

Summary
The Zika epidemic in the Americas has challenged surveillance and control. As the epidemic appears to be waning, it is unclear whether transmission is still ongoing, which is exacerbated by discrepancies in reporting. To uncover locations with lingering outbreaks, we investigated travel-associated Zika cases to identify transmissions not captured by reporting. We uncovered an unreported outbreak in Cuba during 2017, a year after peak transmission in neighboring islands. By sequencing Zika virus, we show that the establishment of the virus was delayed by a year and that the ensuing outbreak was sparked by long-lived lineages of Zika virus from other Caribbean islands. Our data suggest that while mosquito control in Cuba may initially have been effective at mitigating Zika virus transmission, such measures need to be maintained to be effective. Our study highlights how Zika virus may still be ‘silently’ spreading and provides a framework for understanding outbreak dynamics.
Keywords: Genomic epidemiology, infectious disease genomics, clinical sequencing, travel surveillance, Zika virus, phylogenetics
Graphical Abstract
eTOC Blurb
A combination of travel surveillance and clinical virus genomic sequencing of infected travelers provides a framework for detecting hidden outbreaks, such as an unreported Zika outbreak in Cuba during 2017
Introduction
The recent Zika epidemic in the Americas is a testament to how rapidly mosquito-borne viruses can emerge and spread, and has revealed flaws in our surveillance and response systems (Grubaugh et al., 2018; Morens and Fauci, 2017). Due, in part, to high rates of subclinical infections and overlapping symptoms with infections from dengue and chikungunya viruses (Mitchell et al., 2018), Zika virus was circulating for more than a year and a half before it was first detected in Brazil (Faria et al., 2017). By the time Zika virus was discovered in May of 2015 (Zanluca et al., 2015) and recognized for its ability to cause severe congenital disease (França et al., 2016; Mlakar et al., 2016), the virus had already spread from Brazil to more than 40 countries (Faria et al., 2017; Grubaugh et al., 2017; Metsky et al., 2017; Thézé et al., 2018). By mid 2017, reports from the Pan American Health Organization (PAHO) (PAHO, 2017a, 2017b) revealed Zika virus activity throughout the Americas was waning, prompting predictions for the end of the epidemic (e.g., (O’Reilly et al., 2018)) and the removal of the World Health Organization’s (WHO) “Public Health Emergency of International Concern” status (WHO, 2016a, 2016b). More recently, however, new Zika outbreaks have been described across the world (CDC, 2018), including from Angola, India, Cabo Verde, Vietnam, and Thailand, with some of these resulting from Zika virus introductions from the epidemic in the Americas (e.g., (Hill et al., 2019; Lourenço et al., 2018; Meltzer et al., 2016; Phumee et al., 2019; Ruchusatsawat et al., 2019; Yadav et al., 2019). These observations would suggest that significant transmission of Zika virus in the Americas could still be ongoing, despite case reporting having come close to zero.
Coordinated response efforts during the early stages of the Zika epidemic were ultimately contingent on countries detecting cases and reporting them to international health agencies (Lessler et al., 2016), primarily PAHO (PAHO, 2017a, 2017b). For Zika virus and other Aedes aegypti mosquito-borne viruses - including dengue and chikungunya viruses - that are primarily transmitted in urban settings and disproportionately impact those with limited resources (Braga et al., 2010; Gardner et al., 2018; Netto et al., 2017; Weaver et al., 2018), accurate local reporting is especially problematic. Not only are people in poor living conditions more likely to be exposed to infected mosquitoes, but such communities often have less access to adequate healthcare, resulting in more cases going undetected (Hotez, 2016; LaBeaud, 2008). Pockets of virus transmission that occur in countries with inadequate reporting can therefore facilitate ′hidden′ outbreaks, increasing the risk of infected travelers causing outbreaks in new regions of the world. Thus, underreported or unrecognized local outbreaks may prolong epidemics, and hinder global efforts aimed at halting virus spread.
Infectious disease surveillance of international travelers (‘travel surveillance’) has been an effective method for detecting pathogens circulating in resource-limited areas (Hamer et al., 2017; Harvey et al., 2013; Leder et al., 2013, 2017; Wilder-Smith et al., 2012). In contrast to local case reporting, travel surveillance relies on diagnosing patients that have acquired infections while traveling outside the country of diagnosis. More recently, approaches of ‘genomic epidemiology’ using pathogen sequencing of infected patients have also been used to reconstruct the timing, scale, and dynamics of infectious disease outbreaks (Grubaugh et al., 2019a; Ladner et al., 2019). As many regions in the Americas affected by the Zika epidemic attract large volumes of international visitors from countries with stronger surveillance systems (Wilder-Smith et al., 2018), we hypothesized that by creating a framework integrating local case reporting and travel surveillance with genomic epidemiology, we would be able to uncover potentially still-ongoing Zika outbreaks.
In this study, we combined travel surveillance, local case reporting, and clinical sequencing of Zika virus from infected travelers, to detect virus outbreaks that had previously been missed. We discovered a large Zika outbreak in Cuba that was not reported to PAHO (PAHO, 2017c) or other public health agencies, and thus went undetected to the international community. We show that the outbreak in Cuba peaked in 2017, when the epidemic in the rest of the Americas was waning (PAHO, 2017a, 2017b), and estimate that it was at least as large as those in neighboring countries. By recalibrating the dynamics of recent Zika, dengue, and chikungunya outbreaks across the Caribbean, we show that, surprisingly, the Zika outbreak in Cuba was delayed by a full year, which could have been caused by a country-wide vector control campaign. By sequencing Zika virus directly from infected travelers, we show that the establishment of the virus in Cuba itself was delayed, with multiple introductions of Zika virus from other Caribbean islands later fueling the outbreak. Overall, our study creates a combined framework for how travel surveillance and genomic epidemiology can be used as a future surveillance network for detecting ‘hidden’ outbreaks of worldwide emerging infections, and reconstruct transmission dynamics when local reporting is absent, withheld, or otherwise insufficient.
Results
Uncovering an unreported Zika outbreak in Cuba
Zika virus was first detected in Brazil in May, 2015 (Zanluca et al., 2015) and had spread to 48 countries by 2016, with case numbers peaking later that year (PAHO, 2017a, 2017b). By mid 2017, new Zika cases were no longer being reported to the international community (PAHO, 2017a, 2017b). Despite the reach and size of the epidemic, however, studies have shown that the epidemic likely started at least one and a half years prior to its discovery (Faria et al., 2017). Due to widespread surveillance gaps and inconsistent reporting (Grubaugh et al., 2018), we therefore hypothesized that local Zika outbreaks could still be occurring in the Americas, despite not being captured by the international community.
To investigate whether Zika virus transmission is still ongoing, we used travel surveillance to reveal that local outbreaks were still occurring in 2017, despite relatively few cases being reported (Figure 1). Our data demonstrate that the vast majority of Zika cases during 2017 were the result of an unreported Zika outbreak in Cuba, which occurred while public data suggested the epidemic was nearing its end in the Americas (PAHO, 2017a, 2017b) (Figure 1).
Figure 1. International travel cases reveal unreported Zika outbreak in Cuba in 2017.
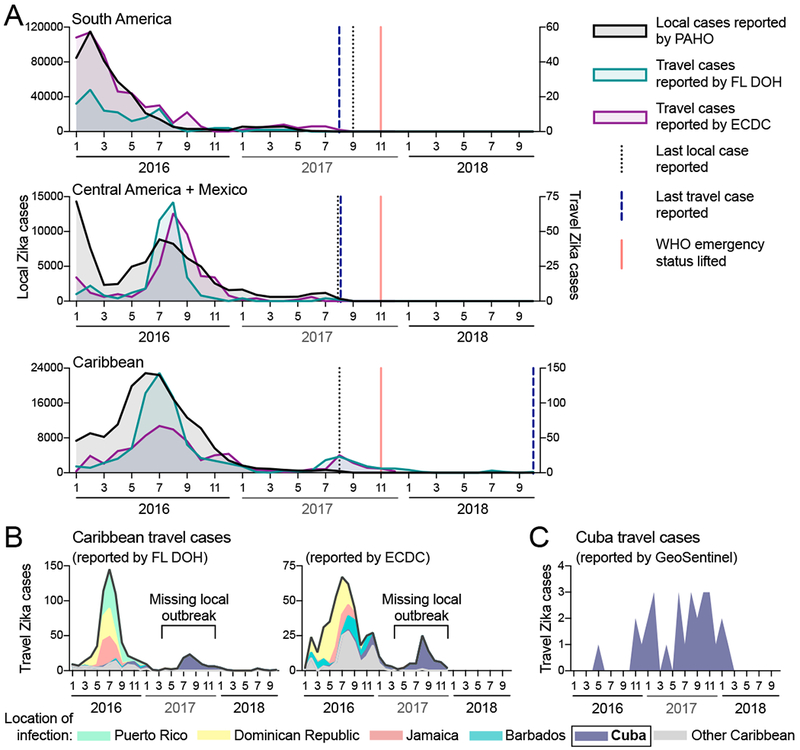
Local and travel-associated Zika cases were used to determine if outbreaks were still occuring during 2017. (A) Monthly local Zika cases (left y-axis) reported by the Pan American Health Organization (PAHO) and monthly travel-associated Zika cases (right y-axis) reported by the Florida Department of Health (FL-DOH) and the European Centre for Disease Prevention and Control (ECDC) were sorted by origin of exposure. The vertical lines represent the months the last local and travel cases were reported per region, and the month that the World Health Organization (WHO) Public Health Emergency of International Concern status was lifted for the Zika epidemic (November, 2017). In each region, travel cases and local cases were correlated (Pearson r range = 0.542-0.976, each comparison can be found in Supplemental File 1). (B) The total number of Zika cases reported by the FL-DOH and the ECDC associated with travel originating in the Caribbean are shown (black line) and are shaded by the top 5 origin locations (all other placed in the ′Other Caribbean′ category). (C) Zika cases associated with travel from Cuba, diagnosed by the GeoSentinel Surveillance Network, were sorted by month of clinic visit. Travel cases diagnosed by the GeoSentinel Surveillance Network originating from other parts of the Americas are not shown. See also Figure S1. The data used for this figure can be found in Supplemental File 1.
To determine whether Zika case reports from international travelers could reveal outbreaks not captured by local case reports, we compared the temporal distribution of local and travel surveillance Zika cases from 2016 to 2018 (Figure 1). We obtained monthly suspected and confirmed Zika cases locally reported by individual countries and territories from PAHO. We obtained reports of international travel-associated Zika cases from the Florida Department of Health (FL-DOH) and the European Centre for Disease Prevention and Control (ECDC). We constructed Zika epidemic (epi) curves based on either local or travel surveillance cases and found that they were in strong agreement from South America (Pearson r = 0.917 and 0.976 using FL-DOH and ECDC data, respectively) and the Caribbean (Pearson r = 0.828 and 0.856), and to a smaller extent Central America and Mexico (Pearson r = 0.542 and 0.583). For South America and Central America, we also found concordance for when the last local and travel cases were reported, which was in August and September, 2017 (Figure 1A).
We found that the last local case from the Caribbean was also reported in August, 2017. However, we observed a spike in Zika cases from travelers returning from this region during the summer of 2017 that were not captured by local reports (Figure 1A), and Zika virus infected travelers from the Caribbean were reported until the end of our reporting period from the ECDC (December, 2017) and FL-DOH (October, 2018; Figure 1A). By examining potential source locations for the travel-associated Zika cases in 2017, we found that between June, 2017 and October, 2018 more than 98% of them came from Cuba (90 of 91 Zika diagnoses in Florida, 63 of 64 Zika diagnoses in Europe; Figure 1B). To further confirm the timing of a Zika outbreak in Cuba, we obtained travel surveillance data from the U.S. CDC GeoSentinel Surveillance Network (Hamer et al., 2017; Leder et al., 2017) and found that 76% of the U.S. Zika cases associated with travel from Cuba were diagnosed in 2017 (22 of 29; Figure 1C). By analyzing the total air travel volumes from Cuba to Florida and European countries, we found that the increase in passengers from 2016 to 2017 (1.2-1.5×; Figure S1) does not account for the increase in travel Zika cases from Cuba reported by those countries (5.3-7.2×; Figure S1). While our travel surveillance show that a Zika outbreak peaked in Cuba in 2017 with waning transmission continuing into 2018, during this time period no local Zika cases were reported by Cuba to PAHO or other international public health agencies (PAHO, 2017c).
The Zika outbreak in Cuba was as large as those on other Caribbean islands
Having uncovered an unreported Zika outbreak in Cuba, we next investigated its size. We created a model using relationships between the local and travel Zika incidence rates and found that it was likely as large as Zika outbreaks on other Caribbean islands that peaked a year prior (Figure 2).
Figure 2. The Zika outbreak in Cuba during 2017 was similar in size to others during 2016.
