

Abstract
Transcriptional regulation of the HOXA genes is thought to involve CTCF-mediated chromatin loops and the opposing actions of the COMPASS and Polycomb epigenetic complexes. We investigated the role of these mechanisms at the HOXA cluster in AML cells with the common NPM1c mutation, which express both HOXA and HOXB genes. CTCF binding at the HOXA locus is conserved across primary AML samples, regardless of HOXA gene expression, and defines a continuous chromatin domain marked by COMPASS-associated histone H3 trimethylation in NPM1-mutant primary AML samples. Profiling of the three-dimensional chromatin architecture in primary AML samples with the NPM1c mutation identified chromatin loops between the HOXA cluster and loci in the SNX10 and SKAP2 genes, and an intergenic region located 1.4Mbp upstream of the HOXA locus. Deletion of CTCF binding sites in the NPM1-mutant OCI-AML3 AML cell line reduced multiple long-range interactions, but resulted in CTCF-independent loops with sequences in SKAP2 that were marked by enhancer-associated histone modifications in primary AML samples. HOXA gene expression was maintained in CTCF binding site mutants, indicating that transcriptional activity at the HOXA locus in NPM1-mutant AML cells may be sustained through persistent interactions with SKAP2 enhancers, or by intrinsic factors within the HOXA gene cluster.
Introduction
The HOX genes encode developmentally regulated transcription factors that are highly expressed in acute myeloid leukemia (AML) and are important drivers of malignant self-renewal in this disease. Previous studies have shown that expression of HOX family members in AML is nearly always restricted to specific genes in the HOXA and/or HOXB clusters (HOXC and HOXD genes are rarely expressed), and that expression patterns correlate with recurrent AML mutations (1). HOX expression is most closely associated with AMLs with MLL rearrangements, which exclusively express HOXA genes, and AMLs with the recurrent NPM1c mutation, which nearly always express both HOXA and HOXB genes (1). The high prevalence of NPM1 mutations make the combined HOXA/HOXB expression pattern the most common HOX phenotype in AML patients. However, the regulatory mechanisms that drive this expression pattern are poorly understood.
Studies of HOX gene regulation in model organisms have established that colinear expression of each HOX cluster is mediated by COMPASS/Trithorax and Polycomb group proteins, which promote gene activation and repression and perform methylation of histone H3 at lysine 4 (H3K4me3) and 27 (H3K27me3), respectively (2,3). These regulatory pathways are also involved in HOX gene regulation in AML cells, and are best understood for the HOXA cluster in AMLs with MLL rearrangements. MLL1 (KMT2A) is a component of the COMPASS complex, and MLL fusion proteins bind to the HOXA locus in AML cells and recruit the non-COMPASS histone H3 methyltransferase DOT1L, which is required for HOXA activation and AML development in MLL-rearranged leukemia models (4–7). Regulatory DNA elements that control three-dimensional chromatin architecture also play a role in HOXA gene regulation in AML cells. Specifically, the HOXA and HOXB clusters contain multiple binding sites for the chromatin organizing factor CTCF, and chromatin conformation experiments suggest these events mediate local chromatin loops in AML cells with MLL rearrangements (8). In addition, heterozygous deletion of a single CTCF binding site in the HOXA cluster in MLL-rearranged AML cells resulted in altered chromatin structure and reduced HOXA gene expression (9). These studies suggest that MLL fusion proteins directly activate the HOXA locus in ways that require specific CTCF binding events or their associated chromatin structures.
While these mechanistic insights have provided valuable information about HOXA regulation in MLL-rearranged AML, this molecular subtype accounts for <5% of all AML patients and only 25% of AMLs that express HOXA genes (1). Although AMLs with the NPM1 mutations nearly always express HOXA genes, it is unclear whether HOXA expression in these cells shares similar regulatory factors and chromatin structures that appear to be critical for HOXA expression in MLL-rearranged AML cells. In this study, we investigated histone modifications and chromatin interactions at the HOXA locus in NPM1-mutated AML samples vs. other AML subtypes, and used a NPM1-mutant AML cell line model to determine whether CTCF at the HOXA locus is required to maintain HOXA expression and chromatin structure.
Materials and Methods
Primary samples and cell lines.
Primary AML samples and normal hematopoietic cells were obtained from diagnostic AML and normal bone marrow aspirates, respectively, following informed consent using protocol (201011766) approved by the Human Research Protection Office at Washington University as described previously ((10,11); Table S1). All experiments with primary AML samples used bulk cells after estimating the leukemic purity (38). OCI-AML3 cells obtained from the DSMZ cell repository were cultured at 0.5–1 × 106 cell/mL in MEM-alpha with 20% FBS and 1% penicillin-streptomycin. NPM1c was verified in the OCI-AML3 line by targeted sequencing and in RNA-seq data from wild type and mutant clones. Kasumi-1 (received as a gift from T. Ley), IMS-M2 (received as a gift from L. Brunetti), and MOLM13 (received as a gift from J. Dipersio) cell lines were cultured in RPMI-1640 with 1% penicillin-streptomycin and FBS (20% for Kasumi-1 and MOLM13, 10% for IMS-M2).
Statistical analysis.
Hypothesis testing was performed using the indicated parametric statistics after verification of normality. Sample sizes for genomic studies using read count data were selected to provide >80% power to detect a fold-change of at least 2.
ChIP-seq.
ChIP-seq was performed using ChIPmentation (12) with the following antibodies: CTCF (2899S), H3K27me3 (9733S), and H3K27ac (8173S) from Cell Signaling Technology and H3K4me3 (ab1012) from Abcam. Sequencing was performed on a NovaSeq 6000 (Illumina, San Diego, CA) to obtain ~50 million 150 bp paired-end reads. Data were analyzed via adapter trimming with trimgalore and alignment to GRCh38 using bwa mem (13). Normalized coverage for visualization and analysis used the deeptools ‘bamCoverage’ tool (14), and peaks were called with macs2 (15) for CTCF and epic2 (16) for histone marks. Statistical comparisons with DESeq2 (17) used raw fragment counts at peak summits and visualizations were prepared with Gviz (18).
Targeted deletion of CTCF binding sites.
Deletions were generated using CRISPR/Cas9 with guide RNAs from the UCSC genome browser (19,20) that overlapped CTCF ChIP-seq peaks (Table S2). Mutagenesis used either inducible Cas9-expressing OCI-AML3 cells (Lenti-iCas9-neo vector; Addgene 85400) with lentiviral sgRNA expression (Addgene 70683), or transfection with Cas9 protein and tracrRNA/crRNA hybrids (Alt-R system, IDT, Coralville, IA). For the latter, RNAs were complexed with Cas9 protein using the manufacturer’s protocol with 1 million cells and 28 μM of Cas9/RNA for either transfection (CRISPRMAX; Thermofisher Scientific, Waltham, MA) or nucleofection (SG Amaxa Cell Line 4D-Nucleofector Kit, Lonza, Basel, Switzerland). Mutation efficiency was assessed in bulk cultures via DNA extraction, PCR with tailed primers (Table S2), and sequencing to obtain 2×250 bp reads on an Illumina MiSeq instrument and analysis of minimap2-based alignments (21). Sorting into 96 well plates via FACS was used for expansion of individual clones. Cells from single wells were screened via direct lysis by proteinase K (P8107S; NEB) in 20 μl of single cell lysis buffer (10 mM Tris-HCl pH 7.6, 50 mM NaCl, 6.25 mM MgCl2, 0.045% NP40, 0.45% Tween-20), PCR, and gel electrophoresis; clones with evidence for deletions were sequenced, and clones with deletions were expanded for analysis.
RNA analysis.
RNA extractions were performed on ~1 million cells with the Quick-RNA MicroPrep Kit (Zymo Research, Irvine, CA). One hundred nanograms of RNA was used for cDNA synthesis (Applied Biosystems, Foster City, CA) and qPCR was done in duplicate in a StepOnePlus PCR System (Applied Biosystems) for HOXA9 exons 1–2 with a GUSB control (IDT). RNA-seq libraries were generated using 300 ng of RNA with the KapaHyper stranded RNA library kit for Illumina (Roche) following the manufacturer’s instructions and then sequenced on a NovaSeq 6000 to obtain ~50 million 2×150 bp reads. Reads were trimmed with trimgalore, aligned with STAR (22), and transcript-level expression values in TPM were obtained with stringtie (23).
In situ Hi-C.
