour cells and adipocytes, we determined the mRNA expression of cytokines and chemokines in ID8 and SP−/− and SP+/+ adipocytes in mono- and co-cultures. We found that the association of ID8 cells with adipocytes significantly up-regulated the expression of IL-6, CCL2, TNFα, VEGF and leptin transcripts in both cell types compared to mono-cultures (Figure 3A–B). The expression of the transcripts of the aforementioned adipokines were significantly higher in SP−/− adipocytes than in SP+/+ (1.9-, 1.8-, 2-, 2.5- and 1.7-fold for IL-6, CCL2, VEGF, TNFα and leptin, respectively). Co-culture with ID8 cells significantly up-regulated the expression of each factor in SP−/− adipocytes over SP+/+ by 2.6–3 folds. Concomitantly, ID8 cells co-cultured with SP−/− adipocytes exhibited ~2–2.7-fold induction of these transcripts, compared to co-culture with SP+/+ adipocytes (Figure 3A–B).
Figure 3: Adipocyte-SPARC suppresses the reciprocal transcriptional activity of pro-inflammatory/adipogenic factors.

A. Schematic illustration of the experimental design and the trans-well assays for the single and co-cultures. B. The levels of adipokines in SP+/+ and SP−/− omental adipocytes and ID8 cells in single and in co-cultures for 24h was determined by qRT-PCR. C. Transcriptional activity of cEBP, NFκB and AP-1 in primary SP+/+ and SP−/− omental adipocytes and ID8 cells in single and in co-cultures was determined by measuring the luciferase reporter activity in each cell type. Results were normalized to fold-change of DNA content of each cell type measured before and after the experiment as determined by CyQuant assay. Bars represent mean ± SEM from one of three experiments, performed in triplicates. *P<0.05 student’s t-test comparing SP+/+ and SP−/− adipocytes; #p<0.05 student’s t-test comparing adipocytes in single to co-cultures with ID8 cells; and **p<0.05 student’s t-test comparing ID8 cells in single to co-culture with SP+/+ and SP−/− adipocytes.
In silico analysis of the common transcriptional regulation of the aforementioned chemokines using (http://opossum.cisreg.ca/cgi-bin/oPOSSUM3/), predicted common transcription machinery regulated by cEBPβ, NFκB and AP-1. These transcription factors (TFs) are not only implicated in adipocyte differentiation but are also recognized as oncogenes and markers of inflammation and aggressiveness of many cancers including OvCa21, 30–34. To monitor the concomitant changes in OvCa cells and adipocytes in co-culture, we co-cultured ID8 cells on 0.4μm inserts on top of 3D SP−/− and SP+/+ omental adipocytes and determined the changes in NFkB, AP-1 and cEBPβ promoter activity in both cell types using luciferase-reporters35, 36. Co-culture of SP−/− and SP+/+ adipocytes with ID8 cells elicited profound significant increase in the activity of these TFs over single cell culture, with SP−/− adipocytes exhibiting significantly higher levels than the SP+/+. ID8 co-cultured with SP−/− adipocytes exhibited significantly higher luciferase reporter activation than those co-cultured with SP+/+ adipocytes (Figure 3C). Consistently, human OvCa cell lines co-cultured with omental adipocytes depleted of SPARC (Supplement Figure 3A) exhibited significant increase in the activation of the 3 TFs in co-cultures compared to those in mono-cultures. Conversely, overexpression of SPARC in adipocytes, inhibited the activation of the 3 transcription factors in both OvCa cells and adipocytes (Supplement Figure 3B). Of note is that genetic manipulation of SPARC in primary human adipocytes (hAdi) by overexpression and knockdown significantly inhibited/increased the luciferase activity of NFκB and AP-1 but not cEBP prompter reporters in mono-cultures, respectively. Importantly, treating OvCa cells and adipocytes in single and co-culture with rSPARC phenocopied the effect of SPARC overexpression in adipocytes and significantly inhibited the promoter activity of the 3 TFs in OvCa cells and adipocytes (Supplement Figure 3B). It is noteworthy that the significantly reciprocated increase in promoter activity of the three TFs reported above is due to increase in activation and not due to increased cell proliferation as we determined changes cell proliferation of in mono- and co-cultures before (0h) and after 24h (end of the assay), and normalized the luciferase reporter activity in each cell type to the fold change in cell proliferation during the experiment. Differentiated human and murine adipocytes did not exhibit changes in proliferation as determined by CyQuant assay (data not shown). However, monoculture of ID8 cells exhibited 1.5-fold increase in proliferation, whereas, in co-culture with SP+/+ and SP−/− adipocytes, ID8 exhibited 2- and 3.8-fold change in proliferation (Supplement Figure 4A). Similarly, mono-cultures of SKOV3, OVCAR3 and CAOV3 exhibited 1.3-, 1.2- and 1.7-fold change in proliferation in 24h, respectively; whereas, in co-cultures with human adipocytes depleted, the 3 cell lines exhibited ~2–2.5-fold increase proliferation (Supplement Figure 4B–C). OvCa cells in co-cultures, with adipocytes overexpressing SPARC exhibited 1.3–1.4-fold increase in their numbers after 24h; whereas co-cultures with adipocytes depleted of SPARC exhibited 3–4-fold increase in cell proliferation (Supplement Figure 4B–C).
Consistently, treatment of OvCa cells and primary omental adipocytes in mono-and co-cultures with rSPARC downregulated the transcript levels of IL-6, CCL2, VEGFA, TNFa and LEP, all are downstream target genes of the 3 transcription factors in both OvCa cells and hAdi (Supplement Figure 5A–B).
Effect of SPARC on the activation of NFkB, AP-1, and cEBPβ in OvCa cells and adipocytes in vivo.
To determine the effect of SPARC on the three TFs in OvCa-adipocytes interactions in vivo, we first determined the levels of total and phosphorylated proteins in lysates of syngeneic ID8 omental tumours that developed after ip injection in SP−/− and SP+/+ mice. We found marked increase in phosphorylation of cJun (S73), p65RelA (S276) as well as cEBPβ (T235) in SP−/− tumor lysates compared to SP+/+ tumors (Figure 4A). Immunostaining of ID8 omental tumors revealed significant increase in frequency of nuclear p65RelA, cJun and cEBPβ in both tumor cells and the juxtra-tumoral adipocytes in SP−/− tumours compared to the SP+/+ tumours (Figure 4B–C). Importantly, immunostaining of human OvCa specimens from OvCa tissue microarray (TMA, Figure 5A–B) revealed significant increase in the expression of nuclear transcription factors in advanced stage tumours,T3 and T4 (referred to thereafter as T3+), compared to early stage tumours, T1 and T2 (referred to thereafter as T1+T2, Figure 5A–B). Importantly, we found significant negative correlation between tumour SPARC with nuclear expression of the p65RelA, cJun and cEBPβ in advanced stage (T3+) OvCa specimens (Figure 5C) in tissue microarrays where only tumour cores are present. In an independent cohort of stage T3+ human OvCa specimens with adjacent omental tissue, we found significant decrease of SPARC expression in the cancerous compartment with distinctive expression in the stroma (Figure 5D). In this cohort, SPARC expression in tumour cells negatively correlated with the nuclear expression of the 3 transcription factors. In addition, SPARC expression in the juxtra-tumoral adipocytes negatively correlated with nuclear expression of the transcription factors in the adipocytes (Figure 5D–E). These data further confirm the negative correlation between SPARC protein expression and the activation of these TFs in tumour cells and adipocytes. It is noteworthy that in the human tissues, adipocytes exhibit detectable expression levels of SPARC protein with increasing intensity of expression as the juxta-tumoral adipocyte become smaller in size (Supplement Figure 6). Together with our earlier reports that SPARC is required for stromal cell differentiation and acquisition of cancer-associated phenotype36, we sought to determine the effect of SPARC on adipocyte differentiation and acquisition of CAA phenotype.
