

Abstract
Crystallographic models of biological macromolecules have been ranked using the quality criteria associated with them in the Protein Data Bank (PDB). The outcomes of this quality analysis have been correlated with time and with the journals that published papers based on those models. The results show that the overall quality of PDB structures has substantially improved over the last ten years, but this period of progress was preceded by several years of stagnation or even depression. Moreover, the study shows that the historically observed negative correlation between journal impact and the quality of structural models presented therein seems to disappear as time progresses.
Keywords: PDB, structure quality, X-ray crystallography, proteins, nucleic acids
Graphical Abstract
Quality criteria are proposed to rank the macromolecular models in the Protein Data Bank (PDB) and the results are correlated with time and with the journals that published papers based on those models. The overall quality of PDB structures has substantially improved over the last decade and the negative correlation between journal impact factor and the quality of structural models presented therein seems to disappear as time progresses.
Introduction
Structural biology has fulfilled a history changing mission in science at the interface of physics, chemistry and biology when for over six decades it has maintained its leading role in providing the structural basis for our understanding of life [1–4]. Its results were always regarded as exceptionally solid, and created a gold standard in biological research, almost unattainable in many other areas of life sciences. This view has largely persisted until today, in part even fortified by the incredible technical advances in the generation and detection of X-rays, progress in computer software development, revolution in biotechnology, and innovations in crystallogenesis. However, with the expansion of the Protein Data Bank (PDB) [5] from merely seven structures at its inception in 1971 to ~160,000 today, it is inevitable that some of the macromolecular models will be subpar and sometimes even incorrect. Unfortunately, suboptimal structures have a tangible negative impact on biomedical research that relies on structural data [6]. However, crystallographers, who have always been in the forefront of structural biology, also in this regard seem to be setting example of how to deal with suboptimal or irreproducible science. The protein crystallographic community has been made painfully aware of these problems [7–10], partly in the rising wave of concern about irreproducibility of scientific research and of biomedical research in particular [11]. This awareness has led to positive outcomes, such as, for example, development of structure validation criteria and protocols, or development of tools for the detection and correction of model errors [12].
The PDB itself, who is the chief custodian of the structural treasury amassed by structural biologists, has been developing tools and standards for the assessment of the quality of the structural models deposited in its archives [13,14]. Similarly, more and more journals are starting to require structural validation reports generated by the PDB upon manuscript submission. However, in some opinions these actions are still insufficient and many problems could be better checked at source rather than being tracked in time-delayed model-correction actions [15,16], when the ripple effect of structural errors may have already taken its toll. Objectively speaking, however, in view of the immense scale of the PDB, one should be in fact grateful for all the effort already taken and the plans proposed for the future of the data bank. In particular, the PDB has been developing a consistent and informative set of quality indicators, which now accompany each new crystal structure deposition. These indicators have been recently used to assess the evolution of the quality of the PDB deposits with time [17].
However, it is not only the PDB that has the responsibility for maintaining high standard of the structural information generated by structural biology. The prime burden is of course on the authors, but this is usually the weakest link: rarely because of ill-intention or fraud and more frequently because of haste, lack of training, lack of supervision, or the delusive belief that the incredible recent progress has converted crystallography to a very easy and almost completely automatic analytical method. An important deal of responsibility rests with the referees and editors of the journals that publish those results, as the ripple effect of error and fatal contamination of science is most efficiently propagated through cited literature [18]. More than a decade ago Brown & Ramaswamy (hereinafter B&R) published a survey of the quality of crystallographic models of biological macromolecules [19], and correlated the results with the journals in which those models had been published. The results came as a bit of a shock to many because it turned out that the journals usually regarded as the most prestigious were found to publish worse than average structures when compared with other journals. The FEBS Journal was one of the first requesting structure validity reports and thus in the ranking list of B&R was among the top journals. Similar questions have been raised by Read & Kleywegt (hereinafter R&K), albeit using different statistical tools [20]. In contrast to the B&R study, R&K reported very small quality differences between structures published in high-impact journals and in other venues.
Nearly 13 years after the B&R study and with the PDB expanded nearly four times we decided to conduct a similar analysis to see if the community at large, or at least its journals, have improved. In our approach, we used the statistical methods of data imputation and Principal Component Analysis (PCA) of the model quality indicators recommended by the PDB. In contrast to previous studies, which focused on protein structures only, our analysis comprises all crystallographic structures in the PDB, i.e. also includes nucleic acids. Moreover, we also consider models marked as To be published, which were not analyzed by B&R or R&K. Although the scope of data and the statistical tools we are using are different from those used by B&R in 2007, we are still able to compare the journal rankings of the two surveys because our approach may be easily adapted to a retrospective analysis of data from past versions of the PDB. It is important to clarify that omission of NMR and Cryo-EM structures was intentional. Considering the difficulties connected with estimation of quality of NMR and Cryo-EM structural models, and also the very small contribution of both these methods to the characterization of structures that contain ligands (and are thus most interesting and important), we decided to focus on models provided by X-ray crystallography, which represent 89% of all models currently deposited in the PDB.
Our results show that the overall quality of PDB structures has substantially improved over the last ten years. However, our study also shows that this period of improvement was preceded by several years of stagnation or, if one considers the improvement of software and hardware over time, even depression. Finally, the observation made by B&R that journal impact factor (reputation) is frequently negatively correlated with structure quality is no longer true.
Results
Measure of Overall Model Quality and Missing Data Imputation
The analysis included all X-ray structures available in the PDB as of December 10, 2019, totaling 141,154 deposits dating back as far as 1972. To assess the quality of structures published in particular journals, we initially attempted to use the Q1p measure proposed by Shao et al. [17]. Q1p is a measure of overall protein structure quality that combines into one number five different indicators: Rfree, RSRZ (normalized Real Space R-factor) outliers, Ramachandran outliers, Rotamer outliers, and Clashscore [21] using the following formula:
| (1) |
where PRfree, P%RSRZ, and PPC1(geometry) are ranking percentiles (the higher the better), characterizing for a given structural model, respectively, its Rfree, percentage of RSRZ outliers, and the first principal component of the PCA of Ramachandran outliers, Rotamer outliers and Clashscore (see Methods section for details). Once Q1p is calculated, each PDB deposit is ranked within the population to obtain its final ranking percentile PQ1p, with the lowest (worst) value of Q1p at 0% and highest (best) at 100% [17]. We note, that in this paper we took an averaging approach to percentiles, i.e., a group of tied Q1p values was assigned the same percentile rank, one that is the average rank of the group. By combining five distinct quality measures, PQ1p provides a simple way of comprehensive comparison and ranking of many structural models.
The PQ1p metric was originally designed to assess protein structures only. For nucleic acid structures, which are also present in the PDB, Q1p cannot be used directly because the notions of Ramachandran and Rotamer outliers are not applicable to those structures. However, for proteins both missing elements are implicitly contained in PPC1(geometry). Therefore, for nucleic acids we calculated analogous Q1n without the use of PCA, but applying the following simplified formula:
| (2) |
where PClashscore is the ranking percentile of Clashscore. In the following analysis, Q1p and Q1n (and, consequently, PQ1p and PQ1n) were computed separately for proteins and nucleic acids, respectively. This way, the percentiles used for Q1p and Q1n rank structures of the respective type. Protein-nucleic acid complexes were assigned to the protein group, since it is possible to calculate all quality metrics for such structures.
Since averaging of multiple quality metrics might potentially blur the spotlight on models with serious problems, an alternative aggregation method could involve taking only the minimum percentile of all the metrics used. In this approach, a structure is considered as good as its weakest feature, according to the following formulas:
| (3) |
| (4) |
In the remainder of the paper, we will focus mainly on the averaging approach using Eq. 1 and 2, but will also compare it with the minimum approach based on Eq. 3 and 4.
It must be emphasized that PQ1p can be computed only for those PDB structures that have all five (or in the case of PQ1n all three) component measures attached to them. The PDB has done an excellent job of calculating these metrics for most of the deposits, but not all structures have all the necessary data to perform these calculations. Overall, 12.7% of all considered deposits are missing at least one quality metric, with RSRZ being the dominating missing value (Table 1). Leaving this situation as is would effectively limit the analysis to structures published after 1992, i.e., to the time after Rfree was introduced [22]. To circumvent this dilemma and to perform a study encompassing the entire timespan of the PDB, we have developed a protocol for the estimation of the missing values based on a machine learning data imputation method.
Table 1.
Quality metric means, standard deviations, and fractions of missing values in the PDB.
| Metric | Mean | Standard deviation | Missing values [%] |
|---|---|---|---|
| Clashscore | 8.05 | 9.11 | 0.10 |
| Ramachandran outliers [%] | 0.49 | 1.26 | 1.69 |
| Rotamer outliers [%] | 3.26 | 3.65 | 1.72 |
| RSRZ outliers [%] | 4.05 | 4.06 | 9.56 |
| Rfree [%] | 23.35 | 3.82 | 4.29 |
| Supporting metrics | |||
| R [%] | 19.31 | 3.25 | 2.39 |
| Resolution [Å] | 2.13 | 0.56 | 0 |
| Year of deposition | - | - | 0 |
The validity of the data imputation procedure was assessed on the complete portion of the PDB, to which artificially missing (i.e. deliberately removed) values were introduced at random following the missing data proportions of each metric. The missing values were then replaced using either the metric’s mean, median or by an iterative method called Multiple Imputation by Chained Equations (MICE) [23,24] with Bayesian linear regression [25]. MICE builds regression functions for subsequent metrics based on non-missing values from other variables. The variables used to aid imputation involved all the metrics in question, plus three supporting variables, not used in the assessment protocol: the R factor, data resolution (dmin), and year of deposition (Table 1). The results of 100 random experiments testing the imputation methods are presented in Table 2.
Table 2. Evaluation of data imputation methods.