Hi-C libraries were prepared as described (24) using 4–5 million cells. Libraries were first assessed by sequencing ~1 million reads on a MiSeq instrument (using metrics recommended by Rao et al., (24)); passing libraries were sequenced to obtain 2×150 bp reads on a NovaSeq 6000. Hi-C data were analyzed on GRCh38 with juicer (25). All analyses used contact matrices (mapping quality >30), and chromatin loops and contact domains identified using HICCUPS and arrowhead, respectively (25). Loops from wild type and mutant OCI-AML3 cells were merged using bedtools ‘pairToPair’ function (26) with 5,000 bp overlap. Pairwise and joint comparisons of chromatin loops were performed with hicCompare (27) and multiHicCompare (28), respectively, using raw contact counts from chromosome 7 obtained from the juicer hic file at 25 kbp resolution. Statistical comparisons used chromosome-wide FDR corrections for multiple hypothesis testing. Visualizations used the HiTC (29), GenomicInteractions, and Gviz R packages.
Data availability.
All processed sequence data from this study (e.g., bigwig files for ChIP-seq, TPM values for RNA-seq, and Hi-C contact matrices) are available for public download at the following site: https://wustl.box.com/v/ghasemiCTCFHOXA. Raw sequence files for cell lines are available upon request.
Results
CTCF defines dynamic chromatin domains at the HOXA locus in primary AML cells
We previously showed that specific regions in the HOXA cluster have accessible chromatin in primary AML samples that coincide with CTCF binding sites in other human cell types (1). To confirm the presence of CTCF at these loci in primary AML cells, we performed CTCF ChIP-seq on primary AML patients with the NPM1c insertion mutation in NPM1, t(9;11) and t(11;19) MLL rearrangements, and t(8;21) creating the RUNX1-RUNX1T1 gene fusion, which displayed the expected HOXA and HOXB, HOXA only, and no HOX expression patterns, respectively (Table S1, Figure S1A). This confirmed CTCF binding in chromatin-accessible regions between HOXA6 and HOXA7 (“CTCF Binding Site CBSA6/7”), and HOXA7 and HOXA9 (“CBSA7/9”), near the transcriptional start site of HOXA10 (“CBSA10”), and in the 5’ UTR of HOXA13 (“CBSA13”) (Figure 1A). All four CTCF peaks were present in all AML samples regardless of HOXA expression status, and quantification of the CTCF signal demonstrated similar occupancy across the AML types (Figures 1B–D).
Figure 1. CTCF is bound to chromatin accessible sites at the HOXA locus in primary AML samples.
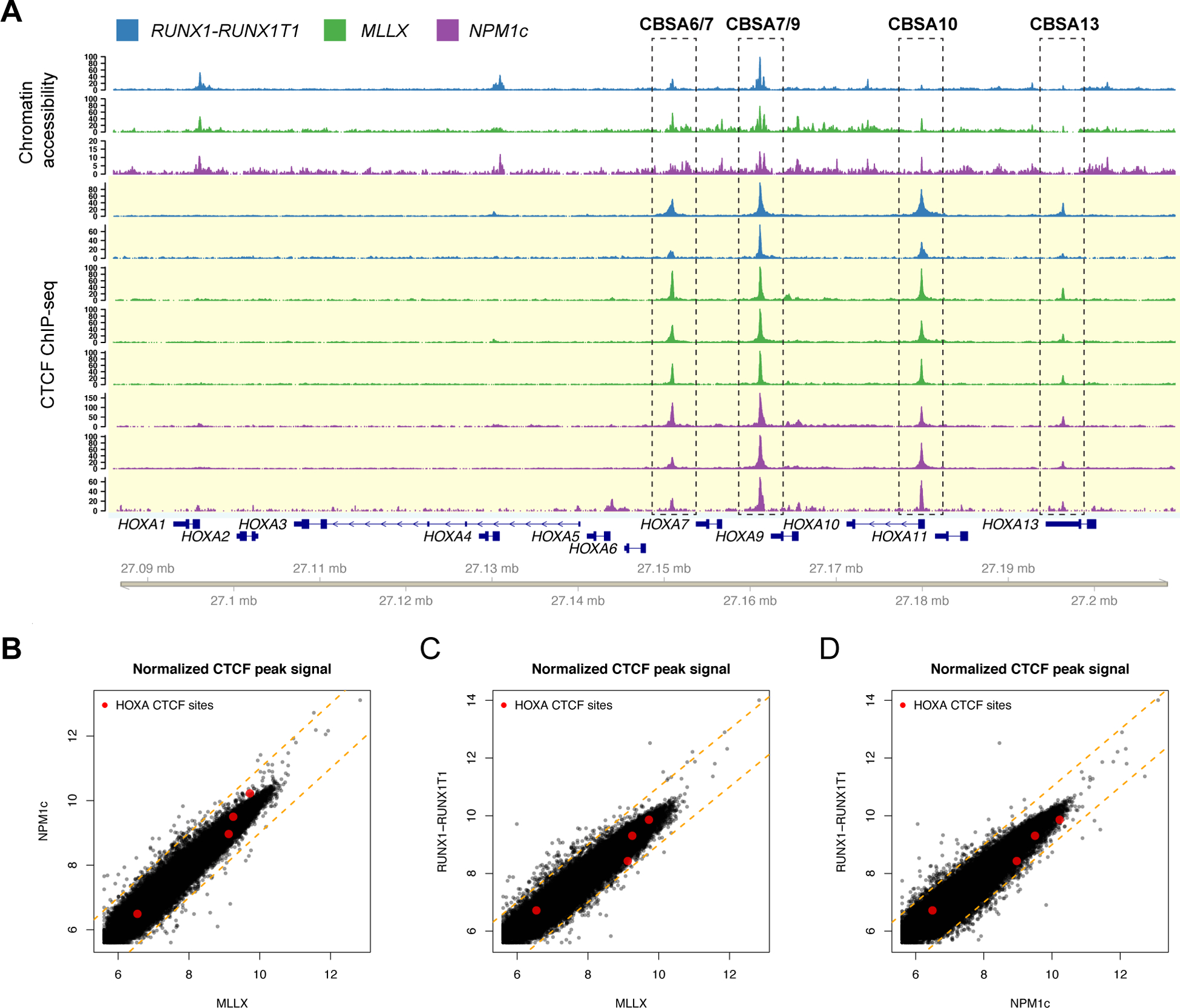
A. Chromatin accessibility by ATAC (top tracks highlighted in white; N=1 from each mutation category, from ref. 1) and ChIP-seq for CTCF (bottom tracks highlighted in yellow) from primary AML samples with either t(8;21) creating the RUNX1-RUNX1T1 gene fusion (blue; N=2), MLL rearrangements (green; N=3), or a normal karyotype and NPM1 mutation (purple; N=3). CTCF sites CBSA6/7, CBSA7/9, CBSA10, and CBSA13 are indicated by the dashed boxes. B-D. Scatter plots comparing the CTCF peak summit counts between each AML type in log2 normalized read counts (mean of 3 samples for NPM1c and MLL-rearranged samples, mean of 2 for RUNX1-RUNX1T1). Red points indicate ChIP-seq signal for the four CTCF binding sites highlighted in panel A, which are similar across AML samples. Dashed orange lines indicate a two-fold change between the samples.
We next determined whether these CTCF binding events defined chromatin domains in primary AML samples by performing ChIP-seq for H3K4me3 and H3K27me3 to measure active and repressed chromatin, respectively. This identified a region of active chromatin in the center of the HOXA cluster that overlapped CBSA6/7 and CBSA7/9, and was conserved in the MLL-rearranged and NPM1-mutant AML samples, and normal CD34+ cells (which also express HOXA and HOXB genes) (Figures 2A, S2A). The H3K4me3 signal was continuous across this interval, including non-promoter sequences, and was also marked with H3K27ac in primary AML samples (Figure S2B). Adjacent regions were marked with repressive H3K27me3 in all AML samples and in CD34+ cells, which correlated with the expression levels of the overlapping genes (Figures S1A, S2A). ChIP-seq using AML samples with the RUNX1-RUNX1T1 gene fusion and low HOXA expression showed that the active chromatin domain is dynamic, with little H3K4me3 signal and increased H3K27me3 in this AML type (Figure 2B). Similar repressive histone methylation patterns were observed in ChIP-seq data from FACS-purified normal promyelocytes and mature neutrophils (10) that also do not express HOXA genes (Figures 2B, S2A).