Figure 4: SPARC suppresses pro-inflammatory/adipogenic factors in ID8 intra-peritoneal tumors.

A. Immunoblots of total and phosphorylated cJun, p65NFκB, in lysates from ID8 intra-peritoneal tumors growing in SP−/− and SP+/+ mice. Tubulin was used as a loading control. B. Immunostaining of cEBPβ, NFκB and cJun in tumors dissected from SP+/+ and SP−/− (magnification, 200x). C. Box plots of the expression scores of the nuclear transcription factors in tumor cells and adipocytes of both genotypes. *p<0.05, Mann-Whitney test.
Figure 5: Correlation of tumor SPARC and cEBPβ, NFkB and AP1.

A. photomicrograph of stage III HGSC specimens (CHTN) showing distinctive compartmentalization of SPARC in the cancerous vs stromal compartments and the expression of cEBPβ, NFkB and cJun. B. Scatter plots of the expression scores of SPARC, cEBPβ, NFkB and cJun. P<0.05, Mann-Whitney test. C. Spearman’s correlation of the expression scores of tumor-SPARC and nuclear transcription factors. D. Immunofluorescence staining of the expression of omental metastases of HGSC specimens, showing the expression of cEBPβ, RelA, cJun and SPARC in the tumor (T)-adipocyte (A) interface. E. Spearman’s correlation of the nuclear expression of tumor and adipocyte cEBPβ, RelA, and cJun with tumor and adipocyte SPARC. Scale bars 100μm.
Effects of SPARC on reciprocal crosstalk between adipocytes and OvCa cells.
Earlier reports demonstrated that tumor-adjacent adipocytes undergo phenotypic changes into CAAs to support cancer cells growth and survival. In addition to expression of pro-inflammatory cytokines and transcription factors37, CAA release their lipids through lipolysis3, 4. Using the heterotypic cultures of ID8 and adipocytes, we found increased FFA production in CM of co-cultures compared to single cell cultures with higher FFA of in co-cultures including SP−/− adipocytes (Figure 6A). This effect was inhibited by treating co-cultures of ID8 cells with SP+/+ and SP−/− adipocytes with inhibitors of NFkB, AP-1 and cEBPβ (Supplement Figure 7) inhibited co-culture-induced FA release from adipocytes with modest effect on the adipocytes in monoculture (Supplement Figure 8). Consistently, SP−/− adipocytes expressed higher levels of adipose triglyceride lipase (ATGL) and total and phospho-hormone-sensitive lipase (HSL), the rate-limiting enzymes in the breakdown lipids and mobilization of FFA from adipocytes38, compared to SP+/+ adipocytes (Figure 6B). ID8 cells exposed to the same co-culture conditions with adipocytes exhibited higher levels of intracellular FA and larger lipid droplets in co-cultures with SP−/− compared to SP+/+ adipocytes as determined by bodipy staining and electron microscopy (Figure 6C–E). To further confirm whether these observations were due to a direct effect of SPARC, we treated human OvCa cells SKOV3 and OVCAR3 with increasing concentrations of rSPARC and found that SPARC exhibited concentration-dependent inhibition of FA uptake in both cell lines (Figure 6F). Moreover, treating SKOV3 and OVCAR3 with rSPARC, significantly decreased the expression of FA transporters FABP4 and CD36 as revealed by immunofluorescence staining and quantification of the fluorescence intensity (Figure 6G–H). To determine whether the increased FA uptake in ID8 cells was associated with changes in FA metabolism, we found that the transcript levels of enzymes involved in β-oxidation of FA as carnitine palmitoyltransferase I (Cpt1a, b), acetyl-CoA acetyltransferase 2 (Acat2) a cholesterol acyltransferase, hydroxyacyl-CoA dehydrogenase (Hadh), medium and short chain FA dehydrogenase, carnitine O-octanoyl transferase (Crot), acetyl-CoA acyl-transferase 1 (Acaa1), ketoacyl-CoA thiolase 2 (Acaa2), and solute carrier family 25 (carnitine/acylcarnitine translocase member 20, Slc25a20) are increased in ID8 cells co-cultured with SP−/− adipocytes compared to either ID8 cultured alone or with SP+/+ adipocytes (Figure 6I).
Figure 6: Effect of SPARC on metabolic programing of adipocytes and OvCa cells.

A. Schema of the co-culture (upper), Bars represent means ±SEM of FFA release in media of co-cultures for 48h. *p<0.05, Student’s t-test. B. WB showing increased lipases in adipocytes in mono- and co-cultures with ID8 cells. C. Schema showing co-cultures of ID8 and adipocytes. D. Photomicrographs of FA uptake by ID8 cells visualized by bodipy staining of in co-cultures (upper, 40x; scale bar 50μm) and electron microscopy (lower, 6800x; scale bar, 500nm). E. Quantification of bodipy fluorescence intensity in the in ID8 cells in mono- and co-cultures with adipocytes. The number and size of lipid droplets (lower) was quantified in 10 cells/condition. Plots represent means±SEM of the fluorescent intensity quantified in 10 fields/experimental condition (n=3 experiments), and unpaired t-test. F. Effect of SPARC on FA uptake by human OvCa cells. p<0.05, one way ANOVA. G. Immunofluorescence showing the effect of SPARC (5μg/ml) on FABP4 and CD36 expression in SKOV3 and OVCAR3 cells. Scale bars, 10μm. H. Bars report means ± SEM of fluorescence intensity of FABP4 and CD36 quantified in 10 fields/experimental condition, Student’s t-test. I. mRNA expression of enzymes involved in FA oxidation of ID8 in mono- and co-culture with SP+/+ and SP−/− adipocytes. Bars report means ±SEM of a representative of 3 independent experiments each in triplicates. *P<0.05. Student’s t-test. J. Bars depict mean ± SEM of the changes in the transcript levels (n=3/genotype). P values are determined by unpaired Student’s t-test.
Effect of loss of stromal-SPARC on lipid metabolism in ID8 omental nodules.
The enhanced growth and metabolic adaptation of OvCa that grow in adipose-rich peritoneal TME have been attributed to unconventional metabolism characterized by increased rates of the oxidation (β-oxidation) of FA released from adipocytes’ lipolysis3. FA and glycerol released from adipocytes are taken up by tumour cells, where FAs are used for β-oxidation, whereas glycerol may either be converted to glucose through gluconeogenesis or directly feeds into the glycolytic pathway providing energy for cellular metabolism3, 39. Therefore, we determined the effect of host-SPARC on the metabolic adaptation of ID8 omental tumour nodules developing in SP−/− and SP+/+ mice. We found significant upregulation in the above mentioned enzymes involved in β-oxidation with exception of Cpt1 in ID8 tumors growing in SP−/− mice compared to those in the SP+/+ (Figure 6J). Concomitantly, tumors growing in SP−/− mice exhibited low levels of acyl carnitine conjugated lipids as palmitoyl carnitine and stearoyl carnitine suggesting reduced substrate availability due to enhanced lipid oxidation by the aforementioned enzymes. Consistently, significantly increased levels of 3-dehydrocarnitine, an intermediate of carnitine degradation were observed in in SP−/− tumors consistent with a tumor metabolic phenotype. In addition, multiple monoacyl glycerols including 1-oleoylglycerol accumulated in SP−/− tissues indicating a change in complex lipid hydrolysis evidenced by elevated levels of glycerol (Supplement Figure 9). These data suggest a novel significant role of adipocyte SPARC on metabolic reprograming of OvCa cells in the unique peritoneal milieu.