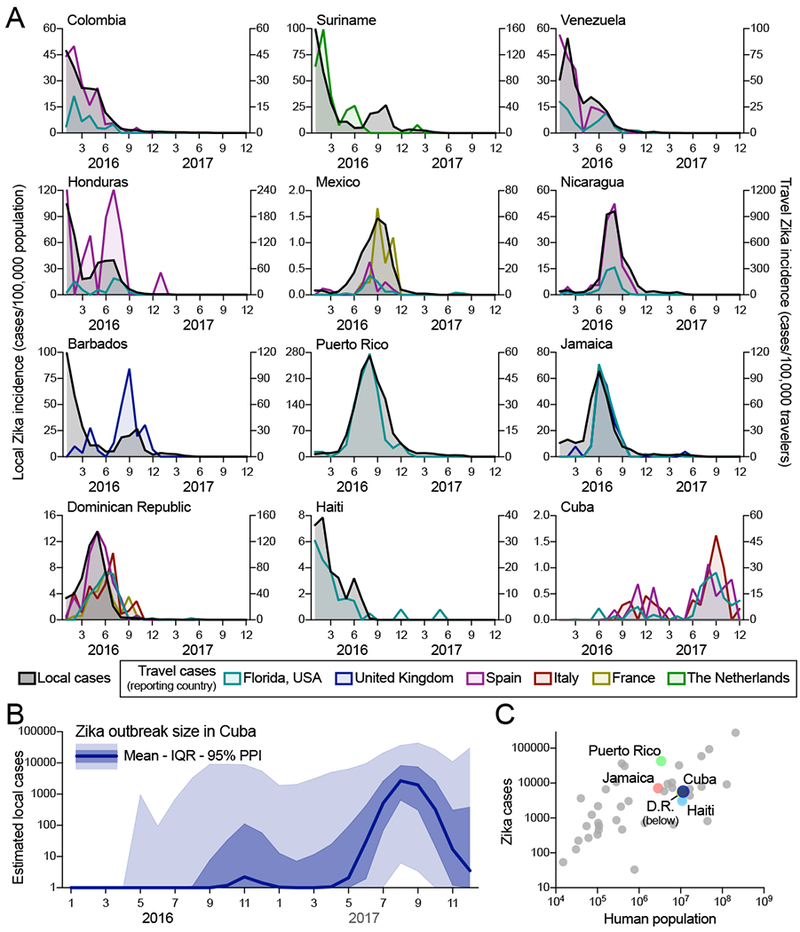
Infections of international travelers were used to estimate the size of the Zika outbreak in Cuba. (A) The local Zika virus incidence rates for each country/territory were calculated by the number of locally reported cases per month per 100,000 population. The travel Zika virus incidence rates for each country/territory of presumed exposure origin and reporting country (i.e., travel destination) pair were calculated by the number of travel-associated cases per month per 100,000 air passenger travelers entering the destination country from the origin. When there were at least 20 travel-associated Zika cases (Figure S2), there was a positive correlation between travel and local incidence for all exposure origin and reporting country (i.e., travel destination) pairs (mean Pearson r = 0.769, range = 0.121-0.984; Supplemental File 1). (B) The number of Zika cases per month (mean, interquartile range, and 95% posterior predictive interval [PPI]) in Cuba during 2016-2017 were estimated by using fitted relationships between estimated local and travel incidence rates in countries with both sets of data to estimate what the local incidence rate in Cuba would have been if local data was available (Figures S4, S5). This local incidence rate was then used to estimate local per capita incidence rates and subsequent number of Zika cases per month in Cuba. (C) The estimated number of Zika cases from Cuba (mean from B) and the total reported number of Zika cases during 2016-2017 from all countries/territories in the Americas with Zika virus transmission were plotted with the human population size from each region. Highlighted are the other large Caribbean countries/territories (D.R. = Dominican Republic). See also Figures S2–S5. The data used for this figure can be found in Supplemental File 1.
In the absence of local case reporting, studies have demonstrated that travel surveillance can be used to infer aspects of local virus transmission dynamics (Cauchemez et al., 2014a; Fraser et al., 2009; Meltzer et al., 2008). Only 187 laboratory-confirmed Zika cases were reported by Cuba in 2016, and none were reported in 2017-2018 (PAHO, 2017c). These reports are inconsistent with the outbreak dynamics that we detected using travel surveillance (Figures 1B, 2A). To estimate the number of cases that likely went unreported in Cuba in 2016 and 2017, we first investigated if travel surveillance accurately reflected the dynamics of known local Zika outbreaks for individual countries and territories outside Cuba. For this purpose, we estimated travel incidence by the number of diagnosed travel Zika cases by the air travel volume between all locations, and compared that the local incidence reported by (PAHO, 2017a, 2017b) (Fig 2A). We found that in places with at least 20 travel-associated Zika cases reported (Figure S2), epi curves constructed from travel surveillance were in agreement with epi curves generated from local data reporting (mean Pearson r = 0.769, range = 0.121-0.984; Supplemental File 1). We also analyzed cruise ship travel volume, but found that it was minimal compared to air travel (Figure S3).
To approximate the size of the Zika outbreak in Cuba, we next constructed a Bayesian model. We used the mean posterior estimates of the proportion of local to travel incidence from 23 countries throughout the Americas (Figure S4), each individually multiplied by the mean posterior estimates of the Cuba travel incidence rate (Figure S5). Taking the population size of Cuba into account, we estimated that 5,707 Zika cases (interquartile range: 1,071 to 22,611) likely went unreported in this country (Figure 2B), with the majority of these cases (>99%) having occurred in 2017. Our results therefore suggest that the 2017 Zika outbreak in Cuba was comparable in size to the known 2016 outbreaks in countries with similar population sizes, such as Haiti (3,103 reported cases), Dominican Republic (5,305 reported cases), and Jamaica (7,165 reported cases; Figure 2C).
A one year delay of the Zika outbreak in Cuba was unusual
Our analyses show that the Zika outbreak in Cuba was delayed by approximately a year compared to those elsewhere in the Caribbean (Figure 1). To investigate if such a delay was unexpected, we reconstructed recent Caribbean outbreaks caused by chikungunya virus and found that a one-year delay of the 2017 Zika outbreak in Cuba was unusual (Figure 3).
Figure 3. The chikungunya outbreak in Cuba during the 2014 epidemic was not delayed.
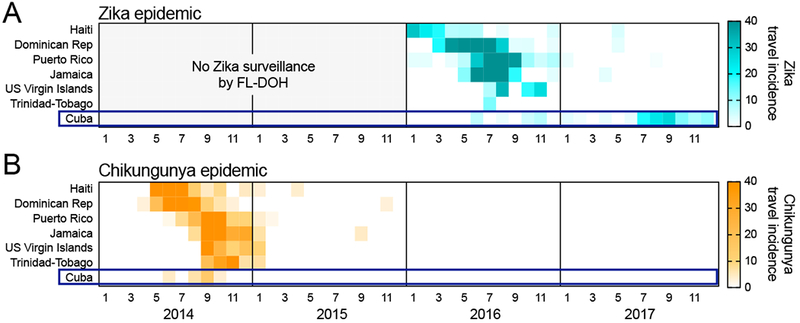
Travel-associated chikungunya cases were used to investigate if the delayed Zika outbreak in Cuba should have been expected. Travel (A) Zika and (B) chikungunya virus incidence rates were calculated by the number of travel-associated cases reported by the FL-DOH per month per 100,000 air passenger travelers entering Florida, USA from the origin. FL-DOH surveillance for Zika cases did not start until January, 2016. Shown are the six largest Caribbean Islands by population plus the U.S. Virgin Islands. All of the data used for this figure can be found in Supplemental File 1.
Like Zika virus, chikungunya virus is primarily transmitted by Ae. aegypti mosquitoes and is thus governed by similar epidemiological factors (Patterson et al., 2016; Weaver et al., 2018), resulting in ‘wave-like’ outbreaks that are comparable to Zika (Cauchemez et al., 2014b; Grubaugh et al., 2018). To reconstruct recent outbreaks of chikungunya in the Caribbean, we used the same framework as we did for Zika (Figures 2, 3A), and analyzed travel surveillance cases reported by the FL-DOH to create chikungunya epi curves (Figure 3B). We found that the chikungunya outbreak in Cuba occured at the same time (2014) as elsewhere in the Caribbean (Figure 3B). This syncrony of chikungunya outbreaks in the Caribbean is in contrast to the 2017 Zika outbreak in Cuba, which was delayed only in this country, and not on any of the other Caribbean islands, as they all experienced outbreaks in 2016 (Figure 3A). These findings suggest that the delay of the Zika outbreak in Cuba could have been caused by events that were specific to this country during the 2015-2016 Zika epidemic.
The establishment of Zika virus in Cuba was delayed and caused by multiple introductions from other Caribbean islands
Local Zika outbreaks are caused by introductions of the virus from outside areas, with later establishment in resident Ae. aegypti mosquitoes. Having shown that the delay in the outbreak in Cuba could have been caused by specific factors in this country, we used genomic epidemiology to investigate the timing and origin of the introduction and establishment of Zika virus in Cuba by sequencing the virus directly from infected travelers. Our phylogenetic analyses showed that the delayed Zika outbreak in Cuba was caused by a delay in the establishment of the virus itself, as opposed to a delay in outbreak dynamics. We also found that the 2017 outbreak in Cuba was caused by multiple introductions of the virus, primarily from outbreaks in other Caribbean islands during the summer of 2016 (Figure 4).
Figure 4. The establishment of Zika virus from other Caribbean islands was delayed in Cuba.
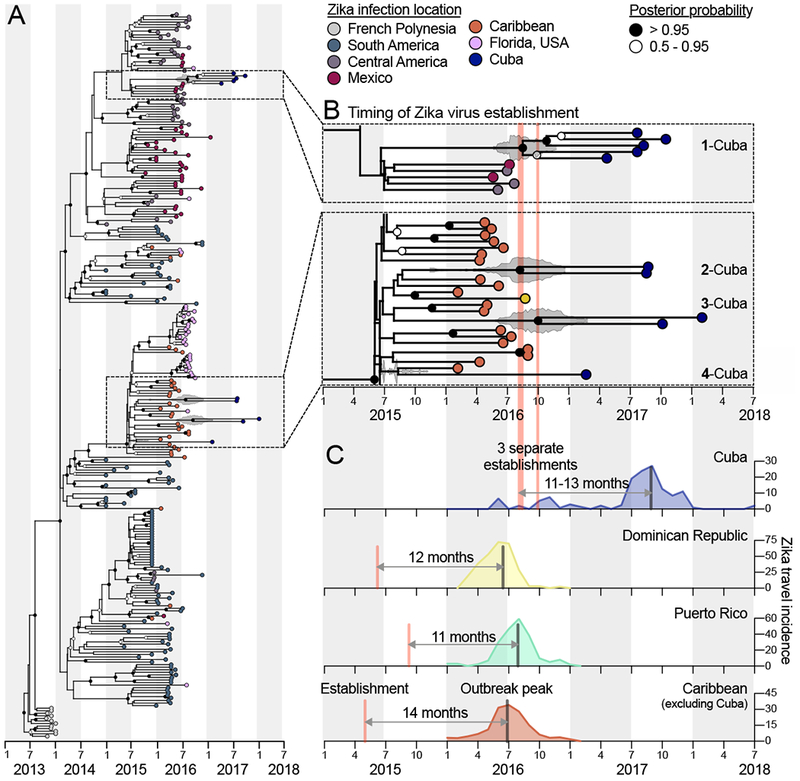
Genomics approaches were used to determine the timing and sources of the Zika virus introductions into Cuba. (A) A time-resolved maximum clade credibility (MCC) tree was constructed using 283 near complete Zika virus protein coding sequences, including 10 sequences from travelers returning from Cuba during 2017-2018. (B) The zooms show the likely times of Zika virus establishment (i.e., tMRCAs) for each of the Cuba clades, as well as potential introduction sources (i.e., locations of the sequences basal on the tree). The fill color on each tip represents the probable location of infection, the clade posterior probabilities at each node are indicated by white circles filled with black relative to the level of posterior support, and the grey violin plot indicates the 95% HPD interval for each tMRCA. The mean tMRCA for clade 1-Cuba was August, 2016 (95% HPD = May-November, 2016), the mean tMRCA for clade 2-Cuba was July, 2016 (95% HPD = March-December, 2016), and the mean tMRCA for clade 3-Cuba was September, 2016 (95% HPD = May, 2016-February, 2017). Clade ‘Cuba-4’ does not have a tMRCA estimate because it consists of a single sequence. A maximum likelihood tree and a root-to-tip molecular clock are shown in Figure S6. (C) The three separate estimated Zika virus establishment times with tMRCA estimates into Cuba are shown with the Zika virus travel incidence rates (travel cases/100,000 travelers, as calculated for Figures 2, 3). The estimated earliest Zika virus establishment times (based on the MCC tree in A) and travel incidence rates for the Dominican Republic, Puerto Rico, and the Caribbean as a whole (minus Cuba) are shown to compare the times from establishment of the virus to outbreak peak. See also Figure S6. GenBank access numbers of Zika virus genomes sequenced during this study can be found in Supplemental File 2, the data used to create (A and B) can be found in Supplemental File 3, and the data used to create (C) can be found in Supplemental File 1.