Figure 2. CTCF defines chromatin boundaries in AML samples and normal CD34+ cells with HOXA gene expression.
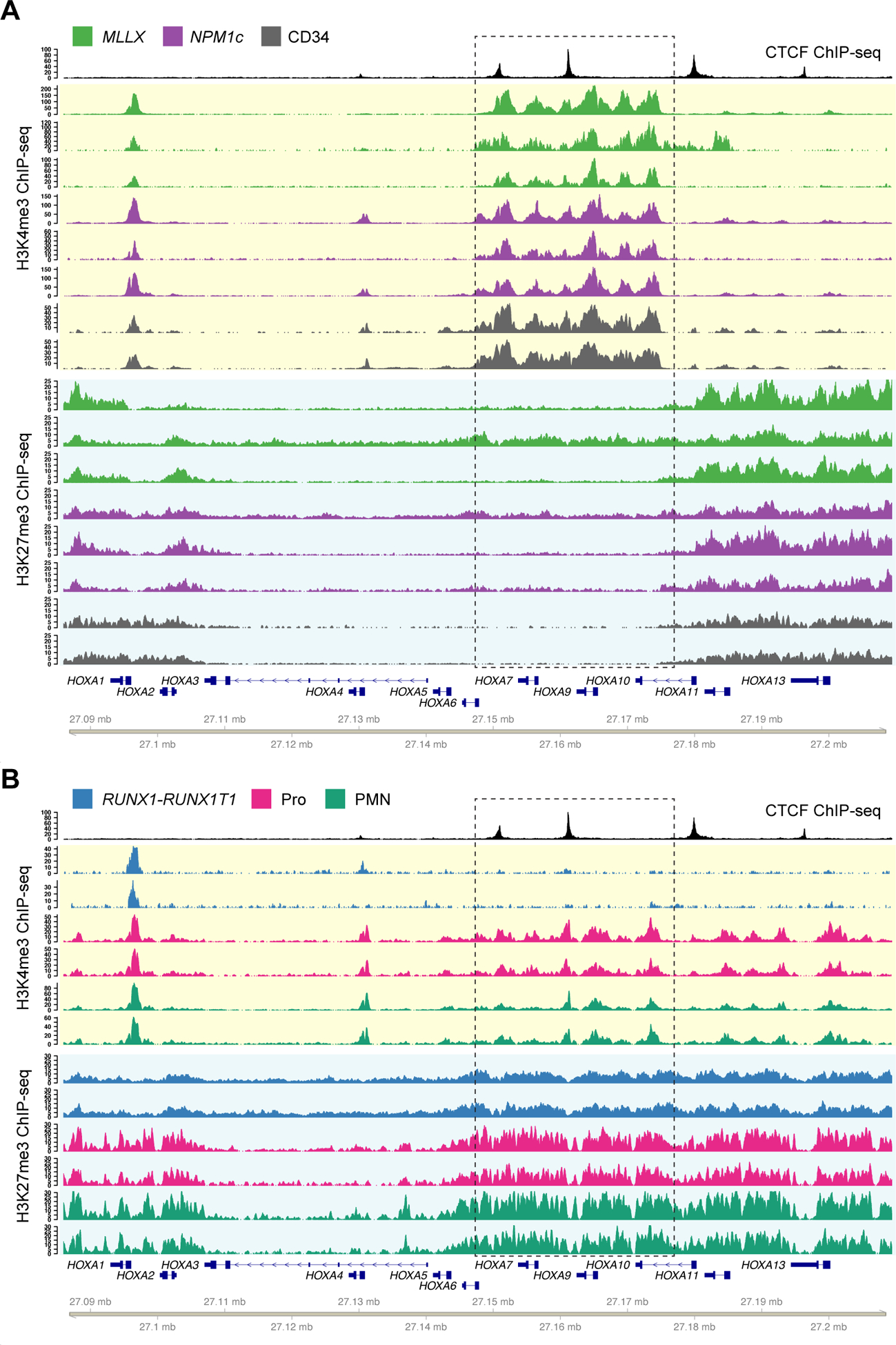
A. Top track shows CTCF ChIP-seq from a NPM1-mutant primary AML sample. Tracks highlighted in yellow show ChIP-seq for H3K4me3 from primary samples with high HOXA expression, including AML samples with MLL rearrangements (green; N=3) and NPM1 mutations (purple; N=3), and primary CD34+ hematopoietic stem/progenitor cells (HSPCs) purified from normal donor bone marrow samples (gray; N=2, from GSE104579). Tracks highlighted in blue show H3K27me3 ChIP-seq from the same set of samples. B. H3K4me3 and H3K27me3 ChIP-seq from primary samples with no HOXA expression, including AML samples with t(8;21) creating the RUNX1-RUNX1T1 fusion (blue; N=2), and normal promyelocytes (CD14-, CD15+, CD16 low; magenta, N=2) and neutrophils (CD14-, CD15+, CD16 high; cyan, N=2) from healthy donor individuals. Dashed box indicates the region of dynamic chromatin that correlates with HOXA gene cluster expression.
Targeted deletions at the HOXA locus eliminate CTCF binding but do not affect viability in NPM1-mutant OCI-AML3 cells
We sought to determine whether loss of these DNA elements has functional consequences in NPM1-mutant AML cells. To this end, we used the OCI-AML3 cell line with a canonical NPM1 insertion mutation and that expresses MEIS1 and both HOXA and HOXB genes (30). This pattern was not observed in cell lines with other mutation-associated HOX expression phenotypes, including MLL-rearranged MOLM13 cells that expressed only HOXA genes, and the RUNX1-RUNX1T1-containing Kasumi-1 cell line, which had low HOX expression (Figure 3A). ChIP-seq for CTCF using OCI-AML3 cells identified the four conserved CTCF sites observed in primary AML samples (Figures 3B, S3A), as well as two smaller peaks in the anterior HOXA cluster. ChIP-seq for H3K4me3 and H3K27me3 demonstrated an active chromatin domain between HOXA9 and HOXA13 (Figure 3B), which was consistent with the patterns of gene expression in this cell line, but different from chromatin domain boundaries in primary AML samples. However, the histone modifications between CBSA7/9 and CBSA10 were shared between OCI-AML3 cells and NPM1-mutated primary AML samples, suggesting that some of the regulatory activities in this region are conserved between primary AML samples and OCI-AML3 cells.
Figure 3. Targeted deletions eliminate CTCF binding in the NPM1-mutant OCI-AML3 cell line.