Inhibitory Effect of SPARC on adipogenic differentiation.
The above described data prompted us to investigate the effect of SPARC on omental adipocytes’ differentiation and plasticity especially that the anti-adipogenic function of SPARC was earlier reported25, 29, 40, 41. Omental tissues isolated from age-matched SP−/− and SP+/+ female mice, did not exhibit discernible macroscopic or microscopic differences. Omental adipocytes were isolated from the buoyant layer after digestion and centrifugation of omental tissues (Supplement Figure 1A). SP−/− adipocytes exhibited increased lipid droplets compared to SP+/+ (Supplement Figure 1A). Pelleted stromal cells were composed mainly of adipocytes, pre-adipocytes and vascular smooth muscle cells29 and exhibited fibrobalstoid phenotype in standard culture as stained with α-smooth muscle actin (Supplement Figure 1C). When confluent monolayers of SP+/+ and SP−/− omental stromal cells were exposed to adipogenic growth medium (ADM) for 7days D7, SP−/− exhibited faster adipogenic differentiation, with increased accumulation of intracellular lipids as determined by oil O red (ORO, Figure 7A), and electron microscopy with significantly larger numbers and size of lipid droplets (Figure 7B–C). Fully differentiated SP−/− adipocytes exhibited significantly higher levels of adipogenic transcription factors PPARγ, and c/EBPβ than SP+/+ adipocytes (Figure 7D). SPARC protein exhibited differential expression in differentiating SP+/+ adipocytes being highest after 3 days (D3), in adipogenic medium and declined to basal levels in fully differentiated adipocytes at D7 (Figure 7E). Exogenous SPARC suppressed the expression of adipogenic factors when added to differentiating SP−/− adipocytes (Figure 7F). Independently, treating 3T3-L1 pre-adipocytes with rSPARC (5μg/ml) for 10 days significantly inhibited the accumulation of intracellular lipids as determined by oil red O (Figure 7G) and bodipy fluorescent staining (Figure 7H). Consistently, SPARC inhibited the expression of adipogenic transcription factors PPARγ, cEBPα and cEBPβ as well as markers of adipogentic differentiation HSL, ATGL, and FABP4 (Figure 7I). Interestingly, consistent with our observation with SP+/+ adipocytes, SPARC protein expression increased in differentiating 3T3L adipocytes by D3 then decreased to basal levels by D7 and D10 (Figure 7J). In addition, treatment with rSPARC decreased the mRNA levels of the master regulator of differentiation Pparg and Cebp isoforms as well as markers of early and late differentiation as Fasn, Adipoq, lep, Cd36, Acly, Scd1, Acaca, Pnpla2/Atgl1, Lipe/Hsl, and Screbp-1c (Supplement Figure 10). These data further support the effect of SPARC inhibiting omental adipocyte differentiation and in accord with earlier reports of the anti-adipogenic effect of SPARC on different adipocyte and mesenchymal stem cell niches. Importantly, when these adipocytes are challenged by OvCa cells, SPARC suppresses their interaction with cancer cells and consequently inhibits the acquisition of CAA phenotype that fosters OvCa omental dissemination and colonization.
Figure 7: SPARC exerts anti-adipogenic effect on omental adipocytes and 3T3L1 cells.

A. Oil O Red staining of differentiating SP+/+ and SP−/− pre-adipocytes (10x magnification). B. SP−/− adipocytes exhibit increased size and number of fat droplets day10 (D10) post-differentiation as shown by Oil Red O (20x magnification) and electron microscopy (6800x). C. Plots indicate mean ± SEM of the number (upper) and size (lower) of lipid droplets quantified in EM images (10 cells/experimental condition). D. Western blots showing the kinetics of expression of adipogenic transcription factors during differentiation of SP+/+ and SP−/− pre-adipocytes. E. The expression of SPARC protein during differentiation of SP+/+ adipocytes. F. Effect of SPARC (5μg/ml in PBS-0.4%BSA) on the expression of the adipogenic transcription factors during differentiation of SP+/+ adipocytes. Tubulin was used as loading control. G. Confluent 3T3-L1 pre-adipocytes (D0) were differentiated either in the presence or absence of 5μg/ml rSPARC up to day 10. The cells were harvested at day 0, 3, 7, and 10. Intracellular lipids were stained with Oil Red O. Bars (right) represent means± SEM of the quantification of Oil Red O stained cells solubilized with 100% isopropanol for 5 min and measuring the absorbance A492 nm. *p<0.05, two-way ANOVA with Sidak post-hoc test. G. Images of bodipy fluorescent staining of intracellular lipids in differentiating 3T3-L1 pre-adipocytes in the presence or absence of 5μg/ml rSPARC. H. Western blots showing the expression of SPARC and the adipogenic differentiation markers in differentiating 3T3-L1 cells in the presence or absence of SPARC.
Discussion
In the present study, we expand our knowledge on the biological roles of SPARC in OvCa by investigating its role in regulating the interactions between OvCa cells and omental adipocytes. Our findings in the present study indicate that SPARC exerts a tumour suppressor effect on OvCa cells in part through inhibiting their interactions with omental adipocytes; the main site of metastasis of OvCa3, 42, 43, and the most common cause of mortality in OvCa patients43.
We used multipronged approach employing human and murine OvCa cell lines as well as primary human and murine omental adipocytes in single and co-culture. We confirmed the inhibitory effect of SPARC by using primary adipocytes from SP−/− and SP+/+ mice in single- and co-cultures with murine ID8 cell line, a model system that is extensively used in syngeneic models of OvCa in normal and transgenic mice C57B6 background and is well-characterized as has been reported in ~145 publications. However, ID8 cell line may not be considered a faithful representation of HGSC because it was originally derived by spontaneous transformation of high passage murine ovarian surface epithelium44. To circumvent this limitation of the mouse model, and to ascertain that our findings are not limited to the phenotype of one cell line or model system, we complemented our mechanistic and phenotypic studies with 3 human ovarian cancer cell lines in mono- and co-cultures with human omental (pre)adipocytes as well as human tumor specimens.as well as genetic manipulation of SPARC in human primary omental (pre)adipocytes and human OvCa cells. We further demonstrated the role of SPARC in omental adipocyte differentiation as SP−/− adipocytes exhibited accelerated adipogenic differentiation and significantly accumulated more lipids than SP+/+ adipocytes. This finding is consistent with earlier report of increased tendency of SP−/− bone marrow cells for adipogenic differentiation25, 29, 40.