We sequenced Zika virus genomes from nine infected Florida travelers arriving from Cuba during 2017-2018 and obtained one Cuban Zika virus genome from GenBank (MF438286). In addition to our previous Zika virus sequences from the 2016 outbreak (Grubaugh et al., 2017), we also sequenced four additional genomes from Florida to demonstrate that the Zika virus lineages from Cuba were distinct from those in Florida, and thus bonafide travel-associated (Figure 4A). We openly shared all our sequences as they were generated (https://andersen-lab.com/secrets/data/zika-genomics/), and combined them with other publicly available sequences for a final dataset of 283 Zika virus genomes (Figure 4 and Supplemental File 2).
We created phylogenetic trees using time-resolved Bayesian inference (Figure 4A and Supplemental File 3) and maximum likelihood reconstruction (Figure S6 and Supplemental File 3). We found that, as expected, the Zika virus lineages in Cuba clustered with other virus genomes from the Americas (Figure 4A), showing that the outbreak in Cuba was a continuation of the epidemic in the Americas, as opposed to introductions from ongoing Zika outbreaks in Asia (Lim et al., 2017; Watts et al., 2018). Based on the placement of the Zika virus genomes, we found evidence for one introduction into Cuba from Central America (Figure 4B, clade ‘1-Cuba’) and three from the Caribbean (Figure 4B, clades ‘2-Cuba’, ‘3-Cuba’, and ‘4-Cuba’). These findings suggest that the outbreak in Cuba was primarily fueled by introductions of the virus from other Caribbean islands, which is similar to our observations from the 2016 Zika outbreak in Florida (Grubaugh et al., 2017).
We next investigated the timing of the establishment of Zika virus in Cuba by estimating the time to the most recent common ancestor (tMRCA) using our time-resolved phylogenetic trees (Figure 4A, B). The tMRCAs indicate the coalescence points for each clade, and thus estimate the earliest times of establishment of the sequenced virus lineages in Ae. aegypti mosquitoes in Cuba that later gave rise to human cases. We found that all our tMRCA estimates for the Cuban Zika virus clades were between July and September, 2016 (Figure 4B), corresponding to the peak of the Zika outbreaks in other Caribbean islands (Figures 1A and 4C).
By comparing our tMRCA estimates from Cuba to those of other Caribbean islands, we found that the establishment of the virus in Cuba was itself delayed by a year (third quarter, 2016 versus mid 2015; Figure 4C). We then compared our tMRCAs to epi curves created from our travel surveillance, and found that the peak of the 2017 Zika outbreak in Cuba occurred 11–13 months after the virus became locally established, which was in agreement to what we found for the 2016 Zika outbreaks in the Dominican Republic, Puerto Rico, and the Caribbean as a whole (excluding Cuba; Figure 4C). Combined, these findings show that Zika outbreaks across the Caribbean peaked a year after successful establishment of the virus in each location, and that the delay in the outbreak in Cuba was likely caused by a delay in the establishment of the virus itself.
Mosquito control may have delayed the Zika outbreak in Cuba
Having shown that the postponement of the Cuban outbreak was likely the result of delayed local establishment of the virus (Figure 4), we next investigated what factors may have been responsible for this delay. We explored three primary hypotheses: (1) fewer opportunities for Zika virus introductions into Cuba in 2015 when the virus was becoming established elsewhere in the Caribbean (Figure 4C), (2) environmental conditions in Cuba from 2015 to 2016 that were unsuitable for Ae. aegypti-borne virus outbreaks, and (3) Ae. aegypti surveillance and control campaigns (Castell-Florit Serrate and Más-Bermejo, 2016; Castro et al., 2017; Gorry, 2016; Reardon, 2016) that limited virus establishment and transmission. To investigate the likelihood of each hypothesis, we examined international travel patterns, yearly transmission of dengue virus (also vectored by Ae. aegypti), local temperature conditions, and news reports. Comparing all three hypotheses, we found that travel patterns and environmental conditions in Cuba could likely have supported a large Zika outbreak in 2016, but that virus establishment, and hence the outbreak, may have been delayed by a country-wide Ae. aegypti control campaign (Figure 5).
Figure 5. Aggressive Aedes aegypti control may have delayed Zika outbreak in Cuba.
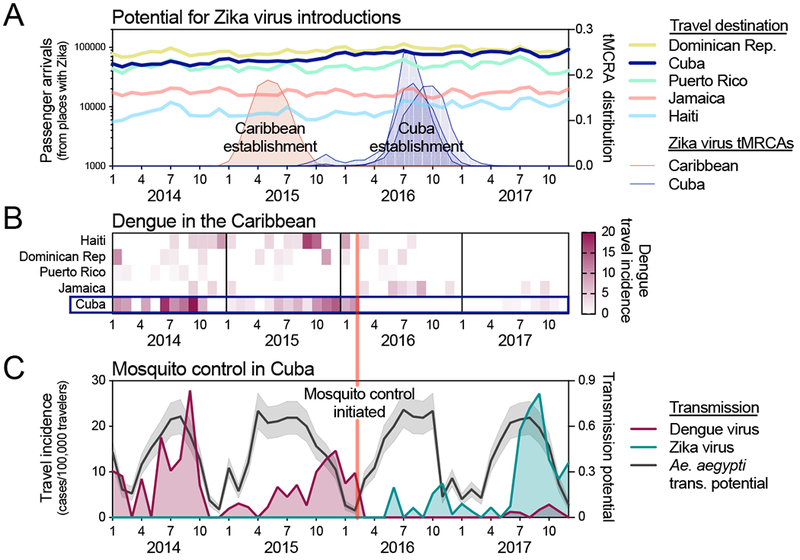
(A)The potential for Zika virus introductions was assessed by total airline passenger arrivals into each of the listed countries per month from 2014-2017 coming from regions in the Americas known to support local Zika virus transmission (excluding the continental United States because the outbreaks were relatively small), along the distribution of likely establishment times (i.e., tMRCAs) of the initial (known) Zika virus establishment in the Caribbean (tMRCA January - September, 2015) and three separate establishments in Cuba (tMRCAs March, 2016 - February, 2017). (B) Monthly dengue virus travel incidence rates (travel cases/100,000 travelers), as reported by the FL-DOH, for Cuba and other large Caribbean Islands were shown to compare Ae. aegypti-borne virus outbreaks. (C) Analysis of dengue and Zika virus incidence, Ae. aegypti transmission potential, and the timing of a reported vector control campaign were used to investigate the delayed Zika outbreak in Cuba. Monthly dengue and Zika virus travel incidence rates (travel cases/100,000 travelers), as reported by the FL-DOH, and relative Ae. aegypti-borne virus transmission potential, determined by a temperature-sensitive model (Mordecai et al., 2017) and monthly temperature from Havana, Cuba, were used to judge the impact of the aggressive Ae. aegypti mosquito control program that was reported to have begun in Cuba during February, 2016. News reports of Zika and dengue cases and the mosquito control campaign in Cuba are summarized in Supplemental File 4. The data used for this figure can be found in Supplemental File 1.
Outbreaks of Ae. aegypti-bome viruses, including Zika virus, require opportunities for virus introductions and conducive conditions to support establishing transmission. As air travel is the main source of long-distance virus dispersion (Khan et al., 2014; Nunes et al., 2014; Semenza et al., 2014), we analyzed air travel patterns to determine if Cuba had fewer opportunities for virus introductions early during the Zika epidemic, potentially delaying the outbreak (Figure 5A). Using monthly airline passenger arrivals coming from all 48 countries and territories in the Americas known to have local Zika virus transmission from 2014-2017, we did not detect any large deviations in air traffic to Cuba during 2015 when Zika virus was becoming established elsewhere in the Caribbean (Figure 5A). Moreover, air travel volumes were higher into Cuba than neighboring islands with large outbreaks in 2016, including Puerto Rico and Jamaica (Figure 5A). These findings suggest that the delayed outbreak in Cuba was not the result of the country having fewer opportunities for Zika virus introductions than other Caribbean islands early in the epidemic.
It is possible that conditions in Cuba from 2015 to 2016, unlike other Caribbean islands, were not conducive for Zika virus establishment and large Ae. aegypti-borne virus outbreaks. To explore this scenario, we examined Caribbean outbreaks of another Ae. aegypti-borne virus, dengue virus, using travel surveillance that we performed based on data reported by the FL-DOH. We found that dengue outbreaks in the Caribbean were more varied than the Zika and chikungunya outbreaks (Figures 3, 5B). Importantly, however, whereas Cuba had dengue virus transmission in 2014, 2015, and 2017, it did not have an outbreak of dengue in 2016 (Figure 5B, C, see also (FL DOH, 2018; Pentón, 2018)), which is similar to what we observed for Zika (Figure 1B). This was despite other Caribbean islands having both dengue and Zika virus transmission in 2016 (Figures 3A, 5B). These findings suggest that, unlike other Caribbean islands, Cuba was not conducive for large Ae. aegypti-borne virus outbreaks in 2016; however, they do not reveal the underlying cause.
We next investigated if environmental factors could have been responsible for making the conditions in Cuba non-conducive for Ae. aegypti-borne virus outbreaks in 2016. Temperature is the primary seasonal factor driving Ae. aegypti-borne virus transmission, as it influences mosquito development, survival, reproduction, biting rates, and vector competence (Caminade et al., 2016; Mordecai et al., 2017; Siraj et al., 2017). To determine if weather conditions in 2016 could have delayed Zika virus establishment, we used a model that estimated when transmission was most likely to occur based on favorable temperature ranges for mosquito-borne transmission (Mordecai et al., 2017). Using temperature data for Cuba, we found that Ae. aegypti transmission potential was as high in 2016 as it was during prior dengue outbreaks, and the Zika outbreak in 2017 (Figure 5C). These findings suggest that environmental factors were likely not responsible for delaying the Zika outbreak in Cuba.
We previously demonstrated that mosquito control campaigns can reduce Ae. aegypti populations and human Zika virus infections (Grubaugh et al., 2017). Cuba has a long history of successful Ae. aegypti control (Gubler, 1989; Guzmán and Kourí, 2009; Toledo et al., 2007), and following the detection of the Zika outbreak in Brazil, the country implemented a “National Zika Action Plan” for aggressive Ae. aegypti mosquito surveillance and control (Castell-Florit Serrate and Más-Bermejo, 2016; Castro et al., 2017; Gorry, 2016; Reardon, 2016). To investigate if mosquito control may have played a role in delaying the Zika outbreak in Cuba, we compared the reported start of the mosquito control campaign to the Zika and dengue outbreaks in Cuba (Figure 5B). We found that immediately following the reported implementation of mosquito control in February, 2016, our travel surveillance showed minimal transmission of both dengue and Zika viruses throughout the year (Figure 5B, C). By searching news articles for Zika and dengue in Cuba from 2015-2018, we found that Cuban officials reported that the mosquito control program was successful (Supplemental File 4), although we were unable to obtain empirical data to support this claim. The timing of the mosquito control campaign, followed by a decrease in both dengue and Zika cases (Figure 5B, C) - despite high transmission potential (Figure 5C) - suggests that mosquito control efforts may have been responsible for delaying the establishment of Zika virus in Cuba, thereby leading to a postponement of the outbreak.
Potential for global spread from unrecognized local outbreaks
Unrecognized and delayed outbreaks have the risk of ‘silently’ spreading viruses to other parts of the world. Using our travel surveillance framework and global suitability for Ae. aegypti mosquitoes, we identified several regions where Zika virus could have been spread from an unrecognized outbreak in Cuba during 2017.
Based on the occurrence of travel-associated Zika cases reported by the FL-DOH and the ECDC, we found that Zika virus transmission in Cuba was the most intense between June-December of 2017 (Figure 6A). We then used this time period to assess where local mosquito-borne Zika virus transmission could have been established from Cuba using global air travel data from Cuba and previously estimated world-wide Ae. aegypti suitability (Kraemer et al., 2015) (Figure 6B). Out of a total of ~4 million air travelers departing Cuba between June and December of 2017, we found 18 countries and U.S. states that received >20,000 travelers, with >100,000 arriving in Florida, Canada, Mexico, and Spain (Figure 6B). Based on environmental suitability for Ae. aegypti of the 18 areas with >20,000 travelers from Cuba, we estimated that Florida, Mexico, Panama, Venezuela, and Colombia were most at risk of Zika virus having been introduced from Cuba during June-December, 2017 (Figure 6B). Indeed, four local Zika cases were reported in Florida during 2017 linked to their partners having recently returned from Cuba (FL DOH, 2017a, 2017b, 2018). Despite these findings, however, beyond a few cases, no Zika outbreaks were reported in these 18 regions in 2017, perhaps due to existing herd immunity (Netto et al., 2017; Zambrana et al., 2018). These results show the global connectedness of Zika endemic areas, and with Zika cases associated with travel from the Americas ongoing as of October, 2018 (Figure 1), continued surveillance is required to detect potential further spread.
Figure 6. Risk of ‘silent’ Zika virus spread from the outbreak in Cuba during 2017.
