

Abstract
Reliable signal transduction via G-protein coupled receptors requires proper receptor inactivation. For example, signals originating from single rhodopsin molecules vary little from one to the next, requiring reproducible inactivation of rhodopsin by phosphorylation and arrestin binding. We determined how reduced concentrations of rhodopsin kinase (GRK1) and/or arrestin1 influenced the kinetics and variability of the single-photon responses of mouse rods. These experiments revealed that arrestin, in addition to its role in quenching rhodopsin's activity, can tune the kinetics of rhodopsin phosphorylation by competing with GRK1. This competition influenced the variability of rhodopsin's active lifetime. Biasing the competition in favor of GRK1 revealed that rhodopsin remained active through much of the single-photon response under the conditions of our experiments. This long-lasting rhodopsin activity can explain the characteristic time course of single-photon response variability. Indeed, explaining the late time-to-peak of the variance required an active lifetime of rhodopsin about twice that of the G-protein transducin. Competition between arrestins and kinases may be a general means of influencing signals mediated by G-protein coupled receptors, particularly when activation of a few receptors produces signals of functional importance.
Keywords: GPCR, Phosphorylation, Kinase, Photoreceptor, Vision, signal transduction
G-protein coupled receptors (GPCRs) regulate numerous biological processes essential for life. These receptors convert extracellular stimuli into intracellular signals by catalyzing G-protein activation. GPCRs regulate our heart rate (Premont and Gainetdinov, 2007), generate the signals that give rise to our sense of smell, taste, and sight (Buck, 2000; Maeda et al., 2003; Chandrashekar et al., 2006; Kobilka and Deupi, 2007), and are the targets of about half of modern therapeutic drugs (Kristiansen, 2004; Eglen et al., 2007). Because GPCRs act as catalysts, the sensitivity and reliability of the signals they mediate rely on the regulation of receptor lifetime. Yet we lack a quantitative understanding of GPCR inactivation.
Here we investigate the inactivation of rhodopsin, a GPCR that converts the absorption of a single photon into an amplified electrical response in rod photoreceptors (Figure 1A) (Baylor et al., 1979; Field et al., 2005). A single activated rhodopsin molecule (Rh*) catalyzes the activation of multiple copies of the G-protein transducin, which in turn activate phosphodiesterase and lead to the closure of cGMP-gated channels in the rod plasma membrane. Amplification persists as long as rhodopsin remains active. As a consequence, the low variability of the rod's single-photon response requires tight control of rhodopsin's active lifetime (Rieke and Baylor, 1998; Whitlock and Lamb, 1999; Field and Rieke, 2002a; Hamer et al., 2003).
Figure 1.

Biochemical readout and control of rhodopsin activity. (A) Schematic of phototransduction cascade in mammalian rods. Rhodopsin (Rh) is activated by the absorption of a photon. Activated rhodopsin (Rh*) leads to the sequential activation of transducin (T) and phosphodiesterase (PDE). PDE* hydrolyzes cGMP and leads to the closure of cGMP-gated channels in the membrane of the rod outer segment. Inactivation of Rh* requires multiple phosphorylations of rhodopsin's C terminus by GRK1 followed by arrestin1 quenching. Guanylate cyclase (GC) maintains cytoplasmic cGMP. (B) Schematic diagram of the molecular reactions involved in the inactivation of a Rh* molecule. The duration of each rhodopsin phosphorylation event depends on the competition between arrestin1 and GRK1 binding, and the time required for phosphate attachment. Reactions 1 to 3 cycle until a sufficient number of phosphates (Pn) are incorporated to the C terminus. Wild-type mouse rhodopsin has 6 potential phosphorylation sites. The final shutoff step is provided by arrestin1. Arrestin competition (reaction 1) slows Rh* shutoff while the binding of arrestin1 to Rh*-Pn (reaction 4) terminates Rh* activity. (C) Simulation of the combined effect of arrestin competition and quenching on rhodopsin inactivation. Rhodopsin inactivation was described as a series of 6 phosphorylation steps as in 1-3 of B, followed by arrestin1 binding (see Materials and Methods for details). The rate constants β and γ in B scaled linearly with arrestin1 concentration. The net effect of decreasing arrestin1 concentration can be to shorten Rh* activity. (D) Decreasing the arrestin1 concentration would lengthen Rh* activity if arrestin's only role is to quench Rh*. Simulations were identical to C except β did not depend on arrestin1 concentration.
As with other GPCRs, the lifetime of Rh* is controlled by a GPCR kinase (GRK1) and arrestin (arrestin1) (Figure 1A and B). Rh* becomes phosphorylated multiple times and then inactivated completely following arrestin1 binding (Wilden and Kuhn, 1982; Kuhn et al., 1984; Ohguro et al., 1995; Wilden, 1995; Chen et al., 1995; Chen et al., 1999; Xu et al., 1997; Mendez et al., 2000; Kennedy et al., 2001; Maeda et al., 2003; Makino et al., 2003). Rh* phosphorylation involves GRK1 binding to Rh* (reaction 2 in Figure 1B) and the attachment of a phosphate group to the C terminus (reaction 3). Arrestin1 then quenches Rh* activity (reaction 4) by binding to phosphorylated Rh* at multiple sites (Gurevich and Benovic, 1993; Gurevich and Gurevich, 2004). The arrestin binding rate depends on the concentration of arrestin1 in the rod outer segment, while the efficacy of quenching depends on the number of phosphates attached to the C terminus of Rh* (Kuhn et al., 1984; Wilden, 1995; Xu et al., 1997; Gibson et al., 2000; Ling et al., 2004; Vishnivetskiy et al., 2007).
In addition to its established role in quenching rhodopsin activity, Figure 1B depicts a new hypothesized role of arrestin1: competition with GRK1 for Rh* binding sites. Several observations motivate this competition hypothesis. First, GRK1 and arrestin1 share partially overlapping binding sites on rhodopsin's cytoplasmic domains (Kelleher and Johnson, 1990; Palczewski et al., 1991; Krupnick et al., 1994; Shi et al., 1995; Raman et al., 1999; Raman et al., 2003). Second, both in vitro and in vivo studies show that arrestin1 is capable of high affinity binding to phosphorylated rhodopsin and low affinity binding to unphosphorylated rhodopsin (Gurevich and Benovic, 1992; Krupnick et al., 1994; Gurevich and Benovic, 1995; Raman et al., 1999; Vishnivetskiy et al., 1999; Gurevich and Gurevich, 2004; Burns et al., 2006). Together, these studies suggest that competition between arrestin1 and GRK1 might regulate GRK1 binding to rhodopsin and thus modulate rhodopsin's active lifetime. Here we test this hypothesis by measuring how alterations in the concentration of GRK1 and/or arrestin1 affect the kinetics and variability of the rod's single-photon responses.
Materials and Methods
Animals
All animal procedures were approved by the Administrative Panel on Laboratory Animal Care at the University of Washington. Control C57BL/6 mice (5 weeks old) were purchased from Harlan Sprague Dawley. Arr1−/− and GRK1−/− mice were obtained from Dr. Jeannie Chen (USC, Los Angeles, CA). Arr1+/− and GRK1+/− mice were generated by crossing Arr1−/− and GRK1−/− with C57BL/6 mice. Some GRK1+/− mice used for the initial recordings were provided by Dr. Ching-Kang Chen (VCU, Richmond, VA). Arr1−/− mice were crossed with GRK1−/− to generate double heterozygotes (GRK1+/−Arr1+/−). Mice were genotyped using PCR primers specific to the mutations.
To control for the possibility that differences in genetic background could produce differences in response kinetics, we compared responses of rods from sibling Arr1+/− and Arr1+/+ mice generated from crosses of Arr1+/− parents. Single photon responses of Arr1+/− were smaller and faster than those of Arr1+/+ rods. Data from siblings and nonsiblings were similar and have been pooled in Table 1.
Table 1.
Comparison of response properties of rods with normal and reduced GRK1 and arrestin1 concentrations. For dim flashes, the amplitude of the mean population single-photon response was determined by dividing the mean response by the number of photoisomerizations, determined by scale factor required to match the time-dependent variance with the square of the mean response. Integration time is the integral of the response normalized by its peak amplitude. CVarea is the standard deviation of the integral of the single-photon response (corrected for dark noise) divided by the mean. tpeak,σ2/tpeak,μ is the time-to-peak of the time-dependent variance divided by the time-to-peak of the mean single-photon response. Each entry represents the mean ± SEM. CVarea and tpeak,σ2/tpeak,μ can only be reliably estimated for isolated single-photon responses.
| Strain | Single-photon response amplitude (pA) | Integration time (ms) | CVarea | tpeak,σ2/tpeak,μ |
|---|---|---|---|---|
| isolated single-photon responses | ||||
| Wild-Type (n = 29) | 1.41 ±0.09 | 510 ± 20 | 0.34 ± 0.01 | 2.00 ± 0.20 |
| GRK1+/− (n = 30) | 1.61 ± 0.06 | 840 ± 30 | 0.40 ± 0.02 | 2.90 ± 0.13 |
| GRK1+/−Arr1+/− (n = 40) | 1.45 ± 0.05 | 630 ± 20 | 0.43 ± 0.01 | 2.60 ± 0.09 |
| Arr1+/− (n = 41) | 1.30 ± 0.03 | 500 ± 20 | 0.43 ± 0.02 | 1.88 ± 0.08 |
| dim flashes, 30°C | ||||
| Wild-Type (n = 38) | 1.02 ±0.03 | 490 ± 20 | – | – |
| GRK1+/− (n = 43) | 1.30 ± 0.04 | 800 ± 20 | – | – |
| GRK1+/−Arr1+/− (n = 39) | 1.11 ± 0.05 | 610 ± 20 | – | – |
| Arr1+/− (n = 36) | 0.76 ± 0.03 | 455 ± 15 | – | – |
| dim flashes, 36°C | ||||
| Wild-Type (n = 15) | 0.98 ±0.02 | 330 ± 20 | – | – |
| Arr1+/− (n = 18) | 0.64 ± 0.01 | 275 ± 20 | – | – |
Electrophysiology
Mice were dark adapted overnight and killed by cervical dislocation. The dissection was carried out under infrared light (> 900 nm) to keep the retina fully dark adapted. Isolated retinas were stored in a light-tight container at 32°C in bicarbonate-buffered Ames solution (Sigma, St. Louis, MO) equilibrated with 5% CO2/95% O2. Photoresponses were recorded using suction electrodes (Baylor et al., 1979; Field and Rieke, 2002a; Doan et al., 2006). A small piece of retina was mechanically shredded, transferred into a recording chamber, and perfused at ∼3.5 ml/min with bicarbonate-buffered Ames solution heated to 30 ± 1°C or 36 ± 1°C. The outer segment currents of single rod photoreceptors were recorded using suction electrodes with optimized geometry. Borosilicate glass capillary tubes were pulled to taper abruptly to an opening of 4 to 6 μm. The tips were polished to an inner diameter (1.6 to 2.2 μm) to encompass snugly the rod outer segment, which was positioned inside the electrode to maximize the dark current. The electrode contained HEPES Ames solution (NaHCO3 replaced with 10 mM Hepes and 15 mM NaCl; pH = 7.40 with NaOH). Most studies of retinal circuitry in mammals use similar recording conditions.