Indeed, the use of the 3D co-cultures of OvCa cells and adipocytes provided a useful tool for mechanistic and functional studies of tumour cells and adipocytes with monitoring concomitant changes in both cell types in response to genetic manipulations and pharmacologic inhibitors. We confirmed the specificity of the inhibitory effect of adipocyte-SPARC on the functional cross-talk between OvCa cells and adipocytes independent of contribution of other cells in the TME and the complex host background. This approach first confirmed the direct autocrine and paracrine effects of SPARC on tumor cell-adipocyte interactions. Adipocyte-SPARC exerted a suppressor effect at multiple levels. First, adipocyte-SPARC decreased homing, adhesion, proliferation of OvCa cells to omental adipocytes and inhibited their invasiveness. We showed that these effects were due to the effects of SPARC mitigating inflammatory secretome as evidenced by functional blocking antibodies. These are in accord with our earlier reports of the anti-inflammatory role of SPARC in OvCa6–8. Second, adipocyte-SPARC as well as rSPARC suppressed the phenotypic switch of omental adipocytes and their acquisition of CAA inflammatory phenotype through suppression of OvCa cell-induced activation of cEBPβ, NFκB, and AP-1 transcription factors and their downstream inflammatory and metabolic effects. Pharmacologic inhibitors of these transcription factors not only phenocopied the effect of rSPARC on FFA release from adipocytes in mono-and co-cultures with OvCa cells, but they also mitigated the increase FFA release from SP−/− adipocytes in co-cultures with ID8 cells. Inflammatory chemokines as TNFα, IL-2 and IL-6, were reported to increase lipolysis and negatively affect metabolic homeostasis15, 45–48. Moreover, the reciprocated transactivation between cEBPβ, NFkB and AP-1 in myriad physiological and pathological contexts including lipid homeostasis, differentiation, inflammation and cancer has been established and involves the transcription of multiple common factors including the enzymes, and chemokines studied herein. Our data using pharmacologic inhibitors of NFkB further supported the transactivation loop between NFkB and cEBPβ that has been earlier reported and confirmed16, 21. The association of these transcription factors with aggressiveness of many cancers including OvCa, have been reported28, 49–51. Our data using patients’ specimens indicated the progressive increase and nuclear localization of these transcription factors as a function of OvCa progression. Importantly, their expression inversely correlated with the expression of SPARC protein in the cancerous compartment as well as in the juxtatumoral adipocytes in 2 independent patients’ cohorts. Survival data curated from The Cancer Genome Atlas (TCGA) revealed that the expression of cEBPβ, cJun, cFos and NFkB1 were associated with poor patients’ survival (Supplement Figure 11). SPARC protein expression in the OvCa cells negatively correlates with nuclear localization of these transcription factors in patients’ specimens and in syngeneic tumours Sparc-deficient mice. In tumour specimens with adjacent omental tissues, we show that the negative correlation between both tumour and adipocyte SPARC with the nuclear localization of these factors in tumour cells and juxta-tumoral adipocytes. SPARC transcript was not correlated with patients’ survival, most probably due to the distinctive compartmentalization of SPARC protein expression in patients’ tumors. In addition, our unpublished data indicated that genetically manipulating SPARC in SPARC-proficient CAOV3 cell line by overexpression or knockdown inversely correlated with malignant phenotype in vitro and peritoneal dissemination and mouse survival when they were injected intraperitoneally in nude mice. Furthermore, we provide evidence that SPARC exerts a tumour suppressor effect in OvCa linking inflammation to metabolic programming, a process termed “metaflammation” that has been implicated in diseases associated with inflammation and perturbed bioenergetics as diabetes, obesity and aging52. Together with our recent report that SPARC inhibits metabolic plasticity and mitochondrial bioenergetics in OvCa53, our data underscore the multi-faceted suppressor role of SPARC in limiting peritoneal dissemination of OvCa.
Using OvCa cell-adipocytes co-cultures, we found that SPARC inhibited the increased lipolysis and FFA release from adipocytes concomitant with inhibition of increased FA uptake by OvCa cells. We show that the effect of SPARC is due to differential effects on adipocytes and OvCa cells; not only inhibiting inflammation-induced lipolysis and FFA release in adipocytes, but also downregulating the surface expression of FA transporters CD36 and FABP4, both have been associated with poor prognosis in patients with OvCa54–56. FABP4 has been recently reported as hypoxia-regulated gene56, and its expression in tumor endothelial cells is associated with increased angiogenesis, especially in low grade stroma-rich tumours57. In addition, FABP4 has been reported as predictor of residual disease in HGSC55 and its overexpression in patients’ tumours is associated with increased metastatic burden and poor survival56.
As a consequence of increase FFA uptake, OvCa cells have been shown to rely on FA oxidation for energy production3. Recent reports58 demonstrated the link between FA oxidation and cancer cell proliferation and survival through activation of salt-inducible kinase–2 (SIK2) in tumor cells co-cultured with omental adipocytes. Activated SIK2 phosphorylated acetyl CoA carboxylase (ACC) and PI3K and, thus, simultaneously regulates both fatty acid oxidation and cancer cell proliferation and survival. Consistently, our recent report53, showed that SPARC inhibits mitochondrial bioenergetics and ATP, glycolysis and ATP production, “metabolic plasticity” through inhibition of activation of adenosine monophosphate kinase (AMPK), ACC as well as mammalian target of rapamycin (mTOR) and its downstream targets; thus linking metabolic programming and mitochondrial dysfunction to cancer cell proliferation and survival. Together with our results herein, it is plausible that the effect of SPARC on cancer cell-adipocyte metabolic programing through an effect on SIK2, or through an effect on enzymatic processes that occur in the mitochondria including FA oxidation, leading to significant reactive oxygen species (ROS) generation and activation of the transcriptional inflammatory oncogenic machinery orchestrated by NFkB, cJun and cEBPβ.
In summary, this is the first investigation of the role of adipocyte-SPARC in regulation of omental adipocytes and its impact on OvCa progression and omental metastasis. Our study provides novel information on the tumour suppressor role of the SPARC in the regulation of OvCa-omental crosstalk and highlights the role of overexpression as well as exogenous recombinant SPARC not only on mitigating the effect of loss of SPARC in adipocytes but on suppressing pro-tumorigenic and metabolic programing, thus making the omental adipocyte niche unfavorable for seeding and colonization of tumor cell. We show the effect of SPARC on activation of the main transcription factors orchestrating inflammation, adipocyte dysfunction and cancer progression. Our data highlights the importance of SPARC protein as a promising therapeutic target in OvCa.
Material and Methods:
Cell culture, plasmid transfections and viral transductions.
Murine (ID8) were earlier described5, 7. Human ovarian cancer (SKOV3, OVCAR3, CAOV3) cell lines were originally from ATCC and were maintained at low passage and were maintained in complete growth media as earlier described5, 7. Cells were tested for Mycoplasma once/month at Wake Forest Baptist Cell and Viral Vector Core Lab (CVVL). Primary human omental pre-adipocytes were obtained from Zen-Bio, Inc. (Raleigh, NC, USA) and were maintained in omental pre-adipocytes medium and differentiated using adipocyte differentiation medium. Transient and stable overexpression of SPARC was done using adenoviral and retroviral vectors as previously described7, 36. Transduction efficiency was optimized to overcome negative selection due to the anti-proliferative effect of SPARC. SPARC depletion was done by transduction using lentiviral vectors with short-hairpin RNA targeting SPARC (shSPARC) with control shRNA targeting non-human target36. All were purchased from Sigma, TRC (TRCN000008710, TRCN000008709 and TRCN000008711). Culture media, supplements, antibiotics and growth factor-reduced matrigel were from Invitrogen (Grand Island, NY) and BD Biosciences (Franklin Lakes, NJ). Recombinant human and murine SPARC were purchased from Peprotech (Rocky Hill, NJ catalog# 120–36) and R&D Systems Inc. (Minneapolis, MN, catalog# 942-SP). Stock solutions of rSPARC from the same lot numbers were reconstituted in DPBS-0.1% BSA (1mg/ml), aliquoted and stored at −08°C till used. The purity of SPARC was validated by SDS-PAGE under reducing and non-reducing conditions, and Commassie blue staining showing the abundance of the protein at the appropriate molecular weight along with the BSA (Supplement Figure 1). Unless otherwise stated, all reagents were purchased from Sigma Aldrich (St. Louis, MO) and ThermoFisher (Pittsburgh, PA).