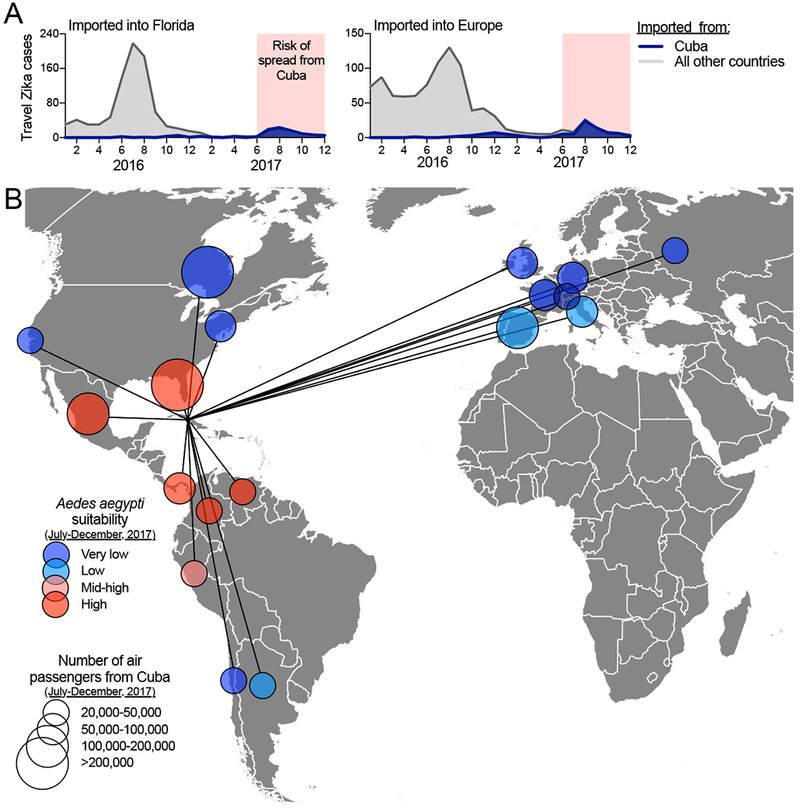
Travel volumes from Cuba and Ae. aegypti suitability were used to address the potential spread of Zika virus from Cuba during the outbreak in 2017. (A) Monthly Zika cases associated with international travel reported by the FL-DOH and the ECDC, sorted by travel origins in Cuba or all other countries/territories in the Americas, were used to demonstrate that >98% of all travel-associated Zika cases during June-December of 2017 came from Cuba. (B) During June-December, 2017, all countries and U.S. states that received > 20,000 airline passengers from Cuba are shown, along with the relative Ae. aegypti suitability, to represent possible destinations for Zika virus spread from Cuba. The data used for this figure can be found in Supplemental File 1.
Discussion
Travel surveillance and genomic epidemiology to detect Zika outbreaks
Using travel surveillance and virus genomics, we discovered a Zika outbreak in Cuba during 2017, a period in which the epidemic was waning across the Americas (PAHO, 2017a, 2017b) (Figures 1, 2). A single report about a Zika outbreak in Cuba made the news in 2017 (Reuters, 2017), but critically, cases were not reported to PAHO (PAHO, 2017c), or other public health agencies, and thus went undetected by the international community. With Zika outbreaks still arising in new locations, including Angola, India, Cabo Verde, Vietnam, and Thailand (Hill et al., 2019; Lourenço et al., 2018; Meltzer et al., 2016; Phumee et al., 2019; Ruchusatsawat et al., 2019; Yadav et al., 2019), it is important to identify and report lingering outbreaks to better prepare for potential future spread (Bogoch et al., 2016; Kraemer et al.,2016).
Epidemiological updates by the WHO (including PAHO) and other public health organizations are the primary methods for disseminating information about infectious disease outbreaks and epidemics. Critically, they rely on accurate case reporting from individual countries and territories, but depending on resources and priorities, reporting of local outbreaks may not be accurate. To overcome some of these limitations, studies based on the detection of infected travelers have been used to fill knowledge gaps about ongoing outbreaks from places that are difficult to sample, as has been previously reported for Zika (Leder et al., 2017; Wilder-Smith et al., 2018). In this study, we built on this framework to not just detect unreported outbreaks (Figure 1), but to include travel patterns, comparisons to local reports from other locations, and virus genomics to reconstruct outbreaks in the absence of local data.
The utility of combining travel surveillance with genomic epidemiology, however, is limited to the travel patterns and destinations of the people included in the surveillance network. By also using travel data from Europe, we were able to capture Zika cases from countries that we could not from Florida (Figure 2A), but we did not detect any infected travelers coming from other known Zika outbreaks in Angola, India, Cabo Verde, Vietnam, and Thailand. Thus, using travelers as sentinels alone cannot provide a complete global picture of ongoing Zika outbreaks, especially when the numbers of cases or travelers are low.
Estimating the size of the Zika outbreak in Cuba
We estimate that the 2017 Zika outbreak in Cuba was similar in size to outbreaks from other Caribbean islands that peaked the year prior (Figure 2). Our analyses utilize the relationships between local cases and travel surveillance from non-Cuba countries, in combination with travel volumes and travel associated cases from Cuba. Other studies have used travelers to estimate local case numbers from influenza (Fraser et al., 2009) and Middle East Respiratory Syndrome outbreaks (Cauchemez et al., 2014a) by assuming that locals and travelers were equally likely to get infected. While that assumption may have been correct for these outbreaks, for Zika, we found that epi curves generated based on local or travel data were correlated, but the incidence rates were different by orders of magnitude (Figure 2A). This suggests that public health systems differ in their ability to detect Zika cases and/or locals and travelers have different risks for Zika infections. For example, because of differences in public health infrastructure and resources, Zika case reporting in Haiti (where our travel incidence is higher than local reporting, Figure 2A) may be less accurate than Puerto Rico (where our travel incidence is lower than local reporting, Figure 2A) (Braga et al., 2017; Dowell et al., 2011). Additionally, location dependent factors such as common tourist behaviors and lengths of stay could also alter the risks for traveler infection (Cauchemez et al., 2014a; Fraser et al., 2009). Thus, our approach of using the relationship between local and travel incidence data from locations with local reporting as a calibration might be a more accurate method for reconstructing Zika outbreaks than using travel incidence alone.
There are limitations to our approaches that may influence our ability to estimate the size of the Zika outbreak in Cuba. First, accurate travel data are necessary to calculate travel incidence rates of Zika cases. This is challenging for Cuba, as travel policies from the United States have repeatedly changed during the past few years (Robles, 2016), and we found that travel volumes between Cuba and Florida increased in 2017 compared to 2016 (Figure S1A). To minimize this issue, we included air travel from both scheduled commercial flights from the International Air Transportation Association (IATA, 2018) and chartered flights from the U.S. Department of Transportation (US DOT, 2018). We also obtained travel surveillance data from Europe, where the travel policies to Cuba have not recently changed, with travel volumes remaining relatively stable (Figure S1A). As we previously showed that cruise ship traffic greatly outnumbered airline traffic during the 2016 Zika outbreak in Florida (Grubaugh et al., 2017), we also investigated cruise ships to Cuba, however, we found their travel volume to be much smaller than airlines for this country (Figure S3). Second, our size estimates are based on averaging across all regions, some of which may be more, or less, representative of the Zika outbreak in Cuba. While we found a strong correlation between epi curves generated from travel associated Zika cases and local reporting, variability in the ratio between local and travel incidence among countries resulted in a wide interquartile range (1,071 to 22,611) on our mean estimate of 5,707 unreported Zika cases in Cuba. Our mean estimate, however, is consistent with the only two public reports from the outbreak in Cuba of 187 cases in 2016 reported by PAHO (PAHO, 2017c) and 1,847 cases in 2017 reported by the news agency Reuters (Reuters, 2017). Zika outbreaks from other locations in the Americas with comparable population sizes to Cuba were also reported to be similar in size, which would still be true even at the lower end of our interquartile range (Figure 2C).
Zika outbreaks peak a year after virus establishment
By sequencing Zika virus genomes from travelers infected in Cuba, we demonstrate that the 2017 outbreak peaked 11-13 months after the virus was established, a time frame that was consistent for other Zika outbreaks in the Caribbean (Figure 4C). Other studies from Brazil, Central America, and Mexico have found similar patterns (Faria et al., 2017; Thézé et al.,2016). This suggests that the amount of time necessary for newly established Zika virus lineages to intensify in transmission to reach peak outbreak size will often require maintenance of the virus during seasons when mosquito abundance is low (e.g., by vertical transmission (Thangamani et al., 2016)). While the factors supporting virus maintenance are still unclear, it is plausible that Zika virus may survive low mosquito abundance through a combination of low level mosquito-to-human transmission, vertical transmission in mosquitoes (da Costa et al., 2018; Thangamani et al., 2016), and, to a lesser extend, human sexual transmission (Althaus and Low, 2016). Considering that all known large Zika outbreaks in the Americas, including Cuba, may have involved prolonged virus maintenance, a better understanding of how Zika virus is maintained during low mosquito abundance could lead to novel vector control and outbreak mitigation strategies.
Factors responsible for delaying the establishment of Zika virus in Cuba
By reconstructing other Ae. aegypti vectored outbreaks, analyzing climatic conditions, investigating news reports, and modeling mosquito abundance, our study suggests that the establishment of Zika virus in Cuba may have been delayed by an Ae. aegypti control campaign (Figure 5C). This accomplishment highlights the value of mosquito control for limiting transmission (Grubaugh et al., 2017), as Cuba may have been able to reduce the local burden of both dengue and Zika, despite otherwise conducive environmental conditions to support transmission of the viruses (Figure 5B). Publicly available reports indicate that the response to Zika by Cuban authorities intensified in early 2016 and included active and passive surveillance, training of health professionals, communication and mobilization, and the application of adulticides and larvicides for mosquito control (Castell-Florit Serrate and Más-Bermejo, 2016; Castro et al., 2017; Gorry, 2016; Reardon, 2016). This strategy was built upon years of effective dengue control through capacity building and research (Guzmán, 2005; Guzmán and Kourí, 2009). While our observation of the suppression of both dengue virus transmission and Zika virus establishment during periods that we found were otherwise suitable for Ae. aegypti-borne transmission (Figure 5C) suggests that the mosquito control campaign was successful, we were unable to obtain empirical data to confirm these findings. Competition between dengue and Zika viruses - as previous exposure to dengue virus may protect against Zika virus infections in humans (Gordon et al., 2019; Rodriguez-Barraquer et al., 2019) - may also have played a role in delaying the establishment of Zika virus in Cuba. However, how cross-protection, or other epidemiological interactions (Vogels et al., 2019), between these two flaviviruses may impact outbreaks is not currently clear, and such potential virus ‘competition’ would likely not be unique to Cuba. For future studies, accurate data on Zika virus seroprevalence in Cuba and other Caribbean islands would be helpful to address these questions. In addition, the availability of empirical mosquito abundance data would allow for an assessment of year-to-year differences in transmission potential and to specifically test if Ae. aegypti populations were reduced during the control campaign in Cuba, as we previously observed in Florida (Grubaugh et al., 2017). Importantly, vector abundance studies, including making such data publicly available, should be prioritized and more fully supported for future mosquito-borne virus outbreaks (Rund and Martinez, 2017).
Conclusions
During rapidly evolving outbreaks, a lack of access to reliable local data can often limit response efforts. By combining travel surveillance with genomic epidemiology, we were able to reconstruct infectious disease outbreaks when local reporting was insufficient. This framework can be applied more generally to detect hidden outbreaks, and future applications may include the integration of empirical data on vector abundance and population immunity. By combining data and resources across multiple scales, we can thus develop surveillance frameworks to uncover epidemiological dynamics of emerging, re-emerging, and endemic infectious diseases across the world.
STAR★Methods
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for data, resources, and reagents should be directed to and will be fulfilled by the Lead Contact, Kristian G. Andersen (andersen@scripps.edu). This study did not generate new unique reagents, but raw data and code generated as part of this research can be found in the Supplemental Files, as well as on public resources as specified in the Data and Code Availability section below.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Ethical statement
This work was evaluated and approved by the Institutional Review Boards (IRB) at The Scripps Research Institute. This work was conducted as part of the public health response in Florida and samples were collected under a waiver of consent granted by the FL-DOH Human Research Protection Program. The work received a non-human subjects research designation (category 4 exemption) by the FL-DOH because this research was performed with remnant clinical diagnostic specimens involving no more than minimal risk. All samples were de-identified before receipt by the study investigators, and information regarding the age and sex and/or gender were not provided.
METHOD DETAILS
Local cases and incidence rates
PAHO is the primary source for information regarding Zika virus spread in the Americas, as well as suspected and confirmed cases per country and territory (PAHO, 2017a). Weekly case counts, however, are made available as cumulative cases, not the number of new cases per week. These data are often problematic for reconstructing outbreak dynamics because of reporting delays and ‘spikes’ (e.g., more than one week of cases submitted after weeks of no reporting). Curated weekly case counts per country and territory are presented as bar graphs (not as datasheets) (PAHO, 2017a). Therefore, to increase the accuracy of calculating Zika virus incidence rates, we captured screenshots of the 2016-2017 weekly Zika virus case (suspected and confirmed) distributions, and extracted the case counts using Web Plot Digitizer v3.10 (http://arohatgi.info/WebPlotDigitizer), which we previously validated (Grubaugh et al., 2017). Extracted case numbers were recorded in .csv files and aggregated per month for this analysis. Yearly human population numbers were retrieved from the United Nations Population Division (https://population.un.org/wpp/) and were used to calculate monthly local Zika virus incidence rates (suspected and confirmed Zika cases/100,000 population) per country and territory. Monthly Zika cases and incidence rates are available at: https://github.com/andersen-lab/paper_2018_cuba-travel-zika.