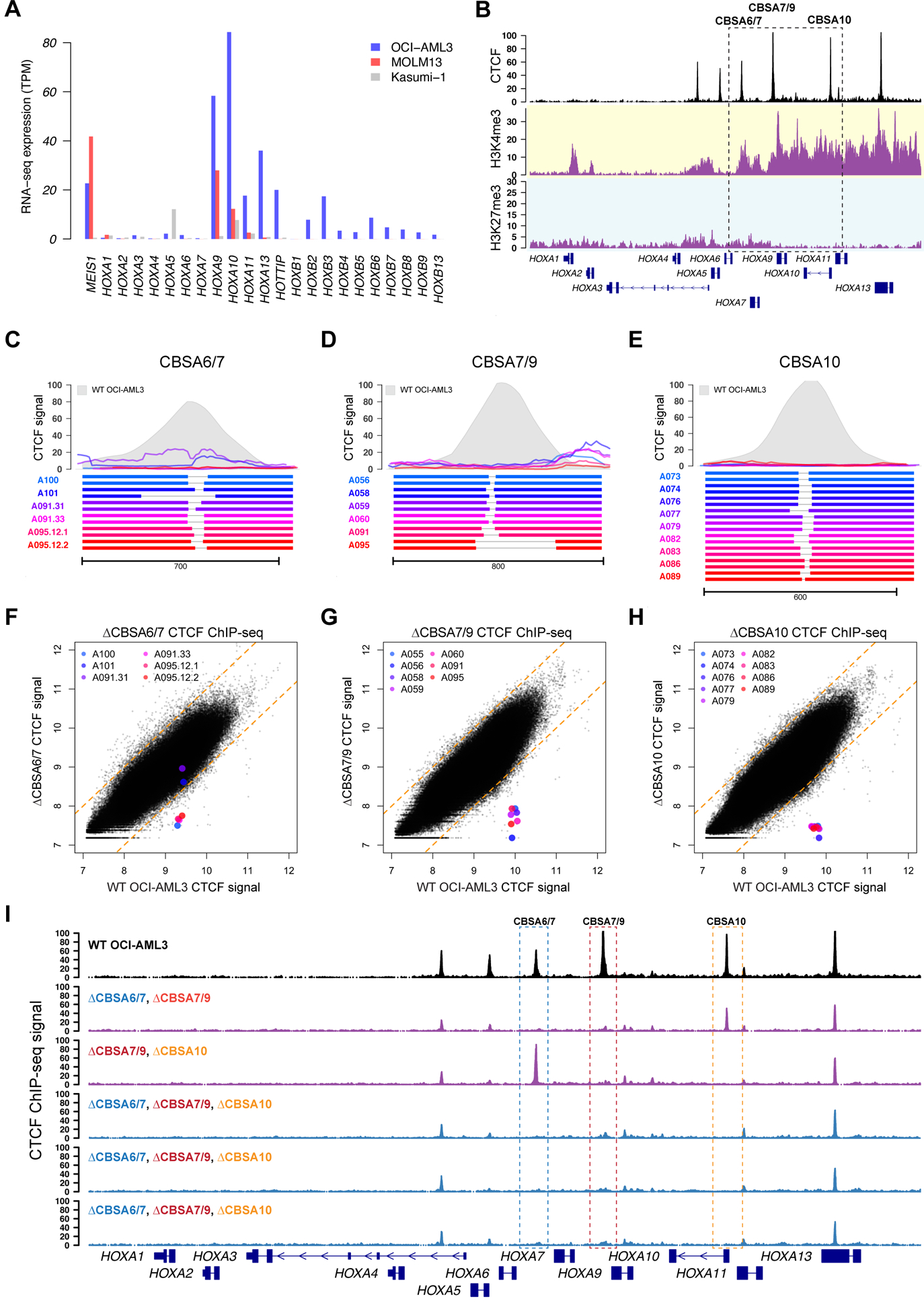
A. RNA-seq expression of HOXA and HOXB genes in OCI-AML3 cells, which display the canonical mutant NPM1-associated HOXA/HOXB expression phenotype. Also shown are the MLL-rearranged MOLM13 cell line that expresses only HOXA genes, and the RUNX1-RUNX1T1-containing Kasumi-1 cell line that has low HOXA and HOXB gene expression. B. ChIP-seq data from OCI-AML3 cells for CTCF (black), H3K4me3 (highlighted in yellow) and H3K27me3 (highlighted in blue), which show conserved CTCF binding sites and distinct regions of active (H3K4me3) and repressed (H3K27me3) chromatin. C-E. Targeted deletions that disrupt CTCF binding in OCI-AML3 cells at sites CBSA6/7 (in C), CBSA7/9 (in D), and CBSA10 (in E). Bottom panels show allele pairs from homozygous or compound heterozygous deletion mutants at each site; top panels show CTCF ChIP-seq signal from these mutant cell lines (multi-colored lines) compared to wild type OCI-AML3 cells (in gray). F-H. CTCF ChIP-seq signal (log2 normalized read counts) for all CTCF peaks from deletion mutants (Y axis) vs. wild type OCI-AML3 cells, showing dramatically reduced CTCF ChIP-seq signal in deletion mutants at all three sites, with the exception of clones A101 and A091.31, which only partially eliminates CTCF signal at site CBSA6/7. I. CTCF ChIP-seq tracks from double (in purple) and triple mutants (in blue), generated via sequential targeted deletion experiments. CTCF ChIP-seq from wild type OCI-AML3 cells is shown in black at the top for reference.
We next used CRISPR/Cas9-mediated editing to delete the three conserved CTCF binding sites in OCI-AML3 cells (CBSA6/7, CBSA7/9 and CBSA10). The resulting mutations did not appreciably alter markers of cell maturation in the edited cells (Figures S3B–C), and the deletion frequency at CBSA7/9 and CBSA10 remained stable after 7 and 14 days; CBSA6/7 deletions were also stable, although the lower deletion efficiency for this site means that subtle fitness defects cannot be excluded (Figure S3D). Single cells were expanded and screened for deletions, which identified at least 5 individual clonal lines with homozygous deletions at each site (Table S3), with deletions as small as 9 bp sufficient to eliminate nearly all CTCF ChIP-seq signal (Figures 3C–H). Additional experiments using single deletion mutants resulted in 10 doubly homozygous mutants and 7 triple mutants with homozygous deletions at all three sites (Figure 3I). None of the mutant OCI-AML3 clones showed overt defects in cell growth (Figure S3E), despite complete loss of CTCF binding in the posterior HOXA cluster.
CTCF binding is not required for maintenance of HOXA gene expression or chromatin boundaries in NPM1-mutant AML cells
We selected 45 OCI-AML3 mutants for HOXA9 expression analysis via RT-qPCR to assess whether loss of CTCF binding affected HOXA9 expression. Surprisingly, HOXA9 expression was essentially unchanged in all deletion lines, with little evidence for consistent reduction in expression in the mutant clones, and no trend toward decreased expression between wild type, heterozygous, and homozygous single mutants (Figure 4A) or multiply mutated clones (Figure 4B); in fact, HOXA9 expression was increased in some mutants. Analysis via RNA-seq showed modest increases in the expression of the anterior HOXA genes HOXA1-HOXA7 in lines with CBSA7/9 deletions, and subtle increases in the posterior HOXA9-HOXA13 genes when CBSA10 was deleted (Figures 4E–D, S4A). However, most mutant lines showed few expression changes. Furthermore, a heterozygous single nucleotide polymorphism (SNP) in the HOXA9 gene showed balanced expression of both alleles in wild type OCI-AML3 cells, and in all mutant lines, except for two with large mutations that involved the posterior HOXA cluster (Figure S4B).
Figure 4. CTCF binding is not required to maintain gene expression or chromatin boundaries in the HOXA gene cluster.
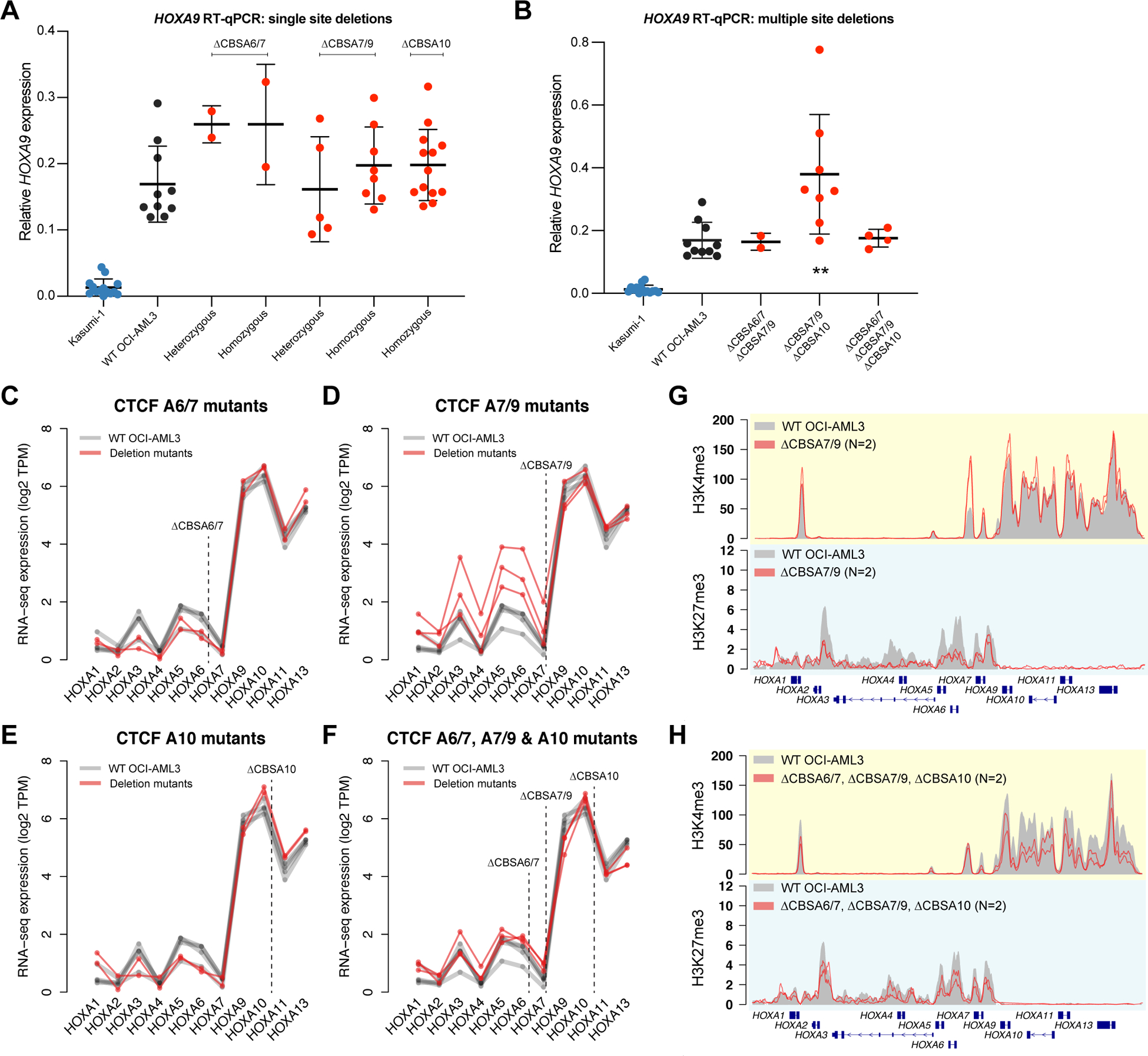
A. RT-qPCR for HOXA9 in single mutants with heterozygous or homozygous deletions at CBSA6/7, CBSA7/9, or CBSA10. HOXA9 expression from the Kasumi-1 cell line is shown in blue as a no-HOXA9 expressing control. No statistically significant differences were identified between wild type OCI-AML3 and homozygous mutants (Bonferroni-corrected P>0.05 using a two-sided unpaired T-test for all comparisons). B. RT-qPCR for HOXA9 in double and triple mutants at the CTCF binding sites indicated. Kasumi-1 cells are included in blue as in panel A. ** denotes a Bonferroni-corrected P<0.01 between wild type OCI-AML3 and CBSA7/9-CBSA10 double mutant clones using a two-sided unpaired T-test. C-F. RNA-seq expression of all HOXA genes in single mutants (C-E) and triple mutants (D). Expression level is shown in log2 transcripts per million (TPM), with lines connecting expression values derived from the same mutant clones and/or the same RNA-seq experiment (for wild type OCI-AML3 cells). G. ChIP-seq for H3K4me3 and H3K27me3 in deletion mutants lacking CTCF at site CBSA7/9 (in red; N=2). Mean ChIP-seq signal from wild type OCI-AML3 cells (N=2) is shown in gray. H. Mean ChIP-seq for H3K4me3 and H3K27me3 in triple mutants (N=2), with ChIP-seq from wild type cells shown in gray, as in panel G.
