

Abstract
The delivery of immunomodulators directly into the tumor potentially harnesses the existing antigen, tumor-specific infiltrating lymphocytes, and antigen presenting cells. This can confer specificity and generate a potent systemic anti-tumor immune response with lower doses and less toxicity compared to systemic administration, in effect an in situ vaccine. Here, we test this concept using the novel combination of immunomodulators anti-CTLA4, -CD137, and -OX40. The triple combination administered intratumorally at low doses to one tumor of a dual tumor mouse model had dramatic local and systemic anti-tumor efficacy in lymphoma (A20) and solid tumor (MC38) models, consistent with an abscopal effect. The minimal effective dose was 10 μg each. The effect was dependent on CD8 T-cells. Intratumoral administration resulted in superior local and distant tumor control compared to systemic routes, supporting the in situ vaccine concept. In a single tumor A20 model, injection close to the tDLN resulted in similar efficacy as intratumoral and significantly better than targeting a non-tDLN, supporting the role of the tDLN as a viable immunotherapy target in addition to the tumor itself. Distribution studies confirmed expected concentration of antibodies in tumor and tDLN, in keeping with the anti-tumor results. Overall intratumoral or peri-tDLN administration of the novel combination of anti-CTLA4, anti-CD137, and anti-OX40, all agents in the clinic or clinical trials, demonstrates potent systemic anti-tumor effects. This immunotherapeutic combination is promising for future clinical development via both these safe and highly efficacious routes of administration.
Keywords: Combination immunotherapy, Intratumoral, Tumor draining lymph node, CD137, OX40, CTLA4
Introduction
Immune checkpoint therapy has been a breakthrough in cancer treatment with survival benefits for several types of advanced malignancies [3, 4]. Unfortunately, single agent immune therapy is not generally curative [4]. Combination of two or more immunotherapeutic agents may have synergistic efficacy by targeting multiple immune pathways [5, 6]. However, with single agent immune checkpoint inhibitors, toxicity in the form of immune-related adverse events can be severe, and for combination therapy, the potential for toxicity is even higher [5, 7–9].
One approach that may enable combination immune therapy but reduce systemic exposure and thereby toxicity is through intratumoral administration of immune agents, also referred to as tumor-directed immunotherapy [10, 11]. The premise of tumor-directed immunotherapy is to generate an in situ vaccine effect by harnessing the pre-existing tumor-specific immune cells and tumor antigen present in the tumor microenvironment, thus generating a systemic anti-tumor immune response. Treating locally to induce systemic therapeutic results is referred to as the abscopal effect [10, 11]. The rationale for tumor-directed immunotherapy may extend to the tumor draining lymph node (tDLN), which has also been shown to harbor tumor-specific T-cells [12, 13].
There are many possible combinations of immune therapies that could be tested. Three drugs have emerged as good candidates as single agents: anti-(α)CD137 (41BB), αOX40, and αCTLA4. CD137 and OX40 are both members of the TNFR superfamily, expressed on T-cells, including tumor-infiltrating lymphocytes, as well as other immune cell subsets [14, 15]. Ligation of these receptors delivers a costimulatory signal to T-cells resulting in activation [16, 17]. Both agents have been assessed in the early clinical trials, showing promising efficacy. However, Urelumumab, one of the αCD137 monoclonal antibodies (mAbs) assessed in clinical trials induced liver toxicity requiring dose reduction for subsequent trials [18]. Targeting both CD137 and OX40 may be synergistic, as OX40 engagement can activate T-cells and result in upregulation of CD137 [6]. αCTLA4 is an immune checkpoint inhibitor, best known for its activity and survival effects in metastatic melanoma, as well as other solid tumors [19, 20]. However, when given alone, clinical benefits are limited to only a subset of patients and is associated with significant immune-related toxicity [7, 9]. Immune checkpoint and costimulatory pathways have fundamentally different mechanisms with potential for therapeutic synergy as a ‘release the brakes and push the gas pedal’ approach [21].
In situ vaccination targeting the tumor or tDLN with low doses of immune activating agents has the potential to promote an abscopal effect while circumventing excessive toxicity, allowing the use of synergistic combinations, and improving clinical responses. Here, we test this concept using the novel immunotherapeutic combination αCD137, αOX40, and αCTLA4 in two mouse tumor model systems.
Materials and methods
Mice
BALB/C and C57BL/6 female mice age 6–8 weeks were purchased from The Jackson Laboratory and housed in the Comparative Medicine Pavilion at the Stanford University Medical Center. All experiments were approved by the Stanford Administrative Panel on Laboratory Animal Care and conducted in accordance with Stanford University and NIH guidelines.
Monoclonal antibodies
Anti-mouse OX40 (CD134) clone OX86, CD137 (clone LOB12.3), CTLA4 (clone 9H10), and depleting antibodies anti-mouse CD8α (clone 2.43), and CD4 (clone GK 1.5) were purchased from BioXCell (West Lebanon, NH). Anti-mouse CD28 (clone 37.51) and CD3e (clone 145–2C11) were purchased from BD Biosciences (San Jose, CA). For flow cytometry, we used anti-mouse CD4-efluor 450 (clone GK1.5), CD8α-PerCP-Cy5.5 (clone 53–6.7), CD44-APC-efluor 780 (clone 1M7), and FOXP3-PE (clone XMG1.2) (eBioscience, San Diego CA); anti-mouse IFN-γ-PE (clone XMG1.2) (BD Biosciences); and anti-mouse CD25-APC/CY7 (clone PC61) (Biolegend; San Diego, CA). Live/Dead fixable aqua dead cell kit (Thermo Fisher Scientific, Waltham, MA) was used to identify dead cells.
Tumor studies
A20, a mouse B-cell lymphoma-derived cell line, was obtained from American Type Culture Collection (Manassas, VA). Tumor cells were cultured in complete medium (RPMI 1640; Cellgro) containing 10% fetal bovine serum (HyClone), 100 U/mL penicillin, 100 mg/mL streptomycin, and 50 mM 2-ME (Gibco). MC38, a mouse colon cancer cell line, was donated by Bavarian Nordic Inc. (formerly in Mountain View, California). MC38 cells were cultured in Dulbecco’s modified Eagle’s medium containing 10% fetal bovine serum (HyClone), 100 U/mL penicillin, 100 mg/mL streptomycin, and 50 mM 2-ME (Gibco).
Tumor cells were harvested from culture, while in the exponential phase of growth, counted, and then washed twice with PBS. Cells were injected subcutaneously bilaterally on the flanks to create two tumors. For some experiments, a single tumor (left flank) model was used. For A20, 10 × 106 cells were used for each site, and for MC38, 1 × 106 cells were injected for each site. Treatment started approximately 8 days after tumor cell inoculation, when tumors reached approximately 7–10 mm diameter. The MC38 model was administered a total of 6 (2 per week) intratumoral injections of the triple combination (αCTLA4, αCD137, and αOX40) at 30 μg each to the left tumor only (Fig. 1a). For A20, 4 biweekly injections were given at doses and routes of administration as specified for each experiment.
Fig. 1.