The Zika-Epidemiological Report for Cuba (PAHO, 2017c), which was last updated on 25 September 2017, states: “Between EW [epidemiological week] 1 and EW 52 of 2016, a total of 187 laboratory confirmed cases of autochthonous Zika virus disease were reported. No information is available on the distribution of cases by epidemiological week. No new information was provided since EW 52 of 2016.” This shows that Zika cases from Cuba were not reported to PAHO in 2017.
Travel-associated cases and incidence rates
Weekly cumulative travel-associated Zika, dengue, and chikungunya case numbers were collected from 2014-2018, and are publically available from the FL DOH (FL DOH, 2018). The cases reported on the FL DOH database include those that were confirmed by both PCR and serological assays, and within and without symptoms onset dates (note that many of the pregnant women that were serologically positive for Zika virus were asymptomatic). A travel history was also recorded for most patients. For this study, we only included PCR positive cases with a known date for the onset of symptoms and who only traveled to one international location within the 2 weeks prior to symptoms onset so we could more accurately sort the temporal and spatial distribution of travel-associated cases. We also excluded cases with sexual or congenital exposure. We aggregated the data by month of symptoms onset and by location of likely exposure (i.e., travel origin). Of the travel-associated Zika cases diagnosed in Florida (n = 1,333), 49% were visiting friends and relatives, 17% were refugees or immigrants, 17% were traveling for tourism, 3% were traveling for business, and 14% were traveling for unknown or other reasons. Of the travel-associated dengue virus cases where the questionnaire was given (only started for dengue in 2016, n = 88), 67% were visiting friends and relatives, 25% were traveling for tourism, and 8% were traveling for other reasons.
We also requested travel-associated Zika cases from the ECDC European Surveillance System (TESSy) (ECDC, 2017). We requested all travel-associated Zika cases reported to the ECDC during 2016-2017, sorted by month of symptoms onset, reporting country, and location of likely exposure. The data was provided by Austria, Belgium, Czech Republic, Denmark, Finland, France, Greece, Hungary, Ireland, Italy, Luxembourg, Malta, The Netherlands, Norway, Portugal, Romania, Slovakia, Slovenia, Spain, Sweden, and the United Kingdom, and released by ECDC. The raw travel-associated case counts from Europe has not been published, was obtained through specific request from the ECDC, and we do not have permission to make it public. In addition, the views and opinions that we expressed herein do not necessarily state or reflect those of ECDC. The accuracy of our statistical analysis and the findings we report are not the responsibility of ECDC. ECDC is not responsible for conclusions or opinions drawn from the data provided. ECDC is not responsible for the correctness of the data and for data management, data merging, and data collation after provision of the data. ECDC shall not be held liable for improper or incorrect use of the data.
Data on travelers to Cuba diagnosed at GeoSentinel sites were also analyzed. The GeoSentinel Global Surveillance Network consists of 72 specialized travel and tropical medicine clinics in 32 countries, and is staffed by specialists in travel and tropical medicine (http://www.istm.org/geosentinel). The GeoSentinel clinics provide routine clinical care to ill travelers and contribute de-identified demographic, travel, and clinical surveillance data on patients with travel-related illnesses to a centralized database (Harvey et al., 2013; Leder et al., 2013). Patient records with Cuba listed as the country of exposure and a diagnosis of mosquito-acquired Zika virus infection were extracted from the GeoSentinel database for the time period January 1, 2016 to November 12, 2018. Only confirmed cases were included in this analysis; these were defined as Zika virus PCR-positive in serum or urine, or Zika virus-specific IgM in serum and Zika virus antibody titers greater than four-fold higher than antibody titers for dengue or other flaviviruses or a four-fold rise in anti-Zika virus IgG and Zika virus antibody titers greater than four-fold higher than antibody titers for dengue or other flaviviruses (Hamer et al., 2017).
Monthly travel incidence rates from all exposure (origin) and reporting (destination) combinations were calculated by number of travel-associated cases per 100,000 airline passengers (from origin to destination/month). Exposure-reporting combinations that accounted for less than 20 imported cases were not included in analysis. Air travel data was obtained as described below.
Though we previously hypothesized cruise ships may have an underrecognized role in Zika virus spread (Grubaugh et al., 2017), we did not use data from Zika virus infections that may have been associated with cruise travel, and thus did not collect cruise ship data for this study. First, there were very few infections linked to cruise travel in our dataset, which may be because these cases would more likely be tourists diagnosed elsewhere (and just visiting Florida for the cruise departure). Second, many of the reported cruise-related Zika infections were associated with more than one site for potential exposure, making it difficult to estimate local incidence rates (we removed all travel cases with multiple locations of potential exposure from our analyses). Third, scheduled cruise ship passengers arriving in Florida that stopped in Cuba are predicted to be substantially fewer (11,675/month scheduled for 2019; crawled from CruiseMapper: https://www.cruisemapper.com/) than air travel passengers from Cuba to Florida (80,366/month in 2017) (Figure S3). Cruise travel between Cuba and Florida only began in 2016 (Vora, 2016), and thus there would have been even fewer passengers during our primary study period between 2016-2017.
The travel incidence rates derived from data collected from the FL DOH and ECDC and the curated travel-associated cases from Florida are available at: https://github.com/andersen-lab/paper_2018_cuba-travel-zika.
Air passenger volumes
We collected air passenger volumes to calculate Zika, dengue, and chikungunya virus travel incidence rates, to assess the potential for Zika virus importations into Cuba, and to investigate potential Zika virus spread from Cuba. From the IATA (IATA, 2018), we obtained the number of passengers traveling by air between all destinations in the Americas, plus to all global destinations from Cuba, from 2010-2017. IATA data consists of global ticket sales which account for true origins and final destinations, and represents 90% of all commercial flights. The remaining 10% of trips are modeled using airline market intelligence. One limitation of IATA data is it does not include chartered flights, which through our investigations, was only an issue for flights to and from the United States and Cuba. To make up for this, we obtained chartered flight data from Cuba to Florida during 2014-2017 from the U.S. DOT (US DOT, 2018). The US DOT publicly reports the number of passengers on all commercial and chartered flights departing and arriving in airports in the United States and includes origin and destination. Summarized air passenger volumes are available at: https://github.com/andersen-lab/paper_2018_cuba-travel-zika.
Estimated local Zika cases in Cuba
We used two data types—locally acquired cases by country and Florida travel cases by country—to inform estimates of per capita local incidence in Cuba on a scale comparable to local incidence in other countries. We limited our analysis of countries besides Cuba to those with a correlation between monthly local and travel cases >0.25 (n=27), which appeared to be a natural breakpoint in the distribution of correlations. For each, we used the fda (https://cran.r-project.org/web/packages/fda/index.html) package in R to model per capita local incidence of Zika over time with univariate cubic B-spline functions with four knots per year for two years (2016-2017) described by parameters ·. We assumed that incidence among travelers from each country followed the same temporal pattern as local incidence but the two differed in magnitude by a factor ·, which could be due to differences in exposure or health-seeking behavior between international travelers and the general population. To estimate · and · for each of the 27 countries, we modeled local and travel incidence for each month as independent binomial random variables, with incidence as the number of “successes” and country population and number of travelers, respectively, as the number of “trials.” Logit-transformed values of the spline functions informed the probability of success in each trial. Based on this likelihood formulation and with non-informative priors, we estimated · and · for each country using a Metropolis-Hastings implementation of Markov chain Monte Carlo (MCMC). We assessed convergence by calculating Gelman-Rubin statistics on five replicate chains, and we performed posterior predictive checks on cumulative local incidence (Figure S4) and travel incidence (Figure S5) (Thompson Hobbs and Hooten, 2015). On the basis of Bayesian p-values < 0.05 on these posterior predictive checks, we removed four countries from subsequent analyses (leaving n=23 countries). To estimate per capita local incidence in Cuba, we first estimated · for Cuba in a similar manner, but based on travel data only. We then took 104 values of · drawn randomly from the posteriors of · pooled across 23 countries and multiplied them by random samples from the posterior of per capita travel incidence curves from Cuba to obtain a set of 104 predictions of per capita local incidence curves for Cuba. R code and posterior samples are available at: https://github.com/andersen-lab/paper_2018_cuba-travel-zika.
Zika virus sequencing
Zika virus RNA was sequenced using a highly multiplexed PCR approach, called PrimalSeq, that we previously described (Grubaugh et al., 2019b; Quick et al., 2017). Detailed protocols, including the primer scheme “ZIKV - Asia/America - 400bp” we used here to amplify Zika virus, can be found online (http://grubaughlab.com/open-science/amplicon-sequencing/ and https://andersen-lab.com/secrets/protocols/). In brief, virus RNA (2 μL) was reverse transcribed into cDNA using Invitrogen Superscript IV VILO (20 μL reactions). Virus cDNA (2 μL) was amplified in 35 × ~400 bp fragments from two multiplexed PCR reactions using Q5 DNA High-fidelity Polymerase (New England Biolabs). Virus amplicons from the two multiplex PCR reactions were purified and combined (25 ng each) prior to library preparation. The libraries were prepared using the Kapa Hyper prep kit (Kapa Biosystems, following the vendor’s protocols but with 1/4 of the recommended reagents) and NEXTflex Dual-Indexed DNA Barcodes (BIOO Scientific, diluted to 250 nM). Mag-Bind TotalPure NGS beads (Omega) were used for all purification steps. The libraries were quantified and quality-checked using the Qubit (Thermo Fisher) and Bioanalyzer (Agilent). Paired-end 250 nt reads were generated using the MiSeq V2 500 cycle kits (Illumina).
Our open source software package, iVar (Grubaugh et al., 2019b), was used to process the Zika virus sequencing data and call the consensus sequences. Source code and detailed documentation for iVar can be found at https://github.com/andersen-lab/ivar. In brief, BWA (Li and Durbin, 2009) was used to align the paired-end reads to a reference genome (GenBank KX087101). The primer sequences were trimmed from the reads using a BED file, with the primer positions, followed by quality trimming. The consensus sequence was called by the majority nucleotide at each position with >10x coverage. All alignments and consensus sequences were visually inspected using Geneious v9.1.5 (Kearse et al., 2012). The Zika virus sequences generated from Cuba can be found using the NCBI Bioproject PRJNA438510 and Genbank IDs can be found in Supplemental File 2.
Phylogenetic analyses
All available complete or near complete Zika virus genomes of the Asian genotype from the Pacific and the Americas were retrieved from GenBank in August, 2018. A total of 283 Zika virus genomes collected between 2013 and 2018 from Cuba (n = 10, including 9 generated in this study) and elsewhere from the Pacific and the Americas (n = 273, including 4 generated in this study from Florida, USA) were codon-aligned together using MAFFT (Katoh and Standley, 2013) and inspected manually.
To determine the temporal signal of the sequence dataset, a maximum likelihood (ML) phylogeny was first reconstructed with RAxML (Stamatakis, 2014) using the general time-reversible (GTR) nucleotide substitution model and gamma-distributed rates amongst sites (Guindon and Gascuel, 2003; Yang, 1994). The resulting tree was rooted on Zika virus sequence KX369547 (French Polynesia). Then, a correlation between root-to-tip genetic divergence and date of sampling was conducted in TempEst (Guindon and Gascuel, 2003; Rambaut et al., 2016; Yang, 1994). Time-scaled phylogenetic trees were reconstructed using the Bayesian phylogenetic inference framework available in BEAST v1.10.2 (Suchard et al., 2018). Accommodating phylogenetic uncertainty, we used an HKY+Γ4 nucleotide substitution model for each codon position, allowing for relative rates between these positions to be estimated, and an uncorrelated relaxed molecular clock model, with an underlying lognormal distribution (Drummond et al., 2006), a non-parametric Skygrid demographic prior (Gill et al., 2013) and otherwise default priors in BEAUti v1.10.2 (Suchard et al., 2018). The MCMC analysis was run for 1 billion iterations, sampling every 100,000th iteration, using the BEAGLE library v2.1.2 to accelerate computation (Ayres et al., 2012). MCMC performance was inspected for convergence and for sufficient sampling using Tracer v.1.7.1 (Rambaut et al., 2018). After discarding the first 200 million iterations as burn-in, virus diffusion over time and space was summarised using a maximum clade credibility (MCC) tree using TreeAnnotator (Suchard et al., 2018). Tree visualizations were generated with the Phylo (Talevich et al., 2012) module from Biopython and matplotlib (Hunter, 2007). Raw MAFFT codon alignment data, PhyML tree, BEAST XML file, and BEAST MCC time-structured phylogeny can be found in Supplemental File 3 and at: https://github.com/andersen-lab/paper_2018_cuba-travel-zika.