ChIP-seq for H3K4me3 and H3K27me3 was also performed on multiple mutants to determine whether loss of CTCF binding altered chromatin boundaries. H3K4me3 signal was reduced specifically at the CBSA7/9 site, but was otherwise intact. H3K27me3 was also still present, but was modestly decreased across the anterior HOXA genes in CBSA7/9 mutants; few changes and no statistically different peaks were present in other mutant lines, including triple mutants (Figures 4G–H, S4C–G). Other histone modifications were also analyzed, including H3K79me2 and H3K27ac, which were intact in the mutant lines compared to wild type OCI-AML3 cells (Figure S4H).
CTCF deletions result in compensatory HOXA chromatin loops
Given the role of CTCF in regulating chromatin architecture, we used in situ Hi-C (24) to define the chromatin interactions at the HOXA locus in wild type OCI-AML3 cells, CBSA7/9 single mutants, double mutants with deletions of CBSA6/7 and CBSA7/9 or CBSA7/9 and CBSA10, and two triply mutant lines (Table S4). Analysis of the chromatin contacts from these cells identified a mean of 9,797 chromatin loops and 5,160 contact domains at 10 kbp resolution (ref. 25; Table S4). The HOXA cluster on chromosome 7p was located at a contact domain boundary (Figure 5A), with centromeric loops connecting HOXA13 to the promoter of TAX1BP1 and intronic sequences of HIBADH and JAZF1. The telomeric loops involved the remaining HOXA genes (HOXA1-HOXA11), which contacted regions in the SKAP2, and SNX10 genes, and an intergenic region 1.4 Mbp upstream with no associated gene annotations. Similar chromatin loops were observed in Hi-C data generated from the MLL-rearranged MOLM13 cell line, and in previously reported data from normal human HSPCs (31) (Figures S5A–B), which both express HOXA genes.
Figure 5. CTCF-mediated chromatin architecture at the HOXA locus in NPM1-mutant OCI-AML3 cells.
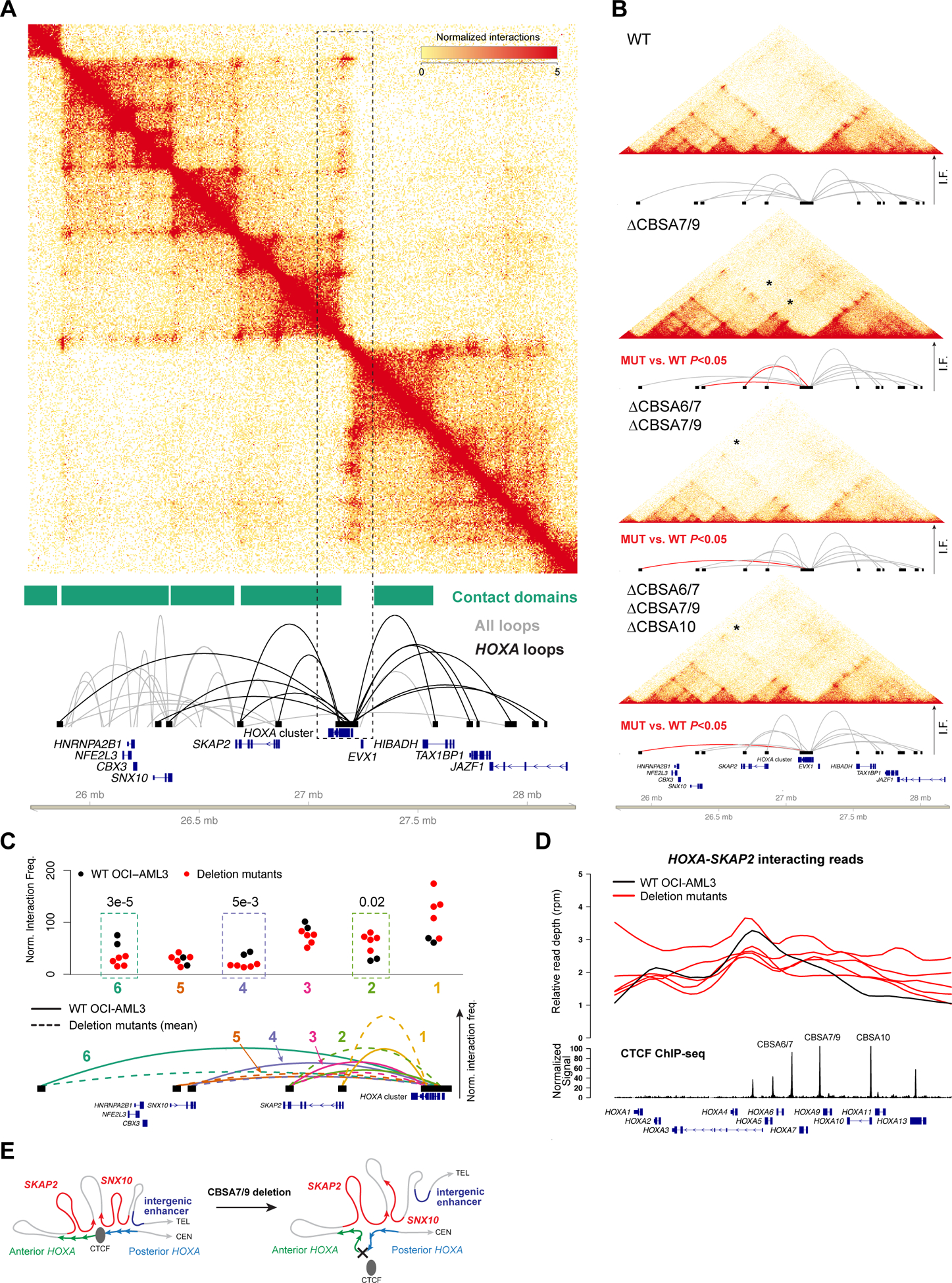
A. Contact matrix, contact domains, and chromatin loops for a 2.5 Mbp region of chromosome 7p that contains the HOXA gene cluster from in situ Hi-C using wild type OCI-AML3 cells. Top panel shows the KR-normalized contact matrix at 5kb resolution. Tracks below the matrix show the contact domains and statistically supported chromatin loops identified using established methods (25). Loops shown in black are anchored within the HOXA gene cluster. B. Contacts and HOXA region loops from in situ Hi-C from wild type OCI-AML3 cells and homozygous (biallelic) single mutants at CBSA7/9, double mutants at CBSA6/7 and CBSA7/9, and triple mutants at CBSA6/7, CBSA7/9, and CBSA10. Loops in red indicate statistically significant differences in pairwise comparisons between each mutant and wild type cells with a chromosome-wide FDR<0.05. C. Normalized interaction frequencies for all datasets for 6 telomeric loops. Top panel shows normalized interaction frequencies for individual Hi-C libraries from wild type OCI-AML3 cells (N=2), single mutants at CBSA7/9, CBSA6/7-CBSA7/9 and CBSA7/9-CBSA10 double mutants, and two CBSA6/7-CBSA7/9-CBSA10 triple mutant lines (N=5 total mutant lines). Dashed boxes highlight loops that were statistically different in comparisons of all mutant libraries vs. two wild type OCI-AML3 libraries (chromosome-wide FDR<0.5). Mean interaction frequencies for chromatin loops from wild type and mutant lines are shown graphically in the lower panel in solid and dashed lines, respectively, and are numbered so they correspond with the normalized frequencies shown in the top panel. D. Relative read depth (reads per million) across the HOXA locus for reads that interact between HOXA and SKAP2. Depth for wild type OCI-AML3 cells is shown in black and deletion mutants are shown in red. CTCF ChIP-seq signal from wild type OCI-AML3 cells and HOXA genes are shown in the bottom tracks. CTCF site deletions result in enhanced interactions between the posterior HOXA cluster (genes HOXA9-HOXA13) with the SKAP2 gene. E. Graphical representation of HOXA chromatin loops in wild type OCI-AML3 cells (left) and deletion mutants lacking HOXA CTCF binding sites (right).
