

Abstract
Pregnane X receptor (PXR) is activated by chemicals to transcriptionally regulate drug disposition and possibly decrease drug efficacy and increase resistance, suggesting therapeutic value for PXR antagonists. We previously reported the antagonist SPA70 and its analog SJB7, which unexpectedly is an agonist. Here, we describe another unexpected observation: mutating a single residue (W299A) within the PXR ligand-binding domain converts SPA70 to an agonist. After characterizing wild-type and W299A PXR activity profiles, we used molecular dynamics simulations to reveal that in wild-type PXR, agonists stabilize the activation function 2 (AF-2) helix in an “inward” position, but SPA70 displaces the AF-2. In W299A, however, SPA70 stabilizes the AF-2 “inward,” like agonists. We validated our model by predicting the antagonist SJC2 to be a W299A agonist, which was confirmed experimentally. Our work correlates previously unobserved ligand-induced conformational changes to PXR cellular activity and, for the first time, reveals how PXR antagonists work.
Keywords: Nuclear receptor, drug metabolism, corepressor, coactivator, cytochrome P450, xenobiotics
INTRODUCTION
Human pregnane X receptor (PXR) is a major nuclear receptor involved in drug metabolism and disposition. PXR is modulated by many diverse chemicals and transcriptionally regulates drug metabolizing enzymes and transporters to control xenobiotic disposition and endobiotic homeostasis [1–5]. Genes regulated by PXR include those encoding cytochrome P450 (CYP) enzymes such as CYP3A4 [5], CYP3A5 [6], and CYP2B6 [7], phase II metabolizing enzymes such as UDP glucuronosyltransferases [8], transporters such as multidrug resistance protein 1 [9,10], and many others. PXR has great promiscuity in its ligand-binding profile and is activated by many xenobiotics, possibly causing drug resistance, toxicity, or drug-drug interactions. Well-known compounds that activate PXR include the antibiotic rifampicin (Rif) [5], the chemotherapy agent paclitaxel [11], the antiviral ritonavir [12], and hyperforin from the herb St. John’s Wort [13]. While many PXR agonists have been identified, there are few known antagonists, and these chemicals are known to have nonspecific effects unrelated to PXR [14]. However, we recently reported SPA70 as a potent and highly selective PXR antagonist, and this compound may serve as a tool for investigating the roles of PXR in cell and disease biology [15,16]. The chemical structures of compounds used in the current study are presented in Figure 1.
Fig 1. Chemical structures of compounds used in this study.
As other nuclear receptors, PXR is composed of an N-terminal DNA-binding domain (DBD) and a C-terminal ligand-binding domain (LBD) connected by a flexible hinge region [17]. Upon ligand-induced activation, PXR forms heterodimers with retinoid X receptor (RXR) to bind to hexanucleotide direct and inverted repeats at the promoters of target genes [2,1,3,5]. The transcriptional activity of PXR is enhanced by coactivators, such as steroid receptor coactivator 1 (SRC-1), and repressed by corepressors, such as nuclear receptor corepressor (NCoR) [18]. Based on knowledge gained from related nuclear receptors, binding of coactivators or corepressors is dependent upon positioning of the C-terminal activation function 2 (AF-2) helix, which in turn is affected by ligand binding [19]. PXR’s large, flexible ligand-binding pocket, with a size of 1200–1600 Å3, is believed to mediate ligand promiscuity, allowing accommodation of compounds with varying sizes and structures [19]. However, species-specific PXR ligand preferences suggest that binding pocket size is not the sole factor in providing promiscuity. Indeed, mutating just one or a few amino acids can alter PXR’s response to human-specific Rif or SR12813 and rodent-specific pregnenolone-16α-carbonitrile [20–24]. Furthermore, we previously showed that structurally similar compounds bind with similar affinity to PXR but have opposing cellular outcomes, suggesting that subtle structural changes in PXR ligands may have dramatically different functional consequences [Figure 1; SJB7 (agonist) and SPA70 (antagonist)] [16].
A previously reported computational approach based on crystal structures of apo and ligand-bound PXR LBD identified five “hot spots” that are major contributors to ligand binding [25]. The hot spot consisting of the cross-species conserved residues F288, W299, and Y306 interacts with all co-crystallized PXR ligands. While there is currently no crystal structure of antagonist-bound PXR, our docking studies coupled with hydrogen-deuterium exchange have shown that the antagonist SPA70 may bind PXR similarly to SPA70’s structurally related agonists such as SJB7 that contact the F288-W299-Y306 hot spot [16], but it is unclear why SPA70 is an antagonist but SJB7 is an agonist. We previously studied the impacts of mutating W299 on the cellular activities of ligand binding and found that mutants have differential effects on PXR activation by the agonists Rif, SR12813, and T0901317 (T0) [26]. Additionally, phenobarbital was recently shown to directly activate PXR in a W299-dependent manner [27]. These studies reveal a critical role of W299 in the cellular activities of ligand binding.
In the current study, we used W299 mutants to study ligand-mediated PXR activation and inhibition. We carefully characterized W299A PXR and found that it lacks the basal activity that is present with WT PXR and disproportionately reduces Rif and T0 potency; both Rif and T0, however, fully activate the mutant at high concentrations. Unexpectedly, SPA70, the antagonist of WT PXR, became an agonist of W299A. Further analyses confirmed the antagonistic behavior of SPA70 for WT PXR and its opposing agonistic behavior for W299A PXR, such as differential DNA binding and cofactor interactions. Because the mutation functionally switches SPA70, we used the W299A LBD as a platform to study the mechanisms of PXR activation vs. inhibition by small molecules. Molecular dynamics (MD) simulations of the PXR-ligand systems suggest that the positioning of the AF-2 helix in WT LBD is differentially affected by agonists [(“inward”) for both T0 and SJB7] and antagonist SPA70 (“outward”), providing the first explanation for the mechanism of PXR antagonism. Consistent with our cellular data showing that SPA70 activates W299A PXR, MD simulations revealed that the AF-2 helix is positioned in an agonistic mode in W299A LBD with SPA70 (“inward”), similar to that in both WT and W299A with agonists T0 and SJB7. We further used our model on the two distinct ligand-induced AF-2 conformations (“inward” for agonist and “outward” for antagonist) to predict a known PXR antagonist (SJC2) [16] to be an agonist for W299A, which was subsequently confirmed in cell-based assays. Our results correlate distinctligand-induced conformations to the opposing cellular activities of agonist and antagonist and provide insights into mechanisms of PXR activation and inhibition by small molecules.
RESULTS
W299A PXR has reduced basal and agonist-induced activities
W299 is a cross-species conserved residue in the PXR ligand-binding pocket (Supplementary Figure 1), and we have previously reported that PXR activation by agonists is affected by W299 mutations [26]. To further investigate these effects, we generated dose response curves using HepG2 cells (clone C3A, henceforth referred to as HepG2) co-transfected with pcDNA3 (empty vector, EV) or WT or mutant PXR expression vector and a reporter plasmid encoding firefly luciferase under the control of a PXR-regulated CYP3A4 promoter and treated with various compounds (Supplementary Figure 2). Whereas some mutants (i.e., W299F) resembled WT, some were completely inactive (i.e., W299D – used as a negative control for the remainder of this study), and others showed decreased basal activity and shifts in agonist potency, but retained inducibility (i.e., W299A). The observation that W299A lacked basal activity but was still agonistinducible, together with previous reports that it reduced [26] or abolished [27] the activity of agonists, prompted us to fully characterize this mutant. Consistent with our previous findings, W299A reduced the activation of PXR by T0 to a larger extent than that by Rif; however, at high concentrations, T0 and Rif both activated W299A equally to WT (Figure 2a). Furthermore, we confirmed that W299A PXR lacked the basal activity present in the WT protein (Figure 2b).
Fig 2. PXR activation by Rif and T0 is differentially affected by W299 mutations.
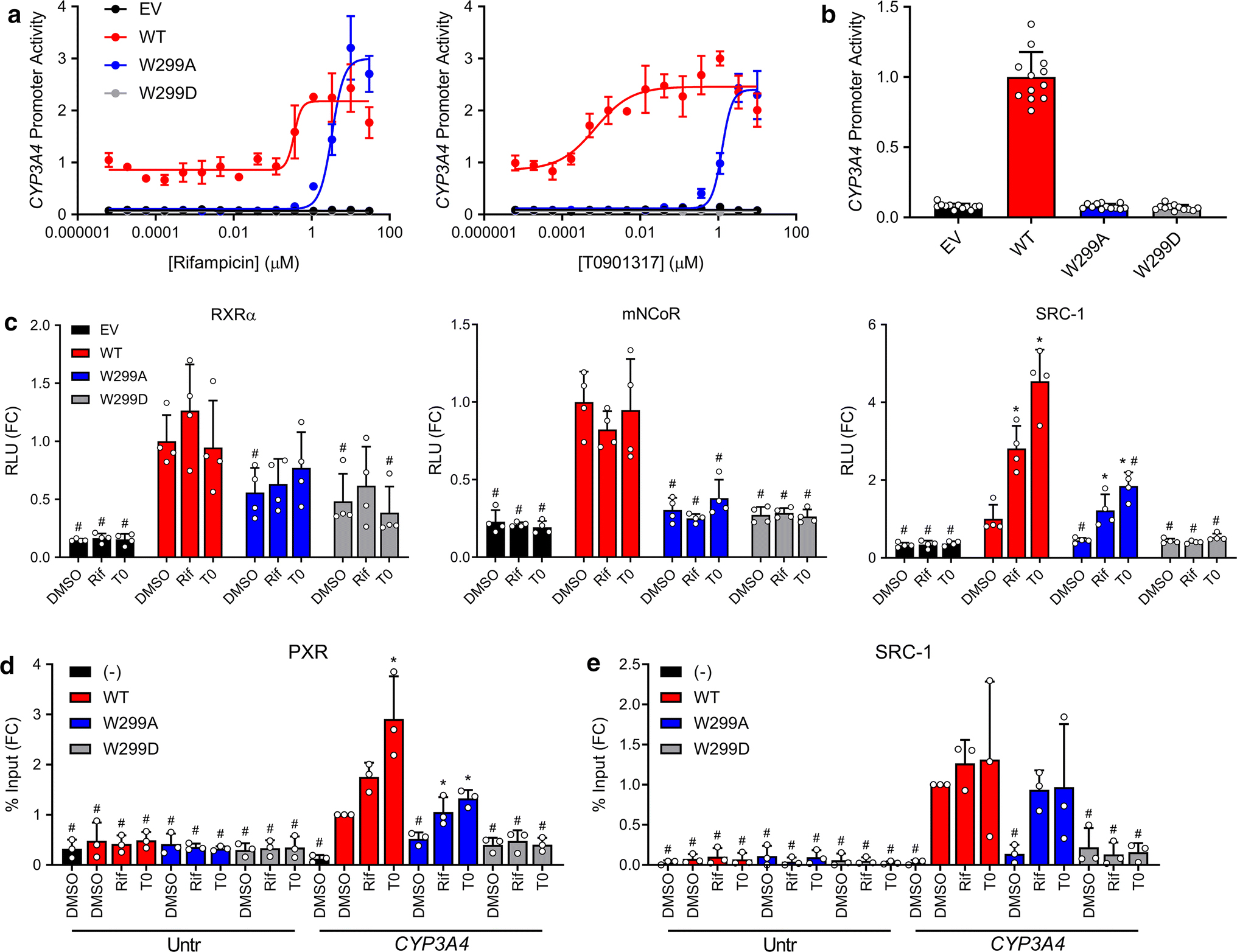
(a) HepG2 cells were co-transfected with empty vector (pcDNA3, EV), WT, W299A, or W299D PXR and a plasmid encoding firefly luciferase under the control of a PXR-responsive CYP3A4 promoter. Cells were treated with Rif or T0 for 24 h and assayed for luciferase activity. (b) The DMSO controls from (a) are plotted. The symbols represent all DMSO wells from 3 independent experiments. (c) Mammalian two-hybrid assays were performed in HepG2 cells. Cells were co-transfected with an empty pACT vector or pACT-PXR LBD plasmid (WT, W299A, or W299D), a coregulator plasmid (pBIND-RXRα, pBIND-mNCoR, or pBIND-SRC-1), and a GAL4 firefly luciferase reporter plasmid. Cells were treated with 10 μM Rif or 5 μM T0 for 24 h and assayed for luciferase activity. The symbols represent measurements from 4 independent experiments. Data in panels a-c are presented as fold change (FC) relative to the WT PXR DMSO control. For c, significance was assessed with one-way ANOVA followed by Dunnett’s test for each sample compared to the DMSO control of its group [p ≤ 0.05 (*)] and for each sample compared to the DMSO control for WT PXR [p ≤ 0.05 (#)]. (d) HepG2 cells were transfected with untagged PXR (−) or FLAG-PXR (WT, W299A, or W299D). Cells were treated with 10 μM Rif or 5 μM T0 for 24 h, and ChIP was performed with anti-FLAG. Primers directed at an untranscribed genomic region (Untr) and the CYP3A4 PXRE were used for qPCR. (e) HepG2 cells were co-transfected with FLAG-PXR plasmid (WT, W299A, or W299D) and either EV (−) or a plasmid expressing SRC-1. Cells were treated with 10 μM Rif or 5 μM T0 for 24 h, and ChIP was performed with anti-SRC-1. Data for d-e are presented as FC relative to the CYP3A4 qPCR from the WT PXR DMSO sample, and symbols represent measurements from 3 independent experiments. Significance was assessed with one-way ANOVA followed by Dunnett’s test for each sample compared to the DMSO control of its group [p ≤ 0.05 (*)] and for each sample compared to the DMSO control for WT PXR CYP3A4 [p ≤ 0.05 (#)].