Our recorded rod responses showed systematically slower kinetics and higher sensitivity than other published work (Xu et al., 1997; Mendez et al., 2000; Calvert et al., 2001; Krispel et al., 2006). The main differences in conditions are the solutions used for retina storage and recording. Thus in a few control experiments we made the following substitutions: (1) we stored the retina in oxygenated L-15 medium (Invitrogen) mixed with 0.1 mg/ml BSA (Fraction V, Sigma) and 10 mM glucose; (2) we perfused the recording chamber with bicarbonate-buffered Locke's solution (112.5 mM NaCl, 3.6 mM KCl, 2.4 mM MgCl2, 1.2 mM CaCl2, 20 mM NaHCO3, 10 mM HEPES, 0.02 mM EDTA, 3 mM succinate, 0.5 mM L-glutamate, with 10 mM glucose, vitamin and amino acid supplements (Sigma), 0.1 mg/mL BSA added on the day of experiment) saturated with 5%CO2/95% O2 and heated to 36 ± 1°C; and (3) we filled the suction electrode with HEPES-buffered Locke's solution (130 mM NaCl, 3.6 mM KCl, 2.4 mM MgCl2, 1.2 CaCl2, 10 mM HEPES, 0.02 EDTA, 0.02 mM EDTA, 3 mM succinate, 0.5 mM L-glutamate, with 10 mM glucose, 0.1 mg/mL BSA added on the day of experiment, pH = 7.4). The kinetics and sensitivity of the responses recorded under these conditions differed substantially from our standard conditions (Supplementary Figure 1). Specifically, estimated single-photon responses in L15/Locke's reached a peak amplitude of 0.2-0.3 pA at a time 110-120 ms after the flash, compared to 1 pA and 190-200 ms in Ames. Similarly Pepperberg time constants (see below) in L15/Locke's were ∼200 ms, compared to ∼320 ms in Ames. The properties of responses measured in L15/Locke's resembled those reported previously (Xu et al., 1997; Mendez et al., 2000; Calvert et al., 2001; Krispel et al., 2006). The origin of these differences is unclear, but will be an important target for future work.
Light stimuli
Light delivered from a light-emitting diode with peak output at 470 nm was directed to the cell via a light guide and focused with an 60× microscope objective (Nikon, Kanagawa, Japan; 0.95 NA). For the Pepperberg time constant experiments, light from the LED was focused to illuminate uniformly a square area of 65 μm on a side, centered on the recorded cell. For isolation of single-photon responses, light stimuli were restricted to a slit (width < 1 μm), positioned perpendicular to the long axis of the outer segment about halfway along its length. For dim flash response experiments, light from the LED uniformly illuminated a circular area 650 μm in diameter centered on the recorded cell. All light stimuli were 10 ms in duration. Calibrated light intensities were converted to photon flux (photons/μm2/sec) at 500 nm (where rod sensitivity is maximal) using the measured LED spectral output and the rod's spectral sensitivity function.
Pepperberg time constant
To estimate the rate of decay of light-activated PDE activity, we used bright flashes, 30 ms in duration, to suppress the rod's dark current for at least 200 ms and allow [Ca2+]i to fall to a minimum. Saturation time was defined as the period over which the flash suppressed 80% of the dark current (Zhang et al., 2007). Assuming that light-activated PDE activity decays exponentially and that guanylate cylase activity is maximal and equal for the duration of the saturated response, the slope of the saturation time as a function of the natural log of the flash strength provides an estimate of the time constant for the decay of PDE activity (τPDE; Pepperberg et al., 1992). Since the decay of PDE activity requires both rhodopsin and transducin shutoff, τPDE could reflect the decay time of rhodopsin, transducin or both.
Dim flash responses
We estimated average single-photon responses from responses to a repeated dim flash. All cells with low baseline noise and a dark current of > 8 pA at 30 ± 1°C and > 12 pA at 36 ± 1°C were retained for this analysis. The mean single-photon response was estimated by dividing the average response to a dim flash by the number of Rh* produced by the flash, calculated from the scale factor required to match the first 200-300 ms of the time-dependent variance and the square of the mean response. The estimated collecting areas (conversions between photons/μm2 and Rh*) were, on average, ∼0.5 μm2 (Field and Rieke, 2002b).
Isolation of single-photon responses
Characterizing variability of the single-photon response requires separating responses to single absorbed photons from those to 0 or multiple photons. Isolation of single-photon responses was more reliable at 30 °C and hence most of our recordings were at this temperature. We used 4 criteria to determine whether to collect data from a recorded cell: (1) a dark current exceeding 8 pA at 30 ± 1°C; (2) qualitatively low baseline dark noise; (3) discernable single-photon responses; and (4) stable response kinetics. About 20% of the recorded cells met all 4 criteria. From this population of selected cells, ∼20 to 30% yielded discrete amplitude histograms (see below) and were retained for further analysis. Recorded currents were low-pass filtered at 30 Hz (8 pole Bessel), digitized at 1 kHz, and digitally filtered at 5 Hz prior to analysis. Periodic checks of response stability followed previous procedures (Field and Rieke, 2002a; Doan et al., 2006).
The procedure used to identify single-photon responses and check for errors in identification is described in detail elsewhere (Field and Rieke, 2002a). Briefly, the amplitudes of the responses to a repeated dim flash were determined from the vertical scale factor required to fit each response with the average dim-flash response. The fit was restricted to the initial 450-500 ms of the response, which included the rising phase but not the recovery phase. This procedure was chosen to minimize errors in identifying single-photon responses; all subsequent analysis used the entire response time course.
Histograms of the response amplitudes were fitted assuming that dark noise and noise from the single-photon responses were independent and additive. In this case, the number of responses with an amplitude between A − ΔA/2 and A + ΔA/2 is
| (1) |
where Ā is the mean single-photon response amplitude and σA is its standard deviation, n is the number of photoisomerizations (Rh*) produced by the flash and n̄ is its mean, and σD is standard deviation of the dark current noise. Thresholds used to identify single-photon responses were set at 3Ā/2 and near Ā/2, or lower if σD was small. This procedure was repeated for 3 different flash strengths in each cell. Repeating this procedure at multiple flash strengths allowed the accuracy of the isolation procedure to be determined by checking that the isolated single-photon responses did not depend on the strength of the flash used to elicit them (Field and Rieke, 2002a; Doan et al., 2006).
The isolation procedure described above produced a bias in favor of rods that produced large single-photon responses since small responses could not be reliably distinguished from responses to 0 or multiple photons. For wild-type, GRK1+/− and GRK1+/− Arr1+/− cells this bias was restricted to the response amplitude, while both amplitude and kinetics of the responses of Arr1+/− were effected (see Table 1).
Markov model for single rhodopsin phosphorylation
To provide a quantitative picture of arrestin competition, we implemented a single phosphorylation step in the kinetic scheme of Figure 1B as a Markov chain. We assumed that each phosphorylation step followed the same sequence — i.e. that GRK1 dissociates after phosphate attachment and the cycle starts anew. Two issues controlled the ability of arrestin competition to control the rate of phosphorylation: the fraction of time rhodopsin spends bound to arrestin1, and the time required for GRK1 binding compared to that for phosphate attachment. These issues depend on the ratios β/α and γ/σ, and hence the goal of the model was to estimate these ratios from the data.
Activated rhodopsin could exist in four states: (1) bound to arrestin1 (Rh*-Arr1); (2) unbound (Rh*); (3) bound to GRK1 (Rh*-GRK1); or (4) phosphorylated (Rh*-P). A state vector s⃗(t) captured the probability of each state at time t. Every time step ΔT, s⃗(t) was updated using a transition matrix M containing the relevant transition rates from Figure 1B— i.e. s⃗(t+ΔT) = M × s⃗(t). This procedure was iterated to determine the mean phosphorylation time (i.e. the time to transition from state 2 to state 4) for a given set of rate constants (α, β, γ, σ).
To compare predictions of the Markov model for wild-type and mutant rods, we assumed that the rate constants for arrestin1 and GRK1 binding to rhodopsin scaled with their concentrations — e.g. β/β0 = [Arr1]/[Arr1]WT, where β0 is the arrestin1 association rate in wild-type rods (see below for quantification of protein concentrations). This assumption will hold if the concentration of arrestin1 monomer in the outer segment (the active form) scales linearly with the total arrestin1 concentration. A similar logic applies to the GRK1 concentration. Two issues could effect this assumption. First, a small fraction of arrestin1 is in the outer segment under dark-adapted conditions (Broekhuyse et al., 1985; Peterson et al., 2003) and this fraction may differ for wild-type, Arr1+/−, GRK1+/− and GRK1+/− Arr1+/− mice (Hanson et al., 2007a). Second, self-association of arrestin1 will cause the monomer concentration to change less than the total arrestin1 concentration; the magnitude of this effect ranges from ∼20 to 60% (Imamoto et al., 2003; Hanson et al., 2007b). Because of the uncertainties associated with these effects we did not include them in the Markov model. Nonetheless, the central conclusions of the model — that rhodopsin spends most of its time bound to arrestin1, and that GRK1 binding is fast compared to phosphate attachment — held for estimates of the changes in arrestin1 concentration that included self association.
Transducin also competes with arrestin1 and GRK1 for rhodopsin binding (Wilden et al., 1986; Krupnick et al., 1997). Thus a complete description of arrestin competition will need to include its effect on transducin binding. This effect will depend on both the rate constant for transducin binding to free rhodopsin and the rate constant for GDP-GTP exchange once transducin has bound. We did not include this effect in the Markov model because of the uncertainties associated with the relevant rate constants. Nonetheless, the impact of arrestin competition on transducin activation is likely to be important and will need to be incorporated in extensions of models like that used here.
Phototransduction cascade model
We used a stochastic model for rhodopsin inactivation (Rieke and Baylor, 1998; Field and Rieke, 2002a; Doan et al., 2006) to investigate how the time-dependent variance of the single-photon response depends on the relative inactivation rates of rhodopsin and transducin. Rhodopsin's catalytic activity was assumed to shut off through a series of phosphorylation events, each composed of two steps (Figure 1B): GRK1 binding and phosphate attachment. The simulations assumed a GRK1 binding rate (γ) 8 times larger than the rate of phosphate attachment (σ) (see Results). Each phosphorylation cycle, on average, controlled an equal fraction of rhodopsin's total catalytic activity; this assumption insures that multiple phosphorylation cycles contribute maximally to reducing single-photon response variability. This constraint was met by assuming that each step produced an equal reduction in activity (see Figure 5C), and that the rate constants for phosphorylation decreased linearly with the number of attached phosphates. Such a progression of rate constants might be produced, at least in part, as rhodopsin is phosphorylated and the affinity of the interaction between arrestin1 and rhodopsin increases (Wilden, 1995; Gibson et al., 2000; Gurevich and Gurevich, 2004; Vishnivetskiy et al., 2007). This increased affinity should cause arrestin1 to compete more effectively with GRK1 and hence delay its binding to rhodopsin. Arrestin could terminate rhodopsin's activity after any number of phosphorylation events, but the rate constant for arrestin quenching increased exponentially with the number of completed phosphorylation cycles (Hamer et al., 2003; Vishnivetskiy et al., 2007).
Figure 5.