We next analyzed Hi-C data from the OCI-AML3 deletion mutants to determine whether loss of CTCF binding altered chromatin architecture at the HOXA gene cluster. Although the general contact domain and chromatin loop structure remained intact, loops involving the HOXA locus were altered in the deletion mutants (Figure 5B). Pairwise comparisons of normalized chromosome 7 interaction frequencies between each mutant line vs. wild type cells identified decreased long-range interactions and increased interactions with the more proximal SKAP2 gene (Figures 5B, S5C). These findings were confirmed via statistical analysis of joint normalized contact frequencies from all samples, which demonstrated that mutant cells had significantly reduced interaction frequencies between the HOXA cluster and SNX10 and the intergenic region, while interactions with two SKAP2 loci that were present in wild type cells were increased (Figure 5C, Table S5). To define the specific HOXA genes involved in these changes, we mapped the positions of HOXA-SKAP2 interacting reads within the HOXA locus. This showed that SKAP2-HOXA loops involved the anterior HOXA genes and intron 1 of SKAP2 in wild type cells (Figure 5D); however, in mutant cells there were increased interactions between the posterior HOXA genes HOXA9-HOXA13 and intron 11 of SKAP2 (Figure 5D). Interacting reads with SNX10 and the intergenic locus showed the opposite pattern, and were decreased in the posterior HOXA cluster (Figure S5D), indicating that loss of CTCF binding resulted in ‘spreading’ of the proximal SKAP2 interactions to include the posterior HOXA genes (Figure 5E).
Posterior HOXA chromatin loops correlate with expression and involve potential enhancer loci in the SKAP2 gene in primary AML samples
To determine the relationship between transcriptional activity and chromatin loops involving the posterior HOXA genes, we performed in situ Hi-C on primary AML samples with the NPM1c mutation and with high HOXA expression (AML 507202) and a sample with the RUNX1-RUNX1T1 gene fusion and low HOXA gene expression (AML 275786; Figure S1A). Hi-C libraries from these samples captured between 284 and 494 million contacts, of which at least 42% were long range (Table S4). These samples displayed similar overall patterns of chromatin interactions in the HOXA region, which included loops between the HOXA cluster and introns 1 and 11 SKAP2, the SNX10 gene, as well as the distal enhancer locus (Figures 6A–B). However, there were clear differences in the number of loops, and the specific locations of the loop anchors in the HOXA cluster, which correlated with HOXA expression. There more statistically supported chromatin loops in the NPM1-mutant AML sample compared to the RUNX1-RUNX1T1 sample (5 vs. 3), and the region of the HOXA cluster that interacted with intron 1 of SKAP2 involved posterior HOXA genes in the NPM1c sample, but was restricted to the anterior HOXA1 gene in the RUNX1-RUNX1T1 sample (Figure 6C). Similar findings were present in Hi-C data from AML cell lines with the same mutations, including OCI-AML3 and IMS-M2 cells with the NPM1c mutation and the Kasumi-1 cell line with the RUNX1-RUNX1T1 gene fusion, which showed that NPM1c containing cells had more loops and a shift in the loop anchors towards the posterior HOXA cluster (Figures S6A–C). Further analysis the interactions between SKAP2 intron 1 and the HOXA cluster in the primary AML samples demonstrated that reads supporting this loop were most abundant near the CTCF binding site CBSA7/9 in the NPM1c sample, compared to the HOXA1 gene in the sample with RUNX1-RUNX1T1 (Figure 6D).
Figure 6. Chromatin interactions with the posterior HOXA genes correlate with gene expression in primary AML samples and involve loci with enhancer-associated histone acetylation.
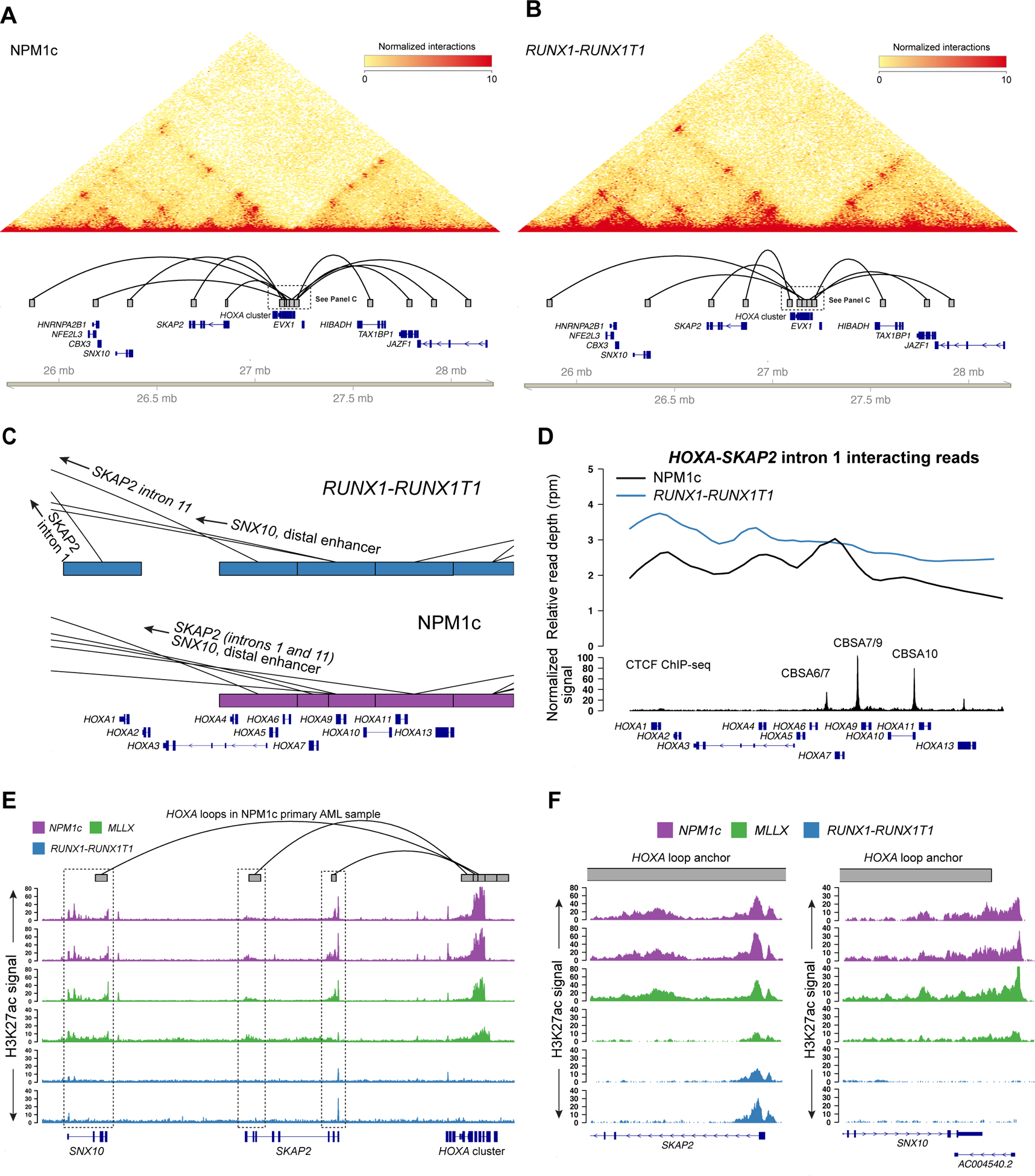
A. Chromatin interactions at chromosome 7p from primary AML sample 507202 with the NPM1c mutation and high HOXA gene expression (see Figure S1A). Heatmap shows the contact matrix for this sample at 10 kbp resolution. Tracks in the lower panel show statistically supported chromatin loops that involve the HOXA gene cluster. B. Chromatin interactions for a primary AML sample with t(8;21)/RUNX1-RUNX1T1 and no HOXA gene expression (see Figure S1A). Panel components are the same as panel (A). C. Focused view of HOXA chromatin loops in the RUNX1-RUNX1T1 (top, in blue) and NPM1c (bottom, in purple) primary AML samples, which highlight the differences in chromatin loop structure and the locations of the loop anchors, which were shifted to the posterior HOXA cluster in the NPM1c-positive sample. D. Relative read depth in reads per million of Hi-C reads between the HOXA locus and intron 1 of SKAP2 in the primary samples with NPM1c and RUNX1-RUNX1T1 black and blue, respectively. Interacting reads in the sample with RUNX1-RUNX1T1 are localized to HOXA1 in the anterior HOXA cluster, compared to the NPM1c sample where interacting reads in map to CBSA7/9 in the posterior HOXA locus. E. Enhancer-associated histone H3 lysine 27 acetylation (H3K27ac) at HOXA interacting loci from primary AML samples with and without HOXA gene expression (including the samples shown in panels A and B). Primary AML samples include patients with NPM1c (purple; N=2 distinct patients; the first track is AML 507202 from panel A) and MLL rearrangements (t(9;11) and t(11;19)) (green; N=2 distinct patients) with high HOXA expression, and samples with t(8;21)/RUNX1-RUNX1T1 (N=2 distinct patients; the first track is AML 275786 from panel B). Regions highlighted in the dashed boxes interact with the HOXA cluster in the NPM1c-containing primary sample 507202 (loop track, top), and display H3K27ac signal suggesting they may represent functional genomic elements. E. High-resolution view of two loop anchor regions in SKAP2 intron 1 and downstream of the SNX10 gene, which possess the enhancer-associated H3K27ac mark in HOXA-expressing AML samples but not in samples with no HOXA expression.