We further tested whether W299A also reduced PXR activation by ligand cocktails. It was previously reported that the pesticide trans-nonachlor (TNC) and the synthetic estrogen 17α-ethinylestradiol (EE2) synergistically activate PXR by binding concomitantly, with TNC forming nonpolar interactions with the F288-W299-Y306 hot spot [28]. As shown in Supplementary Figure 3, whereas TNC or EE2 alone moderately activated WT PXR, their combination potently activated the receptor. The W299A mutation severely diminished the potency of TNC and EE2, either alone or combined, but full activation was still attained at high compound concentrations, similar to T0 and Rif, and the combination of TNC and EE2 appeared to retain its synergy in W299A activation. Together, our data show that W299 is critical for PXR activation by both single agonists and compound mixtures.
WT and W299A PXR have varying cofactor recruitment profiles
To determine if the loss of basal and agonist-induced activities in W299A was simply due to reduced protein expression or altered subcellular localization, we performed western blot and immunofluorescence of FLAG-PXR-transfected cells. The PXR constructs had relatively equal expression, but treatment with 5 μM T0 increased the level of WT PXR, consistent with previous observations that ligands can stabilize PXR (Supplementary Figure 4) [16]. Neither mutation nor compound treatment altered the subcellular distribution of PXR (Supplementary Figure 5). These data suggest that the activity differences are not due to expression or localization changes.
We next assessed the interactions of PXR LBD (fused to the VP16 activation domain) with the heterodimeric partner RXRα, the corepressor mouse NCoR (mNCoR), and the coactivator SRC-1 (fused to the GAL4 DNA-binding domain) by mammalian two-hybrid assays (Figure 2c). HepG2 cells were co-transfected with vectors as indicated, treated with DMSO, Rif (10 μM), or T0 (5 μM), and assayed for luciferase activity. The concentrations of compounds were chosen to fully activate both WT and W299A based on Figure 2a. The W299A and W299D mutations slightly decreased PXR LBD-RXRα interaction compared to WT, but the signals were still higher than empty vector control (EV) background. Only WT PXR LBD interacted with mNCoR, suggesting that the reduced or abolished activity of the PXR mutants is not due to increased corepressor interactions. Unliganded WT PXR LBD interacted with SRC-1, and interaction was enhanced by Rif and T0, as expected. W299A PXR LBD, however, did not interact with SRC-1 in the absence of agonist, but interaction increased with agonist treatment. These data are consistent with our observation that W299A lacks basal activity but is still inducible by Rif and T0. W299D PXR LBD did not show SRC-1 binding under any condition, consistent with the reporter assays showing that this mutant is completely inactive.
Results from the two-hybrid assays suggested that the altered activity of W299A may be caused by altered interaction with coactivator but not with RXR or corepressor. Therefore, we investigated recruitment of PXR and SRC-1 directly to the endogenous CYP3A4 promoter. We transfected HepG2 cells with FLAG-tagged PXR, treated with DMSO, Rif (10 μM), or T0 (5 μM), and performed chromatin immunoprecipitation (ChIP) with anti-FLAG antibody. Binding was assessed at the endogenous CYP3A4 PXR response element (PXRE) and at an unrelated untranscribed genomic sequence (Untr) as previously described (Figure 2d) [29]. WT PXR bound the CYP3A4 PXRE in the absence of ligand, and binding was increased with the addition of Rif or T0. W299A PXR had reduced CYP3A4 PXRE occupancy in the absence of ligand but showed interaction when Rif or T0 was added. Binding of the Untr negative control region was minimal and unaffected by mutation or ligand.
Next, HepG2 cells were co-transfected with WT or mutant PXR and SRC-1 expression constructs, and ChIP assays were performed with anti-SRC-1 antibody (Figure 2e). With WT PXR, SRC-1 was abundant at the CYP3A4 PXRE without ligand, and occupancy increased slightly, but not significantly, with the addition of agonist; this small increase is likely due to low inducibility of endogenous CYP3A4 by PXR ligands in HepG2. No SRC-1 was present at the PXRE in W299A PXR-transfected cells, but binding increased with Rif and T0. SRC-1 was absent from the promoter in W299D PXR-transfected cells under all conditions. These results are consistent with results from the CYP3A4 reporter, mammalian two-hybrid, and FLAG-PXR ChIP assays, and they show that SRC-1 recruitment to the CYP3A4 promoter is dependent on and correlates with PXR activation.
W299A PXR has altered ligand-binding properties
Due to the apparent importance of the W299 residue in determining the cellular outcome of agonist binding, we wished to test the ligand-binding affinity of WT and mutant PXR. We were unable to obtain pure, soluble W299A protein, so we resorted to cell lysate-based experimentation. GST-tagged WT, W299A, and W299D PXR LBD proteins were expressed by in vitro transcription/translation, and the resulting lysate was subjected to our previously reported time-resolved fluorescence resonance energy transfer (TR-FRET) binding assay that measures binding of a fluorescent PXR ligand (BODIPY FL vindoline) to GST-PXR bound by a terbium-labeled GST antibody [30]. W299A and W299D had reduced TR-FRET signal (Supplementary Figure 6), indicating that the mutants have drastically reduced affinity for the BODIPY FL vindoline probe, and this assay could therefore not be used to assess direct and competitive binding of T0 and Rif to the mutants.
We then turned to a cellular thermal shift assay (CETSA) [31,32] using split Nano Luciferase [33] in 293T cells transfected with PXR LBD expression constructs with N-terminal HiBiT peptides. WT LBD was stabilized by T0, as exhibited by dose-responsive shifts in the melting curves (Supplementary Figure 7a–c). T0 did not stabilize W299A or W299D LBD, indicating that the mutants may have decreased affinity for T0. Importantly, the thermal stability of LBD was unaffected by mutation alone (Supplementary Figure 7d). The CETSA was markedly less sensitive (IC50 for T0 with WT PXR was 1.8 ± 0.4 μM) than our previously reported competitive TR-FRET assay (IC50 was 102 nM) [30], and we were therefore unable to observe T0 stabilization of W299A LBD. It is also possible that T0 binds the mutant proteins but does not induce stabilizing conformational changes. Unfortunately, we were unable to obtain results with Rif because the compound inhibited a reaction component, reducing luminescence equally with all proteins (Supplementary Figure 7e). Nevertheless, the CETSA and TR-FRET results (in the context of technical limitations such as low assay sensitivity) together suggest that the W299A mutation alters PXR ligand-binding properties and/or ligand-induced conformational changes. The observation that at high concentrations both T0 and Rif activated W299A equally to WT (Figure 2a), however, indicates that both ligands can induce the proper active conformation of the mutant receptor, provided that they are used at sufficient concentrations.
Because the W299A mutation altered ligand-binding properties of PXR LBD, we asked whether the lack of W299A basal activity (Figure 2b) is due to loss of binding and activation by an endogenous chemical. We made use of a triple mutant of PXR (S208W, S247W, and C284W, henceforth referred to as 3W) previously reported to be fully active without exogenous ligand due to the mutations mimicking ligand binding [34]. As expected, the 3W was fully constitutively active (Supplementary Figure 8, WT vs. 3W). 3W dramatically increased the basal activity of W299A (W299A vs. 3W W299A), but not that of W299D, further confirming the complete loss of function by a charged residue at position 299, and suggesting that either 1) lack of W299A basal activity is due to lack of binding of a weak endogenous agonist or 2) W299 itself has a role in weakly activating the receptor. Interestingly, T0 enhanced the activity of 3W W299A, but not 3W, suggesting that it adopts a new pose in the W299A mutant that is not occluded by the additional 3W mutations, further supporting the notion that W299A alters PXR’s ligand-binding properties and ligand-induced conformational changes.
SPA70 is an antagonist of WT PXR but an agonist of W299A PXR
After characterizing the role of W299 in modulating agonist activity, we were curious how the W299A mutant would function in the presence of an antagonist such as SPA70. Consistent with our previous characterization of SPA70, SPA70 alone decreased the activity of WT PXR in the CYP3A4 reporter assay, suggesting that this compound may be an inverse agonist for WT PXR (Figure 3a) [16]. Unexpectedly, however, we found that SPA70 was an agonist of W299A (Figure 3a). Because W299C and W299V showed activation profiles similar to that of W299A, and W299F activation was like WT (Supplementary Figure 2), we tested these mutants with SPA70 (Supplementary Figure 9) and found that SPA70 was an inverse agonist of W299F (like WT) and an agonist of W299C and, to a lesser extent, W299V (like W299A). To investigate the switch of SPA70 from antagonist to agonist, we performed mammalian two-hybrid assays with WT or W299A PXR LBD in the presence of 10 μM Rif or SPA70 (Figure 3b). SPA70 slightly increased the interaction of WT PXR LBD with SRC-1, but to a lesser extent than Rif (3.2-fold for Rif vs. 2.0-fold for SPA70 compared to DMSO). Rif and SPA70 both increased SRC-1 interaction with W299A PXR LBD to the same extent (3.0-fold for Rif and 3.1-fold for SPA70 compared to DMSO). Consistent with our previously reported data [16], SPA70 enhanced WT PXR LBD interaction with mNCoR, while Rif did not (1.1-fold for Rif vs. 3.0-fold for SPA70 compared to DMSO). However, the SPA70-induced interaction between W299A PXR LBD and mNCoR was significantly less (1.6-fold for SPA70 compared to DMSO). Together, these data indicate that SPA70 more robustly recruits corepressor to WT PXR, but coactivator to W299A PXR, consistent with the expected behavior of an inverse agonist and an agonist.
Figure 3. SPA70 is an antagonist of PXR WT but an agonist of PXR W299A.