The time-dependent variance of the single-photon responses in wild-type, Arr1+/−, GRK1+/− and GRK1+/−Arr1+/− rods. Left panels superimpose 10 isolated single-photon responses from a wild-type and a Arr1+/− rod (A), a GRK1+/− rod (B), and a GRK1+/−Arr1+/− rod (C). Right panels compare the squared mean (thin trace) and the time-dependent variance (thick trace) of wild-type rods (gray, n = 29) with Arr1+/− (red, n = 41) rods (A), GRK1+/− (blue, n = 30) rods (B), and GRK1+/−Arr1+/− (green, n = 40) rods (C). The responses in each cell were normalized by the amplitude and time-to-peak of the cell's average single-photon response to facilitate comparison of the time course of the variance.
The time course of the activity of a single simulated rhodopsin molecule was converted to a change in current through either a linear or nonlinear approximation to the behavior of the post-rhodopsin components of the phototransduction cascade. In the linear case, the cascade was approximated by a filter with a Fourier transform given by (Rieke and Baylor, 1998)
| (2) |
where ω is the temporal frequency in radians/s, GD is the dark cGMP concentration, ϕ is the rate constant for PDE activity, θ is the rate constant for removal of Ca2+ from the outer segment by Na+/K+, Ca2+ exchange, PD is the dark PDE activity, and F̃(ω) = ∫ exp(iωt)F(t)dt. The inverse of ϕ is a measure of the average active lifetime of transducin, τT*. Equation 2 assumes that the cooperativity of the Ca2+-dependence of guanylate cyclase activity is 4 (Koch and Stryer, 1988; Burns et al., 2002). GD was determined from the measured dark current, ID, assuming with k = 0.0026 pA/μM3. Modeled single-photon responses used the following parameters: θ = 10 s−1 (Calvert et al., 2001; Burns et al., 2002), PD = 1 s−1 (Tamura et al., 1991), and ϕ = 5 s−1 (Krispel et al., 2006). In the nonlinear case, the filtered response was passed through a compressive nonlinearity, so that the resulting current was given by
| (3) |
where R is the time course of rhodopsin's activity, η controls the degree of compression and in the absence of compression the mean current response had a peak amplitude of 1. Compression reduced the sensitivity of the measured current to variations in rhodopsin's activity, particularly near the peak of the current response. Such a compression could be produced by local saturation of the transduction cascade (although see Field and Rieke, 2002a for experiments that limit the extent of local saturation).
The behavior of the general model described above is determined by the number of phosphorylation events, the rate of arrestin quenching and the extent of nonlinear compression. The choice of these model parameters is constrained by the essential properties of the measured single-photon responses: their low variability and the late time-to-peak and the near symmetric shape of the time-dependent variance (see Field and Rieke, 2002a). We focused on two models that satisfied these general constraints. In the first, each phosphorylation site contributed equally to rhodopsin shutoff and on average 5.6 phosphorylation cycles completed before rhodopsin was inactivated by arrestin. No compression was required for this model. In the second case, rhodopsin shut off through a maximum of 4 phosphorylation events, with an average of 3.7 events before arrestin binding. Compression (η = 0.6) was required to reduce response variability to experimental levels. This model is a simplified version of a comprehensive biophysical model introduced recently (Bisegna et al., 2008; see also Field and Rieke, 2002a).
In the Results we emphasize the first model above because it has fewer free parameters. However, the central conclusion of the modeling — that the late time-to-peak of the time-dependent variance requires that rhodopsin inactivates more slowly than transducin — held for both models. More generally, this conclusion held for multiple variants of the basic model described above in which each step in rhodopsin inactivation decreased its catalytic activity. This included models in which only phosphate attachment, and not GRK1 binding, decreased rhodopsin's catalytic activity.
Quantification of protein expression
Retinas from wild-type mice, aged 5-8 weeks, and heterozygous knockout mice, aged 4-6 weeks, were harvested in ice-cold HEPES Ames buffer (pH = 7.40). Retinas were immediately frozen on dry ice and stored at -80°C until use. Frozen retinas were thawed and homogenized using a handheld homogenizer with PBS/0.1 mM DTT and a mixture of protease inhibitors (Roche Diagnostics). Homogenized tissues were solubilized in 1× SDS lysis buffer (62.5 mM Tris·HCl, pH 6.8/2% SDS/10% glycerol/0.005% bromophenol blue/5% 2-mercaptoethanol) and left at room temperature for 15 minutes. Retinal equivalent dilutions were separated by 10% SDS/PAGE gel electrophoresis, transferred overnight onto nitrocellulose membranes (Millipore, Bedford, MA), and then blocked in Odyssey blocking buffer (LI-COR Biosciences, Lincoln, NE).
To quantify GRK1 and arrestin1 expression, the membranes were sequentially incubated with a different set of primary and secondary antibodies and imaged. The membranes were first probed with a polyclonal anti-arrestin antibody (1:1,000, PA1-732, Affinity BioReagents, CO) diluted in Odyssey blocking buffer containing 0.01% (vol/vol) Tween-20, washed 4 times with PBS containing 0.01% (vol/vol) Tween-20, and incubated with IRDye 680 goat anti-rabbit secondary antibody (1:5,000, LI-COR Biosciences) in Odyssey blocking buffer containing 0.01% (vol/vol)Tween-20. The membranes were then washed as above and the fluorescent signals were detected with the Odyssey Infrared Imaging System (LI-COR Biosciences) using the 700 nm channel. To probe for GRK1 and β-tubulin, the same membranes were incubated with monoclonal anti-rhodopsin kinase 1a antibody (1:1,000, MA1-720, Affinity BioReagents) and monoclonal anti-β tubulin antibody (1:10,000, MA1-20246, Affinity BioReagents). Membranes were washed, incubated with IRDye 800CW goat anti-mouse secondary antibody (LI-COR BioSciences), and the fluorescent signals were detected using the 800 nm channel.
To quantify PDEαβ and PDEγ, the membranes were probed with a cocktail of PDE (MOE, 1:4000, CytoSignal, Irvine, CA) and anti-β tubulin antibodies, washed, and then incubated with IRDye 680 goat anti-rabbit and IRDye 800CW goat anti-mouse secondary antibodies.
RGS9-1 expression level was determined by probing the membrane with a cocktail of G4695 (anti-RGS9c; 1:4,000, generous gift from Dr. Ted Wensel) and anti-β tubulin antibodies, washed, and incubated with IRDye 800CW donkey anti-goat and IRDye680 donkey anti-mouse secondary antibodies. All blocking and incubation steps were for 1 hour at room temperature. Odyssey 2.1 software was used to quantify protein expression.
To control for loading, the protein signals (integrated fluorescence intensity) of each sample were normalized to the sample's β-tubulin signal. The corrected integrated intensity values of the Arr1+/−, GRK1+/−, and GRK1+/−Arr1+/− samples were then normalized to the corrected integrated intensity value of the wild-type sample on the same membrane to determine the relative protein expression (Figure 2). Each sample was run in duplicate on a different membrane and often in two different dilutions. For each genetic background, the concentrations of arrestin1 and GRK1 determined from the two dilutions were within 20%, with no evidence for systematic bias.
Figure 2.
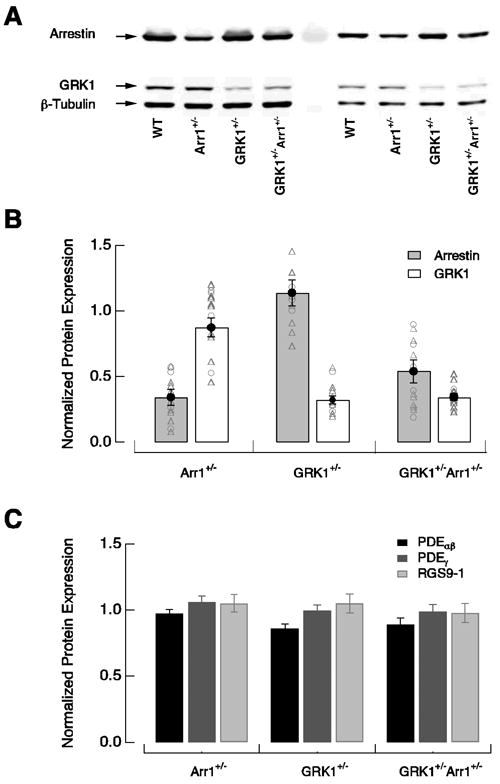
Protein expression in wild-type, Arr1+/−, GRK1+/−, and GRK1+/−Arr1+/− rods. (A) Representative immublot analysis of arrestin and GRK1 expression in retinas of wild-type and heterozygous knockout mice. The blot was probed with polyclonal anti-arrestin, monoclonal anti-rhododopsin kinase 1a, and monoclonal anti-β tubulin antibodies. Two different dilutions of the same samples were run on the same blot. (B) Levels of arrestin1 and GRK1 expression in heterozygous knockout retinas normalized to the levels in wild-type retinas. Integrated fluorescence intensity values were used for all analyses. Variations in sample loading were corrected for by normalizing the intensity value of each protein band to the intensity value of β-tubulin in the same lane. Open circles and triangles show the results from two different dilutions. Normalized ratios of arrestin1 and GRK1 expression compared to wild-type values for Arr1+/− were 0.34 ± 0.06 and 0.87 ± 0.07 (mean ± SEM, n = 5); 1.14 ± 0.10 and 0.32 ± 0.03 for GRK1+/− (n = 4); and 0.54 ± 0.09 and 0.34 ± 0.03 for GRK1+/−Arr1+/− (n = 5). (C) Levels of PDEαβ, PDEγ and RGS9-1 expression in heterozygous retinas normalized to the levels in wild-type retinas. Normalized ratios of PDEαβ, PDEγ and RGS9-1 compared to wild-type values for Arr1+/− were 0.97 ± 0.03, 1.06 ± 0.05, and 1.05 ± 0.07 (n = 4); 0.86 ± 0.03, 0.99 ± 0.04, and 1.05 ± 0.07 for GRK1+/− (n = 4); 0.89 ± 0.05, 0.99 ± 0.05, and 0.98 ± 0.07 for GRK1+/−Arr1+/− (n = 4).
We did not measure expression levels of recoverin (Rv), which could control GRK1 availability. Previous work showed that Arr1+/− mice had normal Rv expression levels (Xu et al., 1997). Furthermore, the effect of lowering Rv on the single-photon response differs substantially from the effects observed here, as the responses of rods from Rv−/− and wild-type mice diverge only well after the responses have reached their maximal amplitude (Makino et al., 2004; Sampath et al., 2005).
Statistics
Reported p values were based on two-sample t-tests.
Results
Competition between arrestin1 and GRK1 controls kinetics of rhodopsin shutoff
Figure 1B illustrates the arrestin competition hypothesis in the context of a kinetic model for a single phosphorylation event. Phosphorylation at a minimum involves two steps — GRK1 binding to active rhodopsin (reaction 2) and the subsequent attachment of a phosphate (reaction 3). For simplicity we assume that GRK1 can only dissociate after phosphate attachment (i.e. we consider only the effective GRK1 binding rate — the product of the true binding rate and the fraction of binding events that lead to phosphate attachment). The effective GRK1 binding rate depends on the fraction of time rhodopsin is available. Our hypothesis is that, in addition to quenching rhodopsin activity, the binding of arrestin1 to rhodopsin (reaction 1) controls rhodopsin availability and hence the effective GRK1 binding rate.