We next analyzed ChIP-seq data for H3K27ac from primary AML samples (including those analyzed with Hi-C above) to determine whether loci that interact with HOXA genes may have enhancer properties. Indeed, intron 1 of SKAP2 (and to some extent intron 11) displayed clear H3K27ac signal in NPM1-mutant AML samples, as well as MLL-rearranged samples that also express HOXA genes (Figures 6E–F). Loci near the SNX10 gene also possessed this mark, including a non-coding RNA downstream of the 3’ UTR of SNX10 (Figure 6F), but H3K27ac was not present at other regions that formed contacts with the HOXA cluster, including the distal intergenic locus (Figure S6D). Although the enhancer modifications observed at interacting regions had relatively low signal, they were clearly absent in the RUNX1-RUNX1T1 samples, which lack HOXA expression. This provides evidence that these specific regions interact with the HOXA cluster in primary AML samples that express HOXA genes, and may therefore contribute to HOXA gene regulation.
Discussion
HOX transcription factors are drivers of self-renewal in AML cells and are highly expressed in AMLs with NPM1c mutations. In this study, we demonstrated that the chromatin organizing factor CTCF is bound equally to specific sites at the HOXA locus in NPM1-mutant primary AML samples compared to other AML subtypes, and define a dynamic chromatin domain in primary AML samples and normal hematopoietic cells. Targeted deletions in the NPM1-mutant OCI-AML3 cell line eliminated CTCF binding, but surprisingly did not disrupt HOXA gene expression. This was true when multiple binding sites were deleted individually, or in combination, and was supported by independent mutant clones that showed little evidence for consistent decreases in HOXA gene expression, or changes to the histone modifications we measured. However, loss of CTCF binding did result in clear alterations to the chromatin loops involving the posterior HOXA genes, including HOXA9 and HOXA10. Specifically, long-range loops were diminished, and were replaced by compensatory interactions with regions of the SKAP2 gene, which also appear to directly interact with active HOXA genes in primary AML samples with the NPM1c mutation. Some of these candidate enhancers have been reported in other studies (32), but have not previously been shown to be active in hematopoietic cells. Further investigation of these sequences, and their associated regulatory proteins, may shed light into factors that promote HOXA gene activation in normal and malignant myeloid cells.
The central observation in this study is that CTCF binding at the HOXA cluster is not absolutely required for maintenance of HOXA expression in NPM1-mutated AML cells. Deletion of CTCF site CBSA7/9 was previously shown to affect HOXA expression in the MLL-rearranged MOLM13 cell line (9). These discrepant findings may be due to fundamental differences in how HOX genes are regulated in MLL-rearranged vs. NPM1-mutant AML cells. Indeed, the HOX expression phenotype of these AML subtypes is strikingly different: MLL rearrangements are associated with only HOXA gene expression, whereas both HOXA and HOXB genes are expressed in NPM1-mutant AML cells. HOXA and HOXB genes are simultaneously downregulated during normal myeloid maturation (1), which implies that the clusters may be controlled by common factors that may function in different ways in NPM1-mutant vs. MLL-rearranged AML cells. We also observed differences in HOXA cluster expression patterns in MOLM13 vs. OCI-AML3 cells, including higher expression of HOXA11, HOXA13, and the HOTTIP long noncoding RNA in OCI-AML3 cells. HOTTIP has an established role in HOXA gene regulation (33,34), and its higher expression may influence the requirement for certain CTCF-mediated chromatin architectures, thereby making these CTCF binding sites dispensable for steady-state expression of posterior HOXA genes in NPM1-mutant AML cells.
Three-dimensional architecture is an important component of the regulatory control of HOXA genes during development. Our analysis of Hi-C data from OCI-AML3 cells and NPM1-mutant primary AML samples identified long-range loops between the active posterior HOXA genes HOXA9-HOXA13 and sequences with enhancer-associated epigenetic marks in the SNX10 and SKAP2 genes. Additional loops were identified at other loci, but these did not possess active chromatin modifications in primary AML samples. AML cells without HOXA expression did not display the same interactions, and lacked active histone marks at the interacting loci, which provides evidence that these contacts may be functionally relevant for HOXA gene expression in AML. Interestingly, loss of CTCF within the HOXA cluster revealed the plasticity of these interactions, implying that multiple loci may serve as exchangeable HOXA enhancers, which is reminiscent of other well-studied, complex regulatory systems, and has been implicated in developmental HOXA gene regulation (35,36).
Our data showed that transcriptional activity and active histone modifications remained highly localized to the posterior HOXA cluster in OCI-AML3 cells, even after elimination of key CTCF boundaries. In fact, there was a tendency for deletion mutants to show increased HOXA gene expression levels compared to wild type cells, which is consistent with recent evidence suggesting that reduction of CTCF levels could enhance HOXA gene expression (37). These observations further suggest the posterior HOXA cluster possesses intrinsic properties that maintain its active state. It is unclear whether this activity is caused by the compensatory interactions we observed, or if these chromatin loops are consequences of persistent gene expression that is mediated by autonomous, sequence-specific factors that recruit transcriptional machinery. The HOXA cluster contains many highly conserved noncoding elements beyond CTCF binding sites. Additional studies will be required to better understand whether these elements provide key signals that promote and maintain HOXA gene expression in NPM1-mutant AML cells.