Aedes aegypti transmission potential
Temperature is an important predictor of Ae. aegypti-borne virus transmission, as it affects mosquito population sizes (i.e., mosquito development, survival, and reproduction rates), interactions between mosquitoes and human hosts (i.e., biting rates), and mosquito transmission competence (i.e., mosquito infection and transmission rates) (Caminade et al., 2016; Mordecai et al., 2017; Siraj et al., 2017). Virus transmission by Ae. aegypti can occur between 18–34°C and peaks at 26–29°C (Mordecai et al., 2017). To assess yearly and seasonal variations in Ae. aegypti transmission potential for dengue and Zika virus, we used a temperature-dependent model of transmission using a previously developed R0 framework (Mordecai et al., 2017). By focusing this analysis on Havana, we controlled for spatial drivers of transmission and thereby isolated a representative example of temporal patterns in transmission potential. Using hourly temperature data obtained from OpenWeatherMap (https://openweathermap.org/), we calculated monthly mean temperature and used it to calculate monthly R0 as estimated by Mordecai et al. (Mordecai et al., 2017) (https://figshare.eom/s/b79bc7537201e7b5603f). Doing so for 5,000 samples from the posterior of temperature-R0 relationships and normalizing between 0 and 1 yielded a description of relative Ae. aegyptitransmission potential per month in Havana, Cuba during 2014-2017. Aggregated monthly temperature data for and model outputs are available at: https://github.com/andersen-lab/paper_2018_cuba-travel-zika.
Relative global Aedes aegypti suitability
To investigate the potential for Zika virus transmission and establishment, we used previously generated Ae. aegypti suitability maps (Kraemer et al., 2015) based on the statistical relationships between mosquito presence and environmental correlates (Bogoch et al., 2016). Maps were produced at a 5-km × 5-km resolution for each calendar month and then aggregated to the level of the U.S. states, countries, and territories, as used previously (Gardner et al., 2018). Relative Ae. aegypti suitability (i.e., very low, low, mid-high, and high) was then derived by using the mean aggregated values for each U.S. state, country, and territory, and also the mean value for the study period (June-December, 2017). The U.S. state, country, and territory suitability means and standard deviations can be found at: https://github.com/andersen-lab/paper_2018_cuba-travel-zika.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses were performed using BEAST and R and are described in the Figure legends and in the Method Details.
DATA AND CODE AVAILABILITY
The Zika virus sequences generated here can be found using the NCBI BioProject PRJNA438510 and individual Genbank IDs can be found in Supplemental File 2. Data used to create the figures can be found in the supplemental files. The raw data and results for our analyses can be found at: https://github.com/andersen-lab/paper_2018_cuba-travel-zika.
ADDITIONAL RESOURCES
Data analysis methods: https://github.com/andersen-lab/paper_2018_cuba-travel-zika Sequencing protocols: http://grubaughlab.conn/open-science/amplicon-sequencing/ and https://andersen-lab.com/secrets/protocols/.
Sequencing bioinformatic software: https://github.com/andersen-lab/ivar
Supplementary Material
Figure S1. Increase in travel Zika cases from Cuba is not due to an increase in travel, related to Figure 1. To assess if the 2016 to 2017 increase in Zika cases reported from travelers who recently visited Cuba were due to a Zika outbreak in Cuba or an increase in air travel passengers leaving Cuba, we compared the yearly travel volumes from Cuba to Florida, Spain, and Italy to the travel Zika cases from Cuba reported by the same counties during 2016 and 2017. The greater increase from 2016 to 2017 of travel cases compared to travel volume suggests that a Zika outbreak in Cuba was the cause.
Supplemental File 3, related to Figure 4. Raw MAFFT codon alignment data, PhyML tree, and BEAST MCC time-structured phylogeny.
Supplemental File 4, related to Figure 5. Timeline of selected news articles from 2015-2017 related to Zika and dengue virus transmission in Cuba.
Figure S2. Relationship between the number of travel-associated Zika cases and the correlation between local and travel incidence rates, related to Figure 2. To determine the number of travel-associated infections needed to infer the shape of a local outbreak, we compared the total travel-associated Zika cases from each exposure-reporting country/territory combination (x-axis) with Pearson correlation between the local and travel incidence rates corresponding to the combination (y-axis). The travel-associated Zika cases were totaled from 2016-2017. For the Pearson correlations between local-travel incidence rates, monthly incidence values from 2016-2017 were compared. When there were >20 travel-associated cases, the local-travel Pearson r was >0.5, indicating a strong positive correlation and that the travel cases can help determine the shape of the local outbreak. The lone exception to that finding was from travelers from Barbados diagnosed in the United Kingdom (UK) because the travel cases miss the locally reported Zika virus peak during January-February, 2016, but they correlate with the second local peak during July-October, 2016 (Figure 2A). In our dataset, there were 25 Zika virus infections diagnosed in Italy with recent travel to Cuba, 30 diagnosed in Spain, and 98 diagnosed in Florida. These totals are all within the range of strong positive correlations between local and travel incidence, justifying their use to infer the local Zika outbreak dynamics in Cuba (Figure 2B).
Figure S3. Cruise ship travel from Cuba to Florida is minimal compared to air travel, related to Figure 2. Cruise ships may be a mechanism for infected individuals to travel to other countries, thus we investigated if we should incorporated cruise ship passenger volumes for calculating travel Zika incidence. The ~7× smaller volume of scheduled monthly cruise ship travel from 2019 compared to air travel in 2017 suggested that this would minimally impact our estimates and the data was not included in our final analyses.
Figure S4. Posterior predictions of estimated total travel incidence from origin country into Florida, related to Figure 2. These distributions were used to inform the joint distribution between travel incidence and local incidence that was used to estimate local incidence in Cuba. Empirical total travel cases per country indicated by red vertical line. All of the countries shown above, besides Cuba, had >0.25 correlation between local incidence and travel incidence and had the observed value fall within the 95% posterior predictive interval of the distribution.
Figure S5. Posterior predictions of estimated total local incidence from origin country into Florida, related to Figure 2. These distributions were used to inform the joint distribution between travel incidence and local incidence that was used to estimate local incidence in Cuba. Empirical total local cases per country indicated by red vertical line. Estimated local incidence of Cuba indicated in blue with no empirical value. All of the countries shown above, besides Cuba, had >0.25 correlation between local incidence and travel incidence and had the observed value fall within the 95% posterior predictive interval of the distribution.
Figure S6. Maximum likelihood tree and root-to-tip regression of Zika virus genomes from Cuba and the epidemic in the Americas, related to Figure 4. (A) Maximum likelihood tree of publicly available Zika virus sequences (n = 269) and sequences generated in this study (n = 14). Tips are coloured by location. Bootstrap support values are colored at the nodes. Divergence shown as substitutions per site. “1-3 Cuba” represent three independent introductions of Zika virus into Cuba. (B) Linear regression of sample tip dates against divergence from root based on sequences with known collection dates estimates an evolutionary rate for the Zika virus phylogeny of 5.71 × 10−3 nucleotide substitutions per site per year.
Supplemental File 2, related to Figure 4. Zika virus genomes generated during this study.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological Samples | ||
| Clinical samples | FL DOH | NA |
| Critical Commercial Assays | ||
| SuperScript IV VILO Master Mix | ThermoFisher | Cat # 11756050 |
| Q5 High-Fidelity 2X Master Mix | New England BioLabs | Cat # M0492S |
| Qubit High Sensitivity dsDNA kit | ThermoFisher | Cat # Q32851 |
| KAPA HyperPrep kit | Roche | Cat # KK8504 |
| Mag-Bind TotalPure NGS | Omega Bio-Tek | Cat # M1378-01 |
| BIOO Scientific NEXTflex Dual-Indexed DNA Barcodes | PerkinElmer | Cat # NOVA-514160 |
| High Sensitivity DNA Analysis Kit | Agilent | Cat # 5067-5585 |
| MiSeq Reagent Kit v3 | Illumina | Cat # MS-102-3003 |
| Deposited Data | ||
| Local Zika cases | PAHO | (PAHO, 2017b) |
| Travel Zika cases (Florida) | FL DOH | (FL DOH, 2018) |
| Travel Zika cases (Europe) | ECDC | (ECDC, 2017) |
| Travel Zika cases (worldwide) | GeoSentinel | http://www.istm.org/qeosentinel |
| Travel dengue cases (Florida) | FL DOH | (FL DOH, 2018) |
| Travel chikungunya cases (Florida) | FL DOH | (FL DOH, 2018) |
| Air passenger volumes (commercial) | IATA | (IATA, 2018) |
| Air passenger volumes (chartered) | U.S. DOT | (US DOT, 2018) |
| Zika virus genomes | GenBank | https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA438510 |
| Havana, Cuba temperatures | OpenWeatherMap | https://openweathermap.org/ |
| Ae. aegypti suitability | Moritz Kraemer | (Kraemer et al., 2015) |
| Software and Algorithms | ||
| FDA package in R | CRAN | https://cran.r-project.org/web/packages/fda/index.html |
| iVar | Andersen Lab | https://github.com/andersen-lab/ivar |
| Geneious v9.1.5 | https://www.geneious.com/ | (Kearse et al., 2012) |
| MAFFT | https://mafft.cbrc.jp/alignment/software/ | (Katoh and Standley, 2013) |
| RAxML | https://cme.hits.org/exelixis/web/software/raxml/ | (Stamatakis, 2014) |
| TempEST | http://beast.community/tempest | (Rambaut et al., 2016) |
| BEAST v1.10.2 | http://beast.community/ | (Suchard et al., 2018) |
| BEAGLE V2.1.2 | http://beast.community/beagle | (Ayres et al., 2012) |
| Tracer v.1.7.1 | http://beast.community/tracer | (Rambaut et al., 2018) |
| TreeAnnotator | http://beast.community/treeannotator | (Suchard et al., 2018) |
| Phylo | https://biopython.org/wiki/Phylo | (Talevich et al., 2012) |
| Other | ||
| Amplicon sequencing protocol | PrimalSeq | (Quick et al., 2017) |
Highlights.
Travel surveillance and genomics uncovered hidden Zika transmission
An unreported and one-year delayed Zika outbreak was detected in Cuba
Mosquito control may delay, not prevent, Zika virus establishment
A surveillance framework to detect hidden outbreaks was created
Acknowledgements
We thank R. Robles-Sikisaka, M. Pauthner, M.R. Wiley, K. Prieto, S. Taylor, and P. Jack for laboratory support and the ECDC for graciously providing data regarding travel-associated Zika virus infections. The findings and conclusions in this report are those of the authors and do not necessarily represent the official position of the U.S. Army, CDC, ECDC, or the FL-DOH. RJO received support from the Arthur J. Schmitt Leadership Fellowship in Science and Engineering and from an Eck Institute for Global Health Graduate Fellowship. MUGK is supported by The Branco Weiss Fellowship - Society in Science, administered by the ETH Zurich and acknowledges funding from a Training Grant from the National Institute of Child Health and Human Development (T32HD040128). CBFV is supported by NWO Rubicon 019.181EN.004. GeoSentinel, the Global Surveillance Network of the International Society of Travel Medicine, is supported by a cooperative agreement (U50CK00189) from the Centers for Disease Control and Prevention, as well as the International Society of Travel Medicine, and the Public Health Agency of Canada. GB acknowledges support from the Interne Fondsen KU Leuven / Internal Funds KU Leuven under grant agreement C14/18/094. TAP received support from a RAPID award from the National Science Foundation (DEB 1641130) and a Young Faculty Award from the Defense Advanced Research Projects Agency (D16AP0014). SI and SFM are supported by NIH NIAID R01AI099210. KGA is a Pew Biomedical Scholar, and is supported by NIH NCATS CTSA UL1TR002550, NIAID contract HHSN272201400048C, NIAID R21AI137690, NIAID U19AI135995, and The Ray Thomas Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
References
- Althaus CL, and Low N (2016). How Relevant Is Sexual Transmission of Zika Virus? PLoS Med. 13, e1002157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayres DL, Darling A, Zwickl DJ, Beerli P, Holder MT, Lewis PO, Huelsenbeck JP, Ronquist F, Swofford DL, Cummings MP, et al. (2012). BEAGLE: an application programming interface and high-performance computing library for statistical phylogenetics. Syst. Biol 61, 170–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogoch II, Brady OJ, Kraemer MUG, German M, Creatore MI, Brent S, Watts AG, Hay SI, Kulkarni MA, Brownstein JS, et al. (2016). Potential for Zika virus introduction and transmission in resource-limited countries in Africa and the Asia-Pacific region: a modelling study. Lancet Infect. Dis [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braga C, Luna CF, Martelli CM, de Souza WV, Cordeiro MT, Alexander N, de Albuquerque M de FPM, Júnior JCS, and Marques ET (2010). Seroprevalence and risk factors for dengue infection in socio-economically distinct areas of Recife, Brazil. Acta Trop 113, 234–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braga JU, Bressan C, Dalvi APR, Calvet GA, Daumas RP, Rodrigues N, Wakimoto M, Nogueira RMR, Nielsen-Saines K, Brito C, et al. (2017). Accuracy of Zika virus disease case definition during simultaneous Dengue and Chikungunya epidemics. PLoS One 12, e0179725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caminade C, Turner J, Metelmann S, Hesson JC, Blagrove MSC, Solomon T, Morse AP, and Baylis M (2016). Global risk model for vector-borne transmission of Zika virus reveals the role of El Niño 2015. Proc. Natl. Acad. Sci. U. S. A 201614303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castell-Florit Serrate P, and Más-Bermejo P (2016). Cuba: Time to Expand Health in All Policies. MEDICC Rev. 18, 48. [DOI] [PubMed] [Google Scholar]
- Castro M, Pérez D, Guzmán MG, and Barrington C (2017). Why Did Zika Not Explode in Cuba? The Role of Active Community Participation to Sustain Control of Vector-Borne Diseases. Am. J. Trop. Med. Hyg 97, 311–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cauchemez S, Fraser C, Van Kerkhove MD, Donnelly CA, Riley S, Rambaut A, Enouf V, van der Werf S, and Ferguson NM (2014a). Middle East respiratory syndrome coronavirus: quantification of the extent of the epidemic, surveillance biases, and transmissibility. Lancet Infect. Dis 14, 50–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cauchemez S, Ledrans M, Poletto C, Quenel P, de Valk H, Colizza V, and Boëlle PY (2014b). Local and regional spread of chikungunya fever in the Americas. Euro Surveill. 19, 20854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CDC (2018). Zika Travel Information. Https://wwwnc.cdc.gov/travel/page/zika-Travel-Information.