The two roles of arrestin depicted in Figure 1B — rhodopsin quenching and competition with GRK1 — will have opposite effects on the active lifetime of rhodopsin when the arrestin1 concentration is altered. In the absence of any changes in quenching, the arrestin competition hypothesis predicts that reducing the arrestin1 concentration will bias the competition in favor of GRK1, accelerating phosphorylation and Rh* shutoff (Figure 1C; see figure legend for simulation details). In the absence of competition, the role of arrestin in quenching rhodopsin's activity (reaction 4) means that lowering the arrestin1 concentration will prolong the active lifetime of Rh* (Figure 1D). Which of these opposing effects dominates depends on the rate constants describing each of the events depicted in Figure 1B. For example, if phosphate attachment (reaction 3) is much slower than GRK1 binding, then altering the binding rate through arrestin competition should have only a small effect on the overall kinetics of rhodopsin inactivation.
GRK1+/− and GRK1+/−Arr1+/− rods reveal strong competition between arrestin1 and GRK1
Arrestin competition should be prominent in rods expressing a lowered GRK1 concentration (GRK1+/− rods). Lowering the GRK1 concentration should slow GRK1 binding to rhodopsin, prolonging rhodopsin's active lifetime and increasing the sensitivity of the response kinetics to modulation of the GRK1 binding rate. Thus we start by characterizing the response properties of these rods.
Quantitative western blots showed that GRK1+/− rods contained ∼1/3 as much GRK1 as wild-type rods, whereas the concentrations of arrestin1 and other key transduction cascade components were near normal (Figure 2). Figure 3A compares estimated single-photon responses (the average response to a dim flash divided by the flash strength) from wild-type and GRK1+/− rods. These measurements were all made at 30°C, where we could also study variability in the single-photon response (see below). As expected for slower GRK1 binding, decreasing the GRK1 concentration increased the amplitude and slowed the single-photon response (Figure 3A and Table 1). We quantified the change in kinetics by computing the integration time — the area of the response divided by its peak amplitude. Changes in amplitude (p < 0.001) and integration time (p < 0.0001) were both significant.
Figure 3.

Rh* inactivation depends on the relative concentration of arrestin1 and GRK1. (A) Mean estimated single-photon responses for wild-type (black), GRK1+/− (blue), and GRK1+/−Arr1+/− (green) rods at 30 ± 1°C. (B) Representative Pepperberg plot (time spent in saturation as a function of the log of flash intensity) for wild-type, GRK1+/−, and GRK1+/−Arr1+/− rods. PDE activity decay time constants (τPDE) were 420, 630, and 430 ms. (C) τPDE for populations of wild-type, GRK1+/− and GRK1+/−Arr1+/−rods at 30 ± 1°C. τPDE = 370 ± 10 ms for wild-type (mean ± SEM; n = 52); 640 ± 25 ms for GRK1+/− (n = 36); and 360 ± 10 ms for GRK1+/−Arr1+/− (n = 38).
The changes in the single-photon response in GRK1+/− rods are consistent with the expected prolongation of rhodopsin's catalytic activity. To confirm this, we used the Pepperberg analysis to quantify the decay rate of light-activated PDE activity (Pepperberg et al., 1992). Decay of PDE activity depends on the inactivation of both rhodopsin and the G-protein transducin (Figure 1A); the decay rate for PDE activity will reflect primarily the slower of these two inactivation processes. The paradigm involves delivering bright flashes that activate PDE sufficiently strongly to minimize the internal cGMP concentration and suppress all of the rod's circulating dark current. This manipulation should also reduce the internal calcium concentration to a minimal level, causing a constant (and high) rate of cGMP synthesis. The time the response remains in saturation then reflects the time required for the PDE activity to drop to a critical value such that the cGMP synthesis rate exceeds the hydrolysis rate. Assuming that the PDE activity decays with an exponential time course, the slope of the relationship between time in saturation and the logarithm of the flash strength yields an estimate of the time constant for the decay of PDE activity (Pepperberg et al., 1992).
Figure 3B plots the dependence of saturation time on flash strength for single wild-type and GRK1+/− rods. The saturation time for the wild-type rod increased less steeply than that for the GRK1+/− rod across a wide range of flash strengths, reflecting a shorter time constant for PDE activity decay. Typical of such experiments, the relationship between saturation time and flash strength became steeper at high flash strengths. Since our interest is in the events shaping responses to dim lights, we estimated the time constant for the decay of PDE activity (τPDE) from the linear region of the plot at low flash strengths. Figure 3C collects results for multiple rods; τPDE for wild-type rods was considerably smaller than that for GRK1+/− rods (p < 0.0001, Figure 3C).
Figures 3A-C indicate that GRK1 binding to rhodopsin contributes substantially to the decay rate of PDE activity in GRK1+/− rods. In this case, the arrestin competition hypothesis (Figure 1B) predicts that lowering the arrestin1 concentration should speed GRK1 binding to rhodopsin, accelerating PDE shutoff and producing a smaller and more rapid flash response. We used GRK1+/−Arr1+/− rods to test this prediction. GRK1+/−Arr1+/− rods expressed ∼1/3 as much GRK1 and ∼1/2 as much arrestin1 as wild-type rods, whereas the concentrations of other key transduction cascade components were near normal (Figure 2). In agreement with the arrestin competition model, GRK1+/−Arr1+/− rods produced smaller and briefer single-photon responses than GRK1+/− rods (Figure 3A and Table 1; p < 0.01 for difference in amplitude and p < 10−5 for difference in integration time). Further, the PDE activity decay time constant measured from the Pepperberg plot was considerably smaller than that of GRK1+/− rods (Figure 3B and C; p < 0.0001).
In principle, arrestin competition could also regulate the rate of activation of transducin since transducin competes with arrestin1 and GRK1 for active rhodopsin. Such an effect could alter the initial rising phase of the light response. However, responses GRK1+/− and GRK1+/−Arr1+/− rods to both dim and bright flashes followed near identical initial trajectories. Two issues may contribute to a lack of an apparent effect on the initial rate of transducin activation. First, GDP-GTP exchange may be slower than the rate of binding of transducin to rhodopsin. Second, arrestin competition likely increases in strength as rhodopsin is phosphorylated and the affinity of the interaction between arrestin and rhodopsin increases (Wilden, 1995; Gibson et al., 2000; Gurevich and Gurevich, 2004; Vishnivetskiy et al., 2007).
The results of Figure 3A-C indicate that the primary effect of halving the arrestin1 concentration was to increase the GRK1 binding rate rather than to slow Rh* quenching. This suggests that arrestin competition, at least in the presence of reduced GRK1, can play an important role in controlling the kinetics of rhodopsin shutoff.
Arrestin competition reduces the catalytic activity of Rh* in wild-type rods
The above experiments revealed arrestin competition in rods with slowed GRK1 binding to rhodopsin. Does competition between arrestin1 and GRK1 continue at normal GRK1 concentrations, and if so what impact can it have on rhodopsin shutoff? Does arrestin competition persist at physiological temperatures? To answer these questions, we compared the single-photon responses and the rate of decay of PDE activity of Arr1+/− and wild-type rods at both 30°C and 36°C.
Arr1+/− rods expressed ∼1/3 as much arrestin1 as wild-type rods; expression levels of GRK1 and other key transduction cascade components were near normal (Figure 2). Figure 4A compares estimated single-photon responses of wild-type and Arr1+/− rods. Arr1+/− rods generated smaller (p < 0.001, 30°C and p < 0.0001, 36°C) and briefer (p < 0.05, 36°C) single-photon responses than wild-type rods (Table 1). Figure 4B plots the dependence of saturation time on flash strength for single wild-type and Arr1+/− rods. Saturation times for the Arr1+/− rod increased less steeply with flash strength than those of the wild-type rod. Figure 4C shows that Arr1+/− rods had a shorter τPDE than wild-type rods at 30°C (p < 0.0001) and at 36°C (p < 0.05).
Figure 4.

Decreasing arrestin1 concentration shortens Rh* lifetime. (A) Mean estimated single-photon responses from wild-type (black) and Arr1+/− (red) rods at 30 ± 1°C. Responses of GRK1+/− and GRK1+/−Arr1+/− rods from Figure 3 are shown for comparison. (B) Representative Pepperberg plot for a wild-type rod and an Arr1+/− rod. The saturation time for the wild-type rod increased more steeply with flash strength than that of the Arr1+/− rod at every flash strength measured. The time constant for PDE activity decay was 420 ms for the wild-type rod and 310 ms for the Arr1+/− rod. (C) τPDE for populations of wild-type and Arr1+/− rods at 30 ± 1°C and 36 ± 1°C. At 30°C, τPDE was 370 ± 10 ms (mean ± SEM) for wild-type rods (n = 52) and 290 ± 10 ms for Arr1+/− rods (n = 41). At 36°C, τPDE was 320 ± 10 ms (mean ± SEM) for wild-type rods (n = 15) and 250 ± 15 ms for Arr1+/− rods (n = 18).
The reduction in response amplitude, the faster recovery phase, and the shorter τPDE in the Arr1+/− rods are consistent with more rapid rhodopsin shutoff due to decreased competition by arrestin1 as depicted in Figure 1B and C. Thus lowering the arrestin1 concentration apparently increases the fraction of time rhodopsin is available for GRK1 binding and by doing so speeds phosphorylation and rhodopsin inactivation. This competition persists at physiological temperatures.
The decay of light-activated PDE activity reflects the inactivation kinetics of both rhodopsin and transducin. In particular, τPDE depends primarily on the slower of these inactivation processes. Prolonging or maintaining τPDE does not define which reaction is normally the rate-limiting step, but shortening τPDE does (Kennedy et al., 2003; Krispel et al., 2006). Thus, the reduced τPDE in Arr1+/− rods indicates that rhodopsin's active lifetime is relatively long in wild-type rods under the conditions of our experiments. We return to this point below in the context of how variability in rhodopsin shutoff produces variability in the single-photon response.
Relative rate constants of events controlling rhodopsin inactivation
To provide a quantitative description of how competition between arrestin1 and GRK1 influences rhodopsin inactivation, we used our experimental data to estimate the average rate constants controlling steps 1–3 in the kinetic scheme of Figure 1B. This is the simplest kinetic scheme consistent with the data of Figures 3 and 4. We were particularly interested in two issues: (1) How effectively does arrestin1 compete with GRK1 for Rh* binding sites? (2) What are the relative rate constants of GRK1 binding and phosphate attachment — the two events involved in a single phosphorylation cycle (completion of step 3 in Figure 1B)? To answer these questions, we described reactions 1-3 in Figure 1B as a set of differential equations that depend on α, β, γ, and σ. The fraction of time rhodopsin spends bound to arrestin1 in the model is β/(β + α) and the fraction of a phosphorylation cycle consumed by GRK1 binding is γ/(γ + σ). Thus we identified the relative rate constants for arrestin1 association and dissociation (β/α) and for GRK1 binding and phosphate attachment (γ/σ) most consistent with experiment (see Materials and Methods for details).
We made three assumptions to compare the kinetic model to experimental data. First, we assumed that β scaled linearly with the arrestin1 concentration (β = β0[Arr1]/[Arr1]WT) and that γ scaled linearly with the GRK1 concentration (γ = γ0[GRK1]/[GRK1]WT). Concentrations of arrestin1 and GRK1 relative to wild-type rods were taken from quantitative protein analysis (Figure 2B; see Materials and Methods for a discussion of this assumption). Second, we focused on the effective GRK1 binding rate — i.e. we did not include a reverse rate constant in reaction 2 of Figure 1B. Third, we assumed that the integral of the single-photon response (response area) was proportional to the time required for a single phosphorylation step. This assumption is reasonable if rhodopsin's active lifetime is proportional to the time required for each phosphorylation event, and the transduction cascade acts linearly to convert rhodopsin's activity to a change in current.