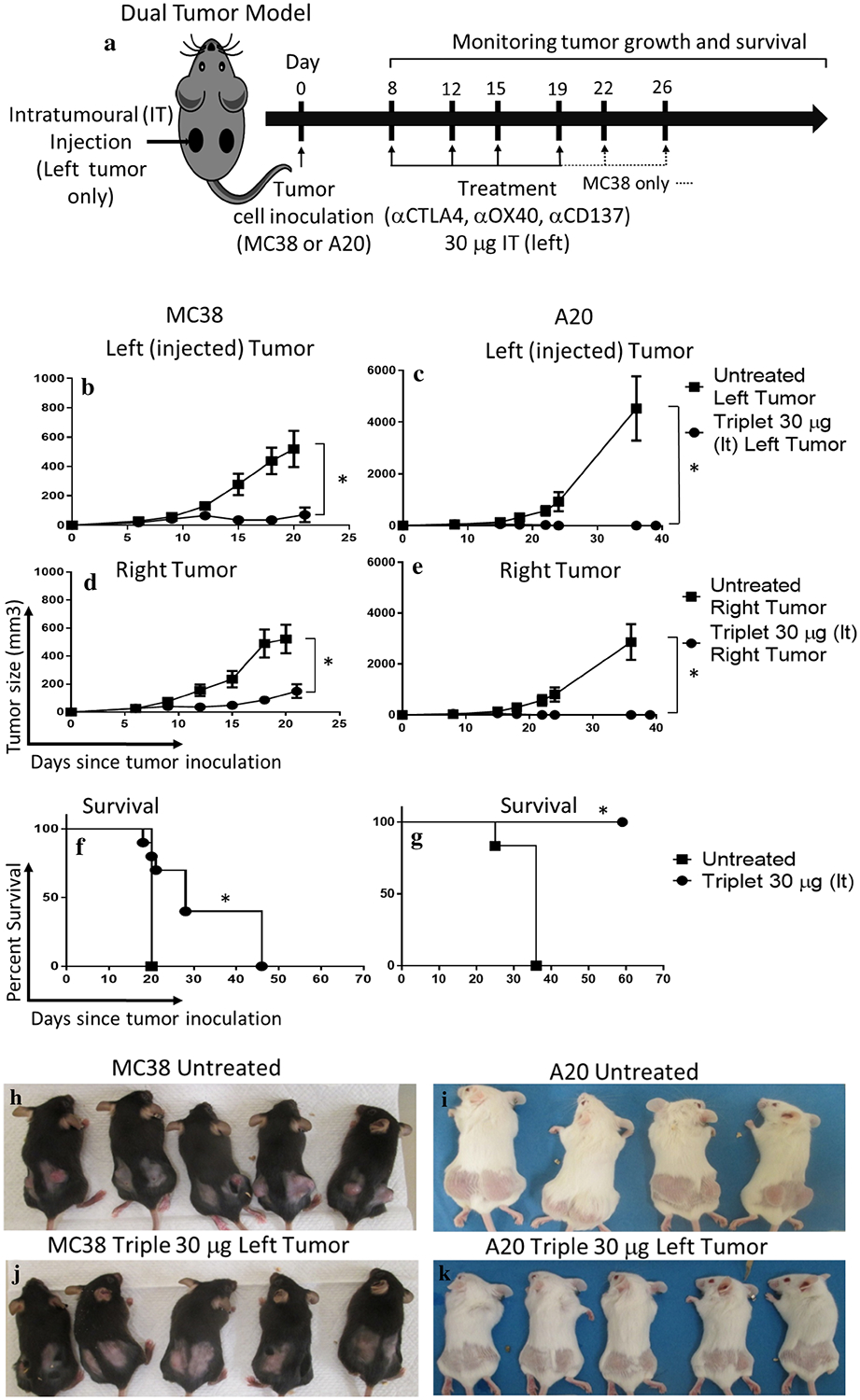
Potent local and systemic anti-tumor effects of low-dose (30 μg) triple combination immunotherapy. a Treatment scheme: a dual tumor mouse model was used for MC38 (colon cancer) and A20 (B-cell lymphoma) models in C57BL/6 and BALB/c mice, respectively. Treatment was administered by intratumoral injection to the left tumor consisting of 6 (MC38) or 4 (A20) biweekly injections of the triple combination αCTLA4, αCD137, and αOX40 at a dose of 30 μg each, started approximately day 8 after tumor inoculation when tumors were 7–10 mm diameter. Tumor growth measure by caliper of MC38 or A20 injected (b, c) or non-injected site (d, e). Survival curves for MC38 (f) or A20 (g) models are shown. h–k Representative photos of the treatment groups at Day 19 of tumor inoculation. ‘*’ denotes p < .05 (unpaired t test for tumor growth and Mantel Cox for survival). Values for b–e are mean ± SEM. n = 10 (MC38) and 7 (A20) per group
Intratumoral injections were delivered to the left tumor only. Subcutaneous injections were to the left or right abdominal wall. Intratumoral (IT) and subcutaneous (SC) injections were 50 μl, intraperitoneal (IP) 100 μl. Cellular vaccines were prepared using 20 × 106 A20 cells per mouse, suspended in 7 mls RPMI, and irradiated with a cobalt irradiator, 50 Gy. Washed cells were reconstituted in 50 μl of Pathclear extracellular matrix (Trevigen Inc, Gaithersburg, MD catalog#3433-005-01) per subcutaneous injection containing 10 μg each of the triple mAb combination.
Tumor measurements by digital caliper were made three times per week. Tumor volume was calculated using the modified ellipsoidal equation (tumor volume = 1/2 (length × width2). As per Stanford University guidelines, mice were sacrificed when tumors reached a maximum diameter of 1.75 cm, or if tumors became ulcerated. The survival endpoint was defined as the point at which mice were sacrificed for tumor size or ulceration.
In vivo T‑cell depletion
αCD4 and/or αCD8 mAbs were injected intraperitoneally in a volume of 100 μl (PBS) for 4 doses—1 day before therapy and immediately prior to the first 3 treatments. All doses were 100 μg, except the first dose of αCD4 was 200 μg. Flow cytometry of peripheral blood confirmed greater than 95% reduction of the respective T-cell subsets.
In vitro T‑cell assay
Single-cell suspensions were made from spleens of treated mice and red cells were lysed with ammonium-chloride-potassium buffer (Quality Biological, Gaithersburg, MD). Splenocytes were cocultured with 1 × 106 irradiated A20 cells or 4T1 cells (irrelevant cell line) for 24 h at 37 °C and 5% CO2 in the presence of 0.5 μg/ml anti-mouse CD28 mAb. αCD28 and αCD3 (0.05 μg/ml) without tumor cells served as a positive control. Monensin (GolgiStop; BD Biosciences, San Jose, CA) and brefeldin A (eBioscience, San Diego, CA) were added for the last 6 h. Intracellular IFN-γ expression was assessed using BD Cytofix/Cytoperm Plus Kit, staining with αCD4, αCD8a, and αIFN-γ and analyzed by flow cytometry.
Blood toxicity analysis
Blood was drawn by submandibular puncture in anesthetized mice. Analysis was performed by the diagnostic laboratory at Stanford University Department of Comparative Medicine.
Biodistribution studies
A dual flank tumor A20 model as described above was used. Mice received one injection of the triple combination (10 μg) by one of several local or systemic routes (Fig. 6a). Volumes were 50 μl for IT and SC injections, and 100 μl for IP injections. There were nine mice per injection route sacrificed in groups of three at time points of 3, 6, and 24 h post-injection. Tissues were collected and flash frozen in liquid nitrogen for later analysis, including both tumors, bilateral inguinal/tDLNs, spleen, and blood.
Fig. 6.
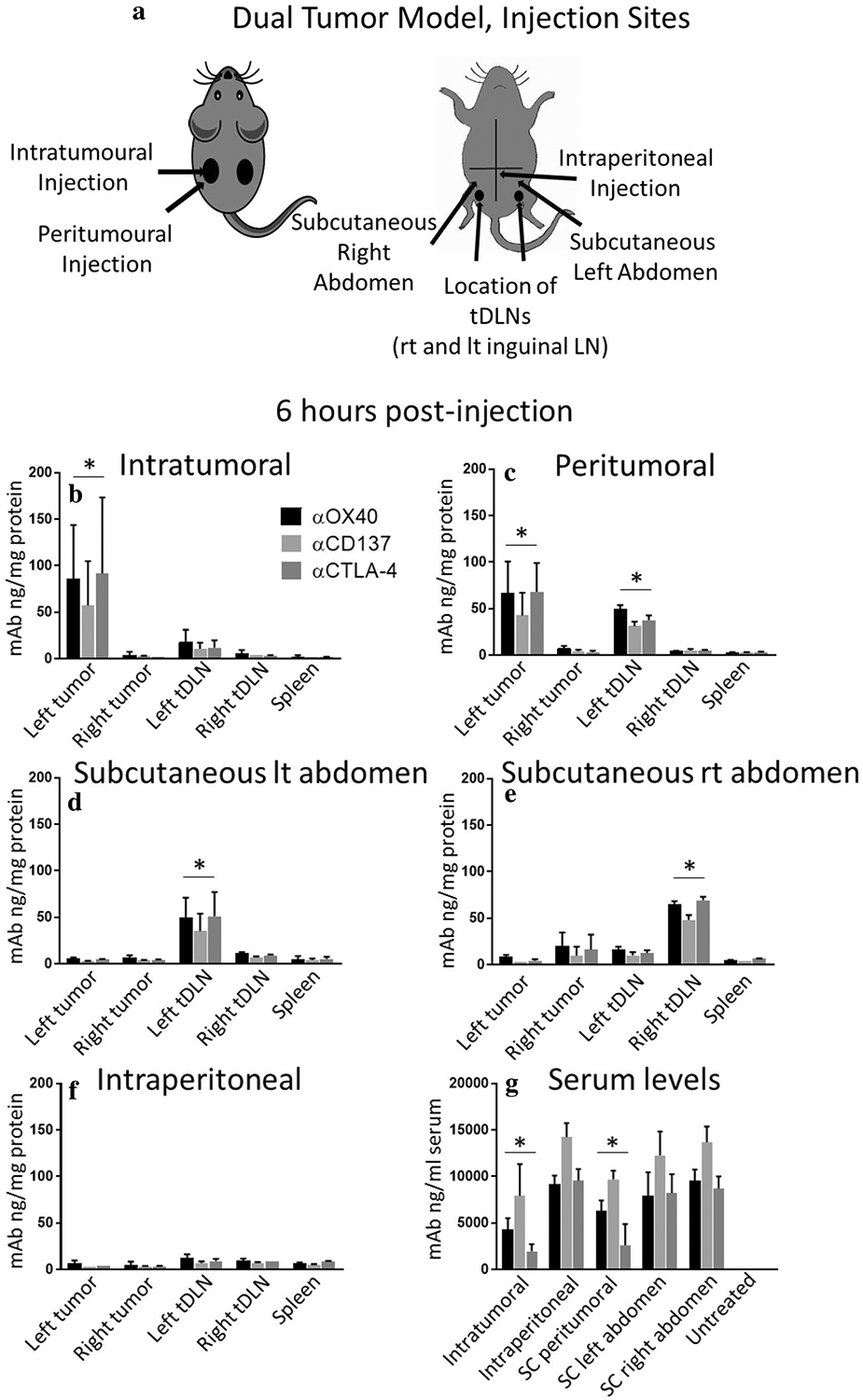
Biodistribution studies: monoclonal antibody (mAb) biodistribution studies, 6 h post-injection, confirm that antibodies concentrate in target structures. a A dual tumor A20 model was used. Mice were injected once with the triple combination (αCD137, αOX40, and αCTLA4) at a dose of 10 μg each. Routes of administration for the different groups are as shown. Mice were sacrificed 6 h post-injection, and tumor, tDLN, non-tDLN, blood, and spleen were collected. b–g Antibody levels in the different compartments were analyzed by ELISA. Values expressed as mean ± SD. n = 3 mice per group per injection site. tDLN tumor draining lymph node; SC subcutaneous. ‘*’ denotes p < .05, Student’s t test. Data for 3 and 24 h time points shown in Supplementary Figure 3
Tissues were homogenized in T-PER® Tissue Protein Extraction Reagent (Thermo Scientific, Waltham, MA, USA), and centrifuged for 5 min at 10,000×g. The supernatant was analyzed for protein content using Pierce™ BCA protein assay kit (Thermo Scientific). Antibody concentration was measured by ELISA. Recombinant murine OX40 fused to human IgG1-Fc (#1256-OX, R&D, Minneapolis MN USA), CD137-human IgG1-Fc (#937–4B, R&D), or CTLA4-murine IgG1-Fc (#59654, Abcam, Cambridge, UK) were coated in PBS into 96-well ELISA plates (# 655074, Greiner Bio-One, Kremsmünster, Austria) blocked with 2% BSA in PBS + 0.05% Tween20. Samples were added in duplicates. HRP-conjugated αrat kappa/lambda light chain (clone MARK-1/MARL-15, Bio-Rad, Hercules, CA, USA) or αsyrian hamster IgG (#107-035-142, Jackson ImmunoResearch, West Grove, PA, USA) was used for detection, followed by SuperSignal Pico ELISA Chemiluminescent Substrate (Thermo Fisher). The luminescence was measured in Fluostar Optima (BMG Labtech, Ortenberg, Germany).