- da Costa CF, da Silva AV, do Nascimento VA, de Souza VC, Monteiro DC da S, Terrazas WCM, Dos Passos RA, Nascimento S, Lima JBP, and Naveca FG (2018). Evidence of vertical transmission of Zika virus in field-collected eggs of Aedes aegypti in the Brazilian Amazon. PLoS Negl. Trop. Dis 12, e0006594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowell SF, Tappero JW, and Frieden TR (2011). Public health in Haiti--challenges and progress. N. Engl. J. Med 364, 300–301. [DOI] [PubMed] [Google Scholar]
- Drummond AJ, Ho SYW, Phillips MJ, and Rambaut A (2006). Relaxed phylogenetics and dating with confidence. PLoS Biol. 4, e88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ECDC (2017). The European Surveillance System (TESSy). Https://ecdc.europa.eu/en/publications-Data/european-Surveillance-System-Tessy.
- Faria NR, Quick J, Claro IM, Thézé J, de Jesus JG, Giovanetti M, Kraemer MUG, Hill SC, Black A, da Costa AC, et al. (2017). Establishment and cryptic transmission of Zika virus in Brazil and the Americas. Nature. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FL DOH (2017a). Sexually Transmitted Case of Zika Identified in Miami-Dade County. http://www.floridahealth.gov/newsroom/2017/11/110317-Zika-Update.html.
- FL DOH (2017b). Single Case of Locally Transmitted Zika Identified in Manatee County. http://www.floridahealth.gov/newsroom/2017/10/101217-Zika.html.
- FL DOH (2018). Mosquito-Borne Disease Surveillance. http://www.floridahealth.gov/diseases-and-Conditions/mosquito-Borne-Diseases/surveillance.html.
- Franç GVA, Schuler-Faccini L, Oliveira WK, Henriques CMP, Carmo EH, Pedi VD, Nunes ML, Castro MC, Serruya S, Silveira MF, et al. (2016). Congenital Zika virus syndrome in Brazil: a case series of the first 1501 livebirths with complete investigation. Lancet 388, 891–897. [DOI] [PubMed] [Google Scholar]
- Fraser C, Donnelly CA, Cauchemez S, Hanage WP, Van Kerkhove MD, Hollingsworth TD, Griffin J, Baggaley RF, Jenkins HE, Lyons EJ, et al. (2009). Pandemic potential of a strain of influenza A (H1N1): early findings. Science 324, 1557–1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner LM, Bóta A, Gangavarapu K, Kraemer MUG, and Grubaugh ND (2018). Inferring the risk factors behind the geographical spread and transmission of Zika in the Americas. PLoS Negl. Trop. Dis 12, e0006194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill MS, Lemey P, Faria NR, Rambaut A, Shapiro B, and Suchard MA (2013). Improving Bayesian population dynamics inference: a coalescent-based model for multiple loci. Mol. Biol. Evol 30, 713–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon A, Gresh L, Ojeda S, Katzelnick LC, Sanchez N, Mercado JC, Chowell G, Lopez B, Elizondo D, Coloma J, et al. (2019). Prior dengue virus infection and risk of Zika: A pediatric cohort in Nicaragua. PLoS Med. 16, e1002726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorry C (2016). Cuba Confronts Zika: All Hands on Deck. MEDICC Rev. 18, 6–10. [DOI] [PubMed] [Google Scholar]
- Grubaugh ND, Ladner JT, Kraemer MUG, Dudas G, Tan AL, Gangavarapu K, Wiley MR, White S, Thézé J, Magnani DM, et al. (2017). Genomic epidemiology reveals multiple introductions of Zika virus into the United States. Nature 90, 4864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grubaugh ND, Faria NR, Andersen KG, and Pybus OG (2018). Genomic Insights into Zika Virus Emergence and Spread. Cell 172, 1160–1162. [DOI] [PubMed] [Google Scholar]
- Grubaugh ND, Ladner JT, Lemey P, Pybus OG, Rambaut A, Holmes EC, and Andersen KG (2019a). Tracking virus outbreaks in the twenty-first century. Nat Microbiol 4, 10–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grubaugh ND, Gangavarapu K, Quick J, Matteson NL, De Jesus JG, Main BJ, Tan AL, Paul LM, Brackney DE, Grewal S, et al. (2019b). An amplicon-based sequencing framework for accurately measuring intrahost virus diversity using PrimalSeq and iVar. Genome Biol. 20, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gubler DJ. (1989). Aedes aegypti and Aedes aegypti-Borne Disease Control in the 1990s: Top Down or Bottom Up. Am. J. Trop. Med. Hyg 40, 571–578. [DOI] [PubMed] [Google Scholar]
- Guindon S, and Gascuel O (2003). A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol 52, 696–704. [DOI] [PubMed] [Google Scholar]
- Guzmán MG (2005). Global voices of science. Deciphering dengue: the Cuban experience. Science 309, 1495–1497. [DOI] [PubMed] [Google Scholar]
- Guzmán MG, and Kourí G (2009). Dengue in Cuba: research strategy to support dengue control. Lancet 374, 1660–1661. [DOI] [PubMed] [Google Scholar]
- Hamer DH, Barbre KA, Chen LH, Grobusch MP, Schlagenhauf P, Goorhuis A, van Genderen PJJ, Molina I, Asgeirsson H, Kozarsky PE, et al. (2017). Travel-Associated Zika Virus Disease Acquired in the Americas Through February 2016: A GeoSentinel Analysis. Ann. Intern. Med 166, 99–108. [DOI] [PubMed] [Google Scholar]
- Harvey K, Esposito DH, Han P, Kozarsky P, Freedman DO, Plier DA, Sotir MJ, and Centers for Disease Control and Prevention (CDC) (2013). Surveillance for travel-related disease--GeoSentinel Surveillance System, United States, 1997-2011. MMWR Surveill. Summ 62, 1–23. [PubMed] [Google Scholar]
- Hill SC, Vasconcelos J, Neto Z, Jandondo D, Zé-Zé L, Aguiar RS, Xavier J, Thézé J, Mirandela M, Cândido ALM, et al. (2019). Emergence of the Zika virus Asian lineage in Angola. bioRxiv 520437. [Google Scholar]
- Hotez PJ (2016). Neglected Tropical Diseases in the Anthropocene: The Cases of Zika, Ebola, and Other Infections. PLoS Negl. Trop. Dis 10, e0004648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter JD (2007). Matplotlib: A 2D Graphics Environment. Comput. Sci. Eng 9, 90–95. [Google Scholar]
- IATA (2018). International Air Transport Association. Https://www.iata.org/pages/default.aspx.
- Katoh K, and Standley DM (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol 30, 772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, et al. (2012). Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan K, Bogoch I, Brownstein JS, Miniota J, Nicolucci A, Hu W, Nsoesie EO, Cetron M, Creatore MI, German M, et al. (2014). Assessing the Origin of and Potential for International Spread of Chikungunya Virus from the Caribbean. PLoS Curr. 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraemer MUG, Sinka ME, Duda KA, Mylne AQN, Shearer FM, Barker CM, Moore CG, Carvalho RG, Coelho GE, Van Bortel W, et al. (2015). The global distribution of the arbovirus vectors Aedes aegypti and Ae. albopictus. Elife 4, e08347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraemer MUG, Brady OJ, Watts A, German M, Hay SI, Khan K, and Bogoch II. (2017). Zika virus transmission in Angola and the potential for further spread to other African settings. Trans. R. Soc. Trop. Med. Hyg 111, 527–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaBeaud AD (2008). Why arboviruses can be neglected tropical diseases. PLoS Negl. Trop. Dis 2, e247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladner JT, Grubaugh ND, Pybus OG, and Andersen KG (2019). Precision epidemiology for infectious disease control. Nat. Med 25, 206–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leder K, Torresi J, Libman MD, Cramer JP, Castelli F, Schlagenhauf P, Wilder-Smith A, Wilson ME, Keystone JS, Schwartz E, et al. (2013). GeoSentinel surveillance of illness in returned travelers, 2007-2011. Ann. Intern. Med 158, 456–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leder K, Grobusch MP, Gautret P, Chen LH, Kuhn S, Lim PL, Yates J, McCarthy AE, Rothe C, Kato Y, et al. (2017). Zika beyond the Americas: Travelers as sentinels of Zika virus transmission. A GeoSentinel analysis, 2012 to 2016. PLoS One 12, e0185689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lessler J, Chaisson LH, Kucirka LM, Bi Q, Grantz K, Salje H, Carcelen AC, Ott CT, Sheffield JS, Ferguson NM, et al. (2016). Assessing the global threat from Zika virus. Science 353, aaf8160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, and Durbin R (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim SK, Lim JK, and Yoon IK (2017). An Update on Zika Virus in Asia. Infect Chemother 49, 91–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lourenço J, de Lourdes Monteiro M, Valdez T, Monteiro Rodrigues J, Pybus O, and Rodrigues Faria N (2018). Epidemiology of the Zika Virus Outbreak in the Cabo Verde Islands, West Africa. PLoS Curr. 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meltzer E, Weld LH, Wilder-Smith A, von Sonnenburg F, Keystone JS, Kain KC, Torresi J, and Freedman DO (2008). Seasonality, Annual Trends, and Characteristics of Dengue among Ill Returned Travelers, 1997-2006. Emerging Infectious Disease 14, 1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meltzer E, Lustig Y, Leshem E, Levy R, Gottesman G, Weissmann R, Rabi DH, Hindiyeh M, Koren R, Mendelson E, et al. (2016). Zika Virus Disease in Traveler Returning from Vietnam to Israel. Emerg. Infect. Dis 22, 1521–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metsky HC, Matranga CB, Wohl S, Schaffner SF, Freije CA, Winnicki SM, West K, Qu J, Baniecki ML, Gladden-Young A, et al. (2017). Zika virus evolution and spread in the Americas. Nature 66, 366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell PK, Mier-Y-Teran-Romero L, Biggerstaff BJ, Delorey MJ, Aubry M, Cao-Lormeau V-M, Lozier MJ, Cauchemez S, and Johansson MA (2018). Reassessing Serosurvey-Based Estimates of the Zika Symptomatic Proportion. Am. J. Epidemiol [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mlakar J, Korva M, Tul N, Popović M, Poljšak-Prijatelj M, Mraz J, Kolenc M, Resman Rus K, Vesnaver Vipotnik T, Fabjan Vodušek V, et al. (2016). Zika Virus Associated with Microcephaly. N. Engl. J. Med 374, 951–958. [DOI] [PubMed] [Google Scholar]
- Mordecai EA, Cohen JM, Evans MV, Gudapati P, Johnson LR, Lippi CA, Miazgowicz K , Murdock CC, Rohr JR, Ryan SJ, et al. (2017). Detecting the impact of temperature on transmission of Zika, dengue, and chikungunya using mechanistic models. PLoS Negl. Trop. Dis 11, e0005568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morens DM, and Fauci AS (2017). Pandemic Zika: A Formidable Challenge to Medicine and Public Health. J. Infect. Dis 216, S857–S859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Netto EM, Moreira-Soto A, Pedroso C, Höser C, Funk S, Kucharski AJ, Rockstroh A, KGmmerer BM, Sampaio GS, Luz E, et al. (2017). High Zika Virus Seroprevalence in Salvador, Northeastern Brazil Limits the Potential for Further Outbreaks. MBio 8, e01390–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunes MRT, Palacios G, Faria NR, Sousa EC, Pantoja JA, Rodrigues SG, Carvalho VL, Medeiros DBA, Savji N, Baele G, et al. (2014). Air Travel Is Associated with Intracontinental Spread of Dengue Virus Serotypes 1-3 in Brazil. PLoS Negl. Trop. Dis 8, e2769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Reilly KM, Lowe R, Edmunds WJ, Mayaud P, Kucharski A, Eggo RM, Funk S, Bhatia D, Khan K, Kraemer MUG, et al. (2018). Projecting the end of the Zika virus epidemic in Latin America: a modelling analysis. BMC Med. 16, 180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PAHO (2017a). Countries and territories with autochthonous transmission of Zika virus in the Americas reported in 2015–2017. https://www.paho.org/hq/index.php?option=com_content&view=article&id=11603:countries-and-Territories-with-Autochthonous-Transmission-of-Zika-Virus-in-the-Americas-Reported-in-2015-2017&Itemid=41696&lang=en.