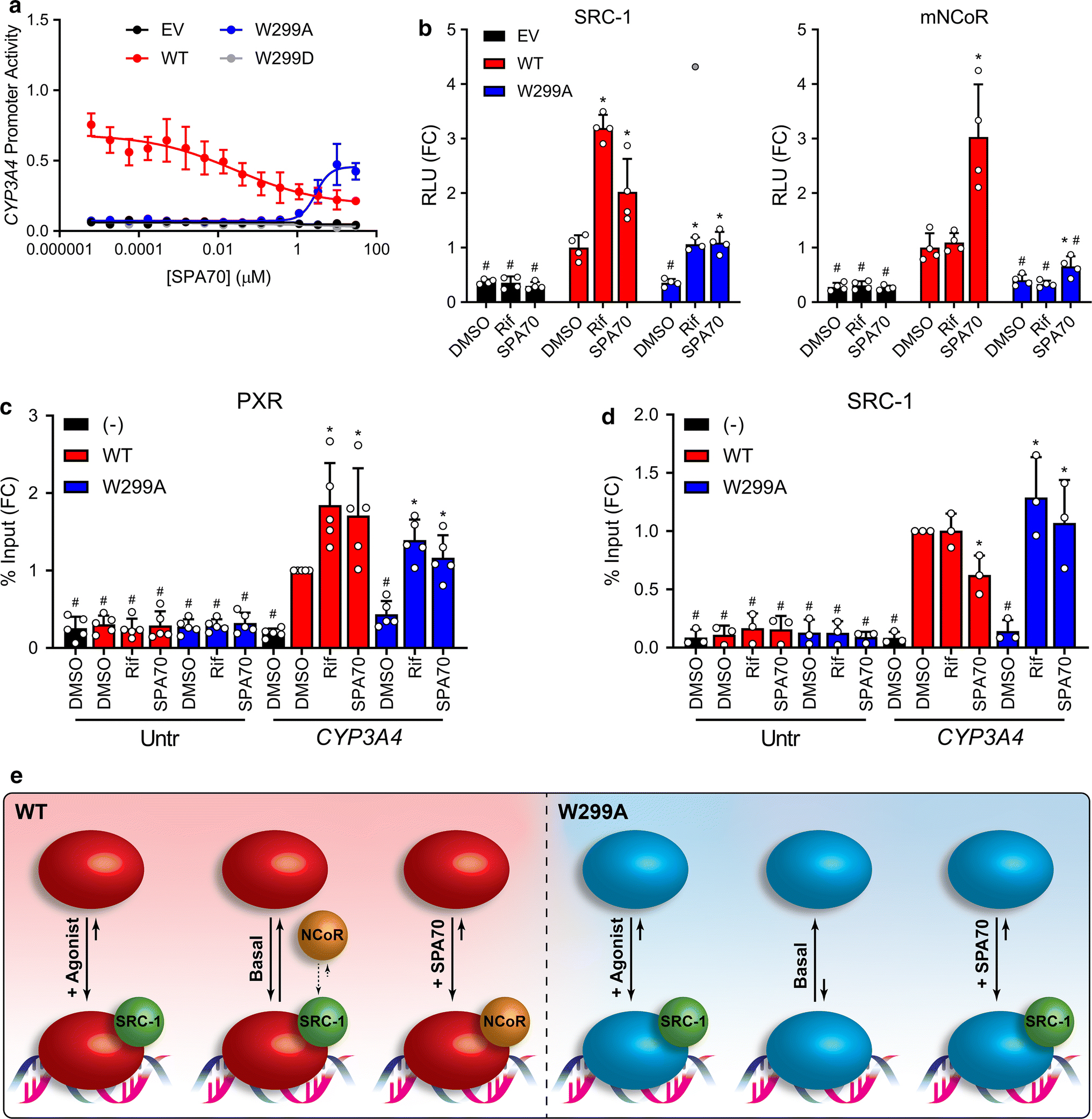
(a) HepG2 cells were co-transfected with empty vector (EV), WT, W299A, or W299D PXR and a plasmid encoding firefly luciferase under the control of a PXR-responsive CYP3A4 promoter. Cells were treated with SPA70 for 24 h and assayed for luciferase activity. (b) Mammalian two-hybrid assays were performed in HepG2 cells. Cells were co-transfected with an empty pACT vector or pACT-PXR LBD plasmid (WT, W299A, or W299D), a coregulator plasmid (pBIND-SRC-1 or pBIND-mNCoR), and a GAL4 firefly luciferase reporter plasmid. Cells were treated with 10 μM Rif or SPA70 for 24 h and assayed for luciferase activity. The symbols represent measurements from 4 independent experiments. The gray point in the W299A + Rif group of the left panel is an excluded outlier. Data in panels a-b are presented as FC relative to the WT PXR DMSO control. For b, significance was assessed with one-way ANOVA followed by Dunnett’s test for each sample compared to the DMSO control of its group [p ≤ 0.05 (*)] and for each sample compared to the DMSO control for WT PXR [p ≤ 0.05 (#)]. (c) HepG2 cells were transfected with untagged PXR (−) or FLAG-PXR (WT or W299A). Cells were treated with 10 μM Rif or SPA70 for 24 h, and ChIP was performed with anti-FLAG. (d) HepG2 cells were co-transfected with FLAG-PXR plasmid (WT or W299A) and either EV (−) or a plasmid expressing SRC-1. Cells were treated with 10 μM Rif or SPA70 for 24 h, and ChIP was performed with anti-SRC-1. Data for c-d are presented as FC relative to the CYP3A4 qPCR from the WT PXR DMSO sample, and symbols represent measurements from at least 3 independent experiments. For panels c-d, significance was assessed with one-way ANOVA followed by Dunnett’s test for each sample compared to the DMSO control of its group [p ≤ 0.05 (*)] and for each sample compared to the DMSO control for WT PXR CYP3A4 [p ≤ 0.05 (#)] (e) The activities of WT and W299A PXR without ligand (basal), with agonist, and with SPA70 are shown graphically. For each condition, the solid arrow lengths and directions depict the qualitative equilibrium of DNA-bound vs. free PXR, and the spheres show cofactor interactions. The small dashed arrows indicate the equilibrium between SRC-1 and NCoR interactions with PXR under the basal condition; the equilibrium is depicted as preferring SRC-1 due to the basal activity present with WT PXR.
Next, we analyzed PXR and SRC-1 recruitment to the endogenous CYP3A4 PXRE by ChIP in HepG2 cells. Surprisingly, both Rif and SPA70 increased WT PXR occupancy at the CYP3A4 PXRE, suggesting that both agonists and antagonists induce PXR binding at responsive promoters (Figure 3c). As in Figure 2d, without ligand, W299A was reduced at the PXRE compared to WT; when Rif or SPA70 was added, W299A PXR at the PXRE increased to near-WT levels. Importantly, SPA70 decreased coactivator SRC-1 recruitment to the CYP3A4 PXRE in the presence of WT PXR, but increased SRC-1 occupancy in the presence of W299A PXR (Figure 3d). Unfortunately, we were unable to obtain ChIP data for NCoR, likely due to the low responsiveness of the CYP3A4 gene to PXR ligands in HepG2 cells. Nevertheless, the PXR and SRC-1 ChIP data together with the two-hybrid results suggest that SPA70 inhibits WT PXR and activates W299A PXR by inducing PXR recruitment to responsive promoters and altering PXR-cofactor interactions (Figure 3e). However, the mechanism responsible for SPA70 being an antagonist for WT PXR but an agonist for W299A PXR is unknown. Interestingly, SPA70 slightly increased PXR LBD interaction with SRC-1 in mammalian two-hybrid, but decreased SRC-1 occupancy at the endogenous CYP3A4 PXRE, suggesting that protein-protein interactions may differ when PXR is free vs. bound to target DNA sequences.
SPA70 antagonist-to-agonist switch correlates to the “outward”-“inward” positioning of AF-2 helix revealed in molecular dynamics (MD) simulations
Having carefully characterized the opposing antagonistic (for WT PXR) and agonistic (for W299A PXR) profiles of SPA70, we wanted to gain insights into ligand-induced structural effects as a result of the W299A mutation and to study the conformational mechanisms by which SPA70 becomes an agonist of W299A PXR. Although several crystal structures for PXR LBD exist, most of these incorporate an SRC-1 peptide for protein solubilization and crystallization; accordingly, no structure yet exists for antagonist-bound PXR. Moreover, though highly informative, crystal structures only provide a snapshot of the total information and lack dynamic detail. Interpreting structural information from proteins as promiscuous and flexible as PXR may be skewed, as crystal structures often force the protein into a limited subset of potential conformations. Since molecular dynamics (MD) simulations have been used to study the binding mechanism of SR12813 to PXR LBD [35], we thought this would be an appropriate approach to gain dynamic structural insights into our systems of interest.
We performed simulations of WT and W299A PXR LBD with no ligand, T0, SPA70, and SJB7 for a total of 8 systems. We first ensured each system had properly equilibrated by measuring the root meat squared deviation (RMSD) and checking that all simulations demonstrated a reasonable plateau and were each under 3 Å (Supplementary Figure 10). Furthermore, we solvated the systems in the same water-based solvent model, ran each system for a total of 200 nanoseconds, and kept all other parameters the same to enable among-simulation comparisons. Because the AF-2 helix has been implicated in the activation mechanism of PXR [19], post-trajectory atomic alignments were performed to capture any relative structural changes in this (or any other) region.
To test and validate the approach, we first analyzed the potent agonist T0 with WT and W299A PXR LBD. No significant secondary structure changes were apparent between the two complexes over the course of the simulation (Figure 4a, Supplementary Figure 11a), but we observed that T0 was able to move deeper into the binding pocket in the W299A mutant to a position that would not be allowed in the WT protein. This observation is consistent with the aforementioned results that T0 was able to activate 3W W299A but not 3W (Supplementary Figure 8). To test whether the lack of the bulky tryptophan residue in W299A was the basis for T0 being able to move deeper into the pocket, we decided to test another ligand. We chose SJB7 because it is an agonist (Supplementary Figure 2) that has high structure similarity to SPA70 (Figure 1) [16]. SJB7 simulations confirmed the observation from T0 simulations: the ligand was able to slide deeper into the binding pocket of W299A to a position that would clash with the W299 residue of WT PXR (Figure 4b). No secondary structure changes between WT and mutant proteins occurred in the SJB7 simulations, including the AF-2 region (Figure 4b, Supplementary Figure 11b). This closely mirrored our results for T0 and prompted us to test if any changes could be observed when using an antagonist as input.
Fig 4. The W299A mutation does not affect agonist-bound PXR conformation.
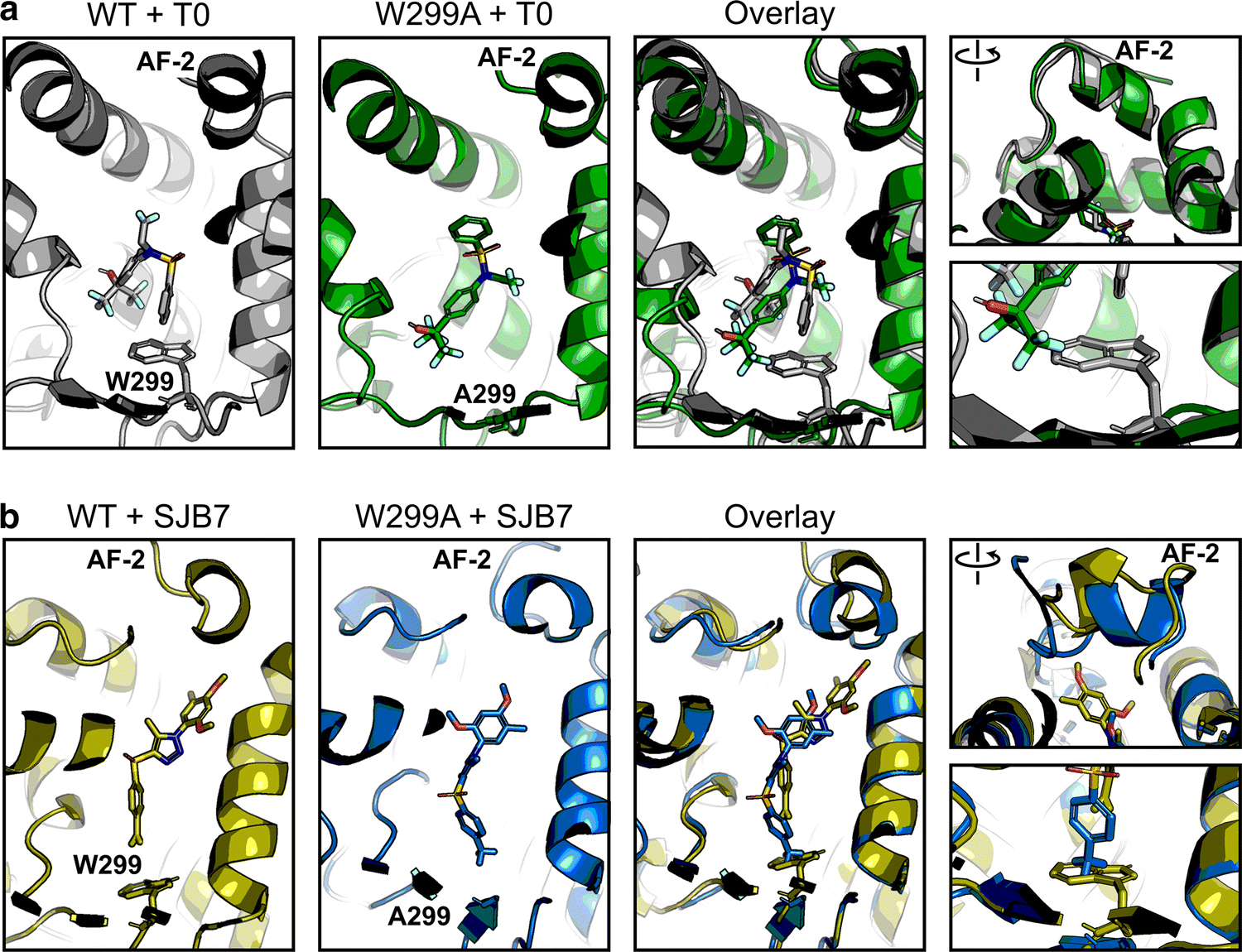
Molecular dynamics simulations were performed for WT and W299A PXR LBD in the presence of (a) T0 or (b) SJB7. Individual and overlaid images are shown for the ligands in the ligand-binding pocket; the right panels focus on the AF-2 helix and W/A299. Each panel is derived from the same frame of its respective simulation. Protein-ligand structures are shown in gray (WT + T0), green (W299A + T0), olive (WT + SJB7), and blue (W299A + SJB7). Ligand heteroatoms are shown in red for oxygen, blue for nitrogen, yellow for sulfur, and teal for fluorine
To obtain insights into how the W299A mutation converts SPA70 to function as an agonist, we simulated WT and W299A proteins with SPA70. As observed with the agonists we tested, W299A permitted SPA70 to slide deeper into the binding pocket, further away from the AF-2 helix (Figure 5a). At the start of the simulation, this was the only difference that could be seen, and, like the agonists, there were no changes in overall protein structure (Supplementary Figure 12a). However, we were surprised that the AF-2 helix quickly adopted a different position in WT PXR LBD relative to W299A. Strikingly, the AF-2 helix in the WT-SPA70 structure was pointed in an outward direction, nearly perpendicular to the W299A-SPA70 AF-2 helix which was facing inward (Figure 5a). We were also surprised that the “outward” vs. “inward” positioning of AF-2 was the only structural difference in SPA70-bound PXR simulations, as all other areas of the protein superimposed clearly (Supplementary Figure 12a). We sought to determine whether differential ligand stability between WT and W299A PXR LBD simulations contributed to the functional switch, since SPA70 moves further away from the AF-2 helix in the W299A mutant. However, the W299A-mediated shift in SPA70’s binding pocket occupancy did not destabilize SPA70 itself, which was an effect also seen in the agonist simulations (Supplementary Figure 13). These data indicate that 1) the mutation of W299 enabled ligands to move deeper into the binding pocket away from the AF-2 helix, 2) the mutation still allowed ligands to bind as stably as the WT simulations, and 3) the AF-2 helix faced outward (“AF-2Out”) in the WT-SPA70 structure but inward (“AF-2In”) in the W299A-SPA70 structure.