Tumor‑infiltrating lymphocyte (TIL) analysis
Mice were sacrificed 48 h after two treatments of the triplet or doublets at 10 μg. Excised tumors were homogenized, and cells stained for flow cytometry.
Flow cytometry
Cells were surface stained in PBS, 1% fetal bovine serum, and 0.01% sodium azide, then fixed in 2% paraformaldehyde, and analyzed by flow cytometry on an LSR II Flow Cytometer (BD Biosciences) at the Stanford FACS Facility. Data were analyzed using FlowJo software (FlowJo LLC, Ashland, OR).
Statistics
Prism software (GraphPad Software, La Jolla, CA) was used for data analysis and graphics. An unpaired student t test was used for tumor growth comparisons and biodistribution studies. The log-rank (Mantel-Cox) test was used to compare survival curves. p values <.05 were considered significant.
Results
Intratumoral triple therapy at low (10 μg) dose maintains clinical efficacy
The triple combination of αCD137, αOX40, and αCTLA4 was delivered intratumorally (IT) to a single tumor in a dual flank tumor model of either colon cancer (MC38; C57BL/6 mice) or B-cell lymphoma (A20; BALB/C mice) (Fig. 1a). Each monoclonal antibody (mAb) was initially dosed at 30 μg. Usual systemic doses are 100–400 μg [22–24]. For both tumor models, tumor regression was observed in all mice for both the injected (left) tumor and right (non-injected) tumor (Fig. 1b–e) with a survival advantage (p < .05, Mantel-Cox; Fig. 1f, g). However, tumor regression was incomplete in the MC38 model, whereas for A20, complete regression of the injected and non-injected tumor was observed in most mice. Figure 1h–k shows representative photos. Thus, the triple combination was effective in the treatment of both the MC38 colon and the A20 lymphoma model, with greater efficacy in the latter.
Next, we examined three different doses (3, 10, and 30 μg each) of the triple combination in the A20 dual tumor model (Fig. 2a). At a 10 or 30 μg dose, anti-tumor efficacy was similar, inducing regression of both the injected (left) and non-injected (right) tumors (Fig. 2c, f, 30 μg not shown). At 3 μg, local anti-tumor effects were observed, however, the untreated tumor was not cleared (Fig. 2d, g). Figure 2h–j shows representative photos. Thus, 10 μg was deemed the optimal dose to achieve local and systemic tumor control. At this dose, 80% of mice achieved a complete response at 35 days, and by 55 days, 70% remained tumor free (n = 20).
Fig. 2.
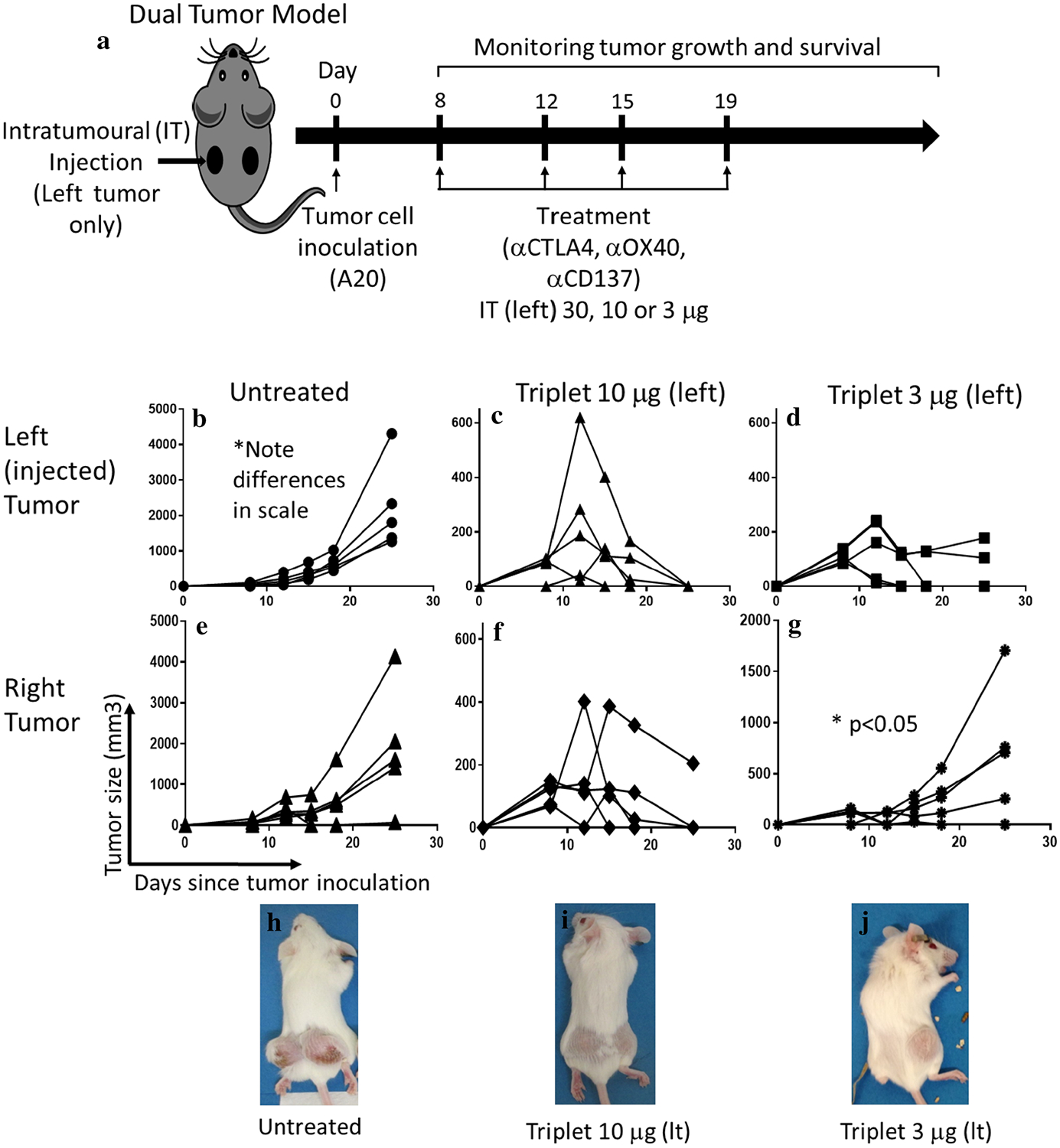
Dose titration: 10 μg IT dose is the lowest dose which maintains local and systemic anti-tumor efficacy. a Mice were treated with four biweekly doses of the triple combination αCD137, αOX40, and αCTLA4 at two dose levels delivered intratumorally to the left tumor of a dual tumor A20 lymphoma model. Tumor growth measure by caliper of untreated (b), triplet 10 μg (c), or 3 μg (d) injected tumor or non-injected tumor (e–g, respectively). Triplet 10 μg was not significantly different than triplet 30 μg (not shown). Representative photos of the mice at day 19 of tumor inoculation are shown in (h–j).’*’ denotes p < .05 by Student’s t test. n = 5 per group
Toxicity is observed at the highest dose level but is minimal at lower dosing regimens
Two mice of a total 137 mice treated with the triple combination during the course of the project died of treatment-related causes. The deaths occurred at the highest dosing regimen for MC38 model, immediately after the fifth and sixth 30 μg (each mAb) injection, respectively. Mice appeared well prior to the injections, tumor size was modest, and they had no overt signs of immune-related toxicity, i.e., no skin, hair, or eye changes, no wasting syndrome [25].