Based on the above assumptions, we determined the ratios of β0/α and γ0/σ that best fit the integrals of the single-photon responses in Arr1+/−, GRK1+/−, and GRK1+/−Arr1+/− rods (Table 2). Because of the third assumption, the estimated ratios represent average values across different phosphorylation events (i.e. unphosphorylated rhodopsin, singly phosphorylated rhodopsin, etc). The fitting procedure is not assured of providing a close correspondence between model and experiment because the model has two free parameters (β0/α and γ0/σ) and is fit to experimentally-determined values (the response areas) of each of the three mutants relative to wild-type. Nonetheless, the model accounted for the measured response areas within the experimental accuracy (Table 2).
Table 2.
Arrestin competition model can capture the changes in response area. Minimizing the MSE between experimental values (middle column) and predicted values (right column) yielded β0/α = 6 and γ0/σ = 8.
| Strain | Area/AreaWT | Area/AreaWT |
|---|---|---|
| isolated single-photon responses | experiment | model |
| GRK1+/− (n = 30) | 1.95 ± 0.08 | 2.02 |
| GRK1+/−Arr1+/− (n = 40) | 1.30 ± 0.07 | 1.34 |
| Arr1+/− (n = 41) | 0.68 ± 0.06 | 0.74 |
Ratios of β0/α ∼ 6 and γ0/σ ∼ 8 minimized the mean-square error between model and experiment. These rate constants make two predictions about the phosphorylation process in wild-type rods (β = β0 and γ = γ0) under the conditions of our experiments. First, active rhodopsin spends ∼85% of its time bound to arrestin1 (reaction 1 in Figure 1B), and only ∼15% of the time is available for GRK1 binding. The large fraction of time rhodopsin spends interacting with arrestin1 is a requirement for arrestin competition to control the effective GRK1 binding rate. Second, GRK1 binding is rapid compared to phosphate attachment (γ0 > σ). This latter observation can explain why arrestin competition was revealed more robustly when GRK1 binding was slowed by reducing the GRK1 concentration.
Implications of arrestin competition for single-photon response variability
The rod's single-photon responses show much less trial-to-trial variability than other signals generated by single molecules (Baylor et al., 1979), such as the charge flowing through an ion channel during a single opening or the signal generated by the binding of an odorant molecule to its cognate GPCR (Bhandawat et al., 2005). Several results indicate that variability in rhodopsin shutoff rather than downstream components of the phototransduction cascade dominates variability in the single-photon response (Rieke and Baylor, 1998; Doan et al., 2006). The model most consistent with experimental observations is that Rh* shuts off through a series of steps (Rieke and Baylor, 1998; Field and Rieke, 2002a; Hamer et al., 2003; Doan et al., 2006; Bisegna et al., 2008). One salient aspect of the measured responses is that most of the variability in the single-photon response occurs well after the response reaches peak (Rieke and Baylor, 1998; Field and Rieke, 2002a; Hamer et al., 2003). This late variance is inconsistent with a short Rh* lifetime (Rieke and Baylor, 1998; Hamer et al., 2003; Krispel et al., 2006), which should cause the responses to vary in amplitude but not in shape.
The low and late variability of the single-photon responses are signatures of the underlying molecular events regulating Rh* activity (Field and Rieke, 2002a; Hamer et al., 2003). We used these characteristics in the context of the arrestin competition hypothesis to test how altering the time constants of known events in Rh* shutoff affects reproducibility, and to resolve the apparent conflict between the late time-dependent variance and the short Rh* lifetime reported previously (Krispel et al., 2006). The experiments and analyses described below indicate that, under the conditions of our experiments, arrestin competition tunes the kinetics of rhodopsin shutoff to minimize variability, and that rhodopsin's active lifetime persists through much of the single-photon response.
Reproducibility of the single-photon responses depends on multiple timely shutoff steps
The left panels in Figure 5A-C superimpose ten isolated single-photon responses from wild-type, Arr1+/−, GRK1+/−, and GRK1+/−Arr1+/− rods. We isolated single-photon responses at 30°C because this minimized contamination from responses to 0 or multiple photons; response variability in wild-type rods was quantitatively similar at 30°C and 36°C (Doan et al., 2006). We characterized the single-photon response variability by measuring the coefficient of variation of the response areas (CVarea, standard deviation of the areas divided by the mean area). The CVarea captures the total variability, independent of the kinetics of rhodopsin shutoff relative to the kinetics of downstream components of the transduction cascade (Field and Rieke, 2002a; Hamer et al., 2003; Doan et al., 2006). The variance attributable to the single-photon response was measured from the difference between the variance of the areas of isolated single-photon responses and equivalent sections of dark record.
The single-photon responses of Arr1+/−, GRK1+/−, and GRK1+/−Arr1+/− rods all varied significantly more than those of wild-type rods (Table 1). Thus manipulations that slow rhodopsin shutoff (via slowed GRK1 binding to rhodopsin in GRK1+/− rods) as well as those that speed rhodopsin shutoff (via more rapid GRK1 binding due to decreased competition between arrestin1 and GRK1 in Arr1+/− rods) increase variability. Surprisingly, decreasing both arrestin1 and GRK1 concentrations (GRK1+/−Arr1+/− rods) increased variability, although the single-photon response kinetics and PDE activity decay rate were near normal (Table 1).
Changes in response variability can be explained in the context of the multi-step shutoff model for rhodopsin inactivation. For multiple shutoff steps to be effective in reducing response variability, each step must control a similar fraction of the total Rh* activity (Rieke and Baylor, 1998; Field and Rieke, 2002a; Hamer et al., 2003; Doan et al., 2006). Altering the arrestin1 or GRK1 concentrations apparently imbalances these steps and increases variability (see Discussion). The increased variability in the GRK1+/−Arr1+/− rods indicates that the absolute, and not just the relative, concentrations of GRK1 and arrestin1 are critical to the generation of reproducible single-photon responses.
The late response variability is consistent with slow rhodopsin shutoff
We characterized the time course of single-photon response variability by comparing the time-dependent variance of isolated single-photon responses with the square of the mean response (Figure 5A-C, right). For responses of wild-type rods, the variance (σ2) took about twice as long to reach its maximum as the mean response (μ) — i.e. tpeak,σ2/tpeak,μ was near 2. The relative time-to-peak of the variance depended on the relative concentrations of arrestin1 and GRK1. Thus tpeak,σ2/tpeak,μ for Arr1+/− rods was similar to that of wild-type rods (Figure 5A, Table 1), while tpeak,σ2/tpeak,μ for both GRK1+/− and GRK1+/−Arr1+/− rods was significantly larger than 2 (Figure 5B and C, Table 1). tpeak,σ2/tpeak,μ for GRK1+/−Arr1+/− rods was significantly less than that for GRK1+/− rods.
The dependence of response variability on arrestin1 and GRK1 concentrations — i.e. on manipulations that should be specific to rhodopsin — supports the idea that variability in rhodopsin shutoff rather than downstream components in phototransduction dominates variability in the single-photon response (also see Doan et al., 2006). In this case, tpeak,σ2/tpeak,μ depends on the lifetime of Rh* (τRh*) relative to that of activated transducin (τT*) (Hamer et al., 2003). If τRh* is short relative to τT*, then most of the variability occurs near the peak of the response as variations in Rh* lifetime primarily produce variations in response amplitude rather than shape. However, if τRh* is long compared to τT*, then most of the variability occurs during response recovery, as variations in Rh* lifetime alter the amplitude and duration of the single-photon responses.
To determine what constraints the measured time-dependent variances place on the ratio of the lifetimes of rhodopsin and transducin, we simulated single-photon responses by passing the modeled stochastic time course of a single rhodopsin molecule through a temporal filter that approximates the action of the phototransduction cascade (Figure 6A; see Materials and Methods for details) (Rieke and Baylor, 1998; Field and Rieke, 2002a). This model assumed τT* = 200 ms (Krispel et al., 2006). Rhodopsin's activity was assumed to be shut off by N first-order and memoryless steps attributed to rhodopsin phosphorylation and arrestin quenching. Each phosphorylation event was a 2-step process (Figure 1B, reactions 1 to 3). Arrestin quenching was modeled as a parallel step with a rate constant that increased with the number of completed phosphorylation events (Hamer et al., 2003; Vishnivetskiy et al., 2007). We used models in which an average of either 3.7 or 5.6 phosphorylation cycles completed before arrestin1 binding (see Materials and Methods). In the first model, saturation in the transduction cascade was required to account for the low response variability. The central conclusions reached below held true for both models; we emphasize the model without saturation since it had fewer free parameters.
Figure 6.

Rhodopsin's lifetime determines the variability and area of the single-photon responses. (A) Multistep shutoff model of rhodopsin's activity. (B) The ratio of the time-to-peak of the time-dependent variance (tpeak,σ2) to the time-to-peak of the mean (tpeak,μ) of the single-photon responses depends on rhodopsin's lifetime (τRh*). The solid black line is fit to the predicted changes (open circles) in tpeak,σ2/tpeak,μ as a function of τRh*. τT* was kept constant at 200 ms. The experimental points (closed circles) are located on the fit at a y-axis location according to the measured tpeak,σ2/tpeak,μ ratio; the resulting x-axis position estimates τRh*. (C) τRh* controls the area of the single-photon response. Solid circles are the experimentally-determined normalized response areas (Table 3). These values were positioned on the x-axis using the predicted τRh* determined in (B). The solid black line is fit to the predicted changes in response area as a function of τRh*. Insets show the average single-photon response of 1,000 simulated trials using the τRh* predicted from the model for each recorded mouse. The model captures the relative differences in amplitude and duration of the isolated single-photon responses in wild-type and mutant rods. Error bars are SEM.
The multi-step shutoff model was first applied to characterize the late time-to-peak of the time-dependent variance. Figure 6B illustrates the predicted dependence of the relative time-to-peak of the variance (tpeak,σ2/tpeak,μ) on the relative rhodopsin lifetime (τRh*/τT*). Consistent with the intuitive argument given above, tpeak,σ2/tpeak,μ increased steadily as τRh*/τT* increased. We used the predictions of the model in Figure 6B to estimate the relative rhodopsin lifetime for wild-type rods and rods with altered arrestin1 and GRK1 concentrations. The experimental points (filled circles) in Figure 6B are located along the line fit through the model prediction (open circles) according to the measured tpeak,σ2/tpeak,μ ratio (y-axis). The resulting x-axis position of the experimental points provides an estimate of the relative rhodopsin lifetime. Accounting for the late time to peak of the wild-type single-photon responses required τRh*/τT* ≈ 2.4 (Figure 6B, black circle).
To test the validity of the model, we used the rhodopsin lifetimes estimated from the time-dependent variance to predict the relative areas of the single-photon responses. The model predicts that the response area depends linearly on rhodopsin's lifetime (Figure 6C, open circles and line). The position of the experimental points in Figure 6C (closed circles) are determined from the measured areas (y-axis) and the estimated τRh*/τT* from Figure 6B (x-axis). Insets show predicted single-photon responses from the model (see Figure 3A and 4A for experimental results). The model, fit only to the time-to-peak of the variance, does a reasonable job accounting for the changes in response area and time course produced by alterations in arrestin1 and GRK1 concentrations.