Mean results of 100 random experiments with standard deviations given in parentheses, in units of the last significant digit of the mean. MAD: Median Absolute Deviation, MAE: Mean Absolute Error, RMSE: Root-Mean-Square Error.
| Error | Method | Clashscore | RSRZ outliers [%] | Ramachandran outliers [%] | Rotamer outliers [%] | R free [%] |
|---|---|---|---|---|---|---|
| MAD | MICE | 2.5(3) | 2.01(2) | 0.21(1) | 1.23(3) | 1.02(2) |
| Mean | 4.4(3) | 2.30(2) | 0.49(1) | 2.11(4) | 2.50(4) | |
| Median | 2.9(3) | 1.86(2) | 0.04(1) | 1.39(4) | 2.50(4) | |
| MAE | MICE | 3.7(4) | 2.54(3) | 0.41(2) | 1.77(4) | 1.31(2) |
| Mean | 5.7(6) | 2.74(3) | 0.61(2) | 2.56(6) | 3.00(3) | |
| Median | 5.1(7) | 2.60(3) | 0.49(3) | 2.34(7) | 3.00(3) | |
| RMSE | MICE | 5.7(15) | 3.84(15) | 0.84(11) | 2.64(10) | 1.77(3) |
| Mean | 8.9(23) | 4.04(14) | 1.24(16) | 3.65(14) | 3.82(4) | |
| Median | 9.3(24) | 4.17(14) | 1.32(16) | 3.85(15) | 3.82(4) |
It can be seen that MICE is superior to mean/median replacement for all metrics according to the Mean Absolute Error (MAE) and Root-Mean-Square Error (RMSE), and for all but two metrics according to the Median Absolute Deviation (MAD). All the differences between MICE and the remaining methods are statistically significant according to the Friedman and Nemenyi post-hoc tests [26] (p < 0.001). In terms of absolute values, the mean absolute error of MICE is usually two to four times smaller than the standard deviation of a given quality metric (Table 1, Supplementary Fig. S11). The results are particularly good for Clashscore and Rfree, owing to the small number of missing values and high correlation with R, respectively. In the remaining part of the paper, we discuss results obtained for the full PDB dataset with missing values imputed using the MICE method. We want to stress that in doing so our goal is to give an approximate overview of the average quality of structures in the early years of the PDB, and not to provide a way to assess individual deposits with missing quality metrics or to create nonexistent data.
Model quality at the Time of Deposition
Fig. 1 shows that PQ1p and PQ1n tend to gradually improve over the years. Almost identical trends can be noticed when looking at deposits without imputed data (Supplementary Fig. S2) and when using the minimum approach (Supplementary Fig. S3). Obviously, this trend is correlated with the advances in the generation of X-rays and in data collection procedures, with better computer hardware and software, with heightened structure validation standards, and with progress in crystallogenesis. If one were to use PQ1 (i.e., PQ1p or PQ1n depending on structure type) calculated over all the analyzed years to rank journals, then journals with longer history would be at a disadvantage because they contain old, quality-wise inferior structures. Thus, even though a structure might have been refined to an impressively high standard in its time, today it might be treated as a poorly refined case. One could, of course, recalculate the percentiles separately for each decade or even shorter time periods, but this might not be enough to cure this problem (see the rapid improvement in quality over the last 10 years) or could drastically reduce the data volume and effectively make journal comparisons impossible. Therefore, we introduce here a new, time (t)-dependent PQ1(t) parameter, which corresponds to PQ1 calculated at the time of structure deposition. For example, the 1990 PDB deposition 2RSP [27] achieves an overall quality percentile PQ1 of 36%, meaning that it is better than only 36% protein deposits that are currently held in the archive. Should the structure be ranked against the 416 structures deposited prior to 2RSP, it achieves PQ1(t) of 69%, meaning that it was significantly above-average at the time of its deposition.
Figure 1. PQ1 analysis.
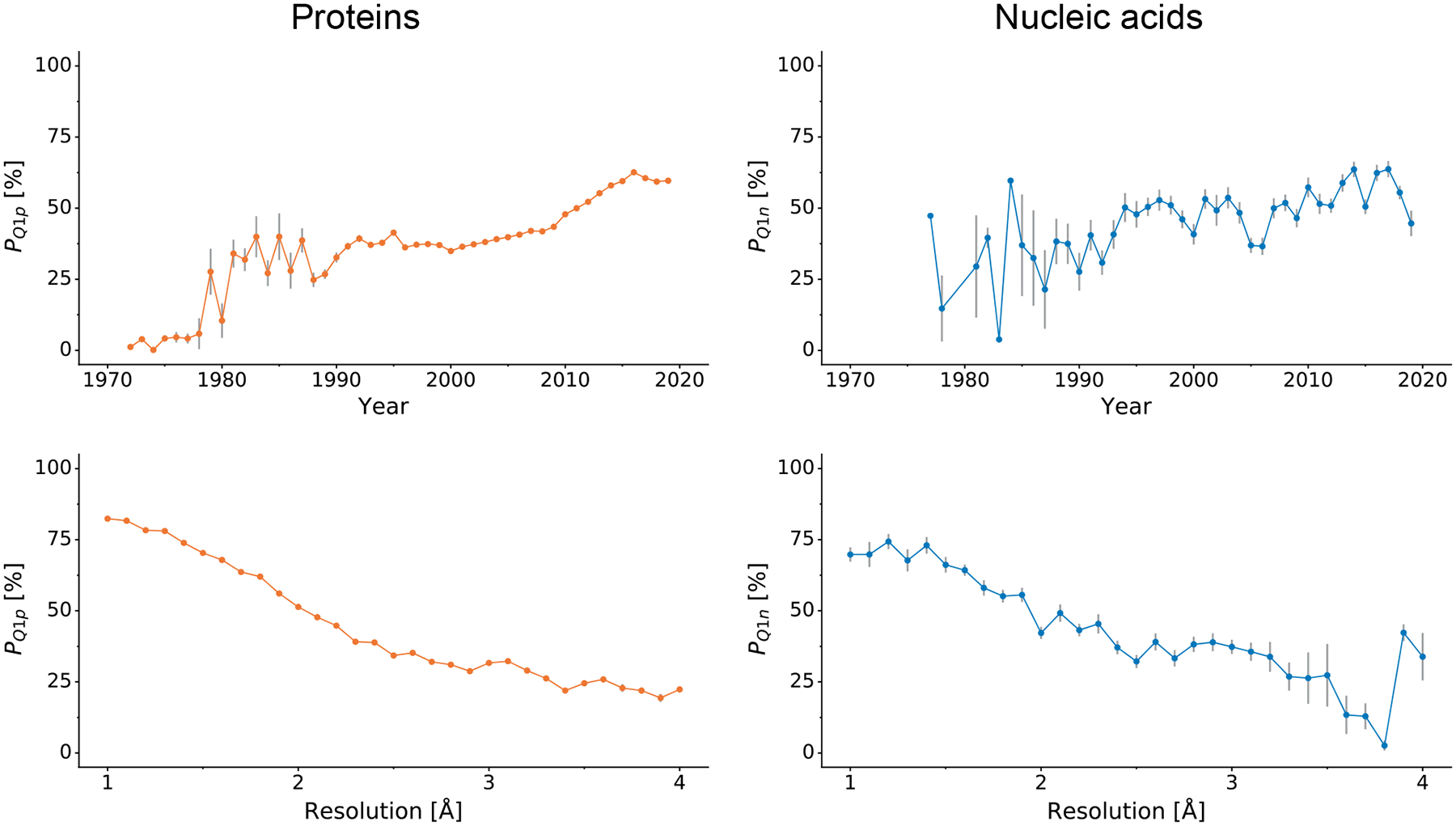
Variation in the mean PQ1 percentile (higher is better) over time (top) and as a function of resolution (bottom) for proteins (left) and nucleic acids (right). Error bars indicate estimated unbiased standard errors of the mean.
Moreover, in view of the very high correlation between quality and resolution (Fig. 1, Supplementary Fig. S2 and S3), we propose yet another measure, called PQ1(t,d). PQ1(t,d) is the Q1 percentile calculated at the time of structure deposition (t) for a given resolution interval (d), where the resolution is rounded to the nearest 0.1 Å and capped at 1 Å and 4 Å. The 2RSP structure from the previous example scores a PQ1(t,d) of 75%. The advantage of using PQ1(t,d) is that data resolution will not affect the journal ranking list.
Using PQ1p(t,d) and PQ1n(t,d) one can assess the quality of protein and nucleic acid models over time. The average PQ1p(t,d) for proteins in the PDB is 58.7%, whereas nucleic acids have the average PQ1n(t,d) of 59.9%. Fig. 2 shows how the model quality at the time of deposition of these two types of macromolecules has evolved over the years. For many years in the past, newly deposited nucleic acid models were usually of better quality than newly deposited protein models, especially between 1993 and 2004. However, the steady improvement of the quality of protein models in the last decade has made them currently to be on a par, if not better, than currently deposited nucleic acids models. Similar trends were observed using PQ1pmin(t,d) and PQ1nmin(t,d), i.e. the minimum approach (Supplementary Fig. S4).
Figure 2. Comparison of PQ1(t,d) of protein and nucleic acid structures over time.