Blood samples were analyzed for toxicity assessment from A20 model mice after 4 biweekly injections of either 30 μg (n = 3) or 10 μg (n = 2) (Supplementary Table 1). There were no clinically significant alterations in renal or liver function. There was mild neutropenia in 2 mice (<35% reduction; 1 at each dose level) and moderate neutropenia in one (45% reduction; 30 μg dose) [26]. There were no observed infectious complications in any of the treated mice. Eosinophilia was noted in 2 of 5 mice, both at the 30 μg dose. No overt manifestations of immune-related toxicity were observed in any of the treated mice.
The double therapy combinations as well as αOX40 alone did induce anti-tumor effects. The triple combination was significantly better as measured by tumor size than any of the doublets or αOX40 alone (p < .05, student t test; Supplementary Figure 1). We conclude that the 10 μg dose of triple therapy is effective with minimal toxicity.
CD8 T‑cells are required for anti‑tumor efficacy of the triple combination
The requirement of T-cells for therapeutic effect of the triple combination was assessed by in vivo depletion of CD4, CD8, or both subsets of T-cells (Fig. 3a–e). For immunologically intact mice, the triple combination IT (left tumor) resulted in bilateral tumor regression (Fig. 3f, g), with 2 of 5 mice having residual right-sided tumors. Depletion of CD8 T-cells completely abrogated the anti-tumor effects of the 10 μg triple combination. CD4-depletion had no discernible effect on treatment response. Dual (CD4/CD8)-depletion resulted in significantly more aggressive tumor growth than in untreated mice (p < .05, student t test) (Fig. 3f, g). Survival was significantly better for treated mice that were immunologically intact or CD4-depleted than for CD8-depleted, dual-depleted, and untreated mice (p < .05, Mantel-Cox; Fig. 3h). Thus, CD8 T-cells are required for treatment effect, while CD4 cells appear to have a lesser role.
Fig. 3.
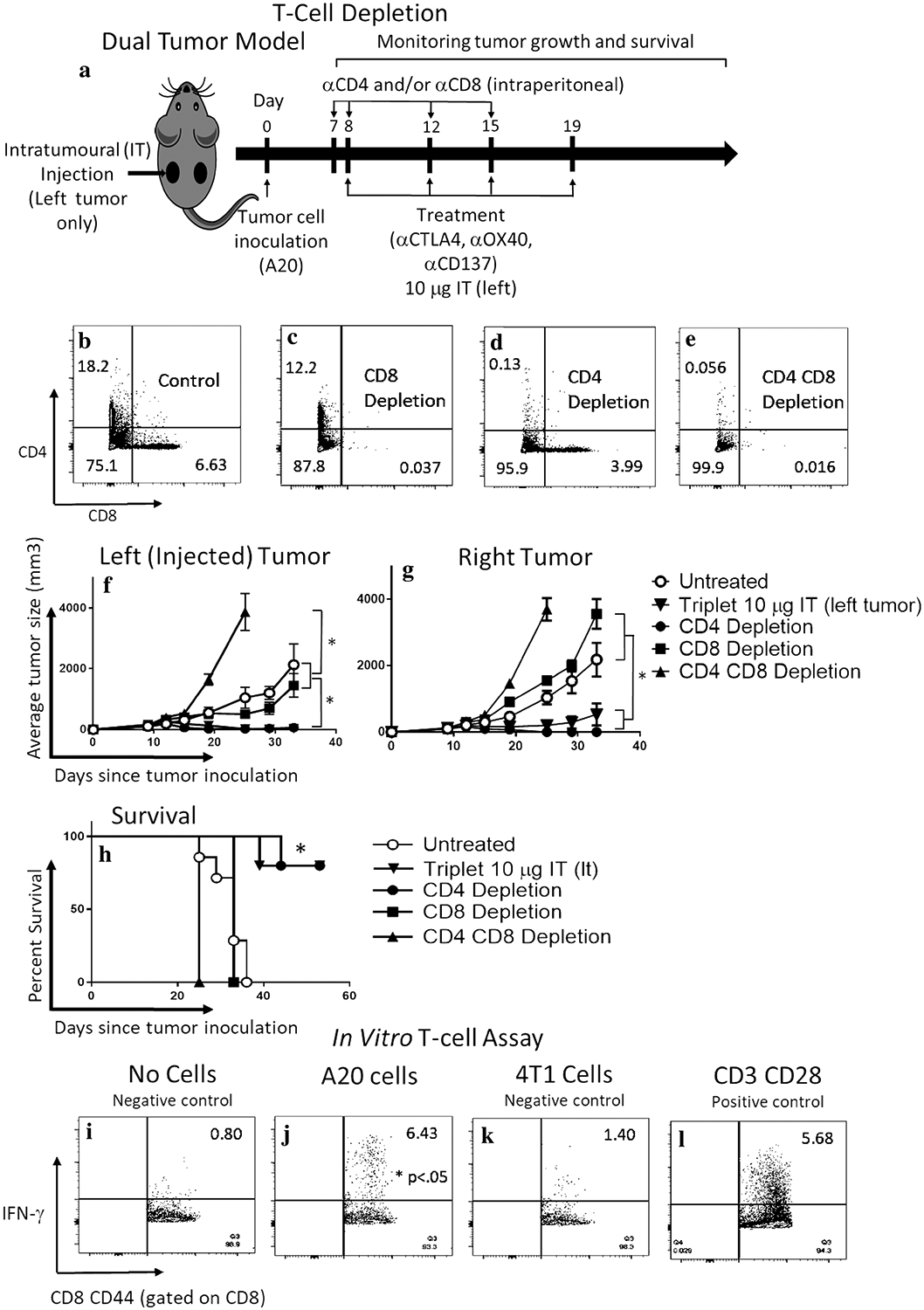
CD8 T-cell is required for efficacy, and CD8CD44hi memory cells generate a specific anti-tumor response ex vivo. a In vivo T-cell depletion. Mice were treated with four biweekly doses of the triple combination αCD137, αOX40, and αCTLA4 at the 10 μg dose delivered intratumorally to the left tumor of a dual tumor A20 lymphoma model. CD8 and/or CD4 cells were depleted by systemic (intraperitoneal) administration of respective antibodies. Dot-plots panels showing peripheral blood staining of CD4 and CD8 T cells in non-depleted (b), CD8 (c), CD4 (d), or CD4 and CD8 (e) depleted mice. Tumor measurement by caliper of injected (f) or non-injected (g) mice after no depletion, CD4, CD8, CD4 and CD8 depleted and non-depleted mice treated with the triple combination. h Survival curve. ‘*’ denotes p < .05 (Student’s t test for tumor growth and Mantel-Cox for survival). Values are expressed as mean ± SEM. n = 5 per group. Splenocytes from mice treated with the triplet combination that had cleared their tumors were incubated overnight with i CD28 alone (negative control), j CD28 + A20 cells, k CD28 + 4T1 breast cancer cells—an irrelevant syngeneic cancer cell line (negative control), or l CD28 + CD3 (positive control). IFN-γ production by CD8CD44 T cells was evaluated by flow cytometry. Numbers in upper right corner represent percentage of positive cells. ‘*’ denotes p < .05 (Student’s t test), n = 3 per group
To assess for a specific T-cell response, splenocytes from fully treated mice (triple therapy 10 μg × 4 doses) were incubated overnight with target A20 cells. This induced significant IFN-γ production by CD8 CD44hi cells (6.43% of cells, Fig. 3j) compared to incubation with the irrelevant cell line 4T1 (1.4%, Fig. 3k) or no cells (0.8%, Fig. 3i, p < .05 Student’s t test), and was similar to positive controls (5.68%, Fig. 3l). These results indicate a specific memory T-cell response to target A20 cells induced by the IT triple combination.