Fig 5. Structural mechanism of SPA70 antagonist-to-agonist switch conferred by the W299A mutation.

Molecular dynamics simulations were performed for WT and W299A PXR LBD in the presence of no ligand, T0, or SPA70. (a) Individual and superimposed images of the ligand-binding pocket are shown for SPA70-bound WT (teal) and W299A (raspberry) PXR LBD; the right panels focus on the AF-2 helix and W/A299. The dashed lines indicate the orientation of each AF-2 helix; arrows point from the N- to the C-terminus of the helix. The AF-2 is labeled “In” or “Out” in reference to the proximity of the C-terminus to the ligand-binding pocket. (b) Overlaid images of T0-bound WT (gray) and SPA70-bound WT (teal) PXR LBD. (c) Overlaid images of T0-bound WT (gray) and SPA70-bound W299A (raspberry) PXR LBD. (d) WT or W299A simulations without ligand, with T0, and with SPA70 are overlaid. Left panel: The AF-2 is in the “In” position for all simulations except SPA70-bound WT. Right panel: The AF-2 cannot adopt the “In” position in WT PXR LBD when SPA70 is present because the helix would clash with the ligand; therefore, the helix in held in the “Out” orientation. (e) Quantification of the “In” vs. “Out” AF-2 orientations between simulations. The distance from the AF-2 residue F429 to the Helix 3 residue F251 was measured for the duration of each simulation (using α-carbon atoms for measurements)
Upon observing the differences in AF-2 positioning between SPA70-bound WT and W299A PXR LBD, we postulated that this positioning could be a structural mechanism for the antagonist-to-agonist switch between the WT and W299A proteins. Therefore, we directly compared the most potent agonist (T0) and antagonist (SPA70) of WT PXR to see if any differences in the AF-2 helix could be discerned in the context of WT PXR LBD alone. Remarkably, while SPA70 induced WT PXR LBD to adopt the AF-2Out conformation as stated above, T0 induced the AF-2In mode (Figure 5b). The consistent observations in Figure 5a–b suggested that the AF-2Out position could be indicative of PXR in an antagonistic conformation while AF-2In might confer agonistic mode. When we compared W299A-SPA70 to WT-T0, we observed the AF-2In position for both complexes (Figure 5c), consistent with our cell-based activity data showing that both of these conditions are agonistic (Figures 2–3). By superimposing the structures of WT and W299A PXR LBD with no ligand, T0, or SPA70 (6 simulations total), we clearly demonstrated that the only system known to be in functionally antagonistic mode (SPA70-bound WT PXR LBD) was also the only simulation resulting in the AF-2Out conformation (Figure 5d, left panel). After closely examining and overlaying each ligand from these simulations, it was apparent that SPA70 prevents the AF-2In conformation only in WT PXR LBD, as a steric clash would occur (Figure 5d, right panel). Conversely, all other systems (including SPA70-bound W299APXR LBD) held the ligands far enough away from the AF-2 that it could adopt the “In” conformation.
While the AF-2In vs. AF-2Out difference was clear visually, we sought to quantify the AF-2 “In-ness” vs. “Out-ness”. To do so, we chose the same C-terminal residue of the AF-2 (F429) and measured its distance to the helix 3 (H3) residue F251. H3 was selected because its structure was very well conserved across all simulations (Supplementary Figure 14a–b), so it could provide a reference point from which AF-2In vs. AF-2Out could be easily distinguished. We observed a dramatiĉ5 Å difference between the “In” vs. “Out” positions of the AF-2 (Figure 5e). The AF-2Out conformation only resulted from SPA70-bound WT PXR and was maintained for the course of the simulation (Figure 5e). Furthermore, the median and distribution values for each collective simulation reinforced this striking difference in AF-2 positioning (Supplementary Figure 14c). When the AF-2 regions of WT-SPA70, WT-T0, and WT-SJB7 simulations were superimposed, although secondary structure of the helix was partially lost in SJB7 simulations (Figure 4), it was clear that SJB7 induced the AF-2In conformation (Supplementary Figure 15). Changes in specific residues involved in ligand-protein interactions were observed between the systems, but no clear pattern could be discerned to explain the antagonist-to-agonist switch of SPA70 (Supplementary Figure 16), suggesting that there may be multiple interaction modes that can lead to PXR modulation.
Comparing the combinations of PXR LBD (WT and W299A) with chemical modulators (agonists and antagonists), our collective results indicate that the AF-2 adopts distinct positions for agonists vs. antagonists. Interestingly, the AF-2 conformation in response to ligands could be manipulated by a mutation (W299A) far across the ligand-binding pocket, but the mutation itself did not appear to affect protein conformation (Supplementary Figure 12b). Mutating tryptophan 299 to alanine allowed for all ligands to bind further from the AF-2, and in the case of SPA70, the ligand became far enough away to permit the AF-2In conformation. In WT PXR LBD, SPA70 was stabilized and held up by the W299 residue in such a way that it prevented the AF-2 helix from folding in, forcing it to remain in the AF-2out conformation.
MD simulations predict an antagonist-to-agonist switch
In our previous work, we reported 3 compounds in addition to SPA70 that exhibited antagonistic activity toward PXR [16]. Of these 3 compounds, SJC2 was the most potent. To test whether the AF-2Out to AF-2In switch was specific to SPA70, we simulated WT and W299A PXR with SJC2. Interestingly, we found that, like with SPA70, SJC2-bound WT AF-2 adopted the “Out” position and SJC2-bound W299A AF-2 adopted the “In” position (Figure 6a). When overlaid with SPA70 simulations, the positionings of the AF-2 were very similar between the respective simulations (Figure 6b), and quantification of the AF-2 to H3 distances supported these observations, although the WT-SPA70 AF-2 was slightly further “Out” than the WT-SJC2 AF-2 (Figure 6c). These data suggested that the AF-2Out conformation is unique to antagonists and predicted that the W299A mutation converts SJC2 to an agonist, like SPA70.
Fig 6. MD predicts an antagonist-to-agonist switch.
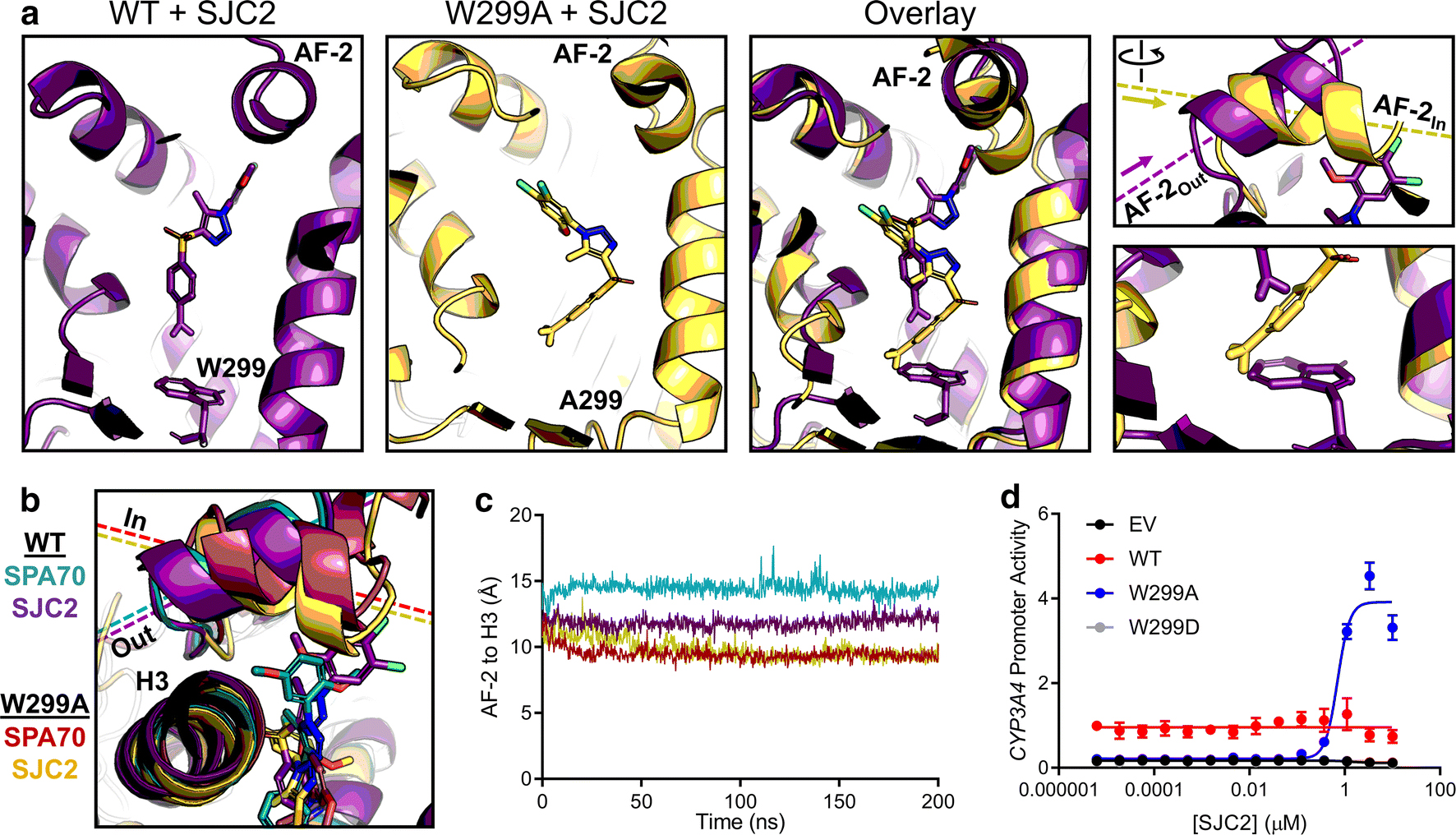
(a) Molecular dynamics simulations were performed for WT and W299A PXR LBD in the presence of SJC2. Individual and superimposed images of the ligand-binding pocket are shown for SJC2-bound WT (violet) and W299A (yellow) PXR LBD; the right panels focus on the AF-2 helix and W/A299. (b) WT or W299A simulations with SJC2 and SPA70 are overlaid. The AF-2 is in the “Out” position for WT simulations and in the “In” position for W299A simulations. (c) Quantification of the “In” vs. “Out” AF-2 orientations between simulations. The distance from the AF-2 residue F429 to the Helix 3 residue F251 was measured for the duration of each simulation (using α-carbon atoms for measurements). (d) HepG2 cells were co-transfected with empty vector (EV), WT, W299A, or W299D PXR and a plasmid encoding firefly luciferase under the control of a PXR-responsive CYP3A4 promoter. Cells were treated with SJC2 for 24 h and assayed for luciferase activity
To determine whether the MD simulations had accurately predicted an antagonist-toagonist switch of SJC2, we tested SJC2 in the CYP3A4 promoter reporter assays with WT and W299A PXR. First, we combined SJC2 with 10 μM Rif and found that SJC2 antagonizes Rif activation of WT PXR, as expected (Supplementary Figure 17). SJC2 alone had no effect on the basal activity of WT PXR but was indeed an agonist of W299A (Figure 6d), demonstrating that our computational approach successfully predicted the functional switch of an antagonist to an agonist. The antagonistic effect of SJC2 on Rif-mediated WT PXR activation and the lack of any effect of SJC2 alone suggested that SJC2 acts by a mechanism distinct from that of SPA70. In mammalian two-hybrid assay, SPA70, but not SJC2, induced corepressor interaction with WT PXR LBD (Supplementary Figure 18). These results suggest that SJC2 may be a neutral antagonist of WT PXR, unlike SPA70, which has characteristics of an inverse agonist.