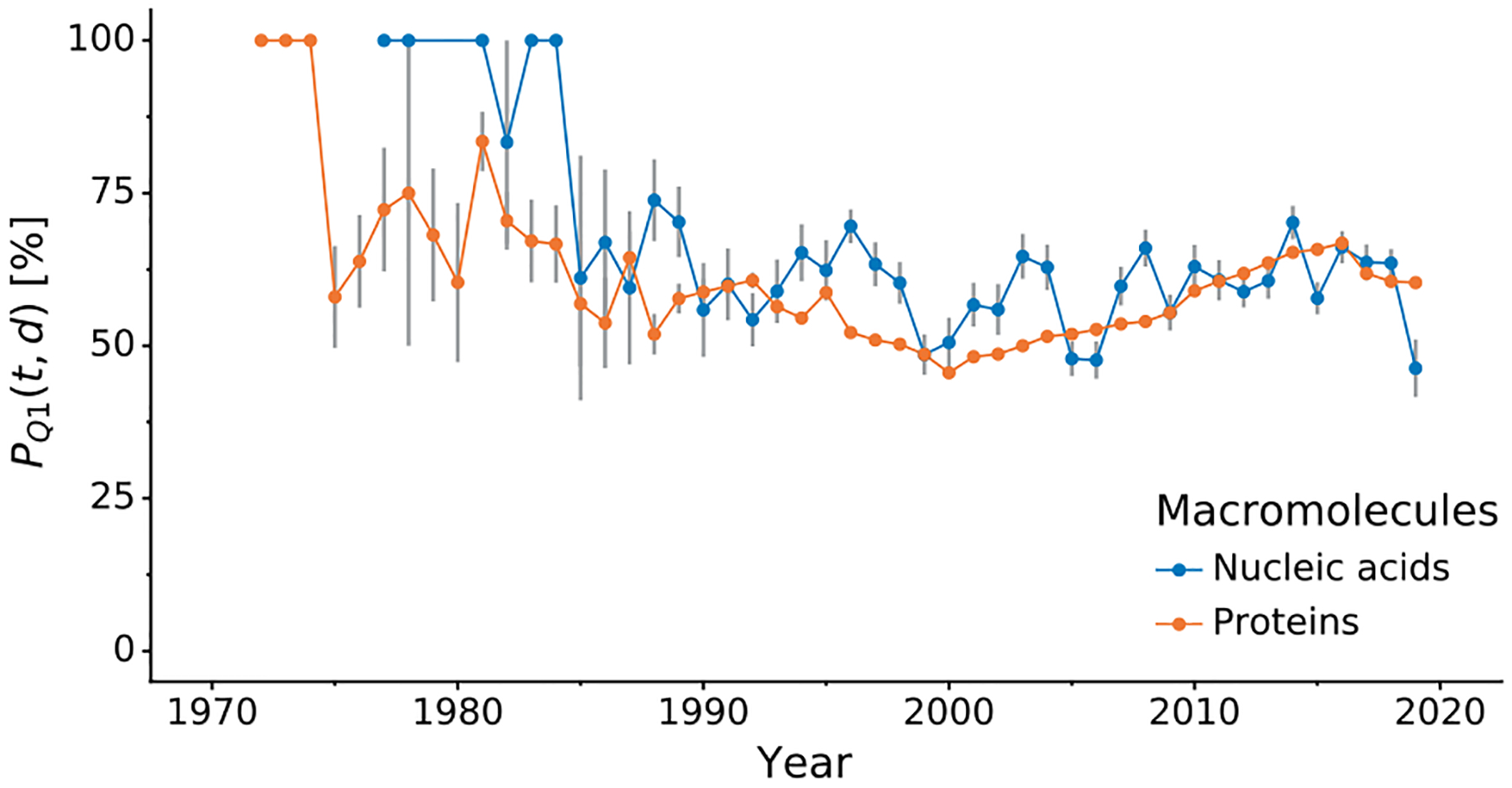
Variation in mean PQ1(t,d) quality percentile (y-axis, higher is better), comparing nucleic acid and protein structures (color) over time (x-axis). Error bars indicate estimated unbiased standard errors of the mean.
In the following subsections, we will focus on ranking structures and their corresponding journals according to PQ1(t,d). The rankings associated with PQ1(t), PQ1min(t,d) and PQ1min(t) are available in the online supplementary materials for this publication. For the purposes of ranking journals, the percentiles for proteins and nucleic acids will be combined, and denoted jointly as PQ1(t,d) or PQ1(t).
All-time Journal Ranking
Out of 800 unique journals being the primary citations for the 141,154 deposits found in the PDB, we selected those that published papers presenting at least 100 macromolecular structures. We decided to limit the list of journals to such a subset, as we believe that it may be too early to assess journals with less than 100 described structures. The resulting 91 journals were ranked according to average PQ1(t,d) (Table 3) as well as PQ1(t), PQ1min(t,d) and PQ1min(t) (Supplementary Tables S1–S3).
Table 3. All-time journal ranking according to PQ1(t,d).
The ranking includes all the journals that had at least 100 primary citations of structures in the PDB. PQ1(t,d) higher than 50% means that the structures published in a given journal were, on average, better than 50% of structures of similar resolution present in the PDB at the time of deposition. Journals with more than 1000 structures are highlighted in gray. The most frequent venue (To be published) is highlighted in bold.
| Rank | Journal | Mean PQ1(t,d) [%] | Mean resolution [Å] | G-Mean resolution [Å] | V-Mean resolution [Å] | Structure count |
|---|---|---|---|---|---|---|
| 1 | TUBERCULOSIS (EDINB)* | 87.38 | 2.02 | 2.00 | 1.92 | 132 |
| 2 | EUR J MED CHEM* | 71.05 | 2.03 | 1.97 | 1.81 | 418 |
| 3 | ACS CATAL* | 69.25 | 1.94 | 1.88 | 1.70 | 241 |
| 4 | ACS INFECT DIS* | 68.32 | 1.95 | 1.90 | 1.78 | 153 |
| 5 | CHEMBIOCHEM* | 68.30 | 1.92 | 1.87 | 1.71 | 527 |
| 6 | IUCRJ* | 67.76 | 1.95 | 1.88 | 1.69 | 281 |
| 7 | ORG BIOMOL CHEM | 67.42 | 1.88 | 1.82 | 1.64 | 167 |
| 8 | MBIO | 66.72 | 2.24 | 2.15 | 1.91 | 169 |
| 9 | ARCH BIOCHEM BIOPHYS* | 66.13 | 2.08 | 2.03 | 1.89 | 276 |
| 10 | INT J MOL SCI | 65.77 | 2.12 | 2.04 | 1.81 | 103 |
| 11 | CHEMISTRY | 65.66 | 1.82 | 1.77 | 1.62 | 242 |
| 12 | BIOCHEM J* | 65.42 | 2.11 | 2.06 | 1.89 | 1,033 |
| 13 | FEBS J* | 65.11 | 2.02 | 1.97 | 1.81 | 1,539 |
| 14 | CELL HOST MICROBE | 64.85 | 2.50 | 2.43 | 2.20 | 107 |
| 15 | PLOS PATHOG* | 64.72 | 2.25 | 2.17 | 1.96 | 656 |
| 16 | NAT MICROBIOL | 64.69 | 2.32 | 2.25 | 2.04 | 111 |
| 17 | CHEM COMMUN | 64.58 | 1.81 | 1.75 | 1.59 | 280 |
| 18 | VIROLOGY | 64.38 | 2.44 | 2.37 | 2.18 | 126 |
| 19 | NAT CHEM BIOL* | 64.12 | 2.19 | 2.12 | 1.92 | 1,013 |
| 20 | APPL ENVIRON MICROBIOL | 64.00 | 2.01 | 1.96 | 1.83 | 112 |
| 21 | ACS CHEM BIOL* | 63.83 | 2.04 | 1.98 | 1.83 | 1,104 |
| 22 | ACTA CRYST F* | 63.59 | 2.09 | 2.02 | 1.81 | 1,466 |
| 23 | SCI REP* | 63.39 | 2.16 | 2.09 | 1.88 | 1,847 |
| 24 | ANGEW CHEM* | 63.25 | 1.92 | 1.85 | 1.64 | 1,065 |
| 25 | J INORG BIOCHEM | 62.82 | 1.78 | 1.73 | 1.57 | 171 |
| 26 | J BIOL INORG CHEM | 62.61 | 1.88 | 1.82 | 1.64 | 265 |
| 27 | ACS OMEGA | 62.60 | 1.81 | 1.77 | 1.64 | 102 |
| 28 | J COMPUT AIDED MOL DES | 62.39 | 1.88 | 1.86 | 1.77 | 115 |
| 29 | CHEM SCI | 61.98 | 1.87 | 1.83 | 1.68 | 268 |
| 30 | MABS | 61.91 | 2.35 | 2.29 | 2.14 | 115 |
| 31 | J SYNCHROTRON RADIAT | 61.75 | 1.79 | 1.73 | 1.56 | 147 |
| 32 | PLOS ONE* | 61.57 | 2.15 | 2.09 | 1.92 | 2,057 |
| 33 | GLYCOBIOLOGY | 61.48 | 1.96 | 1.90 | 1.74 | 188 |
| 34 | CHEMMEDCHEM | 61.40 | 1.94 | 1.88 | 1.69 | 556 |
| 35 | TO BE PUBLISHED* | 61.39 | 2.03 | 1.98 | 1.81 | 22,421 |
| 36 | NAT COMMUN* | 61.37 | 2.21 | 2.11 | 1.86 | 3,538 |
| 37 | FEBS LETT | 61.30 | 2.10 | 2.04 | 1.85 | 814 |
| 38 | NAT CHEM | 61.11 | 1.95 | 1.85 | 1.65 | 173 |
| 39 | ANTIMICROB AGENTS CHEMOTHER | 61.10 | 1.96 | 1.88 | 1.65 | 309 |
| 40 | ACS MED CHEM LETT | 61.01 | 2.12 | 2.06 | 1.88 | 1,062 |
| 41 | RNA | 60.95 | 2.44 | 2.32 | 1.97 | 245 |
| 42 | J AM CHEM SOC | 60.89 | 2.00 | 1.93 | 1.74 | 2,369 |
| 43 | PROTEIN ENG DES SEL | 60.59 | 2.05 | 2.00 | 1.84 | 294 |
| 44 | CELL CHEM BIOL | 60.17 | 2.08 | 2.03 | 1.88 | 902 |
| 45 | ACTA CRYST D | 60.06 | 1.99 | 1.91 | 1.70 | 4,952 |
| 46 | MOL PHARMACOL | 59.99 | 2.35 | 2.27 | 2.02 | 129 |
| 47 | BMC STRUCT BIOL | 59.56 | 2.08 | 2.02 | 1.84 | 228 |
| 48 | BIOCHEMISTRY | 59.45 | 2.05 | 2.00 | 1.84 | 8,896 |
| 49 | MOL MICROBIOL | 59.41 | 2.16 | 2.09 | 1.87 | 412 |
| 50 | BIOCHIMIE | 59.04 | 2.07 | 2.02 | 1.81 | 128 |
| 51 | J BIOCHEM | 59.01 | 2.05 | 2.01 | 1.88 | 279 |
| 52 | FASEB J | 58.88 | 2.03 | 1.98 | 1.86 | 161 |
| 53 | NUCLEIC ACIDS RES | 58.77 | 2.28 | 2.21 | 1.98 | 2,127 |
| 54 | STRUCTURE | 58.53 | 2.20 | 2.11 | 1.86 | 5,348 |
| 55 | PROTEIN SCI | 58.46 | 2.07 | 2.02 | 1.85 | 2,235 |
| 56 | J MED CHEM | 58.29 | 2.07 | 2.02 | 1.86 | 5,525 |
| 57 | J VIROL | 58.24 | 2.34 | 2.26 | 2.06 | 957 |
| 58 | PLANT CELL | 58.00 | 2.21 | 2.17 | 2.05 | 138 |
| 59 | BIOCHIM BIOPHYS ACTA | 57.99 | 2.09 | 2.04 | 1.87 | 600 |
| 60 | SCI ADV | 57.95 | 2.33 | 2.22 | 1.85 | 182 |
| 61 | J BIOL CHEM | 57.91 | 2.12 | 2.06 | 1.89 | 11,055 |
| 62 | J STRUCT BIOL | 57.49 | 2.15 | 2.08 | 1.90 | 1,038 |
| 63 | J BACTERIOL | 57.37 | 2.22 | 2.16 | 2.01 | 371 |
| 64 | BIOORG MED CHEM | 57.28 | 2.10 | 2.04 | 1.88 | 659 |
| 65 | PROTEINS | 57.09 | 2.07 | 2.01 | 1.84 | 1,999 |
| 66 | CELL REP | 56.55 | 2.52 | 2.42 | 2.16 | 399 |