Tumors were collected from 6 mice that had been treated twice with the triple combination (n = 2), CD137-OX40 doublet (n = 2) and OX40-CTLA4 doublet (n = 2) (10 μg) as well as three untreated mice. Flow cytometry analysis showed that both CD4 and CD8 T-cells were increased relative to total tumor cells in treated mice, in both injected and uninjected tumors (p < .05, Student’s t test; Supplementary Figure 2a). The ratio of CD4/CD8 was normalized or reversed (relative increase in CD8 T-cells) in the injected tumor of the triple combination group, and OX40-CD137, but not OX40-CTLA4. Tregs (CD4/CD25/FOXP3+) were present in tumors of untreated and treated mice, and did not appear significantly different between groups (Supplementary Figure 2b). These results support the findings of CD8 T-cell-mediated anti-tumor effects, and do not suggest significant Treg depletion.
Intratumoral triple combination therapy is more effective than systemic administration
Low-dose tumor-directed immunotherapy should, by definition of in situ vaccination, be more specific and effective than systemic delivery. Thus, we compared anti-tumor efficacy of the intratumoral delivery of low-dose (10 μg each) triple combination therapy to two common systemic routes—subcutaneous (SC) to the abdominal wall, and intraperitoneal (IP) (Fig. 4a). The IT route was significantly more efficacious than IP in terms of tumor growth for both injected (left) and non-injected (right) tumors (Fig. 4b, c, e, f) (p < .05, Student’s t test), demonstrating superior local and systemic anti-tumor efficacy, and improved survival (p < .05, Mantel-Cox; Fig. 4h). Comparing IT and SC routes, tumor control was significantly better for the IT injected (left) tumor (p < .05, student’s t test; Fig. 4b, d), however, not statistically better for the right tumor (Fig. 4e, g) or survival (Fig. 4h). We hypothesized that antibodies delivered by the SC route may act on the tumor draining LN, resulting in an in situ vaccine effect. This was further explored in the next experiment (Fig. 5). Overall, these results support the concept of the in situ vaccine/tumor-directed immunotherapy approach.
Fig. 4.
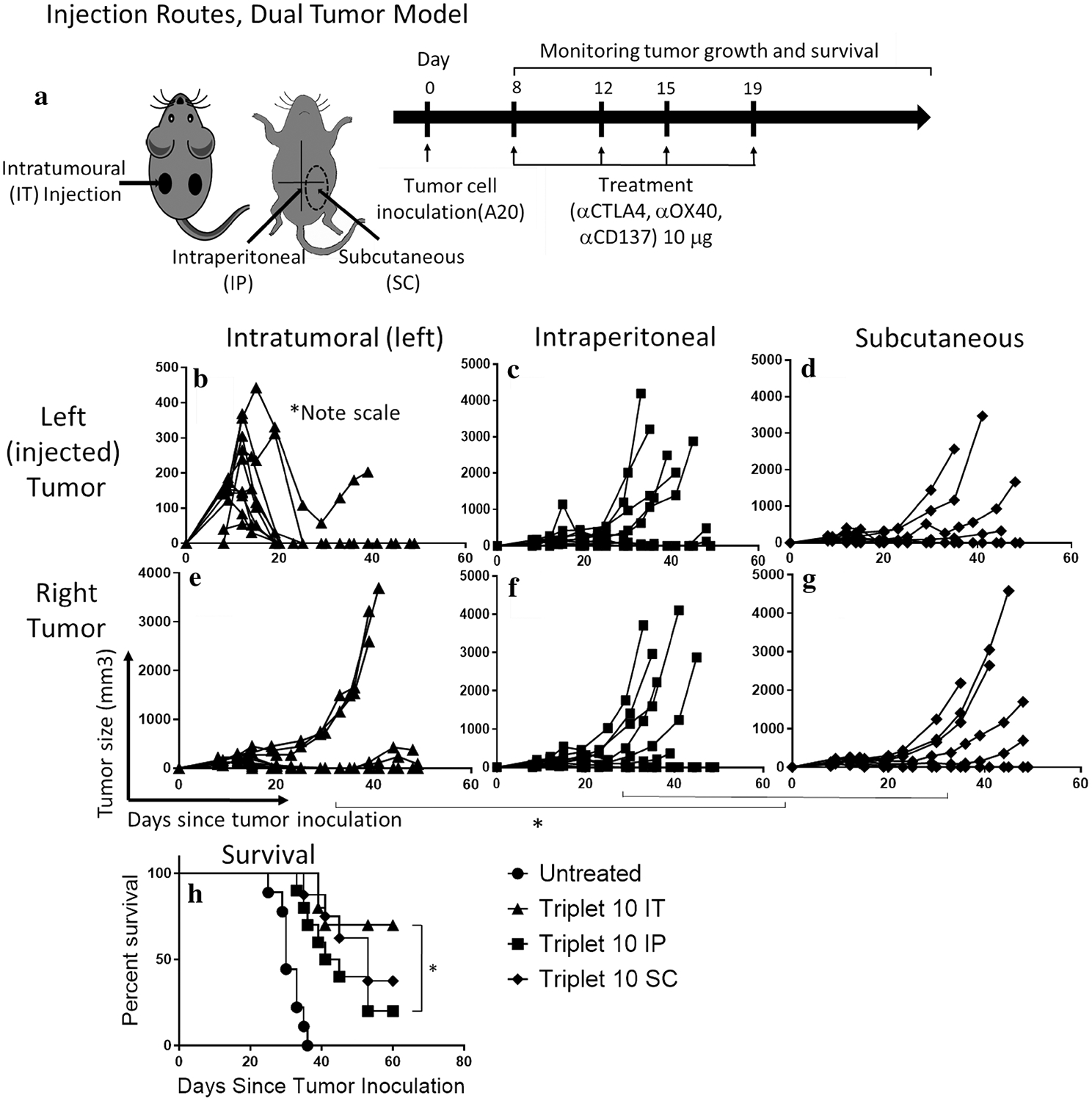
Intratumoral (IT) route of administration is more effective than systemic administration by subcutaneous or intraperitoneal routes at low doses. a Mice were treated with four biweekly doses of the triple combination αCD137, αOX40, and αCTLA4 at a dose of 10 μg each delivered intratumorally (IT) to the left tumor in a dual tumor A20 model, intraperitoneal (IP), or subcutaneous (SC) on the abdominal wall. The dotted circle indicates the general location of SC injections. Tumor growth measured by caliper of the left (injected) side (b–d) or the right tumor (e–g) in IT, IP, or SC injected mice with the triple combination. (h) Survival curve. ‘*’ denotes p < .05, Mantel-Cox test, n = 10 per group
Fig. 5.