DISCUSSION
Previous studies have reported the dependency of PXR agonist activity on W299 [27,26], but we wished to characterize the role of W299 more extensively and expand the investigation into multi-ligand and antagonistic systems. In our initial screen, we observed that W299 mutations can cause loss of basal activity or complete loss of function (Supplementary Figure 2). Substitutions with large hydrophobic residues (L or F) retained WT-like activity, but mutants with small, polar, or charged residues (D, G, or S) lacked activity. The A, C, and V substitutions had an intermediate phenotype, lacking basal activity but retaining inducibility. These findings suggest that size and hydrophobicity are integral dictators of ligand interaction at this hot spot and of general ligand binding. As suspected, W299A and W299D exhibited reduced fluorescent probe binding and ligand-mediated PXR stabilization (Supplementary Figures 6 and 7), and it is possible that these deficiencies reflect decreased ligand-binding affinity of the mutants. The decreased ligand affinity could contribute to loss of PXR basal activity in the W299A mutant by decreasing endogenous chemical binding, and our observation that the basal activity of W299A can be rescued by mutations in the ligand-binding pocket that mimic ligand binding supports this notion (Supplementary Figure 8). On the other hand, at high concentrations, both T0 and Rif activated W299A equally to WT (Figure 2a), indicating adequate binding of T0 and Rif in cells, and support the idea that W299A alters ligand-induced conformational changes. As expected, our observations of PXR activity differences can be attributed to differential cofactor interactions and PXR recruitment to the CYP3A4 promoter (Figure 2). Although WT, W299A, and W299D proteins were equally expressed and had similar subcellular localization (Supplementary Figures 4 and 5), we cannot rule out the potential for misfolding of the mutants. For example, the introduction of a charged residue into the hydrophobic ligand-binding pocket in W299D could destabilize the LBD, leading to the observed lack of activity. W299 itself may play a role in protein stability, and mutation to smaller residues could disrupt protein structure. Because W299A lacked basal activity and was unable to interact with mNCoR or SRC-1 in the absence of ligand (Figures 2 and 3), it is possible that this mutant is conformationally distinct from the WT protein. Another possibility is that due to the altered ligand binding properties of W299A, the mutant protein loses binding of endogenous chemicals that contribute to basal activity and protein-protein interaction equilibria. However, if protein structure is disrupted by W299A mutation, a WT-like conformation is rescued by exogenous ligands, as shown by the ability of Rif and T0, when used at sufficient concentrations, to fully activate the mutant and induce coactivator binding.
Surprisingly, we found that the WT PXR antagonist SPA70 is an agonist of W299A, W299C, and, to a lesser extent, W299V PXR (Figure 3 and Supplementary Figure 9), and, based on our cell-based and MD data, we have proposed a model to explain both the antagonistic activity of SPA70 for WT PXR and the antagonist-to-agonist switch conferred by the W299A mutation (Figure 7). As we previously discussed [16], the reduction of WT PXR basal activity by SPA70 in the cell-based reporter assay (Figure 3a) and the ability of SPA70 to enhance the interaction of WT PXR LBD with the corepressor mNCoR (Figure 3b) suggest that SPA70 functions as an inverse agonist of WT PXR. With W299A PXR LBD, however, SPA70 induced interaction with the coactivator SRC-1 and not mNCoR. Our ChIP results showed that SPA70 enhanced both WT and W299A PXR recruitment to the CYP3A4 promoter (Figure 3c); however, SPA70 reduced SRC-1 recruitment to the promoter in cells with WT PXR but increased SRC-1 recruitment to the promoter in cells with W299A PXR (Figure 3d). While we were not able to obtain corepressor ChIP results, we can surmise from our data that SPA70 functions by increasing the presence of an inhibitory WT PXR-corepressor complex at responsive promoters.
Fig 7. Model of mutational and chemical PXR modulation.
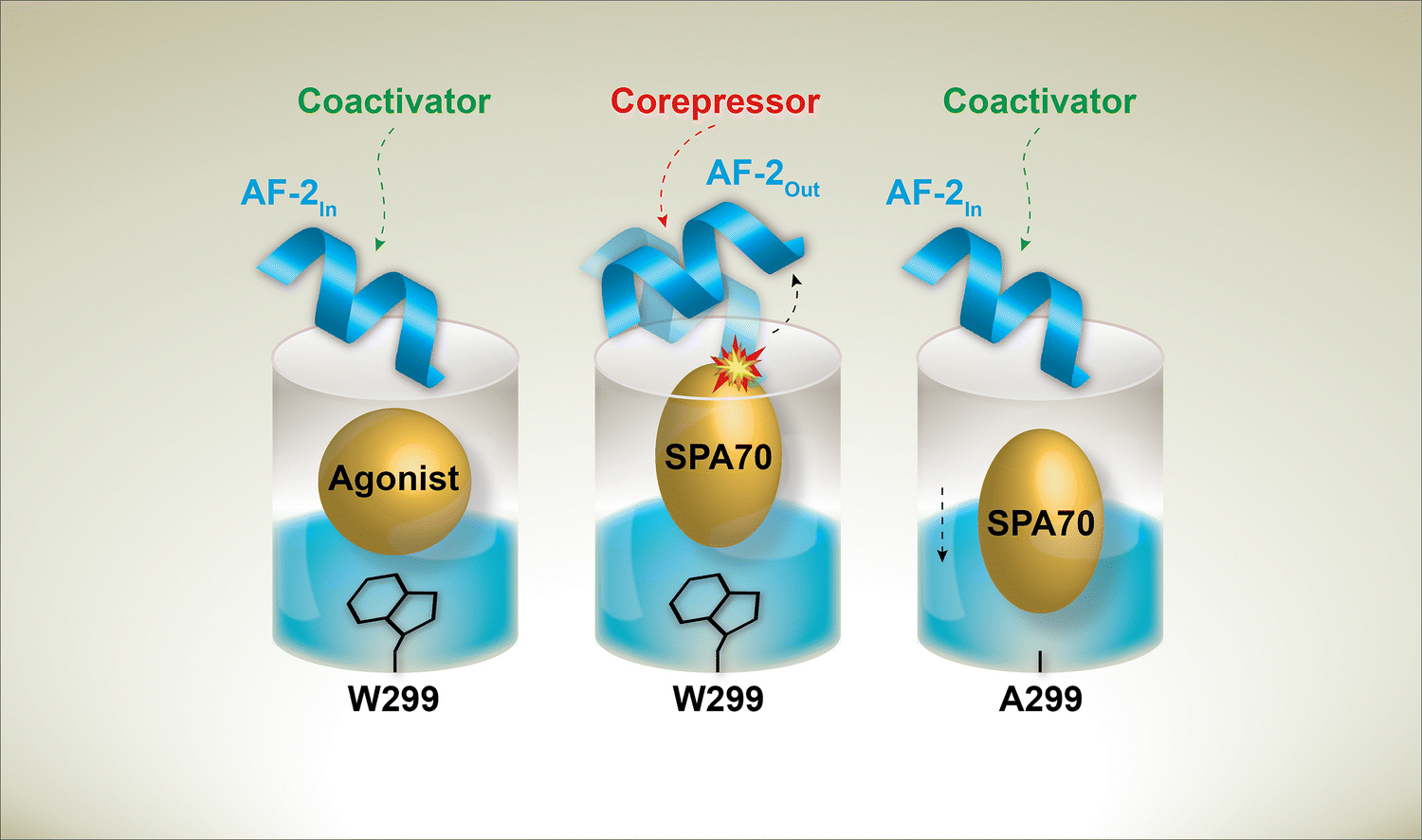
Agonists stabilize the “In” AF-2 position, allowing coactivator to bind. Antagonists, like SPA70, stabilize an “Out” AF-2 position that favors corepressor binding. The W299A mutation allows SPA70 to bind away from the AF-2 and stabilize the “In” AF-2 conformation
Our MD simulations revealed conformational dynamics that were remarkably consistent with our cell-based results. Simulations of WT PXR LBD with T0 and W299A PXR LBD with T0 or SPA70 all displayed similar AF-2 positioning (AF-2In), consistent with our observation that these are all active complexes (agonistic mode); therefore, we can likely refer to this AF-2 conformation as the “active” conformation. The only inhibitory complex, WT PXR LBD in the presence of SPA70, had an AF-2 nearly perpendicular to the positions of the AF-2 in the other simulations, and this complex was the only one to adopt this configuration (AF-2 Out). The AF-2 in the W299A PXR LBD with SPA70 was able to move to the active position due to SPA70 shifting away from the AF-2 toward the F288-W299-Y306 hot spot. Our findings mirror structural insights from related nuclear receptors in that agonists stabilize the AF-2 in a position suitable for coactivator binding; as a likely inverse agonist of PXR, SPA70 not only clashes with the active AF-2 position but may stabilize the AF-2 in a conformation that promotes corepressor binding.
Interestingly, we were able to computationally predict the antagonist-to-agonist switch of a SPA70 analog, SJC2 (Figure 6). These results suggest that our data are not specific to a single compound, and we can propose that the mechanism of PXR antagonism directly correlates with the AF-2 positioning induced by the antagonist. Quantification of “In-ness” vs. “Out-ness” showed that the AF-2 of WT PXR LBD bound by SPA70 was more “Out” than of WT PXR LBD bound by SJC2. It is possible that the degree of “Out-ness” can reflect compound potency, as SPA70 is more potent than SJC2 and is able to decrease basal activity of WT PXR. However, due to the availability of only a small number of PXR antagonists and the large computational requirements for MD, we were unable to test this hypothesis at a larger scale. Unfortunately, it is not possible to simulate the complex cellular environment surrounding PXR, so MD gives only information about PXR alone without additional cellular factors like protein partners and endogenous ligands. It is possible that W299 mutants could behave differently in various cellular systems or when faced with diverse DNA response elements. Our PXR transactivation assays were performed with a reporter construct containing the CYP3A4 promoter and PXRE with ER6 and DR3, respectively. Protein-protein interaction assays showed that W299A had reduced interaction with RXRα, SRC-1, and mNCoR, and the loss of these protein interactors could potentially reflect gain of new interactors that alter DNA-binding specificity.
Our previous characterization of SPA70 and related analogs suggested that subtle chemical modifications of SPA70 (i.e., compounds such as SJB7) can dramatically change the cellular activity of the compounds and that the cellular outcome of ligand binding is likely determined by conformational changes induced by specific ligands [16]. Our current work shows that the inverse is true as well: modifications of PXR structure (e.g., by mutating W299) can dramatically change the cellular activity of compounds. We have taken advantage of these two unexpected observations on PXR-ligand structure activity relationship (i.e., the cellular activity of a ligand can be reversed by either subtle chemical modifications of the ligand, or modifications of the receptor), carefully characterized the cellular activities, and, together with MD simulations, built a model with validated predictive power. Although naturally occurring W299 mutations have not been reported (Supplementary Table 1), mutations at this position may be a powerful tool to study PXR regulation and chemical modulation. This suggestion is illustrated by our observation that a single mutation regulates PXR modulation by SPA70, and we have used this fortuitous finding to study the cellular and structural mechanisms by which SPA70 acts on PXR. We have demonstrated the use of this system in cellular and computational methods, and the approach will be invaluable to future cellular, computational, biochemical, and structural studies of PXR regulation by small molecules. Based on our data, we postulate not only that W299 is a critical residue that dictates ligand binding, but also that this amino acid is a master switch that dictates the cellular activity of ligand binding.
MATERIALS AND METHODS
Sequence alignments
The UniProt IDs for the aligned PXR sequences were rhesus macaque (Q8SQ01), dog (F1Q075), human (O75469), zebrafish (B0V1H8), horse (F6Y2P7), rat (Q9R1A7), cat (M3W595), chimpanzee (A0A2I3S3V5), mouse (O54915), cow (A2VDU4), and pig (A5J0K7). The UniProt ID for human CAR was Q14994. Sequences were aligned in Jalview using the MAFFT module [36], and aligned sequences were annotated for sequence conservation and secondary structure with ESPript 3 [37]. Secondary structure information was extracted from our previously reported structure of the PXR LBD with the agonist SJB7 (PDB ID: 5X0R) [16].