| 67 | J EXP MED | 56.35 | 2.34 | 2.28 | 2.08 | 121 |
| 68 | BIOCHEM BIOPHYS RES COMMUN | 56.28 | 2.18 | 2.11 | 1.92 | 976 |
| 69 | PNAS* | 55.79 | 2.27 | 2.19 | 1.94 | 7,376 |
| 70 | INT J BIOL MACROMOL | 55.51 | 2.05 | 2.00 | 1.80 | 168 |
| 71 | PLOS BIOL | 55.43 | 2.35 | 2.25 | 2.01 | 336 |
| 72 | ELIFE | 55.40 | 2.41 | 2.30 | 2.03 | 869 |
| 73 | BIOPHYS J | 55.36 | 1.92 | 1.85 | 1.66 | 199 |
| 74 | J MOL BIOL* | 55.25 | 2.13 | 2.06 | 1.88 | 9,507 |
| 75 | CELL RES | 54.60 | 2.42 | 2.36 | 2.20 | 189 |
| 76 | J STRUCT FUNCT GENOM | 54.30 | 2.07 | 2.03 | 1.92 | 168 |
| 77 | NATURE* | 53.84 | 2.52 | 2.42 | 2.12 | 3,060 |
| 78 | PROTEIN CELL | 53.44 | 2.26 | 2.21 | 2.04 | 178 |
| 79 | GENES DEV | 53.44 | 2.41 | 2.33 | 2.11 | 279 |
| 80 | SCIENCE* | 53.15 | 2.50 | 2.39 | 2.10 | 1,949 |
| 81 | NAT IMMUNOL | 52.94 | 2.52 | 2.45 | 2.22 | 119 |
| 82 | EMBO REP | 52.92 | 2.35 | 2.28 | 2.09 | 211 |
| 83 | J IMMUNOL | 52.10 | 2.26 | 2.19 | 2.02 | 296 |
| 84 | NEURON | 51.35 | 2.63 | 2.41 | 2.08 | 149 |
| 85 | IMMUNITY | 51.10 | 2.44 | 2.37 | 2.18 | 265 |
| 86 | COMMUN BIOL | 51.01 | 2.39 | 2.31 | 2.00 | 104 |
| 87 | MOL CELL* | 50.38 | 2.45 | 2.37 | 2.14 | 1,599 |
| 88 | BIOORG MED CHEM LETT* | 50.19 | 2.19 | 2.16 | 2.05 | 1,590 |
| 89 | NAT STRUCT MOL BIOL* | 49.78 | 2.40 | 2.31 | 2.07 | 2,915 |
| 90 | CELL* | 49.38 | 2.54 | 2.45 | 2.20 | 1,563 |
| 91 | EMBO J* | 49.15 | 2.37 | 2.30 | 2.10 | 1,910 |
Denotes journals that have average PQ1(t,d) significantly different than the average PQ1(t,d) of the entire PDB, according to Welch’s t-test with Bonferroni correction at significance level α=0.001. Mean denotes the arithmetic mean, G-mean denotes the geometric mean (log-average), V-mean denotes the mean in Å−3.
Surprisingly, the first place in all versions of the ranking is occupied by Tuberculosis, a venue that is not well known as a structural journal. However, this place is well earned since Tubeculosis has over 16 percentage points of advantage over the second ranked journal in terms of PQ1(t,d) and 12 percentage points of advantage in the PQ1(t) ranking. A closer inspection of the structures published in Tuberculosis reveals that the vast majority of structures refer to one publication titled “Increasing the structural coverage of tuberculosis drug targets” [28]. The publication and its corresponding structures are the result of the joint effort of various departments working in the Seattle Structural Genomics Center for Infectious Disease. This finding is in accordance with the conclusions of B&R [19] that structural genomics initiatives usually deposit structures of above-average quality [29,30]. Indeed, taking into account all 12,494 deposits attributed to structural genomics projects, they achieve a mean PQ1(t,d) of 63.7% and PQ1(t) of 64.3%, substantially above the average of the entire PDB (58.6% and 57.7%, respectively). These differences are statistically significant according to Welch’s t-test (p < 0.001) and are much more prominent than those reported in the R&K study [20]. This discrepancy most probably stems from the fact that in our study we used a relative measure that combines several quality metrics, and had 2.3 times more structural genomics deposits at our disposal and 6.1 times more structures overall.
When looking at the most popular journals, i.e. those with more than 1000 structures (Table 3, gray rows), the top three spots are occupied by Biochemical Journal, FEBS Journal, and Nature Chemical Biology. At the other end of the spectrum, we have EMBO Journal, Cell, and Nature Structural & Molecular Biology, which were ranked last according to PQ1(t,d). It is worth noting that the latter three journals are the only journals that have average PQ1(t,d) below 50%. This means that, on average, at the time of deposition, the structures presented in these journals were already worse than over 50% of PDB structures of similar resolution. A similar ranking was obtained using PQ1(t) (Supplementary Table S1), the main difference being that journals publishing structures at superior resolution, such Chemistry or Acta Crystallographica D, achieved much higher positions in the journal ranking. Table 3 and Supplementary Table S1–S3 also identify journals whose average PQ1(t,d), PQ1(t), PQ1min(t,d), and PQ1min(t) are significantly different from the expected values of the entire PDB population.
It should be noted that the ranking presented in Table 3 takes into account over 45 years of structural data. This means that the ranking averages the entire lifespans of journals, which in their own individual history might have evolved over time. That is why in the following section we analyze how the ranking of the most popular journals has changed over the years.
Quality of Journals’ Structures Over Time
Owing to the fact that PQ1(t,d) assesses structures at the time of deposition, we also analyzed rankings of journals as a function of time. Fig. 3 presents the ranking of 25 all-time most popular journals in periods of five years. To minimize the effect of noise on the ranking, journals were assigned to a given five-year period only when they contained primary citations to at least 30 structures within that period.
Figure 3. Journal ranking over time according to PQ1(t,d).
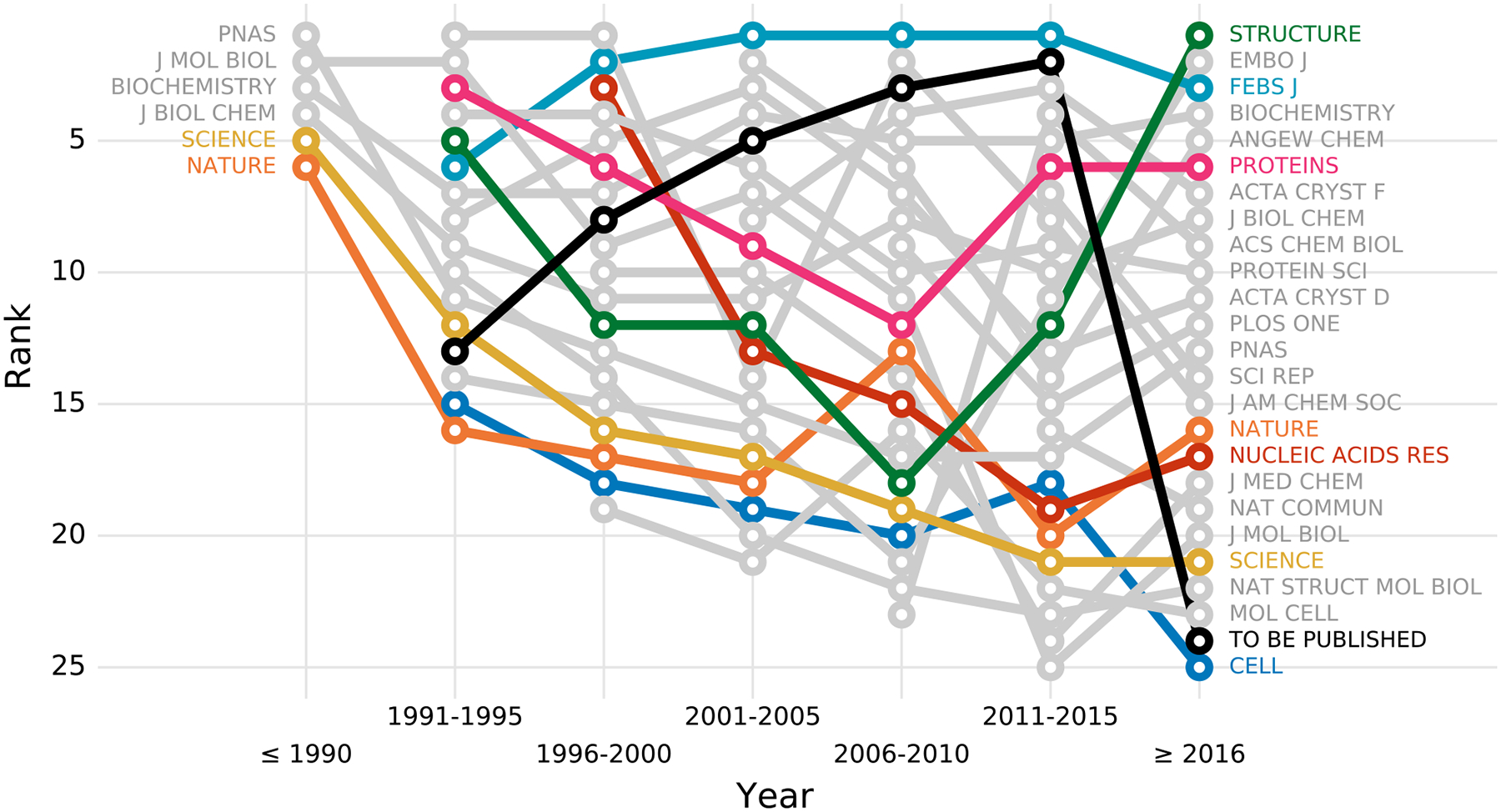
The plot shows the journal’s rank (y-axis) in a given time period (x-axis). The ranking includes 25 most popular journals, i.e. journals with most structures, ranked based on structures deposited within 5-year windows. A point appears only if a journal published at least 30 structures in a given 5-year interval.