The analysis in Figure 6 indicates that, in the context of the multi-step shutoff model, the late time-to-peak of the time-dependent variance requires that rhodopsin remain active for much of the duration of the single-photon response — i.e. that the active lifetime of rhodopsin exceeds that of transducin. A short rhodopsin lifetime would both fail to predict the time course of the time-dependent variance in wild-type rods and the change in variance produced by alterations in arrestin1 and GRK1 concentrations.
Discussion
We studied how changes in the concentration of GRK1 and arrestin1 affected the rod's single-photon responses. Our experiments indicate that arrestin1 competes with GRK1 for binding sites on rhodopsin and in doing so can slow both rhodopsin inactivation and facilitate low variability of the active lifetime of single rhodopsin molecules and of the resulting single-photon responses. A similar tuning of the kinetics of phosphorylation through arrestin competition may be a general feature of how GPCR activity is controlled, particularly for GPCRs in which arrestin and kinase bind to overlapping domains of the receptor.
Arrestin competition modulates single-photon response kinetics
Arrestins play a well known role in quenching the activity of G-protein coupled receptors (Kuhn et al., 1984; Wilden, 1995; Xu et al., 1997; Makino et al., 2003; Gurevich and Gurevich, 2004; Burns et al., 2006). Our results indicate that arrestins can play a second, previously unappreciated, role in regulating receptor activity through competition with kinases. Thus, the concentration of arrestin1 in the rod outer segment regulates the amount of time that rhodopsin is available for GRK1 binding and hence the rate of rhodopsin inactivation. Fitting the simple kinetic scheme in Figure 1 to properties of the measured responses indicated that active rhodopsin spends most of its time bound to arrestin1 and unavailable for GRK1 binding. This competition likely is initially weak and increases as phosphates are attached to rhodopsin (Wilden, 1995; Gibson et al., 2000; Gurevich and Gurevich, 2004; Vishnivetskiy et al., 2007). Phosphorylation is also likely to enhance competition between arrestin and transducin and hence slow transducin activation. Fits to the kinetic scheme of Figure 1 also indicated that GRK1 binding, even when slowed by arrestin competition, is more rapid than the subsequent phosphate attachment step.
Microcalorimetry experiments (Langlois et al., 1996) do not show a speeding of rhodopsin inactivation with lowered arrestin1 concentrations; these experiments instead support the role of arrestin1 in quenching rhodopsin's activity. However, these experiments and other biochemical assays of arrestin1 binding (Wilden et al., 1986; Wilden, 1995; Zhang et al., 1997; Brannock et al., 1999) use arrestin1 concentrations 10-1000 times lower than physiological levels (Hamm and Bownds, 1986; Schubert et al., 1999). Under these conditions, the arrestin quenching interaction is likely to dominate the competition interaction. For example, the kinetic model in Figure 1B predicts that lowering the arrestin1 concentration 10-fold from physiological levels would increase the fraction of time rhodopsin is available for GRK1 binding from ∼15% to ∼60%. A further 2-fold reduction in arrestin1 concentration would increase this to ∼75%, producing a ∼15% increase in the rate of GRK1 binding to rhodopsin. The same calculation, applied to physiological arrestin1 levels, predicts a ∼60% increase in the GRK1 binding rate. Thus when the arrestin1 concentration is substantially less than normal, the slowing of rhodopsin quenching by a further reduction in arrestin1 concentration may dominate any speeding of rhodopsin inactivation by decreased competition between arrestin1 and GRK1.
Arrestin competition minimizes variability in rhodopsin inactivation
A long-standing and puzzling observation about the rod's single-photon responses is their low variability (Baylor et al., 1979). This low variability requires tight regulation of rhodopsin's active lifetime. Several lines of evidence support a model in which the activity of a single rhodopsin molecule is terminated through a series of steps or transitions (Rieke and Baylor, 1998; Field and Rieke, 2002a; Hamer et al., 2003; Doan et al., 2006). The effectiveness of this model depends on the near equal distribution of rhodopsin's total activity among these steps — e.g. if a single step turns off the majority of rhodopsin's activity, then variability in the duration of this step will dominate variability in rhodopsin's effective lifetime. Our results suggest that competition between arrestin1 and GRK1 is important in producing a uniform distribution of rhodopsin's activity across shutoff steps.
Decreasing the concentration of arrestin1 and/or GRK1 increased single-photon response variability. This suggests that the distribution of rhodopsin's activity among steps is made less uniform when arrestin competition is altered. Indeed, we could capture the increase in variability and the change in time-dependent variance for the Arr1+/−, GRK1+/−, and GRK1+/−Arr1+/− rods when we altered the relative timing of the steps in the stochastic model. We did not include this model because it introduced unconstrained parameters. Mechanistically, a disruption of uniform timing of steps in rhodopsin shutoff suggests that the competition between arrestin1 and GRK1 depends on how many phosphates are already attached to rhodopsin's C-terminus. Consistent with this interpretation, the affinity of the interaction between arrestin1 and rhodopsin increases as rhodopsin is phosphorylated (Wilden, 1995; Gibson et al., 2000; Gurevich and Gurevich, 2004; Vishnivetskiy et al., 2007).
The native concentrations of arrestin1 and GRK1 are well suited to maintain low variability in rhodopsin shutoff. This adds to a perplexing functional question: why has the rod gone to such lengths to produce responses with such low variability? One suggestion is that low variability is required for accurate encoding of the time of photon absorption (Rieke and Baylor, 1998). The identification of mice with subtle changes in response variability could provide a needed tool to test this hypothesis.
Rhodopsin's activity persists through much of the single-photon response
The relative lifetimes of rhodopsin and the G-protein transducin has been a debated issue in phototransduction. Recent work has provided good evidence, at least under some experimental conditions, that the lifetime of the activated state of transducin is long compared to that of rhodopsin (Krispel et al., 2006). This issue has implications for both the site of amplification in the system and the underlying mechanism regulating the variability of the single-photon responses. A short rhodopsin lifetime implies that only ∼20 transducin molecules are activated, on average, while rhodopsin is active (Krispel et al., 2006). A brief rhodopsin lifetime also predicts that variability in rhodopsin's activity primarily alters the amplitude but not the duration of the single-photon responses. Measured single-photon responses in toad, mouse, guinea pig, and primate rods, however, vary little in amplitude but more substantially in response recovery (Rieke and Baylor, 1998; Field and Rieke, 2002a; Hamer et al., 2003). As discussed below, differences in experimental conditions appear to explain this discrepancy.
Several results reported here support a relatively long active rhodopsin lifetime. First, we found that the lifetime of light-activated PDE decreased when the arrestin1 concentration was decreased. Manipulations that prolong PDE decay do not define which reaction is normally the rate-limiting step, while manipulations that shorten PDE decay do (Kennedy et al., 2003; Krispel et al., 2006). Second, we showed that the systematic changes in the response kinetics with alterations in arrestin1 and/or GRK1 concentration are consistent with changes in the GRK1 binding rates predicted by the arrestin competition model. Finally, we demonstrated that the conserved characteristic time-dependent variance of the single-photon responses can be explained by a long rhodopsin lifetime (Figure 6).
We estimated that the active lifetime of rhodopsin is ∼2 times longer than the previously estimated active lifetime of transducin (Krispel et al., 2006) but similar to the ratio of ∼1.5 predicted from modeling by Hamer and colleagues (Hamer et al., 2003). The relative active lifetimes of rhodopsin and transducin appear to depend on experimental conditions (see Materials and Methods). Specifically, we found that dim flash responses measured in Locke's solution, as in the experiments of Krispel and colleagues, were faster and less sensitive than those measured in our experiments using Ames solution (Supplementary Figure 1). Decay of PDE activity in Locke's solution is also ∼2 times faster than that in Ames solution. These differences are consistent with slower rhodopsin inactivation in Ames solution. The underlying factors contributing to these differences are not understood.
The dependence of rod responses on recording conditions is striking and unexplained. It is not clear what conditions most closely resemble those in vivo. The kinetics of rod responses measured in Locke's solution resemble the rod component of the electroretinogram (Lyubarsky and Pugh, 1996; Hetling and Pepperberg, 1999): the time-to-peak of the estimated responses to sub-saturating flashes (∼110-130 ms for single rod responses vs ∼80-90 ms for the electroretinogram) and the rate of decay of PDE activity (∼180-250 ms for single rod responses vs ∼190-230 ms for the electroretinogram) are similar. However, the sensitivity of rod responses recorded in Ames is in closer agreement with the electroretinogram (half saturating flash strengths are ∼9 Rh* for single rod responses in Ames, ∼20 Rh* in Locke's and ∼10 Rh* for the electroretinogram; see Supplementary Materials and Hetling and Pepperberg, 1999). Furthermore, the isolated rod component of the electroretinogram shows a prominent contribution from the rod inner segment (Green and Kapousta-Bruneau, 1999; Nymark et al., 2005; Kang Derwent et al., 2007), complicating comparison with outer segment transduction currents. Ames and Locke's solutions are of course only two possibilities, and it is likely that in vivo conditions differ from both. Resolving this issue will be an important aspect of future work.
Regulating the concentrations of proteins involved in receptor shutoff as a mechanism to control GPCR-mediated signals
Controlling the relative concentrations of critical regulatory factors is a common approach used by biological systems to modulate cellular activities, including cellular differentiation, cell cycle checkpoints, organ development, inflammation, and chemotaxis (Parent and Devreotes, 1999; Pinsky et al., 2006; Metaye et al., 2006; Vroon et al., 2006; Ashe and Briscoe, 2006; Charest and Firtel, 2006). Our finding that arrestin competition has profound effects on the kinetics and variability of rhodopsin inactivation provides further evidence for this mechanism in GPCR systems (Detwiler et al., 2000; Wang et al., 2004). Specifically, the observation that changes in the arrestin1 concentration in dark-adapted rods result in altered and variable single-photon responses suggests that arrestin1 translocation yields an appropriate concentration of arrestin1 in the outer segment to compete with GRK1 and enable reliable signal transduction.
In other GPCR-mediated systems, such as the heart or the immune system, minute and prolonged perturbations in GRK concentrations are pathological (Hansen et al., 2006; Metaye et al., 2006; Vroon et al., 2006). An increase in GRK2 is associated with congestive heart failure and hypertension while decreases in GRK2, 3 and 6 are involved in improper inflammatory responses (Lombardi et al., 2001; Vroon et al., 2006; Lymperopoulos et al., 2007). Current therapeutic interventions, such as β-blockers in the treatment of heart failure and hyptertension, directly target GPCRs. If a fundamental strategy in regulating GPCR activity is through a careful balance of arrestin and GRK concentrations, then tissue-specific modulations of GRK and/or arrestin activity might be a new and useful therapeutic intervention for GPCR-related diseases.
Supplementary Material
Acknowledgments
We are grateful to Lars Holzhausen, Visvanathan Ramamurthy, and Florent Houdart for invaluable help with the protein quantification; Anna Mendez, Jeannie Chen, and Ching-Kang Chen for generously providing Arr1−/− and GRK1−/− mice; Ted Wensel for the generous gift of the α-RGS9c antibody; Clint Makino for the detailed protocol for experiments using Locke's solutions; Bertil Hille, Peter Detwiler, Charles Asbury, William Zagotta, Ben Pinsky, Jill Jensen, Felice Dunn, and Gabe Murphy for critical reading of the manuscript; the Rieke, Hurley and Perkel lab members for many helpful discussions; Paul Newman, Richard Alquist, Eric Martinson and Jonathan Linton (supported through the Vision Core Grant EY01730) for excellent technical support. Support was provided by the NIH through grants EY-11850 (F.R.), T32EY-07031(T.D.), T32GM-07270 (A.W.A), EY06641 (J.B.H); Poncin Scholarship (T.D.); and the Howard Hughes Medical Institute (F.R.).