Cell culture
HepG2/C3A and 293T cells were obtained from the American Type Culture Collection (ATCC) and authenticated by short tandem repeat (STR) DNA profiling. HepG2/C3A were maintained in Eagle’s Minimum Essential Medium (ATCC) with 10% FBS (HyClone), and 293T were cultured in Dulbecco’s Modified Eagle Medium (DMEM, Thermo Fisher Scientific) with 10% FBS. Cells were incubated in a humidified atmosphere at 37°C with 5% CO2 and routinely verified to be mycoplasma free by using the MycoProbe Mycoplasma Detection Kit (R&D Systems). Cell counts were obtained with a Countess II Automated Cell Counter using trypan blue staining.
Compounds and oligonucleotides
DMSO, rifampicin, T0901317, SR12813, 17α-ethinylestradiol (EE2), and trans-nonachlor were purchased from Sigma-Aldrich. SPA70, SJB7, and SJC2 were obtained from WuXi AppTec [16]. All oligonucleotides were from Integrated DNA Technologies and are presented in Supplementary Table 2.
Plasmids
Construction of the pcDNA3.1-FLAG-PXR expression plasmid was described previously [38] using pcDNA3.1 from Thermo Fisher Scientific. Mutants of this plasmid were generated by Codex BioSolutions, Inc [39]. The pcDNA3.1-PXR vector containing untagged PXR has been described [38]. Construction of pGL3-CYP3A4-luc containing firefly luciferase under the control of a PXR-responsive CYP3A4 promoter has also been described [40,38]. The CheckMate mammalian two-hybrid system (Promega) contained the pACT, pBIND, and pG5luc vectors. Use of pACT containing full-length PXR (pACT-PXR) and pBIND containing steroid receptor coactivator-1 (amino acids 621–765, pBIND-SRC-1), mouse nuclear receptor corepressor (amino acids 1958–2401, pBIND-mNCoR), or retinoid X receptor alpha (RXRα, pBIND-RXRα) has been described [41,42,39,26,16,43]. For this study, pACT-PXR LBD containing residues 139–434 of PXR LBD was used in mammalian two-hybrid experiments and was generated with the Q5 Site-Directed Mutagenesis Kit (New England Biolabs) using primers P1 and P2 and the previously described pACT-PXR as template. The pACT-PXR LBD W299A mutant was constructed with the Q5 Site-Directed Mutagenesis Kit using primers P3 and P4. The pACT-PXR LBD W299D mutant was constructed with the QuikChange II XL Site-Directed Mutagenesis Kit (Agilent Technologies) using primers P5 and P6. PCR3.1hSRC-1A encoding full-length SRC-1 was a gift from Dr. Bert W. O’Malley (Baylor College of Medicine) [44]. The bacterial GST-PXR-LBD expression vector pGEX-6P1-hPXR-LBD was a gift from Sridhar Mani (Addgene plasmid # 67769) [45]. This vector was not used directly in this study but was used as template for further engineering; the Q5 Site-Directed Mutagenesis Kit and primers P7 and P8 were used to delete the 3C cleavage site and 32 N-terminal residues from the PXR LBD, generating pGEX-6P1-hPXR-LBD-2. The W299A and W299D mutations were made in pGEX-6P1-hPXR-LBD-2 as above, and, after failing to obtain soluble protein from bacterial expression, these vectors were used as templates to generate pcDNA3.1-FLAG-GST-PXR LBD (WT, W299A, and W299D) by the following method: pcDNA3.1-FLAG-PXR (WT, W299A, and W299D) was amplified with primers P9 and P10, and GST-PXR LBD was amplified from pGEX-6P1-hPXR-LBD-2 with primers P11 and P12, and the resulting fragments were assembled with NEBuilder HiFi DNA Assembly Master Mix (New England Biolabs). The pcDNA3.1-FLAG-GST control vector was generated with the Q5 Site-Directed Mutagenesis Kit using primers P13 and P14 with pcDNA3.1-FLAG-GST-PXR LBD as template. The pcDNA3.1-HiBiT-PXR LBD vectors were constructed with the Q5 Site-Directed Mutagenesis Kit using primers P15 and P16 with pcDNA3.1-FLAG-PXR (WT, W299A, and W299D) as template; this strategy replaced the FLAG tag in the original vectors with the 11 amino acid HiBiT peptide flanked by Gly-Ser as described previously [33]. To make pcDNA3.1-FLAG-PXR 3W vectors containing the S208W, S247W, and C284W mutations, pcDNA3.1-FLAG-PXR (WT, W299A, or W299D) was amplified with P17 and P18 and assembled with a gBlock containing the 3 mutations (Integrated DNA Technologies, P19) using NEBuilder HiFi DNA Assembly Master Mix.
Antibodies
Antibodies used in this study were mouse anti-FLAG M2 (Sigma-Aldrich, cat. # F3165), rabbit anti-β-actin (Cell Signaling Technology, cat. # 4967S), rabbit anti-SRC-1 (Abcam, cat. # ab2859), goat anti-mouse IRDye 800CW (LI-COR, cat. # 926–32210), goat anti-rabbit IRDye 680LT (LI-COR, cat. # 926–68021), and goat anti-mouse Alexa Fluor 488 (Thermo Fisher Scientific, cat. # A-11001). Antibodies were used at assay-specific concentrations noted in the relevant method subsections.
PXR transactivation assays
Assays were performed similarly as previously described [16,43]. HepG2 cells (600,000/well) were plated in 6-well tissue culture-treated plates. The following day, cells were co-transfected with pGL3-CYP3A4-luc (2 μg/well) and 100 ng/well of either empty vector (pcDNA3.1) or pcDNA3.1-FLAG-PXR (WT or W299 mutant) using Lipofectamine 3000 (Thermo Fisher Scientific). Twenty-four hours after transfection, cells were trypsinized and suspended in phenol red–free DMEM (Thermo Fisher Scientific) supplemented with 5% charcoal/dextran-treated FBS (HyClone), and 15,000 cells/well were added to white 384-well plates. Compounds were added at indicated concentrations, and the final DMSO concentration was 0.3% (0.6% for the TNC + EE2 combination experiments and the SJC2 antagonist assay). After 24 h, a luciferase assay was performed using the steadylite plus Reporter Gene Assay System and EnVision microplate reader (PerkinElmer).
Western blot analysis
HepG2 cells (600,000/well) were plated in 6-well tissue culture-treated plates. The following day, cells were transfected with indicated plasmids (2 μg/well) using Lipofectamine 3000. Twenty-four hours after transfection, compounds were added at the indicated concentrations in phenol red-free DMEM supplemented with 5% charcoal/dextran-treated FBS. The final DMSO concentration was 0.25%. After an additional 24 h, cells were trypsinized, pelleted by centrifugation, washed with PBS, and lysed in radioimmunoprecipitation assay (RIPA) buffer [50 mM Tris (pH 8.0), 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS]. Protein in the lysate was quantified with the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific), and 25 μg was loaded with NuPAGE LDS Sample Buffer (Thermo Fisher Scientific) into NuPAGE 4–12% Bis-Tris gels (Thermo Fisher Scientific). Separated proteins were transferred to nitrocellulose membranes using the iBlot 2 Dry Blotting System (Thermo Fisher Scientific). Membranes were blocked with TBST [50 mM Tris (pH 7.4), 150 mM NaCl, 0.1% Tween 20] containing 5% milk for 1 h at room temperature (RT). Mouse anti-FLAG M2 (1:2,500 dilution) and rabbit anti-β-actin (1:2,000 dilution) were bound overnight at 4°C in TBST containing 5% milk. Membranes were washed with TBST 3 times for 10 min each, and anti-mouse or anti-rabbit IRDye antibodies (1:10,000 dilution) were added in TBST containing 5% milk for 1 h at RT. Membranes were washed as above, imaged with an Odyssey CLx imaging system (LI-COR), and quantified with Image Studio software (LI-COR).
Immunofluorescence
PerkinElmer ViewPlate-96 Black plates were coated with 2% collagen for 10 min at RT (100% collagen I rat protein was obtained from Thermo Fisher Scientific and diluted to 2% in UltraPure water). Wells were washed with PBS, and 10,000 HepG2 cells were plated per well. The following day, cells were transfected with indicated plasmids (100 ng/well) using Lipofectamine 3000. Twenty-four hours after transfection, compounds were added at the indicated concentrations in phenol red–free DMEM supplemented with 5% charcoal/dextran-treated FBS. The final DMSO concentration was 0.25%. After an additional 24 h, cells were washed with PBS, crosslinked with 4% formaldehyde in PBS at RT for 20 min, washed 3x for 5 min with PBS, permeabilized with 0.1% Triton X-100 in PBS for 10 min, washed 3x for 5 min with PBS, blocked with 5% BSA in PBST (PBS, 0.1% Tween 20) for 1 h, rinsed 1x with PBS, stained with mouse anti-FLAG M2 (1:500 dilution) in PBST with 1% BSA at RT for 1 h, washed 3x for 5 min with PBST, stained with goat anti-mouse Alexa Fluor 488 (1:500 dilution) in PBST with 1% BSA at RT for 1 h, stained with 1 μg/mL DAPI in PBS for 15 min, washed 3x for 5 min with PBST, and stored in PBS. Images were taken with an IN CELL Analyzer 6000 (GE Healthcare) using a 20× objective. CellProfiler was used for quantification [46–48].
Mammalian two-hybrid assays
The CheckMate mammalian two-hybrid system (Promega) was used to assess PXR-cofactor interactions similarly to previously described [41]. HepG2 cells (600,000/well) were plated in 6-well tissue culture-treated plates. The following day, cells were transfected with pG5luc (2 μg/well), indicated pBIND vectors (100 ng/well) and indicated pACT vectors (100 ng/well) using Lipofectamine 3000. Twenty-four hours after transfection, cells were trypsinized and suspended in phenol red–free DMEM (Thermo Fisher Scientific) supplemented with 5% charcoal/dextran-treated FBS (HyClone), and 15,000 cells/well were added to white 384-well plates. Compounds were added at indicated concentrations, and the final DMSO concentration was 0.5%. After an additional 24 h, a luciferase assay was performed using the steadylite plus Reporter Gene Assay System and EnVision microplate reader.
Chromatin immunoprecipitation assays
ChIPs were performed according to Abcam’s protocol, with minor modifications. HepG2 cells (600,000/well) were plated in 6-well tissue culture-treated plates. The following day, cells were transfected with indicated plasmids (2 μg/well for single transfections or 3 μg/well total for co-transfections at 1:1 ratios) using Lipofectamine 3000. Twenty-four hours after transfection, compounds were added at the indicated concentrations in phenol red–free DMEM supplemented with 5% charcoal/dextran-treated FBS. The final DMSO concentration was 0.25%. After an additional 24 h, cells were crosslinked by addition of formaldehyde directly to the media to 1% final concentration and incubation at RT for 10 min. The reaction was quenched by addition of glycine to 125 mM and incubation at RT for 5 min. The liquid was aspirated, and PBS was added to the wells. Cells were scraped, transferred to 15 mL tubes, and centrifuged at 500 × g for 5 min. Two wells were combined for each ChIP sample. The supernatant was removed, and pellets were stored at −80°C. Cell pellets were thawed on ice and lysed with 1 mL of 50 mM HEPES (pH 7.9), 140 mM NaCl, 1 mM EDTA, 0.25% Triton X-100, 0.5% NP-40, and 10% glycerol on ice for 10 min. The lysate was centrifuged at 500 × g for 5 min to pellet nuclei. Nuclei were washed twice with 10 mM Tris (pH 8.0), 200 mM NaCl, 1 mM EDTA, and 0.5 mM EGTA and twice with shearing buffer [10 mM Tris (pH 8.0), 1 mM EDTA, 0.1% SDS]. The pellet was resuspended for each wash followed by centrifugation at 500 × g for 5 min. The final pellets were resuspended in 1 mL shearing buffer and transferred to 1 mL milliTUBEs with AFA fibers (Covaris). Chromatin was sheared in a Covaris E220 Focused-ultrasonicator for 8 min using the manufacturer’s recommended standard settings to yield an average fragment size of 300 bp. Sheared chromatin was centrifuged at 16,000 × g for 10 min to pellet insoluble material. 50 μL of supernatant was saved as input. 800 μL of supernatant was added to 200 μL of 21 mM Tris (pH 8.0), 167 mM NaCl, 1 mM EDTA, 1.1% Triton X-100, and 0.1% SDS with 20 μL of Dynabeads Protein G (Thermo Fisher Scientific) and 2 μg of appropriate antibody and incubated overnight at 4°C with rotation. Beads were washed for 3 min each at 4°C with rotation with low salt wash [20 mM Tris (pH 8.0), 150 mM NaCl, 2 mM EDTA, 1% Triton X-100, 0.1% SDS], high salt wash [20 mM Tris (pH 8.0), 500 mM NaCl, 2 mM EDTA, 1% Triton X-100, 0.1% SDS], lithium chloride wash [10 mM Tris (pH 8.0), 250 mM LiCl, 1 mM EDTA, 1% NP-40, 1% sodium deoxycholate], and twice with TE [10 mM Tris (pH 8.0), 1 mM EDTA]. Bound complexes were eluted twice with 125 μL of 100 mM sodium bicarbonate, 1% SDS for 15 min at 37°C. 1 U of proteinase K (New England Biolabs, cat. # P8107S) was added to the inputs and eluates with 40 mM Tris (pH 8.0), 200 mM NaCl, and 10 mM EDTA, and samples were incubated at 65°C for 2 h. DNA was purified with the QIAquick PCR Purification Kit (QIAGEN). After purification, DNA (2 μL in a 10 μL reaction volume) was subjected to amplification by denaturation at 95°C for 2 min, followed by 40 cycles of denaturation at 95°C for 15 s and annealing/extension at 60°C for 1 min using PowerUp SYBR Green Master Mix (Thermo Fisher Scientific) in an Applied Biosystems 7500 Fast Real-Time PCR System. The primers used in this study have been reported previously [29] (Supplementary Table 2); primers for the untranscribed negative control region (Untr) were P20 and P21, and primers for the CYP3A4 PXRE were P22 and P23. Results are expressed as percent input normalized to the WT PXR DMSO control in each experiment; because transfection efficiencies varied between experiments, we found that normalizing each experiment reduced assay variability.