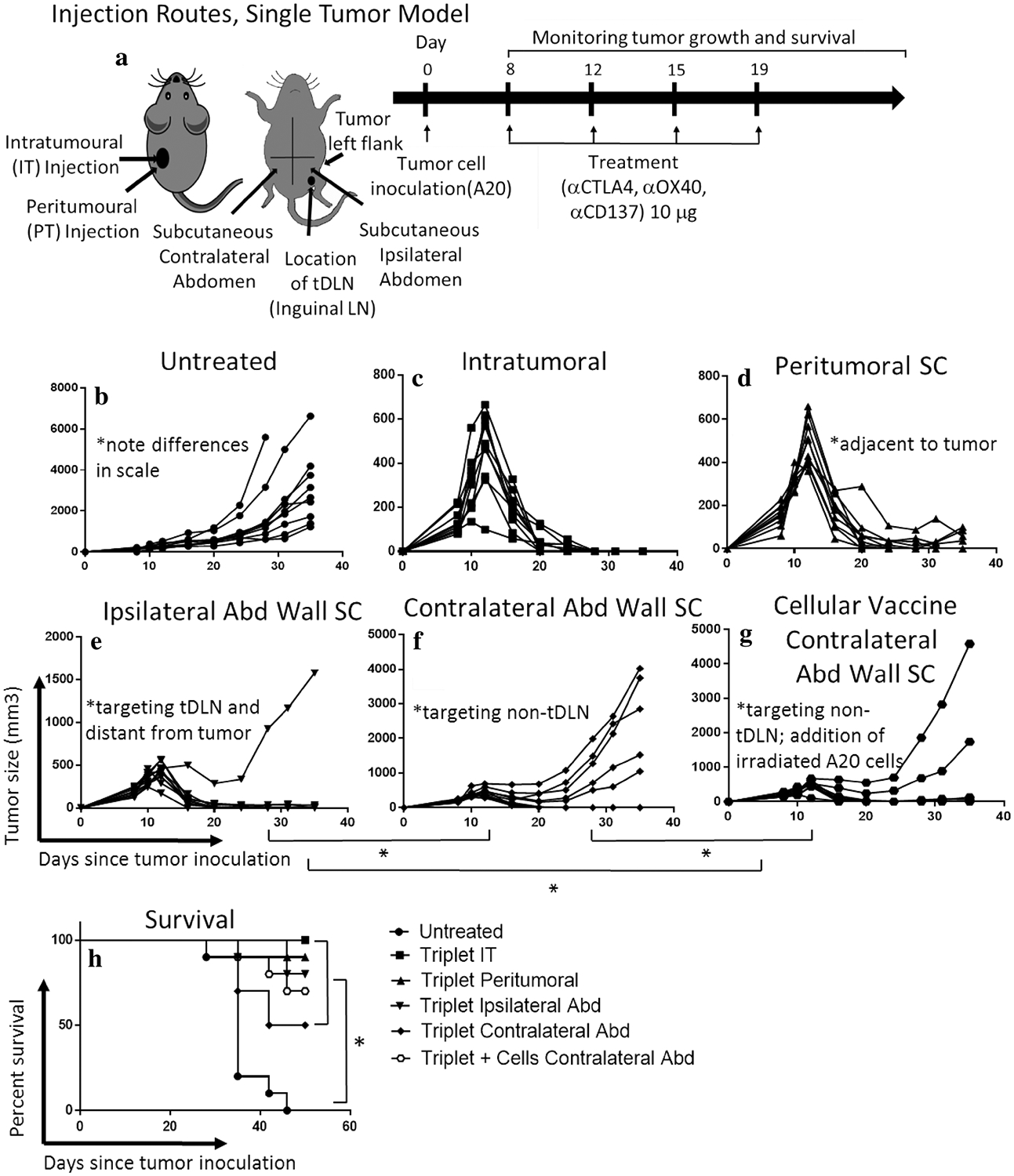
Targeting the tumor draining lymph node (tDLN) produces an abscopal effect; tumor antigens (cellular vaccine) in the absence of tumor/tDLN microenvironment are not sufficient to generate a full anti-tumor response. a Using a single tumor A20 model, mice were treated with four biweekly doses of the triple combination αCD137, αOX40, and αCTLA4 at a dose of 10 μg each delivered IT, or subcutaneous (SC) peri-tumoral, ipsilateral abdominal wall (targeting the tDNL) or contralateral abdominal wall (targeting the non-tDLN). A cellular vaccine (irradiated A20 cells plus the triple combination 10 μg) was delivered on the contralateral wall (SC) × four biweekly doses. Tumor growth measured by caliper (b–g) and survival curve (h). ‘*’ denotes p < .05 Student’s t test (tumor growth) or Mantel-Cox test (survival). n = 10/group
The tumor draining lymph node is a potential target for in situ vaccination
Next, we assessed whether targeting the tDLN would produce an abscopal effect. A single tumor (left flank) A20 model was used in which the left inguinal LN was the tDLN, and the right inguinal lymph node was not associated with tumor (non-tDLN). IT injection of the triple combination (10 μg) was compared to subcutaneous (SC) injection at several sites relative to the tumor and tDLN (Fig. 5a). Comparing average tumor size for the IT route (Fig. 5c) at day 35, both SC peri-tumoral (Fig. 5d) and injection proximal to the tDLN (ipsilateral abdominal wall, Fig. 5e) had pro-found anti-tumor effect not significantly different than IT. Interestingly, injection proximal to a non-tDLN (contralateral abdominal wall, Fig. 5f) resulted in significantly inferior anti-tumor efficacy compared to IT or targeting the tDLN (p < .05, student t test). At day 35, when all mice were alive for comparison, 100% of the IT group attained a complete response, compared to 80% for PT, and 90% for ipsilateral abdominal wall (tDLN). By contrast, targeting the non-tDLN (right inguinal) produced only a 50% complete response rate. This demonstrates that the tDLN is potentially an important target for in situ vaccination, and efficacy does not seem to differ significantly from directly targeting the tumor.
To investigate if the tumor microenvironment was important for the in situ vaccine effect, or if the tumor basically served as a source of tumor antigens, we assessed a cellular vaccine consisting of irradiated A20 cells and the triple combination (10 μg) administered SC in proximity to the non-tDLN (Fig. 5g). The cellular vaccine resulted in 60% complete response at 35 days. Average tumor size was smaller than the triple combination alone targeting the non-tDLN and larger than the IT/tDLN route (p < .05, student t test). This suggests that cells in the tumor/tDLN microenvironment are critical for the full effect of the treatment. Survival differences between treatment groups did not reach statistical significance (Fig. 5h).
Biodistribution studies confirm that antibodies reach the intended target
To confirm that antibodies actually concentrated in the intended target tissue, biodistribution studies were performed in a dual tumor A20 model post a single triple combination (10 μg) injection, via one of the established routes (Fig. 6a). Mice were sacrificed 6 h post-injection with additional time points at 3 and 24 h (Supplementary Figure 3), and tissues collected for analysis. The IT route led to the highest concentration of antibodies in the injected tumor, as expected, with lower concentrations in the tDLN and none detected in the non-injected tumor (p < .05, Student’s t test; Fig. 6b). IT also resulted in the longest persistence in the target tissue (Supplementary Figure 3b, p < .05). Peritumoral injection resulted in accumulation in both the tumor and tDLN (p < .05; Fig. 6c). With injection SC proximal to the tDLN, as predicted antibodies concentrated in the tDLN but not in the tumor (p < .05, Fig. 6d, e). Intraperitoneal injection produced the highest peak serum levels (at 3 h; p < .05; Supplementary Figure 3g), no detected accumulation in tumor, and low-level symmetric accumulation in lymph nodes (Fig. 6f, Supplementary Figure 3f). Serum levels were lowest for IT at 3, 6 h (IT versus peri-tumoral did not reach statistical significance at 6 h) and overall (p < .05; Fig. 6g, Supplementary Figure 3g). By 24 h, serum levels were similar for all routes (although modestly less for IT; Supplementary Figure 3g). While each route resulted in concentration of antibodies in the target tissue, when quantified, the bulk of antibodies was found in serum (Supplementary Table 2). Thus, the biodistribution studies demonstrate that antibodies accumulate in the intended target tissues, further demonstrating the importance of achieving high concentrations of therapeutic immune modulating antibodies in or near the tumor.
Discussion
In this study, we demonstrate that the novel immunotherapy combination αCD137, αOX40, and αCTLA4 has potent, although differential anti-tumor efficacy in two mouse tumor models. Furthermore, we show that this combination is a powerful in situ vaccine—by injecting directly into the tumor which we can use low doses to produce an abscopal effect with regression and elimination of a distant tumor. Moreover, our results suggest that injection in proximity to a draining lymph node can also produce an in situ vaccine effect. Our findings demonstrate the potential for this immunotherapy combination delivered into the tumor or to the tumor draining lymph node to be a highly effective approach for the treatment of cancer. The intratumoral approach to immunotherapy is an area of active clinical exploration (e.g. NCT02254772 and NCT02857569).
Systemic immunotherapy is effective, but limited by toxicity in the form of immune-related adverse events [5, 7, 9]. Our results are consistent with recent studies that suggest that in situ vaccination or tumor-directed immunotherapy can be effective in inducing tumor regression, but also reducing systemic exposure and toxicity [27–29]. A previous study has shown that a triple combination of intratumorally delivered CpG, αCTLA4, and αOX40 produced an abscopal effect with minimal toxicity [28]. However, direct intratumoral injection was an absolute requirement. This contrasts to the triple combination used in our study, where injection in proximity to the tDLN was also effective. Furthermore, the quadruple combination αCD137/PD-1/CTLA4/CD19 demonstrated local and abscopal effects, but with a relatively high dose of 250 μg each and associated toxicity [25].
In our study, we showed that intratumoral administration of the triple combination, αCD137, αOX40, and αCTLA4 10 μg each, was more effective than systemic administration, supporting the in situ vaccine concept. The ability to use such low doses, compared to typical doses of 100–400 μg for these agents [22–24], is key to reducing toxicity.
Hematologic toxicity in treated mice (4 doses of triple combination) showed mild-to-moderate neutropenia and eosinophilia (at 30 μg). No infectious complications were observed during the course of the project. Eosinophilia may reflect non-specific hypersensitivity to the foreign protein antibodies, or alternatively a target-mediated effect—potentially toxic or therapeutic [30, 31]. There were two sudden deaths at the highest dose of six 30 μg injections after the fifth and sixth doses. The timing and acuity were in keeping with an anaphylactic reaction, possibly secondary to repeated injection of foreign (rat/hamster) protein. However, we cannot rule out a target-mediated toxic effect. The treatment-related deaths occurred in 2 of a total of 137 mice treated with the triple combination during the project, after a total dose of 150, or 180 μg of each antibody, compared to our optimal dosing of four doses of 10 μg (40 μg total) each. There were no overt signs of treatment-related toxicity in any of the other mice. Thus, toxicity was observed at the highest dose levels explored in the project, but at intermediate and the lowest optimal dose, the triple combination appeared safe with mild hematological toxicity.