As Fig. 3 shows, only six of the 25 journals published at least 30 structures before 1991, however, these six journals were the primary reference for 482 out of 666 PDB deposits from this period. Biochemistry remains one of the top journals in terms of structure quality to date, PNAS and J. Biol. Chem. are in the middle of the ranking, whereas Nature, Science, and J. Mol. Biol. occupy the bottom half of the ranking. A journal that has steadily remained at the top of the ranking list for most of the years is FEBS Journal. Apart from Biochemistry and FEBS Journal, Proteins can also pride itself with a solid presence in the top 10 of the ranking throughout the years. It is worth noting that these three journals were also highly ranked in the study of B&R [19].
Disappointingly, the relatively poor ranks of highly reputable venues are not a new concern, but rather have been a steady trend for many years. It must be noted, however, that the overall structure quality of practically all 25 of the most popular journals has greatly improved in the last ten years, with Science and Nature noting the most positive trends (Supplementary Fig. S5). Similar observations were made when the journals were ranked according to PQ1(t) (Supplementary Fig. S6, S7).
A separate comment is required for the “venue” To be published, most frequently found in PDB deposits. This category of PDB entries, omitted in the studies of B&R [19] and R&K [20], presents a very interesting pattern over the years. For several decades, unpublished structures (because staying in the “to be published” state for several years in practice means “unpublished”) noted a steady upward trend, coming as far as the second place among the most popular venues between 2011 and 2015. The low position in the latest ranking period (2016-today) may stem from the fact that many of the recently deposited To be published structures from this time range still have a chance of being published and are under peer review. The retrospective pattern is that structures in the To be published category have higher PQ1(t,d) (61.4%) than structures that are presented in concrete journals (58.0%) (Welch’s t-test, p < 0.001). Moreover, 8,543 out of 12,494 structural genomics structures remain unpublished even as structure notes, constituting over 66% and 33% of all unpublished structures in the 2005–2010 and 2011–2015 periods, respectively. With fewer structural genomics depositions in the last four years (only 15% of all unpublished structures for this period), the To be published category currently has a lower ratio of high-quality structures. This, combined with the observed constant improvement of published structures, may further contribute to the drop of the To be published category in the ranking.
Retrospective comparison with the results of Brown & Ramaswamy
The journal rankings presented in this work were inspired by the study of Brown and Ramaswamy (B&R) [19]. Although the methodologies used in these two analyses are different (most notably because of incorporation of nucleic acids and the use of data imputation in the present work), it is worth verifying how the two approaches compare, and what has changed since the original B&R study. To help answer these questions, Table 4 presents the journal ranking reported by B&R in 2007 together with two lists of the same journals ranked according to PQ1(t,d): based on PDB deposits available in 2007, and based on all currently available data.
Table 4. Comparison of journal ranking by Brown and Ramaswamy [19] with rankings of the same journals created using PQ1(t,d).
Numbers of structures considered from a given journal are shown in parentheses. The top three journals according to B&R are highlighted in green, the bottom three journals are highlighted in red.
| B&R ranking [19] (Year < 2007) |
Ranking according to PQ1(t,d) (Year < 2007) |
Ranking according to PQ1(t,d) (current) |
|---|---|---|
| FEBS J*(159) | J BIOL INORG CHEM (114) | BIOCHEM J* (1033) |
| PROTEIN ENG DES SEL (96) | FEBS J*(356) | FEBS J*(1539) |
| BIOCHEMISTRY*(3346) | PROTEIN ENG DES SEL (173) | J BIOL INORG CHEM (265) |
| CELL CHEM BIOL (154) | ACTA CRYST D* (1766) | FEBS LETT* (814) |
| PROTEINS (398) | BIOCHEM J (165) | J AM CHEM SOC* (2369) |
| J MOL BIOL (3855) | FEBS LETT (232) | PROTEIN ENG DES SEL (294) |
| ACTA CRYST D (1074) | CELL CHEM BIOL (204) | CELL CHEM BIOL (902) |
| PROTEIN SCI (771) | PROTEIN SCI* (1104) | ACTA CRYST D* (4952) |
| BIOORG MED CHEM LETT (195) | BIOCHEMISTRY*(4564) | BIOCHEMISTRY*(8896) |
| J STRUCT BIOL (83) | J MOL BIOL* (5497) | NUCLEIC ACIDS RES (2127) |
| BIOPHYS J (71) | BIOPHYS J (95) | STRUCTURE (5348) |
| J BIOL INORG CHEM (81) | PROTEINS (993) | PROTEIN SCI (2235) |
| BIOCHEM J (67) | J AM CHEM SOC (459) | J MED CHEM (5525) |
| J BIOL CHEM (2849) | BIOCHEM BIOPHYS RES COMMUN (167) | J VIROL (957) |
| J AM CHEM SOC (324) | NUCLEIC ACIDS RES (288) | J BIOL CHEM (11055) |
| STRUCTURE (1412) | J BACTERIOL (141) | J STRUCT BIOL (1038) |
| FEBS LETT (173) | BIOORG MED CHEM (59) | J BACTERIOL (371) |
| J BACTERIOL (111) | J BIOL CHEM (4090) | BIOORG MED CHEM (659) |
| BIOORG MED CHEM (53) | J STRUCT BIOL (117) | PROTEINS (1999) |
| J MED CHEM (450) | J MED CHEM (605) | BIOCHEM BIOPHYS RES COMMUN (976) |
| NAT STRUCT MOL BIOL (768) | STRUCTURE (2017) | PNAS (7376) |
| PNAS (1324) | PNAS* (1839) | BIOPHYS J (199) |
| J VIROL (86) | J VIROL (141) | J MOL BIOL* (9507) |
| BIOCHEM BIOPHYS RES COMMUN (103) | SCIENCE*(712) | NATURE* (3060) |
| EMBO J* (768) | EMBO J* (1135) | SCIENCE*(1949) |
| NUCLEIC ACIDS RES (199) | NAT STRUCT MOL BIOL* (1342) | MOL CELL*(1599) |
| NATURE* (807) | NATURE* (976) | BIOORG MED CHEM LETT* (1590) |
| MOL CELL*(422) | BIOORG MED CHEM LETT* (323) | NAT STRUCT MOL BIOL* (2915) |
| SCIENCE*(571) | CELL*(647) | CELL*(1563) |
| CELL*(488) | MOL CELL*(571) | EMBO J* (1910) |
Denotes journals whose quality was determined to be significantly different from the average quality of structures the entire PDB, at significance level α=0.001.
It can be noticed that the rankings bare several similarities, although they are not identical. Journals that were at the top of the B&R ranking generally remain highly ranked according to PQ1(t,d). Similarly, the bottom regions of the rankings are occupied by the same group of journals. However, there are some notable differences. For example, Bioorg. Med. Chem. Lett. is ranked 19 places lower according to PQ1(t,d), whereas J. Biol. Inorg. Chem., FEBS Lett. and Nucleic Acids Res. are ranked 11 places higher. These differences may be the result of the number of structures taken into account by each ranking. Compared to the time of the B&R study, significantly more precomputed quality metrics are now available, even for older PDB deposits. Moreover, the methodology proposed in this work imputes missing values, allowing for inclusion of 12.7% additional structures. As a result, the rankings based on PQ1(t,d) were compiled using much more data, occasionally changing a journal’s rank substantially.
Correlation between structure quality and journal impact
The low-ranking of high impact journals in the current study raises the question of whether structure quality is negatively correlated with journal impact. The study of B&R [19] strongly suggested that this was the case, whereas the slightly more recent work of R&K [20] showed that the differences in structure quality between high-impact and other venues were relatively small. However, both studies manually categorized journals as high- or low-impact venues rather than investigating actual impact metrics for a large set of journal titles.
In this study, we decided to measure journal impact quantitatively and correlate it with our quantitative measure of structure quality. For this purpose, we used two metrics: Impact Per Publication (IPP) and Source Normalized Impact per Paper (SNIP) [31]. IPP is calculated the same way as 3-year impact factor (IF3) but using only publications that are classified as articles, conference papers, or reviews in Scopus. SNIP is a modification of IPP that corrects for differences in citation practices between scientific fields [31]. Both journal metrics have 20 years (1999–2018) of publicly available statistics and are based on the same source data.
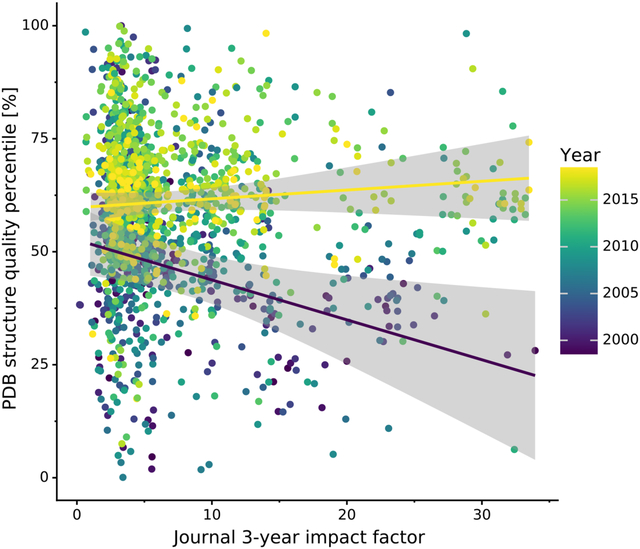
Fig. 4 shows the relation between PQ1(t,d) and the journal impact over time (separate plots for each year are presented in Supplementary Fig. S8). It is evident that structure quality has substantially improved over the last decade and that the negative correlation between journal impact and the quality of structural models presented therein seems to disappear as time progresses. This observation is confirmed when the relation between journal impact (IPP, SNIP) and structure quality (PQ1(t,d)) is gauged using Spearman’s rank correlation coefficient. Fig. 5 shows that even though structure quality and journal impact were indeed negatively correlated 20 years ago, currently there is no correlation between these two criteria.
Figure 4. Scatterplot of mean journal PQ1(t,d) and the journal’s impact over time.