References
- Ashe HL, Briscoe J. The interpretation of morphogen gradients. Development. 2006;133:385–394. doi: 10.1242/dev.02238. [DOI] [PubMed] [Google Scholar]
- Baylor DA, Lamb TD, Yau KW. Responses of retinal rods to single photons. J Physiol. 1979;288:613–634. [PMC free article] [PubMed] [Google Scholar]
- Bhandawat V, Reisert J, Yau KW. Elementary response of olfactory receptor neurons to odorants. Science. 2005;308:1931–1934. doi: 10.1126/science.1109886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisegna P, Caruso G, Andreucci D, Shen L, Gurevich VV, Hamm HE, DiBenedetto E. Diffusion of the second messengers in the cytoplasm acts as a variability suppressor of the single photon response in vertebrate phototransduction. Biophys J. 2008;94:3363–83. doi: 10.1529/biophysj.107.114058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brannock MT, Weng K, Robinson PR. Rhodopsin's carboxyl-terminal threonines are required for wild-type arrestin-mediated quench of transducin activation in vitro. Biochemistry. 1999;38:3770–3777. doi: 10.1021/bi982419w. [DOI] [PubMed] [Google Scholar]
- Broekhuyse RM, Tolhuizen EF, Janssen AP, Winkens HJ. Light induced shift and binding of s-antigen in retinal rods. Curr Eye Res. 1985;4:613–618. doi: 10.3109/02713688508999993. [DOI] [PubMed] [Google Scholar]
- Buck LB. The molecular architecture of odor and pheromone sensing in mammals. Cell. 2000;100:611–618. doi: 10.1016/s0092-8674(00)80698-4. [DOI] [PubMed] [Google Scholar]
- Burns ME, Mendez A, Chen CK, Almuete A, Quillinan N, Simon MI, Baylor DA, Chen J. Deactivation of phosphorylated and nonphosphorylated rhodopsin by arrestin splice variants. J Neurosci. 2006;26:1036–1044. doi: 10.1523/JNEUROSCI.3301-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns ME, Mendez A, Chen J, Baylor DA. Dynamics of cyclic GMP synthesis in retinal rods. Neuron. 2002;36:81–91. doi: 10.1016/s0896-6273(02)00911-x. [DOI] [PubMed] [Google Scholar]
- Calvert PD, Govardovskii VI, Krasnoperova N, Anderson RE, Lem J, Makino CL. Membrane protein diffusion sets the speed of rod phototransduction. Nature. 2001;411:90–94. doi: 10.1038/35075083. [DOI] [PubMed] [Google Scholar]
- Chandrashekar J, Hoon MA, Ryba NJ, Zuker CS. The receptors and cells for mammalian taste. Nature. 2006;444:288–294. doi: 10.1038/nature05401. [DOI] [PubMed] [Google Scholar]
- Charest PG, Firtel RA. Feedback signaling controls leading-edge formation during chemotaxis. Curr Opin Genet Dev. 2006;16:339–347. doi: 10.1016/j.gde.2006.06.016. [DOI] [PubMed] [Google Scholar]
- Chen CK, Burns ME, Spencer M, Niemi GA, Chen J, Hurley JB, Baylor DA, Simon MI. Abnormal photoresponses and light-induced apoptosis in rods lacking rhodopsin kinase. Proc Natl Acad Sci U S A. 1999;96:3718–22. doi: 10.1073/pnas.96.7.3718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Makino CL, Peachey NS, Baylor DA, Simon MI. Mechanisms of rhodopsin inactivation in vivo as revealed by a COOH-terminal truncation mutant. Science. 1995;267:374–377. doi: 10.1126/science.7824934. [DOI] [PubMed] [Google Scholar]
- Detwiler PB, Ramanathan S, Sengupta A, Shraiman BI. Engineering aspects of enzymatic signal transduction: photoreceptors in the retina. Biophys J. 2000;79:2801–2817. doi: 10.1016/S0006-3495(00)76519-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doan T, Mendez A, Detwiler PB, Chen J, Rieke F. Multiple phosphorylation sites confer reproducibility of the rod's single-photon responses. Science. 2006;313:530–533. doi: 10.1126/science.1126612. [DOI] [PubMed] [Google Scholar]
- Eglen RM, Bosse R, Reisine T. Emerging concepts of guanine nucleotide-binding protein-coupled receptor (GPCR) function and implications for high throughput screening. Assay Drug Dev Technol. 2007;5:425–451. doi: 10.1089/adt.2007.062. [DOI] [PubMed] [Google Scholar]
- Field GD, Rieke F. Mechanisms regulating variability of the single photon responses of mammalian rod photoreceptors. Neuron. 2002a;35:733–747. doi: 10.1016/s0896-6273(02)00822-x. [DOI] [PubMed] [Google Scholar]
- Field GD, Rieke F. Nonlinear signal transfer from mouse rods to bipolar cells and implications for visual sensitivity. Neuron. 2002b;34:773–785. doi: 10.1016/s0896-6273(02)00700-6. [DOI] [PubMed] [Google Scholar]
- Field GD, Sampath AP, Rieke F. Retinal processing near absolute threshold: from behavior to mechanism. Annu Rev Physiol. 2005;67:491–514. doi: 10.1146/annurev.physiol.67.031103.151256. [DOI] [PubMed] [Google Scholar]
- Gibson SK, Parkes JH, Liebman PA. Phosphorylation modulates the affinity of light-activated rhodopsin for G protein and arrestin. Biochemistry. 2000;39:5738–5749. doi: 10.1021/bi991857f. [DOI] [PubMed] [Google Scholar]
- Green DG, Kapousta-Bruneau NV. A dissection of the electroretinogram from the isolated rat retina with microelectrodes and drugs. Vis Neurosci. 1999;16:727–741. doi: 10.1017/s0952523899164125. [DOI] [PubMed] [Google Scholar]
- Gurevich VV, Benovic JL. Cell-free expression of visual arrestin. Truncation mutagenesis identifies multiple domains involved in rhodopsin interaction. J Biol Chem. 1992;267:21919–21923. [PubMed] [Google Scholar]
- Gurevich VV, Benovic JL. Visual arrestin interaction with rhodopsin. Sequential multisite binding ensures strict selectivity toward light-activated phosphorylated rhodopsin. J Biol Chem. 1993;268:11628–11638. [PubMed] [Google Scholar]
- Gurevich VV, Benovic JL. Visual arrestin binding to rhodopsin. Diverse functional roles of positively charged residues within the phosphorylation-recognition region of arrestin. J Biol Chem. 1995;270:6010–6016. doi: 10.1074/jbc.270.11.6010. [DOI] [PubMed] [Google Scholar]
- Gurevich VV, Gurevich EV. The molecular acrobatics of arrestin activation. Trends Pharmacol Sci. 2004;25:105–111. doi: 10.1016/j.tips.2003.12.008. [DOI] [PubMed] [Google Scholar]
- Hamer RD, Nicholas SC, Tranchina D, Liebman PA, Lamb TD. Multiple steps of phosphorylation of activated rhodopsin can account for the reproducibility of vertebrate rod single-photon responses. J Gen Physiol. 2003;122:419–444. doi: 10.1085/jgp.200308832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamm HE, Bownds MD. Protein complement of rod outer segments of frog retina. Biochemistry. 1986;25:4512–4523. doi: 10.1021/bi00364a010. [DOI] [PubMed] [Google Scholar]
- Hansen JL, Theilade J, Aplin M, Sheikh SP. Role of G-protein-coupled receptor kinase 2 in the heart–do regulatory mechanisms open novel therapeutic perspectives? Trends Cardiovasc Med. 2006;16:169–177. doi: 10.1016/j.tcm.2006.03.004. [DOI] [PubMed] [Google Scholar]
- Hanson SM, Gurevich EV, Vishnivetskiy SA, Ahmed MR, Song X, Gurevich VV. Each rhodopsin molecule binds its own arrestin. Proc Natl Acad Sci U S A. 2007a;104:3125–3128. doi: 10.1073/pnas.0610886104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson SM, Van Eps N, Francis DJ, Altenbach C, Vishnivetskiy SA, Arshavsky VY, Klug CS, Hubbell WL, Gurevich VV. Structure and function of the visual arrestin oligomer. EMBO J. 2007b;26:1726–1736. doi: 10.1038/sj.emboj.7601614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetling JR, Pepperberg DR. Sensitivity and kinetics of mouse rod flash responses determined in vivo from paired-flash electroretinograms. J Physiol. 1999;516:593–609. doi: 10.1111/j.1469-7793.1999.0593v.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imamoto Y, Tamura C, Kamikubo H, Kataoka M. Concentration-dependent tetramerization of bovine visual arrestin. Biophys J. 2003;85:1186–95. doi: 10.1016/S0006-3495(03)74554-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang Derwent JJ, Saszik SM, Maeda H, Little DM, Pardue MT, Frishman LJ, Pepperberg DR. Test of the paired-flash electroretinographic method in mice lacking b-waves. Vis Neurosci. 2007;24:141–9. doi: 10.1017/S0952523807070162. [DOI] [PubMed] [Google Scholar]
- Kelleher DJ, Johnson GL. Characterization of rhodopsin kinase purified from bovine rod outer segments. J Biol Chem. 1990;265:2632–2639. [PubMed] [Google Scholar]
- Kennedy MJ, Lee KA, Niemi GA, Craven KB, Garwin GG, Saari JC, Hurley JB. Multiple phosphorylation of rhodopsin and the in vivo chemistry underlying rod photoreceptor dark adaptation. Neuron. 2001;31:87–101. doi: 10.1016/s0896-6273(01)00340-3. [DOI] [PubMed] [Google Scholar]
- Kennedy MJ, Sowa ME, Wensel TG, Hurley JB. Acceleration of key reactions as a strategy to elucidate the rate-limiting chemistry underlying phototransduction inactivation. Invest Ophthalmol Vis Sci. 2003;44:1016–1022. doi: 10.1167/iovs.02-0692. [DOI] [PubMed] [Google Scholar]
- Kobilka BK, Deupi X. Conformational complexity of G-protein-coupled receptors. Trends Pharmacol Sci. 2007;28:397–406. doi: 10.1016/j.tips.2007.06.003. [DOI] [PubMed] [Google Scholar]
- Koch KW, Stryer L. Highly cooperative feedback control of retinal rod guanylate cyclase by calcium ions. Nature. 1988;334:64–66. doi: 10.1038/334064a0. [DOI] [PubMed] [Google Scholar]
- Krispel CM, Chen D, Melling N, Chen YJ, Martemyanov KA, Quillinan N, Arshavsky VY, Wensel TG, Chen CK, Burns ME. RGS expression rate-limits recovery of rod photoresponses. Neuron. 2006;51:409–416. doi: 10.1016/j.neuron.2006.07.010. [DOI] [PubMed] [Google Scholar]
- Kristiansen K. Molecular mechanisms of ligand binding, signaling, and regulation within the superfamily of g-protein-coupled receptors: molecular modeling and mutagenesis approaches to receptor structure and function. Pharmacol Ther. 2004;103:21–80. doi: 10.1016/j.pharmthera.2004.05.002. [DOI] [PubMed] [Google Scholar]