In vivo syngeneic model. SP+/+ and SP−/− mice are maintained on C57BL/6 background for at least ten backcrosses. Mice were housed in a specific pathogen-free (SPF) facility. All animal experiments were approved by IACUC of the University of Virginia (IACUC# 3879) and Wake Forest University Schools of Medicine (IACUC# A16–165). ID8 (4×106/100μl sterile PBS) intraperitoneal (ip) injections in SP+/+ and SP−/− mice was previously described5, 7. Dissected tumor tissues were either snap frozen in liquid nitrogen then stored at −80°C till used or fixed in 10% neutral zinc formalin and embedded in paraffin. For in vivo homing experiments, ID8-GFP cells (4×106) were injected ip in 5-weeks old female SP+/+ and SP−/− mice5. Mice were euthanized at the time points indicated in figure legends, omenta were dissected, placed in wells of 6-well plates, and ID8-GFP cell adhesion was determined using a fluorescent inverted microscope as earlier described36. GFP-fluorescence intensity was quantified in 5 images/mouse omentum using Image J software. Background fluorescence was normalized to the fluorescence of images of omenta isolated from sham (PBS)-injected mice. For functional blocking experiments, mouse IL-6 (MAB406; 1μg/ml), mouse TNFα (MAB4101, 1μg/ml), CCL2/MCP-1 (AB-479-NA; 30 μg/ml), mouse anti-leptin (AF 498, 0.3μg/ml), mouse CCL3/MIP1 (AF450, 0.3μg/ml), mouse IL2 (MAB702, 1μg/ml), and VEGF (AF-493-NA; 1μg/ml), were injected ip in SP+/+ and SP−/− mice 30 min before ID8 injection. Normal isotype controls included rat anti-mouse IgG1 (MAB005), goat anti-mouse IgG (AB-108-C) injected in the same dose. All neutralizing antibodies were from R&D Systems. In some experiments mice, athymic nude mice (Jackson Labs, Bar Harbor, ME) were injected subcutaneously (SC) with murine ID8 cells mixed with SP+/+ or SP−/− adipocytes in a ratio of 1:2 (ID8: adipocytes). SC tumors were measured twice weekly by caliper for 6 weeks5, 7, 36.
Isolation of primary murine omental adipocytes.
Omenta were isolated from euthanized 6–8-week-old female mice and were digested by collagenase/dispase for 2h at 37°C with gentle agitation25. Slurries were allowed to separate into floating mature adipocytes (top layer) (Supplement Figure 1) and pellets mainly consisting of fibroblasts, smooth muscle cells and pre-adipocytes25, 41. Mature adipocytes were immediately used for the assays involving murine cells. Adipocytes were maintained in adipocytes maintenance medium (ADMM, Zen Bio, Raleigh, NC) supplemented with 10% fetal bovine serum (FBS), antibiotic-antimycotic solution. Omental stromal cells were maintained in DMEM/F12 with 10%FBS and antibiotic-antimycotic solution. Adipogenic differentiation was initiated in confluent fibroblastoid omental stromal cells, by adipogenic medium (ADM, Zen Bio) for 3days, after which they were switched to ADMM for further 10–14 days. The differentiation of adipocytes was compared by microscopic examination and stained with Oil Red O staining and bodipy fluorescent staining (Molecular Probes, ThermoFisher)3.
3D omental cultures and in vitro proliferation, adhesion and invasion assays.
The 3D omental culture was assembled by plating 5×105 fresh omental adipocytes in 500 μl of reduced growth factor–matrigel mixed with growth media (1:3) into a 24-well culture plate. For proliferation assays, ID8-GFP cells (5000 cells/100μl SFM-2% FBS) were added on top of adipocytes embedded in matrigel. Green fluorescence at A488 was determined daily after plating for 72h. As controls, parallel experiments were performed using ID8-GFP cells grown under similar conditions in absence of adipocytes and/or matrigel. The number of adipocytes was determined by parallel experiments counting the number of adipocytes after sorting out GFP-labeled cancer cells before, during and at the end of corresponding experiments. For proliferation experiments (~72h), parallel experiments were performed as follows: for SP−/− and SP+/+ adipocytes and ID8-GFP cells, another parallel experiment in which adipocytes in matrigel were separated from ID8-GFP cells by 0.4 μm transwell inserts (ID8 cells added in the top chambers and the inserts were touching adipocytes. ID8 proliferation was determined by measuring A488 fluorescence and cell counting by trypan blue exclusion. Adipocytes in the bottom chambers were trypsinized and counted. Similarly, human omental pre-adipocytes embedded in matrigel were allowed to differentiate and OvCa-GFP expressing cells were added on the top chamber of trans-well inserts (0.4μm) directly placed on the adipocytes plugs, only separated by the membranes. At the time points indicated in the figures and figure legends, OvCa cells were collected and their proliferation were determined by cell counting and trypan-blue exclusion as well as measuring the GFP fluorescence. Adipocytes in the bottom were trypsinized and counted by trypan blue exclusion. We did not find changes in the adipocytes numbers between the experimental conditions up to 72h.
For in vitro homing experiments, ID8-GFP cells (1×105) were added in the top chamber of 8μm-pore trans-well inserts and were allowed to migrate towards SP−/− and SP+/+ primary adipocytes or SP−/− and SP+/+ adipocytes in the bottom chambers for 6h at 37°C. For invasion assays, 1×105 ovarian cancer cells/100μl SFM were added on top of matrigel-coated 8μm-pore trans-well inserts, and incubated at 37°C for 6hr with SPARC-deficient and SPARC-proficient omental adipocytes or their conditioned media (CM) in the bottom chambers of the trans-wells3, 59, 60.
Antibodies, Reagents and Western Blots.
Monoclonal and polyclonal antibodies against total and phosphorylated cEBPβ, PPAR-γ, hormone-sensitive lipase (HSL), p-HSL (ser660), cJun, p65NFκB, adipose triglyceride lipase (ATGL), human and murine SPARC, α-smooth muscle actin (α-sma) and β-tubulin antibodies, were obtained from Cell Signaling Technology (Danvers, MA, USA), Abcam (Cambridge, MA), and Santa Cruz (Santa Cruz, CA). Cells were lysed and protein concentration were determined as earlier described7, 36. Cellular proteins (20μg) were resolved by 4–20% SDS-PAGE, transferred to PVDF membranes (Bio-Rad, Hercules, CA, USA), and probed with primary and the appropriate fluorescent- and HRP-conjugated secondary antibodies (Licor, NE and Abcam). Blots were visualized using Odyssey 3v and Amersham Imagers.
Luciferase reporter assays.
NFκB and AP-1 luciferase reporters were described previously35, 36. The cEBPβ responsive luciferase vector (pGL2–5xcEBPβ-TK-Luc) and matching empty vector control21 were kindly provided by Dr. Xianjun Fang at Medical College of Virginia, Richmond, VA. Differential luciferase reporter assays in co-cultures of OvCa cells and adipocytes were performed as earlier described35, 36. For investigation of the transcription factor reporter activity, ID8 and omental adipocytes were transfected with the luciferase-reporter plasmids using Fugene 6 (Promega, Madison WI) 24h prior to co-culture. As an internal control for transfection efficiency, cells were co-transfected with TK-Reinella-luciferase plasmid (at a ratio of 20:1 firefly:TK-Reinella). Cells were co-cultured for 24h and the luciferase activity was measured using Dual Luciferase Reporter Assay kit (Promega) as per manufacturer’s instruction. Results were further normalized to DNA content of the cells determined by CyQuant assay to correct for changes in cell proliferation/experimental condition35, 36, 59.
In some experiments, the following inhibitors were included in the co-cultures of OvCa cells and adipocytes: NFkB inhibitory peptides for RelA/NFkB p65 (p Ser276, NBP2–26505, 50 μM), that functions as a p65 decoy inhibiting Ser276 phosphorylation of RelA, and NF-kB p50 (NLS, NBP2–29323, 50 μM) inhibitory peptide that blocks p50 nuclear translocation as well as their control Peptide (NBP2–29334, 50 μM). NFkB inhibitory peptides were purchased from (R&D Systems). JNK inhibitor SP600125 was from SelleckChem, Inc (Houston, TX) supplied as10μM in DMSO. Of note is that most of commercially available cEBPβ inhibitors function by virtue of their anti-inflammatory effect61 and were also reported to inhibit p65RelA subunit of NFkB62. Thus, we used NFkB inhibitors and confirmed their inhibitory effects on cEBPβ activity.