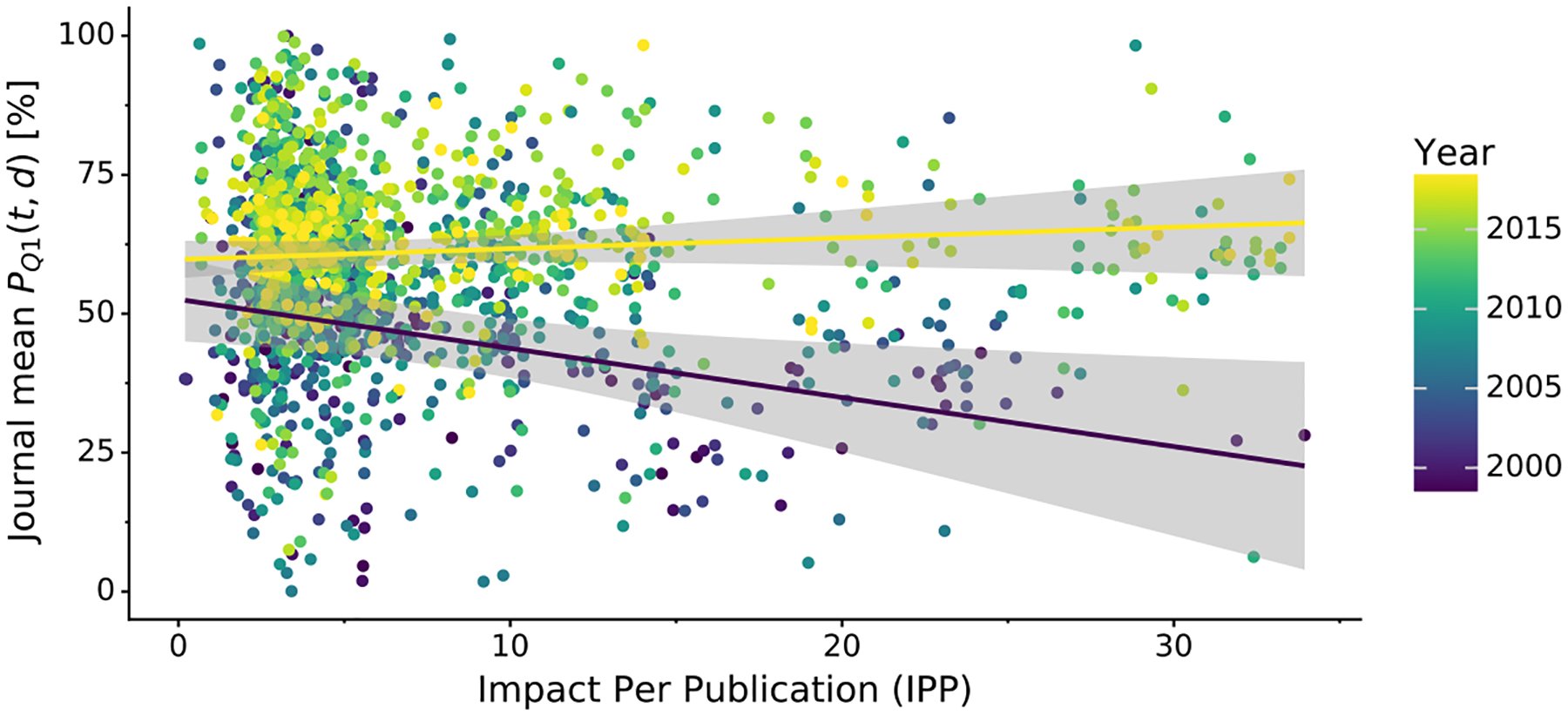
Variation in mean journal PQ1(t,d) (y-axis) in a given year (color) plotted against the journals Impact Per Publication (IPP). IPP uses the same formula as the 3-year Impact Factor, but is based on publicly available Scopus data. The two regression lines show linear trends for 1999 (indigo) and 2018 (yellow) along with 95% confidence intervals (gray areas).
Figure 5. Correlation between structure quality and journal impact.
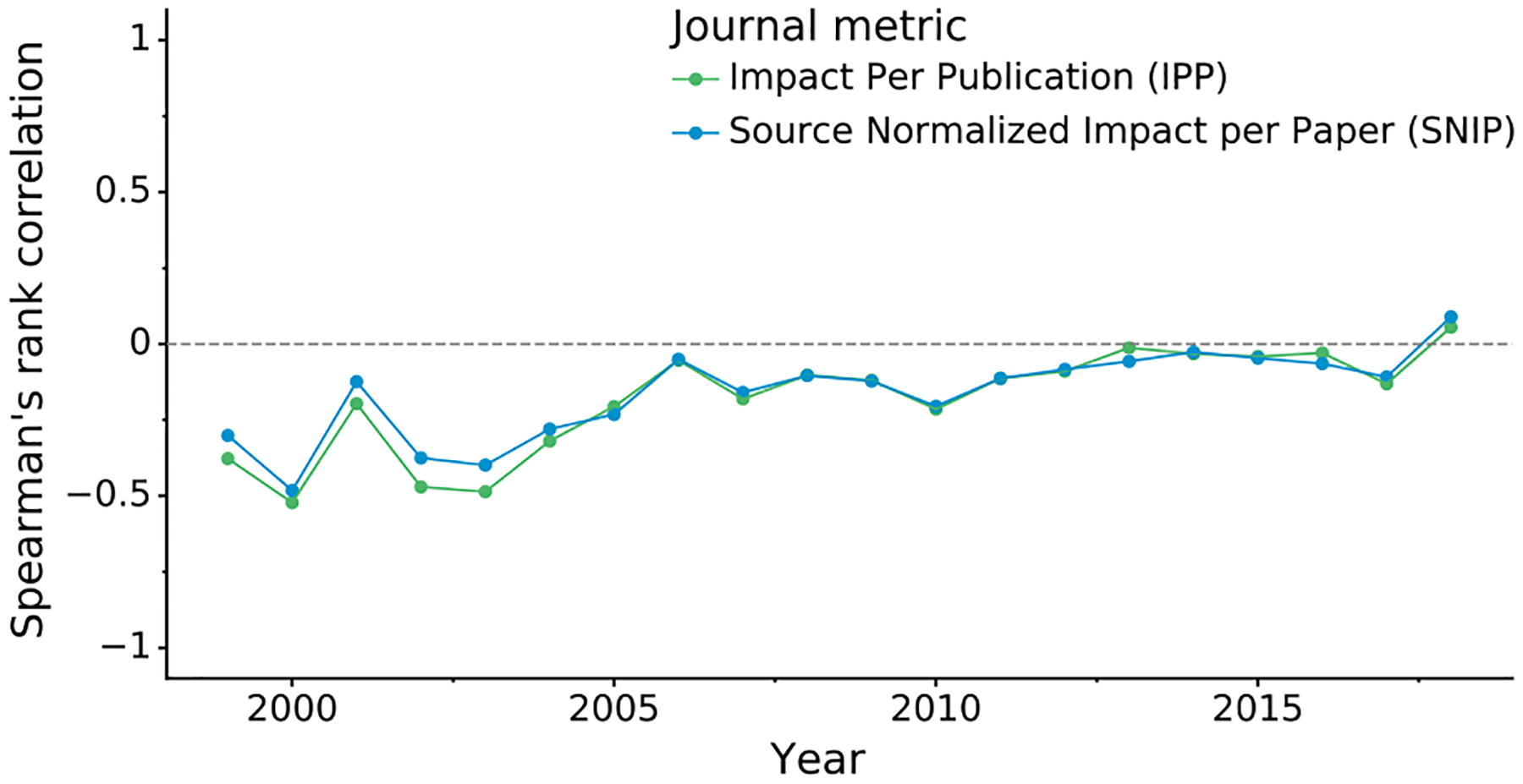
The plot shows Spearman’s rank correlation (y-axis) over time (x-axis) between structure quality measured by PQ1(t,d) and journal impact measured using the IPP and SNIP metrics. IPP uses the same formula as the 3-year Impact Factor but is based on Scopus data, whereas SNIP additionally takes into account the scientific field.
Fig. 4 also shows a very interesting situation in the low-IF range, namely that low-IF journals publish just about anything: the most fantastic work as well as structures beneath contempt. On the other hand, medium-IF journals used to be primary citations of mostly poor structures in the past. At present, however, they are doing a much better job, publishing mostly better-than-average structures.
Discussion
Our analysis confirms recent reports that the quality of crystallographic macromolecular structures has improved over the last years [17]. However, we also found out that at the time of the B&R analysis the quality of PDB structures temporarily stopped improving, and that this is most likely why B&R did not report any correlation between quality and time [19]. In addition to confirming earlier findings, by using a data imputation algorithm we were able to put into context the quality of structural models going back in time as far as 1972. As convincingly illustrated by Figs. 1 and 2, the quality of PDB structures had rapidly improved over the first two decades of the database.
The ability to analyze quality over time using the proposed PQ1(t,d) measure (Fig. 3) shows that there is tight competition among journals as their number increases. Quite interestingly, it is also evident that the PDB treasures many good quality structures that do not have primary citations. The fact that a structure remains To be published indicates that it is getting more and more difficult to publish papers based solely on crystallographic results, even if they are of high quality. Indeed, our study shows that structures without primary citations are on average of higher quality than structures published in many popular journals. Therefore, although many structures do not have any accompanying journal publications, they present a substantial value in their own right. As each PDB deposit has its own digital object identifier (DOI), citation of structures should be acknowledged not only by PDB IDs but also by DOIs. Full implementation of this mechanism would allow for easy estimation of the impact of To be published structures.
The proposed PQ1(t,d) and PQ1(t) measures manifest the overall attitude of authors towards the quality of the PDB: each next deposited structure should be better than the average previous deposit. Each new quality metric [21], visualization technique [32], set of restraints [33], validation algorithm [34], hardware improvement [35] or software update [36], make it easier to produce good quality structures and to avoid simple mistakes. In an effort to promote constant improvement of overall PDB quality it would be a desirable ideal to expect that newly added models are above the current average. However, such a recommendation should be applied judiciously as each case is different and should be always judged in a context-dependent manner. It is gratifying to see that almost all journals publish structures that are, on average, better than most of the previous ones while those that are not at that level yet, seem to be heading in the right direction.
Methods
Data collection and cleaning
To provide a comprehensive analysis of structure quality over time, we examined all X-ray structures available in the PDB that included 141,154 deposits between 1972 and 2019. The data were downloaded by performing an SQL query on PDBj [37,38] as of December 10, 2019.