Supplementary Material
Acknowledgements
This work was supported by the American Society of Hematology (ASH Scholar Award), the American Cancer Society (Institutional Research Grant), and the National Cancer Institute (K08CA190815) to D.H.S. Primary AML samples were provided by the Genomics of AML Program Project (P01CA101937, T. Ley, PI). Technical assistance was provided by the Siteman Cancer Center Tissue Procurement and Cell Sorting cores (NCI Cancer Center Support Grant P30CA91842).
Footnotes
The authors have no conflicts of interest relating to the work described in this manuscript.
References
- 1.Spencer D, Young M, Lamprecht T, Helton N, Fulton R, O’Laughlin M, et al. Epigenomic analysis of the HOX gene loci reveals mechanisms that may control canonical expression patterns in AML and normal hematopoietic cells. Leukemia. 2015;29(6):1279–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hanson R, Hess J, Yu B, Ernst P, van Lohuizen M, Berns A, et al. Mammalian Trithorax and Polycomb-group homologues are antagonistic regulators of homeotic development. Proc National Acad Sci. 1999;96(25):14372–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gould A Functions of mammalian Polycomb group and trithorax group related genes. Curr Opin Genet Dev. 1997;7(4):488–94. [DOI] [PubMed] [Google Scholar]
- 4.Muntean AG, Tan J, Sitwala K, Huang Y, Bronstein J, Connelly JA, et al. The PAF Complex Synergizes with MLL Fusion Proteins at HOX Loci to Promote Leukemogenesis. Cancer Cell. 2010;17(6):609–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Milne TA, Martin M, Brock HW, Slany RK, Hess JL. Leukemogenic MLL Fusion Proteins Bind across a Broad Region of the Hox a9 Locus, Promoting Transcription and Multiple Histone Modifications. Cancer Res. 2005;65(24):11367–74. [DOI] [PubMed] [Google Scholar]
- 6.Daigle SR, Olhava EJ, Therkelsen CA, Basavapathruni A, Jin L, Boriack-Sjodin AP, et al. Potent inhibition of DOT1L as treatment of MLL-fusion leukemia. Blood. 2013;122(6):1017–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Deshpande AJ, Chen L, Fazio M, Sinha AU, Bernt KM, Banka D, et al. Leukemic transformation by the MLL-AF6 fusion oncogene requires the H3K79 methyltransferase Dot1l. Blood. 2013;121(13):2533–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rousseau M, Ferraiuolo MA, Crutchley JL, Wang X, Miura H, Blanchette M, et al. Classifying leukemia types with chromatin conformation data. Genome Biol. 2014;15(4):R60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Luo H, Wang F, Zha J, Li H, Yan B, Du Q, et al. CTCF boundary remodels chromatin domain and drives aberrant HOX gene transcription in acute myeloid leukemia. Blood. 2018;132(8):837–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Spencer DH, Russler-Germain DA, Ketkar S, Helton NM, Lamprecht TL, Fulton RS, et al. CpG Island Hypermethylation Mediated by DNMT3A Is a Consequence of AML Progression. Cell. 2017;168(5):801–816.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cancer Genome Atlas Research Network, Ley TJ, Miller C, et al. Genomic and Epigenomic Landscapes of Adult De Novo Acute Myeloid Leukemia. New Engl J Medicine. 2013;368(22):2059–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schmidl C, Rendeiro AF, Sheffield NC, Bock C. ChIPmentation: fast, robust, low-input ChIP-seq for histones and transcription factors. Nat Methods. 2015;12(10):963–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li H Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv preprint arXiv:1303.3997 2013. March 16. [Google Scholar]
- 14.Ramírez F, Dündar F, Diehl S, Grüning BA, Manke T. deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 2014;42(W1):W187–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, et al. Model-based Analysis of ChIP-Seq (MACS). Genome Biol. 2008;9(9):R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stovner E, Sætrom P. epic2 efficiently finds diffuse domains in ChIP-seq data. Bioinformatics. 2019;35(21):4392–4393. doi: 10.1093/bioinformatics/btz232 [DOI] [PubMed] [Google Scholar]
- 17.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hahne F, Ivanek R. Methods in Molecular Biology. Methods Mol Biology Clifton N J. 2016;1418:335–51. [DOI] [PubMed] [Google Scholar]
- 19.Haeussler M, Schönig K, Eckert H, Eschstruth A, Mianné J, Renaud J-B, et al. Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biol. 2016;17(1):148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Doench JG, Fusi N, Sullender M, Hegde M, Vaimberg EW, Donovan KF, et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat Biotechnol. 2016;34(2):184–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li H Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 2018;34(18):3094–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29(1):15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pertea M, Pertea GM, Antonescu CM, Chang T-C, Mendell JT, Salzberg SL. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol. 2015;33(3):290–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rao S, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, et al. A 3D Map of the Human Genome at Kilobase Resolution Reveals Principles of Chromatin Looping. Cell. 2014;159(7):1665–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Durand NC, Shamim MS, Machol I, Rao SS, Huntley MH, Lander ES, et al. Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell Syst. 2016;3(1):95–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26(6):841–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stansfield JC, Cresswell KG, Vladimirov VI, Dozmorov MG. HiCcompare: an R-package for joint normalization and comparison of HI-C datasets. Bmc Bioinformatics. 2018;19(1):279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stansfield JC, Cresswell KG, Dozmorov MG. multiHiCcompare: joint normalization and comparative analysis of complex Hi-C experiments. Bioinformatics. 2019;35(17):2916–2923. doi: 10.1093/bioinformatics/btz048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Servant N, Lajoie BR, Nora EP, Giorgetti L, Chen C-J, Heard E, et al. HiTC: exploration of high-throughput “C” experiments. Bioinform Oxf Engl. 2012;28(21):2843–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brunetti L, Gundry MC, Sorcini D, Guzman AG, Huang Y-H, Ramabadran R, et al. Mutant NPM1 Maintains the Leukemic State through HOX Expression. Cancer Cell. 2018;34(3):499–512.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jeong M, Huang X, Zhang X, Su J, Shamim M, Bochkov I, et al. A Cell Type-Specific Class of Chromatin Loops Anchored at Large DNA Methylation Nadirs. Biorxiv. 2017;212928. [Google Scholar]
- 32.Su G, Guo D, Chen J, Liu M, Zheng J, Wang W, et al. A distal enhancer maintaining Hoxa1 expression orchestrates retinoic acid-induced early ESCs differentiation. Nucleic Acids Res. 2019;47(13):6737–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Luo H, Zhu G, Xu J, Lai Q, Yan B, Guo Y, et al. HOTTIP lncRNA Promotes Hematopoietic Stem Cell Self-Renewal Leading to AML-like Disease in Mice. Cancer Cell. 2019;36(6):645–659.e8. doi: 10.1016/j.ccell.2019.10.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang KC, Yang YW, Liu B, Sanyal A, Corces-Zimmerman R, Chen Y, et al. A long noncoding RNA maintains active chromatin to coordinate homeotic gene expression. Nature. 2011;472(7341):120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Martin DI, Fiering S, Groudine M. Regulation of β-globin gene expression: straightening out the locus. Curr Opin Genet Dev. 1996;6(4):488–95. [DOI] [PubMed] [Google Scholar]
- 36.Cao K, Collings CK, Marshall SA, Morgan MA, Rendleman EJ, Wang L, et al. SET1A/COMPASS and shadow enhancers in the regulation of homeotic gene expression. Gene Dev. 2017;31(8):787–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang AJ, Han Y, Jia N, Chen P, Minden MD. NPM1c impedes CTCF functions through cytoplasmic mislocalization in acute myeloid leukemia. Leukemia. 2020;34(5):1278–1290. doi: 10.1038/s41375-019-0681-8 [DOI] [PubMed] [Google Scholar]
- 38.Miller CA, White BS, Dees ND, et al. SciClone: inferring clonal architecture and tracking the spatial and temporal patterns of tumor evolution. PLoS Comput Biol. 2014;10(8):e1003665 Published 2014 Aug 7. doi: 10.1371/journal.pcbi.1003665 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All processed sequence data from this study (e.g., bigwig files for ChIP-seq, TPM values for RNA-seq, and Hi-C contact matrices) are available for public download at the following site: https://wustl.box.com/v/ghasemiCTCFHOXA. Raw sequence files for cell lines are available upon request.