CD8 T-cells were required for the anti-tumor effect, and we have demonstrated a specific CD8 CD44hi response to tumor cells in our ex vivo studies. We noted a preferential increase in CD8 cells in the triple therapy injected tumors, in keeping with a CD8-mediated anti-tumor mechanism. Marabelle et al. [28] showed Treg depletion in tumors injected with CpG, αOX40, and αCTLA4; however, our results do not clearly support Treg depletion as a tumor-killing mechanism with the triple combination. Cytotoxic CD8 cells are known to be terminal effector cells responsible for tumor killing [28, 32–34], and our results are in line with the previous studies on monotherapy using antibodies targeting CD137, OX40, and CTLA-4 [17, 35, 36]. These receptors are expressed on CD4 and CD8 T-cells, as well as several other immune cell subtypes [37, 38], and we recognize that other immune cells may be important. The apparent lack of effect of CD4 depletion alone on anti-tumor efficacy of the triple combination does not rule out a role of CD4 cells in CD8 T-cell activation, for example, prior to in vivo depletion [34]. Dual CD4/CD8 depletion led to very aggressive tumor growth despite triple combination treatment, suggesting that CD4 cells do contribute to tumor control, but may be redundant in the presence of CD8 cells.
The triple combination induced bilateral tumor regression in both the A20 and the MC38 tumor models; however, it was more effective in the former than latter. There are a number of potential variables which could account for the difference in efficacy including growth kinetics and immunogenicity of the tumor cells, inherent differences in microenvironment, T-cell repertoire, and differences in immune response between the two mouse strains. Target-related variables such as differential expression of targets on cell subtypes and dynamic changes with time and treatment may also contribute [24, 39–41].
Our results suggest that delivery to the tDLN can be an effective therapy. Similar to the tumor microenvironment, the tDLN has been shown to harbor tumor-specific T-cells, and the tDLN microenvironment is immunosuppressive. [13, 42]. The option of targeting the tDLN increases the versatility of the in situ vaccine approach and may apply in the adjuvant setting where tumors have been treated by radiation, chemotherapy, or surgery.
The biodistribution data clearly show that the antibodies do accumulate in the intended sites, supporting the rationale for IT or tDLN delivery. IT administration resulted in lower maximal and overall serum concentration compared to IP and SC, persistence at the intended site, and a potent abscopal effect, suggesting this route may offer the best combination of safety and efficacy. However, ultimately, it is still only a small fraction that ends up in the tumor/draining lymph nodes (Supplementary Table 2), and most of the antibodies can be found in the serum. Despite this, there are clearly significant differences in the anti-tumor response, supporting tumor-directed immunotherapy/in situ vaccination approaches.
To assess the relative role of tumor antigens and the microenvironment, we circumvented the microenvironment using a cellular vaccine of irradiated tumor cells and our triple mAb combination, and injected remotely from the tumor/tDLN. The vaccine demonstrated greater efficacy than the triple combination alone, but less than IT/peri-tDLN injection, suggesting that elements of the tumor/tDLN microenvironment are, indeed, required for full effect of the triple therapy. These agents have previously been reported individually to have vaccine adjuvant activity [43–45].
Although mouse models of immunotherapy have been invaluable to clinical translation, ultimately, a human clinical trial will be required to confirm safety and efficacy of our triple combination administered as an in situ vaccine. Mouse models may not be an accurate representation of toxicity in humans. However, it is a reasonable assumption that lower doses and less systemic exposure of immune activating agents via the in situ vaccination approach will result in less human toxicity.
This study contributes to the growing body of evidence that combination tumor-directed immunotherapy is a powerful tool in the treatment of cancer. The novel combination of αCTLA4, αOX40, and αCD137 delivered intratumorally to one tumor in a dual tumor model has dramatic local and abscopal anti-tumor effects. Delivery to the tDLN appears similarly effective, potentially extending the in situ vaccine approach to patients lacking an accessible tumor injection site. With this approach, immune-related toxicities can be attenuated and combinations of three or more agents may be used with greater specificity, safety, and promise for lasting anti-tumor immune responses. Overall, tumor or tDLN-directed immunotherapy using the novel combination αCTLA4, αCD137, and αOX40, all agents in clinic or clinical trials, is promising for future clinical development via both these safe and highly efficacious routes of administration.
Supplementary Material
Acknowledgements
The authors would like to dedicate this study to the memory of Dr. Holbrook Kohrt, who conceptualized and inspired this work but sadly passed away before its completion. He was a great scientist, physician, and human being. He continues to inspire. Also thanks to Dr. Idit Sagiv-Barfi and Dr. Suparna Dutt for their advice and expertise during the course of the project.
Funding This project was funded by NIH grants R01 CA170378, U01 CA188383, and R01 CA184384 (D. W. Felsher) and Alligator Biosciences.
Abbreviations
- α
Anti
- ELISA
Enzyme-linked immunosorbent assay
- IFN-γ
Interferon gamma
- IP
Intraperitoneal
- IT
Intratumoral
- LN
Lymph node
- mAb
Monoclonal antibody
- μg
Micrograms
- PBS
Phosphate-buffered saline
- PT
Peritumoral
- SC
Subcutaneous
- tDLN
Tumor draining lymph node
- TNFR
Tumor necrosis factor receptor
Footnotes
Preliminary work on this project was published in abstract form, at the American Society of Hematology 57th Annual Meeting in 2015 (Orlando, Florida) and also in part at the 58th meeting in 2016 (San Diego, California) [1, 2].
Electronic supplementary material The online version of this article (doi:10.1007/s00262-017-2059-y) contains supplementary material, which is available to authorized users.
Conflict of interest D. Felsher and J. Hebb received research funding from Alligator Biosciences. P. Ellmark and A. Rosén are employees of Alligator Biosciences. The remaining authors have no conflicts of interest to declare.
References
- 1.Hebb J, Kohrt H (2015) Systemic antitumor effects of intratumoral administration of the novel immunotherapeutic combination anti-CTLA4, anti-CD137, and anti-OX40 in mouse models of lymphoma and solid tumor [abstract]. In: American Society of Hematology 57th Annual Meeting; 2015 Dec 5–8; Orlando, FLA. Washington (DC): ASH; 2015. Abstract nr 1552. Blood, vol 126, no 23 [Google Scholar]
- 2.Hebb J, Mosley A, Vences Catalan F, Ellmark P, Norlen P, Felsher D (2016) Intratumoral administration of the immunotherapeutic combination anti-ctla4, anti-cd137 and anti-ox40: comparison to systemic administration, peri-draining lymph node injection, and cellular vaccine in a mouse lymphoma model [abstract]. In: American Society of Hematology 58th Annual Meeting; 2016 Dec 3–6; San Diego, CA. Washington (DC): ASH; 2016. Abstract nr 4172. Blood, vol 128, no 22 [Google Scholar]
- 3.Couzin-Frankel J (2013) Breakthrough of the year 2013. Cancer immunotherapy. Science 342(6165):1432–1433 [DOI] [PubMed] [Google Scholar]
- 4.Postow MA, Callahan MK, Wolchok JD (2015) Immune checkpoint blockade in cancer therapy. J Clin Oncol 33(17):1974–1982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Melero I, Grimaldi AM, Perez-Gracia JL, Ascierto PA (2013) Clinical development of immunostimulatory monoclonal antibodies and opportunities for combination. Clin Cancer Res 19(5):997–1008 [DOI] [PubMed] [Google Scholar]
- 6.Linch SN, McNamara MJ, Redmond WL (2015) OX40 agonists and combination immunotherapy: putting the pedal to the metal. Front Oncol 5:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Michot JM, Bigenwald C, Champiat S, Collins M, Carbonnel F, Postel-Vinay S, Lambotte O (2016) Immune-related adverse events with immune checkpoint blockade: a comprehensive review. Eur J Cancer 54:139–148 [DOI] [PubMed] [Google Scholar]
- 8.Khalil DN, Smith EL, Brentjens RJ, Wolchok JD (2016) The future of cancer treatment: immunomodulation, CARs and combination immunotherapy. Nat Rev Clin Oncol 13(5):273–290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Postow MA (2015) Managing immune checkpoint-blocking antibody side effects. Am Soc Clin Oncol Educ Book 35:76–83 [DOI] [PubMed] [Google Scholar]