In order to perform an analysis of structure quality in correlation with the primary citations, journal names had to be extracted from PDB files, cleaned and unified. Initially, the dataset contained 1,342 unique values describing the primary citation journal. After eliminating typos, unifying punctuation and ISSNs, and taking into account that some journals have changed their titles over time, the number of unique journal names dropped down to 800.
Bibliometric indicators of journals (IPP, SNIP) were downloaded from the Leiden University CWTS website (https://www.journalindicators.com/) and joined with the PDB data using ISSNs. Both indicators were calculated based on the Scopus bibliographic database produced by Elsevier.
Missing data imputation
To fill in missing data, three approaches were tested: filling missing values with the metric’s mean value, the metric’s median, and using the Multiple Imputation by Chained Equations algorithm (MICE) [23,24] with Bayesian ridge regression [25] as the predictor.
To see how well each of the three methods performed, the non-missing (i.e. complete) portion of the PDB data was used as the basis for creating a test set. We randomly introduced missing values to the complete portion of the data in the same proportions as those present in the actual dataset. As a result, the test dataset had the same proportion of deposits with at least one missing value and the same percentage of missing values per metric as the original (full) dataset. Next, these randomly introduced missing values were imputed and compared against the values originally present in the dataset. To quantify the imputation error we used the median absolute deviation (MAD), mean absolute error (MAE), and root-mean-square error (RMSE) [39]. The procedure was repeated 100 times with different subsets of values randomly eliminated from the complete dataset in each run. Imputed missing values were clipped when they were outside the range of possible values of a given metric.
Principal component analysis
The principal component analysis (PCA) required to calculate Q1p was performed as described by Shao et al. [17]. The PCA was performed on three quality metrics: Clashscore, Ramachandran outliers, and Rotamer outliers. Since Ramachandran outliers and Rotamer outliers are meaningful only for proteins, the PCA analysis was performed for protein structures only. In the assessment of the quality of nucleic acid structures, the PCA step was not needed, as Clashscore was the only geometry-related quality index.
Upon visual inspection of the metrics’ values (Fig. S9), structures were marked as outliers and removed when the following criteria were reached: Rotamer outliers > 50% or Ramachandran outliers > 45% or Clashscore > 250. In total, 16 structures were marked as outliers: 1C4D, 1DH3, 1G3X, 1HDS, 1HKG, 1HPB, 1PYP, 1SM1, 2ABX, 2GN5, 2OGM, 2Y3J, 3ZS2, 4BM5, 4HIV, 5M2K. These structures were temporarily removed prior to PCA to decrease the effect of outlying values on the principal components, but they were not removed from the quality analysis. After removal of outstanding outliers, the input data for PCA were standardized by setting the mean to be 0 and standard deviation to 1. Running PCA on the standardized data resulted in three principal components: PC1, PC2 and PC3, explaining 78%, 14% and 8% variance, respectively. The coefficients of PC1 were: 0.60, 0.58, 0.56, indicating nearly equal contribution of Clashscore, Ramachandran outliers and Rotamer outliers. The explained variance of each principal component and the coefficients of PC1, were practically identical to those reported by Shao et al. [17].
As noted by one of the reviewers, the PC1 coefficients (0.60, 0.58, 0.56) are almost identical and roughly equal , making the respective weights of these contributions near equal for all three of them. This means that approximately the Q1p measure could be presented as:
| (5) |
The above formula provides a simple metric that can be used without performing PCA. However, this approximate formula assumes that the relations between Clashscore, Ramachandran outliers and Rotamer outliers are fixed and will not change. For this reason, we chose to use the exact formula (1) as proposed by Shao et al. [17]. Nevertheless, the approximate formula (5) may be considered a simpler solution for less technical studies.
Computation
Data were extracted directly from PDBj using its SQL interface. All computations were performed with Python 3.7 using the scipy [40] and scikit-learn [41] libraries. The SQL query used, the resulting dataset, and fully reproducible analysis scripts in the form of a Jupyter notebook are available at https://github.com/dabrze/pdb_structure_quality.
Supplementary Material
Table S1. All-time journal ranking according to PQ1(t). The ranking includes all the journals that had at least 100 primary citations of structures in the PDB. PQ1(t) higher than 50% means that the structures published in a given journal were, on average, better than 50% of structures present in the PDB at the time of deposition (in this case regardless of resolution). Journals with more than 1000 structures are highlighted in gray.
Table S2. All-time journal ranking according to PQ1min(t,d). The ranking includes all the journals that had at least 100 primary citations of structures in the PDB. PQ1min(t,d) higher than 50% means that the structures published in a given journal were, on average, better than 50% of structures present in the PDB at the time of deposition. Journals with more than 1000 structures are highlighted in gray.
Table S3. All-time journal ranking according to PQ1min(t). The ranking includes all the journals that had at least 100 primary citations of structures in the PDB. PQ1min(t) higher than 50% means that the structures published in a given journal were, on average, better than 50% of structures present in the PDB at the time of deposition (in this case regardless of resolution). Journals with more than 1000 structures are highlighted in gray.
Figure S1. Histograms of quality metric values of structures found in the PDB. The histograms show the distribution of Clashscore, percent of Ramachandran outliers, percentage of rotamer outliers, percentage of RSRZ outliers and Rfree values. The y-axis shows the number of depositions with a given metric value displayed on the x-axis.
Figure S2. PQ1 analysis without imputed values. Variation in the mean PQ1 percentile (higher is better) for deposits with all quality metrics over time (top) and resolution (bottom) for proteins (left) and nucleic acids (right). Error bars indicate the estimated unbiased standard error of the mean.
Figure S3. PQ1min analysis (minimum approach). Variation in the mean PQ1min percentile (higher is better) over time (top) and resolution (bottom) for proteins (left) and nucleic acids (right). Error bars indicate the estimated unbiased standard error of the mean.
Figure S4. Comparison of PQ1min(t,d) of protein and nucleic acid structures over time. Variation in mean PQ1min(t,d) quality percentile (y-axis, higher is better), comparing nucleic acid and protein structures (color) over time (x-axis). Error bars indicate estimated unbiased standard errors of the mean.
Figure S5. Average PQ1(t,d) of popular journals for each year. The plot shows yearly (x-axis) means of PQ1(t,d) values (y-axis) for each of the 25 most popular journals (panels). Regression lines computed using the locally estimated scatterplot smoothing (LOESS) method.
Figure S6. Journal ranking over time according to PQ1(t). The plot shows the journal’s rank (y-axis) in a given time period (x-axis). Only the 25 most popular journals with at least 30 deposits in a given period were plotted.
Figure S7. Journal quality over time according to PQ1(t). The plot shows yearly (x-axis) means of PQ1(t) values (y-axis) for each of the 25 most popular journals (panels). Regression lines computed using the locally estimated scatterplot smoothing (LOESS) method.
Figure S8. Scatterplots of mean journal PQ1(t,d) and the journal’s impact. Variation in mean journal PQ1(t,d) (y-axis) in a given year (panel, color) plotted against the journals Impact Per Publication (IPP). IPP uses the same formula as the 3-year Impact Factor, but is based on publicly available Scopus data. The two regression lines show linear trends with 95% confidence intervals (gray areas).
Figure S9. Scatterplots of the values of Clashscore, Ramachandran outliers, and Rotamer outliers found in the PDB. Each point represents a PDB deposit.
Acknowledgments
The authors would like to thank the anonymous reviewers of this paper for their extremely helpful comments and suggestions. DB acknowledges the support of the Polish National Agency for Academic Exchange, grant no. PPN/BEK/2018/1/00058/U/00001. Funding for this research was also provided by NIH grant GM132595.
Abbreviations:
- PDB
Protein Data Bank
- RSRZ
Real-Space R-value Z-score
- MICE
Multiple Imputation by Chained Equations
- MAE
Mean Absolute Error
- RMSE
Root-Mean-Square Error
- MAD
Median Absolute Deviation
- IPP
Impact Per Publication
- SNIP
Source Normalized Impact per Paper
Footnotes
Data accessibility: Supplementary tables and figures are available at figshare.com: 10.6084/m9.figshare.11366222. Datasets and reproducible experimental scripts are available at GitHub: https://github.com/dabrze/pdb_structure_quality.
Conflict of interest
The authors declare no conflict of interest.
Supplementary tables and figures are available at figshare.com: 10.6084/m9.figshare.11366222.