Time-resolved fluorescence resonance energy transfer assays
GST (pcDNA3.1-FLAG-GST) or GST-PXR LBD [pcDNA3.1-FLAG-GST-PXR LBD (WT, W299A, and W299D)] were expressed using the TNT Quick Coupled Transcription/Translation System (Promega); 1 μg of plasmid DNA was used for each 50 μL reaction, and the reactions were incubated at 30°C for 90 min. The TR-FRET binding assay has been described previously [30], and the protocol was modified for the in vitro protein expression method. Reactions (20 μL) contained 50 mM Tris (pH 7.5), 50 mM NaCl, 0.1 mg/mL bovine serum albumin, 5 nM LanthaScreen Tb-anti-GST Antibody (Thermo Fisher Scientific), 2 μL of protein expression reaction product, 1% DMSO, and the indicated concentrations of BODIPY FL vindoline. Reactions were incubated at RT in black 384-well low-volume assay plates for 30 min, and a PHERAstar FS plate reader (BMG Labtech) was used to detect the TR-FRET signals with the following instrumentation settings: a 340 nm excitation filter, a 100 μs delay time, and a 200 μs integration time. The TR-FRET ratio was expressed as 10,000 × 520 nm/490 nm, and values were normalized by subtracting the GST control signal at each point from the corresponding GST-PXR LBD signals.
Split Nano Luciferase cellular thermal shift assays
The use of the split Nano Luciferase technique for CETSA has been described previously [33]. 293T cells (600,000/well) were plated in 6-well tissue culture-treated plates. The following day, cells were transfected with pcDNA3.1-HiBiT-PXR LBD (WT, W299A, or W299D) plasmids (2 μg/well) using Lipofectamine 3000. The media was changed after 24 h, and cells were cultured for an additional day. Cells were washed with PBS, trypsinized, centrifuged at 500 × g for 5 min, washed with phenol red–free DMEM (without FBS), centrifuged at 500 × g for 5 min, and resuspended in phenol red–free DMEM (without FBS) at 106 cells/mL. For full melt curves, cell suspension (38 μL) was added to PCR plates containing 2 μL of DMSO or T0 dilution to yield 5% DMSO and the indicated concentrations of T0. The plates were sealed, incubated at 37°C in the cell culture incubator for 1 h, and heated in an Applied Biosystems ProFlex PCR System for 3.5 min at the indicated temperatures. 20 μL of suspension was added to white 384-well plates containing 5 μL of 5% NP-40 in DPBS, 62.5 nL LgBiT protein, and 125 nL HiBiT Lytic Substrate, the plates were shaken for 10 min at RT, and luminescence was read on an EnVision microplate reader. The LgBiT protein and HiBiT Lytic Substrate are components of the Nano-Glo HiBiT Lytic Detection System (Promega). The same procedure was used for compound dose responses except that the cell suspensions with indicated compounds and 5% DMSO were heated at 46°C for 3.5 min; this temperature was chosen based on the signal-to-background ratio of T0 vs. DMSO in Supplementary Figure 7a. For experiments assessing DMSO effects, 36 μL of cell suspension was added to 4 μL of DMSO at 10x final concentration; this modification was made to allow a highest final DMSO concentration of 10%.
Ligand docking and molecular dynamics simulations
The published structure of the PXR ligand binding domain (LBD) bound to the agonist SJB7 was obtained from the Protein Data Bank (PDB: 5X0R, Chain B) [16] and loaded into Maestro software (Schrödinger Release 2019–3). To prepare the protein for docking and simulations, the protein preparation wizard was used to assign bond orders, add hydrogens, create disulfide bonds, and fill in missing side chains and loops. Default parameters were used for optimization of hydrogen bond assignment (sampling of water orientations and use of pH 7.0). Waters beyond 5 Å of het groups or with less than 3 hydrogen bonds to non-waters were removed. Restrained energy minimization was then applied using the OPLS3e forcefield [49]. Once the wild-type PXR protein structure was prepared, tryptophan 299 was mutated to alanine, and the resulting mutant structure was prepared using the same method to ensure proper handling of the A299 residue. Prepared protein systems were further checked by Ramachandran plots and ensuring there were no steric clashes. To generate receptor grids, SJB7 was selected as the grid-defining ligand (for both wild-type and mutant systems). Default Van der Waals radius scaling parameters were used (scaling factor of 1, partial charge cutoff of 0.25). For docking T0, SPA70, and SJB7, each of the 3D structures were first obtained from the PubChem database (www.pubchem.ncbi.nlm.nih.gov). For SJC2, we used Maestro’s 2D sketch function to draw the ligand before preparation. The virtual screening workflow panel was used to prepare each ligand (by generating possible states at pH 7.0 ± 2.0 and retaining specified stereochemical properties) for docking into the WT and W299A PXR LBD receptor grids in parallel. The most stringent docking mode (extra precision, “XP”) of Glide [50] was used, with the following parameters: dock flexibly, perform post-docking minimization, and keep 100% of scoring compounds. For molecular dynamics simulations, systems were built each for ligand-free, T0-bound, SPA70-bound, SJB7-bound, and SJC2-bound WT and W299A structures using the system builder panel of Desmond (Schrödinger Release 2019–3). The SPC solvent model was used, and the forcefield was set to OPLS3e. Solvated systems were loaded into the workspace using the molecular dynamics panel. The total simulation time for each system was set to 200 nanoseconds, with 200 picosecond trajectory recording intervals. The system energy was set to the default value of 1.2, and the ensemble class used was NPT. Simulations were set to run at 300.0 K and at 1.01325 bar. The option to relax model systems before simulations was selected.
Plotting and statistical analyses
All plots were made in GraphPad Prism 8 and reflect the fold change compared to the WT PXR control in each experiment. Results are expressed as the mean ± standard deviation from at least 3 independent experiments, and basic dose response curves were fitted as needed. Significance was assessed with one-way ANOVA followed by Dunnett’s test for multiple comparisons [p ≤ 0.05 (*), p ≥ 0.05 (non-significant)]. The “additive” lines in the TNC + EE2 combination plot show the theoretical activation curves for the additive combination of EE2 and TNC calculated using the Bliss independence model [51].
Supplementary Material
ACKNOWLEDGEMENTS
We thank all members of the Chen research laboratory and the High-Throughput Bioscience Center for valuable discussions of the paper, the Hartwell Center at St. Jude Children’s Research Hospital for DNA sequencing, and Drs. Bert W. O’Malley and Sridhar Mani for plasmids. This work was supported in part by ALSAC and by the National Institutes of Health (grants R35-GM118041 [to TC] and P30-CA21765 [to the St. Jude Cancer Center to support Cancer Center shared resources]). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
DECLARATION OF INTERESTS
Authors Taosheng Chen and Wenwei Lin have filed the following patent: Chen T, Lin W, Wang Y. PCT/US2017/021949: 1,4,5-Substituted 1,2,3-Triazole Analogues as Antagonists of the Pregnane X Receptor. International Patent Application published September 28, 2017 as WO/2017/165139; US Patent Application published March 14, 2019 as US 2019/0077770 A1.
REFERENCES
- 1.Blumberg B, Sabbagh W Jr., Juguilon H, Bolado J Jr., van Meter CM, Ong, Evans RM (1998) SXR, a novel steroid and xenobiotic-sensing nuclear receptor. Genes Dev 12 (20):3195–3205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bertilsson G, Heidrich J, Svensson K, Asman M, Jendeberg L, Sydow-Backman M, Ohlsson R, Postlind H, Blomquist P, Berkenstam A (1998) Identification of a human nuclear receptor defines a new signaling pathway for CYP3A induction. Proc Natl Acad Sci U S A 95 (21):12208–12213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kliewer SA, Moore JT, Wade L, Staudinger JL, Watson MA, Jones SA, McKee DD, Oliver BB, Willson TM, Zetterstrom RH, Perlmann T, Lehmann JM (1998) An orphan nuclear receptor activated by pregnanes defines a novel steroid signaling pathway. Cell 92 (1):73–82 [DOI] [PubMed] [Google Scholar]
- 4.Xie W, Barwick JL, Downes M, Blumberg B, Simon CM, Nelson MC, Neuschwander-Tetri BA, Brunt EM, Guzelian PS, Evans RM (2000) Humanized xenobiotic response in mice expressing nuclear receptor SXR. Nature 406 (6794):435–439. doi: 10.1038/35019116 [DOI] [PubMed] [Google Scholar]
- 5.Lehmann JM, McKee DD, Watson MA, Willson TM, Moore JT, Kliewer SA (1998) The human orphan nuclear receptor PXR is activated by compounds that regulate CYP3A4 gene expression and cause drug interactions. J Clin Invest 102 (5):1016–1023. doi: 10.1172/JCI3703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burk O, Koch I, Raucy J, Hustert E, Eichelbaum M, Brockmoller J, Zanger UM, Wojnowski L (2004) The induction of cytochrome P450 3A5 (CYP3A5) in the human liver and intestine is mediated by the xenobiotic sensors pregnane X receptor (PXR) and constitutively activated receptor (CAR). J Biol Chem 279 (37):38379–38385. doi: 10.1074/jbc.M404949200 [DOI] [PubMed] [Google Scholar]
- 7.Goodwin B, Moore LB, Stoltz CM, McKee DD, Kliewer SA (2001) Regulation of the human CYP2B6 gene by the nuclear pregnane X receptor. Mol Pharmacol 60 (3):427–431 [PubMed] [Google Scholar]
- 8.Chen C, Staudinger JL, Klaassen CD (2003) Nuclear receptor, pregname X receptor, is required for induction of UDP-glucuronosyltranferases in mouse liver by pregnenolone-16 alpha-carbonitrile. Drug Metab Dispos 31 (7):908–915. doi: 10.1124/dmd.31.7.908 [DOI] [PubMed] [Google Scholar]
- 9.Synold TW, Dussault I, Forman BM (2001) The orphan nuclear receptor SXR coordinately regulates drug metabolism and efflux. Nat Med 7 (5):584–590. doi: 10.1038/87912 [DOI] [PubMed] [Google Scholar]
- 10.Geick A, Eichelbaum M, Burk O (2001) Nuclear receptor response elements mediate induction of intestinal MDR1 by rifampin. J Biol Chem 276 (18):14581–14587. doi: 10.1074/jbc.M010173200 [DOI] [PubMed] [Google Scholar]
- 11.Nallani SC, Goodwin B, Maglich JM, Buckley DJ, Buckley AR, Desai PB (2003) Induction of cytochrome P450 3A by paclitaxel in mice: pivotal role of the nuclear xenobiotic receptor, pregnane X receptor. Drug Metab Dispos 31 (5):681–684 [DOI] [PubMed] [Google Scholar]
- 12.Luo G, Cunningham M, Kim S, Burn T, Lin J, Sinz M, Hamilton G, Rizzo C, Jolley S, Gilbert D, Downey A, Mudra D, Graham R, Carroll K, Xie J, Madan A, Parkinson A, Christ D, Selling B, LeCluyse E, Gan LS (2002) CYP3A4 induction by drugs: correlation between a pregnane X receptor reporter gene assay and CYP3A4 expression in human hepatocytes. Drug Metab Dispos 30 (7):795–804 [DOI] [PubMed] [Google Scholar]