Cytokine, Chemokine and Metabolic Assays.
The levels of cytokines/chemokines in the conditioned media (CM) of different experimental conditions were determined using the appropriate species-specific commercial kits from R&D Systems, and RayBiotech Inc., as per the manufacturer’s recommendations. Intracellular fatty acids were determined by Bodipy fluorescent staining (Molecular Probes) and oil red staining. Fatty Acid Fluorometric Assay and uptake assay Kits were from Cayman Chemicals (Ann Arbor, MI) and Abcam.
Human Ovarian Tumour Tissues.
Human ovarian cancer tissue microarray (TMA) was obtained from the University of Virginia Cooperative Human Tumour Network (CHTN). We included results from serous papillary and poorly differentiated subtypes form CHTN TMA. Thirty-five de-identified human ovarian cancer tumour tissues with the adjacent omental tissues were obtained from Wake Forest Tumour Tissue and Pathology Shared Resources (WF-TTPSR, IRB#IRB00036014). The clinical data of the samples are summarized in Supplement Table 1.
Immunohistochemistry.
Monoclonal and polyclonal antibodies against SPARC, p65NFκB, cEBPα, cJun, CD36 and FABP4 and HRP- and fluorescent-labeled secondary antibodies were purchased from sources described in Supplement Table 2. For IHC paraffin, HRP-secondary antibodies were used and signal was developed with ImmPACT DAB Peroxidase (HRP) Substrate (Vector Labs, Burlingame, CA) and counterstained with hematoxylin. For Immunofluorescence staining, appropriate Alexa-fluor 488 and 594 secondary antibodies were used and nuclei were counterstained with prolong anti-fade mounting media (Invitrogen). Nuclear and cytoplasmic expression of the aforementioned proteins in cancer cells and adipocytes were determined as earlier described35, 36, 60. Images were acquired using Aperio Scanscope (Leica Microsystems, Buffalo Grove, IL), and Olympus VS-110 Virtual Imaging System (Life Science Solutions, Center Valley, Pennsylvania). Digital image analysis of protein intensity and frequency was performed using Halo software (Indica Lab, Corrales, NM) and VisioPharm (Broomfield, CO) to segment tumor vs stroma and nuclear vs cytoplasmic and tissue alignment of sections stained with H&E with those stained colorimetric and fluorescent stains. We specifically focused on the areas that include tumor-adipocyte interface and classified the regions containing mainly adipocytes as “stroma” as guided by aligned matching H&E slides. Staining frequency and intensity were assigned arbitrary scores as earlier described35, 36, 60. Composite expression score (H-score) was calculated by multiplying the frequency and intensity scores35, 36, 60. For CD36 and FABP4 immunofluorescence staining, cells were seeded in duplicate wells LabTek 8-well slide chambers and were treated with SPARC (5μg/ml) for 24h. Immunostaining was carried out using rabbit anti-CD36 and FABP4 after fixing cells with 4% paraformaldehyde and permeabilization with Triton X-100. Alexa Fluor 594 secondary antibodies (Invitrogen) were used. Image acquisition was done using Leica AF6000 Modular System confocal microscope (Leica). Image acquisition, deconvolution, and maximum projection analysis were performed with the program LAS AF (Leica). Morphometric analysis was performed by Image J53 in 10 random fields/experimental condition. Each experiment is performed 3 times.
RNA isolation and qRT-PCR.
Total RNA was extracted from cultured cells by RNeasy kits (Qiagen, Valencia, CA) and qRT-PCR was carried out using iScript cDNA Synthesis Kit and iQ Sybr® Green Supermix and BioRad CFX thermal cycler (BioRad, Hercules, CA). The primer sequences for mouse and human genes are summarized in Supplement Tables 3–4. Each experiment was performed in triplicates and repeated 3 times.
Metabolomic profiling.
Snap-frozen dissected omental tumor nodules that developed in SP+/+ and SP−/− mice (n=6/cohort) were prepared as previously described63. Briefly, samples were de-proteinized and protein–associated metabolites were extracted. The resulting extracts were analyzed by ultra-performance liquid chromatography and mass spectroscopy (UPLC-MS/MS) with positive and negative ion mode electrospray ionization, as well as gas chromatography and mass spectroscopy (GC-MS). Samples were analyzed on a Thermo-Finnigan Trace DSQ fast-scanning single-quadrupole mass spectrometer using electron impact ionization (EI) and operated at unit mass resolving power. Raw data was extracted, peak identified, QC processed and normalized using proprietary Metabolon’s hardware and software. Compounds were identified by comparison to library entries of purified standards or recurrent unknown entities63.
Transmission Electron Microscopy (TEM).
Tumor tissue sections were processed for TEM at Wake Forest Baptist Medical Center (WFBMC) Imaging Core Facility according to standard protocols64. Sections were viewed with an FEI Tecnai Spirit TEM operating at 80 kV and images were acquired with an AMT 2Vu CCD camera. Image analysis was performed by counting the number of lipid droplets/cells in 10 cells/experimental condition. Lipid droplet size was measured in images 1800x magnification by ImageJ. The averages of the area of lipid droplets/cell were calculated in 10 cells/experimental condition and was presented as arbitrary units.
Statistical Analysis.
All other data were analyzed by two-tailed unpaired Student’s t test and one- and two-way analysis of variance (ANOVA) with Sidek or Tuckey post-hoc tests. The association of the expression of different proteins were evaluated using the nonparametric Wilcoxon-Mann-Whitney and Kruskal-Wallis tests. Correlation between the expression scores in stained tissues were performed using Spearman’s correlation. Differences were deemed significant at P< 0.05. GraphPad Prism 7.0 (San Diego, CA).
Supplementary Material
ACKNOWLEDGMENT:
This work is supported by Marsha Rivkin Pilot Award, WFBMC Department of Pathology Research Endowment Funds and R01 CA193437 (NS) and P30CA012197 to the Wake Forest Baptist Comprehensive Cancer Center (WFBCCC). The authors wish to acknowledge the support of the Wake Forest Baptist Comprehensive Cancer Center Tumor Tissue and Pathology Shared Resources (TTPSR), Cellular Imaging Shared Resources, Virtual microscopy core, and Cell & Viral Vector Laboratory Shared Resource. The authors also wish to thank members of the University of Virginia Biorepository Tumor Research Facility (BTRF) for providing TMAs and their technical help with immunostaining, and Mr. Michael White for help with immunofluorescence and confocal microscopy.
Funding: This work is supported by Marsha Rivkin Pilot Award, WFBMC Department of Pathology Research Pilot Funds and R01 CA193437 (NS) and P30CA012197 to the Wake Forest Baptist Comprehensive Cancer Center (WFBCCC).
Footnotes
Conflict of Interest: The authors declare no conflict of interest.