- PAHO (2017b). Regional Zika Epidemiological Update (Americas). https://www.paho.org/hq/index.php?option=com_content&view=article&id=11599:regional-Zika-Epidemiological-Update-americas&Itemid=41691&lang=en.
- PAHO (2017c). Zika-Epidemiological Report: Cuba. https://www.paho.org/hq/dmdocuments/2017/2017-Phe-Zika-Situation-Report-Cub.pdf.
- Patterson J, Sammon M, and Garg M (2016). Dengue, Zika and Chikungunya: Emerging Arboviruses in the New World. West. J. Emerg. Med 17, 671–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pentón MJ (2018). If you plan to travel to Cuba, bring repellent: Deadly dengue virus has returned. Https://www.miamiherald.com/news/nation-world/world/americas/cuba/article222627955.html.
- Phumee A, Buathong R, Boonserm R, Intayot P, Aungsananta N, Jittmittraphap A, Joyjinda Y, Wacharapluesadee S, and Siriyasatien P (2019). Molecular Epidemiology and Genetic Diversity of Zika Virus from Field-Caught Mosquitoes in Various Regions of Thailand. Pathogens 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quick J, Grubaugh ND, Pul Ian ST, Claro IM, Smith AD, Gangavarapu K, Oliveira G, Robles-Sikisaka R, Rogers TF, Beutler NA, et al. (2017). Multiplex PCR method for MinION and Illumina sequencing of Zika and other virus genomes directly from clinical samples. Nature Protocols 12, 1261–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambaut A, Lam TT, Max Carvalho L, and Pybus OG (2016). Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol 2, vew007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambaut A, Drummond AJ, Xie D, Baele G, and Suchard MA (2018). Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst. Biol 67, 901–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reardon S (2016). Mosquito guns and heavy fines: how Cuba kept Zika at bay for so long. Nature 536, 257–258. [DOI] [PubMed] [Google Scholar]
- Reuters (2017). Cuba says Zika tally rises to nearly 1,900 cases. Https://in.reuters.com/article/us-Health-Zika-Cuba-idINKCN18E25G.
- Robles F (2016). Scheduled Flights to Cuba From U.S. Begin Again, Now With Jet Engines. The New York Times. [Google Scholar]
- Rodriguez-Barraquer I, Costa F, Nascimento EJM, Nery N, Castanha PMS, Sacramento GA, Cruz J, Carvalho M, De Olivera D, Hagan JE, et al. (2019). Impact of preexisting dengue immunity on Zika virus emergence in a dengue endemic region. Science 363, 607–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruchusatsawat K, Wongjaroen P, Posanacharoen A, Rodriguez-Barraquer I, Sangkitporn S, Cummings DAT, and Salje H (2019). Long-term circulation of Zika virus in Thailand: an observational study. Lancet Infect. Dis [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rund SSC, and Martinez ME (2017). Rescuing Hidden Ecological Data to Tackle Emerging Mosquito-Borne Diseases, https://www.biorxiv.org/content/early/2017/08/09/096875.abstract096875. [DOI] [PMC free article] [PubMed]
- Semenza JC, Sudre B, Miniota J, Rossi M, Hu W, Kossowsky D, Suk JE, Van Bortel W, and Khan K (2014). International dispersal of dengue through air travel: importation risk for Europe. PLoS Negl. Trop. Dis 8, e3278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siraj AS, Oidtman RJ, Huber JH, Kraemer MUG, Brady OJ, Johansson MA, and Alex Perkins T (2017). Temperature modulates dengue virus epidemic growth rates through its effects on reproduction numbers and generation intervals. PLoS Negl. Trop. Dis 11, e0005797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suchard MA, Lemey P, Baele G, Ayres DL, Drummond AJ, and Rambaut A (2018). Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol 4, vey016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talevich E, Invergo BM, Cock PJA, and Chapman BA (2012). Bio.Phylo: a unified toolkit for processing, analyzing and visualizing phylogenetic trees in Biopython. BMC Bioinformatics 13, 209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thangamani S, Huang J, Hart CE, Guzman H, and Tesh RB (2016). Vertical Transmission of Zika Virus in Aedes aegypti Mosquitoes. Am. J. Trop. Med. Hyg 95, 1169–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thézé J, Li T, du Plessis L, Bouquet J, Kraemer MUG, Somasekar S, Yu G, de Cesare M, Balmaseda A, Kuan G, et al. (2018). Genomic Epidemiology Reconstructs the Introduction and Spread of Zika Virus in Central America and Mexico. Cell Host Microbe 23, 855–864. e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson Hobbs N, and Hooten MB (2015). Bayesian Models: A Statistical Primer for Ecologists (Princeton University Press; ). [Google Scholar]
- Toledo ME, Vanlerberghe V, Baly A, Ceballos E, Valdes L, Searret M, Boelaert M, and Van der Stuyft P (2007). Towards active community participation in dengue vector control: results from action research in Santiago de Cuba, Cuba. Trans. R. Soc. Trop. Med. Hyg 101, 56–63. [DOI] [PubMed] [Google Scholar]
- US DOT (2018). TranStats | Bureau of Transportation Statistics. Https://www.transtats.bts.gov/.
- Vogels CBF, Rückert C, Cavany SM, Perkins TA, Ebel GD, and Grubaugh ND (2019). Arbovirus coinfection and co-transmission: A neglected public health concern? PLoS Biol. 17, e3000130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vora S (2016). Carnival Will Begin Cruises to Cuba in May. The New York Times. [Google Scholar]
- Watts AG, Carmen Huber MSA, Bogoch II, Brady OJ, Kraemer MUG, and Khan K (2018). Potential Zika virus spread within and beyond India. J. Travel Med [DOI] [PubMed] [Google Scholar]
- Weaver SC, Charlier C, Vasilakis N, and Lecuit M (2018). Zika, Chikungunya, and Other Emerging Vector-Borne Viral Diseases. Annu. Rev. Med 69, 395–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO (2016a). Fifth meeting of the Emergency Committee under the International Health Regulations (2005) regarding microcephaly, other neurological disorders and Zika virus. Http://www.who.int/en/news-room/detail/18-11-2016-Fifth-Meeting-of-the-Emergency-Committee-under-the-International-Health-Regulations-(2005)-Regarding-Microcephaly-Other-Neurological-Disorders-and-Zika-Virus.
- WHO (2016b). WHO statement on the first meeting of the International Health Regulations (2005) (IHR 2005) Emergency Committee on Zika virus and observed increase in neurological disorders and neonatal malformations. Http://www.who.int/news-room/detail/01-02-2016-Who-Statement-on-the-First-Meeting-of-the-International-Health-Regulations-(2005)-(ihr-2005)-Emergency-Committee-on-Zika-Virus-and-Observed-Increase-in-1\leurological-Disorders-and-Neonatal-Malformations.
- Wilder-Smith A, Renhorn K-E, Tissera H, Abu Bakar S, Alphey L, Kittayapong P, Lindsay S, Logan J, Hatz C, Reiter P, et al. (2012). DengueTools: innovative tools and strategies for the surveillance and control of dengue. Glob. Health Action 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilder-Smith A, Chang CR, and Leong WY (2018). Zika in travellers 1947-2017: a systematic review. J. Travel Med 25. [DOI] [PubMed] [Google Scholar]
- Yadav PD, Malhotra B, Sapkal G, Nyayanit DA, Deshpande G, Gupta N, Padinjaremattathil UT, Sharma H, Sahay RR, Sharma P, et al. (2019). Zika virus outbreak in Rajasthan, India in 2018 was caused by a virus endemic to Asia. Infect. Genet. Evol 69, 199–202. [DOI] [PubMed] [Google Scholar]
- Yang Z (1994). Maximum likelihood phylogenetic estimation from DNA sequences with variable rates over sites: approximate methods. J. Mol. Evol 39, 306–314. [DOI] [PubMed] [Google Scholar]
- Zambrana JV, Bustos Carrillo F, Burger-Calderon R, Collado D, Sanchez N, Ojeda S, Carey Monterrey J, Plazaola M, Lopez B, Arguello S, et al. (2018). Seroprevalence, risk factor, and spatial analyses of Zika virus infection after the 2016 epidemic in Managua, Nicaragua. Proc. Natl. Acad. Sci. U. S. A 115, 9294–9299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanluca C, Melo V.C.A. de, Mosimann ALP, Santos GIVD, Santos CNDD, and Luz K (2015). First report of autochthonous transmission of Zika virus in Brazil. Mem. Inst. Oswaldo Cruz 110, 569–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Increase in travel Zika cases from Cuba is not due to an increase in travel, related to Figure 1. To assess if the 2016 to 2017 increase in Zika cases reported from travelers who recently visited Cuba were due to a Zika outbreak in Cuba or an increase in air travel passengers leaving Cuba, we compared the yearly travel volumes from Cuba to Florida, Spain, and Italy to the travel Zika cases from Cuba reported by the same counties during 2016 and 2017. The greater increase from 2016 to 2017 of travel cases compared to travel volume suggests that a Zika outbreak in Cuba was the cause.
Supplemental File 3, related to Figure 4. Raw MAFFT codon alignment data, PhyML tree, and BEAST MCC time-structured phylogeny.
Supplemental File 4, related to Figure 5. Timeline of selected news articles from 2015-2017 related to Zika and dengue virus transmission in Cuba.
Figure S2. Relationship between the number of travel-associated Zika cases and the correlation between local and travel incidence rates, related to Figure 2. To determine the number of travel-associated infections needed to infer the shape of a local outbreak, we compared the total travel-associated Zika cases from each exposure-reporting country/territory combination (x-axis) with Pearson correlation between the local and travel incidence rates corresponding to the combination (y-axis). The travel-associated Zika cases were totaled from 2016-2017. For the Pearson correlations between local-travel incidence rates, monthly incidence values from 2016-2017 were compared. When there were >20 travel-associated cases, the local-travel Pearson r was >0.5, indicating a strong positive correlation and that the travel cases can help determine the shape of the local outbreak. The lone exception to that finding was from travelers from Barbados diagnosed in the United Kingdom (UK) because the travel cases miss the locally reported Zika virus peak during January-February, 2016, but they correlate with the second local peak during July-October, 2016 (Figure 2A). In our dataset, there were 25 Zika virus infections diagnosed in Italy with recent travel to Cuba, 30 diagnosed in Spain, and 98 diagnosed in Florida. These totals are all within the range of strong positive correlations between local and travel incidence, justifying their use to infer the local Zika outbreak dynamics in Cuba (Figure 2B).
Figure S3. Cruise ship travel from Cuba to Florida is minimal compared to air travel, related to Figure 2. Cruise ships may be a mechanism for infected individuals to travel to other countries, thus we investigated if we should incorporated cruise ship passenger volumes for calculating travel Zika incidence. The ~7× smaller volume of scheduled monthly cruise ship travel from 2019 compared to air travel in 2017 suggested that this would minimally impact our estimates and the data was not included in our final analyses.
Figure S4. Posterior predictions of estimated total travel incidence from origin country into Florida, related to Figure 2. These distributions were used to inform the joint distribution between travel incidence and local incidence that was used to estimate local incidence in Cuba. Empirical total travel cases per country indicated by red vertical line. All of the countries shown above, besides Cuba, had >0.25 correlation between local incidence and travel incidence and had the observed value fall within the 95% posterior predictive interval of the distribution.
Figure S5. Posterior predictions of estimated total local incidence from origin country into Florida, related to Figure 2. These distributions were used to inform the joint distribution between travel incidence and local incidence that was used to estimate local incidence in Cuba. Empirical total local cases per country indicated by red vertical line. Estimated local incidence of Cuba indicated in blue with no empirical value. All of the countries shown above, besides Cuba, had >0.25 correlation between local incidence and travel incidence and had the observed value fall within the 95% posterior predictive interval of the distribution.
Figure S6. Maximum likelihood tree and root-to-tip regression of Zika virus genomes from Cuba and the epidemic in the Americas, related to Figure 4. (A) Maximum likelihood tree of publicly available Zika virus sequences (n = 269) and sequences generated in this study (n = 14). Tips are coloured by location. Bootstrap support values are colored at the nodes. Divergence shown as substitutions per site. “1-3 Cuba” represent three independent introductions of Zika virus into Cuba. (B) Linear regression of sample tip dates against divergence from root based on sequences with known collection dates estimates an evolutionary rate for the Zika virus phylogeny of 5.71 × 10−3 nucleotide substitutions per site per year.
Supplemental File 2, related to Figure 4. Zika virus genomes generated during this study.
Data Availability Statement
The Zika virus sequences generated here can be found using the NCBI BioProject PRJNA438510 and individual Genbank IDs can be found in Supplemental File 2. Data used to create the figures can be found in the supplemental files. The raw data and results for our analyses can be found at: https://github.com/andersen-lab/paper_2018_cuba-travel-zika.