- Krupnick JG, Gurevich VV, Benovic JL. Mechanism of quenching of phototransduction. binding competition between arrestin and transducin for phosphorhodopsin. J Biol Chem. 1997;272:18125–18131. doi: 10.1074/jbc.272.29.18125. [DOI] [PubMed] [Google Scholar]
- Krupnick JG, Gurevich VV, Schepers T, Hamm HE, Benovic JL. Arrestin-rhodopsin interaction. Multi-site binding delineated by peptide inhibition. J Biol Chem. 1994;269:3226–3232. [PubMed] [Google Scholar]
- Kuhn H, Hall SW, Wilden U. Light-induced binding of 48-kDa protein to photoreceptor membranes is highly enhanced by phosphorylation of rhodopsin. FEBS Lett. 1984;176:473–478. doi: 10.1016/0014-5793(84)81221-1. [DOI] [PubMed] [Google Scholar]
- Langlois G, Chen CK, Palczewski K, Hurley JB, Vuong TM. Responses of the phototransduction cascade to dim light. Proc Natl Acad Sci U S A. 1996;93:4677–4682. doi: 10.1073/pnas.93.10.4677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling Y, Ascano M, Robinson P, Gregurick SK. Experimental and computational studies of the desensitization process in the bovine rhodopsin-arrestin complex. Biophys J. 2004;86:2445–2454. doi: 10.1016/S0006-3495(04)74301-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombardi MS, Kavelaars A, Cobelens PM, Schmidt RE, Schedlowski M, Heijnen CJ. Adjuvant arthritis induces down-regulation of G protein-coupled receptor kinases in the immune system. J Immunol. 2001;166:1635–1640. doi: 10.4049/jimmunol.166.3.1635. [DOI] [PubMed] [Google Scholar]
- Lymperopoulos A, Rengo G, Funakoshi H, Eckhart AD, Koch WJ. Adrenal GRK2 upregulation mediates sympathetic overdrive in heart failure. Nat Med. 2007;13:315–323. doi: 10.1038/nm1553. [DOI] [PubMed] [Google Scholar]
- Lyubarsky AL, Pugh EN., Jr Recovery phase of the murine rod photoresponse reconstructed from electroretinographic recordings. J Neurosci. 1996;16:563–571. doi: 10.1523/JNEUROSCI.16-02-00563.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda T, Imanishi Y, Palczewski K. Rhodopsin phosphorylation: 30 years later. Prog Retin Eye Res. 2003;22:417–434. doi: 10.1016/s1350-9462(03)00017-x. [DOI] [PubMed] [Google Scholar]
- Makino CL, Dodd RL, Chen J, Burns ME, Roca A, Simon MI, Baylor DA. Recoverin regulates light-dependent phosphodiesterase activity in retinal rods. J Gen Physiol. 2004;123:729–741. doi: 10.1085/jgp.200308994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makino CL, Wen XH, Lem J. Piecing together the timetable for visual transduction with transgenic animals. Curr Opin Neurobiol. 2003;13:404–412. doi: 10.1016/s0959-4388(03)00091-6. [DOI] [PubMed] [Google Scholar]
- Mendez A, Burns ME, Roca A, Lem J, Wu LW, Simon MI, Baylor DA, Chen J. Rapid and reproducible deactivation of rhodopsin requires multiple phosphorylation sites. Neuron. 2000;28:153–164. doi: 10.1016/s0896-6273(00)00093-3. [DOI] [PubMed] [Google Scholar]
- Metaye T, Perdrisot R, Kraimps JL. GRKs and arrestins: the therapeutic pathway? Med Sci (Paris) 2006;22:537–543. doi: 10.1051/medsci/2006225537. [DOI] [PubMed] [Google Scholar]
- Nymark S, Heikkinen H, Haldin C, Donner K, Koskelainen A. Light responses and light adaptation in rat retinal rods at different temperatures. J Physiol. 2005;567:923–38. doi: 10.1113/jphysiol.2005.090662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohguro H, Van Hooser JP, Milam AH, Palczewski K. Rhodopsin phosphorylation and dephosphorylation in vivo. J Biol Chem. 1995;270:14259–14262. doi: 10.1074/jbc.270.24.14259. [DOI] [PubMed] [Google Scholar]
- Palczewski K, Buczylko J, Kaplan MW, Polans AS, Crabb JW. Mechanism of rhodopsin kinase activation. J Biol Chem. 1991;266:12949–12955. [PubMed] [Google Scholar]
- Parent CA, Devreotes PN. A cell's sense of direction. Science. 1999;284:765–770. doi: 10.1126/science.284.5415.765. [DOI] [PubMed] [Google Scholar]
- Pepperberg DR, Cornwall MC, Kahlert M, Hofmann KP, Jin J, Jones GJ, Ripps H. Light-dependent delay in the falling phase of the retinal rod photoresponse. Vis Neurosci. 1992;8:9–18. doi: 10.1017/s0952523800006441. [DOI] [PubMed] [Google Scholar]
- Peterson JJ, Tam BM, Moritz OL, Shelamer CL, Dugger DR, McDowell JH, Hargrave PA, Papermaster DS, Smith WC. Arrestin migrates in photoreceptors in response to light: a study of arrestin localization using an arrestin-gfp fusion protein in transgenic frogs. Exp Eye Res. 2003;76:553–563. doi: 10.1016/s0014-4835(03)00032-0. [DOI] [PubMed] [Google Scholar]
- Pinsky BA, Kotwaliwale CV, Tatsutani SY, Breed CA, Biggins S. Glc7/protein phosphatase 1 regulatory subunits can oppose the Ipl1/aurora protein kinase by redistributing glc7. Mol Cell Biol. 2006;26:2648–2660. doi: 10.1128/MCB.26.7.2648-2660.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Premont RT, Gainetdinov RR. Physiological roles of g protein-coupled receptor kinases and arrestins. Annu Rev Physiol. 2007;69:511–534. doi: 10.1146/annurev.physiol.69.022405.154731. [DOI] [PubMed] [Google Scholar]
- Raman D, Osawa S, Gurevich VV, Weiss ER. The interaction with the cytoplasmic loops of rhodopsin plays a crucial role in arrestin activation and binding. J Neurochem. 2003;84:1040–1050. doi: 10.1046/j.1471-4159.2003.01598.x. [DOI] [PubMed] [Google Scholar]
- Raman D, Osawa S, Weiss ER. Binding of arrestin to cytoplasmic loop mutants of bovine rhodopsin. Biochemistry. 1999;38:5117–5123. doi: 10.1021/bi9824588. [DOI] [PubMed] [Google Scholar]
- Rieke F, Baylor DA. Origin of reproducibility in the responses of retinal rods to single photons. Biophys J. 1998;75:1836–1857. doi: 10.1016/S0006-3495(98)77625-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampath AP, Strissel KJ, Elias R, Arshavsky VY, McGinnis JF, Chen J, Kawamura S, Rieke F, Hurley JB. Recoverin improves rod-mediated vision by enhancing signal transmission in the mouse retina. Neuron. 2005;46:413–420. doi: 10.1016/j.neuron.2005.04.006. [DOI] [PubMed] [Google Scholar]
- Schubert C, Hirsch JA, Gurevich VV, Engelman DM, Sigler PB, Fleming KG. Visual arrestin activity may be regulated by self-association. J Biol Chem. 1999;274:21186–21190. doi: 10.1074/jbc.274.30.21186. [DOI] [PubMed] [Google Scholar]
- Shi W, Osawa S, Dickerson CD, Weiss ER. Rhodopsin mutants discriminate sites important for the activation of rhodopsin kinase and gt. J Biol Chem. 1995;270:2112–2119. doi: 10.1074/jbc.270.5.2112. [DOI] [PubMed] [Google Scholar]
- Tamura T, Nakatani K, Yau KW. Calcium feedback and sensitivity regulation in primate rods. J Gen Physiol. 1991;98:95–130. doi: 10.1085/jgp.98.1.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vishnivetskiy SA, Paz CL, Schubert C, Hirsch JA, Sigler PB, Gurevich VV. How does arrestin respond to the phosphorylated state of rhodopsin? J Biol Chem. 1999;274:11451–11454. doi: 10.1074/jbc.274.17.11451. [DOI] [PubMed] [Google Scholar]
- Vishnivetskiy SA, Raman D, Wei J, Kennedy MJ, Hurley JB, Gurevich VV. Regulation of arrestin binding by rhodopsin phosphorylation level. J Biol Chem. 2007;282:32075–32083. doi: 10.1074/jbc.M706057200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vroon A, Heijnen CJ, Kavelaars A. GRKs and arrestins: regulators of migration and inflammation. J Leukoc Biol. 2006;80:1214–1221. doi: 10.1189/jlb.0606373. [DOI] [PubMed] [Google Scholar]
- Wang Q, Zhao J, Brady AE, Feng J, Allen PB, Lefkowitz RJ, Greengard P, Limbird LE. Spinophilin blocks arrestin actions in vitro and in vivo at G protein-coupled receptors. Science. 2004;304:1940–1944. doi: 10.1126/science.1098274. [DOI] [PubMed] [Google Scholar]
- Whitlock GG, Lamb TD. Variability in the time course of single photon responses from toad rods: termination of rhodopsin's activity. Neuron. 1999;23:337–351. doi: 10.1016/s0896-6273(00)80784-9. [DOI] [PubMed] [Google Scholar]
- Wilden U. Duration and amplitude of the light-induced cGMP hydrolysis in vertebrate photoreceptors are regulated by multiple phosphorylation of rhodopsin and by arrestin binding. Biochemistry. 1995;34:1446–1454. doi: 10.1021/bi00004a040. [DOI] [PubMed] [Google Scholar]
- Wilden U, Hall SW, Kuhn H. Phosphodiesterase activation by photoexcited rhodopsin is quenched when rhodopsin is phosphorylated and binds the intrinsic 48-kDa protein of rod outer segments. Proc Natl Acad Sci U S A. 1986;83:1174–1178. doi: 10.1073/pnas.83.5.1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilden U, Kuhn H. Light-dependent phosphorylation of rhodopsin: number of phosphorylation sites. Biochemistry. 1982;21:3014–3022. doi: 10.1021/bi00541a032. [DOI] [PubMed] [Google Scholar]
- Xu J, Dodd RL, Makino CL, Simon MI, Baylor DA, Chen J. Prolonged photoresponses in transgenic mouse rods lacking arrestin. Nature. 1997;389:505–509. doi: 10.1038/39068. [DOI] [PubMed] [Google Scholar]
- Zhang H, Li S, Doan T, Rieke F, Detwiler PB, Frederick JM, Baehr W. Deletion of PrBP/delta impedes transport of GRK1 and PDE6 catalytic subunits to photoreceptor outer segments. Proc Natl Acad Sci U S A. 2007;104:8857–8862. doi: 10.1073/pnas.0701681104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Sports CD, Osawa S, Weiss ER. Rhodopsin phosphorylation sites and their role in arrestin binding. J Biol Chem. 1997;272:14762–14768. doi: 10.1074/jbc.272.23.14762. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.