- 10.Ellmark P, Mangsbo SM, Furebring C, Norlen P, Totterman TH (2017) Tumor-directed immunotherapy can generate tumor-specific T cell responses through localized co-stimulation. Cancer Immunol Immunother 66(1):1–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pierce RH, Campbell JS, Pai SI, Brody JD, Kohrt HE (2015) In-situ tumor vaccination: bringing the fight to the tumor. Hum Vaccin Immunother 11(8):1901–1909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shu S, Cochran AJ, Huang RR, Morton DL, Maecker HT (2006) Immune responses in the draining lymph nodes against cancer: implications for immunotherapy. Cancer Metastasis Rev 25(2):233–242 [DOI] [PubMed] [Google Scholar]
- 13.Fransen MF, Arens R, Melief CJ (2013) Local targets for immune therapy to cancer: tumor draining lymph nodes and tumor microenvironment. Int J Cancer 132(9):1971–1976 [DOI] [PubMed] [Google Scholar]
- 14.Ye Q, Song DG, Poussin M, Yamamoto T, Best A, Li C, Powell DJ Jr (2014) CD137 accurately identifies and enriches for naturally occurring tumor-reactive T cells in tumor. Clin Cancer Res 20(1):44–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vetto JT, Lum S, Morris A, Sicotte M, Davis J, Lemon M, Weinberg A (1997) Presence of the T-cell activation marker OX-40 on tumor infiltrating lymphocytes and draining lymph node cells from patients with melanoma and head and neck cancers. Am J Surg 174(3):258–265 [DOI] [PubMed] [Google Scholar]
- 16.Makkouk A, Chester C, Kohrt HE (2016) Rationale for anti-CD137 cancer immunotherapy. Eur J Cancer 54:112–119 [DOI] [PubMed] [Google Scholar]
- 17.Bansal-Pakala P, Halteman BS, Cheng MH, Croft M (2004) Costimulation of CD8 T cell responses by OX40. J Immunol 172(8):4821–4825 [DOI] [PubMed] [Google Scholar]
- 18.Segal NH, Logan TF, Hodi FS, McDermott D, Melero I, Hamid O, Levy R (2017) Results from an integrated safety analysis of urelumab, an agonist anti-CD137 monoclonal antibody. Clin Cancer Res 23(8):1929–1936 [DOI] [PubMed] [Google Scholar]
- 19.Yun S, Vincelette ND, Green MR, Wahner Hendrickson AE, Abraham I (2016) Targeting immune checkpoints in unresectable metastatic cutaneous melanoma: a systematic review and meta-analysis of anti-CTLA-4 and anti-PD-1 agents trials. Cancer Med 5(7):1481–1491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sharma P, Allison JP (2015) The future of immune checkpoint therapy. Science 348(6230):56–61 [DOI] [PubMed] [Google Scholar]
- 21.Tirapu I, Mazzolini G, Rodriguez-Calvillo M, Arina A, Palencia B, Gabari I, Melero I (2002) Effective tumor immunotherapy: start the engine, release the brakes, step on the gas pedal,…and get ready to face autoimmunity. Arch Immunol Ther Exp (Warsz) 50(1):13–18 [PubMed] [Google Scholar]
- 22.Curran MA, Kim M, Montalvo W, Al-Shamkhani A, Allison JP (2011) Combination CTLA-4 blockade and 4–1BB activation enhances tumor rejection by increasing T-cell infiltration, proliferation, and cytokine production. PLoS One 6(4):e19499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leach DR, Krummel MF, Allison JP (1996) Enhancement of antitumor immunity by CTLA-4 blockade. Science 271(5256):1734–1736 [DOI] [PubMed] [Google Scholar]
- 24.Kjaergaard J, Tanaka J, Kim JA, Rothchild K, Weinberg A, Shu S (2000) Therapeutic efficacy of OX-40 receptor antibody depends on tumor immunogenicity and anatomic site of tumor growth. Cancer Res 60(19):5514–5521 [PubMed] [Google Scholar]
- 25.Dai M, Yip YY, Hellstrom I, Hellstrom KE (2015) Curing mice with large tumors by locally delivering combinations of immunomodulatory antibodies. Clin Cancer Res 21(5):1127–1138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schurig JE, Florczyk AP, Bradner WT (1986) The mouse as a model for predicting the myelosuppressive effects of anticancer drugs. Cancer Chemother Pharmacol 16(3):243–246 [DOI] [PubMed] [Google Scholar]
- 27.Sandin LC, Orlova A, Gustafsson E, Ellmark P, Tolmachev V, Totterman TH, Mangsbo SM (2014) Locally delivered CD40 agonist antibody accumulates in secondary lymphoid organs and eradicates experimental disseminated bladder cancer. Cancer Immunol Res 2(1):80–90 [DOI] [PubMed] [Google Scholar]
- 28.Marabelle A, Kohrt H, Sagiv-Barfi I, Ajami B, Axtell RC, Zhou G, Levy R (2013) Depleting tumor-specific Tregs at a single site eradicates disseminated tumors. J Clin Investig 123(6):2447–2463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fransen MF, Sluijter M, Morreau H, Arens R, Melief CJ (2011) Local activation of CD8 T cells and systemic tumor eradication without toxicity via slow release and local delivery of agonistic CD40 antibody. Clin Cancer Res 17(8):2270–2280 [DOI] [PubMed] [Google Scholar]
- 30.Johncilla M, Misdraji J, Pratt DS, Agoston AT, Lauwers GY, Srivastava A, Doyle LA (2015) Ipilimumab-associated hepatitis: clinicopathologic characterization in a series of 11 cases. Am J Surg Pathol 39(8):1075–1084 [DOI] [PubMed] [Google Scholar]
- 31.Carretero R, Sektioglu IM, Garbi N, Salgado OC, Beckhove P, Hammerling GJ (2015) Eosinophils orchestrate cancer rejection by normalizing tumor vessels and enhancing infiltration of CD8(+) T cells. Nat Immunol 16(6):609–617 [DOI] [PubMed] [Google Scholar]
- 32.Rodriguez-Ruiz ME, Rodriguez I, Garasa S, Barbes B, Solorzano JL, Perez-Gracia JL, Melero I (2016) Abscopal effects of radiotherapy are enhanced by combined immunostimulatory mabs and are dependent on CD8 T cells and crosspriming. Cancer Res 76(20):5994–6005 [DOI] [PubMed] [Google Scholar]
- 33.Moran AE, Polesso F, Weinberg AD (2016) Immunotherapy expands and maintains the function of high-affinity tumor-infiltrating CD8 T cells in situ. J Immunol 197(6):2509–2521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Church SE, Jensen SM, Antony PA, Restifo NP, Fox BA (2014) Tumor-specific CD4+ T cells maintain effector and memory tumor-specific CD8+ T cells. Eur J Immunol 44(1):69–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.van Elsas A, Hurwitz AA, Allison JP (1999) Combination immunotherapy of B16 melanoma using anti-cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) and granulocyte/macrophage colony-stimulating factor (GM-CSF)-producing vaccines induces rejection of subcutaneous and metastatic tumors accompanied by autoimmune depigmentation. J Exp Med 190(3):355–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Miller RE, Jones J, Le T, Whitmore J, Boiani N, Gliniak B, Lynch DH (2002) 4–1BB-specific monoclonal antibody promotes the generation of tumor-specific immune responses by direct activation of CD8 T cells in a CD40-dependent manner. J Immunol 169(4):1792–1800 [DOI] [PubMed] [Google Scholar]
- 37.Wang XB, Fan ZZ, Anton D, Vollenhoven AV, Ni ZH, Chen XF, Lefvert AK (2011) CTLA4 is expressed on mature dendritic cells derived from human monocytes and influences their maturation and antigen presentation. BMC Immunol 12:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Moran AE, Kovacsovics-Bankowski M, Weinberg AD (2013) The TNFRs OX40, 4–1BB, and CD40 as targets for cancer immunotherapy. Curr Opin Immunol 25(2):230–237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wakeham J, Wang J, Xing Z (2000) Genetically determined disparate innate and adaptive cell-mediated immune responses to pulmonary Mycobacterium bovis BCG infection in C57BL/6 and BALB/c mice. Infect Immun 68(12):6946–6953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kodumudi KN, Siegel J, Weber AM, Scott E, Sarnaik AA, Pilon-Thomas S (2016) Immune checkpoint blockade to improve tumor infiltrating lymphocytes for adoptive cell therapy. PLoS One 11(4):e0153053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lafreniere R, Borkenhagen K, Bryant LD (1990) MC-38 adenocarcinoma tumor infiltrating lymphocytes: correlation of cytotoxicity with time of tumor harvest after tumor inoculation. J Surg Oncol 43(1):8–12 [DOI] [PubMed] [Google Scholar]
- 42.Zhang M, Graor H, Visioni A, Strohl M, Yan L, Caja K, Kim JA (2015) T cells derived from human melanoma draining lymph nodes mediate melanoma-specific antitumor responses in vitro and in vivo in human melanoma xenograft model. J Immunother 38(6):229–238 [DOI] [PubMed] [Google Scholar]
- 43.Shirwan H, Sharma RK, Srivastava AK, Yolcu ES (2013) Costimulatory tumor necrosis factor ligands as adjuvants for the development of subunit-based anticancer vaccines. Oncoimmunology 2(4):e23440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schreiber TH, Wolf D, Bodero M, Gonzalez L, Podack ER (2012) T cell costimulation by TNFR superfamily (TNFRSF)4 and TNFRSF25 in the context of vaccination. J Immunol 189(7):3311–3318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Duraiswamy J, Freeman GJ, Coukos G (2014) Dual blockade of PD-1 and CTLA-4 combined with tumor vaccine effectively restores T-cell rejection function in tumors–response. Cancer Res 74(2):633–634 (discussion 635) [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.