References
- 1.Baker EN (2018) Crystallography and the development of therapeutic medicines. IUCrJ 5, 118–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Blundell TL (2011) Celebrating structural biology. Nat. Struct. Mol. Biol 18, 1304–1316. [DOI] [PubMed] [Google Scholar]
- 3.Blundell TL (2017) Protein crystallography and drug discovery: Recollections of knowledge exchange between academia and industry. IUCrJ 4, 308–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pomés A, Chruszcz M, Gustchina A, Minor W, Mueller GA, Pedersen LC, Wlodawer A & Chapman MD (2015) 100 Years later: Celebrating the contributions of x-ray crystallography to allergy and clinical immunology. J. Allergy Clin. Immunol 136, 29–37.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN & Bourne PE (2000) The Protein Data Bank. Nucleic Acids Res. 28, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Raczynska JE, Shabalin IG, Minor W, Wlodawer A & Jaskolski M (2018) A close look onto structural models and primary ligands of metallo-β-lactamases. Drug Resistance Updates 40, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dauter Z, Wlodawer A, Minor W, Jaskolski M & Rupp B (2014) Avoidable errors in deposited macromolecular structures - an impediment to efficient data mining. IUCrJ 1, 179–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Minor W, Dauter Z, Helliwell JR, Jaskolski M & Wlodawer A (2016) Safeguarding structural data repositories against bad apples. Structure 24, 216–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Svobodova Verkova R, Horsky V, Sehnal D, Bendova V, Pravda L & Koca J (2017) Quo vadis, biomolecular structure quality. Biophys. J 112, 346a–47a.28122220 [Google Scholar]
- 10.Wlodawer A, Dauter Z, Minor W, Stanfield R, Porebski P, Jaskolski M, Pozharski E, Weichenberger CX & Rupp B (2018) Detect, Correct, Retract: How to manage incorrect structural models. FEBS J. 285, 444–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ioannidis JPA (2005) Why most published research findings are false. PLoS Medicine 2, 696–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Joosten RP, Long F, Murshudov GN & Perrakis A (2014) The PDB_REDO server for macromolecular structure model optimization. IUCrJ 1, 213–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Read RJ, Adams PD, Arendall WB, Brunger AT, Emsley P, Joosten RP, Kleywegt GJ, Krissinel EB, Lütteke T, Otwinowski Z, Perrakis A, Richardson JS, Sheffler WH, Smith JL, Tickle IJ, Vriend G & Zwart PH (2011) A new generation of crystallographic validation tools for the Protein Data Bank. Structure 19, 1395–1412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gore S, Sanz García E, Hendrickx PMS, Gutmanas A, Westbrook JD, Yang H, Feng Z, Baskaran K, Berrisford JM, Hudson BP, Ikegawa Y, Kobayashi N, Lawson CL, Mading S, Mak L, Mukhopadhyay A, Oldfield TJ, Patwardhan A, Peisach E, Sahni G, Sekharan MR, Sen S, Shao C3, Smart OS, Ulrich EL, Yamashita R, Quesada M, Young JY, Nakamura H, Markley JL, Berman HM, Burley SK, Velankar S & Kleywegt GJ (2017) Validation of Structures in the Protein Data Bank. Structure 25, 1916–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shabalin I, Dauter Z, Jaskolski M, Minor W & Wlodawer A (2015) Crystallography and chemistry should always go together: a cautionary tale of protein complexes with cisplatin and carboplatin. Acta Cryst. D71, 1965–1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Raczynska J, Shabalin I, Minor W, Wlodawer A & Jaskolski M (2018) A close look onto structural models and primary ligands of metallo-β-lactamases. Drug Resistance Updates 40, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shao C, Yang H, Westbrook JD, Young JY, Zardecki C & Burley SK (2017) Multivariate analyses of quality metrics for crystal structures in the PDB archive. Structure 25, 458–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rupp B, Wlodawer A, Minor W, Helliwell JR & Jaskolski M (2016) Correcting the record of structural publications requires joint effort of the community and journal editors. FEBS J. 283, 4452–4457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brown EN & Ramaswamy S (2007) Quality of protein crystal structures. Acta Cryst. D63, 941–950. [DOI] [PubMed] [Google Scholar]
- 20.Read RJ & Kleywegt GJ (2009) Case-controlled structure validation. Acta Cryst. D65, 140–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen VB, Arendall WB, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS & Richardson DC (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Cryst. D66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brünger AT (1992) Free R value: a novel statistical quantity for assessing the accuracy of crystal structures. Nature 355, 472–475. [DOI] [PubMed] [Google Scholar]
- 23.Raghunathan TW, Lepkowksi JM, Van Hoewyk J, & Solenbeger P. (2001) A multivariate technique for multiply imputing missing values using a sequence of regression models. Survey Methodology 27, 85–95. [Google Scholar]
- 24.van Buuren S (2007) Multiple imputation of discrete and continuous data by fully conditional specification. Stat. Methods Med. Res 16, 219–42. [DOI] [PubMed] [Google Scholar]
- 25.MacKay DCJ (1992) Bayesian Interpolation. Neural Comput. 4, 415–447. [Google Scholar]
- 26.Demšar J (2006) Statistical Comparisons of Classifiers over Multiple Data Sets. J. Mach. Learn. Res 7, 1–30. [Google Scholar]
- 27.Jaskolski M, Miller M, Rao JK, Leis J & Wlodawer A (1990) Structure of the aspartic protease from Rous sarcoma retrovirus refined at 2-Å resolution. Biochemistry 29, 5889–5598. [DOI] [PubMed] [Google Scholar]
- 28.Baugh L, Phan I, Begley DW, Clifton MC, Armour B, Dranow DM, Taylor BM, Muruthi MM, Abendroth J, Fairman JW, Fox D 3rd, Dieterich SH, Staker BL, Gardberg AS, Choi R, Hewitt SN, Napuli AJ, Myers J, Barrett LK, Zhang Y, Ferrell M, Mundt E, Thompkins K, Tran N, Lyons-Abbott S, Abramov A, Sekar A, Serbzhinskiy D, Lorimer D, Buchko GW, Stacy R, Stewart LJ, Edwards TE, Van Voorhis WC & Myler PJ (2015) Increasing the structural coverage of tuberculosis drug targets. Tuberculosis 95, 142–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Domagalski MJ, Zheng H, Zimmerman MD, Dauter Z, Wlodawer A & Minor W (2014) The Quality and Validation of Structures from Structural Genomics. In: Methods in Molecular Biology 1091, pp. 297–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grabowski M, Niedzialkowska E, Zimmerman MD & Minor W (2016) The impact of structural genomics: the first quindecennial. J. Struct. Funct. Genomics 17, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Waltman L, van Eck NJ, van Leeuwen TN & Visser MS (2013) Some modifications to the SNIP journal impact indicator. Journal of Informetrics 7, 272–285. [Google Scholar]
- 32.Porebski PJ, Sroka P, Zheng H, Cooper DR & Minor W (2017) Molstack-Interactive visualization tool for presentation, interpretation, and validation of macromolecules and electron density maps. Protein Sci. 27, 86–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kowiel M, Brzezinski D & Jaskolski M (2016) Conformation-dependent restraints for polynucleotides: I. Clustering of the geometry of the phosphodiester group. Nucleic Acids Res. 44, 8479–8489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kowiel M, Brzezinski D, Porebski PJ, Shabalin IG, Jaskolski M & Minor W (2019) Automatic recognition of ligands in electron density by machine learning. Bioinformatics 35, 452–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Förster A, Brandstetter S & Schulze-Briese Clemens (2019) Transforming X-ray detection with hybrid photon counting detectors. Philos T. R. Soc. A 377, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liebschner D, Afonine PV, Baker ML, Bunkóczi G, Chen VB, Croll TI, Hintze B, Hung LW, Jain S, McCoy AJ, Moriarty NW, Oeffner RD, Poon BK, Prisant MG, Read RJ, Richardson JS, Richardson DC, Sammito MD, Sobolev OV, Stockwell DH, Terwilliger TC, Urzhumtsev AG, Videau LL, Williams CJ & Adams PD (2019) Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Cryst. D75, 861–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kinjo AR, Bekker G-J, Wako H, Endo S, Tsuchiya Y, Sato H, Nishi H, Kinoshita K, Suzuki H, Kawabata T, Yokochi M, Iwata T, Kobayashi N, Fujiwara T, Kurisu G & Nakamura H (2018) New tools and functions in Data-out activities at Protein Data Bank Japan (PDBj). Protein Sci. 27, 95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kinjo AR, Bekker G-J, Suzuki H, Tsuchiya Y, Kawabata T, Ikegawa Y, Nakamura H (2017) Protein Data Bank Japan (PDBj): Updated user interfaces, Resource Description Framework, analysis tools for large structures. Nucleic Acids Res. 45, 282–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Japkowicz N & Shah M (2011) Evaluating Learning Algorithms: A Classification Perspective, Cambridge University Press. [Google Scholar]
- 40.Oliphant TE (2007) Python for Scientific Computing. Computing in Science & Engineering 9, 10–20. [Google Scholar]
- 41.Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, Blondel M, Prettenhofer P, Weiss R, Dubourg V, Vanderplas J, Passos A, Cournapeau D, Brucher M, Perrot M & Duchesnay E (2011) Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res 12, 2825–2830. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. All-time journal ranking according to PQ1(t). The ranking includes all the journals that had at least 100 primary citations of structures in the PDB. PQ1(t) higher than 50% means that the structures published in a given journal were, on average, better than 50% of structures present in the PDB at the time of deposition (in this case regardless of resolution). Journals with more than 1000 structures are highlighted in gray.
Table S2. All-time journal ranking according to PQ1min(t,d). The ranking includes all the journals that had at least 100 primary citations of structures in the PDB. PQ1min(t,d) higher than 50% means that the structures published in a given journal were, on average, better than 50% of structures present in the PDB at the time of deposition. Journals with more than 1000 structures are highlighted in gray.
Table S3. All-time journal ranking according to PQ1min(t). The ranking includes all the journals that had at least 100 primary citations of structures in the PDB. PQ1min(t) higher than 50% means that the structures published in a given journal were, on average, better than 50% of structures present in the PDB at the time of deposition (in this case regardless of resolution). Journals with more than 1000 structures are highlighted in gray.
Figure S1. Histograms of quality metric values of structures found in the PDB. The histograms show the distribution of Clashscore, percent of Ramachandran outliers, percentage of rotamer outliers, percentage of RSRZ outliers and Rfree values. The y-axis shows the number of depositions with a given metric value displayed on the x-axis.
Figure S2. PQ1 analysis without imputed values. Variation in the mean PQ1 percentile (higher is better) for deposits with all quality metrics over time (top) and resolution (bottom) for proteins (left) and nucleic acids (right). Error bars indicate the estimated unbiased standard error of the mean.
Figure S3. PQ1min analysis (minimum approach). Variation in the mean PQ1min percentile (higher is better) over time (top) and resolution (bottom) for proteins (left) and nucleic acids (right). Error bars indicate the estimated unbiased standard error of the mean.
Figure S4. Comparison of PQ1min(t,d) of protein and nucleic acid structures over time. Variation in mean PQ1min(t,d) quality percentile (y-axis, higher is better), comparing nucleic acid and protein structures (color) over time (x-axis). Error bars indicate estimated unbiased standard errors of the mean.
Figure S5. Average PQ1(t,d) of popular journals for each year. The plot shows yearly (x-axis) means of PQ1(t,d) values (y-axis) for each of the 25 most popular journals (panels). Regression lines computed using the locally estimated scatterplot smoothing (LOESS) method.
Figure S6. Journal ranking over time according to PQ1(t). The plot shows the journal’s rank (y-axis) in a given time period (x-axis). Only the 25 most popular journals with at least 30 deposits in a given period were plotted.
Figure S7. Journal quality over time according to PQ1(t). The plot shows yearly (x-axis) means of PQ1(t) values (y-axis) for each of the 25 most popular journals (panels). Regression lines computed using the locally estimated scatterplot smoothing (LOESS) method.
Figure S8. Scatterplots of mean journal PQ1(t,d) and the journal’s impact. Variation in mean journal PQ1(t,d) (y-axis) in a given year (panel, color) plotted against the journals Impact Per Publication (IPP). IPP uses the same formula as the 3-year Impact Factor, but is based on publicly available Scopus data. The two regression lines show linear trends with 95% confidence intervals (gray areas).
Figure S9. Scatterplots of the values of Clashscore, Ramachandran outliers, and Rotamer outliers found in the PDB. Each point represents a PDB deposit.