- 13.Moore LB, Goodwin B, Jones SA, Wisely GB, Serabjit-Singh CJ, Willson TM, Collins JL, Kliewer SA (2000) St. John’s wort induces hepatic drug metabolism through activation of the pregnane X receptor. Proc Natl Acad Sci U S A 97 (13):7500–7502. doi: 10.1073/pnas.130155097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mani S, Dou W, Redinbo MR (2013) PXR antagonists and implication in drug metabolism. Drug Metab Rev 45 (1):60–72. doi: 10.3109/03602532.2012.746363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xie Y, Xu M, Deng M, Li Z, Wang P, Ren S, Guo Y, Ma X, Fan J, Billiar TR, Xie W (2019) Activation of Pregnane X Receptor Sensitizes Mice to Hemorrhagic Shock-Induced Liver Injury. Hepatology 70 (3):995–1010. doi: 10.1002/hep.30691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lin W, Wang YM, Chai SC, Lv L, Zheng J, Wu J, Zhang Q, Wang YD, Griffin PR, Chen T (2017) SPA70 is a potent antagonist of human pregnane X receptor. Nat Commun 8 (1):741. doi: 10.1038/s41467-017-00780-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang H, LeCluyse EL (2003) Role of orphan nuclear receptors in the regulation of drug-metabolising enzymes. Clin Pharmacokinet 42 (15):1331–1357. doi: 10.2165/00003088-200342150-00003 [DOI] [PubMed] [Google Scholar]
- 18.Oladimeji P, Cui H, Zhang C, Chen T (2016) Regulation of PXR and CAR by protein-protein interaction and signaling crosstalk. Expert Opin Drug Metab Toxicol 12 (9):997–1010. doi: 10.1080/17425255.2016.1201069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Buchman CD, Chai SC, Chen T (2018) A current structural perspective on PXR and CAR in drug metabolism. Expert Opin Drug Metab Toxicol 14 (6):635–647. doi: 10.1080/17425255.2018.1476488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Watkins RE, Wisely GB, Moore LB, Collins JL, Lambert MH, Williams SP, Willson TM, Kliewer SA, Redinbo MR (2001) The human nuclear xenobiotic receptor PXR: structural determinants of directed promiscuity. Science 292 (5525):2329–2333. doi: 10.1126/science.1060762 [DOI] [PubMed] [Google Scholar]
- 21.Ostberg T, Bertilsson G, Jendeberg L, Berkenstam A, Uppenberg J (2002) Identification of residues in the PXR ligand binding domain critical for species specific and constitutive activation. Eur J Biochem 269 (19):4896–4904. doi: 10.1046/j.1432-1033.2002.03207.x [DOI] [PubMed] [Google Scholar]
- 22.Chrencik JE, Orans J, Moore LB, Xue Y, Peng L, Collins JL, Wisely GB, Lambert MH, Kliewer SA, Redinbo MR (2005) Structural disorder in the complex of human pregnane X receptor and the macrolide antibiotic rifampicin. Mol Endocrinol 19 (5):1125–1134. doi: 10.1210/me.2004-0346 [DOI] [PubMed] [Google Scholar]
- 23.Tirona RG, Leake BF, Podust LM, Kim RB (2004) Identification of amino acids in rat pregnane X receptor that determine species-specific activation. Mol Pharmacol 65 (1):36–44. doi: 10.1124/mol.65.1.36 [DOI] [PubMed] [Google Scholar]
- 24.Jones SA, Moore LB, Shenk JL, Wisely GB, Hamilton GA, McKee DD, Tomkinson NC, LeCluyse EL, Lambert MH, Willsona promiscuous xenobiotic receptor that has diverged during evolution. Mol Endocrinol 14 (1):27–39. doi: 10.1210/mend.14.1.0409 [DOI] [PubMed] [Google Scholar]
- 25.Ngan CH, Beglov D, Rudnitskaya AN, Kozakov D, Waxman DJ, Vajda S (2009) The TM, Kliewer SA, Moore JT (2000) The pregnane X receptor: structural basis of pregnane X receptor binding promiscuity. Biochemistry 48 (48):11572–11581. doi: 10.1021/bi901578n [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Banerjee M, Chai SC, Wu J, Robbins D, Chen T (2016) Tryptophan 299 is a conserved residue of human pregnane X receptor critical for the functional consequence of ligand binding. Biochem Pharmacol 104:131–138. doi: 10.1016/j.bcp.2016.02.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li L, Welch MA, Li Z, Mackowiak B, Heyward S, Swaan PW, Wang H (2019) Mechanistic Insights of Phenobarbital-Mediated Activation of Human but Not Mouse Pregnane X Receptor. Mol Pharmacol 96 (3):345–354. doi: 10.1124/mol.119.116616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Delfosse V, Dendele B, Huet T, Grimaldi M, Boulahtouf A, Gerbal-Chaloin S, Beucher B, Roecklin D, Muller C, Rahmani R, Cavailles V, Daujat-Chavanieu M, Vivat V, Pascussi JM, Balaguer P, Bourguet W (2015) Synergistic activation of human pregnane X receptor by binary cocktails of pharmaceutical and environmental compounds. Nat Commun 6:8089. doi: 10.1038/ncomms9089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hariparsad N, Chu X, Yabut J, Labhart P, Hartley DP, Dai X, Evers R (2009) Identification of pregnane-X receptor target genes and coactivator and corepressor binding to promoter elements in human hepatocytes. Nucleic Acids Res 37 (4):1160–1173. doi: 10.1093/nar/gkn1047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lin W, Liu J, Jeffries C, Yang L, Lu Y, Lee RE, Chen T (2014) Development of BODIPY FL vindoline as a novel and high-affinity pregnane X receptor fluorescent probe. Bioconjug Chem 25 (9):1664–1677. doi: 10.1021/bc5002856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Martinez Molina D, Jafari R, Ignatushchenko M, Seki T, Larsson EA, Dan C, Sreekumar L, Cao Y, Nordlund P (2013) Monitoring drug target engagement in cells and tissues using the cellular thermal shift assay. Science 341 (6141):84–87. doi: 10.1126/science.1233606 [DOI] [PubMed] [Google Scholar]
- 32.Jafari R, Almqvist H, Axelsson H, Ignatushchenko M, Lundback T, Nordlund P, Martinez Molina D (2014) The cellular thermal shift assay for evaluating drug target interactions in cells. Nat Protoc 9 (9):2100–2122. doi: 10.1038/nprot.2014.138 [DOI] [PubMed] [Google Scholar]
- 33.Martinez NJ, Asawa RR, Cyr MG, Zakharov A, Urban DJ, Roth JS, Wallgren E, Klumpp-Thomas C, Coussens NP, Rai G, Yang SM, Hall MD, Marugan JJ, Simeonov A, Henderson MJ (2018) A widely-applicable high-throughput cellular thermal shift assay (CETSA) using split Nano Luciferase. Sci Rep 8 (1):9472. doi: 10.1038/s41598-018-27834-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang H, Li H, Moore LB, Johnson MD, Maglich JM, Goodwin B, Ittoop OR, Wisely B, Creech K, Parks DJ, Collins JL, Willson TM, Kalpana GV, Venkatesh M, Xie W, Cho SY, Roboz J, Redinbo M, Moore JT, Mani S (2008) The phytoestrogen coumestrol is a naturally occurring antagonist of the human pregnane X receptor. Mol Endocrinol 22 (4):838–857. doi: 10.1210/me.2007-0218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Motta S, Callea L, Giani Tagliabue S, Bonati L (2018) Exploring the PXR ligand binding mechanism with advanced Molecular Dynamics methods. Sci Rep 8 (1):16207. doi: 10.1038/s41598-018-34373-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Waterhouse AM, Procter JB, Martin DM, Clamp M, Barton GJ (2009) Jalview Version 2--a multiple sequence alignment editor and analysis workbench. Bioinformatics 25 (9):1189–1191. doi: 10.1093/bioinformatics/btp033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Robert X, Gouet P (2014) Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res 42 (Web Server issue):W320–324. doi: 10.1093/nar/gku316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lin W, Wu J, Dong H, Bouck D, Zeng FY, Chen T (2008) Cyclin-dependent kinase 2 negatively regulates human pregnane X receptor-mediated CYP3A4 gene expression in HepG2 liver carcinoma cells. J Biol Chem 283 (45):30650–30657. doi: 10.1074/jbc.M806132200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Banerjee M, Chen T (2013) Differential regulation of CYP3A4 promoter activity by a new class of natural product derivatives binding to pregnane X receptor. Biochem Pharmacol 86 (6):824–835. doi: 10.1016/j.bcp.2013.07.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Goodwin B, Hodgson E, Liddle C (1999) The orphan human pregnane X receptor mediates the transcriptional activation of CYP3A4 by rifampicin through a distal enhancer module. Mol Pharmacol 56 (6):1329–1339 [DOI] [PubMed] [Google Scholar]
- 41.Pondugula SR, Brimer-Cline C, Wu J, Schuetz EG, Tyagi RK, Chen T (2009) A phosphomimetic mutation at threonine-57 abolishes transactivation activity and alters nuclear localization pattern of human pregnane x receptor. Drug Metab Dispos 37 (4):719–730. doi: 10.1124/dmd.108.024695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Banerjee M, Chen T (2014) Thiazide-like diuretic drug metolazone activates human pregnane X receptor to induce cytochrome 3A4 and multidrug-resistance protein 1. Biochem Pharmacol 92 (2):389–402. doi: 10.1016/j.bcp.2014.08.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang YM, Chai SC, Lin W, Chai X, Elias A, Wu J, Ong SS, Pondugula SR, Beard JA, Schuetz EG, Zeng S, Xie W, Chen T (2015) Serine 350 of human pregnane X receptor is crucial for its heterodimerization with retinoid X receptor alpha and transactivation of target genes in vitro and in vivo. Biochem Pharmacol 96 (4):357–368. doi: 10.1016/j.bcp.2015.06.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jenster G, Spencer TE, Burcin MM, Tsai SY, Tsai MJ, O’Malley BW (1997) Steroid receptor induction of gene transcription: a two-step model. Proc Natl Acad Sci U S A 94 (15):7879–7884. doi: 10.1073/pnas.94.15.7879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li H, Redinbo MR, Venkatesh M, Ekins S, Chaudhry A, Bloch N, Negassa A, Mukherjee P, Kalpana G, Mani S (2013) Novel yeast-based strategy unveils antagonist binding regions on the nuclear xenobiotic receptor PXR. J Biol Chem 288 (19):13655–13668. doi: 10.1074/jbc.M113.455485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kamentsky L, Jones TR, Fraser A, Bray MA, Logan DJ, Madden KL, Ljosa V, Rueden C, Eliceiri KW, Carpenter AE (2011) Improved structure, function and compatibility for CellProfiler: modular high-throughput image analysis software. Bioinformatics 27 (8):1179–1180. doi: 10.1093/bioinformatics/btr095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lamprecht MR, Sabatini DM, Carpenter AE (2007) CellProfiler: free, versatile software for automated biological image analysis. Biotechniques 42 (1):71–75. doi: 10.2144/000112257 [DOI] [PubMed] [Google Scholar]
- 48.Carpenter AE, Jones TR, Lamprecht MR, Clarke C, Kang IH, Friman O, Guertin DA, Chang JH, Lindquist RA, Moffat J, Golland P, Sabatini DM (2006) CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol 7 (10):R100. doi: 10.1186/gb-2006-7-10-r100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Harder E, Damm W, Maple J, Wu C, Reboul M, Xiang JY, Wang L, Lupyan D, Dahlgren MK, Knight JL, Kaus JW, Cerutti DS, Krilov G, Jorgensen WL, Abel R, Friesner RA (2016) OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J Chem Theory Comput 12 (1):281–296. doi: 10.1021/acs.jctc.5b00864 [DOI] [PubMed] [Google Scholar]
- 50.Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, Sanschagrin PC, Mainz DT (2006) Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J Med Chem 49 (21):6177–6196. doi: 10.1021/jm051256o [DOI] [PubMed] [Google Scholar]
- 51.Zhao W, Sachsenmeier K, Zhang L, Sult E, Hollingsworth RE, Yang H (2014) A New Bliss Independence Model to Analyze Drug Combination Data. J Biomol Screen 19 (5):817–821. doi: 10.1177/1087057114521867 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.