References:
- 1.Siegel RL, Miller KD, Jemal A (2018). Cancer statistics, 2018. CA: a cancer journal for clinicians 68: 7–30. [DOI] [PubMed] [Google Scholar]
- 2.Lengyel E, Burdette JE, Kenny HA, Matei D, Pilrose J, Haluska P et al. (2014). Epithelial ovarian cancer experimental models. Oncogene 33: 3619–3633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nieman KM, Kenny HA, Penicka CV, Ladanyi A, Buell-Gutbrod R, Zillhardt MR et al. (2011). Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat Med 17: 1498–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yeung TL, Leung CS, Yip KP, Au Yeung CL, Wong ST, Mok SC (2015). Cellular and molecular processes in ovarian cancer metastasis. A Review in the Theme: Cell and Molecular Processes in Cancer Metastasis. American journal of physiology Cell physiology 309: C444–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Said N, Motamed K (2005). Absence of host-secreted protein acidic and rich in cysteine (SPARC) augments peritoneal ovarian carcinomatosis. The American journal of pathology 167: 1739–1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Said N, Najwer I, Motamed K (2007). Secreted protein acidic and rich in cysteine (SPARC) inhibits integrin-mediated adhesion and growth factor-dependent survival signaling in ovarian cancer. The American journal of pathology 170: 1054–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Said N, Socha MJ, Olearczyk JJ, Elmarakby AA, Imig JD, Motamed K (2007). Normalization of the ovarian cancer microenvironment by SPARC. Molecular cancer research : MCR 5: 1015–1030. [DOI] [PubMed] [Google Scholar]
- 8.Said NA, Najwer I, Socha MJ, Fulton DJ, Mok SC, Motamed K (2007). SPARC inhibits LPA-mediated mesothelial-ovarian cancer cell crosstalk. Neoplasia 9: 23–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dirat B, Bochet L, Dabek M, Daviaud D, Dauvillier S, Majed B et al. (2011). Cancer-Associated Adipocytes Exhibit an Activated Phenotype and Contribute to Breast Cancer Invasion. Cancer research 71: 2455–2465. [DOI] [PubMed] [Google Scholar]
- 10.Das SK, Eder S, Schauer S, Diwoky C, Temmel H, Guertl B et al. (2011). Adipose Triglyceride Lipase Contributes to Cancer-Associated Cachexia. Science 333: 233–238. [DOI] [PubMed] [Google Scholar]
- 11.Ebadi M, Mazurak VC (2014). Evidence and mechanisms of fat depletion in cancer. Nutrients 6: 5280–5297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grisouard J, Bouillet E, Timper K, Radimerski T, Dembinski K, Frey DM et al. (2012). Both inflammatory and classical lipolytic pathways are involved in lipopolysaccharide-induced lipolysis in human adipocytes. Innate immunity 18: 25–34. [DOI] [PubMed] [Google Scholar]
- 13.Howe LR, Subbaramaiah K, Hudis CA, Dannenberg AJ (2013). Molecular pathways: adipose inflammation as a mediator of obesity-associated cancer. Clinical cancer research : an official journal of the American Association for Cancer Research 19: 6074–6083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ji C, Chen X, Gao C, Jiao L, Wang J, Xu G et al. (2011). IL-6 induces lipolysis and mitochondrial dysfunction, but does not affect insulin-mediated glucose transport in 3T3-L1 adipocytes. Journal of bioenergetics and biomembranes 43: 367–375. [DOI] [PubMed] [Google Scholar]
- 15.Zhang W, Mottillo EP, Zhao J, Gartung A, VanHecke GC, Lee JF et al. (2014). Adipocyte lipolysis-stimulated interleukin-6 production requires sphingosine kinase 1 activity. The Journal of biological chemistry 289: 32178–32185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang Z, Bryan JL, DeLassus E, Chang L-W, Liao W, Sandell LJ (2010). CCAAT/Enhancer-binding Protein β and NF-κB Mediate High Level Expression of Chemokine Genes CCL3 and CCL4 by Human Chondrocytes in Response to IL-1β. The Journal of biological chemistry 285: 33092–33103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Esteves CL, Kelly V, Breton A, Taylor AI, West CC, Donadeu FX et al. (2014). Proinflammatory Cytokine Induction of 11β-Hydroxysteroid Dehydrogenase Type 1 (11β-HSD1) in Human Adipocytes Is Mediated by MEK, C/EBPβ, and NF-κB/RelA. The Journal of Clinical Endocrinology & Metabolism 99: E160–E168. [DOI] [PubMed] [Google Scholar]
- 18.Rahman SM, Janssen RC, Choudhury M, Baquero KC, Aikens RM, de la Houssaye BA et al. (2012). CCAAT/Enhancer-binding Protein β (C/EBPβ) Expression Regulates Dietary-induced Inflammation in Macrophages and Adipose Tissue in Mice. The Journal of biological chemistry 287: 34349–34360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ramji DP, Foka P (2002). CCAAT/enhancer-binding proteins: structure, function and regulation. Biochemical Journal 365: 561–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang Z, Zhang Z, Kang Y, Hou C, Duan X, Sheng P et al. (2014). Resistin Stimulates Expression of Chemokine Genes in Chondrocytes via Combinatorial Regulation of C/EBPβ and NF-κB. International Journal of Molecular Sciences 15: 17242–17255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Oyesanya RA, Lee ZP, Wu J, Chen J, Song Y, Mukherjee A et al. (2008). Transcriptional and post-transcriptional mechanisms for lysophosphatidic acid-induced cyclooxygenase-2 expression in ovarian cancer cells. FASEB J 22: 2639–2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rozenberg JM, Bhattacharya P, Chatterjee R, Glass K, Vinson C (2013). Combinatorial Recruitment of CREB, C/EBPβ and c-Jun Determines Activation of Promoters upon Keratinocyte Differentiation. PloS one 8: e78179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Said N, Theodorescu D (2013). Secreted Protein Acidic and Rich in Cysteine (Sparc) in Cancer. J Carcinog Mutagen 4: 151. [Google Scholar]
- 24.Delany AM, Hankenson KD (2009). Thrombospondin-2 and SPARC/osteonectin are critical regulators of bone remodeling. Journal of cell communication and signaling 3: 227–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nie J, Sage EH (2009). SPARC functions as an inhibitor of adipogenesis. Journal of cell communication and signaling 3: 247–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Piconese S, Costanza M, Tripodo C, Sangaletti S, Musio S, Pittoni P et al. (2011). The matricellular protein SPARC supports follicular dendritic cell networking toward Th17 responses. Journal of autoimmunity 37: 300–310. [DOI] [PubMed] [Google Scholar]
- 27.Said NA, Elmarakby AA, Imig JD, Fulton DJ, Motamed K (2008). SPARC ameliorates ovarian cancer-associated inflammation. Neoplasia 10: 1092–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kulbe H, Chakravarty P, Leinster DA, Charles KA, Kwong J, Thompson RG et al. (2012). A dynamic inflammatory cytokine network in the human ovarian cancer microenvironment. Cancer research 72: 66–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bradshaw AD, Graves DC, Motamed K, Sage EH (2003). SPARC-null mice exhibit increased adiposity without significant differences in overall body weight. Proceedings of the National Academy of Sciences 100: 6045–6050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ewing SJ, Zhu S, Zhu F, House JS, Smart RC (2008). C/EBPβ represses p53 to promote cell survival downstream of DNA damage independent of oncogenic Ras and p19(Arf). Cell death and differentiation 15: 1734–1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guo L, Li X, Huang JX, Huang HY, Zhang YY, Qian SW et al. (2012). Histone demethylase Kdm4b functions as a co-factor of C/EBPβ to promote mitotic clonal expansion during differentiation of 3T3-L1 preadipocytes. Cell Death and Differentiation 19: 1917–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guo L, Li X, Tang Q-Q (2015). Transcriptional Regulation of Adipocyte Differentiation: A Central Role for CCAAT/Enhancer-binding Protein (C/EBP) β. The Journal of biological chemistry 290: 755–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hernandez L, Hsu S, Davidson B, Birrer MJ, Kohn EC, Annunziata CM (2010). Activation of NF-κB signaling by IKKβ increases aggressiveness of ovarian cancer. Cancer research 70: 4005–4014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.House CD, Jordan E, Hernandez L, Ozaki M, James JM, Kim M et al. (2017). NF-kB promotes ovarian tumorigenesis via classical pathways supporting proliferative cancer cells and alternative pathways supporting ALDH+ cancer stem-like cells. Cancer research. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Al-Hassan NN, Behzadian A, Caldwell R, Ivanova VS, Syed V, Motamed K et al. (2012). Differential roles of uPAR in peritoneal ovarian carcinomatosis. Neoplasia 14: 259–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Said N, Frierson HF, Sanchez-Carbayo M, Brekken RA, Theodorescu D (2013). Loss of SPARC in bladder cancer enhances carcinogenesis and progression. The Journal of clinical investigation 123: 751–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Iyengar P, Combs TP, Shah SJ, Gouon-Evans V, Pollard JW, Albanese C et al. (2003). Adipocyte-secreted factors synergistically promote mammary tumorigenesis through induction of anti-apoptotic transcriptional programs and proto-oncogene stabilization. Oncogene 22: 6408–6423. [DOI] [PubMed] [Google Scholar]
- 38.Schweiger M, Schreiber R, Haemmerle G, Lass A, Fledelius C, Jacobsen P et al. (2006). Adipose Triglyceride Lipase and Hormone-sensitive Lipase Are the Major Enzymes in Adipose Tissue Triacylglycerol Catabolism. Journal of Biological Chemistry 281: 40236–40241. [DOI] [PubMed] [Google Scholar]
- 39.Currie E, Schulze A, Zechner R, Walther Tobias C, Farese Robert V (2013). Cellular Fatty Acid Metabolism and Cancer. Cell Metabolism 18: 153–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Delany AM, Kalajzic I, Bradshaw AD, Sage EH, Canalis E (2003). Osteonectin-null mutation compromises osteoblast formation, maturation, and survival. Endocrinology 144: 2588–2596. [DOI] [PubMed] [Google Scholar]
- 41.Nie J, Bradshaw AD, Delany AM, Sage EH (2011). Inactivation of SPARC enhances high-fat diet-induced obesity in mice. Connective tissue research 52: 99–108. [DOI] [PubMed] [Google Scholar]
- 42.Pradeep S, Kim SW, Wu SY, Nishimura M, Chaluvally-Raghavan P, Miyake T et al. (2014). Hematogenous metastasis of ovarian cancer: rethinking mode of spread. Cancer cell 26: 77–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bowtell DD, Bohm S, Ahmed AA, Aspuria PJ, Bast RC Jr., Beral V et al. (2015). Rethinking ovarian cancer II: reducing mortality from high-grade serous ovarian cancer. Nature reviews Cancer 15: 668–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Roby KF, Taylor CC, Sweetwood JP, Cheng Y, Pace JL, Tawfik O et al. (2000). Development of a syngeneic mouse model for events related to ovarian cancer. Carcinogenesis 21: 585–591. [DOI] [PubMed] [Google Scholar]
- 45.Vaysse C, Lomo J, Garred O, Fjeldheim F, Lofteroed T, Schlichting E et al. (2017). Inflammation of mammary adipose tissue occurs in overweight and obese patients exhibiting early-stage breast cancer. NPJ Breast Cancer 3: 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.De Pergola G, Silvestris F (2013). Obesity as a Major Risk Factor for Cancer. Journal of Obesity 2013: 291546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fontana L, Eagon JC, Trujillo ME, Scherer PE, Klein S (2007). Visceral fat adipokine secretion is associated with systemic inflammation in obese humans. Diabetes 56: 1010–1013. [DOI] [PubMed] [Google Scholar]
- 48.Doerstling SS, O’Flanagan CH, Hursting SD (2017). Obesity and Cancer Metabolism: A Perspective on Interacting Tumor–Intrinsic and Extrinsic Factors. Frontiers in Oncology 7: 216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Barbolina MV, Moss NM, Westfall SD, Liu Y, Burkhalter RJ, Marga F et al. (2009). Microenvironmental regulation of ovarian cancer metastasis. Cancer Treat Res 149: 319–334. [DOI] [PubMed] [Google Scholar]
- 50.Hein S, Mahner S, Kanowski C, Loning T, Janicke F, Milde-Langosch K (2009). Expression of Jun and Fos proteins in ovarian tumors of different malignant potential and in ovarian cancer cell lines. Oncology reports 22: 177–183. [DOI] [PubMed] [Google Scholar]
- 51.Liu D, Zhang X-X, Li M-C, Cao C-H, Wan D-Y, Xi B-X et al. (2018). C/EBPβ enhances platinum resistance of ovarian cancer cells by reprogramming H3K79 methylation. Nature Communications 9: 1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hotamisligil GS (2017). Inflammation, metaflammation and immunometabolic disorders. Nature 542: 177. [DOI] [PubMed] [Google Scholar]
- 53.Naczki C, John B, Patel C, Lafferty A, Ghoneum A, Afify H et al. (2018). SPARC Inhibits Metabolic Plasticity in Ovarian Cancer. Cancers 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ladanyi A, Mukherjee A, Kenny HA, Johnson A, Mitra AK, Sundaresan S et al. (2018). Adipocyte-induced CD36 expression drives ovarian cancer progression and metastasis. Oncogene 37: 2285–2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tucker SL, Gharpure K, Herbrich SM, Unruh AK, Nick AM, Crane EK et al. (2014). Molecular biomarkers of residual disease after surgical debulking of high-grade serous ovarian cancer. Clinical cancer research : an official journal of the American Association for Cancer Research 20: 3280–3288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gharpure KM, Pradeep S, Sans M, Rupaimoole R, Ivan C, Wu SY et al. (2018). FABP4 as a key determinant of metastatic potential of ovarian cancer. Nat Commun 9: 2923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Harjes U, Bridges E, Gharpure KM, Roxanis I, Sheldon H, Miranda F et al. (2017). Antiangiogenic and tumour inhibitory effects of downregulating tumour endothelial FABP4. Oncogene 36: 912–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Miranda F, Mannion D, Liu S, Zheng Y, Mangala LS, Redondo C et al. (2016). Salt-Inducible Kinase 2 Couples Ovarian Cancer Cell Metabolism with Survival at the Adipocyte-Rich Metastatic Niche. Cancer cell 30: 273–289. [DOI] [PubMed] [Google Scholar]
- 59.Said N, Sanchez-Carbayo M, Smith SC, Theodorescu D (2012). RhoGDI2 suppresses lung metastasis in mice by reducing tumor versican expression and macrophage infiltration. The Journal of clinical investigation 122: 1503–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Said N, Smith S, Sanchez-Carbayo M, Theodorescu D (2011). Tumor endothelin-1 enhances metastatic colonization of the lung in mouse xenograft models of bladder cancer. The Journal of clinical investigation 121: 132–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jakobs A, Steinmann S, Henrich SM, Schmidt TJ, Klempnauer K-H (2016). Helenalin Acetate, a Natural Sesquiterpene Lactone with Anti-inflammatory and Anti-cancer Activity, Disrupts the Cooperation of CCAAT Box/Enhancer-binding Protein β (C/EBPβ) and Co-activator p300. The Journal of biological chemistry 291: 26098–26108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Huber R, Pietsch D, Panterodt T, Brand K (2012). Regulation of C/EBPβ and resulting functions in cells of the monocytic lineage. Cellular signalling 24: 1287–1296. [DOI] [PubMed] [Google Scholar]
- 63.Chinnaiyan P, Kensicki E, Bloom G, Prabhu A, Sarcar B, Kahali S et al. (2012). The metabolomic signature of malignant glioma reflects accelerated anabolic metabolism. Cancer research 72: 5878–5888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cheville NF, Stasko J (2014). Techniques in Electron Microscopy of Animal Tissue. Veterinary Pathology 51: 28–41. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.